Corrigendum: Furanodiene Induces Extrinsic and Intrinsic Apoptosis in Doxorubicin-Resistant MCF-7 Breast Cancer Cells via NF-κB-Independent Mechanism
 Zhang-Feng Zhong1,2*†
Zhang-Feng Zhong1,2*† Hai-Bing Yu1†Chun-Ming Wang2Wen-An Qiang3,4Sheng-Peng Wang2
Hai-Bing Yu1†Chun-Ming Wang2Wen-An Qiang3,4Sheng-Peng Wang2 Jin-Ming Zhang2Hua Yu2Liao Cui1Tie Wu1
Jin-Ming Zhang2Hua Yu2Liao Cui1Tie Wu1 De-Qiang Li5*Yi-Tao Wang2*
De-Qiang Li5*Yi-Tao Wang2*- 1Guangdong Key Laboratory for Research and Development of Natural Drugs, School of Public Health, Guangdong Medical University, Zhanjiang, China
- 2State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macao, China
- 3Division of Reproductive Science in Medicine, Department of Obstetrics and Gynecology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 4Center for Developmental Therapeutics, Chemistry of Life Processes Institute, Northwestern University, Evanston, IL, United States
- 5Department of Pharmacy, The Second Hospital of Hebei Medical University, Shijiazhuang, China
Chemotherapy is used as a primary approach in cancer treatment after routine surgery. However, chemo-resistance tends to occur when chemotherapy is used clinically, resulting in poor prognosis and recurrence. Currently, Chinese medicine may provide insight into the design of new therapies to overcome chemo-resistance. Furanodiene, as a heat-sensitive sesquiterpene, is isolated from the essential oil of Rhizoma Curcumae. Even though mounting evidence claiming that furanodiene possesses anti-cancer activities in various types of cancers, the underlying mechanisms against chemo-resistant cancer are not fully clear. Our study found that furanodiene could display anti-cancer effects by inhibiting cell viability, inducing cell cytotoxicity, and suppressing cell proliferation in doxorubicin-resistant MCF-7 breast cancer cells. Furthermore, furanodiene preferentially causes apoptosis by interfering with intrinsic/extrinsic-dependent and NF-κB-independent pathways in doxorubicin-resistant MCF-7 cells. These observations also prompt that furanodiene may be developed as a promising natural product for multidrug-resistant cancer therapy in the future.
Introduction
Multidrug resistance (MDR) is one of the major factors of chemotherapy failure in the clinic, since it predicts poor prognosis and cancer recurrence of patients. Cancer drug resistance includes primary resistance before chemotherapeutic treatment and acquired resistance after chemotherapeutic exposure (Chemo-resistance). A MDR phenotype is defined as the resistance of malignant cells to different drugs with diverse structures and functions after exposure to the drug (Biedler and Riehm, 1970; Kaye, 1988; Vtorushin et al., 2014; Wu et al., 2014). Since a significant proportion of multidrug-resistant cancer cells could survive even after chemotherapy that results in chemo-resistance, disease progression, and recurrence.
Furanodiene is a heat-sensitive sesquiterpene found in the essential oil of Rhizoma Curcumae (Baldovini et al., 2001). In our previous study, furanodiene was confirmed to inhibit breast cancer growth (Zhong et al., 2012a) through altering angiogenesis (Zhong et al., 2012b) and energy metabolism (Zhong et al., 2016b), suppressing migration and invasion (Zhong et al., 2014), and enhancing the anti-cancer effect of doxorubicin and tamoxifen (Zhong et al., 2012c, 2016a) in vitro or in vivo. Additional researchers have also elucidated that cell death was induced by furanodiene in HepG2 cells (Xiao et al., 2007), HL60 leukemia cells (Ma et al., 2008), 95-D lung cancer cells (Xu et al., 2014), and uterine cervical cancer cells (Sun et al., 2009).
Apoptosis is known as a physiological process of cell deletion and is also a process of programmed cell death. It is stimulated by external or internal events of cells (Strasser et al., 2000), namely, the extrinsic pathway mediated by the death receptor and the intrinsic pathway mediated by mitochondria. The death receptors include tumor necrosis factor (TNF) receptors and TNF-related apoptosis-inducing ligand (TRAIL) receptors. As a surface receptor, TNF receptor-1 (TNF-R1) interacts with TNF and forms a receptor-proximal complex, which recruits a series of downstream factors, including Caspase-8, IκB kinase (IKK) α and β, resulting eventually in NF-κB activation (Chen and Goeddel, 2002; Reed, 2003). Otherwise, the mitochondrial pathway involves the Bcl-2 protein family and Caspase activation (Strasser et al., 2000; Czabotar et al., 2014). Among these effectors, Caspase-8 is an initiator caspase and downstream effector of TNF receptors and TRAIL receptors, directly cleaving Caspase-3/7 to propagate apoptosis signaling. Poly (ADP-ribose) polymerase (PARP) is the main cellular substrate of Caspase-3/7 for cleavage (Fulda and Debatin, 2006).
Even though growing evidence claims that furanodiene possesses anti-cancer activities in various types of cancers, the underlying mechanisms against chemo-resistant cancer are not fully clear. The present study aims to evaluate the initial effects of furanodiene on cell apoptosis and the underlying mechanisms in doxorubicin-resistant MCF-7 human breast cancer cells.
Materials and Methods
Chemicals and Reagents
Furanodiene (FUR) was purchased from the National Institutes for Food and Drug Control. The Roswell Park Memorial Institute-1640 (RPMI-1640) was used as the culture medium and was purchased from Gibco (Gaithersburg, MD, United States). Fetal bovine serum (FBS), phosphate-buffered saline (PBS), penicillin-streptomycin (PS), and 0.25% (w/v) trypsin/1 mM EDTA were obtained from Invitrogen (Carlsbad, CA, United States). 3-[4, 5-Dimethyl-2-thiazolyl]-2, 5-diphenyltetrazolium bromide (MTT), 2′, 7′-dichlorodihydrofluorescein diacetate (H2DCF-DA), CellROX®Deep probe, propidium iodide (PI), and Annexin V/PI detection kit were obtained from Molecular Probes (Eugene, OR, United States). Doxorubicin (DOX) and tert-Butyl hydroperoxide (TBHP) solution were supplied by Sigma-Aldrich (St. Louis, MO, United States). TNF-α immunoassay kit was purchased from R&D Systems (Minneapolis, MN, United States). Radioimmunoprecipitation assay (RIPA) lysis buffer and primary antibodies against TNF-R1 and p65 were obtained from Santa Cruz (Santa Cruz, CA, United States). Primary antibodies against p-IKKα/β (Ser176/180), IKKα, IKKβ, Bcl-xL, Bax, Bad, Caspase-7, Caspase-8, PARP, GAPDH, and β-actin, as well as the secondary antibodies were purchased from Cell Signaling (Danvers, MA, United States). siRNA was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, United States).
Cell Culture and Drug Treatment
Doxorubicin-resistant MCF-7 breast cancer cells were cultured as previously reported (Zhong et al., 2016b). The stock solutions of furanodiene (100 mM) and Doxorubicin (2 and 100 mM) dissolved in DMSO were diluted to different concentrations as needed.
Cell Viability Assay
Cell viability was performed by MTT assay as described previously (Zhong et al., 2011). Briefly, exponentially growing cells were seeded in 96-well plates at a corresponding density depending on different cell lines. Following the required incubation period, cell viability was determined by adding 100 μL of MTT (1 mg/mL). The absorbance values at 570 nm were recorded using SpectraMax M5 microplate reader (Molecular Devices, Silicon Valley, CA, United States).
Cell Cytotoxicity Assay
LDH release from the cells was measured with a cytotoxicity detection kit (Roche), according to the manufacturers’ protocol. In brief, supernatants (70 μL) were dispensed to a new 96-well plate. A 50-μL volume of supernatants was taken from each well for the reaction with LDH substrate after centrifugation at 350 g for 5 min. The absorbance values at 490 and 600 nm were recorded using SpectraMax M5 microplate reader (Molecular Devices, Silicon Valley, CA, United States).
Colony Formation Assay
Cells were seeded in 6-well plates at a density of 5 × 102/well. After a 15-day incubation at 37°C for visible colonies, these colonies were fixed with 4% (w/v) paraformaldehyde for 15 min and stained with crystal violet for 5 min. The images with colonies (≥50 cells as a colony) were captured using a microscope (Olympus MVX10, Japan) equipped with a digital camera (ColorView II, Soft Imaging System, Olympus).
CFDA-SE Cell Proliferation Assay
Cell proliferation determination was conducted with the CFDA-SE probe. Briefly, cells were seeded in 6-well plates at a density of 5 × 102/well and stained with CFDA-SE probe according to the manufacturer’s protocol. Then the cells were harvested and washed with PBS following drug treatments as required for 6 days. CFDA-SE fluorescence was detected using flow cytometry (BD FACS CantoTM, BD Biosciences, San Jose, CA, United States) and represented using FlowJo software (TreeStar, Ashland, OR, United States).
JC-1 Assay
Mitochondrial membrane potential (ΔΨm) was performed by JC-1 assay according to the previous report (Zhong et al., 2012a). Briefly, cells were cultured in 6-well plates at a density of 2 × 105/well and in black 96-well plates (with transparent bottom) at a density of 2 × 104/well, respectively. The incubation periods were performed as required. Then the culture medium was removed and the cells were washed twice with PBS. A further incubation with loading dye buffer (containing 2.5 μg/mL JC-1 and 10 mM glucose) was performed for 15 min at 37°C. The cells were harvested and mean fluorescence intensity of FITC was detected using a flow cytometry (BD FACS CantoTM, BD Biosciences, San Jose, CA, United States). Furthermore, JC-1 fluorescence was also observed via fluorescent microscopy and the images were captured using an Axiovert 200 fluorescent microscope (Carl Zeiss) and AxioCam HRC CCD camera (Carl Zeiss).
Caspase Activity Assay
Caspase-Glo assay kit (Promega, Madison, WI, United States) was used to determine caspase activity according to the manufacturer’s instruction. After indicated treatments, culture medium (50 μL) was discarded. Subsequently, caspase assay reagent (50 μL) was added to each well. Incubation protected from light under shaking condition lasted for 30 min. Reaction complex (70 μL) was transferred to white-walled 96-well plates. The luminescence was measured using SpectraMax M5 microplate reader (Molecular Devices, Silicon Valley, CA, United States). Caspase activity was calculated as the percentage of control. All samples were repeated in triplicate.
Western Blotting Assay
Western blotting assay was performed according to the previous studies (Zhong et al., 2012b; Quan et al., 2015). Briefly, cells were harvested and the total proteins were extracted with RIPA lysis buffer after the required treatments. Equal amounts of total proteins were separated by appropriate SDS-PAGE followed by transferring onto a PVDF membrane. After blocking with non-fat milk, the membrane was incubated with specific primary antibodies and the corresponding second antibodies, respectively. The specific protein bands were visualized with an AmershamTM ECLTM advanced western blotting detection kit (GE Healthcare Life Sciences, United Kingdom).
Annexin V/PI Staining Assay
Cells were seeded in 6-well plate at a density of 2 × 105/well and allowed to adhere overnight. Treatment and incubation period were performed as required. Cell apoptosis was determined by Annexin V/PI labeling according to the manufacturer’s protocol (Invitrogen). The early and late apoptotic cells were detected using a flow cytometry (BD FACS CantoTM, BD Biosciences, San Jose, CA, United States) based on the Annexin V and PI staining.
Cellular Reactive Oxygen Species (ROS) and Calcium Generation Assay
Cells were seeded in 6-well plates at a density of 2 × 105/well or in black 96-well plates (with transparent bottom) at a density of 2 × 104/well, respectively. Then the cells were incubated in a humidified incubator for 24 h. The culture medium was replaced with FBS-free RPMI-1640 medium containing 2 μM H2DCF-DA, CellROX® Deep Red probe, or Fluo-4 AM with a 30-min incubation at 37°C to assess the reactive oxygen species (ROS) or calcium levels. Cellular ROS and calcium generation was determined after the drug treatment as required. TBHP treatment was used as a positive control. For quantitative assessment, the cells were collected, re-suspended in PBS and analyzed using flow cytometry (BD FACS CantoTM, BD Biosciences, San Jose, CA, United States). The mean fluorescence intensity of 10,000 analyzed cells (corrected for autofluorescence) of each group was measured for the total ROS or calcium generation, and represented by FlowJo software (TreeStar, Ashland, OR, United States). For direct observation assessment, images were captured using high-content imaging.
Quantitative Immunoassay for TNF-α Release
Cells were seeded in 12-well plates at a density of 1 × 105/well. The conditioned culture medium was collected and the TNF-α release from the cells was determined by immunoassay kit (R&D Systems, Minneapolis, MN, United States) according to the manufacturer’s protocol. The absorbance values at 450 and 570 nm were determined using SpectraMax M5 microplate reader (Molecular Devices, Silicon Valley, CA, United States).
TNF-α-TNF-R1 Binding ELISA
The binding assay was performed according to the previous report (Leung et al., 2012). Briefly, microtiter plates were coated with TNF-α (0.625 μg/mL) in PBS at 4°C overnight. After washing three times with PBS/0.05% Tween 20 (PBST), the wells were blocked with PBST containing 1% BSA (200 μL) for 1 h. A series of concentrations of the test drugs were added into the wells followed by a 20-min incubation. TNF-R1 solution in PBS (50 μL, 0.2 μg/mL) was added into the wells with further incubation for 2 h. The washed wells were incubated with TNF-R1 antibody solutions (1:1000) in 100 μL of PBST containing 1% BSA for 2 h. Then, the washed wells were incubated with horseradish peroxide-conjugated secondary antibody for 2 h. Finally, the washed wells were incubated with TMB solution (100 μL), followed by quenching treatment with stop solution (100 μL, 2N sulfuric acid). The absorbance was measured at 450 nm with SpectraMax M5 microplate reader (Molecular Devices, Silicon Valley, CA, United States).
Cell Density Assay
After the administered drug treatments, cell density was observed and the images were captured using a microscope (Olympus MVX10, Japan) equipped with a digital camera (ColorView II, Soft Imaging System, Olympus), to survey cell density under 100× magnifications. The representative images were from at least three independent experiments.
Dual-Luciferase Reporter Assay
Cells were seeded in a 24-well plate at a density of 5 × 104/well. The cells were co-transfected with 0.8 μg pNF-κB-luc and 0.8 μg pRL-TK as a transfection efficiency control. The plasmids and Lipofectamine agent were diluted in Opti-MEM serum-free medium according to Lipofectamine DNA transfection reagent protocol. The diluted DNA was mixed together with diluted Lipofectamine agent at the ratio of 1:1 followed by a 20-min incubation at 25°C. DNA-Lipofectamine (100 μL) complexes was transferred to each well. After a 4-h incubation, the cells were cultivated with fresh completed medium for 48 h. Cell lysates were collected by using passive lysis buffer according to the dual luciferase assay protocol (Zhong et al., 2015). Sample light output was recorded by using SpectraMax M5 microplate reader (Molecular Devices, Silicon Valley, CA, United States). Data were aligned to pRL-TK values prior to normalization with its control.
Transient Transfection of siRNAs
RNA interference assay was performed using Lipofectamine 2000 agent according to the manufacturer’s protocol. Briefly, cells were seeded in 6-well plates at a density of 2 × 105/well overnight. siRNA and Lipofectamine agent were diluted in Opti-MEM reduced serum medium and mixed gently, respectively. Then, the siRNA-lipofectamine mixtures were transferred to the culture wells, following a 20-min incubation at room temperature. After a 4-h transfection, the cells were refreshed with completed medium. The transfected cells were selected for the further experiments after a 48-h stable incubation.
Statistical Analysis
All data represent the mean of three separately performed experiments, plus or minus standard deviation or standard error of the mean. The significance of intergroup differences was evaluated by one-way ANOVA using the GraphPad Prism software (GraphPad Software, United States). Newman–Keuls multiple comparison tests were performed for post hoc pairwise comparisons. P-values less than 0.05 were considered as significant.
Results
Furanodiene Displayed Anti-cancer Effects through Inhibiting Cell Viability, Inducing Cell Cytotoxicity, and Suppressing Cell Proliferation in Doxorubicin (DOX)-Resistant MCF-7 (MCF-7/DOXR) Cells
As shown in Figure 1A, furanodiene inhibited the viability of MCF-7/DOXR human breast cancer cells in a dose-dependent manner. In addition, compared with the vehicle control group, LDH releases from MCF-7/DOXR cells were increased dose-dependently after 24-h treatment with furanodiene (Figure 1B). However, neither the cell viability nor the cell cytotoxicity was significantly changed after doxorubicin (2 μM) treatment.
FIGURE 1
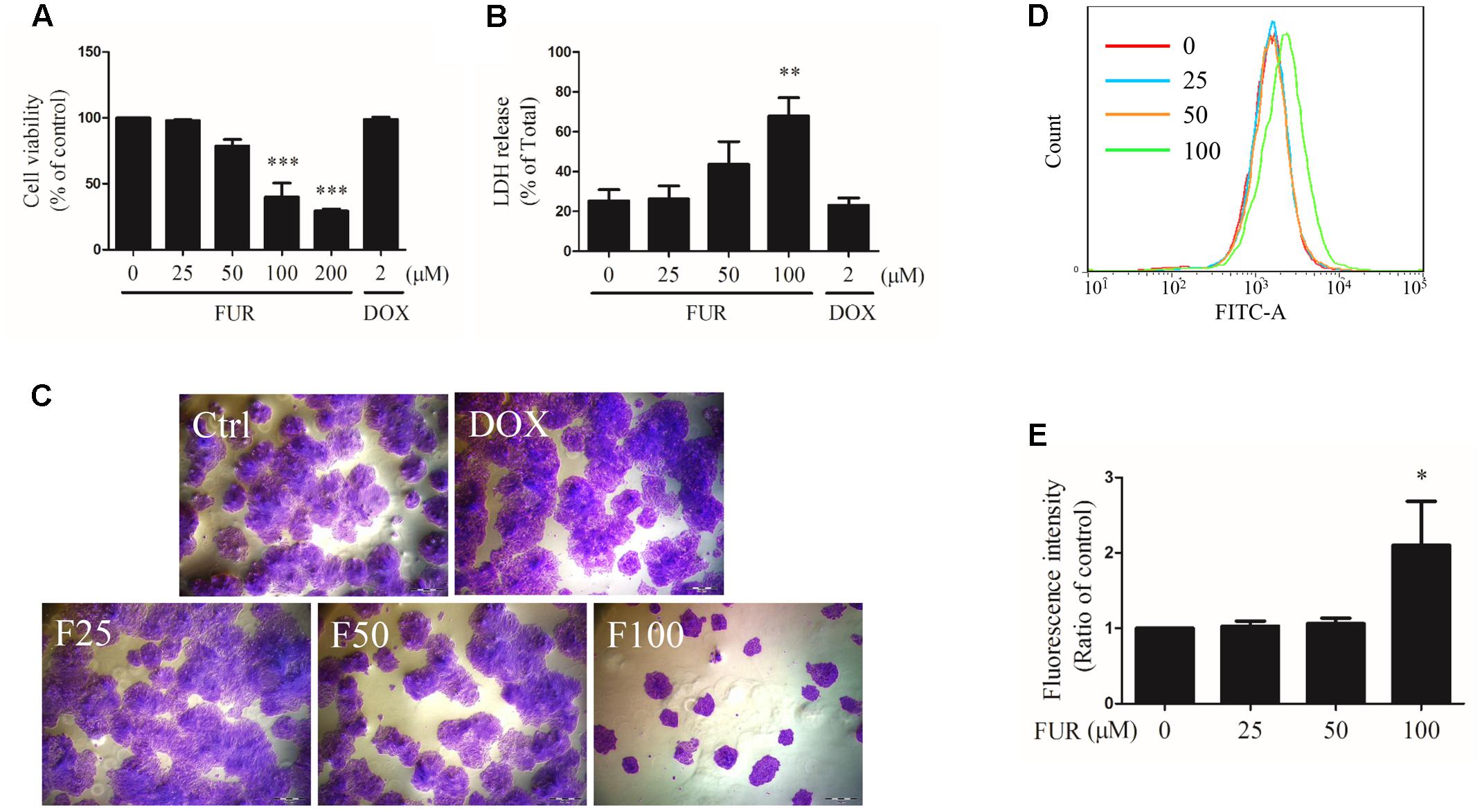
FIGURE 1. Effect of furanodiene (FUR or F) on cell viability, cytotoxicity, and proliferation in doxorubicin (DOX)-resistant MCF-7 breast cancer cells. Cells were treated with furanodiene (0–200 μM) or doxorubicin (2 μM) for 24 h. (A) Cell viability was tested by MTT assay. The values were represented as percentage of the vehicle control. (B) LDH release was detected using a commercial kit. The values were represented as percentage of the total LDH release. (C) Cells were treated with furanodiene (0–100 μM) or doxorubicin (2 μM) for 24 h. Then the cells were harvested, and cultured in complete growth medium at a density of 200 cells per well. After a 2-week incubation, the fixed cells were stained with crystal violet. The colony formation status was visualized at 200× magnifications with an AxioCam HRC CCD phase contrast microscope. (D) Cells were stained with CFDA-SE and then treated with furanodiene (0–100 μM) for 6 days. CFDA-SE fluorescence was detected using flow cytometry. FlowJo software was used for the analysis of flow cytometry data. (E) Statistical result of (D). Ctrl stands for control. Data are expressed as mean ± SEM. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 vs. control.
Cell proliferation induced by furanodiene was assessed by colony formation assay and CFDA-SE assay. In colony formation assay, colony formation was strongly inhibited by furanodiene (100 μM) treatment, and there were almost no alterations observed in doxorubicin (2 μM) treatment compared with vehicle control in MCF-7/DOXR cells (Figure 1C). After furanodiene (25, 50, and 100 μM) treatment for 6 days, the mean fluorescence intensity induced by furanodiene (100 μM) was stronger than the vehicle control, indicating that furanodiene significantly suppressed the proliferation at high concentration in MCF-7/DOXR cells (Figures 1D,E).
Furanodiene Depolarized the Mitochondrial Membrane Potential (ΔΨm) in MCF-7/DOXR Cells
After 4 h of furanodiene treatment in MCF-7/DOXR cells, the JC-1 monomer fluorescence (green) intensity increased dose-dependently, compared with the vehicle control. However, doxorubicin (2 μM) failed to decrease ΔΨm loss in MCF-7/DOXR cells as shown in the flow cytometry results (Figure 2A). For example, furanodiene (100 μM) treatment increased JC-1 monomer fluorescence (green) intensity with sixfold, compared with the vehicle control. tert-Butyl hydroperoxide (TBHP) (300 μM), as a positive control, also significantly depolarized the mitochondrial membrane potential (Figure 2B). Similarly, fluorescent images also showed that furanodiene could decrease the ΔΨm of MCF-7/DOXR cells after a 24-h treatment with furanodiene. The red fluorescence (J-aggregates) intensity is significantly diminished whereas the green fluorescence (monomer) intensity is enhanced after 50 μM treatment with furanodiene. Furthermore, there was only intensive green fluorescence observed after 100 μM treatment with furanodiene, indicating the significant decrease or loss of ΔΨm (Figure 2C).
FIGURE 2
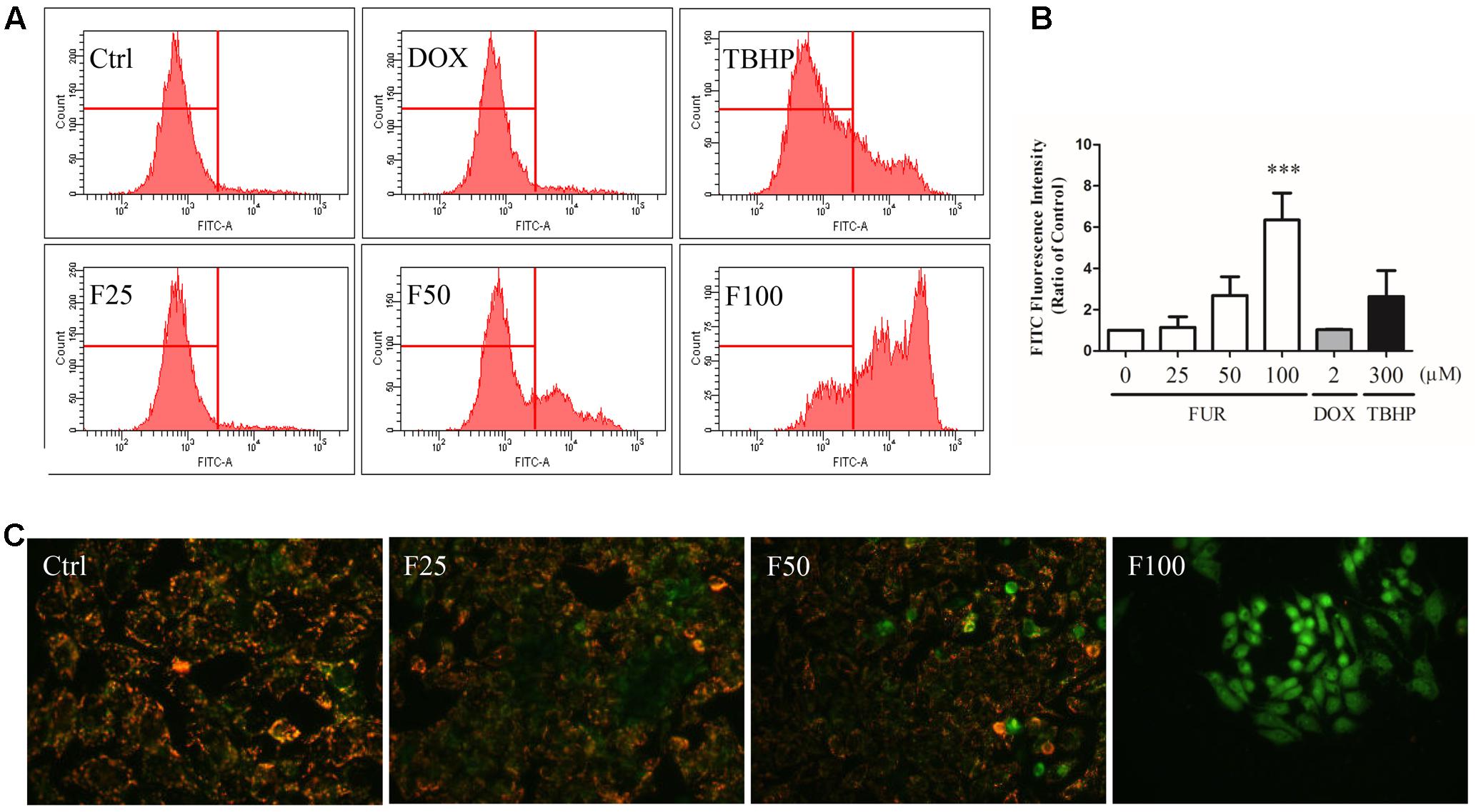
FIGURE 2. Effect of furanodiene (FUR or F) on ΔΨm in doxorubicin (DOX)-resistant MCF-7 breast cancer cells. (A) Cells were treated with furanodiene (0–100 μM), doxorubicin (2 μM), and tert-Butyl hydroperoxide (TBHP) (300 μM) for 4 h, after which, the green fluorescence (FITC) from the JC-1 monomers was monitored with JC-1 using flow cytometry. (B) Statistical result of (A). (C) Cells were exposed to furanodiene (0–100 μM) for 24 h followed by a 30-min incubation with JC-1 monomers. The fluorescence was visualized at 400× magnification using high content imaging. The green fluorescence from the JC-1 monomers was used to represent the cell that lost ΔΨm. TBHP treatment was used as a positive control. Ctrl stands for control. Data are expressed as mean ± SEM. ∗∗∗P < 0.001 vs. control.
Furanodiene Regulated the Mitochondrial Pathway of Cell Death in MCF-7/DOXR Cells
Compared to vehicle control, furanodiene activated Caspase-3/7 in dose- and time-dependent manners (Figures 3A,B). Briefly, 100 μM of furanodiene treatment (24 h) increased the Caspase-3/7 activities with 5.65 folds, compared with the vehicle control. Western blotting results showed that furanodiene treatment decreased Bcl-xL expression, and increased Bad expression, without affecting Bax expression. Meanwhile, furanodiene dose-dependently activated Caspase-7 and PARP, as indicated by the increasing expression of cleaved protein (Figure 3C). Moreover, pre-treatment with Caspase inhibitor (z-VAD-fmk) could reverse the cytotoxic effect of furanodiene with a sharp reduction (from 2.62 to 1.47 folds) in MCF-7/DOXR cells (Figure 3D). These results indicated that furanodiene might induce cell death through the mitochondrial pathway.
FIGURE 3
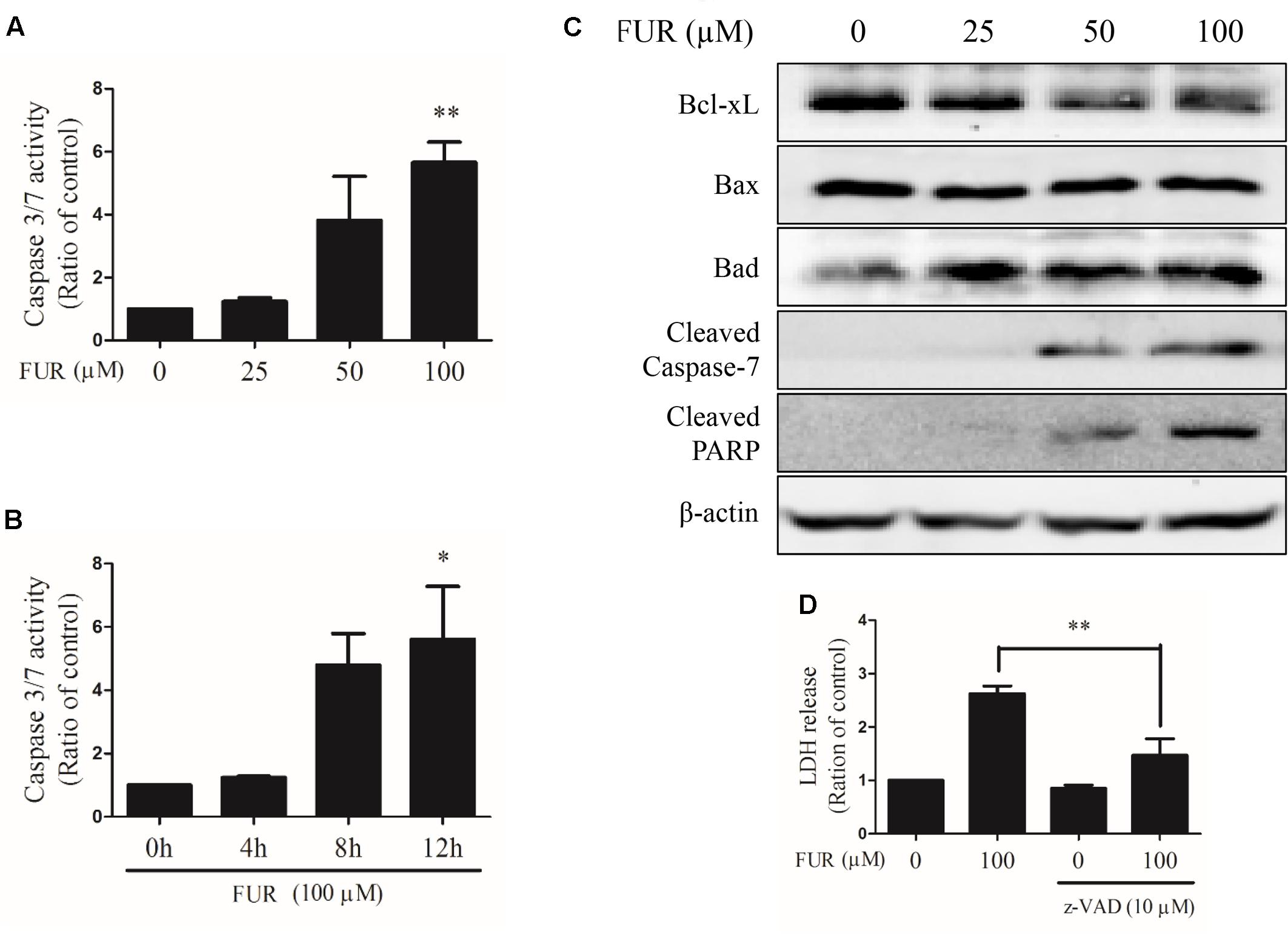
FIGURE 3. Effect of furanodiene (FUR) on the mitochondrial pathway of apoptosis in doxorubicin-resistant MCF-7 breast cancer cells. (A) Cells were treated with different concentrations of furanodiene (0–100 μM) for 24 h. (B) Cells were treated with furanodiene (100 μM) for 0–12 h. Caspase activities were determined with Caspase-Glo assay kit. (C) Cells were treated with different concentrations of furanodiene (0–100 μM) for 24 h. Protein expression was determined by western blotting and β-actin was used as a loading control. (D) Cells were pretreated with z-VAD-fmk (z-VAD) for 1 h before treatment with furanodiene for 24 h. Cytotoxicity was detected by LDH assay. Data are expressed as mean ± SEM. ∗P < 0.05 and ∗∗P < 0.01 vs. control.
Furanodiene Induced Apoptotic Cell Death in MCF-7/DOXR Cells
To confirm that MCF-7/DOXR cells induced by furanodiene undergo programmed cell death, Annexin V/PI staining was performed using flow cytometry analyses. As shown in Figures 4A,C, representative scatter plots of cells showed the differences post treatment with different concentrations of furanodiene used at for different times. The percentage of early/late apoptotic cells was summarized in Figures 4B,D. In vehicle control, the proportion of early and late apoptotic cells was 5.07% and 24-h treatments with furanodiene (25–100 μM) dose-dependently increased the ratio of early and late apoptotic cells from 5.10 to 44.27% (Figure 4B). Moreover, time-dependent treatments (0–12 h) with furanodiene (100 μM) increased the percentage of early and late apoptotic cells up to 28.57% (Figure 4D). The dose- and time-dependent promotion of cell apoptosis induced by furanodiene was also consistent with the outcomes of Caspase-3/7 activation.
FIGURE 4
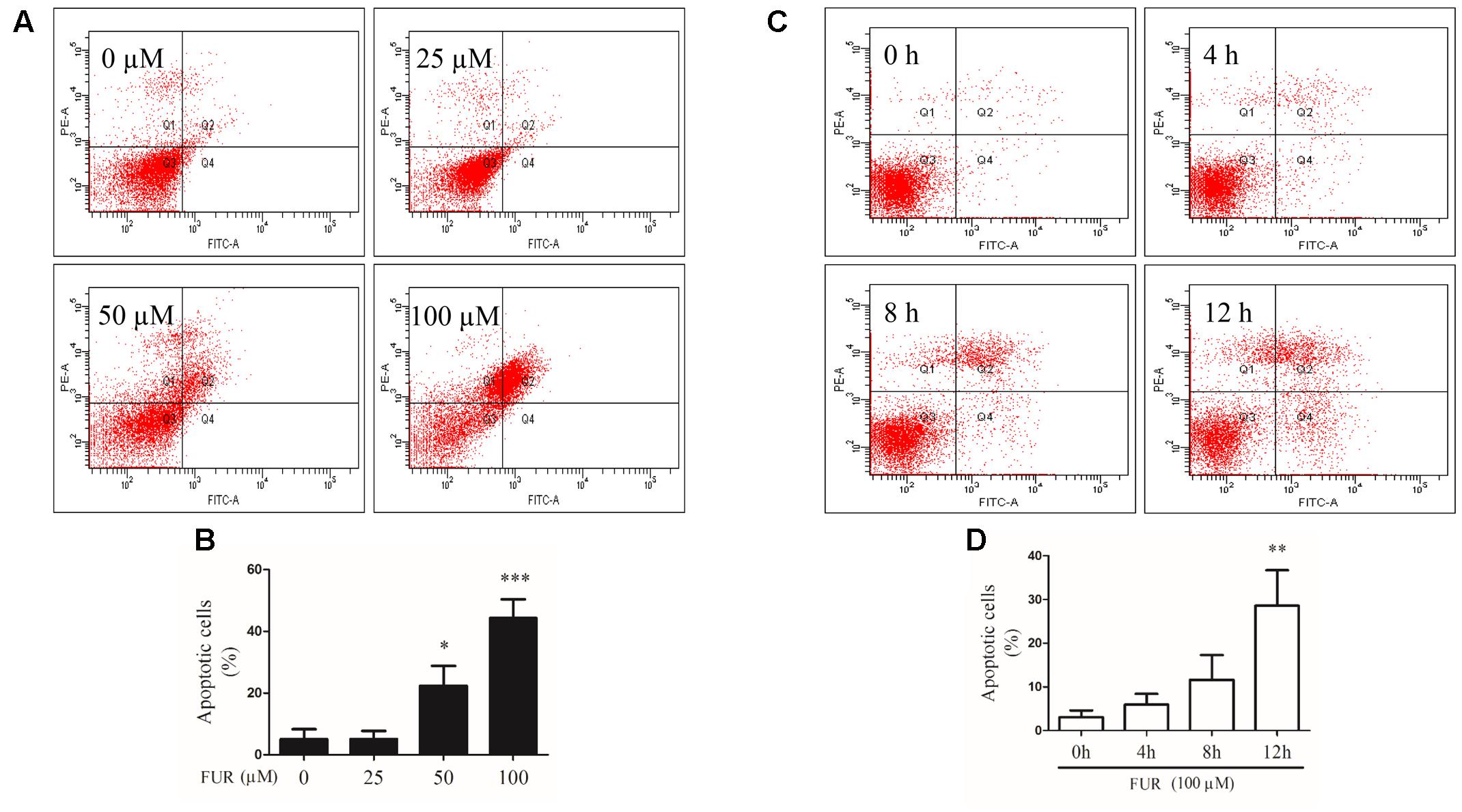
FIGURE 4. Effect of furanodiene (FUR) on cell apoptosis in doxorubicin-resistant MCF-7 breast cancer cells. (A) Cells were treated with different concentrations of furanodiene (0–100 μM) for 24 h. Apoptotic cells were detected by Annexin V/PI staining assay. (B) Statistical result of (A). (C) Cells were treated with furanodiene (100 μM) for 0–12 h. Apoptotic cells were detected by Annexin V/PI staining assay. (D) Statistical result of (C). Data are expressed as mean ± SEM. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 vs. control.
Furanodiene Induced Reactive Oxygen Species (ROS) and Calcium Production in MCF-7/DOXR Cells
Cells were exposed to TBHP and furanodiene for 2 h, and ROS production was detected with H2DCF-DA probe using flow cytometry. As shown in Figures 5A,B, TBHP (300 μM) treatment induced ROS generation in MCF-7/DOXR cells, and furanodiene treatment dose-dependently induced the ROS generation. Long-term effects of furanodiene on ROS levels was investigated with the CellROX® Deep Red probe. Similarly, fluorescence microscopy results confirmed that furanodiene could dose-dependently increase the ROS generation of MCF-7/DOXR cells after a 24-h treatment with furanodiene (Figure 5C). However, addition of NAC (5 mM) or GSH (5 mM) did not attenuate cell viability induced by furanodiene (data not shown). These results indicated that furanodiene inducing cell death might be involved more than ROS-mitochondrial dependent pathway. Moreover, calcium production was also induced by furanodiene (Figures 5D,E).
FIGURE 5
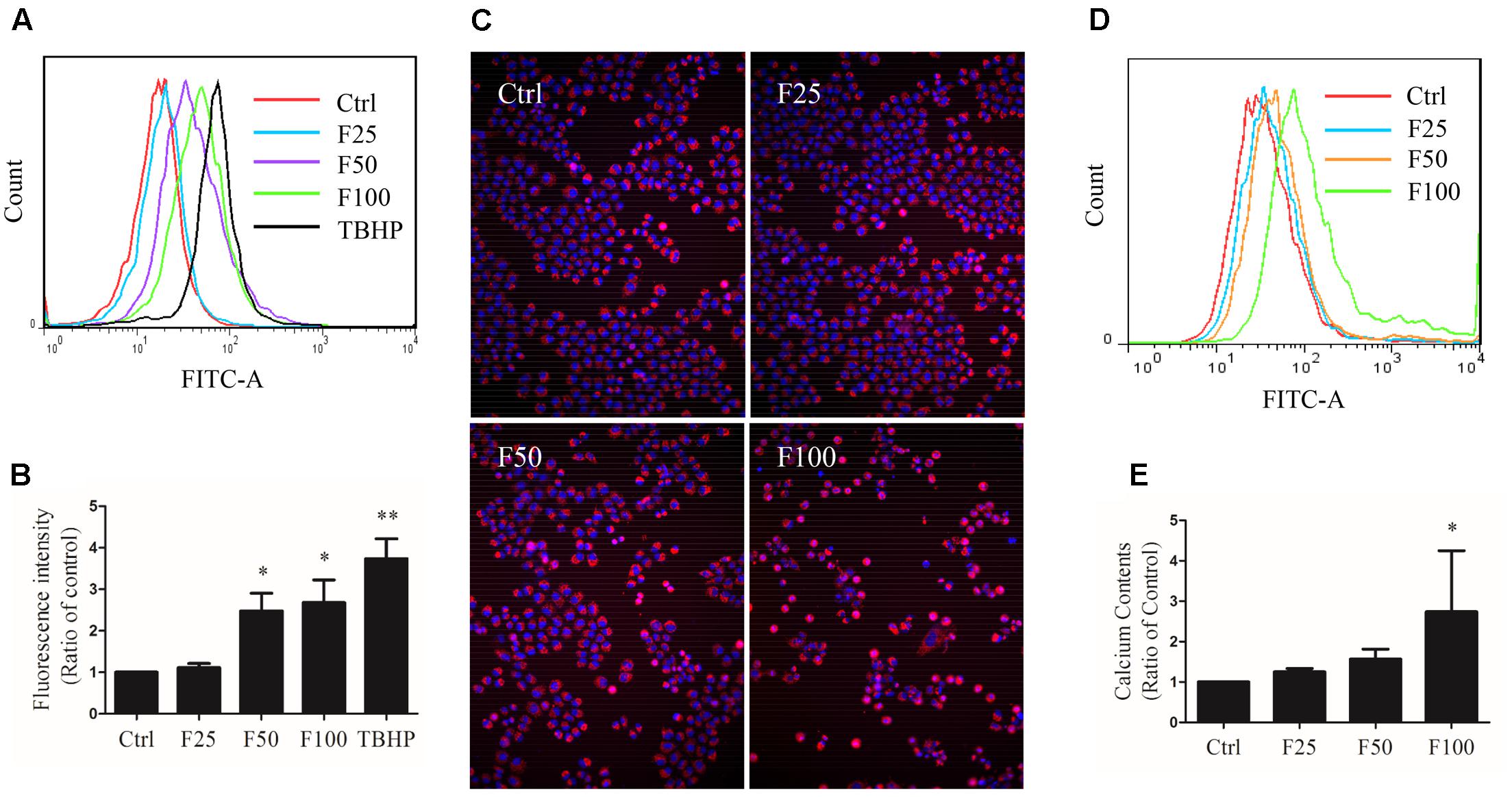
FIGURE 5. Effect of furanodiene (F) on intracellular reactive oxygen species (ROS) and calcium levels in doxorubicin-resistant MCF-7 breast cancer cells. (A) Cells were exposed to furanodiene (0–100 μM) for 2 h after a 30-min pre-incubation with H2DCF-DA. The fluorescence was detected using flow cytometry. tert-Butyl hydroperoxide (TBHP) treatment (300 μM) was used as a positive control. (B) Statistical result of (A). (C) Cells were exposed to furanodiene (0–100 μM) for 24 h followed by a 30-min incubation with CellROX Deep Red reagent and Hoechst 33342. The fluorescence was visualized at 100× magnification using high content imaging. (D) Cells were exposed to furanodiene (0–100 μM) for 2 h after a 30-min pre-incubation with Fluo-4 AM. The fluorescence was detected using flow cytometry. (E) Statistical result of (D). Ctrl stands for control. Data are expressed as mean ± SEM. ∗P < 0.05 and ∗∗P < 0.01 vs. control.
Furanodiene Regulated the Extrinsic Apoptosis Pathway in MCF-7/DOXR Cells
Compared to vehicle control, furanodiene treatment promoted the Caspase-8 activities in a dose-dependent manner (Figure 6A). Additionally, western blotting results also confirmed that Caspase-8 cleavage was induced by furanodiene (Figure 6B). Furthermore, furanodiene treatment induced a substantial TNF-α release from MCF-7/DOXR cells up to 1.5 folds compared with unstimulated cells. Meanwhile, no significant activation of TNF-α release was observed after doxorubicin treatment alone (Figure 6C). As shown in Figure 6B, furanodiene did not affect the protein expression of TNF-R1. A binding assay was performed to show that furanodiene was not a specific inhibitor of TNF-α-TNF-R1 binding (Figure 6D). Then, a TNF-α antagonist SPD304 was used to examine furanodiene-induced apoptotic cell death. Results showed that addition of SPD304 significantly rescued the survivability (Figures 7A,B) and attenuated the cell apoptosis (Figures 7C,D) of MCF-7/DOXR cells induced by furanodiene (100 μM). These findings suggest that furanodiene induces cell apoptosis through activating the extrinsic apoptosis pathway via inducing TNF-α release and activating Caspase-8.
FIGURE 6
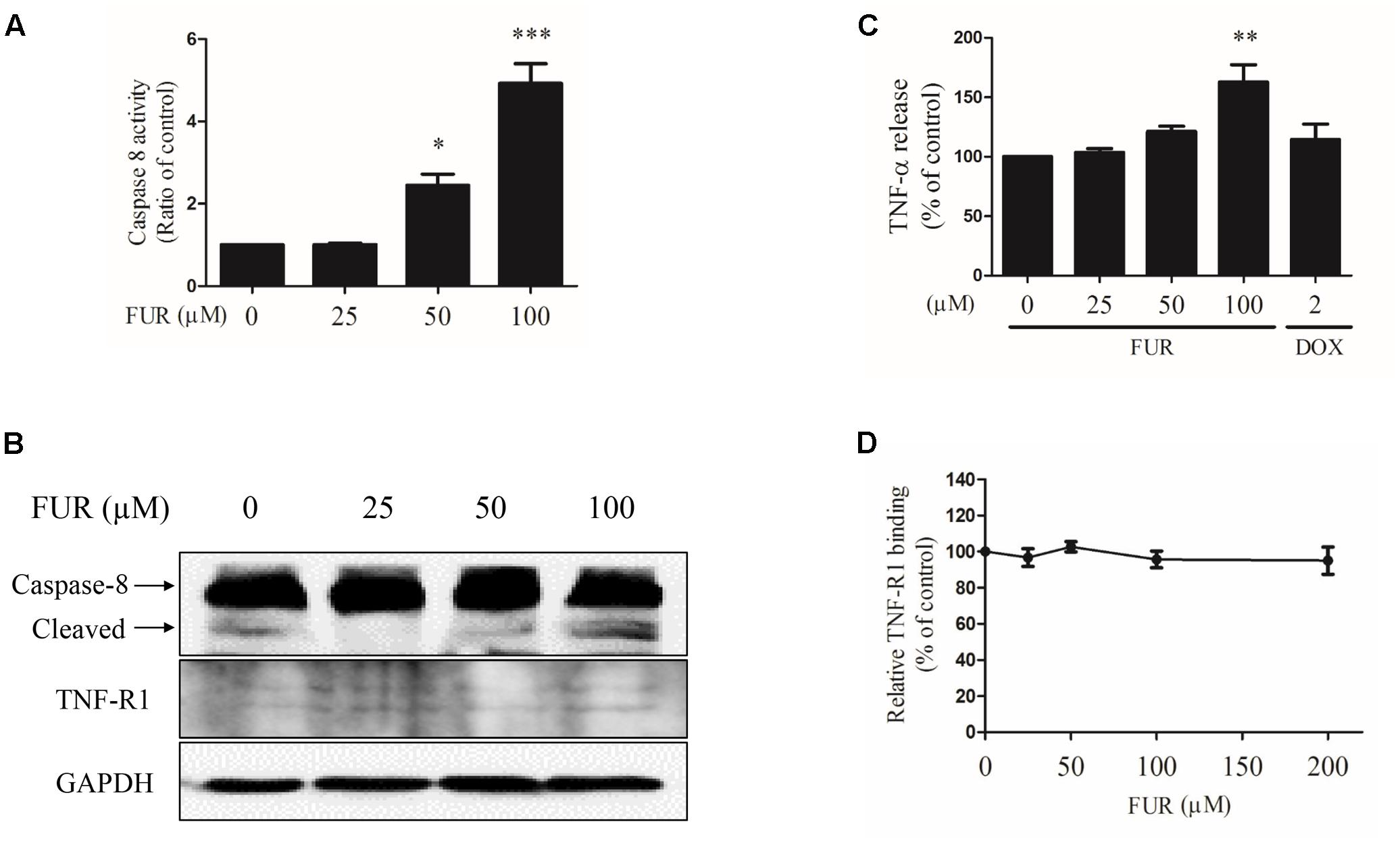
FIGURE 6. Effect of furanodiene (FUR) on the extrinsic pathway to trigger apoptosis in doxorubicin (DOX)-resistant MCF-7 breast cancer cells. Cells were treated with different concentrations of furanodiene (0–100 μM) or doxorubicin (2 μM) for 24 h. (A) Caspase activities were determined with Caspase-Glo assay kit. (B) Protein expression was determined by western blotting and GAPDH was used as a loading control. (C) Cells were treated with different concentrations of furanodiene (0–100 μM) or doxorubicin (2 μM) for 4 h. TNF-α release was determined with ELISA kit. (D) Microtiter plates coated with TNF-α were incubated with TNFR-1 together with furanodiene at the indicated concentrations (0–200 μM). TNF-R1 binding was detected using anti-TNF-R1 antibody and horseradish peroxidase-conjugated secondary antibody. Data are expressed as mean ± SEM. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 vs. control.
FIGURE 7
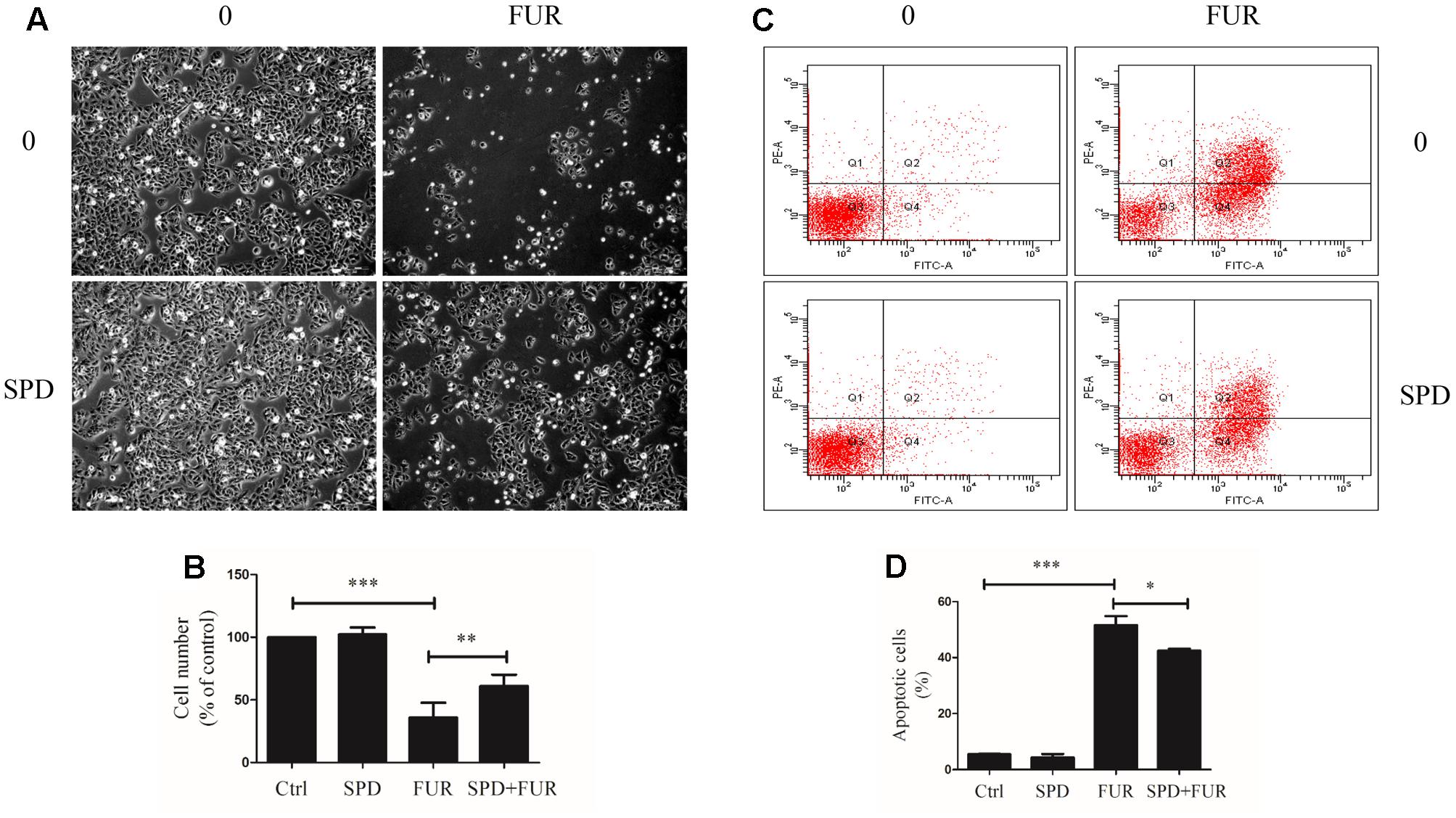
FIGURE 7. Effect of furanodiene (FUR) on TNF-α pathway trigger to cell death in doxorubicin-resistant MCF-7 breast cancer cells. Cells were pretreated with SPD304 (SPD, 5 μM) for 1 h before treatment with 100 μM of furanodiene for 24 h. (A) The morphology alteration and cell number were visualized using microscope at 100× magnifications. (B) Statistical result of (A). (C) Apoptotic cells were detected by Annexin V/PI assay. (D) Statistical result of (C). Ctrl stands for control. Data are expressed as mean ± SEM. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 vs. control.
Furanodiene Regulated the NF-κB Pathway in MCF-7/DOXR Cells
Furanodiene induced a substantial TNF-α release from MCF-7/DOXR cells, which could likely elicit TNF-α activation of downstream in cell apoptosis. Western blotting results showed that furanodiene increased p65 expression and induced IKKα/β phosphorylation, without changing the protein expression levels of IKKα or IKKβ with significance in MCF-7/DOXR cells (Figure 8A). MCF-7/DOXR cells were transiently co-transfected with the NF-κB-responsive luciferase reporter gene and the Renilla luciferase control reporter gene. Results showed that furanodiene (100 μM) treatment increased NF-κB transcriptional activity of the transfected cells up to 2.70 folds compared with vehicle control (Figure 8B). However, doxorubicin (2 μM) treatment just slightly induced NF-κB-driven luciferase reporter activity and IKKα/β phosphorylation without significantly affecting the protein expression levels of p65, IKKα, and IKKβ. Addition of SPD304 significantly attenuated PARP activation, p65 protein expression, and NF-κB transcriptional activity of transfected MCF-7/DOXR cells induced by furanodiene (100 μM) (Figures 8C,D).
FIGURE 8
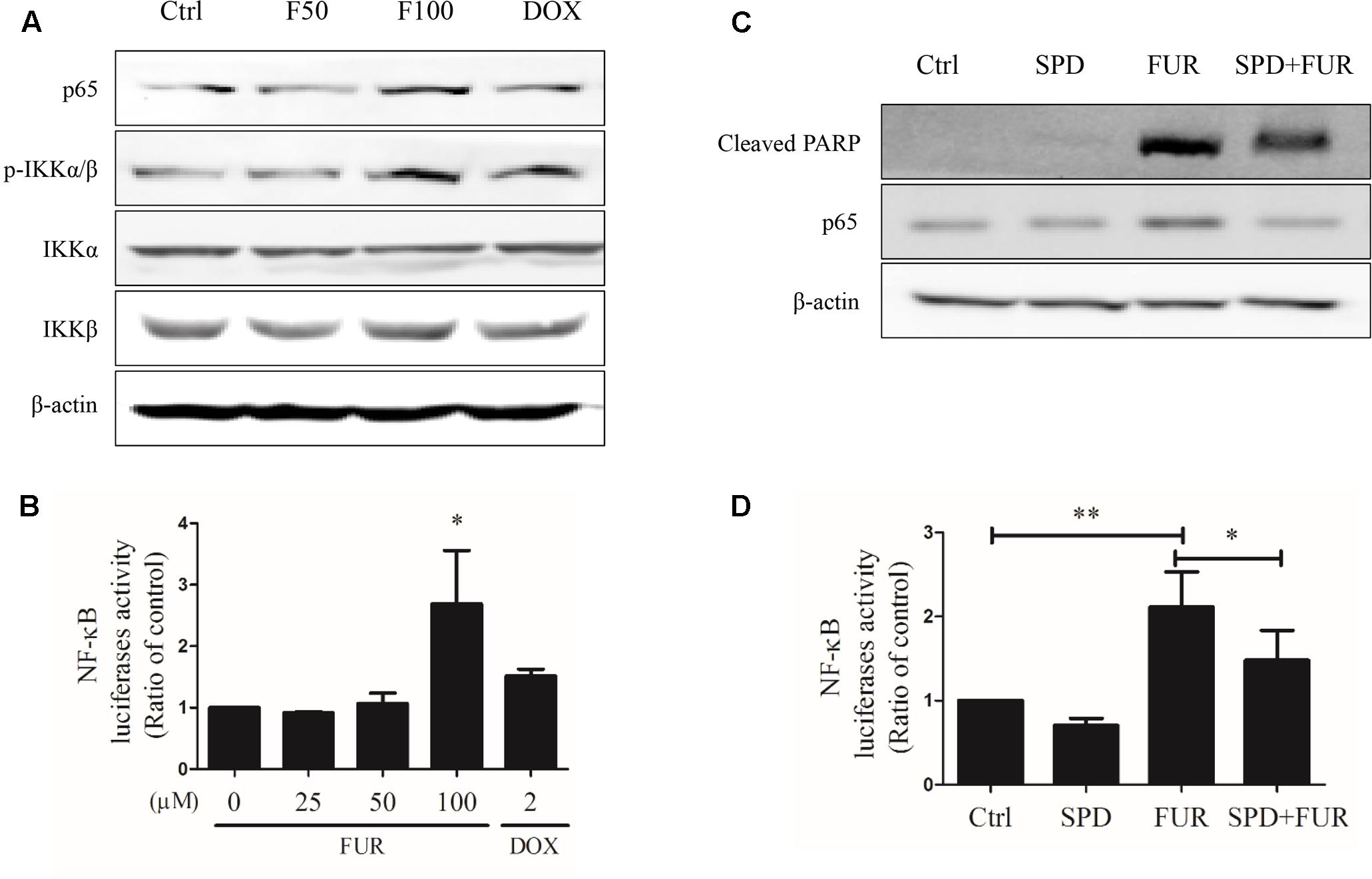
FIGURE 8. Effect of furanodiene (FUR or F) on NF-κB pathway in doxorubicin (DOX)-resistant MCF-7 breast cancer cells. (A) Cells were treated with different concentrations of furanodiene (0–100 μM) or doxorubicin (2 μM) for 24 h. Protein expression was determined by western blotting and β-actin was used as a loading control. (B) Cells transfected with NF-κB luciferase reporter gene were exposed to different concentrations of furanodiene (0–100 μM) or doxorubicin (2 μM) for 24 h. NF-κB transcription activity was detected with the dual-luciferase reporter system. (C) Cells were pretreated with SPD304 (SPD, 5 μM) for 1 h before treatment with furanodiene (100 μM) for 24 h. Protein expression was determined by western blotting and β-actin was used as a loading control. (D) Cells transfected with NF-κB luciferase reporter gene were pretreated with SPD304 (5 μM) for 1 h before treatment with furanodiene (100 μM) for 24 h. NF-κB transcription activity was detected with the dual-luciferase reporter system. Ctrl stands for control. Data are expressed as mean ± SEM. ∗P < 0.05 and ∗∗P < 0.01 vs. control.
Knockdown of NF-κB/p65 Attenuated the Pro-apoptotic Effects of Furanodiene
At the start of this experiment, western blotting results showed that p65 expression was knocked down in MCF-7/DOXR cells transfected with si-NF-κB/p65 compared with si-MOCK. Notably, knockdown of p65 expression could not affect β-actin expression (Figure 9A) and p65-knockdown cells were still resistant to doxorubicin, with an IC50 value of 71.44 μM (Figure 9B). Annexin V/PI staining results confirmed that knockdown of p65 expression significantly enhanced the pro-apoptotic effect of furanodiene (Figures 9C,D). Furanodiene could not induce p65 expression up-regulation, but enhanced PARP activation in MCF-7/DOXR cells transfected with si-NF-KB/p65, compared with the cells transfected with si-MOCK (Figure 9E).
FIGURE 9
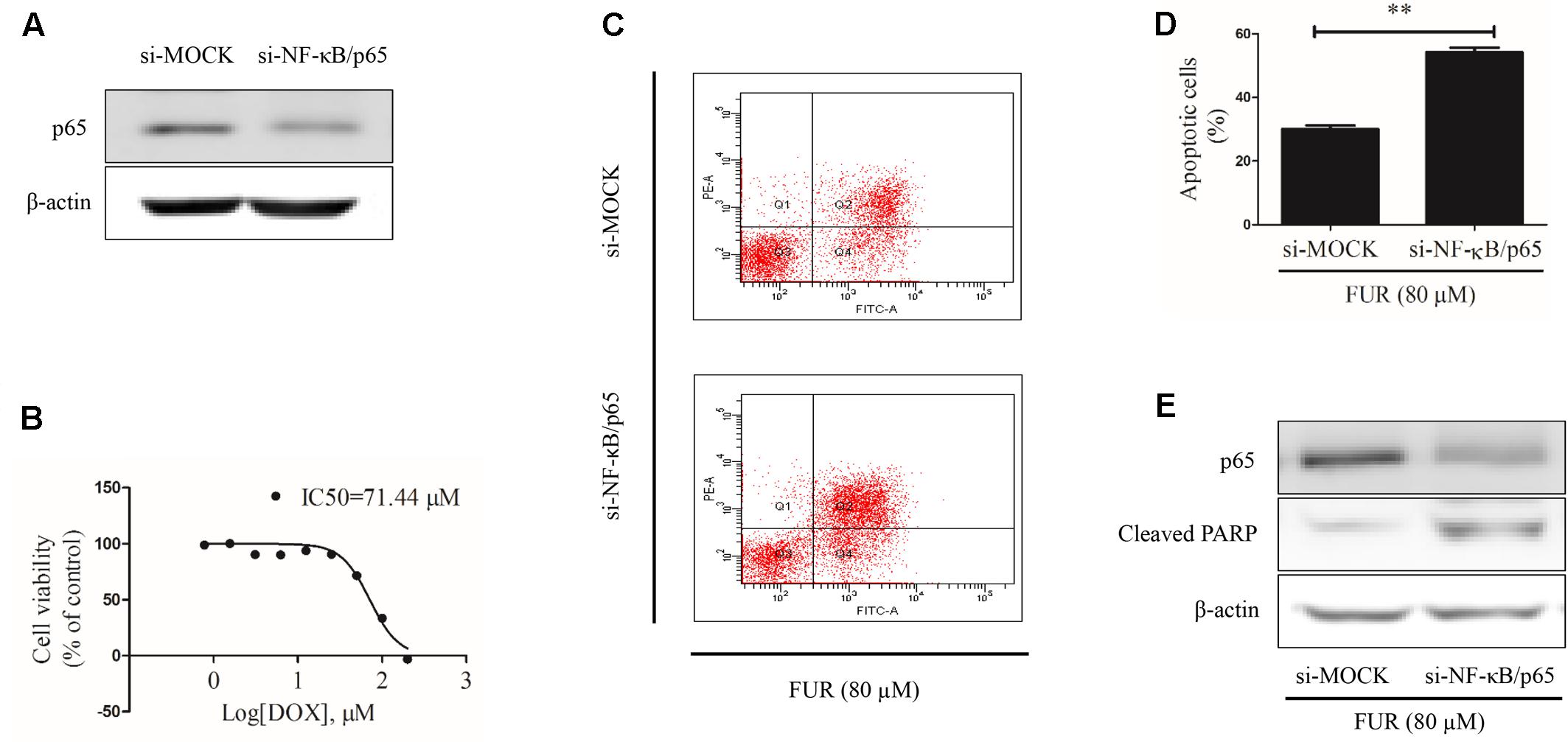
FIGURE 9. Effect of furanodiene (FUR) on cell apoptosis in NF-κB/p65 knockdown doxorubicin (DOX)-resistant MCF-7 breast cancer cells. (A) Cells were transfected with siRNAs using Lipofectamine 2000. The transfection efficacy of siRNAs was confirmed by western blotting. (B) NF-κB knockdown cells were exposed to different concentrations of doxorubicin for 48 h. Cell viability was detected by MTT assay. (C) Transfected cells were exposed to furanodiene (80 μM) for 24 h. Apoptotic cells were detected by Annexin V/PI assay. (D) Statistical result of (C). (E) Cells were treated with furanodiene (80 μM) for 24 h after transfection with siRNAs. Protein expression was measured by western blotting and β-actin was used as a loading control. Data are expressed as mean ± SEM. ∗∗P < 0.01 vs. control.
Discussion and Conclusion
Our results show that furanodiene preferentially causes cancer cell death through inducing cell apoptosis via extrinsic- and intrinsic-dependent pathways and NF-κB-independent pathway in doxorubicin-resistant breast cancer cells (Figure 10). The intrinsic apoptosis pathway is triggered by furanodiene, as demonstrated by alteration of the mitochondrial membrane potential, decrease of anti-apoptotic regulators such as Bcl-xL, increase of pro-apoptotic regulators such as Bad, activation of Caspase-7 and PARP, and promotion of ROS and calcium generation. Inhibition of caspase activations by inactivating caspase components significantly abolish furanodiene-induced cell death. Unexpectedly, addition of anti-oxidants cannot attenuate cell viability induced by furanodiene. We speculate that furandiene also induces cell apoptosis through activation of other pathways, so we focused on the extrinsic apoptosis pathway induced by furanodiene. Our results show that furanodiene-induced cell apoptosis is correlated with activation of Caspase-8 and accumulation of TNF-α release, and is not directly associated with TNF-R1 expression and TNF-α/TNF-R1 binding. Furthermore, addition of TNF-α antagonist significantly rescued cell survival and attenuated cell apoptosis induced by furanodiene. These observations suggest that furanodiene functions on cell death, at least in part, by interfering with the intrinsic and extrinsic apoptosis pathways.
FIGURE 10
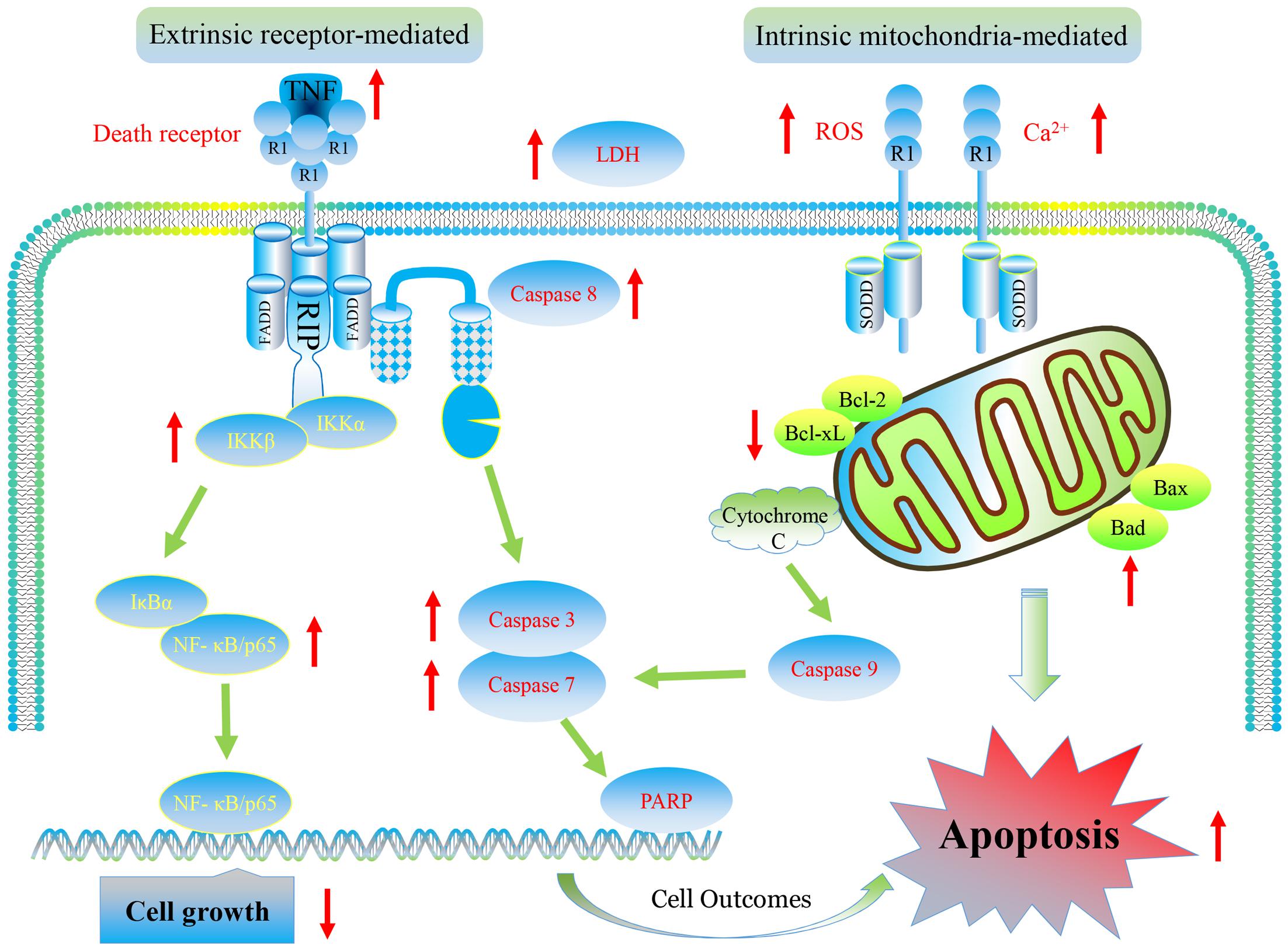
FIGURE 10. Graphic representation of pathways for furanodiene-induced cell apoptosis and for doxorubicin-resistant MCF-7 cells outcomes. Furanodiene preferentially causes cancer cell death through inducing cell apoptosis via extrinsic- and intrinsic-dependent pathways and NF-κB-independent pathway in doxorubicin-resistant MCF-7 breast cancer cells.
The extrinsic apoptosis pathway is triggered by a death ligand binding to a death receptor, such as TNF-α to TNFR. Previous research identified that the binding of TNFR and TNF-α activates NF-κB pathway, which favored both cell survival and apoptosis, depending on the cell type and biological context (Hehlgans and Pfeffer, 2005). In our study, furanodiene treatment increases NF-κB transcriptional activity in MCF-7/DOXR cells, and addition of TNF-α antagonist significantly attenuates NF-κB transcriptional activity induced by furanodiene. We can conclude that NF-κB activation is potentially attributed to furanodiene-induced TNF-α increase in MCF-7/DOXR cells. Many studies have explored the relationship between NF-κB expression and prognosis in solid tumors, but the conclusion remains elusive. Wu’s group performed a meta-analysis to demonstrate that NF-κB expression has strong connections with poor prognosis in most solid tumors irrespective of NF-κB localization (Wu et al., 2015). Interestingly, furanodiene treatment also significantly increases the NF-κB/p65 expression and IKKα/β phosphorylation. Knockdown of NF-κB/p65 significantly sensitizes MCF-7/DOXR cells to furanodiene on cell apoptosis, indicating that furanodiene-induced cell apoptosis is dependent on TNF-α pathway and independent on NF-κB pathway. It also suggests that NF-κB inhibitors can be used to enhance furanodiene-induced cancer cell death. Previous studies indicated that doxorubicin induced drug resistance via up-regulating NF-κB expression in MCF-7 human breast cancer cells (Fang et al., 2014), and many agents targeting NF-κB exerted anti-tumorigenic activity in doxorubicin-resistant MCF-7 breast cancer cells (Tran et al., 2013; Meiyanto et al., 2014). For instance, curcumin and mollugin acquire reversal of chemo-resistance by inhibiting the NF-κB signaling pathway. In contrast, furanodiene failings to inhibit the NF-κB pathway may be attributed to TNF-α activation in the doxorubicin-resistant MCF-7 cells. Therefore, furanodiene, could be combined with NF-κB inhibitors (from natural products or chemical synthesis) to overcome chemo-drug resistance.
Our findings prompt that furanodiene displays anti-cancer effects through inducing cell apoptosis via targeting multiple signaling pathways and may be developed as a promising natural product for multidrug-resistant cancer therapy in the future.
Author Contributions
Z-FZ designed and conducted the experiments, and wrote the manuscript. H-BY and D-QL designed the experiments and revised the manuscript. C-MW, W-AQ, S-PW, J-MZ, HY, LC, and TW gave some data interpretations and provided valuable feedback to this conception. Y-TW organized, conceived, and supervised the study. All authors read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Macao Science and Technology Development Fund (080/2016/A2 and 126/2016/A3), the Research Fund of University of Macau (MYRG2016-00143-ICMS-QRCM and MYRG2016-00031-ICMS-QRCM), the National Natural Science Foundation of China (Nos: 81603066, 81470170, and 81273166), the International Technology Cooperation Projects of Dongguan (20135081520017), the Features Innovative Projects of Key Platform and Major Scientific Research Project of Universities in Guangdong Province (2015KTSCX048), and the Technology Plan Projects of Medical and Health Science in Dongguan City (201605101290). Wen-An Qiang was partially supported by NU-PSOC (U54 CA193419) and Robert H. Lurie Comprehensive Cancer Center OncoSET initiatives at the Northwestern University. We also thank Stacy Ann Kujawa in Northwestern University Feinberg School of Medicine for critical proofreading of the manuscript.
References
Baldovini, N., Tomi, F., and Casanova, J. (2001). Identification and quantitative determination of furanodiene, a heat-sensitive compound, in essential oil by 13C-NMR. Phytochem. Anal. 12, 58–63. doi: 10.1002/1099-1565(200101/02)12:1<58::AID-PCA559>3.0.CO;2-9
Biedler, J. L., and Riehm, H. (1970). Cellular resistance to actinomycin D in Chinese hamster cells in vitro: cross-resistance, radioautographic, and cytogenetic studies. Cancer Res. 30, 1174–1184.
Chen, G., and Goeddel, D. V. (2002). TNF-R1 signaling: a beautiful pathway. Science 296, 1634–1635. doi: 10.1126/science.1071924
Czabotar, P. E., Lessene, G., Strasser, A., and Adams, J. M. (2014). Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63. doi: 10.1038/nrm3722
Fang, X. J., Jiang, H., Zhu, Y. Q., Zhang, L. Y., Fan, Q. H., and Tian, Y. (2014). Doxorubicin induces drug resistance and expression of the novel CD44st via NF-κB in human breast cancer MCF-7 cells. Oncol. Rep. 31, 2735–2742. doi: 10.3892/or.2014.3131
Fulda, S., and Debatin, K. M. (2006). Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25, 4798–4811. doi: 10.1038/sj.onc.1209608
Hehlgans, T., and Pfeffer, K. (2005). The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology 115, 1–20. doi: 10.1111/j.1365-2567.2005.02143.x
Kaye, S. B. (1988). The multidrug resistance phenotype. Br. J. Cancer 58, 691–694. doi: 10.1038/bjc.1988.291
Leung, C. H., Zhong, H. J., Yang, H., Cheng, Z., Chan, D. S., Ma, V. P., et al. (2012). A metal-based inhibitor of tumor necrosis factor-α. Angew. Chem. Int. Ed. Engl. 51, 9010–9014. doi: 10.1002/anie.201202937
Ma, E., Wang, X., Li, Y., Sun, X., Tai, W., Li, T., et al. (2008). Induction of apoptosis by furanodiene in HL60 leukemia cells through activation of TNFR1 signaling pathway. Cancer Lett. 271, 158–166. doi: 10.1016/j.canlet.2008.06.008
Meiyanto, E., Putri, D. D., Susidarti, R. A., Murwanti, R., Sardjiman, Fitriasari, A., et al. (2014). Curcumin and its analogues (PGV-0 and PGV-1) enhance sensitivity of resistant MCF-7 cells to doxorubicin through inhibition of HER2 and NF-κB activation. Asian Pac. J. Cancer Prev. 15, 179–184. doi: 10.7314/APJCP.2014.15.1.179
Quan, Y., Xia, L., Shao, J., Yin, S., Cheng, C. Y., Xia, W., et al. (2015). Adjudin protects rodent cochlear hair cells against gentamicin ototoxicity via the SIRT3-ROS pathway. Sci. Rep. 5:8181. doi: 10.1038/srep08181
Reed, J. C. (2003). Apoptosis-targeted therapies for cancer. Cancer Cell 3, 17–22. doi: 10.1016/S1535-6108(02)00241-6
Strasser, A., O’connor, L., and Dixit, V. M. (2000). Apoptosis signaling. Annu. Rev. Biochem. 69, 217–245. doi: 10.1146/annurev.biochem.69.1.217
Sun, X. Y., Zheng, Y. P., Lin, D. H., Zhang, H., Zhao, F., and Yuan, C. S. (2009). Potential anti-cancer activities of Furanodiene, a Sesquiterpene from Curcuma wenyujin. Am. J. Chin. Med. 37, 589–596. doi: 10.1142/S0192415X09007077
Tran, T. P., Kim, H. G., Choi, J. H., Na, M. K., and Jeong, H. G. (2013). Reversal of P-glycoprotein-mediated multidrug resistance is induced by mollugin in MCF-7/adriamycin cells. Phytomedicine 20, 622–631. doi: 10.1016/j.phymed.2013.01.014
Vtorushin, S. V., Khristenko, K. Y., Zavyalova, M. V., Perelmuter, V. M., Litviakov, N. V., Denisov, E. V., et al. (2014). The phenomenon of multi-drug resistance in the treatment of malignant tumors. Exp. Oncol. 36, 144–156.
Wu, D., Wu, P., Zhao, L., Huang, L., Zhang, Z., Zhao, S., et al. (2015). NF-κB expression and outcomes in solid tumors: a systematic review and meta-analysis. Medicine 94:e1687. doi: 10.1097/MD.0000000000001687
Wu, Q., Yang, Z., Nie, Y., Shi, Y., and Fan, D. (2014). Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 347, 159–166. doi: 10.1016/j.canlet.2014.03.013
Xiao, Y., Yang, F. Q., Li, S. P., Gao, J. L., Hu, G., Lao, S. C., et al. (2007). Furanodiene induces G2/M cell cycle arrest and apoptosis through MAPK signaling and mitochondria-caspase pathway in human hepatocellular carcinoma cells. Cancer Biol. Ther. 6, 1044–1050. doi: 10.4161/cbt.6.7.4317
Xu, W. S., Li, T., Wu, G. S., Dang, Y. Y., Hao, W. H., Chen, X. P., et al. (2014). Effects of furanodiene on 95-D lung cancer cells: apoptosis, autophagy and G1 phase cell cycle arrest. Am. J. Chin. Med. 42, 243–255. doi: 10.1142/S0192415X14500165
Zhong, Z., Chen, X., Tan, W., Xu, Z., Zhou, K., Wu, T., et al. (2011). Germacrone inhibits the proliferation of breast cancer cell lines by inducing cell cycle arrest and promoting apoptosis. Eur. J. Pharmacol. 667, 50–55. doi: 10.1016/j.ejphar.2011.03.041
Zhong, Z., Dang, Y., Yuan, X., Guo, W., Li, Y., Tan, W., et al. (2012a). Furanodiene, a natural product, inhibits breast cancer growth both in vitro and in vivo. Cell Physiol. Biochem. 30, 778–790. doi: 10.1159/000341457
Zhong, Z., Liu, L. J., Dong, Z. Q., Lu, L., Wang, M., Leung, C. H., et al. (2015). Structure-based discovery of an immunomodulatory inhibitor of TLR1-TLR2 heterodimerization from a natural product-like database. Chem. Commun. 51, 11178–11181. doi: 10.1039/c5cc02728d
Zhong, Z., Tan, W., Chen, X., and Wang, Y. (2014). Furanodiene, a natural small molecule suppresses metastatic breast cancer cell migration and invasion in vitro. Eur. J. Pharmacol. 737, 1–10. doi: 10.1016/j.ejphar.2014.04.043
Zhong, Z. F., Hoi, P. M., Wu, G. S., Xu, Z. T., Tan, W., Chen, X. P., et al. (2012b). Anti-angiogenic effect of furanodiene on HUVECs in vitro and on zebrafish in vivo. J. Ethnopharmacol. 141, 721–727. doi: 10.1016/j.jep.2011.08.052
Zhong, Z. F., Li, Y. B., Wang, S. P., Tan, W., Chen, X. P., Chen, M. W., et al. (2012c). Furanodiene enhances tamoxifen-induced growth inhibitory activity of ERa-positive breast cancer cells in a PPARγ independent manner. J. Cell. Biochem. 113, 2643–2651. doi: 10.1002/jcb.24139
Zhong, Z. F., Qiang, W. A., Wang, C. M., Tan, W., and Wang, Y. T. (2016a). Furanodiene enhances the anti-cancer effects of doxorubicin on ERα-negative breast cancer cells in vitro. Eur. J. Pharmacol. 774, 10–19. doi: 10.1016/j.ejphar.2015.11.039
Keywords: furanodiene, multidrug resistance, breast cancer, cell apoptosis, intrinsic/extrinsic apoptosis, NF-κB
Citation: Zhong Z-F, Yu H-B, Wang C-M, Qiang W-A, Wang S-P, Zhang J-M, Yu H, Cui L, Wu T, Li D-Q and Wang Y-T (2017) Furanodiene Induces Extrinsic and Intrinsic Apoptosis in Doxorubicin-Resistant MCF-7 Breast Cancer Cells via NF-κB-Independent Mechanism. Front. Pharmacol. 8:648. doi: 10.3389/fphar.2017.00648
Received: 21 April 2017; Accepted: 31 August 2017;
Published: 14 September 2017.
Edited by:
Kalin Yanbo Zhang, University of Hong Kong, Hong KongReviewed by:
Ouyang Chen, Second Military Medical University, ChinaJohanna Mahwahwatse Bapela, University of Pretoria, South Africa
Copyright © 2017 Zhong, Yu, Wang, Qiang, Wang, Zhang, Yu, Cui, Wu, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi-Tao Wang, ytwang@umac.mo De-Qiang Li, deqli@163.com Zhang-Feng Zhong, zfzhong@aliyun.com
†These authors have contributed equally to this work.