
- 1 Department of Internal Medicine IV, University Heidelberg Hospital, Heidelberg, Germany
- 2 Mass Spectrometry Laboratory, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
It has been long known that hepatic synthesis of phosphatidylcholine (PC) is depressed during acute such as carbon tetrachloride-induced liver injury. Anti-hepatotoxic properties of PC as liposomes have been recognized for treatment of acute liver damage. Ursodeoxycholate (UDCA) is a known hepatoprotectant in stabilizing cellular membrane. For therapeutic management of liver injury, we coupled UDCA with a phospholipid known as ursodeoxycholyl lysophosphatidylethanolamide (UDCA-LPE). UDCA-LPE has been shown to first-in-class hepatoprotectant being superior to UDCA or PC. It inhibits mitochondrial damage and apoptosis, elicits survival signaling pathway, and promotes regeneration of hepatocytes. We herein report that a unique contribution of UDCA-LPE in increasing concentrations of PC in vitro and in vivo. UDCA-LPE-treated hepatocytes contained significantly increased PC levels. UDCA-LPE underwent the hydrolysis to LPE which was not the precursor of the increased PC. The levels of PC in the liver and blood were increased rapidly after intraperitoneally administration UDCA-LPE, and were found to be sustained even after 24 h. Among PC synthesis genes tested, UDCA-LPE treatment of mouse hepatocytes increased transcription of CDP-diacylglycerol synthase 1 which is an enzyme catalyzing phosphatidic acid to generate intermediates for PC synthesis. Thus, UDCA-LPE as a hepatoprotectant was able to induce synthesis of protective PC which would supplement for the loss of PC occurring during acute liver injury. This property has placed UDCA-LPE as a candidate agent for therapy of acute hepatotoxicity such as acetaminophen poisoning.
Introduction
Phospholipids including lecithins and phosphatidylcholine (PC) have been used and tested in humans for treatment of liver disease since the 1970s (Wallnofer and Hanusch, 1973). PC is the major membrane phospholipids and thus their hepatoprotective effects are due to membrane stabilization (Miyazaki et al., 1991) as well as enhancement of cell membrane integrity (Li et al., 2006). Research on phospholipid biochemistry in the past three decades has revealed physiological role of PC which is required for hepatocellular division and survival (Jackowski, 1994), and that alterations of PC homeostasis could result in development of liver diseases (Noga and Vance, 2003; Li et al., 2005).
Hepatoprotective role of PC is applicable not only for chronic liver disease but also for acute liver damage. It was found nearly 50 years ago that carbon tetrachloride (CCl4) intoxication is associated with changes in composition of PC and phosphatidylethanolamine (PE; Sgoutas, 1967; Shimizu, 1969; Sugano et al., 1970). Total phospholipids in hepatic microsomes (Sgoutas, 1967), endoplasmic reticulum (James et al., 1982), and plasma membrane (Camacho and Rubalcava, 1984) are markedly depressed following CCl4 administration of experimental animals. This is due to decreased syntheses of both phospholipids and triglycerides readily observed 4–5 h after treatment (Gebhart and Brabec, 1985). CCl4-induced injury in vivo produces an early peroxidative event from the generation of trichloromethyl radical as a result of cytochrome P450-mediated metabolism of CCl4 (Trudell et al., 1982). These free radicals react with cellular PC forming PC-derived lipid radicals which subsequently react with oxygen to form PC hydroperoxides (Miyazawa et al., 1990). The formation of PC hydroperoxides is indicative of hepatic injury causing alterations in membrane structure. For treatment, it is conceivable that a supplementation of PC during acute liver injury replenishes PC levels which can serve as both structural components, and targets for trichloromethyl free radical binding (Jaeschke et al., 1987). Indeed, administration of PC to experimental animals intoxicated with acetaminophen (Jaeschke et al., 1987), CCl4 (Aleynik et al., 1997), alcohol (Okiyama et al., 2009), or infected with sepsis-induced bacteria (Yan et al., 2004), has been proven to be effective in alleviating liver injury.
To date, therapeutic use of PC to treat liver disease in humans has however not been successful (Lieber et al., 2003). As exogenously added PC can provide only ∼50% of the PC required for membrane biogenesis (Esko et al., 1982), the amount of PC taken up into hepatocytes may be limited thus providing not enough PC needed for membrane protection. For treatment of acute liver injury, antioxidants such as acetylcysteine to treat acetaminophen overdose have been commonly used (Green et al., 2010). Alternatively, manipulation of metabolic pathways to increase PC levels has been adopted for treatment of acute liver injury. These strategies include the use of PC precursors, such as, betaine (Kharbanda et al., 2007), fish oils (Speck and Lauterburg, 1991), dietary saturated and monounsaturated fats (Hwang et al., 2011), and agonists of the peroxisome proliferator-activated receptor (PPAR) alpha (Lee et al., 2004). At least in experimental settings, both administration of PC and the modulation of pathways to increase PC have currently provided positive results in ameliorating acute hepatotoxicity.
We had synthesized a hepatoprotectant ursodeoxycholyl lysophosphatidylethanolamide (UDCA-LPE, Chemical structure shown in Figure 1A), which as an intact compound provides cytoprotection against TNF-alpha-induced apoptosis (Chamulitrat et al., 2009). UDCA-LPE lowers aminotransferases and elicits anti-inflammatory response in mice treated with galactosamine/endotoxin (Pathil et al., 2011a) or fed with high-fat diet (Pathil et al., 2011b). UDCA-LPE as a hepatoprotectant may thus potentially be used to treat acute liver injury. Herein, we provided evidence that UDCA-LPE induced elevation of hepatocellular PC levels under in vitro and in vivo conditions. This is in part due to UDCA-LPE’s ability to elicit transcriptional control of a PC synthesis gene phosphatidate cytidylyltransferase or another name CDP-diacylglycerol synthase 1 (CDS1). The ability of UDCA-LPE to rapidly increase hepatic PC levels renders it suitable to treat acute hepatotoxicity in clinical settings, such as, acetaminophen poisoning.
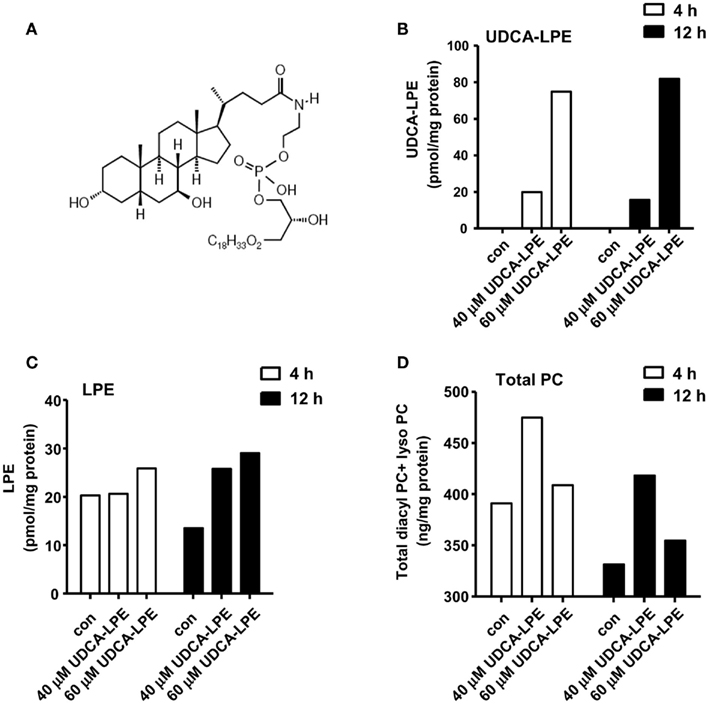
Figure 1. Ursodeoxycholyl lysophosphatidylethanolamide hydrolysis to LPE and increases PC concentrations in mouse hepatocytes. (A) Chemical structure of UDCA-LPE (UDCA-18:1-LPE). (B) Upon treatment with 50 μM UDCA-LPE, UDCA-LPE was taken to hepatocytes over 4–12 h. (C) Upon treatment with 50 μM UDCA-LPE, LPE concentrations were increased after 12 h treatment indicating that UDCA-LPE hydrolysis occurred at 12 h but not at 4 h. (D) Total PC (diacyl PC and lyso PC) concentrations were increased mouse hepatocytes treated with 50 μM UDCA-LPE for 4 and 20 h. “Con” represents control hepatocytes treated with ethanol used as a vehicle. Data are mean ± SD, N = 4; *p < 0.05 versus control.
Materials and Methods
Materials
Ursodeoxycholyl lysophosphatidylethanolamide was synthesized by ChemCon GmbH (Freiburg, Germany). UDCA was purchased from Sigma-Aldrich (St. Louis, MO, USA). Phospholipid standards (1-oleoyl-2-hydroxy-sn-glycero-3-phosphoethanolamine (18:1-lyso PE), 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (16:0-lyso PC), 1-stearoyl-2-hydroxy-sn-glycero-3-phospho-choline (18:0-lyso PC), 1-oleoyl-2-hydroxy-sn-glycero-3-phosphocholine (18:1-lyso PC), 1-arachidonyl-2-hydroxy-sn-glycero-3-phosphocholine (20:4-lyso PC) 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-choline (16:0, 18:1-PC), 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine (16:0, 18:2-PC), 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (16:0, 20:4-PC), 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (18:0, 18:1-PC), 1-stearoyl-2-linoleoyl-sn-glycero-3-phosphocholine (18:0, 18:2-PC), and 1-palmitoyl(d31)-2-oleoyl-sn-glycero-3-phos-phocholine (d31-16:0, 18:1-PC) were obtained from Avanti Lipids, Inc. (Alabaster, AL, USA) and were at least 99% pure. All other solvents and chemicals used were of either high-performance-liquid-chromatography grade or of known analytical purity and obtained from Sigma-Aldrich (St. Louis, MO, USA).
Animal Experiments
Male C57BL/6 mice (Charles River Laboratories, Sulzfeld, Germany) at 10 weeks old received 30 mg/kg UDCA-LPE by intraperitoneal injections. UDCA-LPE was dissolved in 0.5% carboxymethylcellulose. At designated time, each mouse for each time point was sacrificed. Blood, liver, and intestine were collected for lipid extraction and LC–MS/MS analysis. All experiments were approved by the Animal Care and Use Committee of the University Heidelberg.
Hepatocyte Isolation and Cultures
Hepatocytes were isolated from 7- to 10-week-old C57/BL6 mice (Charles Rivers Laboratories, Sulzfeld, Germany) by using two-step collagenase perfusion technique and purified by Percoll gradient centrifugation. Freshly isolated hepatocytes were plated on multi-well collagen-coated plates and cultured for 4 h in M199 medium containing Hank’ salts and L-glutamine (PAA, Cölbe, Germany) supplement with 1% penicillin and streptomycin, 100 nM dexamethasone, 0.5 nM insulin, and 5% neonatal calf serum. Dead hepatocytes were removed, and the adhered cells were treated with serum-free medium. After overnight in culture, hepatocytes were treated with UDCA-LPE in serum-free M199 medium for 4 or 12 h. Ethanol as a vehicle of UDCA-LPE was added in the control groups.
Palmitate-Induced Apoptosis
Pal stock solution in BSA was prepared. Briefly, 250 μL of 200 mM Pal in ethanol was mixed with 4.5 mL of 27% BSA in PBS. The total 5 mL volume was adjusted to pH to 7.4 with 0.1 N NaOH until the mixture became clear. The appropriate control for Pal treatment contained 0.5% BSA and 0.1% ethanol. For apoptosis assays, caspase 3/7glo assay kits (Promega, Mannheim, Germany) were used as previously described (Chamulitrat et al., 2009). Incubation time for apoptosis assay was 20 h.
Preparation of Standards and Samples for LC–MS/MS Analysis
Phosphatidylcholine standards obtained from Avanti Lipids are in chloroform at a concentration of 10 mg/mL. They were diluted to 100 ng/μL with methanol and used as working solutions. Stock solutions of 18:1-lyso PE (1 mg/mL) was prepared in chloroform:methanol (1:4, v/v) and stored at −20°C. UDCA and d4-UDCA stock solution (1 mg/mL) was prepared in methanol and stored at −20°C. Working solutions were prepared by appropriate dilution of the stock solutions with methanol. Calibration curves were constructed for UDCA-LPE, UDCA, and LPE in PBS solution using d4-UDCA as internal standard and curve range was in 0–50 ng. Calibration curve of each individual PC was built using d31-16:0–18:1-PC as an internal standard. The linear response for PC and lyso PC was in the range of 0–200 and 0–100 ng, respectively.
Hepatocytes treated with UDCA-LPE (in 20 μL suspension) was added with 180 μL H2O, and 20 μL of 0.5 ng/μL d4-UDCA, and then extracted with 1 mL CHCl3:MeOH (2:1) twice. The chloroform layer was dried down and reconstituted into 200 μL MeOH, and 20 μL was injected onto the HPLC column. Mouse plasma and urine samples were treated with the same procedure as for hepatocytes. For analyses of tissue samples, homogenates were prepared in PBS to give a final concentration of 1 g/mL d4-UDCA (0.5 ng/μL, 20 μL) and d31-16:0–18:1-PC (2 ng/μL, 20 μL) were added to the homogenates and subsequent lipid extraction with CHCl3:MeOH was performed.
LC–MS/MS Analysis of UDCA-LPE and Phospholipids
LC–MS/MS analysis was carried out on a Waters Quattro Micro API triple quadrupole mass spectrometer (Milford, MA, USA) interfaced with Acquity UPLC system. Nitrogen was used as nebulizer and argon was used as collision gas. Optimized mass spectrometry parameters were determined with individual standard compounds. UDCA-LPE and LPE analysis were performed under negative mode with multiple reactions monitoring (MRM) mass transitions set at 852.5 > 281.3, 478.2 > 281.2, 391.1 > 391.1 respectively. Quantification of diacyl PC and lyso PC were performed under positive mode, monitoring transition pair of the individual protonated parent ions for PCs and their common daughter ion m/z 184. Total PC was the sum of diacyl PC and lyso PC.
Online chromatographic separation was achieved using a 100-mm × 2.0-mm i.d. Luna 3 μ C18 column (Phenomenex, CA, USA). Binary solvents used for analysis were 95% H2O/MeOH with 2 mM ammonium acetate (solvent A) and 95% MeOH/H2O with 2 mM ammonium acetate (solvent B). For analysis of UDCA-LPE and LPE, gradient started with 90% solvent A for 1 min, changed to 100% solvent B in 0.5 min, held for 5 min, and finally switched back to initial condition in 0.1 min. The total analysis time was 8 min. Gradient for PC analysis started with 85% solvent B and was maintained for 1 min. Then rose in 0.5 min linearly to 100% solvent B. The elution was held at 100% B for 10 min, then reversed to 85% methanol in 0.5 min. The 3-min holding at 85% methanol equilibrated the column efficiently. Flow rate for both analyses was 0.2 mL/min.
Gene Expression by Real-Time RT-PCR
Total RNA of treated mouse hepatocytes was isolated using QIAGEN RNeasy Mini kit (Qiagen, Hilden, Germany). cDNA was synthesized from 2 μg RNA using a Maxima First Strand cDNA synthesis kit (Fermentas, St. Leon-Rot, Germany). The mRNA expression was analyzed in quadruplets by real-time PCR using Applied Biosystems TaqMan® gene expression assays with TaqMan® Universal PCR Master Mix (Applied Biosystems) and run on an Applied Biosystem 7500 Fast real-time PCR machine by using assay-on-Demand TaqMan® primers. The expression level of targets was calculated using Δ−Ct transformation method, and showed as ratio after having been normalized to house-keeping gene GAPDH. PCR results were obtained from at least three independent experiments.
Data Analysis
Statistical significance at p > 0.05 was determined with ANOVA or a Student’s t-test using Primer Biostatistics. Results were expressed as mean ± SD.
Results
UDCA-LPE Increases PC Concentrations in Mouse Hepatocytes
Intracellular concentrations of UDCA-LPE (chemical structure shown in Figure 1A) in mouse hepatocytes were increased from 20 to 80 pmol/mg protein with 40 or 60 μM UDCA-LPE (Figure 1B). At the same time, intracellular concentrations of LPE were not yet increased at 4 h but markedly increased after 12 h by ∼15 pmol/mg protein (Figure 1C).
Cellular concentrations of PC were elevated in mouse hepatocytes treated with 40 or 60 μM UDCA-LPE for 4 or 12 h (Figure 1D). Compared with 4 h, the total PC levels were decreased after 12 h incubation likely due to PC export into the medium. It is observed that the increases of total PC (diacyl PC and lyso PC) concentrations at 4 and 12 h were on the order of 52–54 ng/mg protein or ∼150 pmol/mg protein. The increases of LPE observed after 12 h at ∼15 pmol/mg protein did not correspond to the increases of PC because the increased LPE concentrations were ∼10-folds lower than those of the observed increased PC. This excludes the possibility that PC could arise from LPE as a result of UDCA-LPE hydrolysis (to UDCA and LPE). Pathways for LPE conversion to PC include acylation of LPE to PE and subsequent PE metabolism to PC by PE methyltransferase (DeLong et al., 1999). Taken together, increased PC did not arise from UDCA-LPE hydrolysis, and may likely be from the effects of UDCA-LPE on certain metabolic signaling pathways which increase PC abundance.
UDCA-LPE Administration Increases PC Concentrations in vivo
We further confirmed UDCA-LPE effects on PC levels under in vivo conditions. Intraperitoneal injections of UDCA-LPE at 30 mg/kg 1 h prior to the challenge with galactosamine/endotoxin in mice were found to be effective in alleviating acute liver injury observed at 8 h (Pathil et al., 2011a). We therefore performed intraperitoneal injections of 30 mg/kg UDCA-LPE over 2, 4, 8, and 24 h, and determined diacyl PC and total PC (as the sum of diacyl PC and lyso PC). We observed that the levels of PC in plasma were gradually increased after 2 h and stayed elevated even after 24 h (Figure 2A). The levels of PC found in urine were increased significantly 2 h after injection, and quickly decreased to basal levels after 4 h (Figure 2B). As urinary PC arises from PC secreted from tissues, PC pharmacokinetics in urine indicated that PC clearance from tissues was significantly slowed down at the later time, i.e., longer than 4 h.
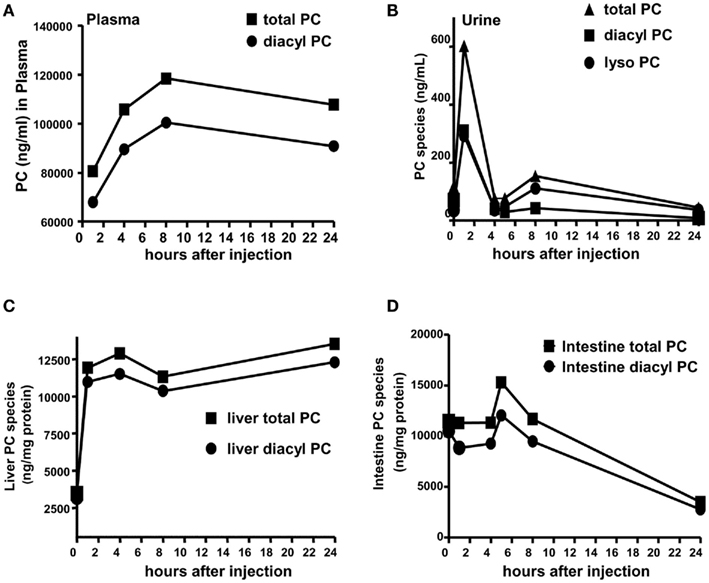
Figure 2. Ursodeoxycholyl lysophosphatidylethanolamide increases PC levels in vivo. Mice were injected with 30 mg/kg UDCA-LPE by an intraperitoneal injection. (A) Concentrations of PC species (total or diacyl PC) in ng/mL in plasma were increased after 2 h and stayed elevated even after 24 h. (B) Concentrations of total PC, diacyl PC, and lyso PC in ng/mL in urine were increased after 2 h and drastically decayed to control levels after 4 h. (C) Concentrations of PC species in nanogram per milligram protein in the liver were increased after 2 h and stayed elevated even after 24 h. (D) Concentrations of total PC, and diacyl PC in ng/mg protein in the intestine were slightly increased after 6 h and drastically decayed to control levels after 8 h.
Similar to plasma, the levels of PC species in the liver were significantly elevated after 2 h and sustained at these concentrations even at 24 h after administration (Figure 2C). However, PC concentrations in the intestine were slightly increased at 6 h, and drastically decreased at 8 and 24 h (Figure 2D). Our data clearly demonstrated sustained increases of PC in blood and liver following UDCA-LPE intraperitoneal injection concomitant with reduced PC clearance from these tissues to the urine. Significant increases of PC were found in the liver but not the intestine indicating UDCA-LPE liver targeting, and hence demonstrating the suitability of UDCA-LPE for treatment of liver disease.
UDCA-LPE Upregulates CDS1 mRNA in Mouse Hepatocytes
To determine possible activation of PC synthesis by UDCA-LPE, we analyzed PC synthesis genes in mouse hepatocytes which had been treated with UDCA-LPE for 4 or 20 h. UDCA-LPE treatment for 4 h did not modulate expression of the genes under investigation (data not shown). These genes included CDP-diacylglycerol synthase 1 (CDS1), choline kinase alpha, CDP-diacylglycerol synthase 2 (CDS2), choline/ethanolamine phosphotransferase 1 (Cept1), choline phosphate cytidylyltransferase 1, alpha isoform (CCTalpha or Pcyt1a), and phosphatidylethanolamine N-methyl-transferase (PEMT). For most genes tested, UDCA-LPE had only marginal effects (Figure 3A). However, UDCA-LPE treatment for 20 h significantly increased mRNA expression of CDS1 by ∼1.4-fold. CDS1 is an enzyme which converts phosphatidic acid to phosphatidylserine (PS) intermediate and then PE which is further metabolized to generate PC by PE methyltransferase (Figure 3B). Under apoptosis induction by palmitate exposure for 20 h, CDS1 mRNA was slightly downregulated by palmitate, and UDCA-LPE again weakly upregulated CDS1 mRNA. (Figure 3C, left). UDCA-LPE co-treatment with palmitate however significantly increased CDS1 mRNA expression by ∼5-folds. UDCA-LPE co-treatment with palmitate did not upregulate other PC synthesis genes listed in Figure 3A (data not shown). The increases of CDS1 mRNA expression were concomitant with UDCA-LPE protection against palmitate-induced apoptosis as determined by caspase 3 activity (Figure 3C, right). Hence UDCA-LPE protection was associated with an upregulation of CDS1 mRNA for increased PC synthesis.
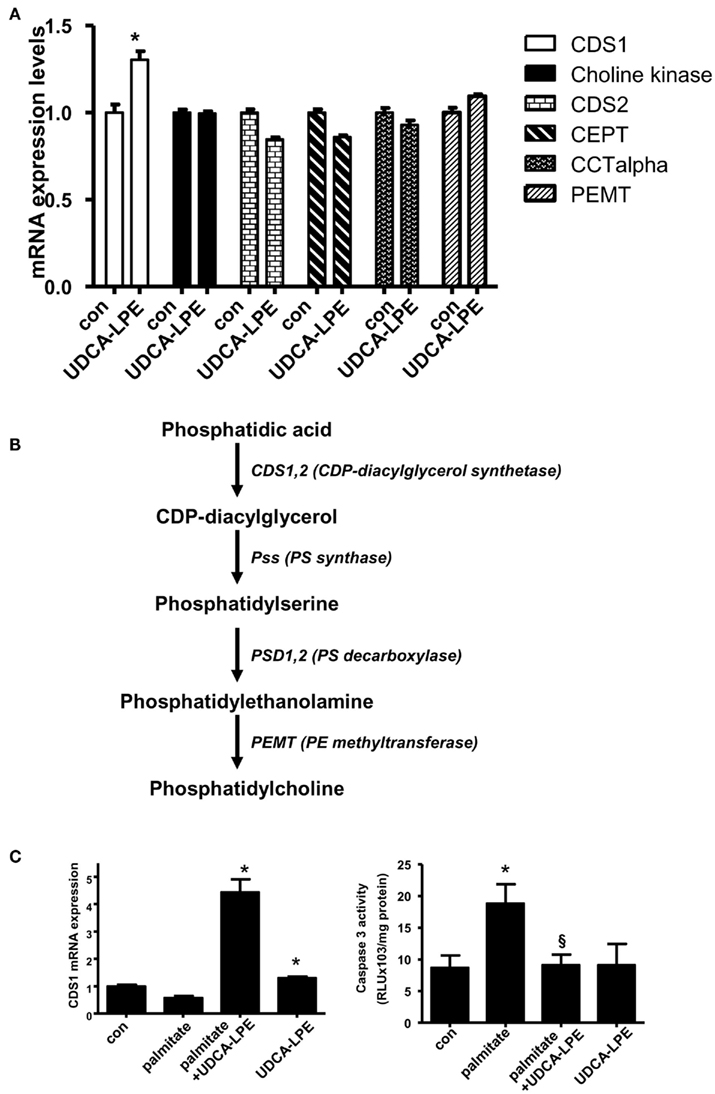
Figure 3. Ursodeoxycholyl lysophosphatidylethanolamide upregulated CDS1 gene concomitant with inhibition of apoptosis induced by palmitate. (A) Treatment of mouse hepatocytes with 60 μM UDCA-LPE for 20 h increased expression of CDS1 but failed to increase other PC synthesis genes including choline kinase, CDS2, CEPT2, CEPT, CCTalpha, and PEMT. Quantitative RT-PCR was performed by TaqMan® RT-PCR with relative expression (ΔRn) of the target gene versus GAPDH mRNA. Data were representative data obtained from two different experiments. Data were mean ± SD, N = 4; *p < 0.05 versus con. (B) A scheme demonstrates metabolism of phosphatidic acid by CDS1. Intermediate lipids were metabolized by PS synthase, PS decarboxylase, and finally PE methyltransferase to result in increased PC abundance. (C) Co-treatment of UDCA-LPE with palmitate markedly upregulated expression of CDS1 after 20 h treatment (left panel). Quantitative RT-PCR was carried out as in (A). Concomitantly, palmitate-induced apoptosis as determined by caspase 3 activity was significantly inhibited by UDCA-LPE (right panel). Data are mean ± SD, N = 6; *p < 0.05 versus control; §p < 0.05 versus palmitate. “Con” represents control hepatocytes treated with 0.5% BSA used as a vehicle.
Discussion
Ursodeoxycholyl lysophosphatidylethanolamide is a first-in-class hepatoprotectant being superior to UDCA or PC capable of inhibiting hepatocellular apoptosis (Chamulitrat et al., 2009) and inflammation in vivo under acute liver injury (Pathil et al., 2011a). PC controls liver homeostasis by membrane stabilization and the loss of PC levels results in an on-set of development of liver disease. It is of a question whether UDCA-LPE could increase PC to replenish PC for cytoprotection. We herein demonstrated that UDCA-LPE enhanced PC concentrations in vitro and in vivo, and these increases were associated with upregulation of CDS1. This ability of UDCA-LPE to increase PC can enhance hepatoprotection and renders it appropriate to treat acute hepatotoxicity.
We have shown that UDCA-LPE is able to induce survival signaling pathway by activating PI3K/Akt and MAPK/ERK1/2 pathways within 0.5–7 h in cultured hepatocytes (Chamulitrat et al., 2009). At the rapid time point of 4 h where increased PC was observed (Figures 1 and 2), it is unlikely that UDCA-LPE could induce PC synthesis genes on the transcriptional and translational levels. In cultured hepatocytes, the early PC response obtainable at 4-h treatment with UDCA-LPE may thus likely due to some unknown signaling pathways triggered by UDCA-LPE which result in increases of PC abundance. For such rapid activation, it is possible that UDCA-LPE may interact with G-protein coupled receptors on plasma membrane or nuclear receptors intracellularly. The immediate PC increases were also observed in vivo 2 h after intraperitoneal injection. Specific signaling pathways in regulating PC metabolism responsible for a rapid rise of PC have not been identified. PPAR alpha and gamma as well as glucocorticoid receptors are among nuclear receptors proposed to be affected by UDCA-LPE. These receptors are known to modulate the syntheses of phospholipids (Lee et al., 2004) including PC (Pan et al., 2006). After prolonged hepatocyte treatment with UDCA-LPE for 20 h, we found that CDS1 mRNA was upregulated. CDS1 generates PC from phosphatidic acid. CDS1 upregulation could explain the increased and sustained PC levels observed even after 24 h under in vivo conditions. The immediate and prolonged increases of hepatic PC levels by UDCA-LPE may provide feasibility for its use in the clinics, for an example, for effective reduction of acute severe hepatotoxicity from acetaminophen overdose. UDCA-LPE may be used as an antidote against hepatotoxins in humans.
Ursodeoxycholyl lysophosphatidylethanolamide is better than UDCA or PC in inhibiting apoptosis and liver injury (Chamulitrat et al., 2009; Pathil et al., 2011a). PC synthesis is known to be downregulated during apoptosis, and that PC supplementation provides protection (Cui and Houweling, 2002). In addition to UDCA-LPE’s ability to inhibit apoptosis, the PC generated by UDCA-LPE by ways of CDS1 upregulation could also be a mechanism for its anti-apoptosis effects (Figure 3C). In support of this notion, the contents of product of CDS1 CDP-diacylglycerol contents are found to be decreased in livers of diabetic rats (Whiting et al., 1977), and increased CDS1 is found during the development of heart failure and identified to be involved in compensatory mechanisms (Saini-Chohan et al., 2009). These data suggest protective function of CDS1 and consistent with UDCA-LPE protection.
In addition to UDCA (Simko and Michael, 1994), other protective bile acids, such as 5 beta-scymnol (Slitt et al., 2004), have been shown to reduce liver injury and acetaminophen toxicity. UDCA-LPE may be hepatoprotective in these settings as UDCA-LPE is superior to UDCA. Furthermore, UDCA-LPE inhibits activities of hepatic phospholipase A2 thus decreasing the levels of cytotoxic lyso PC (Pathil et al., 2011a,b). It has been shown that CDS1 activity is inhibited by lyso PC in vitro (Lin et al., 1991). UDCA-LPE’s ability to lower lyso PC concentrations may lead to increases in CDS1 activity and hence increased PC. Increased CDS1 and PC abundance by UDCA-LPE could also provide dilinoleyl- or polyenyl PC (Aleynik et al., 1997; Lieber et al., 2003) as well as PC-containing medium-chain fatty acids, such as dilauroyl PC, which is a new PC molecule recently identified to elicit protective anti-diabetic effects (Lee et al., 2011). Alternatively, UDCA-LPE may prevent PC export to extracellular space thus allowing more PC for maintaining cell integrity during stress. Further analyses of PC export genes affected by UDCA-LPE are still yet to be determined.
It is possible that increases of PC may indicate increased availability of free fatty acids due to upregulation of lipogenic genes by drug treatment (Anthérieu et al., 2011). This possibility may be applicable for treatment longer than 4 h to allow protein translation. UDCA-LPE’s ability to increase lipogenic genes such as fatty acid synthase and elongase genes has previously been observed (unpublished data). These de novo lipogenic lipids including PC in fact reflect the protection against cell death during palmitate stress after 20 h (Collins et al., 2010; Green and Olsen, 2011). It has been shown that CDS1 expression is concomitantly increased with lipogenesis genes fatty acid synthase during fetal lung development (Zhang et al., 2004), it is surmised that CDS1 may be similarly regulated by de novo lipogenesis transcription factors. As UDCA-LPE elicits lipoprotection after 20 h treatment, UDCA-LPE may be found most effective in treatment of pathological acute hepatotoxicity as to compare with normal physiological conditions. While many cationic amphiphilic drugs such as chlorpromazine and tamoxifen are known to be cytotoxic to hepatocytes by inducing phospholipidosis (Chatman et al., 2009), it is still to be determined whether UDCA-LPE could accumulate phospholipids in lysosomes (which are the organelles for phospholipidosis). As UDCA-LPE targets mitochondria by inhibiting the loss of mitochondrial membrane potentials (Chamulitrat et al., 2009), UCDA-LPE may elicit protection during acute liver injury at this organelle (Pathil et al., 2011a). PC could likely be accumulated in the mitochondrial membrane for protection.
We herein demonstrated that UDCA-LPE rapidly caused accumulation of hepatic PC likely by stimulation of specific signaling pathways of PC metabolism. Further experiments are still yet to be performed to investigate UDCA-LPE protective effects against severe hepatotoxicity induced by drugs such as acetaminophen.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Nenad Katava and Sabine Tuma for technical assistance. This study was supported by DFG (STR216/15-3) to Wolfgang Stremmel.
Abbreviations
BSA, bovine serum albumin; CDS1, CDP-diacylglycerol synthase 1; LC/MS, liquid chromatography mass spectrometry; LPE, 1-18:1 lysophosphatidylethanolamine; Pal, palmitate; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PPAR, peroxisome proliferator-activated receptor; PS, phosphatidylserine; UDCA, ursodeoxycholic acid; UDCA-LPE, ursodeoxycholyl lysophosphatidylethanolamide.
References
Aleynik, S. I., Leo, M. A., Ma, X., Aleynik, M. K., and Lieber, C. S. (1997). Polyenylphosphatidylcholine prevents carbon tetrachloride-induced lipid peroxidation while it attenuates liver fibrosis. J. Hepatol. 27, 554–561.
Anthérieu, S., Rogue, A., Fromenty, B., Guillouzo, A., and Robin, M. A. (2011). Induction of vesicular steatosis by amiodarone and tetracycline is associated with up-regulation of lipogenic genes in HepaRG cells. Hepatology 53, 1895–1905.
Camacho, J., and Rubalcava, B. (1984). Lipid composition of liver plasma membranes from rats intoxicated with carbon tetrachloride. Biochim. Biophys. Acta 776, 97–104.
Chamulitrat, W., Burhenne, J., Rehlen, T., Pathil, A., and Stremmel, W. (2009). Bile salt-phospholipid conjugate ursodeoxycholyl lysophosphatidylethanolamine as a hepatoprotective agent. Hepatology 50, 143–154.
Chatman, L. A., Morton, D., Johnson, T. O., and Anway, S. D. (2009). A strategy for risk management of drug-induced phospholipidosis. Toxicol. Pathol. 37, 997–1005.
Collins, J. M., Neville, M. J., Hoppa, M. B., and Frayn, K. N. (2010). De novo lipogenesis and stearoyl-CoA desaturase are coordinately regulated in the human adipocyte and protect against palmitate-induced cell injury. J. Biol. Chem. 28, 6044–6052.
Cui, Z., and Houweling, M. (2002). Phosphatidylcholine and cell death. Biochim. Biophys. Acta 1585, 87–96.
DeLong, C. J., Shen, Y. J., Thomas, M. J., and Cui, Z. (1999). Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J. Biol. Chem. 274, 29683–29688.
Esko, J. D., Nishijima, M., and Raetz, C. R. (1982). Animal cells dependent on exogenous phosphatidylcholine for membrane biogenesis. Proc. Natl. Acad. Sci. U.S.A. 79, 1698–1702.
Gebhart, A. M., and Brabec, M. J. (1985). Carbon tetrachloride depresses hepatic phospholipid synthesis in rats. Toxicol. Lett. 24, 71–78.
Green, C. D., and Olsen, L. K. (2011). Modulation of lipogenesis in hepatocytes by SCD-1 and Elovol6. Am. J. Physiol. Endocrinol. Metab. 300, E640–E649.
Green, T. J., Sivilotti, M. L., Langmann, C., Yarema, M., Juurlink, D., Burns, M. J., and Johnson, D. W. (2010). When do the aminotransferases rise after acute acetaminophen overdose? Clin. Toxicol. (Phila.) 48, 787–792.
Hwang, J., Chang, Y. H., Park, J. H., Kim, S. Y., Chung, H., Shim, E., and Hwang, H. J. (2011). Dietary saturated and monounsaturated fats protect against acute acetaminophen hepatotoxicity by altering fatty acid composition of liver microsomal membrane in rats. Lipids Health Dis. 10, 184.
Jackowski, S. (1994). Coordination of membrane phospholipid synthesis with the cell cycle. J. Biol. Chem. 269, 3858–3867.
Jaeschke, H., Werner, C., and Wendel, A. (1987). Disposition and hepatoprotection by phosphatidyl choline liposomes in mouse liver. Chem. Biol. Interact. 64, 127–137.
James, J. L., Moody, D. E., Chan, C. H., and Smuckler, E. A. (1982). The phospholipids of the hepatic endoplasmic reticulum. Structural change in liver injury. Biochem. J. 206, 203–210.
Kharbanda, K. K., Mailliard, M. E., Baldwin, C. R., Beckenhauer, H. C., Sorrell, M. F., and Tuma, D. J. (2007). Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J. Hepatol. 46, 314–321.
Lee, J. M., Lee, Y. K., Mamrosh, J. L., Busby, S. A., Griffin, P. R., Pathak, M. C., Ortlund, E. A., and Moore, D. D. (2011). A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature 474, 506–510.
Lee, S. S., Chan, W. Y., Lo, C. K., Wan, D. C., Tsang, D. S., and Cheung, W. T. (2004). Requirement of PPARalpha in maintaining phospholipid and triacylglycerol homeostasis during energy deprivation. J. Lipid Res. 45, 2025–2037.
Li, Z., Agellon, L. B., Allen, T. M., Umeda, M., Jewell, L., Mason, A., and Vance, D. E. (2006). The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 3, 321–331.
Li, Z., Agellon, L. B., and Vance, D. E. (2005). Phosphatidylcholine homeostasis and liver failure. J. Biol. Chem. 280, 37798–37802.
Lieber, C. S., Weiss, D. G., Groszmann, R., and Paronetto, F. II. (2003). Veterans affairs cooperative study of polyenyl-phosphatidylcholine in alcoholic liver disease. Alcohol. Clin. Exp. Res. 27, 1765–1772.
Lin, C. H., Lin, J., and Strickland, K. P. (1991). Bovine brain microsomal CDP-diacylglycerol synthetase: solubilization and properties. Biochem. Int. 25, 299–306.
Miyazaki, M., Bai, L., and Namba, M. (1991). Extending effects of phospholipids, cholesterol, and ethanolamines on survival of adult rat hepatocytes in serum-free primary culture. Res. Exp. Med. (Berl.) 191, 77–83.
Miyazawa, T., Suzuki, T., Fujimoto, K., and Kaneda, T. (1990). Phospholipid hydroperoxide accumulation in liver of rats intoxicated with carbon tetrachloride and its inhibition by dietary alpha-tocopherol. J. Biochem. 107, 689–693.
Noga, A. A., and Vance, D. E. (2003). Insights into the requirement of phosphatidylcholine synthesis for liver function in mice. J. Lipid Res. 44, 1998–2005.
Okiyama, W., Tanaka, N., Nakajima, T., Tanaka, E., Kiyosawa, K., Gonzalez, F. J., and Aoyama, T. (2009). Polyene-phosphatidylcholine prevents alcoholic liver disease in PPARalpha-null mice through attenuation of increases in oxidative stress. J. Hepatol. 50, 1236–1246.
Pan, H. J., Lin, Y., Chen, Y. E., Vance, D. E., and Leiter, E. H. (2006). Adverse hepatic and cardiac responses to rosiglitazone in a new mouse model of type 2 diabetes: relation to dysregulated phosphatidylcholine metabolism. Vascul. Pharmacol. 45, 65–71.
Pathil, A., Warth, A., Chamulitrat, W., and Stremmel, W. (2011a). The synthetic bile acid-phospholipid conjugate ursodeoxycholyl lysophosphatidylethanolamine suppresses TNFα-induced liver injury. J. Hepatol. 54, 674–684.
Pathil, A., Mueller, J., Warth, A., Chamulitrat, W., and Stremmel, W. (2011b). Ursodeoxycholyl lysophosphatidylethanolamine improves steatosis and inflammation in murine models of nonalcoholic fatty liver disease. Hepatology doi: 10.1002/hep.25531. [Epub ahead of print].
Saini-Chohan, H. K., Holmes, M. G., Chicco, A. J., Taylor, W. A., Moore, R. L., McCune, S. A., Hickson-Bick, D. L., Hatch, G. M., and Sparagna, G. C. (2009). Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J. Lipid Res. 50, 1600–1608.
Sgoutas, D. S. (1967). Phospholipid changes during hepatic injury caused by carbon tetrachloride. Metab. Clin. Exp. 16, 382–391.
Shimizu, Y. (1969). Effect of carbon tetrachloride administration on the synthesis of triglycerides and phospholipids in rat liver. J. Lipid Res. 10, 479–486.
Simko, V., and Michael, S. (1994). Effect of ursodeoxycholic acid on in vivo and in vitro toxic liver injury in rats. Aliment. Pharmacol. Ther. 8, 315–322.
Slitt, A. L., Naylor, L., Hoivik, J., Manautou, J. E., Macrides, T., and Cohen, S. D. (2004). The shark bile salt 5 beta-scymnol abates acetaminophen toxicity, but not covalent binding. Toxicology 203, 109–121.
Speck, R. F., and Lauterburg, B. H. (1991). Fish oil protects mice against acetaminophen hepatotoxicity in vivo. Hepatology 13, 557–561.
Sugano, M., Cho, S., Imaizumi, K., and Wada, M. (1970). Hepatotoxicity and lipid metabolism. 3. Changes in phosphatidylcholine and phosphatidylethanolamine during hepatic injury caused by carbon tetrachloride. Biochem. Pharmacol. 19, 2325–2333.
Trudell, J. R., Bösterling, B., and Trevor, A. J. (1982). Reductive metabolism of carbon tetrachloride by human cytochromes P-450 reconstituted in phospholipid vesicles: mass spectral identification of trichloromethyl radical bound to dioleoyl phosphatidylcholine. Proc. Natl. Acad. Sci. U.S.A. 79, 2678–2682.
Wallnofer, H., and Hanusch, M. (1973). Essential phospholipids for therapy in liver diseases. Med. Monatsschr. 27, 131–136.
Whiting, P. H., Bowley, M., Sturton, R. G., Pritchard, P. H., Brindley, D. N., and Hawthorne, J. N. (1977). The effect of chronic diabetes, induced by streptozotocin, on the activities of some enzymes of glycerolipid synthesis in rat liver. Biochem. J. 168, 147–153.
Yan, J. J., Jung, J. S., Lee, J. E., Lee, J., Huh, S. O., Kim, H. S., Jung, K. C., Cho, J. Y., Nam, J. S., Suh, H. W., Kim, Y. H., and Song, D. K. (2004). Therapeutic effects of lysophosphatidylcholine in experimental sepsis. Nat. Med. 10, 161–167.
Keywords: acute liver injury, cytoprotection, drug poisoning, bile acid–phospholipid conjugate, PC homeostasis
Citation: Chamulitrat W, Zhang W, Xu W, Pathil A, Setchell K and Stremmel W (2012) Hepatoprotectant ursodeoxycholyl lysophosphatidylethanolamide increasing phosphatidylcholine levels as a potential therapy of acute liver injury. Front. Physio. 3:24. doi: 10.3389/fphys.2012.00024
Received: 04 January 2012; Accepted: 03 February 2012;
Published online: 20 February 2012.
Edited by:
Ali Canbay, University Hospital Essen, GermanyReviewed by:
Ali Canbay, University Hospital Essen, GermanyMatias Antonio Avila, Universidad de Navarra, Spain
Copyright: © 2012 Chamulitrat, Zhang, Xu, Pathil, Setchell and Stremmel. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Walee Chamulitrat, Forschungsgruppen, Department of Internal Medicine IV, University Heidelberg Hospital, Im Neuenheimer Feld 345, EG, Lab 054, 69120 Heidelberg, Germany. e-mail: walee.chamulitrat@med.uni-heidelberg.de