 Maria Augusta Arruda1,2,3†
Maria Augusta Arruda1,2,3† Leigh A. Stoddart1,2†
Leigh A. Stoddart1,2† Karolina Gherbi1,2
Karolina Gherbi1,2 Stephen J. Briddon1,2*
Stephen J. Briddon1,2* Barrie Kellam4
Barrie Kellam4 Stephen J. Hill1,2*
Stephen J. Hill1,2*- 1Division of Physiology, Pharmacology and Neuroscience, School of Life Sciences, Medical School, Queen's Medical Centre, University of Nottingham, Nottingham, United Kingdom
- 2Centre of Membrane Proteins and Receptors, University of Birmingham and University of Nottingham, The Midlands, United Kingdom
- 3Vice-Diretoria de Ensino, Pesquisa e Inovacao, Farmanguinhos, Fiocruz, Rio de Janeiro, Brazil
- 4Centre for Biomolecular Sciences, School of Pharmacy, University of Nottingham, Nottingham, United Kingdom
Recent advances in fluorescent ligand technology have enabled the study of G protein-coupled receptors in their native environment without the need for genetic modification such as addition of N-terminal fluorescent or bioluminescent tags. Here, we have used a non-imaging plate reader (PHERAstar FS) to monitor the binding of fluorescent ligands to the human adenosine-A3 receptor (A3AR; CA200645 and AV039), stably expressed in CHO-K1 cells. To verify that this method was suitable for the study of other GPCRs, assays at the human adenosine-A1 receptor, and β1 and β2 adrenoceptors (β1AR and β2AR; BODIPY-TMR-CGP-12177) were also carried out. Affinity values determined for the binding of the fluorescent ligands CA200645 and AV039 to A3AR for a range of classical adenosine receptor antagonists were consistent with A3AR pharmacology and correlated well (R2 = 0.94) with equivalent data obtained using a confocal imaging plate reader (ImageXpress Ultra). The binding of BODIPY-TMR-CGP-12177 to the β1AR was potently inhibited by low concentrations of the β1-selective antagonist CGP 20712A (pKi 9.68) but not by the β2-selective antagonist ICI 118551(pKi 7.40). Furthermore, in experiments conducted in CHO K1 cells expressing the β2AR this affinity order was reversed with ICI 118551 showing the highest affinity (pKi 8.73) and CGP20712A (pKi 5.68) the lowest affinity. To determine whether the faster data acquisition of the non-imaging plate reader (~3 min per 96-well plate) was suitable for high throughput screening (HTS), we screened the LOPAC library for inhibitors of the binding of CA200645 to the A3AR. From the initial 1,263 compounds evaluated, 67 hits (defined as those that inhibited the total binding of 25 nM CA200645 by ≥40%) were identified. All compounds within the library that had medium to high affinity for the A3AR (pKi ≥6) were successfully identified. We found three novel compounds in the library that displayed unexpected sub-micromolar affinity for the A3AR. These were K114 (pKi 6.43), retinoic acid p-hydroxyanilide (pKi 6.13) and SU 6556 (pKi 6.17). Molecular docking of these latter three LOPAC library members provided a plausible set of binding poses within the vicinity of the established orthosteric A3AR binding pocket. A plate reader based library screening using an untagged receptor is therefore possible using fluorescent ligand opening the possibility of its use in compound screening at natively expressed receptors.
Introduction
G protein-coupled receptors (GPCRs) represent the largest family of cell surface receptors and account for approximately 4% of the entire protein-coding human genome. There are approximately 700 separate GPCRs of which over 300 are non-olfactory receptors (Kuder and Kieć-Kononowicz, 2014). Based on sequence homology, five distinct families of non-olfactory receptors have been proposed: Family A/Rhodopsin, Family B/secretin, Adhesion GPCRs, Family C/Glutamate, and Family F/frizzled (Guo et al., 2012). Family A contains the largest number of the non-olfactory GPCRs including many of the most widely studied receptors, each of which acts to translate extracellular signals into intracellular effects by activating both heterotrimeric G protein-dependent and -independent signaling cascades (Castro et al., 2005; Guo et al., 2012). Importantly, these family A GPCRs are also currently targeted by a large number of clinically used drugs and are validated targets for a significant number of drug discovery programmes.
Adenosine is one biological transmitter which plays a vital homeostatic role and acts via a family of Class A GPCRs comprising four distinct subtypes: namely the adenosine-A1 receptor (A1AR), A2AAR, A2BAR, and A3AR (Fredholm et al., 2011). Both the A1AR and A3ARs inhibit intracellular cAMP formation by activating inhibitory Gi proteins, whilst the A2AAR and A2BARs generally stimulate cAMP formation via stimulatory Gs proteins. Adenosine-mediated signaling has been implicated in a number of pathological states. For instance, the signaling pathways regulated by these receptors can promote angiogenesis (Headrick et al., 2013) and reduce inflammation (Antonioli et al., 2014). Within this family, the A3AR is a promising molecular target for the control of a range of pathological conditions including cancer (Montinaro et al., 2013; Nakamura et al., 2015; Cao et al., 2017; Joshaghani et al., 2017), inflammation (Cohen et al., 2014; Yoshida et al., 2017), autoimmune diseases (Ravani et al., 2017), ischaemia (Mulloy et al., 2013; González-Fernández et al., 2014; Hussain et al., 2014; Ohana et al., 2016) and chronic neuropathic pain (Little et al., 2015; Tosh et al., 2015), making it an important target for drug development (Borea et al., 2015). As a consequence, identifying new screening methods for discovery of novel chemical scaffolds which bind to the A3AR would be beneficial.
With this in mind, it is of note that recent advances in fluorescent ligand technology have enabled unlabeled GPCRs to be studied in their native environment without any need for genetic modification through the addition of a bioluminescent or fluorescent tag. For instance fluorescent ligands have been used to study various aspects of GPCR pharmacology including ligand binding, receptor-ligand kinetics, receptor localization and trafficking (Stoddart et al., 2015b). Of particular relevance to purinergic drug discovery, Stoddart et al. (2012) developed a competitive binding assay for the human A3AR and A1AR in live cells, using a high content screening (HCS) platform that allowed the screening of small fragment libraries. This assay system was also used to validate the pharmacology of A3AR selective compounds that were identified from virtual screening of homology models (Ranganathan et al., 2015). However, a disadvantage of this technique is that it involves the acquisition and analysis of a large number of images which can impose severe time, data handling and storage limitations at the early stages of drug discovery, particularly in hit discovery, when very large libraries (>100,000 compounds) are used in initial screening campaigns (Tomasch et al., 2012). In this work, we show that such a competitive fluorescent based binding screen is possible on a higher throughput, non-imaging-based platform using two structurally unrelated fluorescent antagonists. The suitability of this assay for higher throughput screens has been demonstrated by screening a library of pharmacological active compounds (LOPAC) against the native human A3AR in living cells, with a view to identifying potential novel scaffolds for A3AR ligands.
Results
Comparison of High Content (HCS) and High Throughput (HTS) Screening Platforms for Measuring Competition Binding to the A3AR
As previously described, competition binding assays have been performed on cells expressing the wild type human A3AR using the fluorescent adenosine receptor antagonist CA200645 by automated image acquisition using an ImageXpress (IX) Ultra confocal imaging plate reader (Stoddart et al., 2012). In order to see if this method could be translated into a faster non-imaging format, we directly compared HCS and plate reader based CA200645 binding by sequentially reading the same samples on the PHERAstar FS (BMG technologies) then the IX Ultra. As shown in the IX Ultra plate image in Figure 1A, binding of 25 nM CA200645 was clearly detected, and was subsequently displaced by increasing concentrations of competing (unlabeled) antagonists. The same 96-well plate was also measured on a standard non-imaging fluorescence plate reader (PHERAstar FS), with 81 separate repeat reads per well to take into account variation in cell density, and a similar pattern of fluorescence was observed (Figure 1B). The montage images from both instruments show that the high affinity A3AR antagonist MRS1220, AV019 (compound 1 in Vernall et al., 2012) and the non-selective adenosine receptor antagonist xanthine amine congener (XAC) caused a concentration-dependent reduction in the fluorescence intensity observed with 25 nM CA200645 alone. Competition binding curves were generated from the quantified data (Figure 1C), and pKi values for the five adenosine receptor antagonists obtained, which were comparable to values reported in the literature (Table 1). Comparison of the affinity values from the HTS platform (PHERAstar) to those from the HCS platform (IX Ultra) showed a high degree of correlation (R2 = 0.94) (Figure 1E) and we have previously shown that affinity values obtained from the HCS platform correlated well with values obtained in a functional assay (Stoddart et al., 2012). In addition to the XAC based fluorescent ligand CA200645, a structurally distinct and highly selective fluorescent A3AR antagonist was also used (AV039; compound 19 in Vernall et al., 2012). As with CA200645, using 5 nM AV039 as label, competition binding experiments measured on the PHERAstar FS produced the expected rank order of antagonist affinity for the A3AR (Figure 1D, Table 1).
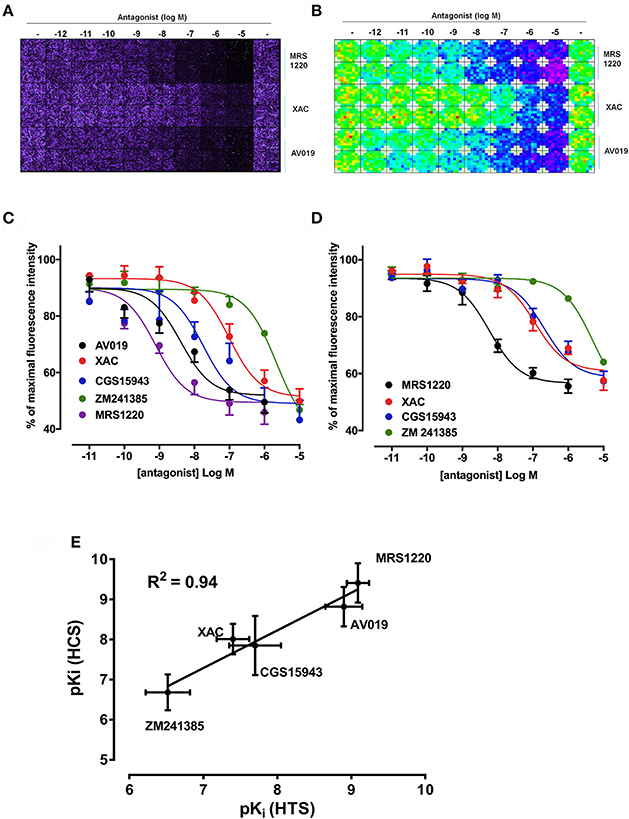
Figure 1. Competition binding at the A3AR using fluorescent ligands. CHO cells expressing the A3AR were incubated with 25 nM CA200645 and increasing concentrations of MRS1220, XAC, or AV019. (A) Four images per well were obtained on the IX Ultra confocal plate reader and resulting images shown as a montage. (B) Montage fluorescence intensity measurement of the same plate obtained using the FS PHERAstar where blue, green, yellow and red pixels represents increasing intensity of fluorescence. (C) Competition curves at the A3AR generated from the total fluorescence intensity measured on the PHERAstar FS microplate reader for five adenosine receptor antagonists. (D) CHO A3AR cells were incubated with increasing concentrations of antagonist and 5 nM AV039 for 1 h, 37°C, washed and fluorescence intensity assessed using the PHERAstar FS. (E) Correlation between pKi values obtained using the IX Ultra (high content screening; HCS) and the PHERAstar FS (high throughput screening; HTS) for the data obtained using CA200645 as fluorescent ligand. Data were normalized to the maximal intensity observed per experiment and each data point represents the mean ± SEM from n number of experiments (See Table 1) performed in triplicate.
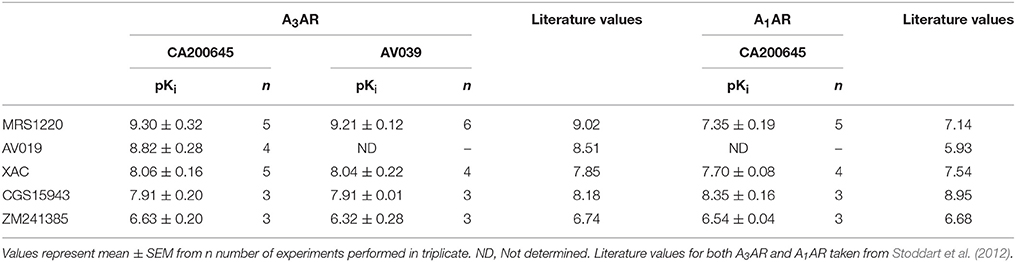
Table 1. Affinity of compounds measured at the A1AR and A3AR: Affinity values from the PHERAstar HTS assay for unlabeled ligands measured on CHO cells expressing the A3AR or the A1AR using 25 nM CA200645 or 5 nM AV039.
Application to A1AR and β-Adrenoceptors
To verify that the experimental approach used for the A3AR was suitable for the study of other GPCRs, we conducted the same experimental design with CA200645 on CHO cells expressing the human A1AR, since this fluorescent ligand also binds with high affinity to this receptor (Stoddart et al., 2012). This is important, since being able to screen for compound selectivity is an important aspect of developing a screening methodology. As with the A3AR, a clear concentration-dependent decrease in fluorescence intensity was detected on the HTS plate reader in the presence of four different adenosine receptor antagonists (Figure 2A). The affinity values from these data were consistent with A1AR pharmacology with CGS 15943 showing the highest affinity and MRS1220 exhibiting a lower affinity than at the A3AR. In addition, ZM241385, an A2AAR selective antagonist showed the expected low affinity at the A1AR (Table 1).
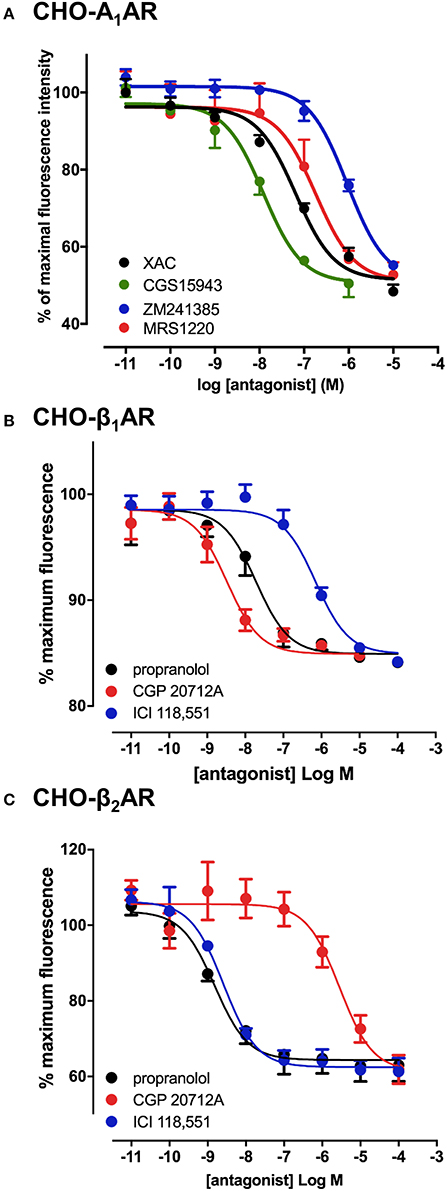
Figure 2. Competition binding assays at the adenosine A1 and β1/β2-adrenoceptors. CHO cell lines stably expressing A1AR (A), β1AR (B) or the β2AR (C) were incubated with 25 nM CA200645 (A1AR) or 10 nM BODIPY-TMR-CGP (β1AR and β2AR), in the absence or the presence of increasing concentrations of antagonists. Fluorescence intensity in each well was monitored using the PHERAstar FS. Values are mean ± SEM from 3–6 independent experiments performed in triplicate.
The confocal based fluorescent ligand binding assay has also been recently applied to study the pharmacology of the β1AR using BODIPY-TMR labeled CGP 12177 (BODIPY-TMR-CGP; Gherbi et al., 2014) and we therefore also tested whether ligand binding to the β1AR and β2AR could also be monitored using the HTS platform in order to develop a counter screen for the A3AR. As shown in Figure 2B, in CHO cells expressing either the β1AR or β2AR, binding of BODIPY-TMR-CGP could be clearly detected, and clear competition binding was observed with all three βAR ligands at both receptors. Importantly, the β1AR selective antagonist CGP 20712A displayed the highest affinity at the β1AR and the β2AR selective antagonist ICI 118551 the lowest (Table 2), whilst this rank order was reversed at the β2AR, with ICI 118551 showing the highest affinity and CGP20712A the lowest affinity (Figure 2C, Table 2).
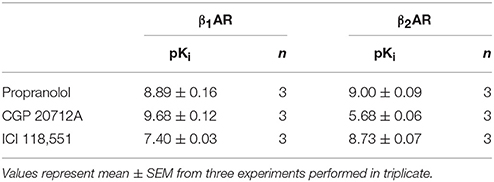
Table 2. Affinity of compounds measured at the β1AR and β2AR: Affinity values for β-adrenoceptor ligands measured in CHO cells expressing the β1AR or the β2AR using 10 nM of BODIPY-TMR-CGP in the HTS format fluorescent ligand binding assay.
Screening of a Focused Library of Pharmacologically Active Ligands at the A3AR
To determine whether the HTS version of the competitive fluorescent binding assay was suitable for the screening of large compound libraries, we chose to screen the Library of Pharmacologically Active Compounds (LOPAC) against the A3AR. The LOPAC library is considered to be a recognized standard for assay validation as it is based on an extensive number of bioactive compounds. Many of these are known to affect targets involved in adenosine receptor signaling (Iturrioz et al., 2010). CHO cells expressing the A3AR were grown to confluency in 96-well plates and incubated with a single concentration (10 μM) of the known A3AR antagonist MRS1220 as a positive control or one of the 1,263 compounds (10 μM) from the LOPAC library and CA200645 (25 nM) and the fluorescence intensity of each well determined on the PHERAstar FS plate reader as described in Experimental Procedures. Hits were defined as those compounds which inhibited the binding of CA200645 by >40%, and of the initial 1263 compounds evaluated, 67 hits were identified (Supporting Information Table 1, Figure 3, Table 3). Inhibition data for all the compounds tested in the initial screen can be found in Supporting Information Table 1. Among the hits, all the compounds within the library with medium to high affinity for the A3AR (pKi ≥6; Figure 3, Table 3) were identified along with four low affinity adenosine-related molecules (1,3-dipropyl-8-p-sulfophenylxanthine, DMPX, etazolate hydrochloride and 2-phenylaminoadenosine; Table 3). This confirmed the utility of this approach to identify compounds with known A3AR binding affinity. Importantly, the assay Z' factor was 0.47 ± 0.03 (mean ± SEM, n = 97), demonstrating its suitability for screening larger libraries in living cells.
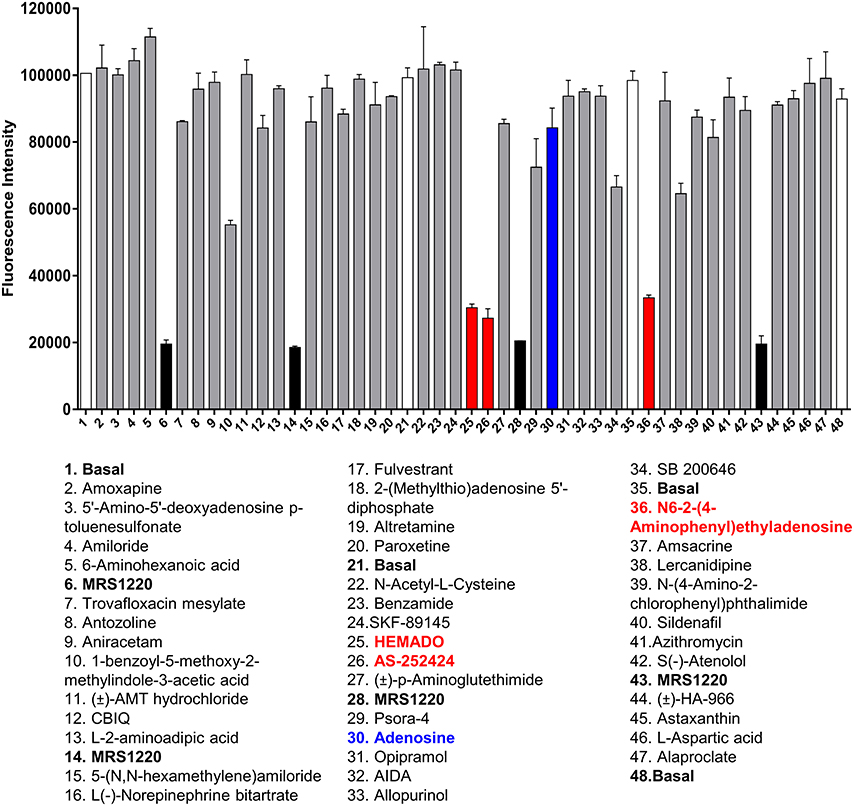
Figure 3. Screening the LOPAC library against the A3AR. Example of the data generated from one plate of compounds from the LOPAC library. Each plate contained 40 compounds (each at 10 μM final concentration) from the LOPAC library in duplicate along with four basal and four MRS1220 (10 μM) controls, also in duplicate. The fluorescence intensities obtained on the PHERAstar FS from this plate are shown as mean and range of duplicates with the hits highlighted in red and adenosine indicated in blue. The plate shown is a representative plate of one of the three experiments performed using these compounds and the inhibition data for all compounds screened can be found in Supporting Information Table 1.
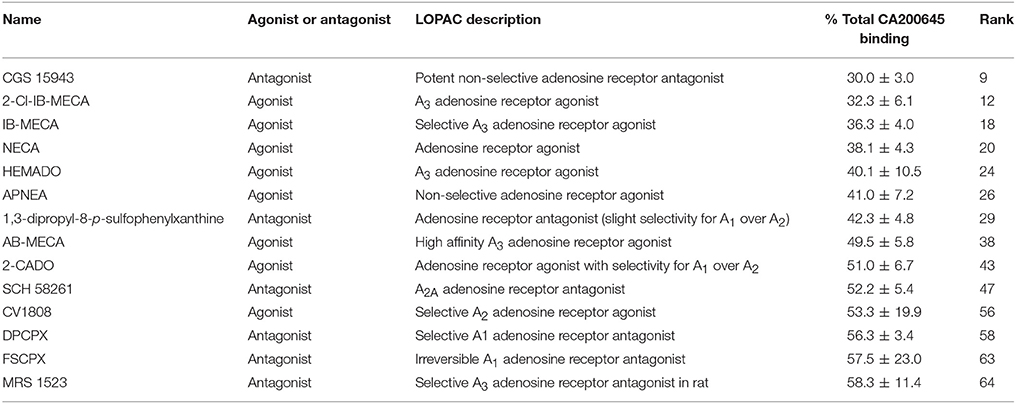
Table 3. Known A3AR ligands in the LOPAC library: Compounds within the LOPAC library that have known activity at adenosine receptors, their rank order in the full screen and the % of 25 nM CA200645 binding in the presence of 10 μM of these compounds.
Ten hits from the initial screen which demonstrated the biggest inhibition of CA200645 binding to the A3AR were investigated further and full inhibition curves for each compound were generated. We were unable to further test reactive blue 2 (position 4 in the full screen) as it is currently not available commercially. As shown in Figure 4, Table 4, four of the top ten compounds showed low- to sub-micromolar affinity for the A3AR. As expected the adenosine receptor antagonist CGS15943 displaced the binding of CA200645 at both the A3AR and A1AR in a concentration-dependent manner with the expected affinity (Figure 4, Table 1). As CGS15943 was one of the top ten hits from the initial screen it was also tested in cells expressing the β2AR and had no effect on the binding of BODIPY-TMR-CGP (Figure 4). Three further compounds, retinoic acid p-hydroxyanilide (fenretinide), K114 and SU 6656, were found to inhibit the binding of CA20065 to the A3AR in a concentration-dependent manner with affinity values in the sub-micromolar range, roughly 10-fold lower than CGS15943 (Figure 4, Table 4). Five further hits (BIO, rottlerin, quercetin, PD173952 and kenpaullone) only displaced the binding of CA200645 at the highest concentration tested (10 μM), prohibiting an accurate affinity determination. For those four compounds showing micromolar affinity, the selectivity of their interaction with the A3AR was determined by investigating their ability to bind to A1AR and β2AR. Both K114 and retinoic acid p-hydroxyanilide inhibited the binding of CA200645 at the A1AR with similar affinity to that observed at the A3AR. SU 6656 only inhibited binding at the highest concentration tested and the affinity was not calculated. None of the other compounds showed any measureable activity at the A1AR. When tested in CHO cells expressing the β2AR, no significant inhibition of BODIPY-TMR-CGP binding was observed for any of the 10 compounds screened but the control β2AR antagonist propranolol had the expected affinity (pKi = 8.72 ± 0.14, n = 3). There was an increase in fluorescence in the presence of 10 μM SU 6656 (128.4 ± 18.4%). However this was small compared to the increase seen with 10 nM BODIPY-TMR-CGP and the large increase in fluorescence in the presence of BIO (pEC50 = 5.84 ± 0.13). This is likely to be due to these compounds interfering with the BODIPY-TMR fluorescence signal, which was not observed when using the more red-shifted BODIPY 630/650 fluorophore in the A1AR and A3AR binding assays.
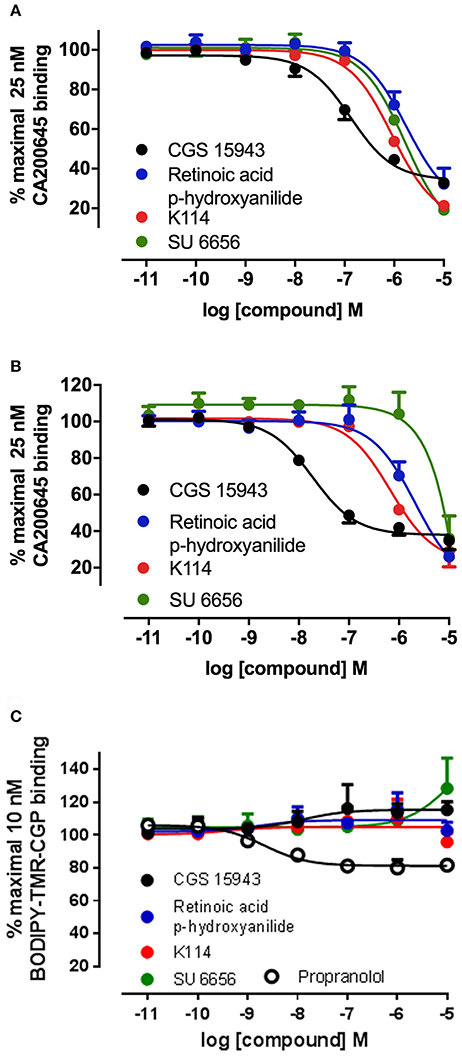
Figure 4. Competition binding curves at the A1AR, A3AR, and β2AR for three hits identified from the LOPAC library. CHO cell lines stably expressing A1AR (A), A3AR (B), or β2AR (C) were incubated with 25 nM CA200645 (A3AR and A1AR) or 10 nM BODIPY-TMR-CGP (β2AR) in the absence or in the presence of increasing concentrations of the indicated compounds. Values are mean ± SEM from three independent experiments performed in triplicate.
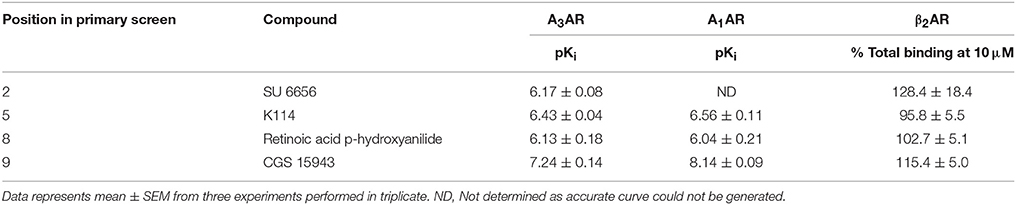
Table 4. Affinity of selected hits from the LOPAC library at the A3AR, A1AR, and β2AR: Compounds were tested on CHO cells expressing the A3AR, A1AR, and β2AR in the HTS format fluorescent ligand binding assay using 25 nM CA200645 as the tracer for A3AR and A1AR and 10 nM of BODIPY-TMR-CGP for β2AR.
Molecular Modeling of Selected LOPAC Hits at the A3AR
Using our previously established homology model of the human A3AR (Vernall et al., 2013) we sought to investigate potential binding poses for the three sub-micromolar compounds (retinoic acid p-hydroxyanilide (fenretinide), K114 and SU 6656) identified in the LOPAC screen which did not have previous literature precedent for interacting with this receptor sub-type. Using the commercially available docking software, CLC Drug Discovery Workbench, ligand and receptor binding pocket preparation was followed by targeted ligand docking. The highest scoring docked poses for K114, SU 6656 and retinoic acid p-hydroxyanilide were selected and are illustrated in Figure 5. All three compounds were able to engage via plausible poses to the A3R within the vicinity of the orthosteric binding pocket of this receptor.
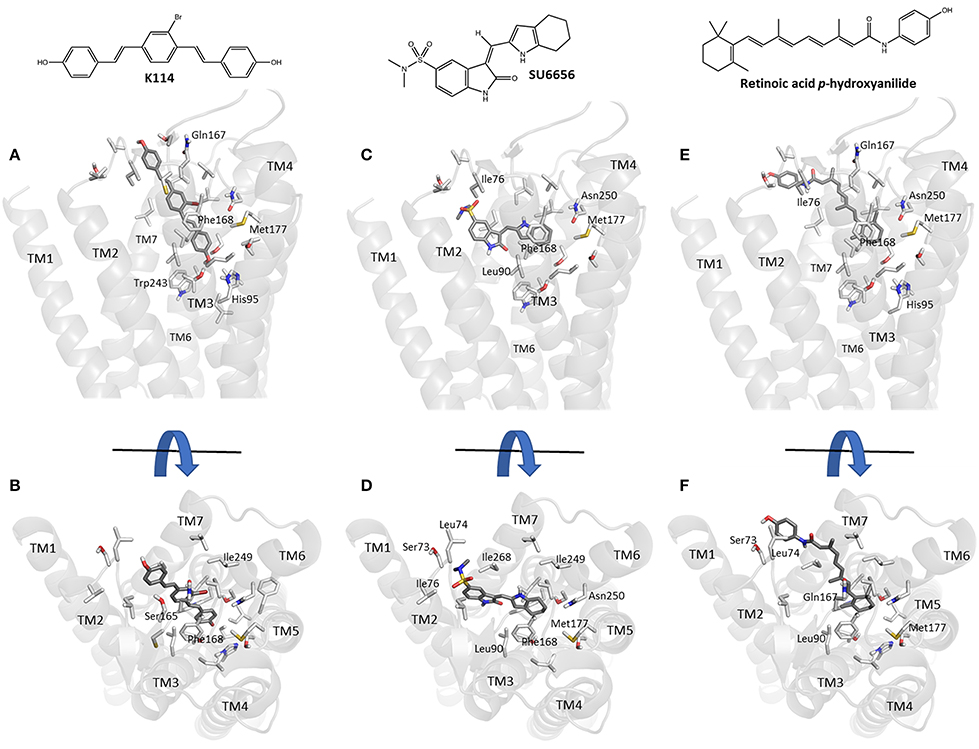
Figure 5. Molecular modeling simulation of K114, SU 6656 and retinoic acid p-hydroxyanilide binding to the A3AR. A side-on (A,C,E) and top-down (B,D,F) view of the top scoring binding poses for K114, SU 6656 and retinoic acid p-hydroxyanilide (dark gray liquorice coloring) respectively, bound into our previously reported A3AR receptor homology model (Vernall et al., 2013). Previously identified amino acid side chain residues associated with the orthosteric binding pocket (Squarcialupi et al., 2013) are represented in light gray liquorice coloring and labeled alongside the TM loop regions for clarity.
Discussion
Fluorescent ligands for GPCRs are a valuable tool in the study of multiple aspects of receptor pharmacology and they are a potential replacement for radiolabelled ligands in saturation and equilibrium binding studies to determine the affinity of labeled and unlabeled ligands (Stoddart et al., 2016). In this study, we aimed to further develop a previously described fluorescence based live cell binding assay that used a HCS system (Stoddart et al., 2012) to an assay that could be performed with un-tagged receptors on a HTS system. To this end, we chose the PHERAstar FS fluorescent plate reader since it allowed the determination of the optimal focal height for the fluorescence read and multiple scans per well. Use of the HTS system to obtain data resulted in a marked reduction in the time each 96-well plate took to process; from around 40 minutes per plate on the confocal HCS system for data collection and analysis to less than 3 minutes for the HTS system. This also produced a significant reduction in the amount of data that needed to be stored; 500 Mb per plate for HCS versus 160 Kb for HTS. Using the A3AR as a model system, we demonstrated that the data generated on the HTS system was in close agreement to that obtained on the HCS system, validating this system as a higher throughput methodology that would be essential for screening large compound libraries using fluorescence-based binding assays in whole cells.
Various methods using fluorescent ligands to measure ligand binding at GPCRs have been recently developed, each using a different approaches to measure the fluorescence of the bound ligand, including flow cytometry (Young et al., 2005; Hara et al., 2009; Kozma et al., 2013), fluorescence polarization (Cornelius et al., 2009; Kecskes et al., 2010) and resonance energy transfer based systems (Zwier et al., 2010; Stoddart et al., 2015a). Each method has advantages and disadvantages, for instance ligand depletion (fluorescence polarization) and the need to tag the receptor of interest (BRET and FRET). One limitation of the simple fluorescent intensity measurement used in the system described here is the potential for a low signal/noise ratio as a result of high levels of non-specific binding and the use of whole cells. As this technique measures total well fluorescence intensity it will be affected by both high levels of non-specific membrane binding and also non-specific uptake of the fluorescent ligand into the cells. As an example of this, for the A3AR the maximal reduction in the levels of CA200645 fluorescence measured in the presence of unlabeled ligands was 60% whilst that with BODIPY-TMR-CGP for the β1AR was only 20% (Figures 1C, 2B). This small signal/noise ratio for this ligand at the β1AR has been observed previously (Gherbi et al., 2014), although it is notable that even under these conditions, the method described here still allowed us to generate robust data within this small signal/noise window. The proximity-based assays (e.g., NanoBRET; Stoddart et al., 2015a) overcome this issue but they obviously require genetic modification of the extracellular N-terminus of the receptor with a fluorescent or luminescent protein, which precludes their use on native receptors—a main aim of the assay developed in this study. What is also clear from this point of view, is that the limit of this signal to noise ratio is likely to be highly dependent on both the pharmacological and photophysical properties of the fluorescent ligand, as we have previously demonstrated (Vernall et al., 2013). To progress the use of this assay to use with endogenously expressed untagged receptors, consideration should also be given to fluorescent ligand selectivity in situations where multiple receptor subtypes are often co-expressed; this is particularly true for adenosine receptors. To this end, the demonstration that this assay also works with a highly A3AR selective ligand, AV039 (Vernall et al., 2012) is important.
To demonstrate the utility of this assay system for compound screening, we investigated if we could identify known ligands for the A3AR within a library of pharmacologically active compounds (LOPAC). Within the LOPAC library there were 37 compounds identified as ligands for adenosine receptors. For the 1,263 compounds screened, we defined a hit as a compound that inhibited more than 40% of the total CA200645 binding. Using these criteria, we identified 67 hits, of which 14 had previously described activity at adenosine receptors (Table 3). Of these, four were the known A3R selective agonists, 2-Cl-IB-MECA (Gallo-Rodriguez et al., 1994), IB-MECA (Klotz et al., 1998), AB-MECA (Klotz et al., 1998) and HEMADO (Klotz et al., 2007), and the A3R selective antagonist MRS1523 (Li et al., 1998). A further five compounds were known to be non-selective at this adenosine receptor subtype [CGS15943 (Ongini et al., 1999), NECA (Gao et al., 2004), APNEA (Gao et al., 2004), 2-CADO (van Galen et al., 1994) and 1,3-dipropyl-8-p-sulfophenylxanthine (Daly et al., 1985)]. The remaining four compounds were SCH 58261, CV1808, DPCPX and FSCPX. SCH 58261 is widely described as an A2A selective and DPCPX as an A1AR-selective antagonist, and both retain affinity in the μM range for the A3AR (Ongini et al., 1999; Stoddart et al., 2012). FSCPX is an irreversible antagonist at the A1AR (van Muijlwijk-Koezen et al., 2001) but to date it had not been tested at other adenosine receptor subtypes. Our data from this screen indicates that FSCPX is likely to retain activity at the A3R at least in the low μM range and this is also true for CV1808 that has been described as an agonist at the A2AAR (Dionisotti et al., 1997). A variety of different compounds that act at different (i.e., non-A3AR) adenosine receptors were included in the library and as expected were not identified as hits in our screen (Supporting Information Table 1). These included A1AR selective agonists and antagonists such as R-PIA (Klotz et al., 1998) and CPT (Dalpiaz et al., 1998), A2AAR selective agonists and antagonists such as CGS 21680 (Klotz et al., 1998) and CSC (Jacobson et al., 1993), and the A2BAR selective antagonist alloxazine (Ji et al., 2001). A variety of low affinity non-selective antagonists and agonists were also present in the library including adenosine, theophylline, caffeine and paraxanthine that have reported affinity at the A3AR in the 13-100 μM range (Jacobson et al., 1999; Fredholm et al., 2001). Due to the concentration of CA200645 (25 nM) used in the primary screen only compounds with an affinity of <10 μM would be expected to be identified as a hit. Overall, the assay performed well at identifying all the compounds with known activity at the A3AR.
We found three compounds in the library that displayed unexpected sub-micromolar affinity at the A3AR (Figure 4, Table 4). These were K114, retinoic acid p-hydroxyanilide and SU 6556. K114 is used to identify amyloid lesions from Aβ peptide, α-synuclien and tau through an increase in its fluorescence upon binding to these lesions. It is has minimal fluorescence in aqueous solution and has emission maxima of 550 nm that is unlikely to interfere with the emission of BY630 at 650 nm (Crystal et al., 2003). In addition, the assay described here monitors a decrease in fluorescence in the presence of inhibitors that would mean it would be more likely to give false-negatives rather than false-positives. Retinoic acid p-hydroxyanilide, also known as fenretinide or 4-HPR, is an analog of retinoic acid and is a potential therapy in the treatment of cancer due to its ability to induce apoptosis (Wu et al., 2001). It is possible that it was causing apoptosis of the cells in our assay system leading to a concurrent decrease in fluorescence but as the presence of retinoic acid p-hydroxyanilide had no effect in cells expressing the β2AR this is unlikely to be the case (Figure 4). SU6556 is a Src kinase inhibitor that has also been found to inhibit a variety of other kinases including Aurora C and AMPK (Bain et al., 2007). It also displayed slight selectivity for the A3AR over A1AR.
Docking of the sub-micromolar compounds identified in the LOPAC screen provided a plausible set of binding poses within the vicinity of the established orthosteric A3AR binding pocket (Figure 5). K114 bound in a fully extended form with one of the terminal phenols optimally positioned to engage in a hydrogen bond interaction with the side-chain of Thr94. Meanwhile, the remaining vinyl-linked aromatic moieties pass through a hydrophobic channel created by Ile76, Val169, Leu90, Leu246, Ile249, Leu264, Ile268, and Phe168; the latter engaging via a face-to-face pi-stacking interaction. SU 6656 favored binding higher up in the orthosteric pocket with the 4,5,6,7-tetrahydroindolyl portion of the molecule engaging in a face-to-face interaction with Phe168, with the hydrophobic interactions predominating with Leu90, Val65, Ile268, and Leu246. Finally, retinoic acid p-hydroxyanilide displayed a binding pose passing through the same hydrophobic channel observed with K114. The 1,3,3-trimethylcyclohex-1-enyl region of the molecule was positioned deepest into the binding pocket engaging in hydrophobic interactions with residues Leu246, Ile249, Met177, and Phe168. The p-hydroxyanilde region of the molecule was positioned in such a way as to allow a face-to-edge interaction with Tyr265 at the top of transmembrane helix 7. With the predominance of aromatic and hydrophobic interactions observed between the receptor and the three ligands discussed, this would seem to correlate well with the experimental binding affinities whilst also offering the potential to undertake productive modifications of these compounds to potentially enhance their overall binding interactions.
In conclusion, we have shown that a simple intensity based fluorescent ligand binding assay can be modified to work in a potentially high throughput format, giving significant advances in both speed and data volume compared to previous high content versions. The assay allows screening of a small compound library in live cells, and can assess binding to the unmodified native receptors. The assays performed well under test conditions, identifying both known adenosine receptor ligands in a focused library as well as novel potential ligand scaffolds. Further work on establishing this assay to screen at endogenous A3AR in a mixed receptor background will be important to allow subsequent screens to be performed under more physiological conditions.
Experimental Procedures
Chemicals
Known GPCR antagonists were purchased from Tocris Bioscience and G418 was obtained from Invitrogen. Fetal calf serum was obtained from PAA Laboratories and L-glutamine from Lonza. All other biological reagents were obtained from Sigma-Aldrich. CA200645 was obtained from CellAura Technologies. BODIPY-TMR-CGP (BODIPY-TMR-(±)-CGP 12177) was purchased from Molecular Probes. AV039 and AV019 were synthesized in house as previously described (Vernall et al., 2012). The LOPAC library was obtained from Sigma-Aldrich.
Cell Culture
CHO-K1 cells stably expressing the human A3AR (Vernall et al., 2012), β1AR (Guo et al., 2012), β2AR (Baker et al., 2002) or the human A1AR (May et al., 2010) were maintained in DMEM/F12 medium containing 10% fetal calf serum and 2 mM L-glutamine at 37°C in a humidified atmosphere of air/CO2 (19:1).
Fluorescence Competition Binding Assay
CHO cells stably expressing the A3AR, A1AR, β1AR or β2AR were seeded into the central 60 wells (for high content confocal analysis) or every well (high throughput analysis) of a 96-well clear-bottomed, black-walled plate (Greiner BioOne) and grown to confluency. On the day of experiment, normal growth medium was removed and cells washed twice with HEPES-buffered saline solution (HBSS; 10 mM HEPES, 10 mM glucose, 145 mM NaCl, 5 mM KCl, 1 mM MgSO4, 2 mM sodium pyruvate, 1.3 mM CaCl2, 1.5 mM NaHCO3, pH 7.4) pre-warmed to 37°C. Fresh HBSS was added to each well followed by the addition of the required concentration of unlabeled compound and the respective fluorescent ligands (25 nM CA200645, 5 nM AV039 or 10 nM BODIPY-TMR-CGP). Cells were incubated for 1h at 37°C/5% CO2. Buffer was then removed from each well, cells washed once in HBSS and fresh HBSS added at room temperature. Plates were then immediately subjected to high content or HTS analysis as detailed below.
High Content Screening
High content analysis was conducted as previously described (Stoddart et al., 2012). Briefly, plates were imaged using an ImageXpress Ultra confocal plate reader, which captured four central images per well using a Plan Fluor 40x NA0.6 extra-long working distance objective. CA200645 was excited at 635 nm and emission collected through a 640–685 nm band pass filter. Total image intensity was obtained using a modified multi-wavelength cell scoring algorithm within the MetaXpress software (MetaXpress 2.0, Molecular Devices).
High Throughput Screening
High throughput analysis was performed using a PHERAstar FS plate reader (BMG Technologies). Fluorescent intensity of each well was assessed by bottom scanning using the following optical modules: excitation 540 nm and emission 590 nm (for BODIPY-TMR-CGP-labeled cells), or excitation 630 nm and emission 650 nm (for the BY630 compounds CA200645 and AV039). Optimal focal height was determined automatically and total fluorescence intensity was assessed by taking 81 reads per well.
Screening of the LOPAC Library of Pharmacological Active Compounds
The LOPAC compound library contained 1263 compounds and each compound was provided as a pre-dissolved solution in 10 mM in DMSO. Compound plates containing 2 μl of compound per well were provided by the University of Nottingham Managed Compound Collection. Each plate contained 40 compounds from the LOPAC library together with positive and blank control samples. For the blank controls, 2 μl of DMSO was added per well and for the positive controls the A3AR antagonist MRS1220 (10 μM final concentration) was used. The compounds were diluted to 100 μM in HBSS prior to assay. Each compound was tested in duplicate at a final concentration of 10 μM on three separate experimental days. Experiment was carried out as detailed above using the A3AR expressing cell line and 25 nM CA200645 as the tracer ligand. Data were normalized on a per plate basis to the fluorescence observed in blank control wells.
The 67 compounds that inhibited by more than 40% the total binding of CA200645 compared to blank controls were classed as hits. From this list 16 compounds were selected for secondary screening to determine their IC50 values and binding affinity. This was achieved by investigating the effect of increasing concentrations of each inhibitor on the specific binding of 25 nM CA200645 or 10 nM BODIPY-TMR-CGP in cells expressing the A3AR, A1AR or β2AR.
Molecular Modeling
Using our previously reported homology model of the human A3AR (Vernall et al., 2013) and the CLC Drug Discovery Workbench software package (Version 3.0.2, Qiagen, Netherlands), the protein target was prepared with no water molecules present. Before setting up the docking experiments, the binding site was generated as a 13 Å sphere centered around the established orthosteric pocket. All small molecules were constructed using ChemDraw Professional 16.0 (CambridgeSoft, Cambridge, MA, USA) and imported into the docking programme using the Balloon PlugIn (http://users.abo.fi/mivainio/balloon) (Vainio and Johnson, 2007) to afford the lowest energy conformer for each ligand. During the docking process, each ligand underwent 1000 individual iterations, with the conformation of each ligand set as flexible, allowing full movement around all rotatable bonds, whilst the protein was held as a rigid structure. The best scoring pose for each ligand was returned using the PLANTSPLP algorithm to determine that docking score (Korb et al., 2009) and the best ranked compounds were selected and their binding residues observed using the CLC Drug Discovery Workbench visualization tool.
Data Analysis
Competition binding curves were fitted to the following equation using GraphPad Prism 5 (GraphPad Software):
where [A] is the concentration of competing drug and IC50 is the molar concentration of ligand required to inhibit 50% of the specific binding of a fixed concentration [L] of the appropriate fluorescent ligand. The IC50 values obtained were converted to Ki values using the following equation:
where [L] is the concentration and KD is the equilibrium dissociation constant of the fluorescent ligand. The KD values for the fluorescent ligands used were 11.0 nM and 3.11 nM for CA200645 at the A1AR and A3AR respectively (Stoddart et al., 2012). KD values for BODIPY-TMR-CGP were taken from Baker et al. (2003).
The Z′ values were calculated on a per plate basis using the following equation:
where μp and σp are the mean and standard deviation from the control wells (DMSO only) and μn and σn are the mean and standard deviation from the MRS1220 treated wells.
Author Contributions
SH, SB, and BK: Conceived the study; MA, LS, SB, BK, and SH: Participated in research design; MA and LS: Performed the experiments and data analysis; KG: Performed the beta receptor screening experiments and analyzed the data; BK: Performed the molecular docking studies; MA, LS, BK, SB, and SH: All wrote or contributed to the writing and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Medical Research Council (grant numbers G0800006, MR/N020081/1).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2017.00908/full#supplementary-material
References
Antonioli, L., Csóka, B., Fornai, M., Colucci, R., Kókai, E., Blandizzi, C., et al. (2014). Adenosine and inflammation: what's new on the horizon? Drug Discov. Today. 19, 1051–1068. doi: 10.1016/j.drudis.2014.02.010
Bain, J., Plater, L., Elliott, M., Shpiro, N., Hastie, C. J., McLauchlan, H., et al. (2007). The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315. doi: 10.1042/BJ20070797
Baker, J. G., Hall, I. P., and Hill, S. J. (2002). Pharmacological characterization of CGP 12177 at the human beta(2)-adrenoceptor. Br. J. Pharmacol. 137, 400–408. doi: 10.1038/sj.bjp.0704855
Baker, J. G., Hall, I. P., and Hill, S. J. (2003). Pharmacology and direct visualisation of BODIPY-TMR-CGP: a long-acting fluorescent β2-adrenoceptor agonist. Br. J. Pharmacol. 139, 232–242. doi: 10.1038/sj.bjp.0705287
Borea, P. A., Varani, K., Vincenzi, F., Baraldi, P. G., Tabrizi, M. A., Merighi, S., et al. (2015). The A3 adenosine receptor: history and perspectives. Pharmacol. Rev. 67, 74–102. doi: 10.1124/pr.113.008540
Cao, H. L., Liu, Z. J., and Chang, Z. (2017). Cordycepin induces apoptosis in human bladder cancer cells via activation of A3 adenosine receptors. Tumour Biol. 39. doi: 10.1177/1010428317706915
Castro, M., Nikolaev, V. O., Palm, D., Lohse, M. J., and Vilardaga, J.-P. (2005). Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc. Natl. Acad. Sci. U.S.A. 102, 16084–16089. doi: 10.1073/pnas.0503942102
Cohen, S., Barer, F., Bar-Yehuda, S., IJzerman, A. P., Jacobson, K. A., and Fishman, P. (2014). A3 adenosine receptor allosteric modulator induces an anti-inflammatory effect: in vivo studies and molecular mechanism of action. Mediators Inflamm. 2014:708746. doi: 10.1155/2014/708746
Cornelius, P., Lee, E., Lin, W., Wang, R., Werner, W., Brown, J. A., et al. (2009). Design, synthesis, and pharmacology of fluorescently labeled analogs of serotonin: application to screening of the 5-HT2C receptor. J. Biomol. Screen. 14, 360–370. doi: 10.1177/1087057109331804
Crystal, A. S., Giasson, B. I., Crowe, A., Kung, M. P., Zhuang, Z. P., Trojanowski, J. Q., et al. (2003). A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. J. Neurochem. 86, 1359–1368. doi: 10.1046/j.1471-4159.2003.01949.x
Dalpiaz, A., Townsend-Nicholson, A., Beukers, M. W., Schofield, P. R., and IJzerman, A. P. (1998). Thermodynamics of full agonist, partial agonist, and antagonist binding to wild-type and mutant adenosine A1 receptors. Biochem. Pharmacol. 56, 1437–1445.
Daly, J. W., Padgett, W., Shamim, M. T., Butts-Lamb, P., and Waters, J. (1985). 1,3-Dialkyl-8-(p-sulfophenyl)xanthines: potent water-soluble antagonists for A1- and A2-adenosine receptors. J. Med. Chem. 28, 487–492.
Dionisotti, S., Ongini, E., Zocchi, C., Kull, B., Arslan, G., and Fredholm, B. B. (1997). Characterization of human A2A adenosine receptors with the antagonist radioligand [3H]-SCH 58261. Br. J. Pharmacol. 121, 353–360.
Fredholm, B. B., IJzerman, A. P., Jacobson, K. A., Linden, J., and Müller, C. E. (2011). International Union of Basic And Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update. Pharmacol. Rev. 63, 1–34. doi: 10.1124/pr.110.003285
Fredholm, B. B., Irenius, E., Kull, B., and Schulte, G. (2001). Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem. Pharmacol. 61, 443–448. doi: 10.1016/S0006-2952(00)00570-0
Gallo-Rodriguez, C., Ji, X. D., Melman, N., Siegman, B. D., Sanders, L. H., Orlina, J., et al. (1994). Structure-activity relationships of N6-benzyladenosine-5'-uronamides as A3-selective adenosine agonists. J. Med. Chem. 37, 636–646.
Gao, Z. G., Mamedova, L. K., Chen, P., and Jacobson, K. A. (2004). 2-Substituted adenosine derivatives: affinity and efficacy at four subtypes of human adenosine receptors. Biochem. Pharmacol. 68, 1985–1993. doi: 10.1016/j.bcp.2004.06.011
Gherbi, K., Briddon, S. J., and Hill, S. J. (2014). Detection of the secondary, low-affinity β1-adrenoceptor site in living cells using the fluorescent CGP 12177 derivative BODIPY-TMR-CGP. Br. J. Pharmacol. 171, 5431–5445. doi: 10.1111/bph.12858
González-Fernández, E., Sánchez-Gómez, M. V., Pérez-Samartín, A., Arellano, R. O., and Matute, C. (2014). A3 Adenosine receptors mediate oligodendrocyte death and ischemic damage to optic nerve. Glia 62, 199–216. doi: 10.1002/glia.22599
Guo, J., Song, L., Liu, M., and Mahon, M. J. (2012). Fluorescent ligand-directed co-localization of the parathyroid hormone 1 receptor with the brush-border scaffold complex of the proximal tubule reveals hormone-dependent changes in ezrin immunoreactivity consistent with inactivation. Biochim. Biophys. Acta 1823, 2243–2253. doi: 10.1016/j.bbamcr.2012.09.010
Hara, T., Hirasawa, A., Sun, Q., Koshimizu, T. A., Itsubo, C., Sadakane, K., et al. (2009). Flow cytometry-based binding assay for GPR40 (FFAR1; Free Fatty Acid Receptor 1). Mol. Pharmacol. 75, 85–91. doi: 10.1124/mol.108.052225
Headrick, J. P., Ashton, K. J., Rose'meyer, R. B., and Peart, J. N. (2013). Cardiovascular adenosine receptors: expression, actions and interactions. Pharmacol. Ther. 140, 92–111. doi: 10.1016/j.pharmthera.2013.06.002
Hussain, A., Gharanei, A. M., Nagra, A. S., and Maddock, H. L. (2014). Caspase inhibition via A3 adenosine receptors: a new cardioprotective mechanism against myocardial infarction. Cardiovasc. Drugs Ther. 28, 19–32. doi: 10.1007/s10557-013-6500-y
Iturrioz, X., Alvear-Perez, R., De Mota, N., Franchet, C., Guillier, F., Leroux, V., et al. (2010). Identification and pharmacological properties of E339-3D6, the first nonpeptidic apelin receptor agonist. FASEB J. 24, 1506–1517. doi: 10.1096/fj.09-140715
Jacobson, K. A., Gallo-Rodriguez, C., Melman, N., Fischer, B., Maillard, M., van Bergen, A., et al. (1993). Structure-activity relationships of 8-styrylxanthines as A2-selective adenosine antagonists. J. Med. Chem. 36, 1333–1342.
Jacobson, K. A., Ijzerman, A. P., and Linden, J. (1999). 1,3-dialkylxanthine derivatives having high potency as antagonists at human A2B adenosine receptors. Drug Dev. Res. 47, 45–53.
Ji, X., Kim, Y. C., Ahern, D. G., Linden, J., and Jacobson, K. A. (2001). [3H]MRS 1754, a selective antagonist radioligand for A(2B) adenosine receptors. Biochem. Pharmacol. 61, 657–663. doi: 10.1016/S0006-2952(01)00531-7
Joshaghani, H. R., Jafari, S. M., Aghaei, M., Panjehpour, M., and Abedi, H. (2017). A3 adenosine receptor agonist induce G1 cell cycle arrest via Cyclin, D., and cyclin-dependent kinase 4 pathways in OVCAR-3 and Caov-4 cell lines. J. Cancer Res. Ther. 13, 107–112. doi: 10.4103/0973-1482.199381
Kecskes, M., Kumar, T. S., Yoo, L., Gao, Z. G., and Jacobson, K. A. (2010). Novel Alexa Fluor-488 labeled antagonist of the A(2A) adenosine receptor: application to a fluorescence polarization-based receptor binding assay. Biochem. Pharmacol. 80, 506–511. doi: 10.1016/j.bcp.2010.04.027
Klotz, K.-N., Falgner, N., Kachler, S., Lambertucci, C., Vittori, S., Volpini, R., et al. (2007). [3H]HEMADO– a novel tritiated agonist selective for the human adenosine A3 receptor. Eur. J. Pharmacol. 556, 14–18. doi: 10.1016/j.ejphar.2006.10.048
Klotz, K. N., Hessling, J., Hegler, J., Owman, C., Kull, B., Fredholm, B. B., et al. (1998). Comparative pharmacology of human adenosine receptor subtypes - characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedebergs Arch. Pharmacol. 357, 1–9.
Korb, O., Stutzle, T., and Exner, T. E. (2009). Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 49, 84–96. doi: 10.1021/ci800298z
Kozma, E., Gizewski, E. T., Tosh, D. K., Squarcialupi, L., Auchampach, J. A., and Jacobson, K. A. (2013). Characterization by flow cytometry of fluorescent, selective agonist probes of the A(3) adenosine receptor. Biochem. Pharmacol. 85, 1171–1181. doi: 10.1016/j.bcp.2013.01.021
Kuder, K. J., and Kieć-Kononowicz, K. (2014). Fluorescent GPCR ligands as new tools in pharmacology-update, years 2008-early 2014. Curr. Med. Chem. 21, 3962–3975. doi: 10.2174/0929867321666140826120058
Li, A. H., Moro, S., Melman, N., Ji, X. D., and Jacobson, K. A. (1998). Structure-activity relationships and molecular modeling of 3,5-diacyl-2,4-dialkylpyridine derivatives as selective A(3) adenosine receptor antagonists. J. Med. Chem. 41, 3186–3201.
Little, J. W., Ford, A., Symons-Liguori, A. M., Chen, Z., Janes, K., Doyle, T., et al. (2015). Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 138, 28–35. doi: 10.1093/brain/awu330
May, L. T., Self, T. J., Briddon, S. J., and Hill, S. J. (2010). The effect of allosteric modulators on the kinetics of agonist-G protein-coupled receptor interactions in single living cells. Mol. Pharmacol. 78, 511–523. doi: 10.1124/mol.110.064493
Montinaro, A., Iannone, R., Pinto, A., and Morello, S. (2013). Adenosine receptors as potential targets in melanoma. Pharmacol. Res. 76, 34–40. doi: 10.1016/j.phrs.2013.07.002
Mulloy, D. P., Sharma, A. K., Fernandez, L. G., Zhao, Y., Lau, C. L., Kron, I. L., et al. (2013). Adenosine A3 receptor activation attenuates lung ischemia-reperfusion injury. Ann. Thorac. Surg. 95, 1762–1767. doi: 10.1016/j.athoracsur.2013.01.059
Nakamura, K., Shinozuka, K., and Yoshikawa, N. (2015). Anticancer and antimetastatic effects of cordycepin, an active component of Cordyceps sinensis. J. Pharmacol. Sci. 127, 53–56. doi: 10.1016/j.jphs.2014.09.001
Ohana, G., Cohen, S., Rath-Wolfson, L., and Fishman, P. (2016). A3 adenosine receptor agonist, CF102, protects against hepatic ischemia/reperfusion injury following partial hepatectomy. Mol. Med. Rep. 14, 4335–4341. doi: 10.3892/mmr.2016.5746
Ongini, E., Dionisotti, S., Gessi, S., Irenius, E., and Fredholm, B. B. (1999). Comparison of CGS 15943, ZM 241385 and SCH 58261 as antagonists at human adenosine receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 359, 7–10.
Ranganathan, A., Stoddart, L. A., Hill, S. J., and Carlsson, J. (2015). Fragment-based discovery of subtype-selective adenosine receptor ligands from homology models. J. Med. Chem. 58, 9578–9590. doi: 10.1021/acs.jmedchem.5b01120
Ravani, A., Vincenzi, F., Bortoluzzi, A., Padovan, M., Pasquini, S., Gessi, S., et al. (2017). Role and function of A(2A) and A3 adenosine receptors in patients with ankylosing spondylitis, psoriatic arthritis and rheumatoid arthritis. Int. J. Mol. Sci. 18:E697. doi: 10.3390/ijms18040697
Squarcialupi, L., Colotta, V., Catarzi, D., Varano, F., Filacchioni, G., Varani, K., et al. (2013). 2-Arylpyrazolo[4,3-d]pyrimidin-7-amino derivatives as new potent and selective human A3 adenosine receptor antagonists. Molecular modeling studies and pharmacological evaluation. J. Med. Chem. 56, 2256–2269. doi: 10.1021/jm400068e
Stoddart, L. A., Johnstone, E. K., Wheal, A. J., Goulding, J., Robers, M. B., Machleidt, T., et al. (2015a). Application of BRET to monitor ligand binding to GPCRs. Nat. Methods 12, 661–663. doi: 10.1038/nmeth.3398
Stoddart, L. A., Kilpatrick, L. E., Briddon, S. J., and Hill, S. J. (2015b). Probing the pharmacology of G protein-coupled receptors with fluorescent ligands. Neuropharmacology 98, 48–57. doi: 10.1016/j.neuropharm.2015.04.033
Stoddart, L. A., Vernall, A. J., Denman, J. L., Briddon, S. J., Kellam, B., and Hill, S. J. (2012). Fragment screening at adenosine-A(3) receptors in living cells using a fluorescence-based binding assay. Chem. Biol. 19, 1105–1115. doi: 10.1016/j.chembiol.2012.07.014
Stoddart, L. A., White, C. W., Nguyen, K., Hill, S. J., and Pfleger, K. D. (2016). Fluorescence- and bioluminescence-based approaches to study GPCR ligand binding. Br. J. Pharmacol. 173, 3028–3037. doi: 10.1111/bph.13316
Tomasch, M., Schwed, J. S., Kuczka, K., Meyer Dos Santos, S., Harder, S., Nusing, R. M., et al. (2012). Fluorescent human EP3 receptor antagonists. ACS Med. Chem. Lett. 3, 774–779. doi: 10.1021/ml300191g
Tosh, D. K., Padia, J., Salvemini, D., and Jacobson, K. A. (2015). Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain. Purinergic Signal. 11, 371–387. doi: 10.1007/s11302-015-9459-2.
Vainio, M. J., and Johnson, M. S. (2007). Generating conformer ensembles using a multiobjective genetic algorithm. J. Chem. Inf. Model. 47, 2462–2474. doi: 10.1021/ci6005646
van Galen, P. J., van Bergen, A. H., Gallo-Rodriguez, C., Melman, N., Olah, M. E., IJzerman, I. J., et al. (1994). A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol. Pharmacol. 45, 1101–1111.
van Muijlwijk-Koezen, J. E., Timmerman, H., van der Sluis, R. P., van de Stolpe, A. C., Menge, W. M., Beukers, M. W., et al. (2001). Synthesis and use of FSCPX, an irreversible adenosine A1 antagonist, as a ‘receptor knock-down’ tool. Bioorg. Med. Chem. Lett. 11, 815–818. doi: 10.1016/S0960-894X(01)00069-5
Vernall, A. J., Stoddart, L. A., Briddon, S. J., Hill, S. J., and Kellam, B. (2012). Highly potent and selective fluorescent antagonists of the human adenosine A(3) receptor based on the 1,2,4-triazolo 4,3-a quinoxalin-1-one Scaffold. J. Med. Chem. 55, 1771–1782. doi: 10.1021/jm201722y
Vernall, A. J., Stoddart, L. A., Briddon, S. J., Ng, H. W., Laughton, C. A., Doughty, S. W., et al. (2013). Conversion of a non-selective adenosine receptor antagonist into A3-selective high affinity fluorescent probes using peptide-based linkers. Org. Biomol. Chem. 11, 5673–5682. doi: 10.1039/c3ob41221k
Wu, J. M., DiPietrantonio, A. M., and Hsieh, T. C. (2001). Mechanism of fenretinide (4-HPR)-induced cell death. Apoptosis 6, 377–388. doi: 10.1023/A:1011342220621
Yoshida, K., Ito, M., and Matsuoka, I. (2017). Divergent regulatory roles of extracellular ATP in the degranulation response of mouse bone marrow-derived mast cells. Int. Immunopharmacol. 43, 99–107. doi: 10.1016/j.intimp.2016.12.014
Young, S. M., Bologa, C., Prossnitz, E. R., Oprea, T. I., Sklar, L. A., and Edwards, B. S. (2005). High-throughput screening with HyperCyt (R) flow cytometry to detect small molecule formylpeptide receptor ligands. J. Biomol. Screen. 10, 374–382. doi: 10.1177/1087057105274532
Keywords: adenosine receptors, fluorescent ligands, adenosine A3 receptor, high throughput screening, LOPAC library
Citation: Arruda MA, Stoddart LA, Gherbi K, Briddon SJ, Kellam B and Hill SJ (2017) A Non-imaging High Throughput Approach to Chemical Library Screening at the Unmodified Adenosine-A3 Receptor in Living Cells. Front. Pharmacol. 8:908. doi: 10.3389/fphar.2017.00908
Received: 17 October 2017; Accepted: 28 November 2017;
Published: 13 December 2017.
Edited by:
Kenneth A. Jacobson, National Institutes of Health (NIH), United StatesReviewed by:
Vittoria Colotta, University of Florence, ItalyEddy Sotelo, Universidade de Santiago de Compostela, Spain
Copyright © 2017 Arruda, Stoddart, Gherbi, Briddon, Kellam and Hill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen J. Briddon, stephen.briddon@nottingham.ac.uk
Stephen J. Hill, stephen.hill@nottingham.ac.uk
†These authors have contributed equally to this work.