
- Cardiovascular Research Group, School of Biomedicine, University of Manchester, Manchester, UK
Age is a significant risk factor for the development of vascular diseases, such as atherosclerosis. Although pharmacological treatments, including statins and anti-hypertensive drugs, have improved the prognosis for patients with cardiovascular disease, it remains a leading cause of mortality in those aged 65 years and over. Furthermore, given the increased life expectancy of the population in developed countries, there is a clear need for alternative treatment strategies. Consequently, the relationship between aging and progenitor cell-mediated repair is of great interest. Endothelial progenitor cells (EPCs) play an integral role in the cellular repair mechanisms for endothelial regeneration and maintenance. However, EPCs are subject to age-associated changes that diminish their number in circulation and function, thereby enhancing vascular disease risk. A great deal of research is aimed at developing strategies to harness the regenerative capacity of these cells. In this review, we discuss the current understanding of the cells termed “EPCs,” examine the impact of age on EPC-mediated repair and identify therapeutic targets with potential for attenuating the age-related decline in vascular health via beneficial actions on EPCs.
Introduction
Despite significant advances in cardiovascular medicine over the past decade, cardiovascular disease (CVD) remains the leading cause of morbidity and mortality throughout the developed world. Aging is a major risk factor for the onset and progression of CVD. The endothelium, which lines the lumen of all blood vessels, plays a pivotal role in maintaining cardiovascular homeostasis. This dynamic interface serves an enormous array of functions including the regulation of coagulation, arterial tone, permeability, vessel growth, and inflammation. As such, maintaining the functional integrity of the endothelial monolayer is of crucial importance for the prevention of CVDs, such as atherosclerosis. However, a characteristic of the aging process is the development of endothelial dysfunction, rendering the vasculature susceptible to the development of atherosclerosis and subsequent cardiovascular events. Although the mechanism of age-related impaired endothelial function remains unclear, an imbalance between the magnitude of vascular injury and the capacity for repair appears to play a role. Accumulating evidence suggests that bone-marrow derived circulating endothelial progenitor cells (EPCs) contribute to vascular repair and regeneration. In settings of tissue ischemia or endothelial damage, EPCs are mobilized from the bone marrow into the circulation and home to sites of vascular injury where they are able to contribute to new blood vessel formation and aid recovery. A deterioration of endogenous EPC function with age may culminate in a decreased capacity for neovascularization of ischemic tissues and/or reduced re-endothelialization of vascular lesions, facilitating the development, progression, and clinical sequelae of atherosclerotic disease.
The Discovery of Circulating EPCs
Asahara et al. (1997) published the first description of a candidate EPC; CD34+ circulating cells capable of differentiating into cells with endothelial characteristics and the ability to improve vascularization in a mouse model of hind limb ischemia. This discovery heralded a paradigm shift in vascular biology as it was recognized that vasculogenesis, a process by which new blood vessels form de novo through the differentiation of progenitor cells, and generally accepted as being restricted to embryonic development, may contribute to vascular repair and regeneration in adult life. The possibility of inducing new blood vessel formation and/or repairing damaged vessels by delivering/recruiting EPCs is an attractive idea that has stimulated much interest among the scientific community.
However, contradictory findings regarding the origin, identification, and contribution of EPCs to the neoangiogenic process has generated much debate within the field. The controversy is largely due to differences in the methodology used to isolate and culture EPCs. Consequently, the term EPC has encompassed a host of different cell types, which unsurprisingly, have demonstrated a mixed ability to contribute to blood vessel formation and repair.
In general, three methods have been described to isolate and expand EPCs from umbilical cord and peripheral blood mononuclear cells (MNCs; Figure 1; reviewed in Hirschi et al., 2008). The first method, originally described by Asahara et al. (1997) involves the plating of MNCs on fibronectin-coated tissue culture surfaces and re-plating the non-adherent cells after 48 h. Colonies form after 4–9 days and have been termed early EPCs. It is accepted that these are myeloid-derived cells that have a limited proliferative capacity and fail to form tube-like structures in vitro. The ability of these cells to augment new blood vessel formation appears to be through the production of angiogenic growth factors, chemokines, and cytokines without direct incorporation into vascular networks (Hur et al., 2004). The second approach involves plating MNCs on fibronectin-coated dishes for 4 days, but this time keeping the adherent cells which give rise to a heterogenous collection of cells termed circulating angiogenic cells (CACs). As with early EPCs, CACs express endothelial cell surface antigens (Vasa et al., 2001) but also retain monocytic characteristics and have a low proliferative potential (Rehman et al., 2003). The third method of EPC isolation involves a longer period of culture of MNCs on type I collagen coated surfaces. A population of cells originate from the adherent cells between 7 and 21 days, and have been referred to as outgrowth endothelial cells “OECs” (Lin et al., 2000; Gulati et al., 2003). OECs uniformly express endothelial markers, such as cluster of differentiation (CD) 31, vascular endothelial (VE)-cadherin, vascular endothelial growth factor receptor-2 (VEGFR-2), and uptake of acetylated low density lipoprotein (AcLDL), but of note, unlike early EPCs and CACs, this cell type is negative for hematopoietic or monocyte/macrophage cell surface antigens CD45, CD14, and CD115. OECs are capable of high rates of proliferation in vitro and vessel-forming activity when implanted in collagen gels in vivo (Ingram et al., 2004; Melero-Martin et al., 2007; Yoder et al., 2007; Au et al., 2008).
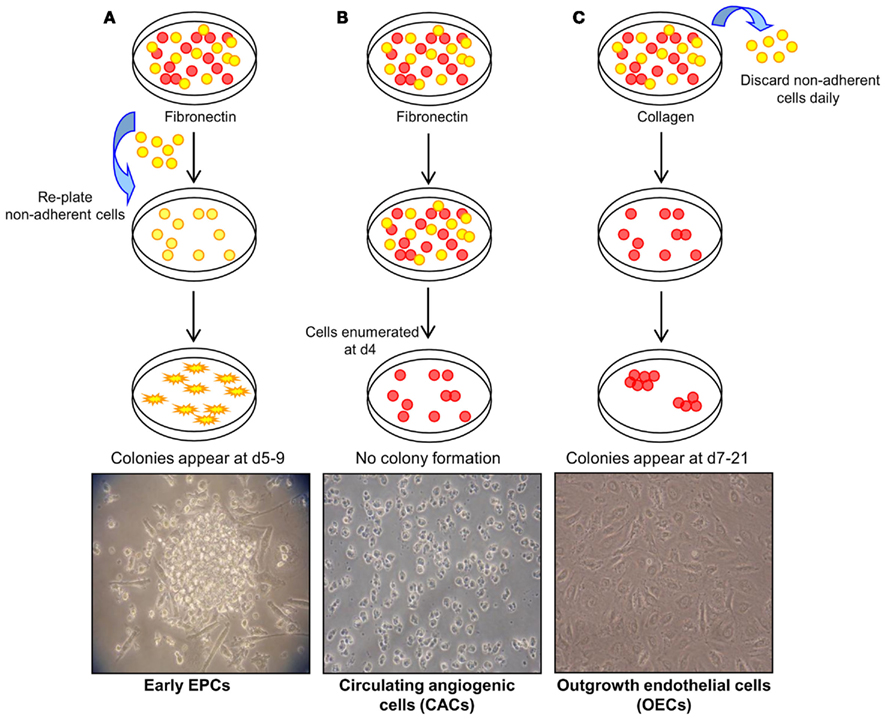
Figure 1. Methods of endothelial progenitor cell (EPC) isolation. (A) Method 1: culture of early EPCs. Non-adherent MNCs are plated on fibronectin-coated tissue surfaces and form colonies after 4–9 days. These colonies have been termed early EPCs and are comprised of round cells surrounded by spindle-shaped cells. Image courtesy of Sahena Haque (×40 magnification). (B) Method 2: culture of circulating angiogenic cells (CACs). MNCs are plated on fibronectin-coated tissue surfaces for 4 days after which the non-adherent cells are removed. The adherent cells have been termed CACs and do not typically display colony formation. Image courtesy of John Reynolds (×20 magnification). (C) Method 3: culture of outgrowth endothelial cells (OECs). MNCs plated on collagen I-coated surfaces form colonies, termed OECs, with a cobblestone morphology after 7–21 days of culture (×20 magnification). Adapted from Hirschi et al. (2008).
Considerable effort has been devoted to distinguishing these different cell types so that a definitive classification of an EPC phenotype can be established (Medina et al., 2010; Richardson and Yoder, 2011). It is important to note, that to gain quantitative data of putative circulating EPCs, flow cytometric analysis, in which EPCs are identified by cell surface phenotype, has been widely used. However, as the field has yet to reach agreement as to the definitive phenotype(s) of an EPC, much debate surrounds the choice of surface markers used in this approach. Indeed, many studies reporting to quantify circulating EPCs have in fact quantified hematopoietic cells with angiogenic capabilities (reviewed by Mund and Case, 2011).
EPCs and Reduced Vascular Repair with Age
The mobilization of EPCs from their bone-marrow niche appears to constitute an integral part of the vascular homeostatic response, since acute ischemic events, such as myocardial infarction (Shintani et al., 2001; Massa et al., 2005), acute coronary syndrome (George et al., 2004), and vascular injury secondary to burns or surgery (Gill et al., 2001) are associated with a significant and rapid increase in the levels of circulating EPCs. Consistent with the notion that EPCs serve an integral role in cardiovascular repair mechanisms, depletion of the circulating EPC pool has been shown to be a marker of cardiovascular damage and an independent predictor of cardiovascular events and death (Schmidt-Lucke et al., 2005; Werner et al., 2005). Whether the decrease in EPC number and impairment of function contributes to a loss of vasculoprotection, and the progression of CVD with age, remains to be fully elucidated. However, the link between age and EPC dysfunction is supported by a number of studies. Heiss et al. compared the number and function of early EPCs isolated from the peripheral blood of healthy young and old (average ages of 25 and 61 years respectively) individuals. Whilst there were no significant differences in the numbers of circulating EPCs (defined as CD34+/KDR+ or CD133+/KDR+) between the two groups, early EPCs from the old subjects were found to be significantly impaired in terms of proliferation, migration, and survival (Heiss et al., 2005). Rauscher et al. (2003) demonstrated that bone-marrow derived EPCs (defined as CD31+ CD45−) from young non-atherosclerotic ApoE−/− mice reduced atherosclerotic plaque size in ApoE−/− recipients despite persistent hypercholesterolemia, an effect not detected when using EPCs from old ApoE−/− mice, thus suggesting that the atheroprotective properties of EPCs are diminished with age. In addition, transplantation of bone-marrow derived EPCs from young, but not old donor mice, was seen to prevent a decline in the angiogenic platelet-derived growth factor (PDGF)-B signaling and cardiac angiogenesis in an aging murine model (Edelberg et al., 2002). It is likely that the age-associated impairment of EPC number and function is due to a variety of environmental changes that impair EPC generation, mobilization from the bone marrow, homing, and function, as well as intracellular alterations within the cells that induce a senescent phenotype (Figure 2).
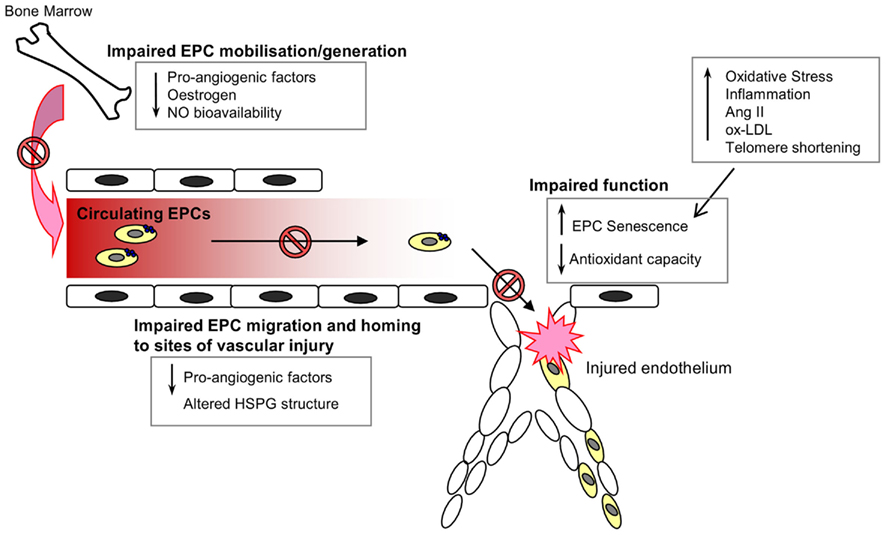
Figure 2. Endothelial progenitor cells in the aging vasculature. A wide range of environmental and internal cellular changes occur with advancing age that impair EPC numbers in circulation and function at the site of injury. The generation, mobilization, and survival of circulating EPCs are dependent on the presence of various pro-angiogenic factors, estrogen, and nitric oxide, all of which are known to decline with increasing age. Furthermore, the angiogenic capacity of EPCs is also impaired with age owing to an increase in oxidative stress, inflammation and the induction of a senescent phenotype.
Increased EPC Senescence with Age
With advancing age most, if not all, mammalian cell types are subject to internal alterations as well as environmental influences that ultimately cause the cell to enter a state of irreversible growth arrest, termed senescence. Internal changes include a reduction in telomere length, a region of repetitive DNA sequences that serve as the protective caps for chromosome ends. Telomere shortening beyond a critical length leads to genomic instability and ultimately cell cycle arrest. Decreased telomere length has been reported in the leukocytes of patients with CVD, such as, chronic heart failure (van der Harst et al., 2007), atherosclerosis (Samani et al., 2001), and ventricular dysfunction (Collerton et al., 2007). A reduction in telomere length with age has been shown in early EPCs of healthy, sedentary men (Kushner et al., 2009) This is, in part, due to an age-related decline in the expression of telomerase, a telomere elongating reverse transcriptase enzyme. A 57% reduction in early EPC telomerase activity was found in older men as compared to younger counterparts (Kushner et al., 2011) and this is further modulated by oxidative stress. EPC senescence induced by telomere shortening may contribute to numerical and functional impairments of EPCs and thus diminished endothelial regeneration in the aging vasculature. Interestingly, overexpression of the human telomerase reverse transcriptase (hTERT) gene in CACs has been shown to conserve telomerase activity, delay cell senescence, and enhance EPC reparative functions in a murine ischemic hind limb model (Murasawa et al., 2002).
EPCs and Pro-Angiogenic Factors
An age-related decline in the expression of pro-angiogenic factors, including growth factors, cytokines, and hormones, is likely to contribute to impaired EPC generation, mobilization, migration, and survival. The mobilization of EPCs from the bone marrow and homing to sites of injury/neovascularization is dependent upon a number of factors, including stromal derived factor-1 (SDF-1), VEGF, granulocyte-colony stimulating factor (G-CSF), and estrogen. Decreased SDF-1 and VEGF expression in aged tissues has been shown to impair EPC (Flk-1+/CD11b−) trafficking to sites of ischemia, and is related to depressed hypoxia-inducible factor-1α (HIF-1α) signaling (Chang et al., 2007).
Premenopausal women have a lower risk of CVD than postmenopausal women or age-matched men (Grady et al., 1992; Grodstein et al., 1997). This is, in part, due to the dramatic decrease in levels of the female reproductive hormone estrogen at the onset of menopause. A number of studies have highlighted the role of estrogen in EPC generation, mobilization from the bone marrow, and incorporation at the sites of ischemia (Strehlow et al., 2003; Hamada et al., 2006; Iwakura et al., 2006; Ruifrok et al., 2009). Ovariectomy of young female mice was shown to significantly decrease the levels of EPCs (Sca-1+/Flk-1+) in the peripheral blood and residing bone marrow, but this effect was prevented by treatment with exogenous estrogen (Strehlow et al., 2003). In addition to augmenting EPC generation and mobilization, estrogen has been shown to inhibit the senescence of CACs in vitro by increasing telomerase activity, and also appears to promote EPC proliferation and their incorporation into vascular networks (Imanishi et al., 2005b).
The activity of a wide array of pro-angiogenic factors, including VEGF, basic fibroblast growth factor (bFGF), and SDF-1 is modulated by cell surface heparan sulfate (HS) proteoglycans (Fuster and Wang, 2010). Work from our laboratory suggests that age-associated changes of HS structure on the surface of OECs correlates with a significant reduction in migratory function (Williamson et al., 2011).
EPCs and Oxidative Stress
The progressive accumulation of oxidative damage with age, due to increased production of reactive oxygen species (ROS) and decreased expression of antioxidant proteins, also promotes senescent cell changes. ROS lead to inactivation of nitric oxide (NO), cause damage to proteins, lipids, and DNA, and alter cellular redox state. Oxidative stress promotes the development of vascular pathologies (Higgins et al., 2011; Sugamura and Keaney, 2011). With increasing age, it is likely that oxidative cellular damage accumulates in EPCs, diminishing function and enhancing vascular disease risk. Early EPCs, CACs, and OECs have been shown to express high levels of antioxidant enzymes and as a result, are more resistant to oxidative stress, as compared with human umbilical vein endothelial cells (HUVECs) and adult microvascular endothelial cells (Dernbach et al., 2004; He et al., 2004, 2009; Cai et al., 2006). Such an enhanced antioxidant system may endow EPCs with improved resistance to oxidative stress and thus the ability to promote vascular regeneration in settings of ischemic injury. However, the antioxidant capacity of EPCs appears to be impaired with age, for example, He et al. (2009) demonstrated that early EPCs derived from old subjects had significantly reduced levels and activity of the antioxidant enzyme glutathione peroxidase-1 (GPx-1) and were more sensitive to oxidative stress-induced apoptosis as compared to EPCs of younger subjects. Together, these factors may reduce EPC survival capacity and their ability to promote endothelial repair in the aging host. Aging is also associated with an upregulation of proatherogenic stimuli including angiotensin II (Ang II) (Baylis et al., 1997; Wang et al., 2003), which has been shown to enhance the production of ROS in early EPCs and thereby accelerate cellular senescence (Imanishi et al., 2005a; Endtmann et al., 2011). Moreover, treatment of coronary artery disease patients with an Ang II type I receptor (AT1-R) antagonist was found to significantly increase the number of circulating EPCs (CD34+/KDR+) (Endtmann et al., 2011).
A Decrease in Nitric Oxide Bioavailability in EPCs with Age
A decline in EPC levels and function may be related to the reduced capacity of the aging endothelium to generate nitric oxide (NO) and the increased production of ROS with age. The importance of endothelial nitric oxide synthase (eNOS) expression, and subsequent NO production, for EPC mobilization, was documented in eNOS null mice by Laufs and colleagues. Here, physical exercise, which increases NO bioavailability, was found to significantly increase EPC numbers (Sca-1+/Flk-1+) while this effect was attenuated in eNOS null mice and wild type mice treated with an NOS inhibitor (Laufs et al., 2004). The accumulation of oxidized low density lipoprotein (ox-LDL) with age may also contribute to a reduction in the number of circulating EPCs. ox-LDL has been shown to impair early EPC survival and function, due to its inhibitory effect on EPC eNOS expression and activity (Ma et al., 2006).
EPCs and Inflammation
Aging is known to be associated with the development of chronic low grade inflammation, which renders the vasculature susceptible to the development of CVD (Csiszar et al., 2008). Among the proinflammatory changes that occur with increasing age, dysregulation of the proinflammatory cytokine tumor necrosis factor-alpha (TNF-α) has been well established (Csiszar et al., 2003, 2004, 2007). This multifunctional proinflammatory cytokine is a key player in the pathogenesis of atherosclerosis. Indeed increased circulating levels of TNF-α is associated with a high prevalence of atherosclerosis in elderly patients (Bruunsgaard et al., 2000). Chronic TNF-α treatment has been found to induce premature senescence of highly proliferative OECs, which was related to an increase in expression of the cell cycle inhibitor p16INK4a (Zhang et al., 2009). Thus, the shift to a proinflammatory state in the aged vasculature may contribute to EPC dysfunction and a reduced regenerative potential.
Concluding Remarks
Although age-related CVD may have multifactorial etiologies, a common element appears to be the deterioration of endogenous EPC function. Understanding EPC biology and identifying strategies to restore the factors that are depleted or impaired with age may provide a protective benefit to the cardiovascular system and limit vascular disease progression in the aging population. Indeed, pharmacological treatments including statins (Landmesser et al., 2004), estrogen (Iwakura et al., 2003, 2006), and peroxisome proliferator-activated receptor gamma agonists (Redondo et al., 2007; Werner et al., 2007) have been shown to augment EPC number and function, by increasing NO bioavailability in EPCs, among other mechanisms of action. Treatment with angiotensin receptor antagonists have been shown to reduce oxidative stress in EPCs and augment function by blocking the effect of AngII (Bahlmann et al., 2005). Other pharmacological strategies to reverse age-related EPC dysfunction include growth hormone treatment to increase insulin-like growth factor-1 (IGF-1) levels, the synthesis of which is attenuated with aging (Thum et al., 2007). Lifestyle modifications, such as regular aerobic exercise or cessation of smoking, are known to promote vascular health in the aging population and have direct effects on EPC number and function. Reports suggest that physical exercise increases the number and re-endothelialization capacity of EPCs in previously sedentary older men (Hoetzer et al., 2007; Xia et al., 2011; Yang et al., 2011), while smoking cessation has been found to induce rapid restoration of EPC levels (Kondo et al., 2004). Although long term follow up is necessary, these findings suggest that it is possible to increase EPC number and function in the aging host by restoring the pathways that govern EPC generation, mobilization, and homing. Such approaches have potential for attenuating the age-related decline in vascular health and improving the outcomes for older individuals with CVD. However, further work is needed to overcome the challenges associated with engraftment of these cells to the damaged tissue and the resulting cross-talk with the host endothelium and medial cells that will ultimately result in the repair of the damaged tissue.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Asahara, T., Murohara, T., Sullivan, A., Silver, M., Van Der Zee, R., Li, T., Witzenbichler, B., Schatteman, G., and Isner, J. M. (1997). Isolation of putative progenitor endothelial cells for angiogenesis. Science 275, 964–967.
Au, P., Daheron, L. M., Duda, D. G., Cohen, K. S., Tyrrell, J. A., Lanning, R. M., Fukumura, D., Scadden, D. T., and Jain, R. K. (2008). Differential in vivo potential of endothelial progenitor cells from human umbilical cord blood and adult peripheral blood to form functional long-lasting vessels. Blood 111, 1302–1305.
Bahlmann, F. H., De Groot, K., Mueller, O., Hertel, B., Haller, H., and Fliser, D. (2005). Stimulation of endothelial progenitor cells: a new putative therapeutic effect of angiotensin II receptor antagonists. Hypertension 45, 526–529.
Baylis, C., Engels, K., Hymel, A., and Navar, L. G. (1997). Plasma renin activity and metabolic clearance rate of angiotensin II in the unstressed aging rat. Mech. Ageing Dev. 97, 163–172.
Bruunsgaard, H., Skinhoj, P., Pedersen, A. N., Schroll, M., and Pedersen, B. K. (2000). Ageing, tumour necrosis factor-alpha (TNF-alpha) and atherosclerosis. Clin. Exp. Immunol. 121, 255–260.
Cai, H., Gehrig, P., Scott, T. M., Zimmermann, R., Schlapbach, R., and Zisch, A. H. (2006). MnSOD marks cord blood late outgrowth endothelial cells and accompanies robust resistance to oxidative stress. Biochem. Biophys. Res. Commun. 350, 364–369.
Chang, E. I., Loh, S. A., Ceradini, D. J., Chang, E. I., Lin, S. E., Bastidas, N., Aarabi, S., Chan, D. A., Freedman, M. L., Giaccia, A. J., and Gurtner, G. C. (2007). Age decreases endothelial progenitor cell recruitment through decreases in hypoxia-inducible factor 1alpha stabilization during ischemia. Circulation 116, 2818–2829.
Collerton, J., Martin-Ruiz, C., Kenny, A., Barrass, K., Von Zglinicki, T., Kirkwood, T., and Keavney, B. (2007). Telomere length is associated with left ventricular function in the oldest old: the Newcastle 85+ study. Eur. Heart J. 28, 172–176.
Csiszar, A., Labinskyy, N., Smith, K., Rivera, A., Orosz, Z., and Ungvari, Z. (2007). Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am. J. Pathol. 170, 388–398.
Csiszar, A., Ungvari, Z., Koller, A., Edwards, J. G., and Kaley, G. (2003). Aging-induced proinflammatory shift in cytokine expression profile in coronary arteries. FASEB J. 17, 1183–1185.
Csiszar, A., Ungvari, Z., Koller, A., Edwards, J. G., and Kaley, G. (2004). Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol. Genomics 17, 21–30.
Csiszar, A., Wang, M., Lakatta, E. G., and Ungvari, Z. (2008). Inflammation and endothelial dysfunction during aging: role of NF-kappaB. J. Appl. Physiol. 105, 1333–1341.
Dernbach, E., Urbich, C., Brandes, R. P., Hofmann, W. K., Zeiher, A. M., and Dimmeler, S. (2004). Antioxidative stress-associated genes in circulating progenitor cells: evidence for enhanced resistance against oxidative stress. Blood 104, 3591–3597.
Edelberg, J. M., Tang, L., Hattori, K., Lyden, D., and Rafii, S. (2002). Young adult bone marrow-derived endothelial precursor cells restore aging-impaired cardiac angiogenic function. Circ. Res. 90, E89–E93.
Endtmann, C., Ebrahimian, T., Czech, T., Arfa, O., Laufs, U., Fritz, M., Wassmann, K., Werner, N., Petoumenos, V., Nickenig, G., and Wassmann, S. (2011). Angiotensin II impairs endothelial progenitor cell number and function in vitro and in vivo: implications for vascular regeneration. Hypertension 58, 394–403.
Fuster, M. M., and Wang, L. (2010). Endothelial heparan sulfate in angiogenesis. Prog. Mol. Biol. Transl. Sci. 93, 179–212.
George, J., Goldstein, E., Abashidze, S., Deutsch, V., Shmilovich, H., Finkelstein, A., Herz, I., Miller, H., and Keren, G. (2004). Circulating endothelial progenitor cells in patients with unstable angina: association with systemic inflammation. Eur. Heart J. 25, 1003–1008.
Gill, M., Dias, S., Hattori, K., Rivera, M. L., Hicklin, D., Witte, L., Girardi, L., Yurt, R., Himel, H., and Rafii, S. (2001). Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ. Res. 88, 167–174.
Grady, D., Rubin, S. M., Petitti, D. B., Fox, C. S., Black, D., Ettinger, B., Ernster, V. L., and Cummings, S. R. (1992). Hormone therapy to prevent disease and prolong life in postmenopausal women. Ann. Intern. Med. 117, 1016–1037.
Grodstein, F., Stampfer, M. J., Colditz, G. A., Willett, W. C., Manson, J. E., Joffe, M., Rosner, B., Fuchs, C., Hankinson, S. E., Hunter, D. J., Hennekens, C. H., and Speizer, F. E. (1997). Postmenopausal hormone therapy and mortality. N. Engl. J. Med. 336, 1769–1775.
Gulati, R., Jevremovic, D., Peterson, T. E., Chatterjee, S., Shah, V., Vile, R. G., and Simari, R. D. (2003). Diverse origin and function of cells with endothelial phenotype obtained from adult human blood. Circ. Res. 93, 1023–1025.
Hamada, H., Kim, M. K., Iwakura, A., Ii, M., Thorne, T., Qin, G., Asai, J., Tsutsumi, Y., Sekiguchi, H., Silver, M., Wecker, A., Bord, E., Zhu, Y., Kishore, R., and Losordo, D. W. (2006). Estrogen receptors alpha and beta mediate contribution of bone marrow-derived endothelial progenitor cells to functional recovery after myocardial infarction. Circulation 114, 2261–2270.
He, T., Joyner, M. J., and Katusic, Z. S. (2009). Aging decreases expression and activity of glutathione peroxidase-1 in human endothelial progenitor cells. Microvasc. Res. 78, 447–452.
He, T., Peterson, T. E., Holmuhamedov, E. L., Terzic, A., Caplice, N. M., Oberley, L. W., and Katusic, Z. S. (2004). Human endothelial progenitor cells tolerate oxidative stress due to intrinsically high expression of manganese superoxide dismutase. Arterioscler. Thromb. Vasc. Biol. 24, 2021–2027.
Heiss, C., Keymel, S., Niesler, U., Ziemann, J., Kelm, M., and Kalka, C. (2005). Impaired progenitor cell activity in age-related endothelial dysfunction. J. Am. Coll. Cardiol. 45, 1441–1448.
Higgins, P., Dawson, J., Lees, K. R., Mcarthur, K., Quinn, T. J., and Walters, M. R. (2011). Xanthine oxidase inhibition for the treatment of cardiovascular disease: a systematic review and meta-analysis. Cardiovasc. Ther. doi: 10.1111/j.1755-5922.2011.00277.x. [EPub ahead of print].
Hirschi, K. K., Ingram, D. A., and Yoder, M. C. (2008). Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler. Thromb. Vasc. Biol. 28, 1584–1595.
Hoetzer, G. L., Van Guilder, G. P., Irmiger, H. M., Keith, R. S., Stauffer, B. L., and Desouza, C. A. (2007). Aging, exercise, and endothelial progenitor cell clonogenic and migratory capacity in men. J. Appl. Physiol. 102, 847–852.
Hur, J., Yoon, C. H., Kim, H. S., Choi, J. H., Kang, H. J., Hwang, K. K., Oh, B. H., Lee, M. M., and Park, Y. B. (2004). Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler. Thromb. Vasc. Biol. 24, 288–293.
Imanishi, T., Hano, T., and Nishio, I. (2005a). Angiotensin II accelerates endothelial progenitor cell senescence through induction of oxidative stress. J. Hypertens. 23, 97–104.
Imanishi, T., Hano, T., and Nishio, I. (2005b). Estrogen reduces endothelial progenitor cell senescence through augmentation of telomerase activity. J. Hypertens. 23, 1699–1706.
Ingram, D. A., Mead, L. E., Tanaka, H., Meade, V., Fenoglio, A., Mortell, K., Pollok, K., Ferkowicz, M. J., Gilley, D., and Yoder, M. C. (2004). Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 104, 2752–2760.
Iwakura, A., Luedemann, C., Shastry, S., Hanley, A., Kearney, M., Aikawa, R., Isner, J. M., Asahara, T., and Losordo, D. W. (2003). Estrogen-mediated, endothelial nitric oxide synthase-dependent mobilization of bone marrow-derived endothelial progenitor cells contributes to reendothelialization after arterial injury. Circulation 108, 3115–3121.
Iwakura, A., Shastry, S., Luedemann, C., Hamada, H., Kawamoto, A., Kishore, R., Zhu, Y., Qin, G., Silver, M., Thorne, T., Eaton, L., Masuda, H., Asahara, T., and Losordo, D. W. (2006). Estradiol enhances recovery after myocardial infarction by augmenting incorporation of bone marrow-derived endothelial progenitor cells into sites of ischemia-induced neovascularization via endothelial nitric oxide synthase-mediated activation of matrix metalloproteinase-9. Circulation 113, 1605–1614.
Kondo, T., Hayashi, M., Takeshita, K., Numaguchi, Y., Kobayashi, K., Iino, S., Inden, Y., and Murohara, T. (2004). Smoking cessation rapidly increases circulating progenitor cells in peripheral blood in chronic smokers. Arterioscler. Thromb. Vasc. Biol. 24, 1442–1447.
Kushner, E. J., Maceneaney, O. J., Weil, B. R., Greiner, J. J., Stauffer, B. L., and Desouza, C. A. (2011). Aging is associated with a proapoptotic endothelial progenitor cell phenotype. J. Vasc. Res. 48, 408–414.
Kushner, E. J., Van Guilder, G. P., Maceneaney, O. J., Cech, J. N., Stauffer, B. L., and Desouza, C. A. (2009). Aging and endothelial progenitor cell telomere length in healthy men. Clin. Chem. Lab. Med. 47, 47–50.
Landmesser, U., Engberding, N., Bahlmann, F. H., Schaefer, A., Wiencke, A., Heineke, A., Spiekermann, S., Hilfiker-Kleiner, D., Templin, C., Kotlarz, D., Mueller, M., Fuchs, M., Hornig, B., Haller, H., and Drexler, H. (2004). Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation 110, 1933–1939.
Laufs, U., Werner, N., Link, A., Endres, M., Wassmann, S., Jurgens, K., Miche, E., Bohm, M., and Nickenig, G. (2004). Physical training increases endothelial progenitor cells, inhibits neointima formation, and enhances angiogenesis. Circulation 109, 220–226.
Lin, Y., Weisdorf, D. J., Solovey, A., and Hebbel, R. P. (2000). Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Invest. 105, 71–77.
Ma, F. X., Zhou, B., Chen, Z., Ren, Q., Lu, S. H., Sawamura, T., and Han, Z. C. (2006). Oxidized low density lipoprotein impairs endothelial progenitor cells by regulation of endothelial nitric oxide synthase. J. Lipid Res. 47, 1227–1237.
Massa, M., Rosti, V., Ferrario, M., Campanelli, R., Ramajoli, I., Rosso, R., De Ferrari, G. M., Ferlini, M., Goffredo, L., Bertoletti, A., Klersy, C., Pecci, A., Moratti, R., and Tavazzi, L. (2005). Increased circulating hematopoietic and endothelial progenitor cells in the early phase of acute myocardial infarction. Blood 105, 199–206.
Medina, R. J., O’Neill, C. L., Sweeney, M., Guduric-Fuchs, J., Gardiner, T. A., Simpson, D. A., and Stitt, A. W. (2010). Molecular analysis of endothelial progenitor cell (EPC) subtypes reveals two distinct cell populations with different identities. BMC Med. Genomics 3, 18. doi:10.1186/1755-8794-3-18
Melero-Martin, J. M., Khan, Z. A., Picard, A., Wu, X., Paruchuri, S., and Bischoff, J. (2007). In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood 109, 4761–4768.
Mund, J. A., and Case, J. (2011). The ontogeny of endothelial progenitor cells through flow cytometry. Curr. Opin. Hematol. 18, 166–170.
Murasawa, S., Llevadot, J., Silver, M., Isner, J. M., Losordo, D. W., and Asahara, T. (2002). Constitutive human telomerase reverse transcriptase expression enhances regenerative properties of endothelial progenitor cells. Circulation 106, 1133–1139.
Rauscher, F. M., Goldschmidt-Clermont, P. J., Davis, B. H., Wang, T., Gregg, D., Ramaswami, P., Pippen, A. M., Annex, B. H., Dong, C., and Taylor, D. A. (2003). Aging, progenitor cell exhaustion, and atherosclerosis. Circulation 108, 457–463.
Redondo, S., Hristov, M., Gumbel, D., Tejerina, T., and Weber, C. (2007). Biphasic effect of pioglitazone on isolated human endothelial progenitor cells: involvement of peroxisome proliferator-activated receptor-gamma and transforming growth factor-beta1. Thromb. Haemost. 97, 979–987.
Rehman, J., Li, J., Orschell, C. M., and March, K. L. (2003). Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation 107, 1164–1169.
Richardson, M. R., and Yoder, M. C. (2011). Endothelial progenitor cells: quo vadis? J. Mol. Cell. Cardiol. 50, 266–272.
Ruifrok, W. P., De Boer, R. A., Iwakura, A., Silver, M., Kusano, K., Tio, R. A., and Losordo, D. W. (2009). Estradiol-induced, endothelial progenitor cell-mediated neovascularization in male mice with hind-limb ischemia. Vasc. Med. 14, 29–36.
Samani, N. J., Boultby, R., Butler, R., Thompson, J. R., and Goodall, A. H. (2001). Telomere shortening in atherosclerosis. Lancet 358, 472–473.
Schmidt-Lucke, C., Rossig, L., Fichtlscherer, S., Vasa, M., Britten, M., Kamper, U., Dimmeler, S., and Zeiher, A. M. (2005). Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation 111, 2981–2987.
Shintani, S., Murohara, T., Ikeda, H., Ueno, T., Honma, T., Katoh, A., Sasaki, K., Shimada, T., Oike, Y., and Imaizumi, T. (2001). Mobilization of endothelial progenitor cells in patients with acute myocardial infarction. Circulation 103, 2776–2779.
Strehlow, K., Werner, N., Berweiler, J., Link, A., Dirnagl, U., Priller, J., Laufs, K., Ghaeni, L., Milosevic, M., Bohm, M., and Nickenig, G. (2003). Estrogen increases bone marrow-derived endothelial progenitor cell production and diminishes neointima formation. Circulation 107, 3059–3065.
Sugamura, K., and Keaney, J. F. Jr. (2011). Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 51, 978–992.
Thum, T., Hoeber, S., Froese, S., Klink, I., Stichtenoth, D. O., Galuppo, P., Jakob, M., Tsikas, D., Anker, S. D., Poole-Wilson, P. A., Borlak, J., Ertl, G., and Bauersachs, J. (2007). Age-dependent impairment of endothelial progenitor cells is corrected by growth-hormone-mediated increase of insulin-like growth-factor-1. Circ. Res. 100, 434–443.
van der Harst, P., Van Der Steege, G., De Boer, R. A., Voors, A. A., Hall, A. S., Mulder, M. J., Van Gilst, W. H., and Van Veldhuisen, D. J. (2007). Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J. Am. Coll. Cardiol. 49, 1459–1464.
Vasa, M., Fichtlscherer, S., Aicher, A., Adler, K., Urbich, C., Martin, H., Zeiher, A. M., and Dimmeler, S. (2001). Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 89, E1–E7.
Wang, M., Takagi, G., Asai, K., Resuello, R. G., Natividad, F. F., Vatner, D. E., Vatner, S. F., and Lakatta, E. G. (2003). Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension 41, 1308–1316.
Werner, C., Kamani, C. H., Gensch, C., Bohm, M., and Laufs, U. (2007). The peroxisome proliferator-activated receptor-gamma agonist pioglitazone increases number and function of endothelial progenitor cells in patients with coronary artery disease and normal glucose tolerance. Diabetes 56, 2609–2615.
Werner, N., Kosiol, S., Schiegl, T., Ahlers, P., Walenta, K., Link, A., Bohm, M., and Nickenig, G. (2005). Circulating endothelial progenitor cells and cardiovascular outcomes. N. Engl. J. Med. 353, 999–1007.
Williamson, K., Stringer, S., Sipos, P., Crocker, I., and Alexander, M. (2011). Age-related modification of heparan sulphate proteoglycans on human endothelial progenitor cells. Heart 97, e7.
Xia, W. H., Li, J., Su, C., Yang, Z., Chen, L., Wu, F., Zhang, Y. Y., Yu, B. B., Qiu, Y. X., Wang, S. M., and Tao, J. (2011). Physical exercise attenuates age-associated reduction in endothelium-reparative capacity of endothelial progenitor cells by increasing CXCR4/JAK-2 signaling in healthy men. Aging Cell.
Yang, Z., Xia, W. H., Su, C., Wu, F., Zhang, Y. Y., Xu, S. Y., Liu, X., Zhang, X. Y., Ou, Z. J., Lai, G. H., Liao, X. X., Jin, Y. F., and Tao, J. (2011). Regular exercise-induced increased number and activity of circulating endothelial progenitor cells attenuates age-related decline in arterial elasticity in healthy men. Int. J. Cardiol.
Yoder, M. C., Mead, L. E., Prater, D., Krier, T. R., Mroueh, K. N., Li, F., Krasich, R., Temm, C. J., Prchal, J. T., and Ingram, D. A. (2007). Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 109, 1801–1809.
Zhang, Y., Herbert, B. S., Rajashekhar, G., Ingram, D. A., Yoder, M. C., Clauss, M., and Rehman, J. (2009). Premature senescence of highly proliferative endothelial progenitor cells is induced by tumor necrosis factor-alpha via the p38 mitogen-activated protein kinase pathway. FASEB J. 23, 1358–1365.
Keywords: endothelial progenitor cells, age, vasculature, nitric oxide, estrogen, senescence, oxidative stress
Citation: Williamson K, Stringer SE and Alexander MY (2012) Endothelial progenitor cells enter the aging arena. Front. Physio. 3:30. doi: 10.3389/fphys.2012.00030
Received: 21 December 2011;
Accepted: 06 February 2012;
Published online: 20 February 2012.
Edited by:
Ana Paula Dantas, Institut d’Investigacions Biomediques August Pi i Sunyer, SpainReviewed by:
Magda Heras, Hospital Clínic de Barcelona, SpainMaria Helena C. Carvalho, Universidade de São Paulo, Brazil
Susana Novella, Research Foundation Hospital Clinico Valencia, Spain
Copyright: © 2012 Williamson, Stringer and Alexander. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: M. Y. Alexander, Cardiovascular Research Group, School of Biomedicine, University of Manchester, 46 Grafton Street, Manchester M13 9NT, UK. e-mail: yvonne.alexander@manchester.ac.uk