 Estela N. B. Busanello1Ana C. Marques1Noelia Lander1
Estela N. B. Busanello1Ana C. Marques1Noelia Lander1 Diogo N. de Oliveira1Rodrigo R. Catharino1
Diogo N. de Oliveira1Rodrigo R. Catharino1 Helena C. F. Oliveira2*
Helena C. F. Oliveira2* Anibal E. Vercesi1*
Anibal E. Vercesi1*- 1Departamento de Patologia Clínica, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, São Paulo, Brazil
- 2Departamento de Biologia Estrutural e Funcional, Instituto de Biologia, Universidade Estadual de Campinas, São Paulo, Brazil
Statins are efficient cholesterol-lowering medicines utilized worldwide. However, 10% of patients suffer from adverse effects specially related to skeletal muscle function. Pro- or anti-oxidant effects of statins have been reported. Here we hypothesized that statins induce muscle mitochondrial oxidative stress leading to mitochondrial permeability transition (MPT) which may explain statin muscle toxicity. Thus, our aims were to investigate the effects of statin chronic treatment on muscle mitochondrial respiration rates, MPT and redox state indicators in the context of hypercholesterolemia. For this purpose, we studied muscle biopsies of the hypercholesterolemic LDL receptor knockout mice (LDLr-/-) treated with pravastatin during 3 months. Plantaris, but not soleus muscle of treated mice showed significant inhibition of respiration rates induced by ADP (–14%), oligomycin (–20%) or FCCP (–40%). Inhibitions of respiratory rates were sensitive to EGTA (Ca2+ chelator), cyclosporin A (MPT inhibitor), ruthenium red (inhibitor of mitochondria Ca2+ uptake) and coenzyme Q10 (antioxidant), indicating that pravastatin treatment favors Ca2+ induced MPT. Diet supplementation with creatine (antioxidant) also protected treated mice against pravastatin sensitization to Ca2+ induced MPT. Among several antioxidant enzymes analyzed, only catalase activity was increased by 30% in plantaris muscle of pravastatin treated mice. Oxidized lipids, but not proteins biomarkers were identified in treated LDLr-/- plantaris muscle. Taken together, the present results suggest that chronic pravastatin administration to a model of familial hypercholesterolemia promotes mitochondrial dysfunctions in plantaris muscle that can be counteracted by antioxidants administered either in vitro (CoQ10) or in vivo (creatine). Therefore, we propose that inhibition of muscle mitochondrial respiration by pravastatin leads to an oxidative stress that, in the presence of calcium, opens the permeability transition pore. This mitochondrial oxidative stress caused by statin treatment also signals for cellular antioxidant system responses such as catalase upregulation. These results suggest that the detrimental effects of statins on muscle mitochondria could be prevented by co-administration of a safe antioxidant such as creatine or CoQ10.
Introduction
Statins are fungal-derived or synthetic cholesterol-lowering medicines that act by inhibiting 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductase, the rate-limiting enzyme in cholesterol synthesis (Endo, 1992; Tobert, 2003). These medicines are the most commonly prescribed worldwide and represent the primary treatment strategy for hypercholesterolemia and prevention of mortality related to atherosclerosis (Naci et al., 2013). In addition to lowering plasma cholesterol levels, statins are claimed to exhibit pleiotropic effects that include an antioxidant action (Carneado et al., 2002; Wassmann et al., 2002; Manfredini et al., 2010). Therefore, it has been suggested that statins could also have beneficial effects in the treatment of oxidative stress associated diseases such as metabolic syndrome, sepsis, neurological conditions and even tumors (Dobesh and Olsen, 2014; Malfitano et al., 2014; Vallianou et al., 2014). On the other hand, approximately 10% of the patients under statin treatment develop a variety of muscle symptoms including myalgia, muscle cramps, and rarely rhabdomyolysis (Bruckert et al., 2005).
While inhibiting cholesterol synthesis, statins also inhibit the production of ubiquinone (CoQ10) and other intermediaries including dolichol and isoprenoids (Sirvent et al., 2008). CoQ10 is a component of the electron transport chain and also displays antioxidant properties in its reduced form (ubiquinol). Although the molecular mechanisms underlying statin-induced myotoxicity are not yet fully understood, a common hypothesis suggests that it is mediated by inhibition of mitochondrial respiration as a consequence of CoQ10 depletion (Päivä et al., 2005; Bookstaver et al., 2012; Larsen et al., 2013). In addition, previous studies propose that statins cause cell death associated with alterations in calcium homeostasis, inhibition of beta-oxidation, inhibition of mitochondrial respiratory complexes I and II followed by mitochondrial oxidative stress (Kaufmann et al., 2006; Oliveira et al., 2008; Costa et al., 2013; La Guardia et al., 2013) and also inhibition of complex III (Schirris et al., 2015). We have previously shown that statins stimulate Ca2+ induced mitochondrial permeability transition (MPT) in mitochondria isolated from murine liver and muscle, and from mice treated with lovastatin (Velho et al., 2006). Ca2+ and reactive oxygen species (ROS) act synergistically in the mechanism of MPT, a non-specific permeabilization of the inner mitochondrial membrane that (Kowaltowski et al., 2000) triggers cell death under a variety of pathological conditions or drug toxicity (Vercesi et al., 2006; Rasola and Bernardi, 2011; Javadov and Kuznetsov, 2013). The close localization of mitochondria and the endoplasmic reticulum (ER) in situ (Hajnóczky and Csordás, 2010) allows for rapid Ca2+ uptake by mitochondria from the ER microdomains. The existence of a redox controlled cross talk between mitochondria and the ER involving NADPH oxidases has been described (Dikalov, 2011). These redox interactions may control MPT and the execution of Ca2+ signaling for cell death (Figueira et al., 2013).
Sacher et al. (2005) reported that simvastatin and lovastatin activate the mitochondrial pathway of apoptosis in primary cultures of human skeletal muscle obtained from healthy individuals. We have further investigated the mechanisms of cell death induced by simvastatin in PC3 prostate cancer cells that underwent necrosis, in a manner sensitive to cyclosporine A (CsA), an MPT inhibitor. The necrotic cell death was preceded by increased cytosolic free Ca2+ concentration, ROS generation, inhibition of respiration and mitochondrial membrane potential disruption (Oliveira et al., 2008). Kwak et al. (2012) showed that simvastatin impairs ADP-stimulated respiration at the level of complex I, increases ROS generation and induces apoptosis in human skeletal muscle primary culture. We have also shown that in rat soleus muscle fibers incubated with simvastatin, the content of CoQ10 was reduced by 40% and addition of CoQ10 in these muscle biopsies prevented the inhibition of respiration at complex I and II levels and MPT, via free radical scavenging properties (Deichmann et al., 2010; La Guardia et al., 2013). Therefore, findings regarding statins redox effects are controversial and include antioxidant (Carneado et al., 2002; Wassmann et al., 2002; Manfredini et al., 2010; Zhou and Liao, 2010) and pro-oxidant actions (Velho et al., 2006; Oliveira et al., 2008; Kwak et al., 2012; La Guardia et al., 2013). In line with our previous works, here we hypothesized that statins induce muscle mitochondrial oxidative stress, which increases susceptibility to MPT. Thus, our aims were to investigate the effects of statin chronic treatment on muscle mitochondrial respiration rates, MPT and redox state indicators in the context of hypercholesterolemia. For this purpose, we used the mouse model that mimics the human disease familial hypercholesterolemia, since statins are used to treat specifically genetic hypercholesterolemic patients. We also chose a therapeutic dose of a hydrophilic statin (pravastatin) and two types of muscles predominantly aerobic (soleus) or anaerobic (plantaris) as target tissues.
Materials and Methods
Animals and Reagents
LDL receptor knockout mice (LDLr-/-) founders were purchased from Jackson Laboratory (Bar Harbor, ME) and the breeding colony was maintained at the Universidade Estadual de Campinas (CEMIB-Unicamp), Campinas, Brazil. LDLr-/- mice had access to standard laboratory rodent chow (AIN 93M, PragSoluções, SP, Brazil), and water ad libitum and were housed at 22 ± 2°C on a 12h light-dark cycle. This study was performed in accordance with the Guide for the Care and Use of Laboratory Animals published by National Academy of Sciences and with the approval of University Committee for Ethics in Animal Experimentation (protocol # 3401-1). Chemicals were purchased from Sigma (St. Louis, MO, USA).
Pravastatin Treatment and Creatine Supplementation
Thirty-day-old male LDLr-/- mice received pravastatin sodium (Medley) diluted in the drinking water (400 mg/L) during 2 or 3 months according to Lorza-Gil et al. (2016). The estimated pravastatin dose of 40 mg/Kg body weight per day was based on average consumption rate measurements (3.5 mL/day). Controls received filtered tap water without pravastatin. Additional groups of mice were treated with 2% creatine supplemented into standard diet (AIN 93M, PragSoluções, SP, Brazil) without alteration of total calories during the last 15 days of pravastatin treatment.
Plasma Cholesterol Analysis
Blood samples were collected with heparin from LDLr-/- mice tail between 8 and 9 am after a 12-h fasting. Samples were centrifuged and plasma was utilized for cholesterol measurement using a standard commercial kit (Roche Diagnostics) according to the manufacturer’s instructions. Plasma cholesterol levels in pravastatin treated LDLr-/- mice were significantly reduced compared to untreated mice (437 ± 57 vs. 390 ± 36, respectively, P < 0.05).
Skeletal Muscle Sample Preparation
Plantaris and soleus muscles were harvested from LDLr-/- mice and placed on ice-cold buffer containing 10 mM Ca-EGTA buffer (2.77 mM of CaK2EGTA + 7.23 mM of K2EGTA, free concentration of calcium 0.1 mmol/L), 20 mmol/L imidazole, 50 mmol/L K+/ 4-morpholinoethanesulfonic acid, 0.5 mmol/L dithiothreitol, 7 mmol/L MgCl2, 5 mmol/L ATP, 15 mmol/L phosphocreatine, pH 7.1. Individual fiber bundles from three to 5 mg of soleus or plantaris skeletal muscle were separated with forceps. Samples were permeabilized in ice-cold buffer containing saponin (50 μg/mL) during 30 min, gently stirred and washed three times with MiR05 medium (60 mmol/L potassium lactobionate, 0.5 mmol/L EGTA, 3 mmol/L MgCl2, 20 mmol/L taurine, 10 mmol/L KH2PO4, 20 mmol/L HEPES, 110 mmol/L sucrose, 1 g/L BSA, pH 7.1) at 4°C. Samples were dried with filter paper and weighted (Kuznetsov et al., 2008; La Guardia et al., 2013).
Oxygen Consumption
Oxygen consumption was evaluated in permeabilized skeletal muscle according to Kuznetsov et al. (2008) and La Guardia et al. (2013) with slight modifications. Permeabilized tissues were added to MiR05 medium without EGTA containing Ca2+ (4.4 μM) at 37°C supported with 10 mM glutamate plus 5 mM malate in a high-resolution oxygraph OROBOROS (Innsbruck, Austria). ADP (400 μM), oligomycin (0.63 μM), and FCCP (0.6 μM) were added during the experiments. Some analyses were evaluated in the presence of EGTA (500 μM), CsA (0.83 mM), ruthenium red (1 μM) or coenzyme Q10 (10 μM). Figure 1A shows the typical experimental respiratory profile.
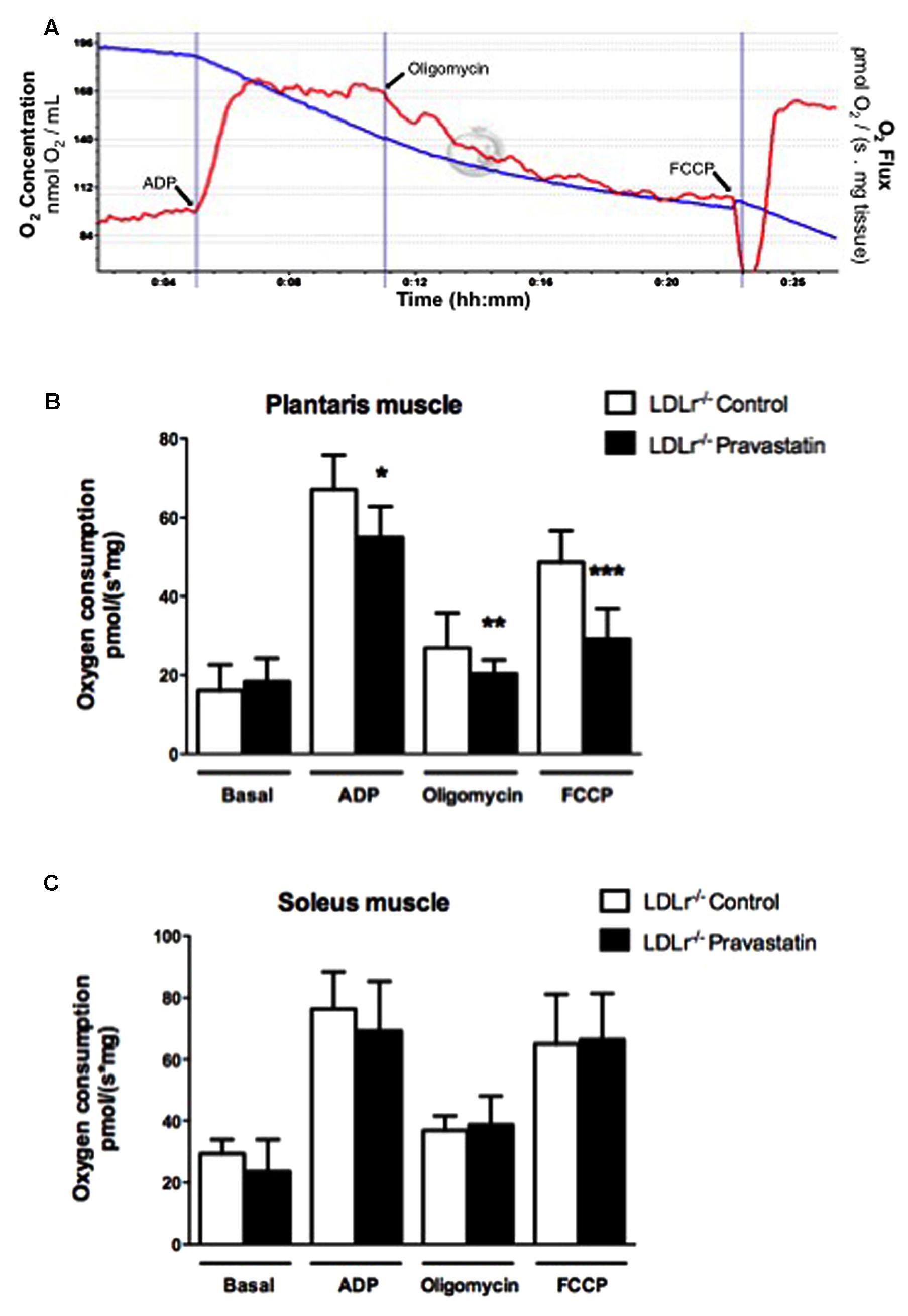
FIGURE 1. Pravastatin treatment inhibits oxygen consumption by plantaris muscle from LDLr-/- mice in the presence of Ca2+. Respiration was evaluated in a medium MiR05 at 37°C containing 10 mM glutamate plus 5 mM malate as substrates in the presence of Ca2+ (4.4 μM). ADP (400 μM), oligomycin (0.63 μM), and FCCP (0.6 μM) were added during the experiments. Representative traces of plantaris respiration where O2 concentration (blue line) is expressed as nmol O2/mL and O2 flux per mass (red line) is expressed as ρmol O2/s. mg tissue (A). Bar graphs show plantaris (B) and soleus muscle (C) from LDLr-/- mice treated or not with pravastatin (40 mg/Kg/day). Values are means ± standard deviation and are expressed as ρmol O2/s. mg tissue. ∗P = 0.0103, ∗∗P = 0.0442, ∗∗∗P = 0.0004 compared to control (Student’s t-test). N = 7–9, at least seven independent experiments.
Tissue Preparation and Enzymatic Activities
Plantaris and soleus muscles were harvested from LDLr-/- mice and homogenized in 9 volumes (1:10, w/v) of 20 mM sodium phosphate buffer, pH 7.4 containing 140 mM KCl. Homogenates were centrifuged at 1000 × g for 10 min at 4°C for nuclei and cell debris removal (Evelson et al., 2001). The pellet was discarded and the total supernatant was used for enzymatic activity determination.
Glutathione peroxidase, glutathione reductase, superoxide dismutase and peroxiredoxin were determined according to Wendel (1981), Carlberg and Mannervik (1985), Marklund (1985), and Kim et al. (2005), respectively. Catalase activity was analyzed by measuring the absorbance decrease at 240 nm according to Aebi (1984) and one unit (U) of the enzyme is defined as the metabolization of 1 μmol of H2O2 per min. The specific activity was calculated and expressed as U/mg protein. The activity of aconitase was measured according to Morrison (1954), following the reduction of NADP+ at wavelengths of excitation and emission of 340 and 466 nm, respectively. Aconitase activity was expressed as nmol NADPH/min/mg protein. Protein content was measured according to Lowry et al. (1951) using bovine serum albumin as standard.
Reverse Transcriptase (RT)-qPCR
Catalase mRNA expression was quantified by RT-qPCR using GAPDH housekeeping gene to normalize each sample. Plantaris muscles were harvested from LDLr-/- mice, total RNA was extracted using TRIzol (Thermo Fisher Scientific) following manufacturer’s instructions. Total RNA was used as template for cDNA synthesis in a reaction with oligo(dT)18 primer (Exxtend Biotecnologia) and SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) at 50°C for 60 min. The enzyme was then inactivated at 70°C for 15 min. Real-time PCR was performed on a Rotor Gene system (Qiagen, Hilden, Germany) using Rotor Gene SYBR Green PCR kit (Qiagen) and the following cycling conditions: 95°C for 5 s and 60°C for 10 s. Data acquisition was performed during the annealing step at 60°C. Primers used in qPCR were as follows: CAT (98 bp), 5′ GTTGAACGAGGAGGAGAGG 3′ (forward) and 3′ GTGAAATTCTTGACCGCTTTC 5′ (reverse); GAPDH (175 bp), 5′ GCACCACCAACTGCTTAGC 3′ (forward) and 3′ ATGCAGGGATGATGTTCTGG 5′ (reverse). CAT and GAPDH mRNA quantification was performed twice in N = 5 animals from each group. Data were analyzed using the Delta CT method of Rotor Gene Q series Software and catalase relative mRNA expression levels were obtained by normalizing against the level of GAPDH from the same sample and conditions. Efficiencies of CAT and GAPDH qPCRs were 1.00 and 0.90, respectively. Standard curves were prepared for each run using known quantities of pGEM-T-easy plasmids (Promega) containing CAT and GAPDH genes.
Sulfhydryl Content
Protein oxidative damage was evaluated by sulfhydryl content measurement according to Aksenov and Markesbery (2001). The reduction of 5,5′-dithio-bis (2-nitrobenzoic acid (DTNB) by thiols present in the sample generates a yellow compound (TNB) whose absorption is measured spectrophotometrically at 412 nm. Briefly, 30 μL of 10 mM DTNB and 980 μL of PBS were added to 50 μL muscle supernatant followed by a 30 min incubation at room temperature in the dark. The absorption measured was proportional to the amount of thiol groups present in the sample. Results were calculated as nmol TNB/mg of protein.
Electrospray Ionization High Resolution Mass Spectrometry (ESI-HRMS) Analysis
Plantaris muscles were removed from LDLr-/- mice and rapidly homogenized with a methanol:H2O (50:50) solution under sonication. Resulting homogenates were filtered through a 0.22 μm nylon membrane; 10 μL of the filtrate were further diluted in methanol:H2O (50:50) solution containing 0.1% formic acid to a final volume of 1 mL. Samples were directly infused in an ESI-LTQ-XL Orbitrap Discovery instrument (Thermo Scientific, Bremen, Germany). Typical operating conditions were as follows: sheath gas at 10 arbitrary units, 4.5 kV and m/z range of 50–1000 in the positive ion mode. Structural elucidation was carried out using mass accuracy as the main parameter, with a mass shift (error) less than 2 ppm. Spectral data were submitted to a partial least squares discriminant analysis (PLS-DA) using MetaboAnalyst 3.0 (Xia et al., 2015) to identify markers for each condition. Data normalization was performed using log transformation and range scaling. The selected ions were then researched in the Lipid Maps database, where oxidized species were identified.
Statistical Analysis
Results are presented as mean ± standard deviation of at least eight mice Data were analyzed using one-way analysis of variance (ANOVA) followed by the post hoc Tukey’s multiple comparison test when F was significant. The Student’s t-test for unpaired samples was also used for two-means comparisons. Differences between groups were rated significant at P < 0.05. All analyses were carried out using the GraphPad software.
Results
Inhibition of Respiration Supported by Site I Substrates in Plantaris Muscle Biopsies from LDLr-/- Mice Treated with Pravastatin
In order to investigate the effects of pravastatin chronic treatment on mitochondrial respiration of soleus and plantaris muscle, LDLr-/- mice received pravastatin (40 mg/kg/day) added to the drinking water during 3 months. Oxygen consumption supported by 10 mM glutamate plus 5 mM malate was evaluated in the presence of Ca2+ (4.4 μM) with the addition of ADP (400 μM), oligomycin (0.63 μM), and FCCP (0.6 μM) during the experiments. Figure 1A shows typical traces of mitochondrial respiration rates in all conditions. Figure 1B shows that pravastatin treatment promoted significant inhibition of mitochondrial respiration in all states: phosphorylating (ADP), resting (oligomycin) and maximal (FCCP) respiration rates of plantaris muscle in the presence of Ca2+. The inhibitions were 14, 24, and 40% for ADP-, oligomycin- and FCCP- stimulated respiration, respectively [n = 8; P = 0.0103; P = 0.0442; P = 0.0004]. The lower rate of FCCP-induced respiration compared to ADP-induced respiration is in agreement with recent data (Ruas et al., 2016) showing that oligomycin treatment previous to FCCP addition leads to an underestimation of maximal respiratory capacity induced by FCCP. In contrast to plantaris, no significant alterations of oxygen consumption rates were observed in soleus muscle (Figure 1C). Furthermore, no differences in oxygen consumption were observed in plantaris after only 2 months of pravastatin treatment (data not shown).
Pravastatin-Induced Inhibition of Mitochondrial Respiration Is Dependent on Mitochondrial Permeability Transition Pore (PTP) Opening
Considering that Ca2+ is essential for PTP opening (Hunter et al., 1976; Kowaltowski et al., 2001), and that toxic effects of statins have been associated with alterations in calcium homeostasis (Sirvent et al., 2005b; Sirvent et al., 2012), our next step was to investigate the role of Ca2+ on oxygen consumption of plantaris muscle of LDLr-/- mice. For this purpose, the Ca2+ chelator EGTA, ruthenium red (a mitochondrial Ca2+ uptake inhibitor) or cyclosporin A (CsA, a permeability transition inhibitor) were added in the reaction medium before oxygen consumption measurements. Figure 2 shows that all these compounds fully reversed the mitochondrial respiration inhibition in plantaris muscle of pravastatin treated LDLr-/- mice.
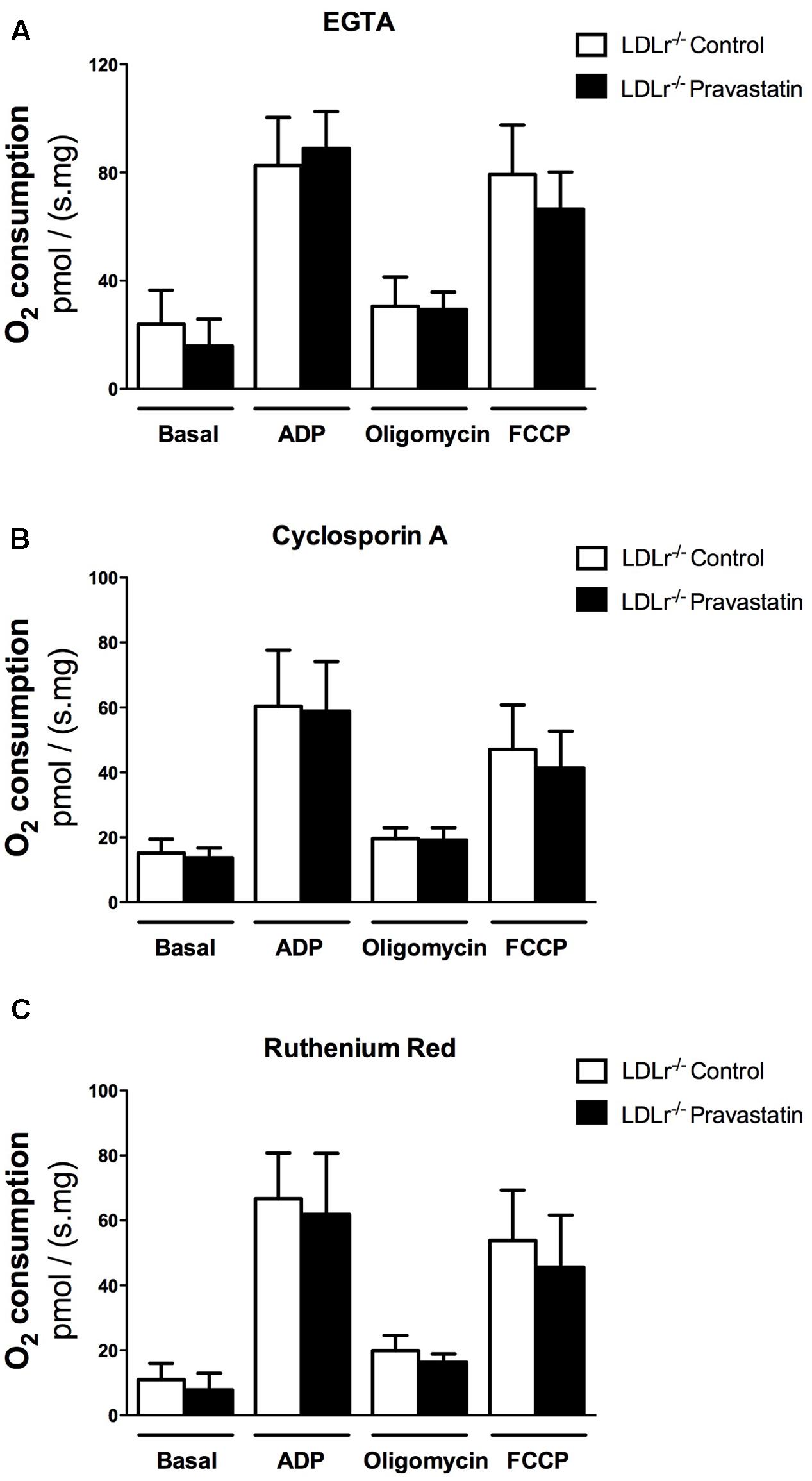
FIGURE 2. Pravastatin treatment does not inhibit oxygen consumption in the presence of EGTA, cyclosporin A or ruthenium red in permeabilized plantaris muscle of LDLr-/- mice. Respiration was evaluated in a medium MiR05 at 37°C containing 10 mM glutamate plus 5 mM malate as substrates in the presence of 500 μM EGTA (A), 0.83 mM cyclosporin A (B) or 1 μM ruthenium red (C) in plantaris muscle from LDLr-/- mice treated or not with pravastatin (40 mg/Kg/day). ADP (400 μM), oligomycin (0.63 μM) and FCCP (0.6 μM) were added during the experiments. Values are means ± standard deviation and are expressed as ρmol O2/s. mg tissue. No significant difference was observed. N = 10–12, at least ten independent experiments.
Both Creatine and Coenzyme Q10 Prevented Mitochondrial Respiratory Inhibition Induced by Pravastatin
Creatine acts directly as antioxidant (Lawler et al., 2002). In addition, creatine supplementation acts on ATP/ADP ratio maintenance due to creatine kinase (CK) activation and CK is part of the protein complex that is involved in MPT regulation (Kowaltowski et al., 2001; Dolder et al., 2003; Meyer et al., 2006). Therefore, we supplemented LDLr-/- mouse chow diet with 2% of creatine during the last 15 days of pravastatin treatment. Figure 3A shows that creatine diet supplementation prevented the inhibitory action of pravastatin on ADP- and FCCP-stimulated oxygen consumption in the presence of Ca2+ in plantaris muscle of LDLr-/- mice [n = 10; P < 0.05].
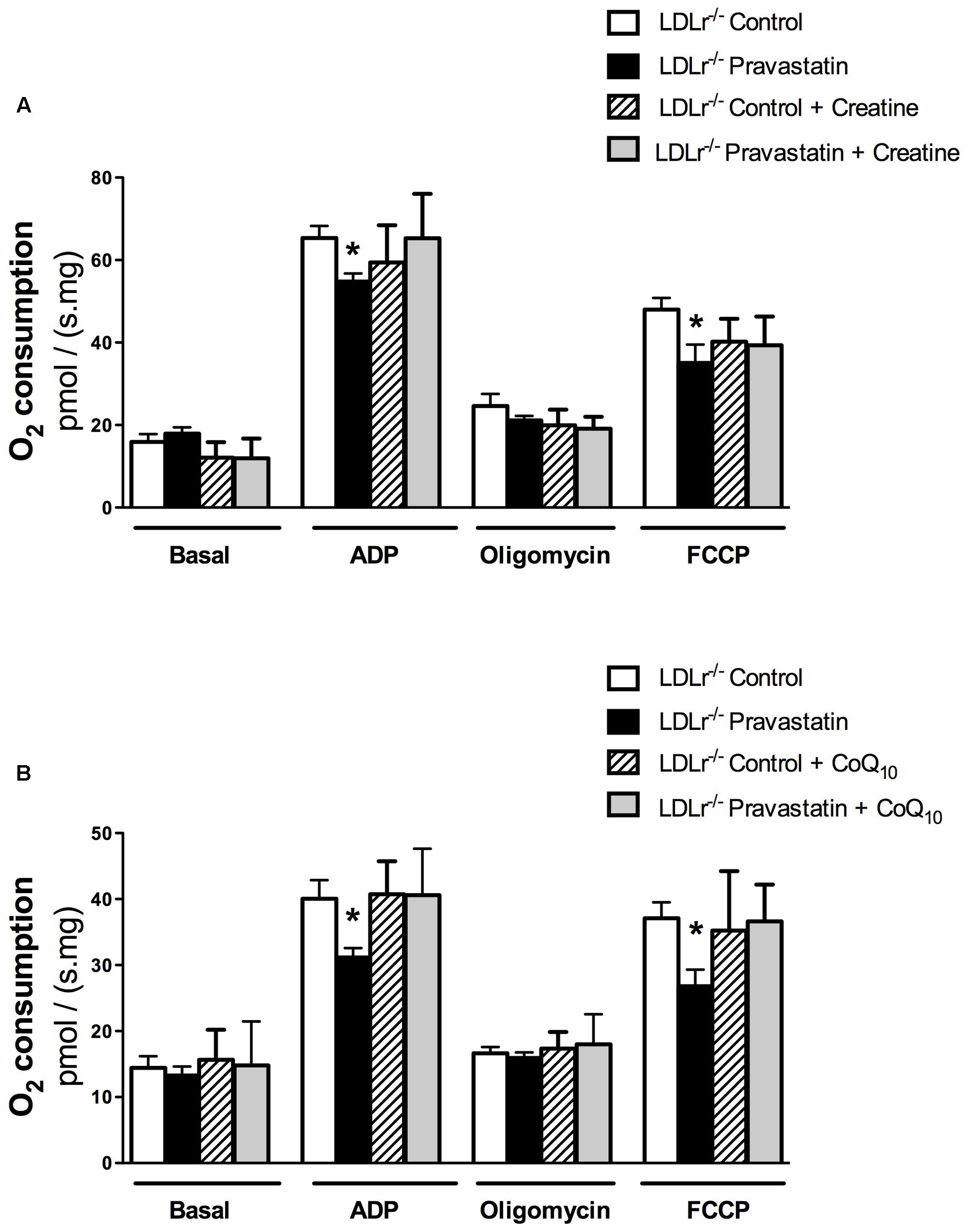
FIGURE 3. Inhibition of oxygen consumption in the presence of Ca2+ is prevented by creatine (A) or Coenzyme Q10 (B) in plantaris muscle of LDLr-/- mice treated with pravastatin (40 mg/kg/day). Respiration was evaluated in a medium MiR05 at 37°C containing 10 mM glutamate plus 5 mM malate as substrates in the presence of Ca2+ (4.4 μM). ADP (400 μM), oligomycin (0.63 μM), and FCCP (0.6 μM) were added during the experiments. Coenzyme Q10 (CoQ10, 10 μM) was added in the reaction medium before the biopsies. Values are means ± standard deviation and are expressed as ρmol O2/s. mg tissue. ∗P < 0.05 compared to control (One-Way ANOVA). N = 9–12, at least nine independent experiments.
CoQ10, which was previously reported by us to protect against mitochondrial dysfunction caused by simvastatin in rat soleus muscle (La Guardia et al., 2013), also showed the same protective effect under the experimental in vitro conditions of mouse mitochondrial phosphorylation (ADP) and maximal respiration (FCCP) rates [n = 10; P < 0.05] (Figure 3B).
Pravastatin Treatment Upregulates Catalase Activity and Induces Lipid Oxidation in Plantaris Muscle
Considering that several studies claim an antioxidant activity of statins due to upregulation of antioxidant defenses (Carneado et al., 2002; Wassmann et al., 2002; Manfredini et al., 2010; Zhou and Liao, 2010), we investigated the activity of antioxidant enzymes in muscle of LDLr-/- mice. Figure 4A shows that pravastatin treatment increased catalase activity up to 30% in plantaris muscle homogenates. In addition, creatine diet supplementation abolished the differences in catalase activity between control and pravastatin treated mice [n = 5, P < 0.05]. This increase is probably the consequence of a pravastatin effect at a post-transcriptional step or on the enzymatic catalysis, since catalase mRNA expression levels were not altered in LDLr-/- mice muscle (Figure 4B). On the other hand, pravastatin treatment caused no differences in superoxide dismutase, glutathione reductase, glutathione peroxidase, peroxiredoxin and glucose-6-phosphate dehydrogenase activities either in plantaris or in soleus muscle (data not shown).
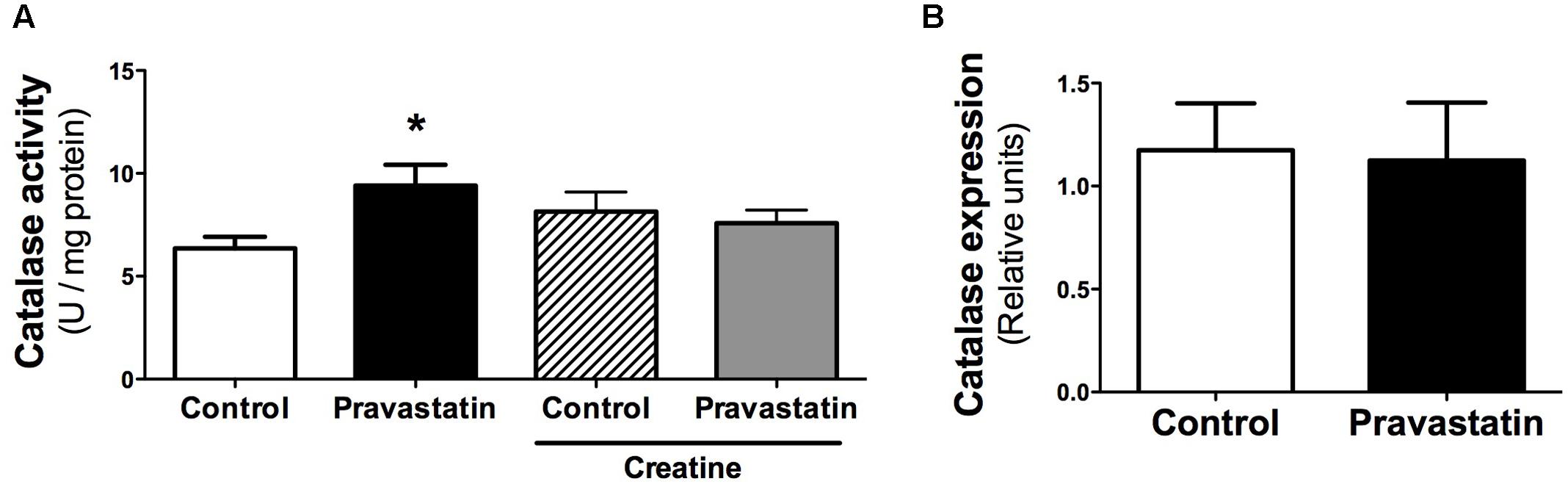
FIGURE 4. Catalase activity (A) and gene expression (B) evaluated in plantaris muscle of LDLr-/- mice. Plantaris muscles of control- and pravastatin-treated (40 mg/kg/day) LDLr-/- mice and both groups with creatine diet supplementation were used for catalase activity. Values are means ± standard deviation and are expressed in U/mg protein for activity and normalized by GAPDH for mRNA levels. ∗P < 0.05 compared to control (One-Way ANOVA and Student’s t-test). N = 12 for catalase activity and N = 5 for mRNA expression.
To verify that ROS production may occur due to pravastatin treatment, we investigated the presence of oxidized lipids in LDLr-/- mice. Using electrospray ionization high-resolution mass spectrometry analysis and a lipidomics approach, we identified oxidized lipid markers, especially phosphatidic acid and derivatives of arachidonic acid in plantaris muscle of LDLr-/- mice under pravastatin treatment (Table 1).
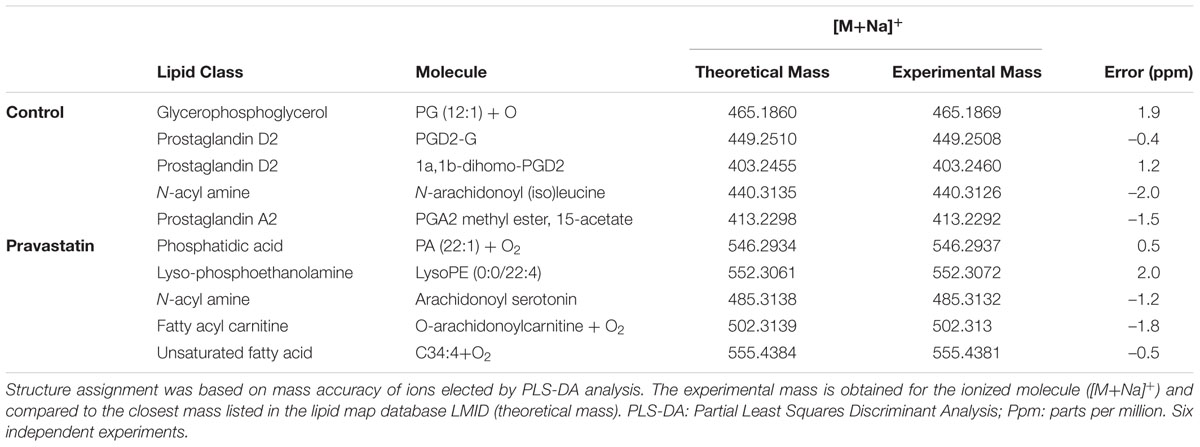
TABLE 1. Lipid markers identified by electrospray ionization high-resolution mass spectrometry in LDLr-/- mice.
To further investigate possible oxidative damage on other cellular components, we evaluated aconitase activity, a ROS-susceptible enzyme (Tretter and Adam-Vizi, 2000), and total sulfhydryl content, a protein oxidative damage marker in plantaris muscle of LDLr-/- mice. Both oxidative stress markers were not altered, suggesting that oxidative damage to proteins is probably not occurring in plantaris muscle of LDLr-/- mice under pravastatin treatment (Supplementary Figure S1).
Discussion
Most literature data on statins toxicity indicate a series of metabolic alterations, such as inhibition of mitochondrial respiration (Kwak et al., 2012; La Guardia et al., 2013), imbalance in calcium homeostasis (Sirvent et al., 2005a; Oliveira et al., 2008), inhibition of β -oxidation (Kaufmann et al., 2006; Costa et al., 2013) and mitochondrial oxidative stress (Velho et al., 2006; Oliveira et al., 2008; Kwak et al., 2012; Abdoli et al., 2013; Costa et al., 2013; La Guardia et al., 2013). However, these data were obtained in normocholesterolemic wild type models or in cultured cells or isolated mitochondria. Here, we investigated the mechanisms underlying mitochondrial dysfunction and MPT in skeletal muscle biopsies of a familial hypercholesterolemic mice model under chronic treatment with therapeutic doses of the hydrophilic pravastatin. The present work provides evidence that plantaris (but not soleus) muscle from LDLr-/- mice treated during 3 months (but not less) with pravastatin presents both inhibition of respiration (40% reduction in maximal respiration rate) and MPT when Ca2+ is present in the incubation medium, a condition that may lead to cell death. The protection from these toxic statin effects by the antioxidants CoQ10 and creatine suggests the participation of ROS in this mechanism, in agreement with previous data (Velho et al., 2006; Manfredini et al., 2010; Abdoli et al., 2013; La Guardia et al., 2013).
Searching for possible oxidative damage signals, several oxidized lipids species were identified in mitochondria of pravastatin treated LDLr-/- plantaris muscle, reinforcing the existence of an oxidative insult. However, since no protein oxidation markers (diminished SH- groups content or aconitase activity) were found, we may conclude that the nature of this oxidative insult must be mild and/or partially counteracted by cell defenses. Upregulation of catalase activity in pravastatin treated LDLr-/- plantaris muscle is one of these cell defense responses to oxidative stress (Kirkman and Gaetani, 2007; Jackson and McArdle, 2011). This suggests the participation of a signaling pathway linking mild mitochondrial oxidative stress to activation of catalase (Zarse et al., 2012; Ristow, 2014). Indeed, it was previously shown that the antioxidant effects of statins are possibly related to their ability to upregulate antioxidant defenses, including catalase expression and activity in vitro and in vivo (Carneado et al., 2002; Wassmann et al., 2002; Manfredini et al., 2010). The minor oxidative signs observed in pravastatin treated LDLr-/- are in line with this homeostatic antioxidant response to a chronic and mild oxidative stress. This is also in accordance with the safety of these drugs and the fact that only 10% of statin-treated hypercholesterolemic patients present adverse effects (Bruckert et al., 2005).
Among the several oxidized lipids found in muscle of pravastatin treated mice, we highlight two species, phosphatidic acid and arachidonic acid derivatives. Phosphatidic acid acts as second messenger that regulates several proteins (Testerink and Munnik, 2005), including mTOR (mammalian target of rapamycin). It is required for the stability and activity of this protein kinase (Steed and Chow, 2001; Foster et al., 2014; Shad et al., 2015; Yoon et al., 2015; Ghim et al., 2016). Thus, we could speculate that the oxidation of phosphatidic acid caused by pravastatin may impair mTOR pathway, affecting the maintenance of muscle mass and protein turnover (Shad et al., 2015). On the other hand, arachidonic acid metabolites, such as prostaglandin and leukotriene are involved in inflammatory muscle pain, and also in myogenesis and muscle repair (Korotkova and Lundberg, 2014). Therefore, oxidized derivatives of arachidonic acid could also impair muscle repair process in LDLr-/- mice under pravastatin treatment.
Previous studies proposed that statin-induced myotoxicity may be mediated by the reduction of ubiquinone content (Sirvent et al., 2008). Accordingly, inhibition of mitochondrial respiration was associated with ubiquinone depletion (Päivä et al., 2005; Bookstaver et al., 2012; Larsen et al., 2013) and ubiquinol treatment protected human rhabdomyosarcoma cells against simvastatin-induced mitochondrial dysfunction and cell death (Vaughan et al., 2013). While several studies propose that ubiquinone depletion by statins may be deleterious due to impairment of mitochondrial respiration (Päivä et al., 2005; Bookstaver et al., 2012; Larsen et al., 2013; Vaughan et al., 2013), we previously provided evidence that the decreased levels of CoQ10 by statin are not enough to limit mitochondrial respiration but rather impair its free radical scavenger action leading to oxidative stress (La Guardia et al., 2013). In addition, the rate of hydrogen peroxide production was increased in the presence of simvastatin and was normalized by CoQ10, reinforcing the involvement of oxidative stress in simvastatin-induced toxicity to skeletal muscle (La Guardia et al., 2013).
Creatine supplementation, widely and safely used by athletes, exerts beneficial effects on muscle growth and strength as well as in rehabilitation (Hespel and Derave, 2007; D’Antona et al., 2014). Creatine also has direct antioxidant properties (Lawler et al., 2002; Sestili et al., 2006), inhibits PTP opening and reduces muscle necrosis (O’Gorman et al., 1996; Dolder et al., 2003). Based on these findings, we evaluated whether creatine diet supplementation would prevent pravastatin-induced myotoxicity. Indeed, creatine treatment reversed mitochondrial dysfunction of plantaris muscle of LDLr-/- mice.
An important finding of the present work is that the mitochondrial respiratory inhibition provoked by chronic pravastatin treatment was sensitive to Ca2+ chelator (EGTA), ruthenium red (an inhibitor of Ca2+ uptake by mitochondria) or CsA (MPT inhibitor). Therefore, mitochondrial permeability transition may explain the occurrence of muscle dysfunctions in patients sensitive to statin toxicity.
It is of note that these pravastatin effects on plantaris muscle were not observed in soleus muscle under the same experimental conditions. These distinct skeletal muscles present different types of metabolism and fiber composition. Plantaris is mainly composed by type II fibers, presenting less mitochondrial content and higher glycolytic activity whereas soleus is rich in type I fibers and presents higher mitochondrial content and oxidative capacity (Cornachione et al., 2011). Results from other studies have also shown distinct sensitivities of different muscles to statins (Waclawik et al., 1993) and insensitivity of soleus to these drugs (Schaefer et al., 2004).
Taken together, the present results provide evidence that chronic pravastatin administration to a murine model of familial hypercholesterolemia promotes mitochondrial dysfunctions in plantaris muscle that can be counteracted by antioxidants administered either in vitro (CoQ10) or in vivo (creatine). Therefore, we propose that inhibition of muscle mitochondrial respiration by pravastatin leads to an oxidative stress that in the presence of calcium opens the PTP. This mitochondrial oxidative stress caused by statin treatment also signals for cellular antioxidant system responses such as catalase upregulation.
Author Contributions
EB performed the experiments, data analyses and interpretation and wrote the manuscript. AM and NL helped with the experiments, data analyses and interpretation. DdO and RC performed the lipidomic analyses and interpretation. HO and AV designed the work, interpreted the data, and wrote the manuscript.
Funding
This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP # 2011/50400-0), Obesity and Co-morbidities Research Center (OCRC/Fapesp # 2013/07607-8) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). EB, NL, and DO are supported by FAPESP fellowships (FAPESP # 2014/11261-2, 2014/00084-2 and 2014/08995-4) and ACM by CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) fellowship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Theo Wallimann (Swiss Federal Institute of Technology, ETH Zurich, Switzerland) for the suggestion of using creatine, Prof. Luis E. S. Netto (University of São Paulo, Brazil) for kindly helping with peroxiredoxin analysis, Dr. Miguel A. Chiurillo for catalase primer design and Prof. Roberto Docampo (University of Georgia, Athens, USA) for carefully reading the manuscript. We also thank Estela Lorza-Gil (Biology Institute, University of Campinas, Brazil) for stablishing pravastatin mice treatment and Roberto Stahl for excellent technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphar.2017.00185/full#supplementary-material
FIGURE S1 | Protein oxidative markers evaluated in plantaris muscle of LDLr-/- mice. Aconitase activity (A) and sulfhydryl content (B) were evaluated in plantaris muscle homogenates of control- and pravastatin- treated (40 mg/kg/day) LDLr-/- mice. Values are means ± standard deviation and are expressed as nmol/mg protein (One-Way ANOVA and Student’s t-test, non-significant). N = 6 for aconitase activity and N = 8 for sulfhydryl content.
References
Abdoli, N., Heidari, R., Azarmi, Y., and Eghbal, M. A. (2013). Mechanisms of the statins cytotoxicity in freshly isolated rat hepatocytes. J. Biochem. Mol. Toxicol. 27, 287–294. doi: 10.1002/jbt.21485
Aebi, H. (1984). Catalase in vitro. Methods Enzymol. 105, 121–126. doi: 10.1016/S0076-6879(84)05016-3
Aksenov, M. Y., and Markesbery, W. R. (2001). Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci. Lett. 302, 141–145. doi: 10.1016/S0304-3940(01)01636-6
Bookstaver, D. A., Burkhalter, N. A., and Hatzigeorgiou, C. (2012). Effect of coenzyme Q10 supplementation on statin-induced myalgias. Am. J. Cardiol. 110, 526–529. doi: 10.1016/j.amjcard.2012.04.026
Bruckert, E., Hayem, G., Dejager, S., Yau, C., and Bégaud, B. (2005). Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients–the PRIMO study. Cardiovasc. Drugs. Ther. 19, 403–414. doi: 10.1007/s10557-005-5686-z
Carlberg, I., and Mannervik, B. (1985). Glutathione reductase. Methods Enzymol. 113, 484–490. doi: 10.1016/S0076-6879(85)13062-4
Carneado, J., Alvarez de Sotomayor, M., Perez-Guerrero, C., Jimenez, L., Herrera, M. D., Pamies, E., et al. (2002). Simvastatin improves endothelial function in spontaneously hypertensive rats through a superoxide dismutase mediated antioxidant effect. J. Hypertens. 20, 429–437. doi: 10.1097/00004872-200203000-00018
Cornachione, A. S., Benedini-Elias, P. C., Polizello, J. C., Carvalho, L. C., and Mattiello-Sverzut, A. C. (2011). Characterization of fiber types in different muscles of the hindlimb in female weanling and adult Wistar rats. Acta Histochem. Cytochem. 44, 43–50. doi: 10.1267/ahc.10031
Costa, R. A., Fernandes, M. P., de Souza-Pinto, N. C., and Vercesi, A. E. (2013). Protective effects of l-carnitine and piracetam against mitochondrial permeability transition and PC3 cell necrosis induced by simvastatin. Eur. J. Pharmacol. 701, 82–86. doi: 10.1016/j.ejphar.2013.01.001
D’Antona, G., Nabavi, S. M., Micheletti, P., Di Lorenzo, A., Aquilani, R., Nisoli, E., et al. (2014). Creatine, L-carnitine, and ω3 polyunsaturated fatty acid supplementation from healthy to diseased skeletal muscle. Biomed. Res. Int. 2014:613890. doi: 10.1155/2014/613890
Deichmann, R., Lavie, C., and Andrews, S. (2010). Coenzyme q10 and statin-induced mitochondrial dysfunction. Ochsner J. 10, 16–21.
Dikalov, S. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033
Dobesh, P. P., and Olsen, K. M. (2014). Statins role in the prevention and treatment of sepsis. Pharmacol. Res. 88, 31–40. doi: 10.1016/j.phrs.2014.04.010
Dolder, M., Walzel, B., Speer, O., Schlattner, U., and Wallimann, T. (2003). Inhibition of the mitochondrial permeability transition by creatine kinase substrates. Requirement for microcompartmentation. J. Biol. Chem. 278, 17760–17766. doi: 10.1074/jbc.M208705200
Endo, A. (1992). The discovery and development of HMG-CoA reductase inhibitors. J. Lipid Res. 33, 1569–1582.
Evelson, P., Travacio, M., Repetto, M., Escobar, J., Llesuy, S., and Lissi, E. A. (2001). Evaluation of total reactive antioxidant potential (TRAP) of tissue homogenates and their cytosols. Arch. Biochem. Biophys. 388, 261–266. doi: 10.1006/abbi.2001.2292
Figueira, T. R., Barros, M. H., Camargo, A. A., Castilho, R. F., Ferreira, J. C., Kowaltowski, A. J., et al. (2013). Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid. Redox Signal. 18, 2029–2074. doi: 10.1089/ars.2012.4729
Foster, D. A., Salloum, D., Menon, D., and Frias, M. A. (2014). Phospholipase D and the maintenance of phosphatidic acid levels for regulation of mammalian target of rapamycin (mTOR). J. Biol. Chem. 289, 22583–22588. doi: 10.1074/jbc.R114.566091
Ghim, J., Chelakkot, C., Bae, Y. S., Suh, P. G., and Ryu, S. H. (2016). Accumulating insights into the role of phospholipase D2 in human diseases. Adv. Biol. Regul. 61, 42–46. doi: 10.1016/j.jbior.2015.11.010
Hajnóczky, G., and Csordás, G. (2010). Calcium signalling: fishing out molecules of mitochondrial calcium transport. Curr. Biol. 20, R888–R891. doi: 10.1016/j.cub.2010.09.035
Hespel, P., and Derave, W. (2007). Ergogenic effects of creatine in sports and rehabilitation. Subcell. Biochem. 46, 245–259. doi: 10.1007/978-1-4020-6486-9_12
Hunter, D. R., Haworth, R. A., and Southard, J. H. (1976). Relationship between configuration, function, and permeability in calcium-treated mitochondria. J. Biol. Chem. 251, 5069–5077.
Jackson, M. J., and McArdle, A. (2011). Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J. Physiol. 589(Pt 9), 2139–2145. doi: 10.1113/jphysiol.2011.206623
Javadov, S., and Kuznetsov, A. (2013). Mitochondrial permeability transition and cell death: the role of cyclophilin d. Front. Physiol. 4:76. doi: 10.3389/fphys.2013.00076
Kaufmann, P., Török, M., Zahno, A., Waldhauser, K. M., Brecht, K., and Krähenbühl, S. (2006). Toxicity of statins on rat skeletal muscle mitochondria. Cell Mol. Life Sci. 63, 2415–2425. doi: 10.1007/s00018-006-6235-z
Kim, J. A., Park, S., Kim, K., Rhee, S. G., and Kang, S. W. (2005). Activity assay of mammalian 2-cys peroxiredoxins using yeast thioredoxin reductase system. Anal. Biochem. 338, 216–223. doi: 10.1016/j.ab.2004.12.008
Kirkman, H. N., and Gaetani, G. F. (2007). Mammalian catalase: a venerable enzyme with new mysteries. Trends Biochem. Sci. 32, 44–50. doi: 10.1016/j.tibs.2006.11.003
Korotkova, M., and Lundberg, I. E. (2014). The skeletal muscle arachidonic acid cascade in health and inflammatory disease. Nat. Rev. Rheumatol. 10, 295–303. doi: 10.1038/nrrheum.2014.2
Kowaltowski, A. J., Castilho, R. F., and Vercesi, A. E. (2001). Mitochondrial permeability transition and oxidative stress. FEBS Lett. 495, 12–15. doi: 10.1016/S0014-5793(01)02316-X
Kowaltowski, A. J., Vercesi, A. E., Rhee, S. G., and Netto, L. E. (2000). Catalases and thioredoxin peroxidase protect Saccharomyces cerevisiae against Ca(2+)-induced mitochondrial membrane permeabilization and cell death. FEBS Lett. 473, 177–182. doi: 10.1016/S0014-5793(00)01526-X
Kuznetsov, A. V., Veksler, V., Gellerich, F. N., Saks, V., Margreiter, R., and Kunz, W. S. (2008). Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat. Protoc. 3, 965–976. doi: 10.1038/nprot.2008.61
Kwak, H. B., Thalacker-Mercer, A., Anderson, E. J., Lin, C. T., Kane, D. A., Lee, N. S., et al. (2012). Simvastatin impairs ADP-stimulated respiration and increases mitochondrial oxidative stress in primary human skeletal myotubes. Free Radic. Biol. Med. 52, 198–207. doi: 10.1016/j.freeradbiomed.2011.10.449
La Guardia, P. G., Alberici, L. C., Ravagnani, F. G., Catharino, R. R., and Vercesi, A. E. (2013). Protection of rat skeletal muscle fibers by either L-carnitine or coenzyme Q10 against statins toxicity mediated by mitochondrial reactive oxygen generation. Front. Physiol. 4:103. doi: 10.3389/fphys.2013.00103
Larsen, S., Stride, N., Hey-Mogensen, M., Hansen, C. N., Bang, L. E., Bundgaard, H., et al. (2013). Simvastatin effects on skeletal muscle: relation to decreased mitochondrial function and glucose intolerance. J. Am. Coll. Cardiol. 61, 44–53. doi: 10.1016/j.jacc.2012.09.036
Lawler, J. M., Barnes, W. S., Wu, G., Song, W., and Demaree, S. (2002). Direct antioxidant properties of creatine. Biochem. Biophys. Res. Commun. 290, 47–52. doi: 10.1006/bbrc.2001.6164
Lorza-Gil, E., Salerno, A. G., Wanschel, A. C., Vettorazzi, J. F., Ferreira, M. S., Rentz, T., et al. (2016). Chronic use of pravastatin reduces insulin exocytosis and increases β-cell death in hypercholesterolemic mice. Toxicology 344–346, 42–52. doi: 10.1016/j.tox.2015.12.007
Lowry, O. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J. (1951). Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275.
Malfitano, A. M., Marasco, G., Proto, M. C., Laezza, C., Gazzerro, P., and Bifulco, M. (2014). Statins in neurological disorders: an overview and update. Pharmacol. Res. 88, 74–83. doi: 10.1016/j.phrs.2014.06.007
Manfredini, V., Biancini, G. B., Vanzin, C. S., Dal Vesco, A. M., Cipriani, F., Biasi, L., et al. (2010). Simvastatin treatment prevents oxidative damage to DNA in whole blood leukocytes of dyslipidemic type 2 diabetic patients. Cell Biochem. Funct. 28, 360–366. doi: 10.1002/cbf.1654
Marklund, S. L. (1985). “Pyrogallol autoxidation,” in Handbook of Methods for Oxygen Radical Research, ed. R. A. Greenwald (Boca Raton, FL: CRC Press).
Meyer, L. E., Machado, L. B., Santiago, A. P., da-Silva, W. S., De Felice, F. G., Holub, O., et al. (2006). Mitochondrial creatine kinase activity prevents reactive oxygen species generation: antioxidant role of mitochondrial kinase-dependent ADP re-cycling activity. J. Biol. Chem. 281, 37361–37371. doi: 10.1074/jbc.M604123200
Morrison, J. F. (1954). The activation of aconitase by ferrous ions and reducing agents. Biochem. J. 58, 685–692. doi: 10.1042/bj0580685
Naci, H., Brugts, J. J., Fleurence, R., Tsoi, B., Toor, H., and Ades, A. E. (2013). Comparative benefits of statins in the primary and secondary prevention of major coronary events and all-cause mortality: a network meta-analysis of placebo-controlled and active-comparator trials. Eur. J. Prev. Cardiol. 20, 641–657. doi: 10.1177/2047487313480435
O’Gorman, E., Beutner, G., Wallimann, T., and Brdiczka, D. (1996). Differential effects of creatine depletion on the regulation of enzyme activities and on creatine-stimulated mitochondrial respiration in skeletal muscle, heart, and brain. Biochim. Biophys. Acta 1276, 161–170. doi: 10.1016/0005-2728(96)00074-6
Oliveira, K. A., Zecchin, K. G., Alberici, L. C., Castilho, R. F., and Vercesi, A. E. (2008). Simvastatin inducing PC3 prostate cancer cell necrosis mediated by calcineurin and mitochondrial dysfunction. J. Bioenerg. Biomembr. 40, 307–314. doi: 10.1007/s10863-008-9155-9
Päivä, H., Thelen, K. M., Van Coster, R., Smet, J., De Paepe, B., Mattila, K. M., et al. (2005). High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin. Pharmacol. Ther. 78, 60–68. doi: 10.1016/j.clpt.2005.03.006
Rasola, A., and Bernardi, P. (2011). Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 50, 222–233. doi: 10.1016/j.ceca.2011.04.007
Ristow, M. (2014). Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat. Med. 20, 709–711. doi: 10.1038/nm.3624
Ruas, J. S., Siqueira-Santos, E. S., Amigo, I., Rodrigues-Silva, E., Kowaltowski, A. J., and Castilho, R. F. (2016). Underestimation of the maximal capacity of the mitochondrial electron transport system in oligomycin-treated cells. PLoS ONE 11:e0150967. doi: 10.1371/journal.pone.0150967
Sacher, J., Weigl, L., Werner, M., Szegedi, C., and Hohenegger, M. (2005). Delineation of myotoxicity induced by 3-hydroxy-3-methylglutaryl CoA reductase inhibitors in human skeletal muscle cells. J. Pharmacol. Exp. Ther. 314, 1032–1041. doi: 10.1124/jpet.105.086462
Schaefer, W. H., Lawrence, J. W., Loughlin, A. F., Stoffregen, D. A., Mixson, L. A., Dean, D. C., et al. (2004). Evaluation of ubiquinone concentration and mitochondrial function relative to cerivastatin-induced skeletal myopathy in rats. Toxicol. Appl. Pharmacol. 194, 10–23. doi: 10.1016/j.taap.2003.08.013
Schirris, T. J., Renkema, G. H., Ritschel, T., Voermans, N. C., Bilos, A., van Engelen, B. G., et al. (2015). Statin-induced myopathy is associated with mitochondrial complex III inhibition. Cell Metab. 22, 399–407. doi: 10.1016/j.cmet.2015.08.002
Sestili, P., Martinelli, C., Bravi, G., Piccoli, G., Curci, R., Battistelli, M., et al. (2006). Creatine supplementation affords cytoprotection in oxidatively injured cultured mammalian cells via direct antioxidant activity. Free Radic. Biol. Med. 40, 837–849. doi: 10.1016/j.freeradbiomed.2005.10.035
Shad, B. J., Smeuninx, B., Atherton, P. J., and Breen, L. (2015). The mechanistic and ergogenic effects of phosphatidic acid in skeletal muscle. Appl. Physiol. Nutr. Metab. 40, 1233–1241. doi: 10.1139/apnm-2015-0350
Sirvent, P., Bordenave, S., Vermaelen, M., Roels, B., Vassort, G., Mercier, J., et al. (2005a). Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem. Biophys. Res. Commun. 338, 1426–1434. doi: 10.1016/j.bbrc.2005.10.108
Sirvent, P., Fabre, O., Bordenave, S., Hillaire-Buys, D., Raynaud De Mauverger, E., Lacampagne, A., et al. (2012). Muscle mitochondrial metabolism and calcium signaling impairment in patients treated with statins. Toxicol. Appl. Pharmacol. 259, 263–268. doi: 10.1016/j.taap.2012.01.008
Sirvent, P., Mercier, J., and Lacampagne, A. (2008). New insights into mechanisms of statin-associated myotoxicity. Curr. Opin. Pharmacol. 8, 333–338. doi: 10.1016/j.coph.2007.12.010
Sirvent, P., Mercier, J., Vassort, G., and Lacampagne, A. (2005b). Simvastatin triggers mitochondria-induced Ca2+ signaling alteration in skeletal muscle. Biochem. Biophys. Res. Commun. 329, 1067–1075. doi: 10.1016/j.bbrc.2005.02.070
Steed, P. M., and Chow, A. H. (2001). Intracellular signaling by phospholipase D as a therapeutic target. Curr. Pharm. Biotechnol. 2, 241–256. doi: 10.2174/1389201013378644
Testerink, C., and Munnik, T. (2005). Phosphatidic acid: a multifunctional stress signaling lipid in plants. Trends Plant Sci. 10, 368–375. doi: 10.1016/j.tplants.2005.06.002
Tobert, J. A. (2003). Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat. Rev. Drug Discov. 2, 517–526. doi: 10.1038/nrd1112
Tretter, L., and Adam-Vizi, V. (2000). Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 20, 8972–8979.
Vallianou, N. G., Kostantinou, A., Kougias, M., and Kazazis, C. (2014). Statins and cancer. Anticancer Agents Med. Chem. 14, 706–712. doi: 10.2174/1871520613666131129105035
Vaughan, R. A., Garcia-Smith, R., Bisoffi, M., Conn, C. A., and Trujillo, K. A. (2013). Ubiquinol rescues simvastatin-suppression of mitochondrial content, function and metabolism: implications for statin-induced rhabdomyolysis. Eur. J. Pharmacol. 711, 1–9. doi: 10.1016/j.ejphar.2013.04.009
Velho, J. A., Okanobo, H., Degasperi, G. R., Matsumoto, M. Y., Alberici, L. C., Cosso, R. G., et al. (2006). Statins induce calcium-dependent mitochondrial permeability transition. Toxicology 219, 124–132. doi: 10.1016/j.tox.2005.11.007
Vercesi, A. E., Kowaltowski, A. J., Oliveira, H. C., and Castilho, R. F. (2006). Mitochondrial Ca2+ transport, permeability transition and oxidative stress in cell death: implications in cardiotoxicity, neurodegeneration and dyslipidemias. Front. Biosci. 11:2554–2564. doi: 10.2741/1990
Waclawik, A. J., Lindal, S., and Engel, A. G. (1993). Experimental lovastatin myopathy. J. Neuropathol. Exp. Neurol. 52, 542–549. doi: 10.1097/00005072-199309000-00012
Wassmann, S., Laufs, U., Müller, K., Konkol, C., Ahlbory, K., Bäumer, A. T., et al. (2002). Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 22, 300–305. doi: 10.1161/hq0202.104081
Wendel, A. (1981). Glutathione peroxidase. Methods Enzymol. 77, 325–333. doi: 10.1016/S0076-6879(81)77046-0
Xia, J., Sinelnikov, I. V., Han, B., and Wishart, D. S. (2015). MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res. 43, W251–W257. doi: 10.1093/nar/gkv380
Yoon, M. S., Rosenberger, C. L., Wu, C., Truong, N., Sweedler, J. V., and Chen, J. (2015). Rapid mitogenic regulation of the mTORC1 inhibitor, DEPTOR, by phosphatidic acid. Mol. Cell. 58, 549–556. doi: 10.1016/j.molcel.2015.03.028
Zarse, K., Schmeisser, S., Groth, M., Priebe, S., Beuster, G., Kuhlow, D., et al. (2012). Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 15, 451–465. doi: 10.1016/j.cmet.2012.02.013
Keywords: pravastatin, muscle mitochondria, mitochondrial permeability transition, catalase, LDL receptor knockout mice
Citation: Busanello ENB, Marques AC, Lander N, de Oliveira DN, Catharino RR, Oliveira HCF and Vercesi AE (2017) Pravastatin Chronic Treatment Sensitizes Hypercholesterolemic Mice Muscle to Mitochondrial Permeability Transition: Protection by Creatine or Coenzyme Q10. Front. Pharmacol. 8:185. doi: 10.3389/fphar.2017.00185
Received: 19 August 2016; Accepted: 22 March 2017;
Published: 05 April 2017.
Edited by:
Eleonore Fröhlich, Medical University of Graz, AustriaReviewed by:
Markus Storvik, University of Eastern Finland, FinlandAndrey V. Kozlov, Ludwig Boltzmann Institute for Traumatology, Austria
Copyright © 2017 Busanello, Marques, Lander, de Oliveira, Catharino, Oliveira and Vercesi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helena C. F. Oliveira, ho98@unicamp.br Anibal E. Vercesi, anibal@unicamp.br