
- 1 Department of Biology, University Roma Tre, Roma, Italy
- 2 Interdepartmental Laboratory of Electron Microscopy, University Roma Tre, Roma, Italy
Besides its crucial role in many physiological events, 17β-estradiol (E2) exerts protective effects in the central nervous system. The E2 effects are not restricted to the brain areas related with the control of reproductive function, but rather are widespread throughout the developing and the adult brain. E2 actions are mediated through estrogen receptors (i.e., ERα and ERβ) belonging to the nuclear receptor super-family. As members of the ligand-regulated transcription factor family, classically, the actions of ERs in the brain were thought to mediate only the E2 long-term transcriptional effects. However, a growing body of evidence highlighted rapid, membrane initiated E2 effects in the brain that are independent of ER transcriptional activities and are involved in E2-induced neuroprotection. The aim of this review is to focus on the rapid effects of E2 in the brain highlighting the specific role of the signaling pathway(s) of the ERβ subtype in the neuroprotective actions of E2.
Introduction
In women at approximately 51 years of age starts a profound physiological change known as menopause, which is accompanied with a dramatic decrease in estrogen levels. Taking into the account that the average of life span has increased from 50 years to over 80 years of age, an increasing number of women are living a larger portion of their lives in a chronically hypo-estrogenic state (Wise et al., 2001). The post-menopausal state of estrogen deficiency leads to different consequences throughout the female body which include the increased risk for osteoporosis and cardiovascular diseases (McEwen, 2002; Gillies and McArthur, 2010). Besides these effects, most studies have recognized that the brain is one of the organs of the body that suffer estrogen deficiency.
In addition to the well-established effects of estrogens on the reproductive behaviors and the associated brain regions (e.g., hypothalamus), 17β-estradiol (E2), the most active estrogen, also serves as a neurotrophic and a neuroprotective agent. Indeed, E2 influences memory formation, cognition, mood, pain sensitivity, motor coordination, neurodevelopment, and neurodegeneration (Wise et al., 2001; McEwen, 2002; Barha and Galea, 2010; Gillies and McArthur, 2010). E2 elicits either organizational effects during embryonic and neonatal development or the activational control of gene expression during the later stages of life cycle by regulating synaptic transmission, neuronal survival, neuronal, and glial differentiation (Garcia-Segura et al., 2001; Woolley, 2007; DonCarlos et al., 2009). In addition, different experimental models of neurodegeneration have shown that E2 acts as a neuroprotective factor promoting neuronal survival and tissue integrity (Maggi et al., 2004; Suzuki et al., 2006; Marin et al., 2009).
Most of these actions of E2 are mediated by estrogen receptors (i.e., ERα and ERβ) belonging to the nuclear receptor super-family (Pettersson and Gustafsson, 2001; Ascenzi et al., 2006). As members of ligand-regulated transcription factor family, classically, ERs were thought to mediate only the E2 long-term transcriptional effects. However, a growing body of evidence highlighted rapid, membrane initiated E2 effects that are independent of the ER transcriptional control (Raz et al., 2008; Arnold and Beyer, 2009; De Marinis et al., 2011; Roepke et al., 2011).
Although different subtypes of ER have been reported on neuronal membranes, emerging pharmacological and ultra-structural evidences demonstrated that ERα and ERβ are localized at the cell membrane where mediate rapid activation of intracellular brain signaling pathways (Vasudevan and Pfaff, 2007; Raz et al., 2008; De Marinis et al., 2010; Gillies and McArthur, 2010).
This review will highlights the rapid effects of E2 in the brain taking into account the contribution of ERβ intracellular signaling pathway in neuroprotective actions of E2.
Structure and Activity of Estrogen Receptors
Estrogen receptor alpha and ERβ (NR3A1 and NR3A2, respectively) are products of separate genes (ESR1 and ESR2, respectively) present on distinct chromosomes (locus 6q25.1 and locus 14q23-24.1, respectively; Gosden et al., 1986; Ascenzi et al., 2006; Luisi et al., 2006; Zhou et al., 2006).
Like other members of the NR family, ERs contain evolutionarily conserved structurally and functionally distinct domains. In particular, ERs are modular proteins composed of six regions named A/B, C, D, E, and F which participate in the formation of independent but interacting functional domains. DNA-binding domain (DBD or C region), the central and most conserved domain, plays a pivotal role in receptor dimerization and in the binding of specific DNA consensus sequences (i.e., estrogen responsive elements, EREs) recognized by both receptors, whereas ligand binding occurs in the C-terminal multifunctional ligand binding domain (LBD or E region; Ascenzi et al., 2006). ERs transcriptional activation is mediated by two distinct transcription activation functions (AFs). The AF-1 is located at the N-terminus of the receptor and the AF-2 domain resides in the LBD (Ascenzi et al., 2006; Acconcia and Marino, 2011).
Classically, the ER biological activities result from modifications of the expression pattern of specific target genes. In the absence of ligands, ERs cycle on and off their DNA-binding sites (i.e., EREs). The trans-activation activity of ERs initiates after the binding of E2 which stabilizes the binding of receptors to DNA (Metivier et al., 2003; Reid et al., 2003; Picard et al., 2008; Acconcia and Marino, 2011).
In the cytoplasm of the ER-expressing cells, ER is in the monomeric state (inactive state) and forms a multiprotein complex with immunophilins and heat shock proteins (e.g., Hsp90, Hsp70, and Hsp56). Upon E2 binding, Hsps dissociate from ERs which dimerize and translocate to the nucleus. In the nucleus, the E2:ER complexes interact with ERE sequences within the promoter regions of target genes to regulate gene transcription by recruiting cofactors proteins (i.e., coactivators and corepressors) and the components of the transcription machinery (Ascenzi et al., 2006; Acconcia and Marino, 2011). Thus, the ER-macromolecular complexes facilitate the transcriptional activities of E2 responsive genes by triggering chromatin remodeling in target promoters (Ascenzi et al., 2006; Morissette et al., 2008; Acconcia and Marino, 2011).
The consensus palindromic element ERE, 5′-GGTCAnnnTGACC-3′, acts in a direction and distance independent manner, both of which are properties of an enhancer (Ascenzi et al., 2006). Nevertheless, only a fraction of known mammalian ERE displays this consensus, about one-third of the ER-regulated genes in humans do not contain ERE-like sequences (O’Lone et al., 2004; Bjornstrom and Sjoberg, 2005). Indeed, ERs can also regulate gene transcription without binding directly to DNA. This indirect genomic mechanism requires the interaction of ERs with either DNA-bound transcription factors like CREB or with Fos and Jun proteins regulating gene transcription via the activator protein 1 (AP-1) and stimulating protein 1 (Sp1; Kalaitzidis and Gilmore, 2005; Morissette et al., 2008; Acconcia and Marino, 2011).
Since 1967 it has been reported that E2 induces rapid effects in target cells that can not be related to ER transcriptional activities (Szego and Davis, 1967). Rapid effects of E2 have been reported in bone, breast, blood vessels, cancer cells, nervous system, sperm, and maturating oocytes (Nilsson and Gustafsson, 2011). E2 rapid induce the activation of several intracellular pathways (Kelly and Levin, 2001; Levin, 2005; Ascenzi et al., 2006; Acconcia and Marino, 2011). E2-induced rapid responses are important for several neural functions including cognition, behavior, stress responses, and reproduction (Farach-Carson and Davis, 2003).
E2 membrane initiated actions require ER localization at the plasma membrane. Membrane ERs are localized either in caveolae (Razandi et al., 2003) or in other membrane raft structures (Marquez et al., 2006; Acconcia and Marino, 2011). Remarkably, ERα and ERβ can be localized at the plasma membrane through the direct interaction with caveolin-1 to initiate the signal transduction pathway(s) (Acconcia and Marino, 2011; Roepke et al., 2011). Indeed, ERs require a post translational modification by lipid (i.e., S-palmitoylation) to be localized at the plasma membrane, to interact with caveolin-1, and to initiate the E2 rapid signal cascade activation (Acconcia et al., 2005; Galluzzo et al., 2007; Pedram et al., 2007; Marino and Ascenzi, 2008).
In addition, E2 determines rapid membrane initiated effects through other receptors belonging to a protein family completely different from nuclear receptor super-family. This class of receptors includes ER-X and a G-protein coupled seven-transmembrane receptor, named GPR30 (Toran-Allerand et al., 2002; Filardo et al., 2007). ER-X is oriented in caveolar-like microdomains of postnatal, but not adult, mouse neocortical, and uterine plasma membrane. It is functionally distinct from ERα and ERβ, and is re-expressed in the adult brain after ischemic stroke injury, like ERα (Toran-Allerand et al., 2002). GPR30 is expressed in areas of the brain important for spatial learning, memory, and attention and could be an important regulator of basal forebrain cholinergic functions (Prossnitz and Barton, 2011). In vivo studies showed that only pharmacological concentrations of the GPR30 specific ligand G-1 (i.e., 50 μg) could replicate the effects of the physiological concentration of E2 (i.e., 2.2 μg) in promoting neuronal survival following global ischemia in the rodent brain (Etgen et al., 2011). Although GPR30 and its ligand may represent a new pharmacological approach for treating neuronal damage, the role of these receptors in cells from ERα/ERβ homozygous double knockout (DERKO) mice is not yet demonstrated; thus, at present, the possibility that GPR30 and/or ER-X mediate the E2-induced rapid transduction pathways important for brain functions is questionable.
In addition, membrane ERs activate rapid transduction pathway(s) by interacting with either cell surface receptors, such as the growth factor receptors (e.g., the EGF receptor and the IGF-1 receptor) and the metabotropic glutamate receptor, or with other signal proteins including G-proteins, non-growth factor tyrosine kinase (e.g., Src and Ras), and linker proteins (e.g., MNAR and striatin; Hammes and Levin, 2007; Gillies and McArthur, 2010). In Figure 1 a schematic representation of nuclear and extranuclear ER activities is reported.
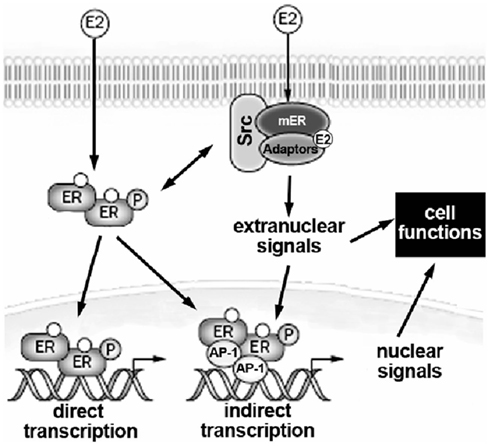
Figure 1. Schematic model illustrating the relationship between extranuclear and nuclear actions of E2 on target cells. E2, 17β-estradiol; mER, estrogen receptor located at the plasma membrane; ER, estrogen receptor; AP-1, activating factor-1. For details, see text.
Estrogen Effects and Action Mechanisms in the Brain
E2 in the brain is either locally synthesized by the precursor testosterone or imported through the blood brain barrier from circulating factors. ERs are expressed in different brain regions such as the bed nucleus of the stria terminalis, the medial amygdala, the preoptic area, and the nucleus of the solitary tract. ERα is predominantly located in the hypothalamus ventromedial nucleus and in the amygdala of humans and rodents (Shughrue et al., 1998; Osterlund et al., 2000a,b,c; Gillies and McArthur, 2010). On the contrary, ERβ is the predominant form expressed in the cerebral cortex, the hippocampus, the dorsal raphe, the substantia nigra, the cerebellum, and the hypothalamic nuclei; also serotonergic and dopaminergic neurons express ERβ (Bodo and Rissman, 2006; Handa et al., 2010). In the mammalian nervous system, ERα and ERβ distribution patterns provide some neuroanatomical evidences for their involvement in specific brain functions. Indeed, ERα, but not ERβ, is crucial for E2-induced neuroreproductive functions (Ogawa et al., 1998), however many of the non-reproductive functions of E2 in the brain can be explained only through ERβ-mediated effects (Kudwa et al., 2006; Antal et al., 2008).
Although E2 is the predominant circulating sex steroid hormone after puberty in females, it also plays a pivotal role in the male brain, E2 being synthesized from steroid precursors (e.g., testosterone by P450 aromatase enzyme). Testosterone produced during the critical developmental window in male is a key factor in the masculinization/defeminization process (Huhtaniemi, 1994). Morphological, cellular, and molecular differences exist in diverse male and female brain regions important for cognition and memory (e.g., the hippocampus, the amygdala, the cortex, and the regions controlling sensorimotor and reward systems; see Gillies and McArthur, 2010).
Functional differences in male and female brain explain the diverse responses to environmental challenges and different vulnerabilities to behavioral and neurological disorders. Striking differences between sexes have been reported concerning the symptoms, the prevalence, the progression, and the severity of several neurodegenerative diseases. Indeed, pre-menopausal women seem to be less prone to Alzheimer’s, Parkinson’s, and Huntington’s diseases than males or post-menopausal women (Amantea et al., 2005; Morissette et al., 2008 and literature cited therein). Moreover, the pivotal role of E2 on higher brain functions including mood, anxiety, fear, learning, and memory have been confirmed by epidemiological data which indicate that pre-menopausal women seem to be more vulnerable that men to develop anxiety or depression behavior (Schneier et al., 1992; Kessler et al., 1994; Breslau et al., 1995; Seeman, 1997; Luine, 2008; Watson et al., 2010).
E2 as a Neurotrophic Factor
E2 exerts multiple and diverse actions in the brain throughout the life span from development to senescence. E2 exerts neurotrophic and neuroprotective effect in adult brain regulating the synthesis and the secretory patterns of neurotransmitters, neuropeptides, and their receptors, influencing the sexual behavior as well as gonadotropin and prolactin secretion (Pfaff et al., 1994). Moreover, as it does during development.
E2 is a potent trophic factor that influences brain development and differentiation, plasticity and cell survival both during fetal life and in the early postnatal period. These trophic effects include the modulation of cell migration, neuronal growth, formation, and elimination of synapses, and neurogenesis (Pfaff et al., 1994; Garcia-Segura et al., 1996; Wise et al., 2001; Gillies and McArthur, 2010).
In nearly all mammalian species, including humans, adult neurogenesis has been observed in two brain regions characterized by neuronal progenitor cells (NPCs): the dentate gyrus of the hippocampus and the lateral walls of lateral ventricles (Gould et al., 1997, 2001; Eriksson et al., 1998; Amrein et al., 2004; Handa et al., 2010). E2 can promote the proliferation of hippocampal neuron progenitor cells in vitro and neurogenesis in rat hippocampus in vivo, both under physiological and pathological conditions (Tanapat et al., 1999, 2005; Brannvall et al., 2002, 2005; Suzuki et al., 2007). In human NPCs, E2-induced proliferation seems to be mediated only by ERβ (Wang et al., 2008), which represent the predominant ER subtype in these cells (Fried et al., 2004; Hong et al., 2004). Interestingly, both E2 and the ERβ selective-ligand diarylpropionitrile (DPN) are able to induce in vitro human NPCs proliferation through the activation of ERβ that in turn leads to a rapid increase in kinase levels (Squires et al., 2002; Ussar and Voss, 2004; Hata et al., 2005; Chambard et al., 2007; Wang et al., 2008; Handa et al., 2010).
Neurogenesis is involved in hippocampus-dependent learning and memory, it is highly correlated with the chronic antidepressant treatment of male and female rodents affected by the depressive-like behavior; the antidepressant treatment increases also cell proliferation (Malberg and Duman, 2003; Vollmayr et al., 2003; Green and Galea, 2008). Moreover, neurogenesis could represent a neuroprotective action of E2 in neurodegenerative conditions such as Alzheimer’s disease, which is accompanied by a decline of neurogenesis (Wang et al., 2007, 2010; Rodriguez et al., 2008; Demars et al., 2010). In addition to the reported effects, E2 in the hippocampus enhances learning and memory promoting the formation of new dendritic spines and new excitatory synapses, increasing the expression of NMDA receptors (Adams et al., 2004), and enhancing long-term potentiation (LTP; Smith and McMahon, 2006; Liu et al., 2008).
Dramatic changes in E2 levels, due to surgery and/or to menopause, are associated with changes in incidence and symptomatology of anxiety and depression, in memory ability, and performance in visual–spatial tasks (Hampson and Kimura, 1988; Phillips and Sherwin, 1992). E2 replacement can improve mood score (Wise et al., 2001; Walf and Frye, 2006) and counteract memory loss in both verbal and non-verbal memory test and attention (Campbell and Whitehead, 1977; Halbreich, 1997; Rhodes and Frye, 2004; Sherwin, 2005). Further confirmation of the E2 involvement in higher brain functions derives from animal models. In fact, an increased anxiety-related behavior has been observed in both rats and mice as a consequence of E2 decline levels (Gupta et al., 2001; Morgan and Pfaff, 2001, 2002; Bodo and Rissman, 2006; Inagaki et al., 2010; Jacome et al., 2010).
Mechanisms Underlying E2-induced Effects on Neurotrophism
The hippocampus seems to represent the main brain area that mediate E2 effects on the affective behavior and the cognitive function. Together with the amygdala area, hippocampus is considered an important component of the limbic system and a regulator of the hypothalamic–pituitary–adrenal axis (HPA; Walf and Frye, 2006). Although the proper mechanism by which E2 acts in hippocampus is openly debated, growing evidence indicates that E2 effects are rapid and could require a membrane-associated receptor (Walf and Frye, 2007, 2008).
Although rapid membrane-mediated effects of E2 have been identified in the hippocampus and in some hypothalamic nuclei (Ronnekleiv and Kelly, 2005; Roepke et al., 2009), they may occur throughout the whole central nervous system. Membrane initiated E2 signaling involves the rapid activation of various protein kinase cascades, including protein kinase C (PKC), protein kinase A (PKA), phosphatidylinositol-3 kinase (PI3K), protein kinase B (or Akt), and mitogen-activated protein kinase (MAPK), which in turn modulate protein phosphorylation, cation channel activity, and gene expression (Roepke et al., 2011). Furthermore, E2-activated signaling pathways modulate the intracellular Ca2+ levels regulating the activation of protein kinases including the Ca2+-calmodulin-dependent protein kinase II (CaMKII; Gillies and McArthur, 2010).
A rapid induction of the cAMP/PKA pathway, mediating the effect of E2 on the change of K+ fluxes in GnRH neurons (Gu and Moss, 1996; Malyala et al., 2005), has been demonstrated in hippocampal neurons (Chen et al., 1998; Ascenzi et al., 2006). Moreover, E2 can attenuate the ability of μ-opioid and GABA receptor-mediated activation of G-protein-gated inwardly rectifying K+ channels (Kelly et al., 1992). Notably, PKA is involved in this E2-induced pathway, in fact forskolin, a PKA activator, and cAMP can mimic the E2 effects (Lagrange et al., 1997). Several in vitro reports, using hippocampal primary cell culture, have demonstrated that E2 can activate intracellular signaling pathway(s) to indirectly affect the genomic activity through transcriptional regulators such as the cAMP response element binding protein (CREBP; Kelly and Levin, 2001; Wade and Dorsa, 2003; Lee et al., 2004). Notably, CREBP is regulated through phosphorylation by several signaling kinases, including Akt, CaMKII, and MAPK (Brunet et al., 2001; Sawai et al., 2002). The E2-dependent phosphorylation of CREBP has been shown to be a critical step in neuronal survival, synaptic plasticity, and learning as well as particularly in processes that lead to the “generation” of new dendritic spines in dissociated hippocampal neurons (Murphy and Segal, 1996, 1997).
The regulation of AMPA receptors is a key mechanism for the potentiation of synaptic strength in LTP, which is considered a cellular model of learning and memory (Bliss and Collingridge, 1993; Malinow and Malenka, 2002; Malenka and Bear, 2004). Treatment of hippocampal slices with the ERβ agonist WAY-200070 induces a PKA-dependent phosphorylation of the GLUR1 subunit, which leads to the increased expression of AMPAR at the cell surface. Furthermore, ERβ activation induces morphological changes in hippocampal neurons in vivo, including increased branching and density of spines (Liu et al., 2008).
E2 as a Neuroprotective Agent
E2 plays a significant neuroprotective role in the brain, this explains its ability to ameliorate symptoms and to decrease the risk of neurodegenerative events and of ischemic stroke, Alzheimer’s, Parkinson’s, and Huntington’s diseases (Amantea et al., 2005). In particular, E2 displays neuroprotection against Aβ amyloid toxicity (Fitzpatrick et al., 2002; Guerra et al., 2004) and oxidative stress-induced neuronal death (Bae et al., 2000; Wang et al., 2006; De Marinis et al., 2010) in many neuronal cell lines and tissue preparations. Moreover, E2 seems to protect neurons against apoptosis by reducing reactive oxygen species (ROS) production (Razmara et al., 2007; Irwin et al., 2008; Numakawa et al., 2011), inhibiting the neurotoxic effect of oxidized LDL and glutamate (Bhavnani, 2003), maintaining the Ca2+ homeostasis (Ba et al., 2004a), regulating pro-apoptotic caspase activities (Bao et al., 2011), and maintaining mitochondrial membrane integrity (Simpkins and Dykens, 2008; Arnold and Beyer, 2009; Wang et al., 2011). Furthermore, E2 displays neuroprotection against ischemic injuries (Dubal et al., 2001; Carswell et al., 2004). Remarkably, the inhibition of cell death by E2 has been demonstrated in neuronal cell lines, such as NT2 neurons, PC12, mouse hippocampal T22 neurons, as well as mouse and human neuroblastoma cells exposed to glutamate, hydrogen peroxide, and AMPA (Zaulyanov et al., 1999; Ba et al., 2004b; Sribnick et al., 2004; Amantea et al., 2005; De Marinis et al., 2010).
Both nuclear and membrane initiated mechanisms of action are required for the neuroprotective role of E2. ERs can interact directly with the MAPK pathway resulting in the activation of a wide variety of transcription factors involved in neuronal survival (Morissette et al., 2008). Notably, MAPK signaling is accompanied by the activation of CREBP, the induction of Bcl-2 expression, and the inhibition of the apoptotic cascade (Azcoitia et al., 2011). In particular, the E2 neuroprotective effects against the quinolonic acid-induced toxicity in rat hippocampus cells is abolished by pre-treatment with U0126, an inhibitor of MAPK/ERK kinase (Kuroki et al., 2001).
Estrogen receptors can also interact with the PI3K signaling pathway leading to activation of the effector protein kinase Akt. Activated Akt, in turn, can modulate the expression of apoptosis inhibitors (i.e., Bcl-2 and Bcl-x) or inducers (i.e., Bax and Bad), thus influencing cell death. Similarly, phosphorylation of either caspase-9 or the fork head transcription factors by Akt could further block the apoptosis induction in neurons. Therefore, Akt promotes cell survival opposing to the apoptosis by a variety of routes. In particular, the PI3K/Akt pathway is shown to be a critical survival mechanism in the neuroprotective activities of E2 in dopamine neurotransmission in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice model of Parkinson’s disease. In this model, Akt activates anti-apoptotic proteins and inactivates glycogen synthase kinase-3β (GSK3β) activity by increasing the phosphorylation level of Ser9 and Ser21. GSK3β is highly expressed in the central nervous system; the activation of GSK3β facilitates neuronal death whereas its inactivation promotes cellular mechanisms involved in neuronal survival pathways. Furthermore, evidence shows that GSK3β could play a key role in the pathogenesis of the Alzheimer’s disease (Morissette et al., 2008).
E2 rapidly promotes the phosphorylation and the subsequent inactivation of the voltage dependent anion channel (VDAC), a mitochondrial porin also found at the neuronal membrane, where it appears to be involved in redox regulation, extrinsic pathway, and Aβ amyloid neurotoxicity. This E2 effect requires the activation of PKA and Src-kinase which may be relevant to maintain this channel inactivated (Herrera et al., 2011).
ERβ-Signal Transduction Pathways Involved in E2-Induced Neuroprotection
Whereas ERα is a critical link in the mechanism of E2 neuroprotection, the role of ERβ is less clear. By now there is growing evidence from in vitro and in vivo studies which shows the involvement of ERβ-mediated rapid signaling in neuroprotection. Despite both ERs are expressed by hippocampal neurons, E2 effects on hippocampus seem to be mediated by ERβ which is more prevalent than ERα (Shughrue et al., 1996, 1998; Laflamme et al., 1998; Hileman et al., 1999; Mitra et al., 2003). Different studies, based on knockout animal models, have been performed to determine whether ERβ in the hippocampus is a E2 target. Notably, E2-treated wild-type mice exhibit a less depressive-like behavior than controls, this E2 effect is lost in ERβ knockout mice βERKO (Rocha et al., 2005; Weiser et al., 2008). Furthermore, ovariectomized female rats treated with the ERβ agonist DPN show a decrease of the anxiety-type behavior (Weiser et al., 2009) and the administration of ERβ-selective ligands directly in the hippocampus decreases the depressive-like behavior suggesting the pivotal role of this brain area (Walf and Frye, 2007). All together, these results indicate that the ERβ-mediated mechanism(s) is pivotal for the E2-induced synaptic plasticity modulation in the hippocampus and, ultimately, for E2-induced learning and memory.
Exposure of human SK-N-BE neuroblastoma cell to H2O2 (=50 μM) induces the increase of ROS in cells (followed by oxidation of proteins, lipids, and DNA), glutathione depletion, mitochondria dysfunction, intracellular Ca2+ increase, and caspase-3 activation followed by apoptotic cell death (Wang et al., 2006). Recently, cells pre-treatment with physiological E2 concentration has been demonstrated to decrease cell death and to reduce the activation of the pro-apoptotic cascade (i.e., caspase-3 activation and PARP cleavage; De Marinis et al., 2010).
Although SK-N-BE cells express different level of both ER subtypes, the E2-protective effects against H2O2-induced neuron toxicity required rapid ERβ-activities (De Marinis et al., 2010). In fact, cell pre-treatment with the specific ERβ-inhibitor THC completely prevents E2 effects (De Marinis et al., 2010). Interestingly, the monomeric globin neuroglobin (Ngb), displaying a protective function in the brain (Fago et al., 2008; Burmester and Hankeln, 2009; Yu et al., 2009; De Marinis et al., 2010, 2011; DellaValle et al., 2010), is pivotal in ERβ-mediated effects on neuroprotection (De Marinis et al., 2010). The E2 effect on Ngb expression is rapid (1 h), persistent (24 h), specific, and mediated by the ERβ subtype. In particular, the E2-induction of Ngb increase requires the ERβ-mediated rapid activation of p38/MAPK. Moreover, cell pre-treatment with the transcription inhibitor actinomycin and the translation inhibitor cycloheximide completely prevents the increase of E2-induced Ngb levels. Note that the integration between rapid and genomic ERβ-mediated events is required to guarantee rapid and persistent E2 effects in neuroprotection against H2O2 toxicity (De Marinis et al., 2010). Furthermore, in mouse primary cortical astrocytes, one of the cellular targets of E2 in the brain (Arevalo et al., 2010), E2 stimulation reduces lipopolysaccharide-induced cytokine production only in the presence of ERβ-induced increase of Ngb protein levels (Marino M., unpublished data). As a whole, these data suggest a new membrane initiated signaling pathway involving ERβ, p38/MAPK, and Ngb in E2-induced neuroprotection (Figure 2).
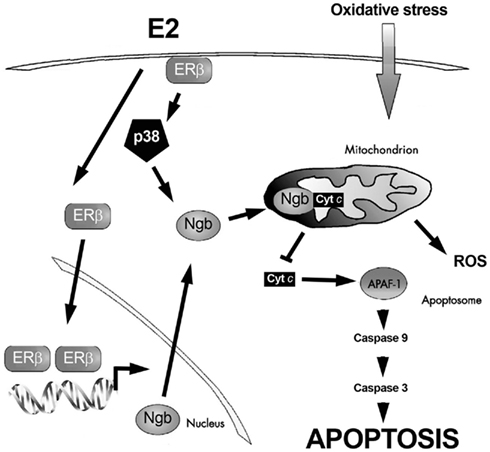
Figure 2. Schematic model illustrating the ERβ-dependent E2-induced neuroprotection against oxidative stress. Exposure to oxidative stress induces cytochrome c (cyt c) release into the cytoplasm. Once in the cytosol cyt c mediates the activation of the adaptor molecule apoptosis–protease activating factor-1 (Apaf-1), generating the apoptosome. Apoptosome can recruit caspase-9 favoring proteinase activation. These events induce the catalytic maturation of caspase-3, which mediates the biochemical and morphological features of apoptosis. E2, via the synergy between extranuclear and nuclear ERβ activities, increases neuroglobin (Ngb) levels, reallocates Ngb at mitochondria, facilitates Ngb-cytochrome c interaction, and prevents apoptosome formation. For details, see text.
However, conflicting data are available on the role played by ERβ in neuroprotection. Several in vivo studies in ERα (αERKO) and ERβ (βERKO) knockout mice model of cerebral ischemia report the pivotal role of ERα, but not of ERβ, in the mechanism(s) associated with the estrogen-mediated neuroprotection in different brain regions, such as the cortex and the striatum (Dubal et al., 2001). Moreover, ERβ seems to be not involved in E2-induced neuroprotection against the Aβ amyloid toxicity in septal-derived SN56 cells and in hippocampal-derived cell line HT22 (Guerra et al., 2004; Marin et al., 2007). On the other hand, a pronounced E2 neuroprotection against β-amyloid peptide in HT22 cells stably transfected with ERβ (HTERβ) and with ERα (HTERα) has been reported. In particular, E2 induced a similar time course of MAPK activation in ERα or ERβ expressing cells (maximal activation at 15 min) which was necessary for neuroprotection against β-amyloid toxicity (Fitzpatrick et al., 2002). Moreover, the loss of ERα did not enhance tissue damage in the female mice after experimental stroke (Sampei et al., 2000). Furthermore, Carswell et al. (2004) showed that administration of the specific ERβ agonist DPN reduced ischemic damage in the striatum and in the CA1 region of the hippocampus in ovariectomized C57B1/6J mice while pre-treatment with the ERα agonist PPT had no effect on the extension of the area affected by neural damage.
Further evidence for the involvement of ERβ-mediated effect(s) in neuroprotection derives from studies on the Mongolian Gerbil global cerebral ischemia model. In this in vivo model, it has been demonstrated that the plant-derived phytoestrogen genistein, a mild-selective agonist for ERβ (with more than sevenfold higher binding affinity for ERβ than ERα; Kuiper et al., 1997), reversed ischemic-induced memory impairment, evaluated through both behavior and object recognition tests, and fully promoted survival of pyramidal cells in the CA1 hippocampal subfield. Moreover, the use of the selective ERβ antagonist PHTTP completely prevented the neuroprotective effect(s) of genistein (Donzelli et al., 2010). These findings confirmed data obtained administrating genistein before injury in focal cerebral ischemia in the mouse (Trieu et al., 1999), and in global cerebral ischemia in rats (Schreihofer et al., 2005) and gerbils (Kindy, 1993).
Conclusion
In mammals, E2 exerts a profound influence on multiple brain circuits involved in reproductive and non-reproductive physiology and behavior. In fact, E2 modulates inflammatory processes, anxiety, depressive-like behaviors, and cognitive functions. In addition, an increasing number of evidence suggests that E2 exerts significant neuroprotective effects against a variety of neurodegenerative pathologies such as the Alzheimer’s and Parkinson’s diseases.
Several studies based on both in vitro and in vivo models demonstrated that E2 can exert its neuroprotective role against multiple types of insults including Aβ-amyloid toxicity, ischemic injuries, and other oxidative stresses. Furthermore, E2 acts in the brain modulating synaptogenesis, neurogenesis, inflammation, and protecting neurons against apoptosis.
In addition to the well-established ERα and ERβ direct transcriptional activities, both ER subtypes are also involved in rapid, extranuclear, E2 actions in the brain. Membrane initiated estrogen signaling involves the rapid activation of different protein kinase pathways, including PKC/PKA/MAPK and PI3K/Akt, to modulate signal transduction, protein phosphorylation, and gene expression suitable for cell survival.
Although the specific role of ERα and ERβ in neuroprotection is openly debated, the studies reviewed here clearly support the idea that E2 acts as neuroprotective factor through direct and indirect genomic mechanisms that converge with rapid membrane initiated effects on signaling proteins in order to regulate cell death and survival factors.
The field of E2-induced neuroprotection is booming in recent years. For many years ERα has been considered a critical link in the mechanism of protection of cell death, but the increasing amount of data propose that also ERβ could play a pivotal role in neuroprotection through the integration between the direct genomic and membrane initiated activities. Thus, a profound knowledge of the membrane ERβ initiated pathways is required in order to identify critical players of these mechanisms that might represent target candidates for therapeutic development in the treatment of post-menopausal neuronal deficiency and neurodegenerative diseases. Further studies are needed to better characterize the modulation and the modes of action of Ngb both in in vitro and in vivo models in order to provide a rational for its pharmacological exploitation as an effective neuroprotective mediator of E2-dependent actions in the brain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Some experimental concepts described in the current paper are based on work conducted in the laboratory of the authors. The Authors wish to thank past and present members of their laboratory who contributed with data and discussions to the ideas presented here. These experimental studies were supported by grants (to Maria Marino) from the Italian Ministry of Health (Strategico, 2008) and University Roma Tre.
References
Acconcia, F., Ascenzi, P., Bocedi, A., Spisni, E., Tomasi, V., Trentalance, A., Visca, P., and Marino, M. (2005). Palmitoylation-dependent estrogen receptor α membrane localization: regulation by 17beta-estradiol. Mol. Biol. Cell 16, 231–237.
Acconcia, F., and Marino, M. (2011). The effects of 17β-estradiol in cancer are mediated by estrogen receptor signaling at the plasma membrane. Front. Physiol. 2:30. doi:10.3389/fphys.2011.00030
Adams, M. M., Fink, S. E., Janssen, W. G., Shah, R. A., and Morrison, J. H. (2004). Estrogen modulates synaptic N-methyl-D-aspartate receptor subunit distribution in the aged hippocampus. J. Comp. Neurol. 474, 419–426.
Amantea, D., Russo, R., Bagetta, G., and Corasaniti, M. T. (2005). From clinical evidence to molecular mechanisms underlying neuroprotection afforded by estrogens. Pharmacol. Res. 52, 119–132.
Amrein, I., Slomianka, L., Poletaeva, I., Bologova, N. V., and Lipp, H. P. (2004). Marked species and age-dependent differences in cell proliferation and neurogenesis in the hippocampus of wild-living rodents. Hippocampus 14, 1000–1010.
Antal, M. C., Krust, A., Chambon, P., and Mark, M. (2008). Sterility and absence of histopathological defects in nonreproductive organs of a mouse ERβ-null mutant. Proc. Natl. Acad. Sci. U.S.A. 105, 2433–2438.
Arevalo, M. A., Santos-Galindo, M., Bellini, M. J., Azcoitia, I., and Garcia-Segura, L. M. (2010). Actions of estrogens on glial cells: implications for neuroprotection. Biochim. Biophys. Acta 1800, 1106–1112.
Arnold, S., and Beyer, C. (2009). Neuroprotection by estrogen in the brain: the mitochondrial compartment as presumed therapeutic target. J. Neurochem. 110, 1–11.
Ascenzi, P., Bocedi, A., and Marino, M. (2006). Structure-function relationship of estrogen receptor α and β: impact on human health. Mol. Aspects Med. 27, 299–402.
Azcoitia, I., Arevalo, M. A., De Nicola, A. F., and Garcia-Segura, L. M. (2011). Neuroprotective actions of estradiol revisited. Trends Endocrinol. Metab. 22, 467–473.
Ba, F., Pang, P. K., and Benishin, C. G. (2004a). The role of Ca2+ channel modulation in the neuroprotective actions of estrogen in β-amyloid protein and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) cytotoxic models. Neurochem. Int. 45, 31–38.
Ba, F., Pang, P. K., Davidge, S. T., and Benishin, C. G. (2004b). The neuroprotective effects of estrogen in SK-N-SH neuroblastoma cell cultures. Neurochem. Int. 44, 401–411.
Bae, Y. H., Hwang, J. Y., Kim, Y. H., and Koh, J. Y. (2000). Anti-oxidative neuroprotection by estrogens in mouse cortical cultures. J. Korean Med. Sci. 15, 327–336.
Bao, Y. J., Li, L. Z., Li, X. G., and Wang, Y. J. (2011). 17β-estradiol differentially protects cortical pericontusional zone from programmed cell death after traumatic cerebral contusion at distinct stages via non-genomic and genomic pathways. Mol. Cell. Neurosci. 48, 185–194.
Barha, C. K., and Galea, L. A. (2010). Influence of different estrogens on neuroplasticity and cognition in the hippocampus. Biochim. Biophys. Acta 1800, 1056–1067.
Bhavnani, B. R. (2003). Estrogens and menopause: pharmacology of conjugated equine estrogens and their potential role in the prevention of neurodegenerative diseases such as Alzheimer’s. J. Steroid Biochem. Mol. Biol. 85, 473–482.
Bjornstrom, L., and Sjoberg, M. (2005). Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 19, 833–842.
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39.
Bodo, C., and Rissman, E. F. (2006). New roles for estrogen receptor β in behavior and neuroendocrinology. Front. Neuroendocrinol. 27, 217–232.
Brannvall, K., Bogdanovic, N., Korhonen, L., and Lindholm, D. (2005). 19-Nortestosterone influences neural stem cell proliferation and neurogenesis in the rat brain. Eur. J. Neurosci. 21, 871–878.
Brannvall, K., Korhonen, L., and Lindholm, D. (2002). Estrogen-receptor-dependent regulation of neural stem cell proliferation and differentiation. Mol. Cell. Neurosci. 21, 512–520.
Breslau, N., Schultz, L., and Peterson, E. (1995). Sex differences in depression: a role for preexisting anxiety. Psychiatry Res. 58, 1–12.
Brunet, A., Datta, S. R., and Greenberg, M. E. (2001). Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 11, 297–305.
Burmester, T., and Hankeln, T. (2009). What is the function of neuroglobin? J. Exp. Biol. 212, 1423–1428.
Campbell, S., and Whitehead, M. (1977). Oestrogen therapy and the menopausal syndrome. Clin. Obstet. Gynaecol. 4, 31–47.
Carswell, H. V., Macrae, I. M., Gallagher, L., Harrop, E., and Horsburgh, K. J. (2004). Neuroprotection by a selective estrogen receptor β agonist in a mouse model of global ischemia. Am. J. Physiol. Heart Circ. Physiol. 287, H1501–H1504.
Chambard, J. C., Lefloch, R., Pouyssegur, J., and Lenormand, P. (2007). ERK implication in cell cycle regulation. Biochim. Biophys. Acta 1773, 1299–1310.
Chen, Z. J., Yu, L., and Chang, C. H. (1998). Stimulation of membrane-bound guanylate cyclase activity by 17-β estradiol. Biochem. Biophys. Res. Commun. 252, 639–642.
De Marinis, E., Ascenzi, P., Pellegrini, M., Galluzzo, P., Bulzomi, P., Arevalo, M. A., Garcia-Segura, L. M., and Marino, M. (2010). 17β-estradiol-A new modulator of neuroglobin levels in neurons: role in neuroprotection against H2O2-induced toxicity. Neurosignals 18, 223–235.
De Marinis, E., Marino, M., and Ascenzi, P. (2011). Neuroglobin, estrogens, and neuroprotection. IUBMB Life 63, 140–145.
DellaValle, B., Hempel, C., Kurtzhals, J. A., and Penkowa, M. (2010). In vivo expression of neuroglobin in reactive astrocytes during neuropathology in murine models of traumatic brain injury, cerebral malaria, and autoimmune encephalitis. Glia 58, 1220–1227.
Demars, M., Hu, Y. S., Gadadhar, A., and Lazarov, O. (2010). Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J. Neurosci. Res. 88, 2103–2117.
DonCarlos, L. L., Azcoitia, I., and Garcia-Segura, L. M. (2009). Neuroprotective actions of selective estrogen receptor modulators. Psychoneuroendocrinology 34(Suppl. 1), S113–S122.
Donzelli, A., Braida, D., Finardi, A., Capurro, V., Valsecchi, A. E., Colleoni, M., and Sala, M. (2010). Neuroprotective effects of genistein in Mongolian gerbils: estrogen receptor-β involvement. J. Pharmacol. Sci. 114, 158–167.
Dubal, D. B., Zhu, H., Yu, J., Rau, S. W., Shughrue, P. J., Merchenthaler, I., Kindy, M. S., and Wise, P. M. (2001). Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc. Natl. Acad. Sci. U.S.A. 98, 1952–1957.
Eriksson, P. S., Perfilieva, E., Bjork-Eriksson, T., Alborn, A. M., Nordborg, C., Peterson, D. A., and Gage, F. H. (1998). Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317.
Etgen, A. M., Jover-Mengual, T., and Zukin, R. S. (2011). Neuroprotective actions of estradiol and novel estrogen analogs in ischemia: translational implications. Front. Neuroendocrinol. 32, 336–352.
Fago, A., Mathews, A. J., and Brittain, T. (2008). A role for neuroglobin: resetting the trigger level for apoptosis in neuronal and retinal cells. IUBMB Life 60, 398–401.
Farach-Carson, M. C., and Davis, P. J. (2003). Steroid hormone interactions with target cells: cross talk between membrane and nuclear pathways. J. Pharmacol. Exp. Ther. 307, 839–845.
Filardo, E., Quinn, J., Pang, Y., Graeber, C., Shaw, S., Dong, J., and Thomas, P. (2007). Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 148, 3236–3245.
Fitzpatrick, J. L., Mize, A. L., Wade, C. B., Harris, J. A., Shapiro, R. A., and Dorsa, D. M. (2002). Estrogen-mediated neuroprotection against beta-amyloid toxicity requires expression of estrogen receptor alpha or beta and activation of the MAPK pathway. J. Neurochem. 82, 674–682.
Fried, G., Andersson, E., Csoregh, L., Enmark, E., Gustafsson, J. A., Aanesen, A., and Osterlund, C. (2004). Estrogen receptor beta is expressed in human embryonic brain cells and is regulated by 17beta-estradiol. Eur. J. Neurosci. 20, 2345–2354.
Galluzzo, P., Caiazza, F., Moreno, S., and Marino, M. (2007). Role of ERβ palmitoylation in the inhibition of human colon cancer cell proliferation. Endocr. Relat. Cancer 14, 153–167.
Garcia-Segura, L. M., Azcoitia, I., and Doncarlos, L. L. (2001). Neuroprotection by estradiol. Prog. Neurobiol. 63, 29–60.
Garcia-Segura, L. M., Duenas, M., Fernandez-Galaz, M. C., Chowen, J. A., Argente, J., Naftolin, F., and Torres-Aleman, I. (1996). Interaction of the signalling pathways of insulin-like growth factor-I and sex steroids in the neuroendocrine hypothalamus. Horm. Res. 46, 160–164.
Gillies, G. E., and McArthur, S. (2010). Estrogen actions in the brain and the basis for differential action in men and women: a case for sex-specific medicines. Pharmacol. Rev. 62, 155–198.
Gosden, J. R., Middleton, P. G., and Rout, D. (1986). Localization of the human oestrogen receptor gene to chromosome 6q24–q27 by in situ hybridization. Cytogenet. Cell Genet. 43, 218–220.
Gould, E., Mcewen, B. S., Tanapat, P., Galea, L. A., and Fuchs, E. (1997). Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J. Neurosci. 17, 2492–2498.
Gould, E., Vail, N., Wagers, M., and Gross, C. G. (2001). Adult-generated hippocampal and neocortical neurons in macaques have a transient existence. Proc. Natl. Acad. Sci. U.S.A. 98, 10910–10917.
Green, A. D., and Galea, L. A. (2008). Adult hippocampal cell proliferation is suppressed with estrogen withdrawal after a hormone-simulated pregnancy. Horm. Behav. 54, 203–211.
Gu, Q., and Moss, R. L. (1996). 17β-estradiol potentiates kainate-induced currents via activation of the cAMP cascade. J. Neurosci. 16, 3620–3629.
Guerra, B., Diaz, M., Alonso, R., and Marin, R. (2004). Plasma membrane oestrogen receptor mediates neuroprotection against beta-amyloid toxicity through activation of Raf-1/MEK/ERK cascade in septal-derived cholinergic SN56 cells. J. Neurochem. 91, 99–109.
Gupta, R. R., Sen, S., Diepenhorst, L. L., Rudick, C. N., and Maren, S. (2001). Estrogen modulates sexually dimorphic contextual fear conditioning and hippocampal long-term potentiation (LTP) in rats(1). Brain Res. 888, 356–365.
Hammes, S. R., and Levin, E. R. (2007). Extranuclear steroid receptors: nature and actions. Endocr. Rev. 28, 726–741.
Hampson, E., and Kimura, D. (1988). Reciprocal effects of hormonal fluctuations on human motor and perceptual-spatial skills. Behav. Neurosci. 102, 456–459.
Handa, R. J., Ogawa, S., Wang, J. M., and Herbison, A. E. (2010). Roles for estrogen receptor β in adult brain function. J. Neuroendocrinol. 24, 160–173.
Hata, T., Furukawa, T., Sunamura, M., Egawa, S., Motoi, F., Ohmura, N., Marumoto, T., Saya, H., and Horii, A. (2005). RNA interference targeting Aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res. 65, 2899–2905.
Herrera, J. L., Fernandez, C., Diaz, M., Cury, D., and Marin, R. (2011). Estradiol and tamoxifen differentially regulate a plasmalemmal voltage-dependent anion channel involved in amyloid-β induced neurotoxicity. Steroids 76, 840–844.
Hileman, S. M., Handa, R. J., and Jackson, G. L. (1999). Distribution of estrogen receptor-β messenger ribonucleic acid in the male sheep hypothalamus. Biol. Reprod. 60, 1279–1284.
Hong, S. H., Nah, H. Y., Lee, Y. J., Lee, J. W., Park, J. H., Kim, S. J., Lee, J. B., Yoon, H. S., and Kim, C. H. (2004). Expression of estrogen receptor-α and -β, glucocorticoid receptor, and progesterone receptor genes in human embryonic stem cells and embryoid bodies. Mol. Cells 18, 320–325.
Inagaki, T., Gautreaux, C., and Luine, V. (2010). Acute estrogen treatment facilitates recognition memory consolidation and alters monoamine levels in memory-related brain areas. Horm. Behav. 58, 415–426.
Irwin, R. W., Yao, J., Hamilton, R. T., Cadenas, E., Brinton, R. D., and Nilsen, J. (2008). Progesterone and estrogen regulate oxidative metabolism in brain mitochondria. Endocrinology 149, 3167–3175.
Jacome, L. F., Gautreaux, C., Inagaki, T., Mohan, G., Alves, S., Lubbers, L. S., and Luine, V. (2010). Estradiol and ERbeta agonists enhance recognition memory, and DPN, an ERβ agonist, alters brain monoamines. Neurobiol. Learn. Mem. 94, 488–498.
Kalaitzidis, D., and Gilmore, T. D. (2005). Transcription factor cross-talk: the estrogen receptor and NF-κB. Trends Endocrinol. Metab. 16, 46–52.
Kelly, M. J., and Levin, E. R. (2001). Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol. Metab. 12, 152–156.
Kelly, M. J., Loose, M. D., and Ronnekleiv, O. K. (1992). Estrogen suppresses μ-opioid- and GABAB-mediated hyperpolarization of hypothalamic arcuate neurons. J. Neurosci. 12, 2745–2750.
Kessler, R. C., Mcgonagle, K. A., Zhao, S., Nelson, C. B., Hughes, M., Eshleman, S., Wittchen, H. U., and Kendler, K. S. (1994). Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch. Gen. Psychiatry 51, 8–19.
Kindy, M. S. (1993). Inhibition of tyrosine phosphorylation prevents delayed neuronal death following cerebral ischemia. J. Cereb. Blood Flow Metab. 13, 372–377.
Kudwa, A. E., Michopoulos, V., Gatewood, J. D., and Rissman, E. F. (2006). Roles of estrogen receptors α and β in differentiation of mouse sexual behavior. Neuroscience 138, 921–928.
Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Haggblad, J., Nilsson, S., and Gustafsson, J. A. (1997). Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology 138, 863–870.
Kuroki, Y., Fukushima, K., Kanda, Y., Mizuno, K., and Watanabe, Y. (2001). Neuroprotection by estrogen via extracellular signal-regulated kinase against quinolinic acid-induced cell death in the rat hippocampus. Eur. J. Neurosci. 13, 472–476.
Laflamme, N., Nappi, R. E., Drolet, G., Labrie, C., and Rivest, S. (1998). Expression and neuropeptidergic characterization of estrogen receptors (ERα and ERβ) throughout the rat brain: anatomical evidence of distinct roles of each subtype. J. Neurobiol. 36, 357–378.
Lagrange, A. H., Ronnekleiv, O. K., and Kelly, M. J. (1997). Modulation of G protein-coupled receptors by an estrogen receptor that activates protein kinase A. Mol. Pharmacol. 51, 605–612.
Lee, S. J., Campomanes, C. R., Sikat, P. T., Greenfield, A. T., Allen, P. B., and Mcewen, B. S. (2004). Estrogen induces phosphorylation of cyclic AMP response element binding (pCREB) in primary hippocampal cells in a time-dependent manner. Neuroscience 124, 549–560.
Levin, E. R. (2005). Integration of the extranuclear and nuclear actions of estrogen. Mol. Endocrinol. 19, 1951–1959.
Liu, F., Day, M., Muniz, L. C., Bitran, D., Arias, R., Revilla-Sanchez, R., Grauer, S., Zhang, G., Kelley, C., Pulito, V., Sung, A., Mervis, R. F., Navarra, R., Hirst, W. D., Reinhart, P. H., Marquis, K. L., Moss, S. J., Pangalos, M. N., and Brandon, N. J. (2008). Activation of estrogen receptor-β regulates hippocampal synaptic plasticity and improves memory. Nat. Neurosci. 11, 334–343.
Luisi, S., Galleri, L., Marini, F., Ambrosini, G., Brandi, M. L., and Petraglia, F. (2006). Estrogen receptor gene polymorphisms are associated with recurrence of endometriosis. Fertil. Steril. 85, 764–766.
Maggi, A., Ciana, P., Belcredito, S., and Vegeto, E. (2004). Estrogens in the nervous system: mechanisms and nonreproductive functions. Annu. Rev. Physiol. 66, 291–313.
Malberg, J. E., and Duman, R. S. (2003). Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology 28, 1562–1571.
Malinow, R., and Malenka, R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126.
Malyala, A., Kelly, M. J., and Ronnekleiv, O. K. (2005). Estrogen modulation of hypothalamic neurons: activation of multiple signaling pathways and gene expression changes. Steroids 70, 397–406.
Marin, R., Diaz, M., Alonso, R., Sanz, A., Arevalo, M. A., and Garcia-Segura, L. M. (2009). Role of estrogen receptor α in membrane-initiated signaling in neural cells: interaction with IGF-1 receptor. J. Steroid Biochem. Mol. Biol. 114, 2–7.
Marin, R., Ramirez, C. M., Gonzalez, M., Gonzalez-Munoz, E., Zorzano, A., Camps, M., Alonso, R., and Diaz, M. (2007). Voltage-dependent anion channel (VDAC) participates in amyloid β-induced toxicity and interacts with plasma membrane estrogen receptor α in septal and hippocampal neurons. Mol. Membr. Biol. 24, 148–160.
Marino, M., and Ascenzi, P. (2008). Membrane association of estrogen receptor α and β influences 17β-estradiol-mediated cancer cell proliferation. Steroids 73, 853–858.
Marquez, D. C., Chen, H. W., Curran, E. M., Welshons, W. V., and Pietras, R. J. (2006). Estrogen receptors in membrane lipid rafts and signal transduction in breast cancer. Mol. Cell. Endocrinol. 246, 91–100.
Metivier, R., Penot, G., Hubner, M. R., Reid, G., Brand, H., Kos, M., and Gannon, F. (2003). Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115, 751–763.
Mitra, S. W., Hoskin, E., Yudkovitz, J., Pear, L., Wilkinson, H. A., Hayashi, S., Pfaff, D. W., Ogawa, S., Rohrer, S. P., Schaeffer, J. M., Mcewen, B. S., and Alves, S. E. (2003). Immunolocalization of estrogen receptor β in the mouse brain: comparison with estrogen receptor α. Endocrinology 144, 2055–2067.
Morgan, M. A., and Pfaff, D. W. (2001). Effects of estrogen on activity and fear-related behaviors in mice. Horm. Behav. 40, 472–482.
Morgan, M. A., and Pfaff, D. W. (2002). Estrogen’s effects on activity, anxiety, and fear in two mouse strains. Behav. Brain. Res. 132, 85–93.
Morissette, M., Le Saux, M., D’Astous, M., Jourdain, S., Al Sweidi, S., Morin, N., Estrada-Camarena, E., Mendez, P., Garcia-Segura, L. M., and Di Paolo, T. (2008). Contribution of estrogen receptors α and β to the effects of estradiol in the brain. J. Steroid Biochem. Mol. Biol. 108, 327–338.
Murphy, D. D., and Segal, M. (1996). Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J. Neurosci. 16, 4059–4068.
Murphy, D. D., and Segal, M. (1997). Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc. Natl. Acad. Sci. U.S.A. 94, 1482–1487.
Nilsson, S., and Gustafsson, J. A. (2011). Estrogen receptors: therapies targeted to receptor subtypes. Clin. Pharmacol. Ther. 89, 44–55.
Numakawa, T., Matsumoto, T., Numakawa, Y., Rizchards, M., Yamawaki, S., and Kunugi, H. (2011). Protective action of neurotrophic factors and estrogen against oxidative stress-mediated neurodegeneration. J. Toxicol. 2011, 405194.
Ogawa, S., Eng, V., Taylor, J., Lubahn, D. B., Korach, K. S., and Pfaff, D. W. (1998). Roles of estrogen receptor-α gene expression in reproduction-related behaviors in female mice. Endocrinology 139, 5070–5081.
O’Lone, R., Frith, M. C., Karlsson, E. K., and Hansen, U. (2004). Genomic targets of nuclear estrogen receptors. Mol. Endocrinol. 18, 1859–1875.
Osterlund, M. K., Grandien, K., Keller, E., and Hurd, Y. L. (2000a). The human brain has distinct regional expression patterns of estrogen receptor α mRNA isoforms derived from alternative promoters. J. Neurochem. 75, 1390–1397.
Osterlund, M. K., Gustafsson, J. A., Keller, E., and Hurd, Y. L. (2000b). Estrogen receptor beta (ERβ) messenger ribonucleic acid (mRNA) expression within the human forebrain: distinct distribution pattern to ERα mRNA. J. Clin. Endocrinol. Metab. 85, 3840–3846.
Osterlund, M. K., Keller, E., and Hurd, Y. L. (2000c). The human forebrain has discrete estrogen receptor α messenger RNA expression: high levels in the amygdaloid complex. Neuroscience 95, 333–342.
Pedram, A., Razandi, M., Sainson, R. C., Kim, J. K., Hughes, C. C., and Levin, E. R. (2007). A conserved mechanism for steroid receptor translocation to the plasma membrane. J. Biol. Chem. 282, 22278–22288.
Pettersson, K., and Gustafsson, J. A. (2001). Role of estrogen receptor β in estrogen action. Annu. Rev. Physiol. 63, 165–192.
Pfaff, D. W., Schwanzel-Fukuda, M., Parhar, I. S., Lauber, A. H., Mccarthy, L. M., and Kow, L. M. (1994). GnRH neurons and other cellular and molecular mechanisms for simple mammalian reproductive behaviors. Recent Prog. Horm. Res. 49, 1–25.
Phillips, S. M., and Sherwin, B. B. (1992). Variations in memory function and sex steroid hormones across the menstrual cycle. Psychoneuroendocrinology 17, 497–506.
Picard, N., Charbonneau, C., Sanchez, M., Licznar, A., Busson, M., Lazennec, G., and Tremblay, A. (2008). Phosphorylation of activation function-1 regulates proteasome-dependent nuclear mobility and E6-associated protein ubiquitin ligase recruitment to the estrogen receptor β. Mol. Endocrinol. 22, 317–330.
Prossnitz, E. R., and Barton, M. (2011). The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 7, 715–726.
Raz, L., Khan, M. M., Mahesh, V. B., Vadlamudi, R. K., and Brann, D. W. (2008). Rapid estrogen signaling in the brain. Neurosignals 16, 140–153.
Razandi, M., Alton, G., Pedram, A., Ghonshani, S., Webb, P., and Levin, E. R. (2003). Identification of a structural determinant necessary for the localization and function of estrogen receptor α at the plasma membrane. Mol. Cell. Biol. 23, 1633–1646.
Razmara, A., Duckles, S. P., Krause, D. N., and Procaccio, V. (2007). Estrogen suppresses brain mitochondrial oxidative stress in female and male rats. Brain Res. 1176, 71–81.
Reid, G., Hubner, M. R., Metivier, R., Brand, H., Denger, S., Manu, D., Beaudouin, J., Ellenberg, J., and Gannon, F. (2003). Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol. Cell 11, 695–707.
Rhodes, M. E., and Frye, C. A. (2004). Estrogen has mnemonic-enhancing effects in the inhibitory avoidance task. Pharmacol. Biochem. Behav. 78, 551–558.
Rocha, B. A., Fleischer, R., Schaeffer, J. M., Rohrer, S. P., and Hickey, G. J. (2005). 17β-estradiol-induced antidepressant-like effect in the forced swim test is absent in estrogen receptor-β knockout (BERKO) mice. Psychopharmacology (Berl.) 179, 637–643.
Rodriguez, J. J., Jones, V. C., Tabuchi, M., Allan, S. M., Knight, E. M., Laferla, F. M., Oddo, S., and Verkhratsky, A. (2008). Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 3, e2935. doi:10.1371/journal.pone.0002935
Roepke, T. A., Qiu, J., Bosch, M. A., Ronnekleiv, O. K., and Kelly, M. J. (2009). Cross-talk between membrane-initiated and nuclear-initiated oestrogen signalling in the hypothalamus. J. Neuroendocrinol. 21, 263–270.
Roepke, T. A., Ronnekleiv, O. K., and Kelly, M. J. (2011). Physiological consequences of membrane-initiated estrogen signaling in the brain. Front. Biosci. 16, 1560–1573.
Ronnekleiv, O. K., and Kelly, M. J. (2005). Diversity of ovarian steroid signaling in the hypothalamus. Front. Neuroendocrinol. 26, 65–84.
Sampei, K., Goto, S., Alkayed, N. J., Crain, B. J., Korach, K. S., Traystman, R. J., Demas, G. E., Nelson, R. J., and Hurn, P. D. (2000). Stroke in estrogen receptor-α-deficient mice. Stroke 31, 738–743.
Sawai, T., Bernier, F., Fukushima, T., Hashimoto, T., Ogura, H., and Nishizawa, Y. (2002). Estrogen induces a rapid increase of calcium-calmodulin-dependent protein kinase II activity in the hippocampus. Brain Res. 950, 308–311.
Schneier, F. R., Johnson, J., Hornig, C. D., Liebowitz, M. R., and Weissman, M. M. (1992). Social phobia. Comorbidity and morbidity in an epidemiologic sample. Arch. Gen. Psychiatry 49, 282–288.
Schreihofer, D. A., Do, K. D., and Schreihofer, A. M. (2005). High-soy diet decreases infarct size after permanent middle cerebral artery occlusion in female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R103–R108.
Seeman, M. V. (1997). Psychopathology in women and men: focus on female hormones. Am. J. Psychiatry 154, 1641–1647.
Sherwin, B. B. (2005). Surgical menopause, estrogen, and cognitive function in women: what do the findings tell us? Ann. N. Y. Acad. Sci. 1052, 3–10.
Shughrue, P. J., Komm, B., and Merchenthaler, I. (1996). The distribution of estrogen receptor-β mRNA in the rat hypothalamus. Steroids 61, 678–681.
Shughrue, P. J., Scrimo, P. J., and Merchenthaler, I. (1998). Evidence for the colocalization of estrogen receptor-β mRNA and estrogen receptor-β immunoreactivity in neurons of the rat forebrain. Endocrinology 139, 5267–5270.
Simpkins, J. W., and Dykens, J. A. (2008). Mitochondrial mechanisms of estrogen neuroprotection. Brain. Res. Rev. 57, 421–430.
Smith, C. C., and McMahon, L. L. (2006). Estradiol-induced increase in the magnitude of long-term potentiation is prevented by blocking NR2B-containing receptors. J. Neurosci. 26, 8517–8522.
Squires, M. S., Nixon, P. M., and Cook, S. J. (2002). Cell-cycle arrest by PD184352 requires inhibition of extracellular signal-regulated kinases (ERK) 1/2 but not ERK5/BMK1. Biochem. J. 366, 673–680.
Sribnick, E. A., Ray, S. K., Nowak, M. W., Li, L., and Banik, N. L. (2004). 17β-estradiol attenuates glutamate-induced apoptosis and preserves electrophysiologic function in primary cortical neurons. J. Neurosci. Res. 76, 688–696.
Suzuki, S., Brown, C. M., and Wise, P. M. (2006). Mechanisms of neuroprotection by estrogen. Endocrine 29, 209–215.
Suzuki, S., Gerhold, L. M., Bottner, M., Rau, S. W., Dela Cruz, C., Yang, E., Zhu, H., Yu, J., Cashion, A. B., Kindy, M. S., Merchenthaler, I., Gage, F. H., and Wise, P. M. (2007). Estradiol enhances neurogenesis following ischemic stroke through estrogen receptors α and β. J. Comp. Neurol. 500, 1064–1075.
Szego, C. M., and Davis, J. S. (1967). Adenosine 3’,5’-monophosphate in rat uterus: acute elevation by estrogen. Proc. Natl. Acad. Sci. U.S.A. 58, 1711–1718.
Tanapat, P., Hastings, N. B., and Gould, E. (2005). Ovarian steroids influence cell proliferation in the dentate gyrus of the adult female rat in a dose- and time-dependent manner. J. Comp. Neurol. 481, 252–265.
Tanapat, P., Hastings, N. B., Reeves, A. J., and Gould, E. (1999). Estrogen stimulates a transient increase in the number of new neurons in the dentate gyrus of the adult female rat. J. Neurosci. 19, 5792–5801.
Toran-Allerand, C. D., Guan, X., Maclusky, N. J., Horvath, T. L., Diano, S., Singh, M., Connolly, E. S. Jr., Nethrapalli, I. S., and Tinnikov, A. A. (2002). ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J. Neurosci. 22, 8391–8401.
Trieu, V. N., Dong, Y., Zheng, Y., and Uckun, F. M. (1999). In vivo antioxidant activity of genistein in a murine model of singlet oxygen-induced cerebral stroke. Radiat. Res. 152, 508–516.
Ussar, S., and Voss, T. (2004). MEK1 and MEK2, different regulators of the G1/S transition. J. Biol. Chem. 279, 43861–43869.
Vasudevan, N., and Pfaff, D. W. (2007). Membrane-initiated actions of estrogens in neuroendocrinology: emerging principles. Endocr. Rev. 28, 1–19.
Vollmayr, B., Simonis, C., Weber, S., Gass, P., and Henn, F. (2003). Reduced cell proliferation in the dentate gyrus is not correlated with the development of learned helplessness. Biol. Psychiatry 54, 1035–1040.
Wade, C. B., and Dorsa, D. M. (2003). Estrogen activation of cyclic adenosine 5’-monophosphate response element-mediated transcription requires the extracellularly regulated kinase/mitogen-activated protein kinase pathway. Endocrinology 144, 832–838.
Walf, A. A., and Frye, C. A. (2006). A review and update of mechanisms of estrogen in the hippocampus and amygdala for anxiety and depression behavior. Neuropsychopharmacology 31, 1097–1111.
Walf, A. A., and Frye, C. A. (2007). Administration of estrogen receptor β-specific selective estrogen receptor modulators to the hippocampus decrease anxiety and depressive behavior of ovariectomized rats. Pharmacol. Biochem. Behav. 86, 407–414.
Walf, A. A., and Frye, C. A. (2008). Rapid and estrogen receptor beta mediated actions in the hippocampus mediate some functional effects of estrogen. Steroids 73, 997–1007.
Wang, J. M., Irwin, R. W., Liu, L., Chen, S., and Brinton, R. D. (2007). Regeneration in a degenerating brain: potential of allopregnanolone as a neuroregenerative agent. Curr. Alzheimer Res. 4, 510–517.
Wang, J. M., Liu, L., and Brinton, R. D. (2008). Estradiol-17beta-induced human neural progenitor cell proliferation is mediated by an estrogen receptor β-phosphorylated extracellularly regulated kinase pathway. Endocrinology 149, 208–218.
Wang, J. M., Singh, C., Liu, L., Irwin, R. W., Chen, S., Chung, E. J., Thompson, R. F., and Brinton, R. D. (2010). Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 107, 6498–6503.
Wang, S., Ren, P., Li, X., Guan, Y., and Zhang, Y. A. (2011). 17β-estradiol protects dopaminergic neurons in organotypic slice of mesencephalon by MAPK-mediated activation of anti-apoptosis Gene BCL2. J. Mol. Neurosci. 45, 236–245.
Wang, X., Dykens, J. A., Perez, E., Liu, R., Yang, S., Covey, D. F., and Simpkins, J. W. (2006). Neuroprotective effects of 17β-estradiol and non-feminizing estrogens against H2O2 toxicity in human neuroblastoma SK-N-SH cells. Mol. Pharmacol. 70, 395–404.
Watson, C. S., Alyea, R. A., Cunningham, K. A., and Jeng, Y. J. (2010). Estrogens of multiple classes and their role in mental health disease mechanisms. Int. J. Womens Health 2, 153–166.
Weiser, M. J., Foradori, C. D., and Handa, R. J. (2008). Estrogen receptor β in the brain: from form to function. Brain. Res. Rev. 57, 309–320.
Weiser, M. J., Wu, T. J., and Handa, R. J. (2009). Estrogen receptor-β agonist diarylpropionitrile: biological activities of R- and S-enantiomers on behavior and hormonal response to stress. Endocrinology 150, 1817–1825.
Wise, P. M., Dubal, D. B., Wilson, M. E., Rau, S. W., and Liu, Y. (2001). Estrogens: trophic and protective factors in the adult brain. Front. Neuroendocrinol. 22, 33–66.
Woolley, C. S. (2007). Acute effects of estrogen on neuronal physiology. Annu. Rev. Pharmacol. Toxicol. 47, 657–680.
Yu, Z., Fan, X., Lo, E. H., and Wang, X. (2009). Neuroprotective roles and mechanisms of neuroglobin. Neurol. Res. 31, 122–127.
Zaulyanov, L. L., Green, P. S., and Simpkins, J. W. (1999). Glutamate receptor requirement for neuronal death from anoxia-reoxygenation: an in vitro model for assessment of the neuroprotective effects of estrogens. Cell. Mol. Neurobiol. 19, 705–718.
Keywords: estrogen receptor α, estrogen receptor β, 17β-estradiol, neuroprotective effects, membrane initiated signals
Citation: Fiocchetti M, Ascenzi P and Marino M (2012) Neuroprotective effects of 17β-estradiol rely on estrogen receptor membrane initiated signals. Front. Physio. 3:73. doi: 10.3389/fphys.2012.00073
Received: 30 December 2011; Paper pending published: 24 January 2012;
Accepted: 13 March 2012; Published online: 05 April 2012.
Edited by:
Raquel Marin, Universidad de La Laguna, SpainReviewed by:
Manuel José Villalón, Pontificia Universidad Católica de Chile, ChileDandan Sun, University of Pittsburgh, USA
Copyright: © 2012 Fiocchetti, Ascenzi and Marino. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Maria Marino, Department of Biology, University Roma Tre, Viale Guglielmo Marconi 446, I-00146 Roma, Italy. e-mail: m.marino@uniroma3.it