Perspectives in Super-Resolved Fluorescence Microscopy: What Comes Next?
 Christoph Cremer1,2,3*
Christoph Cremer1,2,3*  Udo Birk1,2
Udo Birk1,2- 1Superresolution Microscopy, Institute of Molecular Biology, Mainz, Germany
- 2Physics Department, University Mainz, Mainz, Germany
- 3Kirchhoff Institute of Physics, and Institute of Pharmacy and Molecular Biotechnology, University Heidelberg, Heidelberg, Germany
The Nobel Prize in Chemistry 2014 has been awarded to three scientists involved in the development of STED and PALM super-resolution fluorescence microscopy (SRM) methods. They have proven that it is possible to overcome the 100 year old theoretical limit for the resolution potential of light microscopy (of about 200 nm for visible light), which for decades has precluded a direct glimpse of the molecular machinery of life. None of the present-day super-resolution techniques have invalidated the Abbe limit for light optical detection; however, they have found clever ways around it. In this report, we discuss some of the challenges still to be resolved before arising SRM approaches will be fit to bring about the revolution in Biology and Medicine envisaged. Some of the challenges discussed are the applicability to image live and/or large samples, the further enhancement of resolution, future developments of labels, and multi-spectral approaches.
Introduction
Novel developments in optical microscopy technology and photophysics [1] made it possible to radically overcome the classical diffraction limit (ca. 200 nm laterally, 600 nm along the optical axis; also called the Abbe-limit [2], Equation 1) of conventional far-field microscopy.
In this formula for the resolution limit, the minimum width d of structural features to be resolved is proportional to the wavelength λ used for imaging, and inversely proportional to the refractive index n and the sine of the half opening angle α, under which light emanating from the sample is detected.
The recent discoveries which promise to revolutionize Biology and Medicine have been honored by the 2014 Nobel Prize in Chemistry [3] to Eric Betzig and William Moerner, for developing single fluorescent molecule detection, which later led to photoactivated localization microscopy (PALM); and to Stefan Hell, for the development of Stimulated Emission Depletion (STED) Microscopy, a “focused nanoscopy” method. Using these approaches, both optical resolution (smallest distance measureable between two adjacent point sources) and structural resolution (smallest structural detail determined based on the density of point sources resolved) can be enhanced very substantially. At the present state of the art, they allow a breathtaking light-optical resolution of biostructures down to about 5 nm [4], corresponding to 1/100th of the excitation wavelength.
The concept of “super-resolution” in microscopy was introduced in a 1955 paper by Toraldo di Francia [5], in which he defined super-resolution as the discrimination of details finer than the Abbe resolution limit. In the following, we shall use the term in this traditional, more general meaning to describe far field microscopy approaches allowing the analysis of structural details smaller than 200 nm [6, 7]. Among the most prominent concepts of super-resolution microscopy, the methods of focused nanoscopy (STED), of Single Molecule Localization Microscopy (SMLM), and of Structure Illumination Microscopy (SIM) are to be mentioned (for an introduction to these techniques see, e.g., [6–10]. To what extent the new super-resolution techniques developed in the labs of the three Nobel laureates and other groups can be applied in the most useful manner to the wealth of questions in Biology and Medicine as well as in the material sciences, depends on the possibility to realize the necessary experimental conditions.
For example, the use of fluorescent proteins usually follows expression of a genetically modified gene, hampering its application in a medical diagnostic context. As a solution of this problem, instead of using fluorescent proteins for localization microscopy, single standard fluorophores excited to “blinking” can also be effectively used [11–13] to obtain super-resolved images of relevant cellular structures, down to the molecular resolution level. However, in order to obtain reasonable frame rates and well separated (isolated) molecule signals, a compromise has to be made for the blinking rate. This problem has been paradigmatically solved in the original PALM technique described by Betzig et al. [14], in which he used 405 nm illumination to control the density of photoactivated Green Fluorescent Protein (GFP) molecules. A variety of solutions have been described for related approaches, according the fluorophore employed [6, 12, 13, 15–19]. So far, all these solutions are mostly based on experimental phenomenology, since the necessary quantum chemical calculations for precise predictions are still lacking.
Future Challenges
In the following, a few perspectives and challenges will be addressed how the fascinating new field of super-resolution fluorescence microscopy (SRM) might be further developed for the benefit of both fundamental research and medical applications.
Live Cell SRM Imaging
Both STED and PALM, as well as localization microscopy techniques using standard fluorophores, allow us to also obtain a super-resolved image of a live cell [20]. However, the road from single “snapshots” of live cells to in vivo observations over a longer period of time is difficult, to say the least, due to the phototoxicity of the illumination intensities required for the “squeezing” of the fluorescent area (in STED) or for photoswitching (in PALM, [d]STORM and related approaches). The two fundamental limitations for imaging (not only live cell) are signal-to-noise (SNR) level and the structure integrity (of the sample), and for localization microscopy additionally the photon statistics of single molecule fluorescence emission [21], i.e., the absolute signal level (Figure 1). Localization accuracy and data acquisition speed are limited by the finite photon emission rate, the finite total number of photons emitted from a dye molecule, the noise level (of the detector and of the background signal), and in localization microscopy also the blinking rate. Super-resolution images are based on composition of thousands of scan points (in STED) or thousands of frames acquired (in single molecule localization microscopy); therefore, live cell experiments with their inherent mobility rely on sufficiently fast data acquisition. However, specifically in Localization Microscopy, in order to detect a fluorescent signal, a number of photons must be registered with the detector. The absorption cross-section, the quantum yield, and the life-time of the excited (fluorescent) state of a dye molecule, as well as the transition to a non-fluorescent state (e.g., the triplet state) fundamentally limit the photon emission rate. Quantum dots exhibit excellent photophysical characteristics for SRM [22, 23], but are challenging to interface with biological systems and structures.
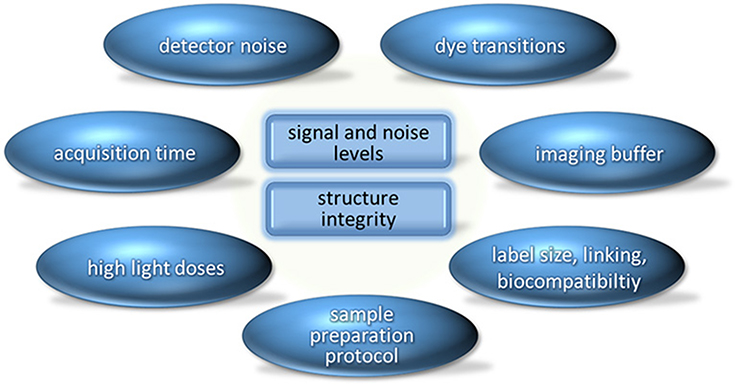
Figure 1. The two fundamental limitations in imaging (signal and noise levels; structure integrity of the sample) and one possible categorization of the most important factors in super-resolution microscopy which have an influence on these limitations. Modern detectors already have high quantum efficiency and low noise levels. Development of novel markers and improved sample preparation and imaging protocols therefore promise greater potential to address the present challenges in SRM.
The label size and solubility gives an indication for the accessibility (and hence the labeling efficiency) as well as for the distance between the measured position and the true location of the labeled target, while larger label sizes facilitate better targeting sensitivity. Additives such as the imaging buffer medium (which is used to control electronic transitions and photobleaching), or binding of the dye itself affect the structural integrity of the biological specimens, as does the incident light dose. In STED, circumventing the Abbe-limit means to apply STED intensities several orders of magnitude above the saturation intensity [3, 7]; in PALM (as described by Betzig et al. [14]), the low intensity illumination with UV light (405 nm) has to be repeated thousands of times (or used continuously); hence, with time also in this technique phototoxic doses of UV light are highly probable to be administered. While the disexcitation STED beam uses wavelengths that are much less phototoxic than the blue or UV wavelength used in PALM, the light doses are still sufficient to potentially inflict serious damage to the sample not only in biological samples, but also in the material sciences. Also in other localization microscopy approaches based on photoswitching of standard fluorophores using illumination wavelengths in the visible range only, phototoxicity remains a severe challenge for multiple frame live cell imaging. New dyes have to be developed with saturation/switching intensities sufficiently low for multiple time-lapse SRM imaging.
As an alternative way (or intermediate solution) to realize prolonged live cell SRM imaging, a combination of methods to circumvent the Abbe-limit of 200 nm should be highly useful. For example, various kinds of structured illumination excitation (SIE) [7] provide images at low or even very low illumination intensities. In structured illumination excitation, the sample illumination features high spatial variation (e.g., a sine-pattern or even a random pattern); thereby, fine object details which are inaccessible in classical microscopy are rendered accessible to the diffraction limited microscopic detection [7, 24–26], and dyes which are photostable can be applied. In this illumination mode, SIE techniques do not completely bypass the Abbe-limit (as the fluorescence response depends linearly on the illumination intensity), they can “stretch” it by a factor of about two to four in the object plane and also along the optical axis: But even a stretching of the 200 nm Abbe limit by a factor of two in all three dimensions can be of substantial importance in Biology and Medicine: In this case the observation volume is smaller by the factor 23 = 8, i.e., the 3D sampling can be enhanced by about one order of magnitude. In many cases, this will already be sufficient to obtain multiple “nanoimages” of live cells; these high resolution live cell observations can be followed by a last “snapshot” image taken with one of the SRM methods indicated above [27, 28]. This is analogous to the observation of a moving animal at a distance, followed by a close look at the animal after it has been captured; peculiarities of movement can be understood by combining the two aspects, the lower resolution time lapse series and the final, further enhanced super-resolution snapshot. Similar considerations also hold for other advanced microscopy approaches replacing the SIE for in vivo observations, such as confocal microscopy or 4Pi microscopy in the low illumination intensity mode. In many cases such as for the size measurement of biomolecular machines (e.g., replication or transcription factories in a cell) under circumstances where they are optically well separated from one another, it is not required to realize either an enhanced optical resolution according to the Abbe criterion (smallest distance that can be measured between two adjacent point sources), or an enhanced structural resolution according to the Nyquist criterion (related to the density of individual point sources detected). In these cases, structured illumination excitation at very low intensities can be used to determine such sizes in the live cell; since structured illumination has been shown to allow size determination down to about 30 nm [29–31], for many biologically relevant problems structured illumination can provide an information equivalent to very advanced STED/PALM/(d)STORM/SPDM etc. techniques. The techniques of structured illumination can be combined with saturation effects [32–34] also known under the term “RESOLFT”; however, the strain on the fluorescent dyes and the lower SNR have rendered its applicability in live cell studies limited [6]; with the developments of novel, bleaching-resistant probes these difficulties may be overcome.
As a long term perspective, localization microscopy methods using a multitude of photostable spectral characteristics (e.g., absorption bands, emission bands, fluorescence life-times, etc.) should facilitate multiple time-lapse SRM acquisitions with an enhanced resolution substantially below 100 nm and very low illumination conditions; this is possible since in the case of photostable fluorophores, a linear dependency on the illumination intensity only is required. In contrast, STED/SMLM still require non-linear absorption or specific (often toxic) chemical environments. When using photostable spectral characteristics, in contrast to the previous SRM methods, scanning or multiple frame registration is not required to obtain an enhanced optical resolution, e.g., in case of different emission wavelengths all fluorescence signals can be registered simultaneously; in the case of different fluorescence life times, ranging, e.g., from 1 ns to 1 ms, only a short registration time are needed. Successful “proof-of-principle” experiments toward such a solution have been reported already in the 1990s [35, 36] and in the beginning of the 2000s [37], indicating an optical resolution down to few tens of nm. If N types of molecules need to be discriminated within the observation volume of the point spread function (PSF), N different spectral signatures allowing independent registration are required, without prior assumptions on the 3D molecule arrangement. In recent years, these approaches to “photostability” based localization microscopy have been neglected in favor of “blinking” based localization microscopy approaches because only these latter promised to allow a highly enhanced structural resolution according to the Nyquist theorem. As discussed below, using new detectors and dyes it should become possible to meet also this challenge: In case photostable 20 molecule types would become distinguishable in a diffracted limited region (Airy disc) of 200 nm diameter, theoretically up to 20/[0.2 μm × 0.2 μm] = 500 = 22 × 22 molecules could be localized per μm2; or 1 molecule per 1000 nm/22 = 45 nm in each direction in the object plane. In real samples, such maximum values will of course not be achieved. But even if the real structural resolution obtained should not be better than about 100 nm, the great advantage compared with structured illumination would be the single molecule resolution capability. This would for example allow to count the molecules and to locate them relative to each other with a localization accuracy down to the few nm range in time and at illumination intensities sufficiently low for multiple time lapse acquisitions of live cells.
Figure 1 has been limited to the fundamental challenges. Each of these problems may be addressed by a large variety of quantitative technical approaches.
Further Enhancement of Resolution
As discussed in the context of live-cell experiments, the signals and noise levels, as well as the structural integrity fundamentally limit the achievable resolution. A particular sample preparation protocol might be necessary to preserve a specific nanostructure, but is not compatible with the imaging buffer protocol required for advanced optical microscopy methods. Furthermore, the high light doses in STED and various types of single molecule localization microscopy approaches lead to formation of chemical radicals and to fast photobleaching. Development of novel probes with low molecular weight and increased photostability in switching-based super-resolution microscopy is currently a major field of research. A combination of structured illumination excitation and other SRM techniques [38] such as “photostability” based localization microscopy at low intensities may be extremely useful also to further enhance substantially the optical resolution [39]: Computer simulations as well as proof-of-principle experiments have shown that the localization of a fluorescent point source by structured illumination excitation may enhance the localization accuracy and hence the optical resolution up to an order of magnitude, depending on the photon yield [40]. The underlying principle is similar to determining the position of the central maximum of the Airy disk from stochastic sampling of the diffraction pattern in 2D localization microscopy [41, 42]. Using an ideal optical system, in the localization microscopy mode a few thousand detected photons would be sufficient to obtain a subnanometer localization accuracy (and hence optical resolution), while under the same assumptions the resolution obtained by homogeneous wide field illumination would be 10 times worse. Since standard fluorophores presently used in localization microscopy may achieve even much higher photon yields, this goal can be realized. This should be possible not only in the object plane but even along the optical axis: Experimental observation using fluorescent point sources and axially structured illumination achieved an axial localization accuracy of 0.7 nm [43]; “virtual microscopy” simulations indicated that a detection of 10,000 photons should be sufficient for a localization accuracy of 0.3 nm (standard deviation) [40], corresponding to a two-point optical resolution around 0.7 nm (Full-Width-at-Half-Maximum/FWHM of a Gaussian shaped localization accuracy distribution). A technical challenge to realize such a microscope with “blinking” fluorophores will be the appropriately fast image acquisition in the structured illumination mode: Typically, the phase of the patterned excitation field has to be moved and rotated in a few milliseconds. This could be achieved already by present technology.
To summarize, from the point of Physics the construction of a combined structured illumination-localization microscope with an optical 3D resolution around 1 nm should become feasible. Such an “Avogadro” microscope (to honor the pioneer of Molecular Physics) would have a 3D resolution millions of times more enhanced than then conventional microscopy technology (Vobsconv/VobsAvogadro = [200 nm × 200 nm × 600 nm]/[1 nm × 1 nm × 1 nm] = 2,4 × 107. For example, this would make possible to use visible light microscopy to look directly into the nanostructure of the biomolecular complexes responsible for the functioning of the cell's machinery, in transcription, splicing, repair, translation, signaling etc.: If one assumes a biomolecular complex of about 20 nm in size, and a typical protein in the 3 nm range, then a resolution of 1 nm combined with appropriate “multispectral” labeling should be sufficient to resolve all its individual proteins and their relative location. Since at this level of resolution, antibodies and even F(ab)2-fragments would be too bulky, standard fluorophores engineered to bind to specific amino acid residues may be used [28], provided that the targeting specificity is not compromised. At the genome sequence level, a resolution of 1 nm would correspond to a very few base pairs/nucleotides only. Compared with X-ray microscopy or high resolution Electron Microscopy, the optical resolution of the envisaged “Avogadro” microscope may still be 10 times worse due to the real imaging conditions (background noise, long acquisition times, fixation artifacts, etc.); however, it would allow us to study such individual complexes and their formation on the single unit level under physiological conditions. Above all, these difficulties exist in the domain of cellular imaging. However, SRM methods may also be applied to isolated macromolecules/complexes, in case of which the imaging conditions can be highly optimized [44]. If fluorophore discrimination based on the triplet state [45] could be realized, e.g., by illumination of very few molecules only within the detection volume of the PSF and by use of photon counting detectors, this would potentially allow tracking of a high number of individual fluorescently labeled targets inside a live cell.
Even without combination of multiple super-resolution microscopy techniques into a single imaging device, strictly enhanced optical resolution might be obtained by defined swelling of the biological structure [46]. This technique termed “expansion” microscopy which acts on the sample side, rather than on the side of the instrumentation, may also be applicable to resolve subcellular or subnuclear biostructures.
“Nanoimaging” of Multiple Molecular Target Types (“multicolor SRM”)
The STED/PALM technology is presently limited to a very small number of different “colors” (i.e., fluorophore combinations); this means that only two or three different molecule types can be distinguished in a given object. In SMLM approaches, this problem is somewhat easier to overcome but remains difficult. The maximum number of compounds simultaneously resolved on the single molecule level by these has been 3–4. The cell is crowded, however, with targets composed of multiple types of macromolecules; their mutual spatial relations are of utmost importance for their function. For example, an important problem in present molecular biology is to study the activity of individual genes on the single cell level. In tissues, this can be done by sequence analysis combined with epigenetic annotation, such as the methylation status of associated histones; to perform this on the single cell level [47] (e.g., for 10,000 individual cells in a tissue section) is extremely difficult in terms of workload as well as expensive in terms of reagents required for sample preparation; but even if this problem should be solved, one would also like to study simultaneously details of transcription, splicing, protein synthesis, transport etc. on the single cell level. Since the products of genes interact with each other, one would like to know these parameters in the functional context on a cell to cell basis, not just the averaged bulk values. For the development of “multiplex” SRM, in particular localization microscopy techniques may be used which are based not on photoactivatable fluorescent proteins or on the very special and hence few molecule types useful for STED but on synthetic fluorophores with different spectral signatures (like differences in the absorption/emission spectrum, or the fluorescence life times).
New multichannel detector devices now allow us to measure the fluorescence life times of individual molecules with a time resolution of few tens of picoseconds [48]; hence many fluorescence life times should become distinguishable even at the same absorption/emission wavelength. If one assumes that 7 molecule types can be distinguished by their absorption/emission spectrum, and that for each of those wavelengths three different fluorescence times could be measured, then 7 × 3 = 21 different reporter molecule types might be discriminated on the single molecule/single cell level. In the case of DNA, for example, short fluorescently labeled oligonucleotides may be used in a combinatorial approach to label a given small section of the genome in an individual cell nucleus [49]. Since with n discrimination possibilities, N = 2n−1 different combinatorial label schemes would be possible, already 10 different labeling possibilities should be sufficient to simultaneously analyze in an individual nuclei about 1000 different genomic sequence targets, or RNA species. Remaining spectral signatures could be used to label various protein types related to gene activity. The functional context provided in this way could very advantageously be combined with other single cell analysis methods, e.g., DNA/RNA sequencing, or proteomics. In the case of “optically isolated” biomolecular machines (i.e., two functional multi-protein complexes such as, e.g., transcription sites having a mutual distance of >200 nm, or nuclear pore complexes), even a few different spectral signatures would be sufficient to obtain highly relevant nanostructural information on the number, orientation, etc. of the constituting proteins [50]. On the other hand, multi- or hyperspectral detection of individual dye molecule emission—while providing great potential to extract additional information, e.g., about the nanoenvironment and multiplying the number of simultaneously usable fluorescent probes—is strictly limited by the number of photons emitted per dye molecules [51–53].
Another resolution challenge is the axial resolution, especially for imaging of a larger field-of-view: In thin specimens such as individual cells located on an object slide, present SRM methods using high Numerical Aperture (NA) objective lenses have achieved an axial optical resolution down to few tens of nm. However, the axial resolution and hence the 3D observation volume rapidly declines with decreasing NA (required for scanning larger areas). For example, at a NA of 0.7 (assumed refraction index 1) the best achievable (confocal) resolution at 500 nm excitation is about 360 nm, while the axial resolution is at best around 1 μm. Since in localization microscopy, the localization accuracy is proportional to the conventional resolution, the axial localization accuracy and hence the optical resolution along the optical axis is several times worse. This problem can be overcome using an “axial tomographic approach”: The image is taken at different angles (views), from all these angles, a 3D image can be calculated with an optical and structural resolution corresponding to the best (lateral) resolution obtained in each of the views [54].
Photophysics of Individual Reporter Molecules for Nanoscopy
To realize a “multiplex” super-resolution microscopy, a much more detailed experimental and theoretical understanding of the spectral characteristics and temporal dynamics of single organic molecules on the level of Molecular Physics is required. Such an advanced understanding will be instrumental to provide a rational basis for the synthesis of novel reporter molecules given the complex biochemical environment inside of cells. Typical fluorescent reporter molecules may undergo thousands of different electronic transitions, may modify their 3D structure, or change their composition. During this time, photoproducts may be formed. These can be a source for formation of radicals, for change in the microenvironment, and for toxicity, influencing both the cell as well as the fluorophore itself. An application scheme could be to develop combinations of reporter molecules which specifically alter the local nanoenvironment and hence have a specific effect on the time dependent spectral behavior of individual fluorophores in “blinking” based localization microscopy. As a second application scheme, for multiplex super-resolution in vivo studies it should be highly desirable to develop new tuples of fluorophores with similar absorption/emission spectra but with highly different fluorescent life times, and—where required—extremely low photoswitching intensities.
For the combination of various SRM techniques, reporter molecules should be extremely useful which are photostable at illumination conditions A, and photoswitchable at illumination conditions B [55]. Such molecules could for example be employed to perform prolonged time lapse nano-imaging of live cells and tissue sections using structured illumination schemes at low excitation intensities (conditions A), and then switch to Abbe-limit breaking super-resolution procedures like STED, PALM, STORM/(d)STORM and other “blinking” based localization microscopy techniques, using conditions B for final snapshots.
Development of Novel Labeling Techniques
Once the specifications for photoswitching and fluorescence output required for super-resolution microscopy in the complex chemical environment of biological samples have been determined, corresponding synthetic fluorescent dye molecules need to be designed with high photon yield and high photostability. Achieving cleaner, narrower emission spectra (for more distinct colors) and controlled narrowed lifetime distributions could be additional benefits. Such enhanced dyes would be preferentially less sensitive to the nanoenvironment, or their sensitivity to the nanoenvironment could be tuned for SMLM in a way such as to use the resulting changes in the blinking behavior as novel contrasting agent [56]. Ideally, these dyes should be of low molecular weight, and carry molecular properties that provide control of their photoswitching behavior. A promising approach might be the direct conjugation of the fluorophore with a reducing group. These probes need to be coupled to the target binding site by means of highly selective, efficient, and stable (or transiently stable) binding. Furthermore, the linkers used to bind the fluorophores to the target binding site need to be small; click-chemistry or “nanobodies” are an important step in this direction [57].
One of the most challenging biological applications of novel labeling techniques will be to realize SRM imaging of specific genomic sites in living cells. To this end, it is envisaged that the recently developed imaging techniques using the bacterial CRISPR/Cas system in combination with green fluorescent proteins [58, 59] will be highly useful and significantly improve the capacity to study the conformation and dynamics of chromatin in living (even human) cells on the nanoscale.
Development of Fast and Reliable Data Evaluation Modes (“Instant Nanoimaging”)
Presently, a variety of evaluation programs is available for all kinds of present super-resolution microscopy approaches. Often even the developers do have difficulties to understand exactly which algorithm should be best for which problem. It is desirable to develop user friendly “plug-in programs” which are capable of automatically suggesting optimized evaluation algorithms based on the specific input dataset and on the quantitative analysis to be performed. As a long term perspective, one can imagine that for specific purposes simple, robust, and miniaturized (and therefore mobile) optical detection devices can be built. After acquisition, the data will be transferred to a remote computer system; which performs the detailed quantitative analysis, and returns the results to the local measurement device. Such a possibility is expected to be highly interesting in many application fields, e.g., the control of organic toxins or viruses and bacteria in the environment.
Sometimes it is reported in the literature that the exact localization of a single molecule from its fluorescence signal is “trivial.” This might be the case under ideal circumstances, especially under well-defined optical conditions and in a well-defined chemical environment; but in many practically relevant problems, extraction of exact molecule positions still remains a major challenge. For example, the background signal originating from the surrounding of a molecule, as well as blinking events above and below the z-plane of the molecule considered has to be taken into account.
In structured illumination approaches, one of the great challenges is to reduce the reconstruction time. For example, in our experience, a high precision multicolor structured illumination experiment may take up to several hours to reconstruct on a normal PC, and requires many Gigabytes of storage available. For use in high throughput analyses, or in medical diagnosis, such long times are unacceptable and have to be reduced by orders of magnitude.
Further challenges of SRM are remote processing and automatic calibration procedures. The first steps in SRM after fluorescence registration are the reconstruction of the super-resolved images, immediately followed by data visualization. STED and localization microscopy have less requirements and hence need less computation time. However, these are just the first steps: Quantitative evaluation and comparison with simulation data (computer models) has to follow. For example, localization microscopy of nuclear genome structure presently provides coordinates of millions of DNA sites per cell [55]; it is anticipated that in the near future, these coordinates can be refined to provide also the 3D coordinates of individual short DNA sequences, RNA sequences (including mi-RNAs and other oligonucleotides), and proteins. As in Astrophysics, this allows for the first time to compare the coordinates obtained and their dynamics with the coordinates obtained by numerical modeling; together with quantitative biochemical data, this will eventually result in truly mechanistic models of nuclear organization and gene regulation, and provide us with a deeper understanding of the emergence of complex biosystems from fundamental laws of Physics and Chemistry.
Super-Resolution of 3D Extended Objects
Presently, the thickness of an object which can be analyzed in 3D with an optical/structural resolution using high NA objective lenses is restricted to about 100 μm. This means that in most cases, individual cells arranged in monolayers on glass substrates are studied. It has been shown, however, that under these conditions cells may behave very differently from cells of the same type embedded in a 3D tissue surrounding. To overcome this problem, light sheet microscopy methods [60] have been developed; with Bessel beam shaped light sheets [61, 62], or in combination with axially structured illumination they already now provide a 3D resolution far beyond the conventional Abbe-limit. Such techniques are expected to be developed further and to be extended to still larger objects. An additional way to achieve this is the combination of structured illumination/localization microscopy with optical projection tomography approaches or with axial tomography (see above). In this way, it is anticipated to achieve single molecule resolution in an object close to 1 mm in thickness. For example, this would make possible to study the distribution of cancer related molecules (such as receptors, miRNAs, specific transcribed genes etc.) in individual cells of small tissue sections or 3D cell spheroids, and thus allow us, e.g., to identify metastatically competent cancer stem cells in their cellular context. Another highly relevant application would be to study on the nanoscale brain sections of unprecedented large thickness.
Development of Large Field-of-View Nanoscopes
In many biomedical applications, a substantial problem of present super-resolution methods is their limitation to a relatively small field–of-view, typically in the order of 100 μm diameter. This allows a vast scope of applications in fundamental biological research. In many medical applications but also in developmental biology, a field-of-view many times larger should be highly desirable (e.g., 1 mm diameter, or even more).
The solution with high NA objective lenses would be to perform multiple acquisitions at different locations which may be very time-consuming and impractical as overlapping acquisitions cannot easily be realized due to irreversible bleaching. While this is doable in rare scientific cases (like the Nobel Prize winning PALM experiment of Betzig [14]), it should be difficult to implement this into cellular pathology, with thousands of samples to be screened in a given lab/year; time lapse imaging of the whole field of view would be impractical.
There are various approaches to overcome this problem. For example, one might use an objective lens with a lower magnification and NA. A more radical solution would be to scan the object by multiple beams, e.g., 10,000 scanning beams, each scanning a field of view of 100 μm; in this case imaging could be parallelized, corresponding to a total field of view of 1 mm2. Such multiple beam scanning devices may be realized by using diffractive elements. In the case of STED beams, first steps in this direction have already been taken [63], and one can obtain even super-resolution; but also with diffraction limited beams like those used in confocal or 4Pi microscopy with laser excitation at constant excitation intensity, the combination with localization microscopy using photostable fluorophores has been shown to be possible [37, 50].
For quantitative analysis, it is essential to notice that in large field-of-view observations, optical aberrations (which can readily be corrected for in observation volumes of a few μm3) will have a more pronounced effect. For this, mapping of the refractive index inside the 3D sample could be a possible solution.
Combination of Light-Optical Nanoscopy and Electron/X-ray Microscopy
Present SRM techniques still offer an optical resolution 10 times worse than high performance Electron microscopy. For example, the distribution of proteins in a nuclear pore complex can now be studied optically, but the exact fitting of the molecules within the individual complex is still difficult due to the limited localization accuracy. Furthermore, the relation of individual molecule locations to structural landmarks is often difficult to establish, e.g., the question whether a given molecule adjacent to the nuclear envelope is located inside (i.e., in the nucleus) or outside the envelope (i.e., resides in the cytoplasm). Such questions are much easier to study in a combination of SRM techniques with EM. A number of attempts have been made to combine structured illumination with EM [64–67]. One way to do this is to perform first SRM and then transfer the specimen to the electron microscope. Another way would be to develop methods for SRM at cryo-temperatures in such a way that the EM image can be registered immediately afterwards of the same field-of-view [68].
Concluding Remarks
SRM methods already now have contributed a wealth of new insights in the life sciences, from the distribution of proteins on the cell membrane and the attachment of viruses; to cytoplasmic nanostructures; to the nuclear boundary; and to molecular machines inside the nucleus and nuclear genome structure. This information is highly complementary to the existing molecular biology and electron microscopy methods. It is anticipated these traditional methods in many applications will be supplanted by the novel microscopy approaches. Major advantages are the inherent simplicity, speed, costs, and versatility of the optical methods. The SRM techniques, especially localization microscopy, do not simply provide nice images of cellular nanostructures, but deliver single molecule coordinates; this opens an entire new field of quantitative structural and molecular biology, including rigorous testing of numerical models of cellular nanostructures. With the further developments outlined above, SRM approaches in combination with molecular biology and biochemistry methods and theoretical Biocomputing efforts will make possible to gain a full quantitative insight into the mechanisms of the single cell and its function in its environment. From the medical application side, the further development of SRM is expected to substantially enhance novel approaches to the diagnosis and therapy of important human diseases and other unfavorable conditions, from cancer to disturbances of normal development to aging.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
UB and CC are also affiliated with LuciaOptics Forschungszentrum, a non-profit company based in Karlsruhe, Germany.
Acknowledgments
In the preparation of this manuscript, we relied on the experience and advice of a large number of collaborators and partners, mainly based at the Universities of Heidelberg, Ludwig-Maximalians University Munich (LMU), and the Institute of Molecular Biology Mainz (IMB) which are gratefully acknowledged here. In particular we want to thank Thomas Cremer (LMU) and Aleksander Szczurek (IMB). This research was supported by the Boehringer Ingelheim Foundation.
References
1. Sydor AM, Czymmek KJ, Puchner EM, Mennella, V. Super-resolution microscopy: from single molecules to supramolecular assemblies. Trends Cell Biol. (2015) 25:730–48. doi: 10.1016/j.tcb.2015.10.004
2. Abbe E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch für Mikrosk Anat. (1873) 9:413–8. doi: 10.1007/BF02956173
3. Ehrenberg M. The Nobel Prize in Chemistry 2014 (Press Release). Stockholm (2014). Available online at: http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2014/advanced-chemistryprize2014.pdf
4. Galbraith CG, Galbraith JA. Super-resolution microscopy at a glance. J Cell Sci. (2011) 124:1607–11. doi: 10.1242/jcs.080085
5. Toraldo di Francia G. Resolving power and information. J Opt Soc Am. (1955) 45:497–9. doi: 10.1364/JOSA.45.000497
6. Cremer C, Masters BR. Resolution enhancement techniques in microscopy. Eur Phys J H. (2013) 38:281–344. doi: 10.1140/epjh/e2012-20060-1
7. Cremer C. Optics far beyond the diffraction limit. In: Springer Handbook of Lasers and Optics. Springer (2012). p. 1359–97. Available online at: http://link.springer.com/chapter/10.1007/978-3-642-19409-2_20
8. Allen JR, Ross ST, Davidson, MW. Structured illumination microscopy for superresolution. Chem Phys Chem. (2014) 15:566–76. doi: 10.1002/cphc.201301086
9. Blom H, Widengren J. STED microscopy—towards broadened use and scope of applications. Curr Opin Chem Biol. (2014) 20:127–33. doi: 10.1016/j.cbpa.2014.06.004
11. Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science (2008) 319:810–3. doi: 10.1126/science.1153529
12. Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee, A, et al. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed. (2008) 47:6172–6. doi: 10.1002/anie.200802376
13. Reymann J, Baddeley D, Gunkel M, Lemmer P, Stadter W, Jegou T, et al. High-precision structural analysis of subnuclear complexes in fixed and live cells via spatially modulated illumination (SMI) microscopy. Chromosome Res. (2008) 16:367–82. doi: 10.1007/s10577-008-1238-2
14. Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science (2006) 313:1642–5. doi: 10.1126/science.1127344
15. Huang B, Jones SA, Brandenburg B, Zhuang X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat Methods (2008) 5:1047–52. doi: 10.1038/nmeth.1274
16. Lemmer P, Gunkel M, Baddeley D, Kaufmann R, Urich A, Weiland Y, et al. SPDM: light microscopy with single-molecule resolution at the nanoscale. Appl Phys B (2008) 93:1–12. doi: 10.1007/s00340-008-3152-x
17. Lemmer P, Gunkel M, Weiland Y, Müller P, Baddeley D, Kaufmann R, Urich A, et al. Using conventional fluorescent markers for far-field fluorescence localization nanoscopy allows resolution in the 10-nm range. J Microsc. (2009) 235:163–71. doi: 10.1111/j.1365-2818.2009.03196.x
18. Thompson MA, Lew MD, Moerner WE. Extending microscopic resolution with single-molecule imaging and active control. Annu Rev Biophys. (2012) 41:321–42. doi: 10.1146/annurev-biophys-050511-102250
19. Steinhauer C, Forthmann C, Vogelsang J, Tinnefeld P. Superresolution microscopy on the basis of engineered dark states. J Am Chem Soc. (2008) 130:16840–1. doi: 10.1021/ja806590m
20. Cox S. Super-resolution imaging in live cells. Dev Biol. (2015) 40:175–81. doi: 10.1016/j.ydbio.2014.11.025
21. Pertsinidis A, Zhang Y, Chu S. Subnanometre single-molecule localization, registration and distance measurements. Nature (2010) 466:647–51. doi: 10.1038/nature09163
22. Lidke KA, Rieger B, Jovin TM, Heintzmann R. Superresolution by localization of quantum dots using blinking statistics. Opt Express (2005) 13:7052–62. doi: 10.1364/OPEX.13.007052
23. Moser F, Hildenbrand G, Müller P, Al Saroori A, Biswas A, Bach M, et al. Cellular uptake of gold nanoparticles and their behavior as labels for localization microscopy. Biophys J. (2016) 110:947–53. doi: 10.1016/j.bpj.2016.01.004
24. Chakrova N, Heintzmann R, Rieger B, Stallinga S. Studying different illumination patterns for resolution improvement in fluorescence microscopy. Opt Expr. (2015) 23:31367–83. doi: 10.1364/oe.23.031367
25. Komis G, Mistrik M, Šamajová O, Ovečka M Bartek J, Šamaj J. Superresolution live imaging of plant cells using structured illumination microscopy. Nat. Protoc. (2015) 10:1248–63. doi: 10.1038/nprot.2015.083
26. Li D, Shao L, Chen B-C, Zhang X, Zhang M, and Moses B, et al. Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science (2015) 349:aab3500. doi: 10.1126/science.aab3500
27. Burnette DT, Sengupta P, Dai Y, Lippincott-Schwartz J, Kachar B. Bleaching/blinking assisted localization microscopy for superresolution imaging using standard fluorescent molecules. Proc Natl Acad Sci USA. (2011) 108:21081–6. doi: 10.1073/pnas.1117430109
28. Cremer C, Kaufmann R, Gunkel M, Pres S, Weiland Y, Müller, P, et al. Superresolution imaging of biological nanostructures by spectral precision distance microscopy. Biotechnol J. (2011) 6:1037–51. doi: 10.1002/biot.201100031
29. Failla AV, Spoeri U, Albrecht B, Kroll A, Cremer C. Nanosizing of fluorescent objects by spatially modulated illumination microscopy. Appl Opt. (2002) 41:7275–7283. doi: 10.1364/AO.41.007275
30. Spöri U, Failla AV, Cremer, C Superresolution size determination in fluorescence microscopy: a comparison between spatially modulated illumination and confocal laser scanning microscopy. J Appl Phys. (2004) 95:8436–43. doi: 10.1063/1.1751633
31. Wagner C, Spöri U, Cremer, C. High-precision SMI microscopy size measurements by simultaneous frequency domain reconstruction of the axial point spread function. Optik Int J Light Electron Opt. (2005) 116:15–21. doi: 10.1016/j.ijleo.2004.10.002
32. Heintzmann R, Jovin TM, Cremer C. Saturated patterned excitation microscopya concept for optical resolution improvement. J Opt Soc Am A. (2002) 19:1599–609, doi: 10.1364/JOSAA.19.001599
33. Gustafsson MGL. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci USA. (2005) 102:13081–6, doi: 10.1073/pnas.0406877102
34. Hofmann M, Eggeling C, Jakobs S, Hell SW. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Aci USA. (2005) 102:17565−9. doi: 10.1073/pnas.0506010102
35. Bornfleth H, Saetzler K, Eils R, Cremer C. High-precision distance measurements and volume-conserving segmentation of objects near and below the resolution limit in three-dimensional confocal fluorescence microscopy. J Microsc. (1998) 189:118–36. doi: 10.1046/j.1365-2818.1998.00276.x
36. Cremer C, Edelmann P, Bornfleth H, Luz H, Kreth G, Münch, H, et al. Principles of spectral precision distance confocal microscopy of molecular nuclear structure. In: Jähne B, Haußecker H, Geißler P, editors. Handbook of Computer Vision and Applications. San Diego, CA; New York, NY: Academic Press (1999). p. 839–57.
37. Esa A, Edelmann P, Kreth G, Trakhtenbrot L, Amariglio N, Rechavi G, et al. (2000). Three-dimensional spectral precision distance microscopy of chromatin nanostructures after triple-colour dna labelling: a study of the bcr region on chromosome 22 and the philadelphia chromosome. J Microsc. 199(Pt 2):96–105. doi: 10.1046/j.1365-2818.2000.00707.x
38. Rossberger S, Best G, Baddeley D, Heintzmann R, Birk U, Dithmar S, et al. Combination of structured illumination and single molecule localization microscopy in one setup. J Opt. (2013) 15:094003. doi: 10.1088/2040-8978/15/9/094003
39. Zanacchi F, Lavagnino Z, Donnorso M, Bue A, Furia L, Faretta M, et al. (2011). Live-cell 3D super-resolution imaging in thick biological samples. Nat. Meth. 8:1047–9. doi: 10.1038/nmeth.1744
40. Albrecht B, Failla AV, Heintzmann R, Cremer C. Spatially modulated illumination microscopy: online visualization of intensity distribution and prediction of nanometer precision of axial distance measurements by computer simulations. J Biomed Opt. (2001) 6:292–9. doi: 10.1117/1.1383293
41. Mortensen KI, Churchman LS, Spudich JA, Flyvbjerg H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Meth. (2010) 7:377–81. doi: 10.1038/nmeth.1447
42. Nieuwenhuizen RP, Stallinga S, Rieger B. Visualization and resolution in localization microscopy. In: Cambi A, and Lidke DS, editors. Cell Membrane Nanodomains: From Biochemistry to Nanoscopy. Boca Raton, FL: CRC Press (2014). p. 409–30.
43. Albrecht B, Failla AV, Schweitzer A, Cremer C. Spatially modulated illumination microscopy allows axial distance resolution in the nanometer range. Appl Opt. (2002) 41:80–7. doi: 10.1364/AO.41.000080
44. Weisenburger S, Jing B, Renn A, Sandoghdar V. Cryogenic localization of single molecules with angstrom precision. (2013) SPIE Proc. 8815:88150D–88150D–9. doi: 10.1117/12.2025373
45. Hell SW, Kroug M. Ground-state-depletion fluorescence microscopy: a concept for breaking the diffraction resolution limit. Appl Phys B. (1995) 60:495–97. doi: 10.1007/BF01081333
46. Chen F, Tillberg PW, Boyden ES. Expansion microscopy. Science (2015) 347:1260088. doi: 10.1126/science.1260088
47. Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, et al. Single-cell hi-C reveals cell-to-cell variability in chromosome structure. Nature (2013) 502:59–64. doi: 10.1038/nature12593
48. Hartig R, Prokazov Y, Turbin E, Zuschratter W. Wide-field fluorescence lifetime imaging with multi-anode detectors - springer. In: Engelborghs Y, Visser AJWG editors. Methods in Molecular Biology (1076). Humana Press (2014). Available online at: http://link.springer.com/protocol/10.1007%2F978-1-62703-649-8_20.
49. Cremer C. Far-field light microscopy. In: Zheng Y, editor. Encyclopedia of Life Sciences (eLS). Chichester: John Wiley & Sons, Ltd. (2014). p. 1–13.
50. Hüve J, Wesselmann R, Kahms M, Peters R. 4Pi microscopy of the nuclear pore complex. Biophys J. (2008) 95:877–85. doi: 10.1529/biophysj.107.127449
51. Moerner WE, Fromm DP. Methods of single-molecule fluorescence spectroscopy and microscopy. Rev Sci Instrum. (2003) 74:3597. doi: 10.1063/1.1589587
52. Ha T, Tinnefeld P. Photophysics of fluorescent probes for single-molecule biophysics and super-resolution imaging. Annu Rev Phys Chem. (2012) 63:595–617. doi: 10.1146/annurev-physchem-032210-103340
53. Zhang Z, Kenny SJ, Hauser M, Li W, Xu K. Ultrahigh-throughput single-molecule spectroscopy and spectrally resolved super-resolution microscopy. Nat Meth. (2015) 12:935–40. doi: 10.1038/nmeth.3528
54. Heintzmann R, Cremer C. Axial tomographic confocal fluorescence microscopy. J Microsc. (2002) 206(Pt 1):7–23. doi: 10.1046/j.1365-2818.2002.01000.x
55. Szczurek AT, Prakash K, Lee H-K, Żurek-Biesiada DJ, Best G, Hagmann M, et al. Single molecule localization microscopy of the distribution of chromatin using hoechst and DAPI fluorescent probes. Nucleus (2014) 5:331–40. doi: 10.4161/nucl.29564
56. Endesfelder U, Heilemann M. Art and artifacts in single-molecule localization microscopy: beyond attractive images. Nat Methods (2014) 11:235–8. doi: 10.1038/nmeth.2852
57. Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci USA. (2008) 105:2415–20. doi: 10.1073/pnas.0712168105
58. Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang, G-W, Li J, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system Cell (2013) 155:1479–91. doi: 10.1016/j.cell.2013.12.001
59. Ma H, Reyes-Gutierrez P, Pederson, T. Visualization of repetitive DNA sequences in human chromosomes with transcription activator-like effectors. Proc Natl Acad Sci USA. (2013) 110:21048–53. doi: 10.1073/pnas.1319097110
60. Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EHK. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science (2008) 322:1065–9. doi: 10.1126/science.1162493
61. Planchon TA, Gao L, Milkie DE, Davidson MW, Galbraith JA, Galbraith CG, et al. Rapid three-dimensional isotropic imaging of living cells using bessel beam plane illumination. Nat Methods (2011) 8:417–23. doi: 10.1038/nmeth.1586
62. Chen B-C, Legant WR, Wang K, Shao L, Milkie DE, Davidson, MW, et al. Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science (2014) 346:1257998. doi: 10.1126/science.1257998
63. Chmyrov A, Keller J, Grotjohann T, Ratz M, d'Este E, Jakobs, S, et al. Nanoscopy with More than 100,000 ‘Doughnuts.’ Nat Methods (2013) 10:737–40. doi: 10.1038/nmeth.2556
64. Kopek BG, Shtengel G, Xu CS, Clayton DA, Hess, HF. Correlative 3D superresolution fluorescence and electron microscopy reveal the relationship of mitochondrial nucleoids to membranes. Proc Natl Acad Sci USA. (2012) 109:6136–41. doi: 10.1073/pnas.1121558109
65. Löschberger A, Franke C, Krohne G, van de Linde S, Sauer M. Correlative super-resolution fluorescence and electron microscopy of the nuclear pore complex with molecular resolution. J Cell Sci. (2014) 127:4351–5. doi: 10.1242/jcs.156620
66. Rouquette J, Genoud C, Vazquez-Nin GH, Kraus B, Cremer T, Fakan, S. Revealing the high-resolution three-dimensional network of chromatin and interchromatin space: a novel electron-microscopic approach to reconstructing nuclear architecture. Chromosome Res. (2009) 17:801–10. doi: 10.1007/s10577-009-9070-x
67. Smeets D, Markaki Y, Schmid VJ, Kraus F, Tattermusch A, Cerase A, et al. Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenet Chromatin (2014) 7:1–27. doi: 10.1186/1756-8935-7-8
Keywords: STED, PALM, super-resolution fluorescence microscopy, live cell imaging, dSTORM, SMLM, Structured illumination, SPDM
Citation: Cremer C and Birk U (2016) Perspectives in Super-Resolved Fluorescence Microscopy: What Comes Next? Front. Phys. 4:11. doi: 10.3389/fphy.2016.00011
Received: 25 August 2015; Accepted: 22 March 2016;
Published: 12 April 2016.
Edited by:
Andrey Vadimovich Kanaev, US Naval Research Laboratory, USAReviewed by:
David Mayerich, University of Houston, USAAndrew G. Edwards, Oslo University Hospital and Simula Research Laboratory, Norway
Copyright © 2016 Cremer and Birk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Cremer, c.cremer@imb-mainz.de; u.birk@imb-mainz.de