 Michelle F. Corrêa1†
Michelle F. Corrêa1† Álefe J. R. Barbosa1†
Álefe J. R. Barbosa1† Larissa B. Teixeira2Diego A. Duarte2
Larissa B. Teixeira2Diego A. Duarte2 Sarah C. Simões2
Sarah C. Simões2 Lucas T. Parreiras-e-Silva2Aleksandro M. Balbino1
Lucas T. Parreiras-e-Silva2Aleksandro M. Balbino1 Richardt G. Landgraf1
Richardt G. Landgraf1 Michel Bouvier3
Michel Bouvier3 Claudio M. Costa-Neto2*
Claudio M. Costa-Neto2* João P. S. Fernandes1*
João P. S. Fernandes1*- 1Departamento de Ciências Farmacêuticas, Universidade Federal de São Paulo, Diadema, Brazil
- 2Departamento de Bioquímica e Imunologia, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, Brazil
- 3Department of Biochemistry and Molecular Medicine, Institute for Research in Immunology and Cancer, University of Montréal, Montréal, QC, Canada
The histamine receptors (HRs) are traditional G protein-coupled receptors of extensive therapeutic interest. Recently, H3R and H4R subtypes have been targeted in drug discovery projects for inflammation, asthma, pain, cancer, Parkinson’s, and Alzheimer’s diseases, which includes searches for dual acting H3R/H4R ligands. In the present work, nine 1-[(2,3-dihydro-1-benzofuran-2-yl)methyl]piperazine (LINS01 series) molecules were synthesized and evaluated as H3R and H4R ligands. Our data show that the N-allyl-substituted compound LINS01004 bears the highest affinity for H3R (pKi 6.40), while the chlorinated compound LINS01007 has moderate affinity for H4R (pKi 6.06). In addition, BRET assays to assess the functional activity of Gi1 coupling indicate that all compounds have no intrinsic activity and act as antagonists of these receptors. Drug-likeness assessment indicated these molecules are promising leads for further improvements. In vivo evaluation of compounds LINS01005 and LINS01007 in a mouse model of asthma showed a better anti-inflammatory activity of LINS01007 (3 g/kg) than the previously tested compound LINS01005. This is the first report with functional data of these compounds in HRs, and our results also show the potential of their applications as anti-inflammatory.
Introduction
Histamine is one of the most important chemical transmitters involved in several biological processes. It has been widely studied since its discovery1-3 due to its role in inflammatory and allergic reactions, but it is also involved in the regulation of gastric acid secretion, sleep, mood, and food intake (Passani and Blandina, 2011). These processes are triggered by the interaction of histamine with its receptors (HRs), named H1R, H2R, H3R, and H4R. HRs are members of the G protein-coupled receptor (GPCR) class A family, which are also known as 7 transmembrane (7TM) receptors (Seifert et al., 2011; Corrêa and Fernandes, 2015).
The H3R is mainly found in CNS, and is involved in the inhibition of histamine release, as well as in the inhibition of other neurotransmitters, such as norepinephrine, acetylcholine, and serotonin (Leurs et al., 2005). Therefore, H3R modulates neurotransmitters release by acting as a presynaptic receptor (auto- and hetero-receptor). It has been previously reported that signal transduction by H3R occurs mainly by activation of Gi/o proteins, leading to decrease of intracellular cAMP concentration and decrease of Ca2+ influx in neurons (Tiligada et al., 2009). As a result of its actions, H3R could be involved in several neurological disorders, including cognitive, convulsive, and sleep–wake disorders, as well as in obesity (Łażewska et al., 2014). Therefore, H3R ligands could be promising drugs to treat these conditions. H3R ligands have been widely explored. Recently, the novel H3R antagonist pitolisant has been approved for treatment of narcolepsy, highlighting the importance of this receptor as a new target for treatment of certain CNS disorders (Sayed, 2016). Ciproxifan and ABT-239 (Figure 1) are other examples of such well-studied compounds.
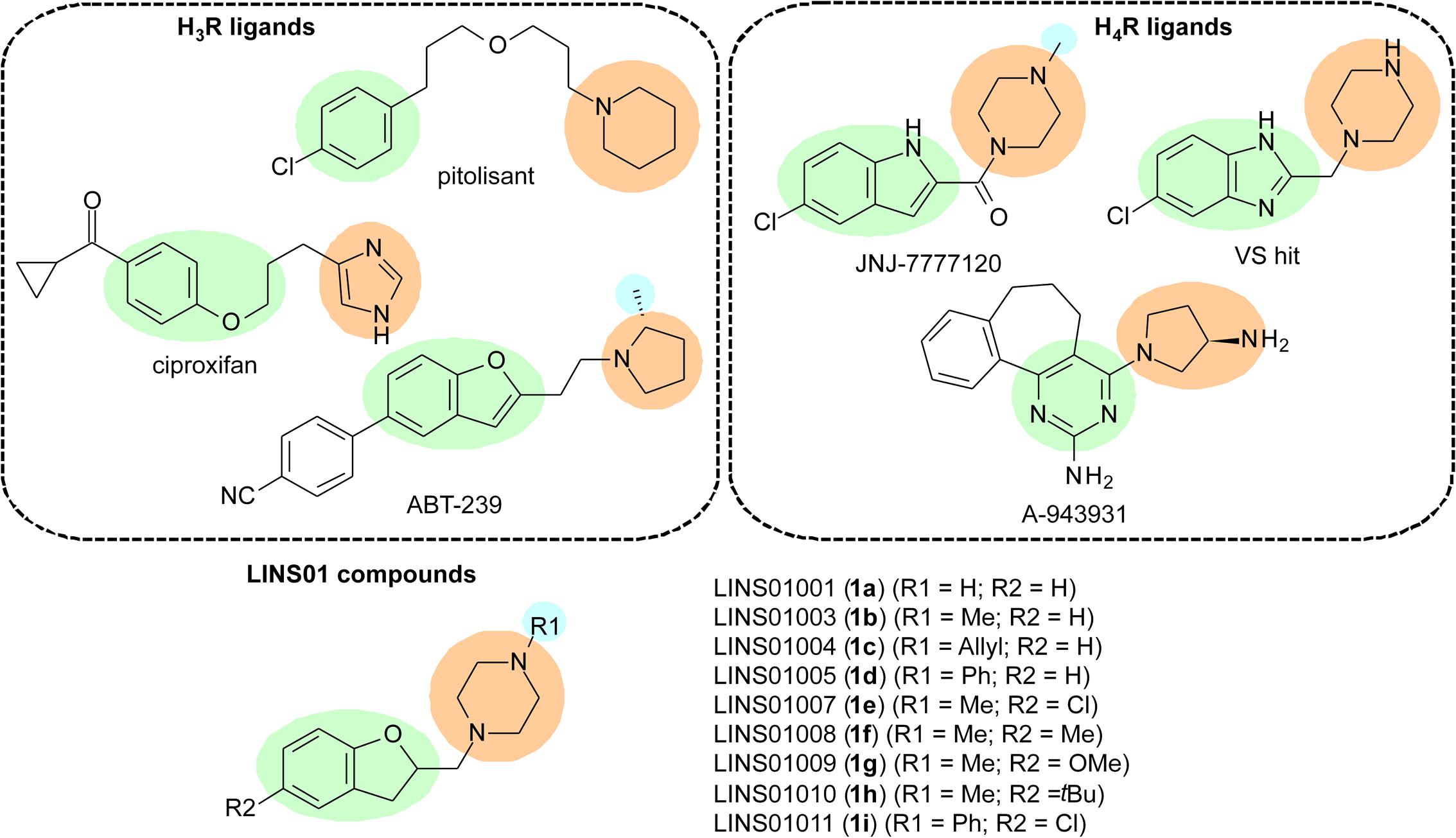
FIGURE 1. Characteristics considered in the design of LINS compounds 1a–i; green – aromatic core; orange – polar group; blue – additional lipophilic group.
In the early 2000s, several research groups described a new isoform of HR, expressed in immune cells (eosinophils, basophils, mast cells, NK cells, DCs, monocytes, and T cells), which is involved in the modulation of chemotaxis as well as other functions (Oda et al., 2000; Liu et al., 2001). This receptor was named H4R, is also coupled to Gi/o proteins (Corrêa and Fernandes, 2015), and bears a considerable sequence identity to H3R (∼31% total, 54% in TM domains) (Oda et al., 2000; Liu et al., 2001). Considering the physico-chemical properties of the different amino acids, this similarity in TM domains can reach 68% (de Esch et al., 2005). Since these cells are involved in immune and inflammatory processes, H4R is considered a promising target to future anti-inflammatory and modulatory immunological drugs. H4R ligands have been designed and evaluated as drug candidates (Figure 1), with the indolecarboxamides (JNJ-7777120) and 2-aminopyrimidines (A-943931) being the most explored chemotypes in this regard. Some molecules lacking the carbonyl group from the indolecarboxamides were also identified as H4R ligands in virtual screening experiments (Christopher et al., 2012).
Due to the high sequence identity between H3R and H4R, it is likely that several compounds that bind to one of them can present considerable affinity for the other. Hence, some pharmacophore templates to H3R ligands can also be applied to H4R ligands (Kottke et al., 2011). In fact, the search for selective ligands that are able to discriminate one of those receptors is frequently reported in the literature, and most medicinal chemists working with HR-ligand research have such selectivity parameter as one of the most important goals. However, dual-acting H3R/H4R ligands may also present therapeutic potential in determined pathological conditions, such as neuropathic pain (Smith et al., 2007), cancer (Medina et al., 2008), and have also been reported in Parkinson’s disease (Shan et al., 2015a,b). Such potential applications were already reported for imidazole-containing H3R/H4R ligands, such as imetit, immepip, clobenpropit, and thioperamide.
To the best of our knowledge, the functional activity of dihydrobenzofuran-containing molecules have not yet evaluated in the H3R and H4R until the present report. The affinities to these receptors were evaluated as well. Considering the chemical similarities observed for several H3R and H4R ligands, we have designed a set of 1-[(2,3-dihydro-1-benzofuran-2-yl)methyl]piperazines (LINS01 series – 1a–i), evaluated their affinity and selectivity for those receptors, and characterized their functional activities. Drug-likeness was also assessed to define some structure-activity relationship (SAR) roles in this set of compounds, and an evaluation of the anti-inflammatory potential of a selected molecule was carried out in a mouse asthma model.
Materials and Methods
Reagents and starting materials were obtained from commercial suppliers (Sigma–Aldrich Co., Saint Louis, MO, United States; LabSynt Co., Diadema, Brazil) and used without further purification. 1H and 13C NMR spectra were recorded in a Bruker Ultrashield 300 spectrometer, operating at 300 and 75 MHz, respectively, using CDCl3 as solvent with TMS as internal standard. Chemical shifts are reported in parts per million (ppm, δ units). Coupling constants (J) are reported in units of hertz (Hz), if applicable. The high resolution mass spectra (HRMS) were obtained through direct injection after electron-spray ionization in positive mode (ESI+) in a MicroTOF from Bruker Daltonics mass spectrometer. Gas chromatography coupled to mass spectrometer (GC-MS) analysis were done in a Shimadzu GC-2010 coupled to mass spectrometer GCMS-QP2010 plus, using helium as carrier gas in a silica capillary column. The low resolution mass spectra (LRMS) were obtained through electron impact ionization (70 eV). The ion-radical and its fragments are reported in mass/charge ratio (m/z). The purity (>95%) for the final compounds 1a–i were determined and confirmed (H2O/MeOH 50%) by HPLC in a C-18 column coupled to UV detector (254 nm). Only compounds with >95% purity were considered to the biological assays. The yields for each step are summarized in Table 1. Spectral data and characterization for the intermediates can be found in Supplementary Information.
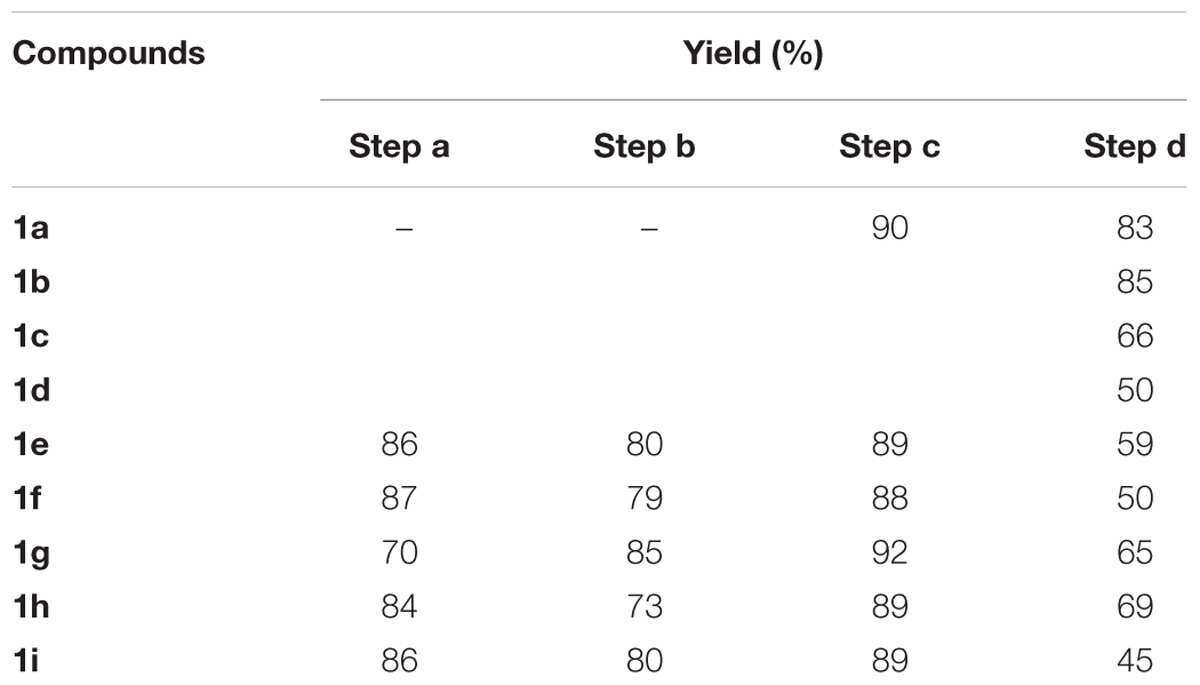
TABLE 1. Yields obtained for the synthesis of compounds 1a–i.
Synthetic Procedure for Intermediates 2–5
The intermediate compounds (Figure 2 and Supplementary Figure S1) were synthesized according to previously reported procedures from our group (Corrêa et al., 2016, 2017). The experimental details can be found in the Supplementary Information.
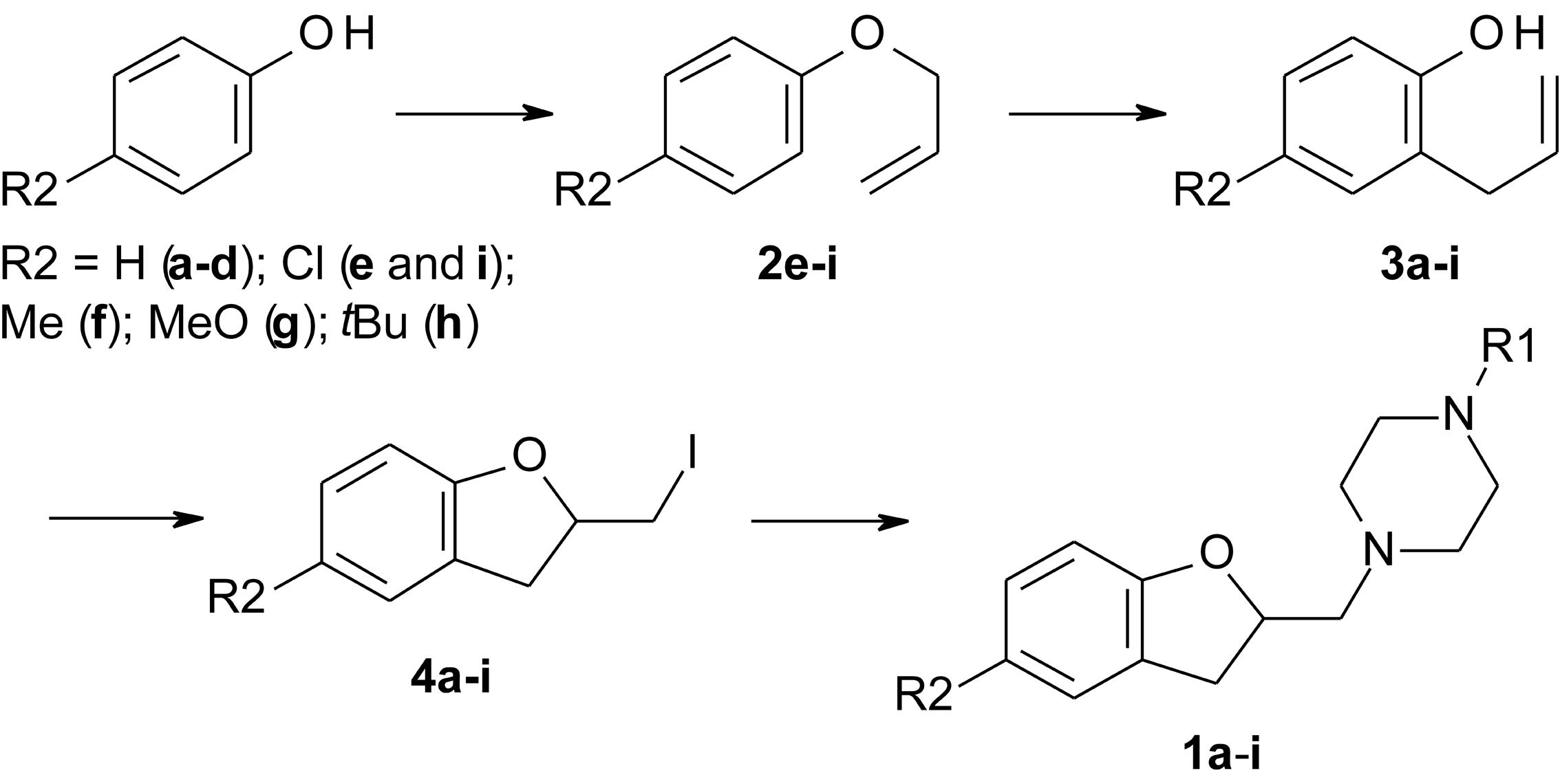
FIGURE 2. Reaction scheme for the synthesis of LINS01 compounds.
General Procedure for the Synthesis of Final Products (1a–i)
In a round-bottom flask, were added 2 mmol of corresponding 2-iodomethyl-2,3-dihydrobenzofuran (4a–h), 2.8 mmol (0.379 g) of K2CO3, and 8 mmol of corresponding 1-substituted piperazine in 15 mL of tetrahydrofuran. The mixture was reacted at 70°C for 18–24 h. The reactional mixture was filtered, the solvent removed under vacuum, and the residue was taken up in 1 M HCl solution (pH < 2), and washed with 2 × 10 mL of hexane. The aqueous phase was alkalinized (pH > 12) with 1 M NaOH solution, and extracted 3 × 10 mL of ethyl acetate. The organic layer was dried with anhydrous Na2SO4 and filtered. The solution was filtered over silica gel to remove any piperazine remaining, and the solvent removed under vacuum. If necessary, column chromatography in silica gel was used to purify the compounds, using dichloromethane:methanol (8:1) as eluent (Figure 2).
1-[(2,3-Dihydro-1-benzofuran-2-yl)methyl]piperazine (1a). Yellowish solid. mp 118–121°C. 1H NMR (CDCl3, 300 MHz): δ 2.45–2.70 (m, 8H), 2.79 (dd, 1H, J = 13.2, 7.7 Hz), 2.88–3.00 (m, 2H), 3.27 (dd, 1H, J = 15.7, 9.3 Hz), 4.89–5.04 (m, 1H), 6.77–6.88 (m, 2H), 7.08–7.20 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 34.3, 51.4, 53.9, 63.4, 80.8, 109.6, 120.3, 124.9, 126.5, 127.9, 159.6. LRMS (EI) m/z (rel %): 218 (15) [M+], 99 (100), 70 (26). HRMS (ESI+) for C13H18N2O [M+H]+: calcd 219.1493; found 219.1523.
1-[(2,3-dihydro-1-benzofuran-2-yl)methyl]-4-methylpiperazine (1b). Yellowish solid. mp 45–49°C. 1H NMR (CDCl3, 300 MHz): δ 2.30 (s, 3H), 2,49 (br.s, 4H), 2.57 (dd, 1H, J = 13.4, 4.4 Hz), 2.63 (br.s, 4H), 2.79 (dd, 1H, J = 13.4, 7.8 Hz), 2.94 (dd, 1H, J = 15.6, 7.8 Hz), 3.26 (dd, 1H, J = 15.6, 9.2 Hz), 4.95 (dq, 1H, J = 9.0, 3.9 Hz), 6.75–6.85 (m, 2H), 7.06–7.20 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 34.2, 46.0, 53.8, 55.0, 63.2, 80.8, 109.6, 120.3, 124.9, 126.4, 128.0, 159.5. LRMS (EI) m/z (rel %): 232 (18) [M+], 113 (95), 70 (100). HRMS (ESI+) for C14H20N2O [M+H]+: calcd 233.1649; found 233.1683.
1-allyl-4-[(2,3-dihydro-1-benzofuran-2-yl)methyl]piperazine (1c). Yellowish oil. 1H NMR (CDCl3, 300 MHz): δ 2.38–2.72 (m, 8H), 2.57 (dd, 1H, J = 13.3, 4.1 Hz), 2.79 (dd, 1H, J = 13.3, 8.1 Hz), 2.94 (dd, 1H, J = 15.6, 8.1 Hz), 3.01 (dq, 2H, J = 6.6, 1.4 Hz), 3.26 (dd, 1H, J = 15.5, 9.1 Hz), 4.95 (dq, 1H, J = 9.1, 4.1 Hz), 5.09–5.26 (m, 2H), 5.78–5.96 (m, 1H), 6.75–6.88 (m, 2H), 7.05–7.21 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 34.2, 53.0, 53.8, 61.8, 63.3, 80.8, 109.7, 118.0, 120.3, 124.9, 126.5, 128.0, 135.1, 159.5. LRMS (EI) m/z (rel %): 258 (23) [M+], 139 (100), 70 (27). HRMS (ESI+) for C16H22N2O [M+H]+: calcd 259.1806; found 259.1840.
1-[(2,3-dihydro-1-benzofuran-2-yl)methyl]-4-phenyl-piperazine (1d). Yellowish solid. mp 47–51°C. 1H NMR (CDCl3, 300 MHz): δ 2.64 (dd, 1H, J = 13.4, 4.2 Hz) 2.69–2.83 (m, 4H), 2.85 (dd, 1H, J = 13.2, 7.7 Hz), 3.05 (dd, 1H, J = 15.5, 7.7 Hz), 3.21–3.35 (m, 5H), 4.95–5.06 (m, 1H), 6.76–6.90 (m, 3H), 6.91–6.97 (m, 2H), 7.07–7.20 (m, 2H), 7.23–7.31 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 34.2, 49.1, 53.9, 63.2, 80.8, 109.7, 116.1, 119.7, 120.4, 124.9, 126.4, 128.1, 129.1, 151.4, 159.5. LRMS (EI) m/z (rel %): 294 (25) [M+], 175 (100), 70 (69). HRMS (ESI+) for C19H22N2O [M+H]+: calcd 295.1806; found 295.1846.
1-[(5-chloro-2,3-dihydro-1-benzofuran-2-yl)methyl]-4-methylpiperazine (1e). Yellow oil. 1H NMR (CDCl3, 300 MHz): δ 2.33 (s, 3H), 2.55 (br.s, 4H), 2.57 (dd, 1H, J = 13.4, 4.2 Hz), 2.65 (br.s, 4H), 2.78 (dd, 1H, J = 13.4, 7.7 Hz), 2.94 (dd, 1H, J = 15.9, 8.0 Hz), 3.24 (dd, 1H, J = 15.9, 9.1 Hz), 4.91–5.03 (m, 1H), 6.68 (d, 1H, J = 8.4 Hz), 7.04 (dd, 1H, J = 8.4, 2.3 Hz), 7.09–7.11 (m, 1H).13C NMR (CDCl3, 75 MHz): δ 34.0, 45.9, 53.5, 54.9, 62.8, 81.5, 110.5, 125.0, 127.9, 128.4, 128.5, 158.2. LRMS (EI) m/z (rel %): 266 (8) [M+], 113 (97), 70 (100). HRMS (ESI+) for C14H19ClN2O [M+H]+: calcd 267.1259; found 267.1231.
1-methyl-4-[(5-methyl-2,3-dihydro-1-benzofuran-2-yl) methyl]piperazine (1f). Yellow oil. 1H NMR (CDCl3, 300 MHz): δ 2.26 (s, 3H), 2.29 (s, 3H), 2.49 (br.s, 4H), 2.55 (dd, 1H, J = 13.4, 4.2 Hz), 2.62 (br.s, 4H), 2.78 (dd, 1H, J = 13.4, 7.7 Hz), 2.90 (dd, 1H, J = 15.5, 8.0 Hz), 3.22 (dd, 1H, J = 15.5, 9.0 Hz), 4.86–4.99 (m, 1H), 6.67 (d, 1H, J = 8.1 Hz), 6.89 (d, 1H, J = 7.9 Hz), 6.96 (s, 1H). 13C NMR (CDCl3, 75 MHz): δ 20.8, 34.3, 46.1, 53.8, 55.0, 63.2, 80.8, 109.1, 125.5, 126.5, 128.3, 129.6, 157.4. LRMS (EI) m/z (rel %): 245 (25) [M+], 113 (100), 70 (85). HRMS (ESI+) for C15H22N2O [M+H]+: calcd 247.1806; found 247.1817.
1-methyl-4-[(5-methoxy-2,3-dihydro-1-benzofuran-2-yl) methyl]piperazine (1g). Yellow oil. 1H NMR (CDCl3, 300 MHz): δ 2.30 (s, 3H), 2.30–2.90 (br.s, 8H), 2.57 (dd, 1H, J = 13.2, 3.8 Hz), 2.78 (dd, 1H, J = 13.2, 8.1 Hz), 2.94 (dd, 1H, J = 15.7, 8.1 Hz), 3.24 (dd, 1H, J = 15.7, 9.0 Hz), 3.75 (s, 3H), 4.95 (dq, 1H, J = 8.4, 3.8 Hz), 6.64 (dd, 1H, J = 8.6, 2.3 Hz), 6.71 (d, 1H, J = 8.6 Hz), 6.75 (d, 1H, J = 2.3 Hz). 13C NMR (CDCl3, 75 MHz): δ 34.7, 46.1, 53.8, 54.9, 56.0, 63.3, 80.8, 109.5, 111.2, 112.6, 127.5, 153.6, 153.9. LRMS (EI) m/z (rel %): 294 (25) [M+], 175 (100), 70 (69). LRMS (EI) m/z (rel %): 262 (28) [M+], 113 (100), 70 (85). HRMS (ESI+) for C15H22N2O2 [M+H]+: calcd 263.1754; found 263.1755.
1-methyl-4-[(5-tert-butyl-2,3-dihydro-1-benzofuran-2-yl) methyl]piperazine (1h). Yellowish solid. mp 90–93°C. 1H NMR (CDCl3, 300 MHz): δ 1.29 (s, 9H), 2.30 (s, 3H), 2.50 (br.s, 4H), 2.58 (dd, 1H, J = 13.4, 4.2 Hz), 2.64 (br.s, 4H), 2.81 (dd, 1H, J = 13.4, 7.8 Hz), 2.95 (dd, 1H, J = 15.5, 8.1 Hz), 3.27 (dd, 1H, J = 15.5, 9.1 Hz), 4.96 (dq, 1H, J = 8.3, 4.1 Hz), 6.72 (d, 1H, J = 8.1 Hz), 7.14 (d, 1H, J = 8.5 Hz), 7.19 (s, 1H). 13C NMR (CDCl3, 75 MHz): δ 31.8, 34.3, 34.5, 46.0, 53.8, 55.0, 63.4, 80.9, 108.8, 121.9, 124.7, 126.0, 143.4, 157.3. LRMS (EI) m/z (rel %): 288 (14) [M+], 113 (100), 70 (65). HRMS (ESI+) for C18H28N2O [M+H]+: calcd 289.2274; found 289.2263.
1-[(5-chloro-2,3-dihydro-1-benzofuran-2-yl)methyl]-4-phenylpiperazine (1i). Yellow oil. 1H NMR (CDCl3, 300 MHz): δ 2.63 (dd, 1H, J = 13.4, 4.2 Hz), 2.75 (q, 4H, J = 4.4 Hz), 2.83 (dd, 1H, J = 13.4, 7.8 Hz), 2.98 (dd, 1H, J = 15.7, 7.8 Hz), 3.20–3.32 (m, 5H), 5.02 (m, 1H), 6.70 (d, 1H, J = 8.4 Hz), 6.71 (d, 1H, J = 8.4 Hz), 6.86 (dt, 1H, J = 7.1, 0.9 Hz), 6.91–6.95 (m, 2H), 7.03–7.08 (m, 1H), 7.10–7.14 (m, 1H), 7.23–7.30 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 34.3, 48.0, 53.3, 61.9, 79.7, 110.6, 116.7, 120.9, 125.1, 125.8, 127.5, 128.2, 129.3, 150.3, 157.6. LRMS (EI) m/z (rel %): 328 (8) [M+], 175 (100), 70 (72). HRMS (ESI+) for C19H21ClN2O [M+H]+: calcd 329.1415; found 329.1413.
Cell Culture and Transfection
HEK293T cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin/streptomycin, at 37°C in 5% CO2; 48 h before the binding assays, cells seeded in 10-cm dishes were transiently transfected with H3R or H4R using polyethylenimine (PEI; 25 kDa linear; Polysciences, Warrington, PA, United States) at a ratio of 3:1 PEI/DNA. For BRET assays, cells were transfected in suspension, using the same PEI/DNA ratio as for attached cells, and directly seeded in 96-well white plates (OptiPlate; PerkinElmer) at a density of 4 × 104 cells/well and grown for 48 h at 37°C in 5% CO2. When needed, total DNA amount was adjusted with salmon sperm DNA (Invitrogen, Carlsbad, CA, United States).
Binding Assays
Competition binding assays were performed in HEK293T cells transiently expressing the H3R or H4R (Santos et al., 2015) in order to assess the affinities of synthesized compounds for each receptor. HEK293T cells transiently expressing H3R or H4R were transferred to 24-well culture plates 24 h after the transfection. One day after plating, the cells were washed once with cold wash buffer (Tris–HCl buffer 25 mM, pH 7.4 containing NaCl 140 mM, MgCl2 5 mM, and 0.1% bovine serum albumin). Cells were incubated with 1.15 nM [3H]-histamine and increasing concentrations of non-radioactive histamine and compounds as a competitor in cold binding buffer [Tris–HCl 25 mM, pH 7.4, including MgCl2 5 mM, 0.1% bovine serum albumin, and 100 μg/mL bacitracin (Sigma–Aldrich, St. Louis, MO, United States)]. Cells were maintained at 4°C for at least 16 h, washed twice and then lysed with lysis buffer (48% urea, 2% NONIDET P-40, acetic acid 3M). Cell lysates were transferred to scintillation vials and 3 mL of scintillation liquid (ScintiSafeTM Econo 1, Fisher Scientific, Santa Clara, CA, United States) were added. Bound radioactivity was quantified on a Tri-Carb 20100TR liquid scintillation counter (PerkinElmer, Waltham, MA, United States). The median inhibitory concentration (IC50) of the tested compounds for displacement [3H]-histamine was obtained from the concentration–response curves, and then using the Cheng–Prusoff equation, the apparent affinities [Ki = IC50/(1 + [ligand]/Kd)] were calculated (Cheng and Prusoff, 1973; Reis et al., 2007). Commercially available compounds JNJ-7777120 and clobenpropit were used as reference ligands of H4R and H3R, respectively.
Analysis of G Protein Activation by BRET Assays
G protein activation was evaluated in HEK293T cells transiently expressing H3R or H4R and a BRET-based biosensor composed of Gαi fused to RLucII (in this case, Gi1-RLucII) and GFP10-Gγ2, in the presence of the untagged Gβ1 subunit (Galés et al., 2005). Cells were transfected in suspension, seeded in 96-well white plates (OptiPlate; PerkinElmer) and grown for 48 h. BRET values were monitored using VictorTM X Light Luminescence microplate reader (PerkinElmer) equipped with BRET400-GFP2/10 filter set (acceptor, 515 ± 20 nm; and donor, 400 ± 70 nm filters), 5 min after the addition of 2.5 μM of coelenterazine 400-a (Biotium, Hayward, CA, United States) and the analyzed ligands. In the antagonism assays, the different ligands were incubated 30 min prior to stimulation with histamine and addition of coelenterazine, according to the procedure described earlier.
Calculated Physicochemical Properties
The molecular properties clogP (calculated lipophilicity), logS (water solubility), molecular weight (MW), topological polar surface area (TPSA) as well as the hydrogen-bond donor (HBD) and acceptor (HBA), and the rotatable bonds (RotB) counts were calculated using the MolSoft online software for drug-likeness and molecular property prediction (La Jolla, CA, United States). The drug-likeness score was also calculated by the software as a prediction of the overall drug-likeness of the molecule based on a MolSofts’s chemical fingerprint. Positive values mean similarity to market drugs.
Ligand Metric Analyses
The metric analysis of a test molecule in a given target can be done using values of ligand efficiency (LE), lipophilic ligand efficiency (LLE), ligand efficiency dependent lipophilicity (LELP), and group efficiency (GE) (Verdonk and Rees, 2008; Hopkins et al., 2014). These metric values are widely employed in drug discovery to indicate whether a potency value derives from a specific target interaction or simply due to many unspecific contacts. These values were calculated using the following equations (1)–(4).
Where HA is the number of heavy (non-hydrogen) atoms in the molecule.
Animals
Male C57Bl/6 mice weighing 20–25 g, 6–8 weeks old, from our own animal facilities were housed in a room with a 12 h light–dark cycle with water and food ad libitum. Animal care and research protocols were in accordance with the principles and guidelines adopted by the Brazilian College of Animal Experimentation (COBEA) and this project was approved by the Ethical Committee for Animal Research of the Federal University of São Paulo (CEUA 1666/99).
Induction of Allergic Asthma and Treatments
Mice (n = 5 per group) were sensitized on days 0 and 7 by an intraperitoneal injection of a mixture containing 50 mg of ovalbumin (OVA; Grade V, Sigma–Aldrich, United States) and 1 mg of Al(OH)3 in PBS (a total volume of 0.2 ml). Mice were challenged by exposure to an aerosol of OVA generated by an ultrasonic nebulizer (ICEL US-800, SP, Brazil) delivering particles of 0.5–10 mm diameter at approximately 0.75 cc/min for 20 min at days 14 and 21. The concentration of OVA in the nebulizer was 2.5% w/v in PBS. The control group consisted of animals immunized as previously described and challenged two times with PBS solution. Compounds 1d and 1e were administered intraperitoneally in two different dosages (5 and 3 mg/kg) to the test groups 30 min before the antigen challenge. Sensitized and control groups were used to assess the anti-inflammatory activity (Corrêa et al., 2017).
Bronchoalveolar Lavage
Mice were euthanized with intraperitoneal ketamine and xylazine (100 and 10 mg/kg, respectively; Agibrands do Brazil, São Paulo, São Paulo, Brazil), 24 h after exposure to the last aerosol challenge. A tracheal cannula was inserted via a mid cervical incision, and the airways were washed twice with 1 ml of phosphate-buffered saline (PBS, pH 7.4 at 4°C).
Total and Differential Cell Counts in the Bronchoalveolar Lavage Fluid
The bronchoalveolar lavage fluid was centrifuged at 170 × g for 10 min at 4°C, the supernatant was removed, and the cell pellet was re-suspended in 1 ml of PBS. One volume of a solution containing 0.5% crystal violet dissolved in 30% acetic acid was added to nine volumes of the cell suspension. The total number of cells was determined by counting in a hematocytometer. Following cytocentrifugation of the bronchoalveolar lavage fluid, cells were stained with hematoxylin-eosin (Hema 3) for determination of the differential cell numbers (Corrêa et al., 2017).
Statistical Analyses
The results from in vivo evaluations are described as the means ± SEM. Statistical evaluation of the data was carried out using the one-way analysis of variance (ANOVA) followed by Tukey’s post-test. A p-value that was lower than 0.05 was considered to be significant. All statistical analyses were performed with the aid of GraphPad software (San Diego, CA, United States).
Results and Discussions
The design of LINS01 molecules was done considering the presence of an aromatic nucleus (aryl or heteroaryl group) linked to a polar moiety (imidazole, pyrrolidine, or piperazine) observed in H3R and H4R ligands. The presence of an extra lipophilic group attached to the polar moiety was also evaluated. In a previous paper from Fernandes et al. (2011), a QSAR study defined the importance of the R2 group for the affinity for H4R in indolecarboxamides (e.g., chlorine in JNJ-7777120). The role of the R2 group is also important for ligand affinity to H3R, which are represented by the cyclopropyl ketone and 4-cyanophenyl groups in H3R ligands such as ciproxifan and ABT-239, respectively (Cowart et al., 2004). Considering this, the present set of molecules includes the substitutions not only in piperazine nitrogen (R1), but also in the dihydrobenzofuran moiety (R2) to determine preliminary SAR data for these compounds. The results show that the R2 substituents can play an important role in the binding affinities, being possibly more important than the substituent R1 to drive the receptor selectivity.
The 2-allylphenol was used as a starting material (Figure 2) for compounds 1a–d, which was converted into the (2-iodomethyl)-2,3-dihydrobenzofuran, as previously described by our group, giving excellent yield (Table 1 and Supplementary Information) (Corrêa et al., 2016, 2017). The iodinated dihydrobenzofuran 4a was then reacted with the corresponding piperazines in considerable excess, giving moderate to good yields. The yields of this step are presented in Table 1. A similar approach was applied to prepare the commercially unavailable 1-allylpiperazine 5 (Supplementary Figure S1) with 40% yield. The 1-methyl and 1-phenyl piperazines are commercially available.
The compounds 1e–i were prepared starting from the corresponding 4-substituted phenol, which was allylated using allyl bromide, and then thermally isomerized through Claisen rearrangement to give the corresponding 2-allylphenols, as reported previously by Corrêa et al. (2017). Both steps resulted in good to excellent yields (Table 1 and Supplementary Information). With the 4-substituted 2-allylphenols, the final compounds were prepared in the same fashion to compounds 1a–d (Corrêa et al., 2016).
The binding analyses were initially performed in the format of a screening, to allow a quick assessment of the compounds presenting relevant affinity for H3R and/or H4R (Supplementary Figure S2). The results show that most of the compounds presented higher affinity for H3R, and that only compounds 1e and 1f displayed considerable binding to H4R. Accordingly, competition binding assays with full concentration–response curves were performed with the selected compounds in the H3R and H4R (Figure 3). The obtained Ki values are in the range of 0.4–10 μM (Table 2).
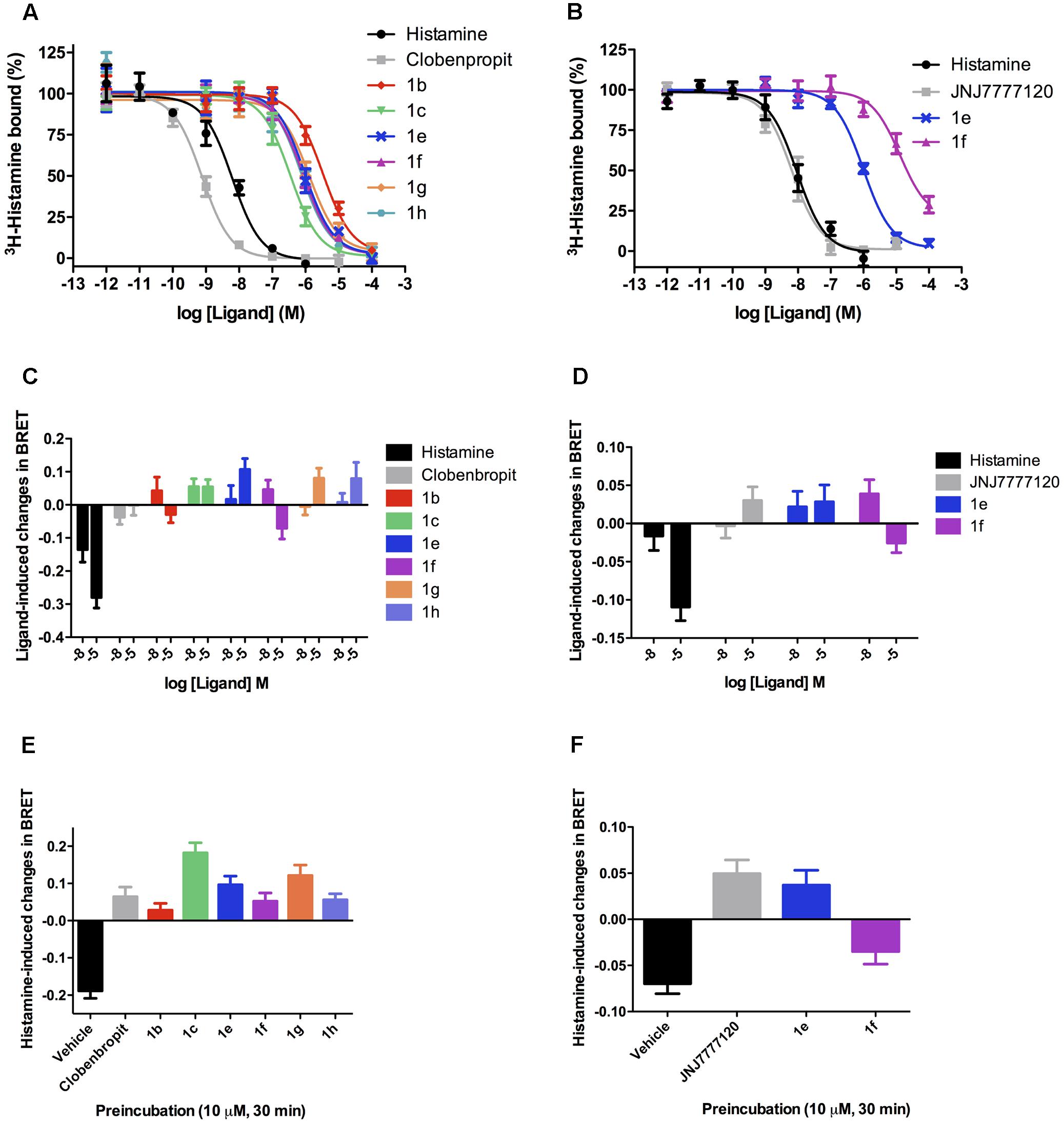
FIGURE 3. Binding and functional characterization of the compounds in the H3R and H4R. Competition dose–response binding curves for selected compounds show that they presented lower affinity for either H3R (A) or H4R (B), as compared with histamine and selective ligands, with Ki values varying from high nanomolar to low micromolar. Functional assays in the H3R (C) reveal that all compounds do not trigger Gi activation, suggesting that they may behave as antagonists in this receptor. In the H4R (D) a similar profile was observed. Antagonistic assays show that all tested compounds when preincubated at 10,000 nM were able to fully block histamine activity in the H3R (E) and H4R (F), except for compound 1f that only partially blocked histamine activity on H4R, probably due to a weak agonistic effect in high concentrations. Data were obtained from at least three independent experiments performed in duplicate. It is also interesting to note that either on H3R or on H4R some compounds displayed a profile that suggests a mild inverse agonist activity, as can be observed by an inverse value of BRET as compared to histamine.
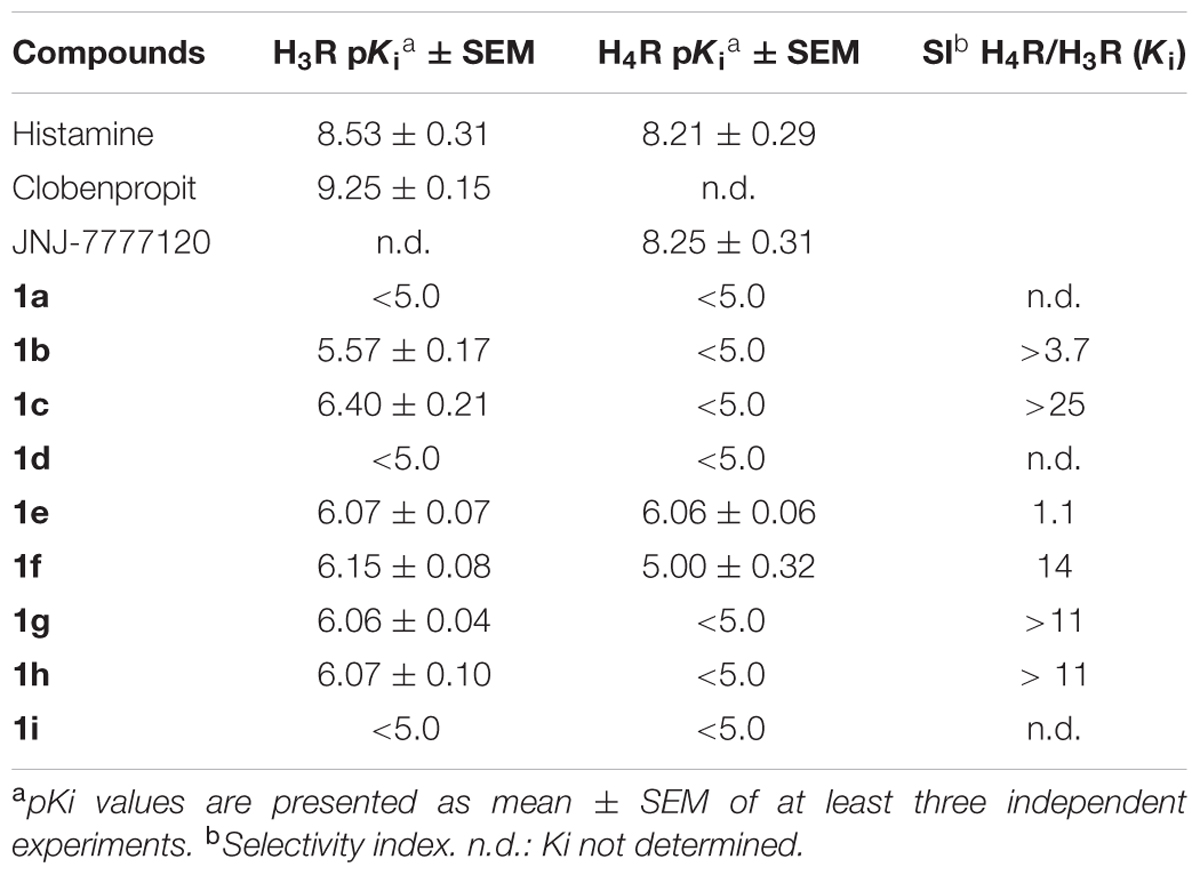
TABLE 2. pKi values of tested compounds for H3R and H4R.
Drug-likeness is a qualitative evaluation of a certain compound for how drug-like it is with respect to some factors such as bioavailability and potency based on the characteristics of market drugs. The drug-likeness of the compounds was assessed through the Lipinski’s rule-of-five (Lipinski et al., 2001) and Veber’s rules (Veber et al., 2002) parameters as well as water solubility. Lipinski’s rule-of-five proposes that a molecule should have MW < 500, clogP < 5, HBD < 5 and HBA < 10 to have good oral bioavailability. Veber’s rules include in these parameters the TPSA < 140 and RotB < 10. The LINS01 compounds fulfill all these parameters (Table 3), suggesting they should have good bioavailability and possibly adequate pharmacokinetic profile. The water solubility also determines the bioavailability and other ADME processes. LINS01 molecules show adequate water solubility, as indicated by the logS values (acceptable values for > -4). With this regard, it is noteworthy the contribution of methoxy group (1g) to enhanced water solubility. The balanced hydrophilic–lipophilic character of LINS01 molecules (as indicated by logP < 5 and logS > -4 values) suggests they may have good pharmacokinetic behavior in vivo. Drug-likeness scores corroborate to this estimation, since only positive values were obtained (Table 3). Considering that market drugs and non-drugs usually present, respectively, positive and negative score values, LINS01 molecules can be considered promising drug-like compounds to further optimization according to this criteria.
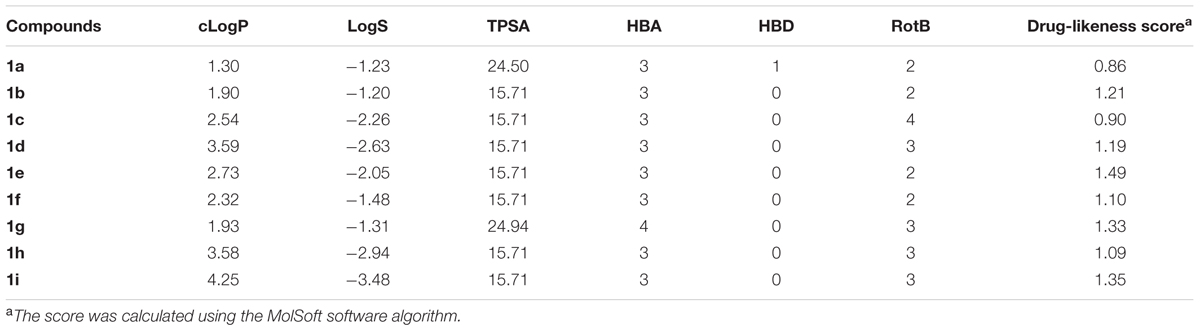
TABLE 3. Drug-likeness calculations for the compounds 1a–1i.
In order to evaluate the potency of these compounds, metric LE, LLE, LELP, and GE values were calculated (Table 4). During drug discovery and development process, the MW and lipophilicity usually increase in consonance with the potency. This can be explained by the contribution of the inserted non-hydrogen atoms to binding energy. LE is considered a weighted value for non-hydrogen atoms contributions to binding energy, which allows to compare affinities of molecules with different sizes (Hopkins et al., 2014). Ligands with LE > 3 are considered promising drug-like compounds. For comparison, the mean value for oral drugs is around 4.5. Considering that lipophilicity also influences on binding affinity, the LLE and LELP values are lipophilicity-weighted LE values. The ideal LLE value for an optimized drug candidate is ∼5–7, while LELP optimal range values are between -10 and 10. GE is a measurement of the binding efficiency for an added functional group in the parent molecule (Verdonk and Rees, 2008). Accordingly, it represents how much the added group contributed to overall affinity of the molecule, considering the number of heavy atoms. For example, a GE = 0.31 should represent at least a gain of 1.7-fold in the potency of a small molecule (MW < 500). The higher the GE value, the higher the contribution of the group to the potency.
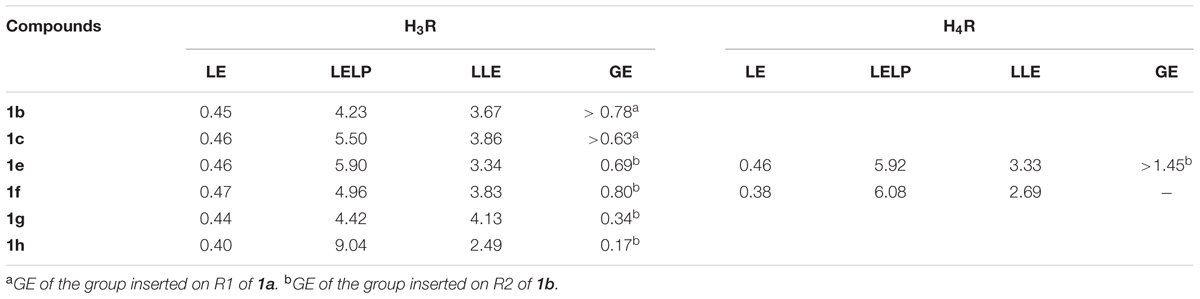
TABLE 4. Ligand metric analysis of the compounds tested for H3R and H4R.
Regarding the H3R, the N-allyl derivative 1c showed the highest affinity in the series, showing a pKi value of 6.40, followed by the chloro, methoxy, and t-butyl derivatives 1e, 1g, and 1h, with pKi values around 6.07, and the methyl derivative 1f with pKi 6.15. The non-alkylated piperazine compound 1a, as well as the N-phenyl derivatives 1d and 1i did not show appreciable affinity for H3R, as mentioned before. The binding efficiencies of the compounds 1b, 1c, and 1e–h are considered adequate, since they present LE > 3 and with exception of compounds 1g and 1h, ≥4.5, indicating that although their low MW, their efficiencies are comparable to a real drug. LELP and LLE values suggest that lipophilicity is relatively high in these molecules, thus suggesting that polar groups (which can improve binding affinity) can be inserted for improvement in a future set of compounds. We believe that H3R may have a considerable different binding pocket than H4R in the R1 and R2 regions, which allow the interaction of bulky hydrophobic groups (such as allyl in R1 and t-butyl in R2). However, some steric hindrance effects might also play a role (especially in the R1 region), limiting the interaction of bulkier groups such as the phenyl group of 1d or 1i, but allows the interaction of the allyl group present in 1c. The results also suggest that the presence of an alkyl group linked to the polar moiety of the molecule increases the affinity for H3R, as can be seen by the high GE values for methyl (1b) and allyl (1c) groups (Table 4).
Several 2-aminoethylbenzofurans have already been evaluated as H3R ligands, showing good affinity (Cowart et al., 2004). These compounds can be viewed as rigid analogs of ciproxifan, such as ABT-239. Hence, the compounds here presented can also be considered rigid derivatives of ciproxifan, but lacking aromaticity in the furan ring. As observed in ABT-239 analogs, small alkyl substituents in the polar moiety increase H3R affinity. This could be the explanation for the poor affinity of 1a and moderate affinities for 1c and the N-methyl derivatives 1e–h (Figure 3 and Table 2).
Since ABT-239 and its analogs possess a 5-aryl substituted benzofuran and better affinity, substituents at the 5-position were explored to evaluate the role of the R2 group in the dihydrobenzofuran derivatives. These data (Table 2) define the importance of the R2 substituent in the binding affinity to H3R of these compounds. Although there are no significant differences between the affinity of compounds 1e–h to H3R, the affinity gain caused by the R2 substitution can be noted by the pKi of the non-substituted molecule 1b (5.57). Although compounds 1e–h present quite similar pKi, the GE is very different among the substituents (Table 4). Methyl group (1f) has shown the higher contribution to binding efficacy in H3R, while t-butyl group (1h) gave lower GE value. The presence of a polar group in R2 (such as methoxy in 1g) also suggests that hydrophobic groups are preferred in this moiety, as indicated by GE = 0.34.
In summary, substitutions in R2 seem to play an important role in H3R binding, since substituting the hydrogen in this position led to improved affinity for the receptor. A rationale SAR for the substitution pattern cannot be defined yet, and further substituents must be explored. However, it is possible to verify that possibly larger volume is tolerated in this region of the molecule, such as the t-butyl group in 1h, which is not tolerated when interacting to the H4R. Possibly, this group presents the same role than the cyanophenyl group in ABT-239 (Figure 4), and maybe it could generate selectivity toward H3R. Previous report from Dastmalchi et al. (2008) suggests that bulkier molecules may display increased affinities for H3R, possibly because there is a hydrophobic pocket nearby the R2 region which can be accessed by these groups, driving the selectivity toward H3R over H4R. With exception of compound 1e, the molecules with substituents in R2 presented over 10-fold selectivity for H3R as compared to H4R (Table 2). Other molecules presenting other bulky groups in R2 are in progress by our group to verify this possibility.
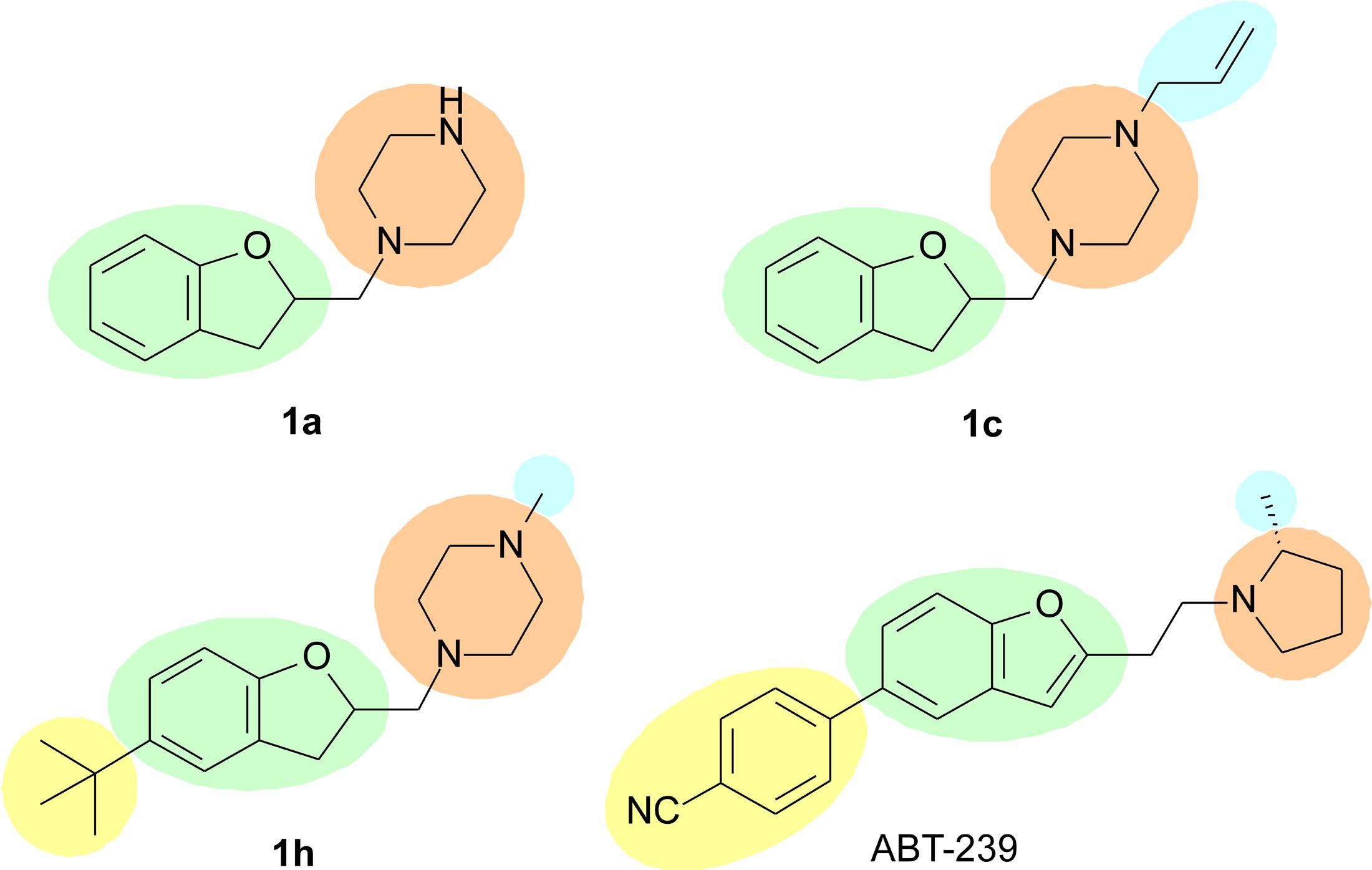
FIGURE 4. Possible characteristics involved in H3R binding; green – aryl or heteroaryl; orange – polar group; blue – lipophilic group; yellow – bulky group. The yellow group may define the selectivity for the H3R.
Regarding the affinity for H4R, compound 1e was the only one to present affinity in the micromolar range (pKi 6.06). Compound 1f has a Ki in the range of ∼10,000 nM. The remaining molecules presented negligible binding affinities, as found in our initial screening approach (see Supplementary Information) and aforementioned. Considering the similarity of the presented compounds with the high affinity H4R antagonist JNJ-7777120, it seems that the absence of the carbonyl group in addition to the change of the indole ring to dihydrobenzofuran is detrimental to the affinity of the ligands for H4R. In fact, the changes in affinity caused by such changes corroborate previous findings in benzofuran compounds (Engelhardt et al., 2012).
Previous reports (Venable et al., 2005; Engelhardt et al., 2012) suggested that a methyl group is the best substituent in the piperazine nitrogen of indolecarboxamide and benzimidazol carboxamide series. Higher homologs or bulkier groups lead to molecules with lower affinity for H4R. In the present series, this pattern was also detected, since compounds 1e and 1f are N-methyl derivatives. The LE values suggest these molecules present good efficacy in the H4R binding affinity. Moreover, LELP and LLE values also suggest that 1e and 1f represent drug-like molecules that can still be optimized regarding the hydrophilic–lipophilic balance. The present results suggest the R2 substituent is more important to the binding affinity than the R1 group, and bulkier groups (such as phenyl in 1d and 1h) lead to low binding affinities (>10,000 nM) when binding H4R. Venable et al. (2005) evaluated several H4R ligands with different substituents in R2 position, with chlorine being the group leading to an important increase in the affinity. It is proposed that this atom performs a key interaction with the binding pocket of H4R. In the present report, this role was also observed, since compound 1e presented the highest affinity in the series for this receptor. The insertion of the chlorine atom resulted in more than 10-fold increase in affinity and a very high GE value (Table 4). This suggests that chlorine atom yielded in improved efficiency for 1e as H4R ligand. In addition, when methyl is present in R2, the affinity drops significantly, and bulkier groups led to total loss of affinity. Accordingly, the H4R seems to present steric hindrance in the R2 region, which is not observed to H3R.
The functional activities of the LINS01 compounds were performed to evaluate their possible actions as agonists of H3R and/or H4R. Both H3R and H4R are GPCRs known to be Gi-coupled. To determine the possible activity of the compounds as agonists of this pathway, we used a BRET biosensor to assess and quantify Gi1 isoform activation.
Regarding H3R, Figure 3C shows that all analyzed compounds were unable to trigger Gi activation even in the highest tested concentration (10,000 nM), likewise clobenpropit, a known H3R antagonist. On the other hand, histamine (used as a positive control) yielded a strong activation signal at 10 and 10,000 nM. The results suggest that the compounds could act as antagonists and block the activation triggered by histamine. This hypothesis was indeed confirmed when cells expressing H3R were incubated with the LINS01 compounds prior to stimulation with histamine. As can be seen in Figure 3E, preincubation with any of the tested compounds at the concentration of 10,000 nM fully blocked histamine agonistic effect. The compounds were equally efficacious in antagonizing histamine effects when compared to clobenpropit, and therefore can be considered H3R antagonists. Interestingly, some compounds displayed a profile of reversal of BRET values, as compared to histamine alone, suggesting an inverse agonist activity.
Analyses of functional activity on H4R showed that similarly to JNJ-777120, a known H4R antagonist, the chlorinated compound 1e did not trigger Gi activation (Figure 3D). Possibly the specific interaction of chlorine with H4R does not lead to activation of the receptor, but when methyl group is present an agonist mode interaction may occur and therefore a halogen-bonding interaction is suggested rather than a hydrophobic interaction to reach the antagonist binding mode. The role of chlorine in H4R ligands was already widely studied, and it has been directly linked to the antagonist activity on Gi as well as β-arrestin activation (Nijmeijer et al., 2013). It is important to stress that histamine itself was a poor stimulant of Gi in H4R (compare BRET values from Figures 3C,D). When evaluating the potential to block histamine activity, preincubation with the compound 1e at the concentration of 10,000 nM fully blocked histamine agonistic effect, similarly to the effect obtained for the H4R antagonist JNJ-777120. The compound 1f, as described above, displays a moderate agonistic effect, and therefore only partially reduced histamine effect.
In order to evaluate the anti-inflammatory potential of the best H4R blocker from the LINS01 set, compound 1e was tested in a mouse asthma model and compared to the previously tested compound 1d in two different doses (3 and 5 mg/kg, Figure 5). Experimental murine asthma was induced by sensitization and provocation with OVA. In a recent report (Corrêa et al., 2017), we verified that 1d showed anti-inflammatory activity only at 5 mg/kg dose.
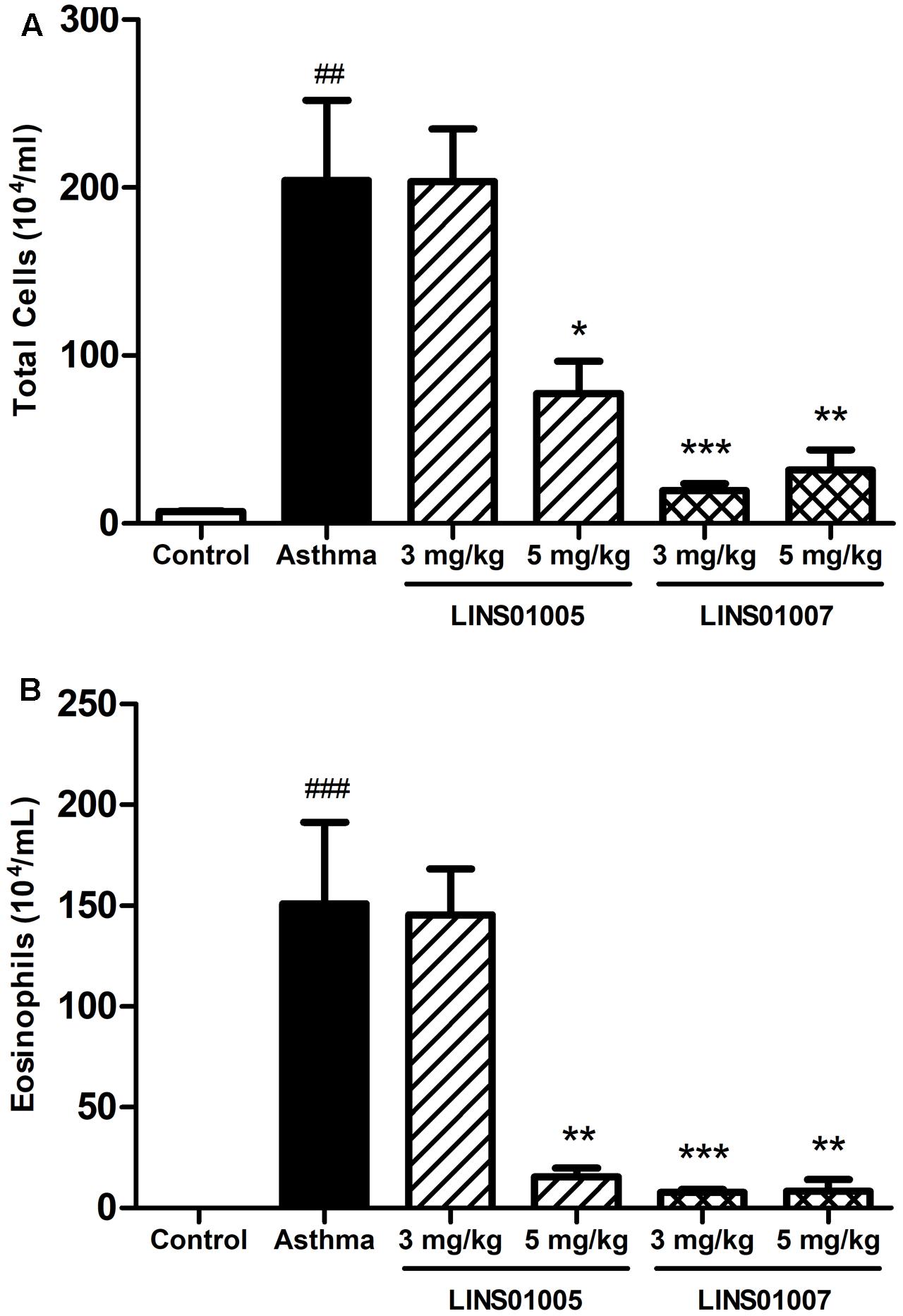
FIGURE 5. Effects of compounds 1d (LINS01005) and 1e (LINS01007) in the (A) total inflammatory cell count and (B) eosinophil count obtained from bronchoalveolar lavage (3 and 5 mg/kg); ##p < 0.01 relative to control group; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 relative to asthma group.
In the present work, we evaluated in parallel the compounds 1e (LINS01007) and 1d (LINS01005) to rodents. The reduction in cell counts in both groups correlates well to the affinity to H4R (see Figure 3B), supporting the involvement of this receptor in the mechanism of the observed anti-inflammatory action. The results presented in Figure 5 also suggest a dose-dependent effect for compound 1d on both total cell and eosinophil counts in bronchoalveolar lavage fluids, since a sounder reduction in cellularity was observed in the highest concentration of 5 mg/kg. This reduction can be considered more relevant due to the important reduction in eosinophilia. On the other hand, compound 1e led to low counts in both doses. Possibly even in the lower dose (3 mg/kg) the maximum efficacy was obtained.
In bronchoalveolar lavage fluids with experimental allergic asthma, enhanced cell counts are commonly observed. In particular, there is considerable increase in eosinophils, which are commonly absent or in low density in the non-sensitized animals. The involvement of H4R in experimental murine asthma model has been demonstrated in several studies, including results with JNJ-7777120, which was capable of reducing the inflammatory infiltrations (especially eosinophils) and other asthma parameters (Neumann et al., 2013). Considering that the pharmacological profile obtained with LINS01 compounds in the total cell and eosinophil counts is very similar to that obtained with the JNJ compound, we believe the infiltration reductions are correlated to H4R. Moreover, the higher activity observed to 1e also supports this conclusion.
Conclusion
In this study, we present nine LINS01 molecules with quite simple structure, which have been evaluated concerning binding affinities for the H3R and H4R, generating corresponding receptor subtype selectivity values. We have also performed functional analyses of Gi activation, which revealed that all tested compounds act as antagonists to different extents on both H3R and H4R. This is the first report regarding functional activity of such compounds. SAR information concerning this chemotype was generated and metric analyses suggest that future derivatives with improved affinity can still be obtained. In vivo evaluation in a mouse asthma model was performed and the potential anti-inflammatory activity of such molecules is exemplified by 1e, which showed to be highly effective at low doses.
Author Contributions
MC and ÁB performed the synthesis of the compounds, and were supervised by JF at LINS-UNIFESP. LT, DD, SS, and LP performed the binding and functional assays at CC-N’s laboratory, their supervisor. AB and RL did the in vivo assays. MB generated the constructions for BRET assays and discussed obtained data. JF and CC-N wrote the manuscript.
Funding
The authors are grateful to FAPESP [Grant Nos. 2013/20479-9, 2016/25028-3, and 2017/02042-3] and to CNPq [Grant No. 455411/2014-0] for the support provided to JF and RL. CC-N’s laboratory is mainly supported by FAPESP [Grant No. 2012/20148-0]. AB thanks to CAPES for the scholarships. MC [Grant No. 2016/23139-2], LT, DD, SS, and LP are recipients of FAPESP scholarships. MB holds the Canada Research Chair in Signal Transduction and Molecular Pharmacology and is supported by the Canadian Institute for Health Research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2017.00825/full#supplementary-material
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Cheng, Y., and Prusoff, W. H. (1973). Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108.
Christopher, F., Thangam, E. B., and Suresh, M. X. (2012). A bioinformatics search for selective histamine H4 receptor antagonists through structure-based virtual screening strategies. Chem. Biol. Drug Des. 79, 749–759. doi: 10.1111/j.1747-0285.2012.01336.x
Corrêa, M. F., Barbosa, A. J. R., Sato, R., Junqueira, L. O., Politi, M. J., Rando, D. G., et al. (2016). Factorial design study to access the “green” iodocyclization reaction of 2-allylphenols. Green Process. Synth. 5, 145–151. doi: 10.1515/gps-2015-0101
Corrêa, M. F., and Fernandes, J. P. d. S. (2015). Histamine H4 receptor ligands: future applications and state of art. Chem. Biol. Drug Des. 85, 461–480. doi: 10.1111/cbdd.12431
Corrêa, M. F., Varela, M. T., Balbino, A. M., Torrecilhas, A. C., Landgraf, R. G., Troncone, L. R. P., et al. (2017). 1-[(2,3-Dihydro-1-benzofuran-2-yl)methyl]piperazines as novel anti-inflammatory compounds: synthesis and evaluation on H3R/H4R. Chem. Biol. Drug Des. 90, 317–322. doi: 10.1111/cbdd.12947
Cowart, M., Pratt, J. K., Stewart, A. O., Bennani, Y. L., Esbenshade, T. A., and Hancock, A. A. (2004). A new class of potent non-imidazole H3 antagonists: 2-aminoethylbenzofurans. Bioorg. Med. Chem. Lett. 14, 689–693. doi: 10.1016/j.bmcl.2003.11.032
Dastmalchi, S., Hamzeh-Mivehroud, M., Ghafourian, T., and Hamzeiy, H. (2008). Molecular modeling of histamine H3 receptor and QSAR studies on arylbenzofuran derived H3 antagonists. J. Mol. Graph. Model. 26, 834–844. doi: 10.1016/j.jmgm.2007.05.002
de Esch, I. J. P., Thurmond, R. L., Jongejan, A., and Leurs, R. (2005). The histamine H4 receptor as a new therapeutic target for inflammation. Trends Pharmacol. Sci. 26, 462–469. doi: 10.1016/j.tips.2005.07.002
Engelhardt, H., de Esch, I. J., Kuhn, D., Smits, R. A., Zuiderveld, O. P., Dobler, J., et al. (2012). Detailed structure-activity relationship of indolecarboxamides as H4 receptor ligands. Eur. J. Med. Chem. 54, 660–668. doi: 10.1016/j.ejmech.2012.06.016
Fernandes, J. P. S., Pasqualoto, K. F. M., Ferreira, E. I., and Brandt, C. A. (2011). Molecular modeling and QSAR studies of a set of indole and benzimidazole derivatives as H4 receptor antagonists. J. Mol. Model. 17, 921–928. doi: 10.1007/s00894-010-0779-4
Galés, C., Rebois, R. V., Hogue, M., Trieu, P., Breit, A., Hébert, T. E., et al. (2005). Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2, 177–184. doi: 10.1038/nmeth743
Hopkins, A. L., Keserü, G. M., Leeson, P. D., Rees, D. C., and Reynolds, C. H. (2014). The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 13, 105–121. doi: 10.1038/nrd4163
Kottke, T., Sander, K., Weizel, L., Schneider, E. H., Seifert, R., and Stark, H. (2011). Receptor-specific functional efficacies of alkyl imidazoles as dual histamine H3/H4 receptor ligands. Eur. J. Pharmacol. 654, 200–208. doi: 10.1016/j.ejphar.2010.12.033
Łażewska, D., Więcek, M., Ner, J., Kamińska, K., Kottke, T., Schwed, J. S., et al. (2014). Aryl-1,3,5-triazine derivatives as histamine H4 receptor ligands. Eur. J. Med. Chem. 83, 534–546. doi: 10.1016/j.ejmech.2014.06.032
Leurs, R., Bakker, R. A., Timmerman, H., and de Esch, I. J. P. (2005). The histamine H3 receptor: from gene cloning to H3 receptor drugs. Nat. Rev. Drug Discov 4, 107–142. doi: 10.1038/nrd1631
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (2001). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26. doi: 10.1016/S0169-409X(00)00129-0
Liu, C., Ma, X., Jiang, X., Wilson, S. J., Hofstra, C. L., Blevitt, J., et al. (2001). Cloning and pharmacological characterization of a fourth histamine receptor (H4) expressed in bone marrow. Mol. Pharm. 59, 420–426. doi: 10.1124/mol.59.3.420
Medina, V., Croci, M., Crescenti, E., Mohamad, N., Sanchez-Jiménez, F., Massari, N., et al. (2008). The role of histamine in human mammary carcinogenesis: H3 and H4 receptors as potential therapeutic targets for breast cancer treatment. Cancer Biol. Ther. 7, 28–35. doi: 10.4161/cbt.7.1.5123
Neumann, D., Beerman, S., Burhenne, H., Glage, S., Hartwig, C., and Seifert, R. (2013). The dual H3/4R antagonist thioperamide does not fully mimic the effects of the ‘standard’ H4R antagonist JNJ 7777120 in experimental murine asthma. Naunyn Schmeidebergs Arch. Pharmacol. 386, 983–990. doi: 10.1007/s00210-013-0898-4
Nijmeijer, S., Vischer, H. F., Sirci, F., Schultes, S., Engelhardt, H., de Graaf, C., et al. (2013). Detailed analysis of biased histamine H4 receptor signalling by JNJ 7777120 analogues. Br. J. Pharmacol. 170, 78–88. doi: 10.1111/bph.12117
Oda, T., Morikawa, N., Saito, Y., Masuho, Y., and Matsumoto, S. (2000). Molecular cloning and characterization of a novel type of histamine receptor preferentially expressed in leukocytes. J. Biol. Chem. 275, 36781–36786. doi: 10.1074/jbc.M006480200
Passani, M. B., and Blandina, P. (2011). Histamine receptors in the CNS as targets for therapeutic intervention. Trends Pharm. Sci. 32, 242–249. doi: 10.1016/j.tips.2011.01.003
Reis, R. I., Santos, E. L., Pesquero, J. B., Oliveira, L., Schanstra, J. P., Bascands, J. L., et al. (2007). Participation of transmembrane proline 82 in angiotensin II AT1 receptor signal transduction. Regul. Pept. 140, 32–36. doi: 10.1016/j.regpep.2006.11.028
Santos, G. A., Duarte, D. A., Parreiras-E-Silva, L. T., Teixeira, F. R., Silva-Rocha, R., Oliveira, E. B., et al. (2015). Comparative analyses of downstream signal transduction targets modulated after activation of the AT1 receptor by two β-arrestin-biased agonists. Front. Pharmacol. 6:131. doi: 10.3389/fphar.2015.00131
Sayed, Y. Y. (2016). Pitolisant: first global approval. Drugs 76, 1313–1318. doi: 10.1007/s40265-016-0620-1
Seifert, R., Schneider, E. H., Dove, S., Brunskole, I., Neumann, D., Strasser, A., et al. (2011). Paradoxical stimulatory effects of the “standard” histamine H4-receptor antagonist JNJ7777120: the H4 receptor joins the club of 7 transmembrane domain receptors exhibiting functional selectivity. Mol. Pharm. 79, 631–638. doi: 10.1124/mol.111.071266
Shan, L., Bao, A., and Swaab, D. F. (2015a). The human histaminergic system in neuropsychiatric disorders. Trends Neurosci. 38, 167–177. doi: 10.1016/j.tins.2014.12.008
Shan, L., Bossers, K., Luchetti, S., Balesar, R., Lethbridge, N., Chazot, P. L., et al. (2015b). Alterations in the histaminergic system in the substantia nigra and striatum of Parkinson’s patients: a postmortem study. Neurobiol. Aging 33, 1488.e1–1488.e13. doi: 10.1016/j.neurobiolaging.2011.10.016
Smith, F. M., Haskelberg, H., and Tracey, D. J. (2007). Role of histamine H3 and H4 receptors in mechanical hyperalgesia following peripheral nerve injury. Neuroimmunomodulation 14, 317–325. doi: 10.1159/000125048
Tiligada, E., Zampeli, E., Sander, K., and Stark, H. (2009). Histamine H3 and H4 receptors as novel drug targets. Expert Opin. Investig. Drugs 18, 1519–1531. doi: 10.1517/14728220903188438
Veber, D. F., Johnson, S. R., Cheng, H. Y., Smith, B. R., Ward, K. W., and Kopple, K. D. (2002). Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623. doi: 10.1021/jm020017n
Venable, J. D., Cai, H., Chai, W., Dvorak, C. A., Grice, C. A., Jablonowski, J. A., et al. (2005). Preparation and biological evaluation of indole, benzimidazole, and thienopyrrole piperazine carboxamides: potent human histamine H4 antagonists. J. Med. Chem. 48, 8289–8298. doi: 10.1021/jm0502081
Keywords: H3R antagonists, H4R antagonists, histamine receptors, dihydrobenzofuran, SAR, anti-inflammatory activity, asthma
Citation: Corrêa MF, Barbosa ÁJR, Teixeira LB, Duarte DA, Simões SC, Parreiras-e-Silva LT, Balbino AM, Landgraf RG, Bouvier M, Costa-Neto CM and Fernandes JPS (2017) Pharmacological Characterization of 5-Substituted 1-[(2,3-dihydro-1-benzofuran-2-yl)methyl]piperazines: Novel Antagonists for the Histamine H3 and H4 Receptors with Anti-inflammatory Potential. Front. Pharmacol. 8:825. doi: 10.3389/fphar.2017.00825
Received: 29 September 2017; Accepted: 30 October 2017;
Published: 14 November 2017.
Edited by:
Antonietta Rossi, University of Naples Federico II, ItalyReviewed by:
Ahmed El-Hashim, Kuwait University, KuwaitDetlef Neumann, Hannover Medical School, Germany
Copyright © 2017 Corrêa, Barbosa, Teixeira, Duarte, Simões, Parreiras-e-Silva, Balbino, Landgraf, Bouvier, Costa-Neto and Fernandes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio M. Costa-Neto, claudio@fmrp.usp.br João P. S. Fernandes, joao.fernandes@unifesp.br
†These authors have contributed equally to this work.