
- Institute of Animal Physiology, Justus Liebig University of Giessen, Giessen, Germany
The vectorial transport of Na+ across epithelia is crucial for the maintenance of Na+ and water homeostasis in organs such as the kidneys, lung, or intestine. Dysregulated Na+ transport processes are associated with various human diseases such as hypertension, the salt-wasting syndrome pseudohypoaldosteronism type 1, pulmonary edema, cystic fibrosis, or intestinal disorders, which indicate that a precise regulation of epithelial Na+ transport is essential. Novel regulatory signaling molecules are gasotransmitters. There are currently three known gasotransmitters: nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S). These molecules are endogenously produced in mammalian cells by specific enzymes and have been shown to regulate various physiological processes. There is a growing body of evidence which indicates that gasotransmitters may also regulate Na+ transport across epithelia. This review will summarize the available data concerning NO, CO, and H2S dependent regulation of epithelial Na+ transport processes and will discuss whether or not these mediators can be considered as true physiological regulators of epithelial Na+ transport biology.
Epithelial Na+ Transport
The vectorial transport of Na+ across epithelia of various organs is a crucial event in the maintenance of general salt and water homeostasis in vertebrates. The molecular nature of transepithelial Na+ transport has been revealed in pioneer studies by Koefoed-Johnsen and Ussing (1958): Na+ ions enter epithelial cells at the apical membrane of the epithelial cells. The Na+ uptake mechanism can be Na+ selective ion channels, such as the amiloride-sensitive epithelial Na+ channel (ENaC), or Na+ coupled transporters (Figure 1). The chemical driving force for the apical influx of Na+ is created by the Na+/K+-ATPase at the basolateral side of the epithelial cells. This enzyme actively pumps three Na+ ions out of the cell – in exchange for two K+ ions. The K+ ions are recycled and leave the cell via K+ channels at the basolateral membrane. Furthermore, the K+-conductance keeps the membrane potential of the epithelial cells negative. Thus, both the Na+/K+-ATPase as well as K+ channels deliver the electrochemical driving force for transepithelial Na+ transport (Figure 1).
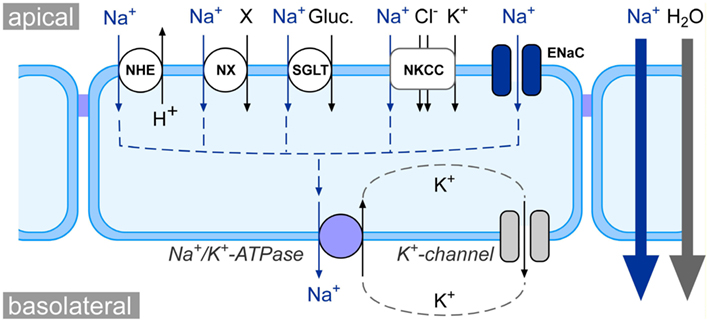
Figure 1. Schematic model of epithelial Na+ transport physiology. At the apical side of epithelia, Na+ enters the cell either via Na+ coupled transporters, such as the Na+/H+ exchanger (NHE), Na+-coupled amino acid or phosphate symporters (NX), Na+/glucose symporters (SGLT), and Na+/K+/2Cl− cotransporters (NKCC), or via Na+ selective ion channels such as the epithelial Na+ channel (ENaC). The electrochemical gradient for apical Na+ uptake is generated at the basolateral membrane via the activity of the Na+/K+-ATPase as well as K+ channels. The net effect of the concerted interplay between channels/transporters and the Na+/K+-ATPase is a movement of Na+ from the apical to the basolateral side of the epithelium (blue arrows). This creates osmotic forces which drive the absorption of water from the apical to the basolateral compartment (gray arrow).
Due to the concerted action of Na+ channels/transporters and the Na+/K+-ATPase, there is a net movement of Na+ ions from the apical to the basolateral side of the epithelium. This results in an osmotic gradient across the epithelium, which eventually drives the transepithelial absorption of water (Figure 1). This mechanism is a basic principle for the physiology of various organs: (i) in the kidneys, transepithelial Na+ and water transport drives the fluid reabsorption from the primary urine and indirectly regulates blood volume and hence blood pressure (Bhalla and Hallows, 2008); (ii) in the lungs, Na+ and water transport across the pulmonary epithelium regulates the volume and viscosity of the airway lining fluid (Hollenhorst et al., 2011) and keeps alveoli free of fluid (Olver et al., 1986; Hummler et al., 1996; Althaus et al., 2011); (iii) in the intestine, Na+ transport facilitates uptake of electrolytes and nutrients (e.g., amino acids) as well as water absorption from the chime.
The importance of Na+ transport processes becomes also evident when one considers pathological conditions which are associated with dysregulated Na+ transport. (i) In the kidneys, a hereditary form of hypertension, Liddle syndrome, is reasoned by hyperabsorption of Na+ from the primary urine due to a mutation which leads to an increased number of ENaCs in the plasma membrane (Shimkets et al., 1994; Schild et al., 1995). Conversely, mutations which lead to ENaC hypoactivity cause the salt-wasting syndrome pseudohypoaldosteronism type 1 (Chang et al., 1996). (ii) In the lung, impaired reabsorption of Na+ and water across pulmonary epithelia can lead to pulmonary edema (Hummler et al., 1996; Althaus et al., 2011), whereas increased Na+ transport can contribute to cystic fibrosis (CF)-like lung disease (Mall et al., 2004; Azad et al., 2009; Rauh et al., 2010). (iii) In the intestine, an inhibition of Na+ absorption, e.g., by viral infections, induces diarrhea (Ousingsawat et al., 2011).
These physiological and pathophysiological examples demonstrate that a precise regulation of transepithelial Na+ transport is crucial for the function of multiple organs. A novel class of regulatory signaling molecules is the class of gasotransmitters (Wang, 2002). Gasotransmitters are endogenously produced as a product of amino acid metabolism and regulate a variety of cell and organ functions. There are currently three molecules which are regarded as gasotransmitters: nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S; Wang, 2002). Recently, there is a growing body of evidence that these molecules may serve as regulators of epithelial Na+ transport processes. The following sections will describe the three known gasotransmitters and review their impact on Na+ transport processes and Na+ transporting molecules in renal, pulmonary, and intestinal epithelia.
Nitric Oxide (NO)
Nitric oxide was the first gasotransmitter that was recognized as a physiological signaling mediator. In 1987, NO was identified to be the “endothelial derived relaxing factor” inducing the relaxation of vascular smooth muscle cells (Ignarro et al., 1987). NO is endogenously produced from the amino acid L-arginine by nitric oxide synthases (NOS). There are three NOS isoforms, NOS1 (neuronal NOS), NOS2 (inducible NOS), and NOS3 (endothelial NOS; Garvin et al., 2011). Apart from a classical role as a regulator of smooth muscle tone, NO has also been demonstrated to play an important role in the regulation of transepithelial Na+ transport.
Effects of NO on Renal Epithelial Na+ Transport
NO reduces Na+ absorption in the kidneys
In the kidneys, NO is generally regarded to facilitate natriuresis and diuresis, which is evident from animal studies using NO-donating molecules. The NO-donor S-nitroso-acetylpenicillamine (SNAP) increased urinary Na+ excretion in anesthetized dogs (Majid et al., 1998). Similarly, the infusion of the NO-donor molecule sodium nitroprusside (SNP) increased natriuresis and diuresis in conscious rats (Grandes et al., 1991). By contrast, the intrarenal arterial infusion of the NO-donor molecule NOC-7 did not affect baseline urinary Na+ excretion in anesthetized rabbits (Adachi et al., 1997). However, angiotensin 2- and norepinephrine-induced reduction in urinary Na+ excretion was blocked by NOC-7 in this model (Adachi et al., 1997). These data suggest that NO may block angiotensin 2- or norepinephrine-activated renal transepithelial Na+ absorption in rabbits. These findings are confirmed in studies with anesthetized rats, where SNP decreased angiotensin 2-stimulated proximal tubular fluid absorption (Eitle et al., 1998). By contrast to the study by Adachi et al. (1997), SNP also decreased basal tubular fluid absorption (Eitle et al., 1998). This discrepancy might be explained by different employed concentrations of SNP (Eitle et al., 1998). These studies indicate that exogenously applied NO-donating drugs increase urinary Na+ excretion – which is likely the result of a decreased transepithelial Na+ transport.
Consistent with studies investigating effects of exogenously applied NO, inhibitors of NO synthesis, such as nitro-L-arginine methyl ester (L-NAME), were demonstrated to exert anti-natriuretic and anti-diuretic effects (Lahera et al., 1991, 1993; Ortiz and Garvin, 2002). Furthermore, NOS3 knock-out mice had a reduced Na+ excretion and urinary volume after acute volume load (Perez-Rojas et al., 2010). The treatment of anesthetized rats with cholesterol also blocked urinary Na+ excretion (Kopkan et al., 2009). There was no such effect in the presence of the NOS inhibitor L-NAME, which suggests that cholesterol prevents NO-mediated natriuresis (Kopkan et al., 2009). Those studies imply that there is a basal tone of NO, which keeps transepithelial Na+ transport and consequently Na+ retention in the kidneys low.
NO-mediated regulation of Na+ transporting molecules along nephrons
Several studies investigated the regulatory impact of NO on ion transporting molecules across nephronic epithelial cells. The focus of the following paragraphs will be on epithelial Na+ transporting molecules. For effects of NO on other renal ion channels and transporters, the reader is referred to an excellent review by Ortiz and Garvin (2002).
The bulk of the fluid and electrolytes which are filtered at the glomeruli are reabsorbed from the primary urine along the proximal tubule. The reabsorption of Na+ occurs primarily by Na+ coupled cotransporters which are located in the apical membrane of the epithelium. The gradient for Na+ uptake is created by the basolaterally located Na+/K+-ATPase. The NO donors SNAP and SNP inhibited the Na+/H+-exchanger (NHE) in rabbit proximal tubules and Caco-2 cells (Roczniak and Burns, 1996; Gill et al., 2002). Furthermore, NO donors also inhibited the Na+/K+-ATPase in an opossum proximal tubule cell line (Liang and Knox, 1999). Consistent with this observation, Linas and Repine (1999) demonstrated that endothelial cells can regulate Na+ transport by proximal tubule epithelial cells via NO synthesis and consequently inhibition of the Na+/K+-ATPase. Taken together, NO is thus likely an inhibitor of Na+ reabsorption in the proximal tubule, which is due to inhibition of NHE as well as the Na+/K+-ATPase.
In the thick-ascending limb, the uptake of Na+ from the primary urine occurs via apical Na+ transporters, such as the Na+/K+/2Cl− cotransporter (NKCC) and NHE, as well as the basolateral Na+/K+-ATPase. The NO donors spermine NONOate and nitroglycerin both decreased NHE activity in isolated and perfused rat thick ascending limbs (Garvin and Hong, 1999). Although Spermine NONOate decreased NKCC activity in the same model, the Na+/K+-ATPase was not affected (Ortiz et al., 2001). These data indicate that in the thick ascending limb, NO rather interferes with apical transport systems than the basolateral Na+/K+-ATPase. The source of NO in thick ascending limbs is likely a production by NOS3, since L-arginine, the substrate for NO production, inhibits thick ascending limb Na+ transport in NOS1 and NOS2, but not in NOS3 knock-out mice (Plato et al., 2000).
Beside the thick ascending limb, there is also Na+ transport across the thin ascending limb or the descending limb. A putative effect of NO on Na+ transport activities in these segments, however, has not been investigated so far (Garvin et al., 2011).
In the cortical-collecting duct (CCD), Na+ is taken up by the epithelium via the concerted action of apically located ENaCs and the basolateral Na+/K+-ATPase. Spermine NONOate and nitroglycerin decreased ENaC activity in rat CCD (Stoos et al., 1995). This effect was also apparent for endothelium-derived NO, which decreased ENaC activity (Stoos et al., 1994, 1995). By contrast, Lu et al. (1997) observed an increase in ENaC activity due to NO. This was likely an indirect effect via activation of basolateral K+ channels (Lu et al., 1997). Thus, whether NO elicits a net-inhibition or activation of ENaC in the CCD remains controversial. Interestingly, there was no effect of NO donors on Na+/K+-ATPase activity in the CCD (Stoos et al., 1994, 1995). The reason why NO inhibits the Na+/K+-ATPase in the proximal tubule but not in the CCD is unknown. However, this discrepancy might be explained by differences in the oxidative state of epithelial cells, which may counter effects of NO on Na+ transport (Yu et al., 2007; Helms et al., 2008).
Taken together, the currently available data speak in favor of a NO-mediated inhibition of Na+ transporting molecules in renal epithelia.
NO-induced signaling events in renal epithelia
The classical mechanism how NO exerts biological effects is the stimulation of soluble guanylate cyclase (sGC). This results in increased cyclic guanosine monophosphate (cGMP) production and downstream effects on protein kinases such as protein kinase G. NO-mediated activation of the sGC/cGMP pathway has been demonstrated to be involved in NO regulation of NHE3 (Roczniak and Burns, 1996; Gill et al., 2002) and the Na+/K+-ATPase (Liang and Knox, 1999; Linas and Repine, 1999) in the proximal tubule. These studies showed that NO-induced production of cGMP is crucial for the regulation of Na+ transport in the proximal tubule. Interestingly, Sasaki et al. (2004) demonstrated that cGMP is produced upon administration of SNAP in proximal tubule cells in humans; however, the resulting decrease in Na+ uptake by the epithelial cells was blocked by probenecid. This finding suggests that export of cGMP by a probenecid-sensitive organic anion transporter is necessary for the regulation of Na+ transport (Sasaki et al., 2004). This may be the result of a downstream activation of protein kinase G or Src tyrosine kinase signaling pathways by extracellular cGMP (Jin et al., 2004; Nascimento et al., 2011). The activation of the Src signaling complex is associated with endocytosis of the Na+/K+-ATPase as well as the NHE3 in renal cells (Liu et al., 2004, 2005; Oweis et al., 2006; Cai et al., 2008; Nascimento et al., 2011). This would provide a molecular link between NO-mediated cGMP production, cGMP export, and downstream inhibition of Na+ transport in the proximal tubule.
The general inhibition of Na+ absorption by NO in the thick-ascending limb is also likely due to cGMP mediated signaling (Ortiz and Garvin, 2001), however, if cGMP and downstream activated mediators specifically decrease NKCC or NHE activity in this segment, remains to be proven.
Aside from the activation of the sGC/cGMP pathway, there are alternative signaling mechanisms that might explain effects of NO on Na+ transporting molecules. For example, higher oxidative derivates of NO, such as NO2 or N2O3, can interact with cysteine-residues of proteins – a mechanism which is termed S-nitrosylation (Hess et al., 2005). This direct protein-modification can regulate the activity of ion channels and transporters, as for example shown for purified Na+/K+-ATPase (Sato et al., 1995). The distinct contribution of S-nitrosylation of Na+ transporting molecules to NO-mediated interference with general Na+ transport in renal epithelia, however, is not sufficiently investigated and needs to be elucidated.
Renal sources of NO
An important question which remains is the precise source for NO in the kidneys. Although inhibitors of NOS show obvious effects on renal Na+ transport (see Section “NO Reduces Na+ Absorption in the Kidneys”), it is hard to conclude from these studies what is the cellular origin of NO. Furthermore, it is noteworthy that in studies with systemic administration of NOS inhibitors or with knock-out mice, a contribution of other physiological parameters, i.e., indirect impact of blood pressure on glomerular filtration and renal function, is difficult to exclude.
Nevertheless, it has been demonstrated that all three NOS isoforms are expressed in nephronic epithelia, which indicates that the epithelial cells are generally capable of producing NO (Cabral and Garvin, 2011). This may imply that NO might be an autocrine regulator of Na+ transport in renal epithelia.
There is also a cross-talk between the epithelium and other cells, such as endothelial cells or neuronal cells. A study by Linas and Repine (1999) showed that endothelial cells can inhibit the Na+/K+-ATPase and Na+ transport via NO release when co-cultured with proximal tubule epithelial cells. As mentioned earlier, endothelium-derived NO can also regulate ENaC activity in the CCD (Stoos et al., 1994, 1995). Furthermore, there is evidence that nervous stimulation can regulate Na+ transport in the kidney. Denervation of the kidney, for example, can abolish the anti-natriuretic effects of L-NAME (Wu et al., 1999; Ortiz and Garvin, 2002). Thus a role for NO as a neurotransmitter with secondary effects on Na+ transport is also likely.
Taken together, although there are still controversies in the sources of NO, the precise signaling mechanisms as well as targets of action of NO, there is a convincing body of evidence that NO impairs Na+ transporting molecules in nephronic epithelia and consequently leads to renal natriuresis and diuresis.
Effects of NO on Pulmonary Epithelial Na+ Transport
General effects of NO on pulmonary Na+ transport
Consistent with the data from renal epithelia, NO is regarded to inhibit Na+ transport across the pulmonary epithelium. There are several studies which investigated the effects of NO on lung fluid clearance (which is driven by transepithelial Na+ transport) in whole-lung models. A study by Nielsen et al. (2000) demonstrated that DETANONOate, a NO-donor, decreased amiloride-sensitive fluid clearance in rabbits, which may indicate a decrease in ENaC-mediated transepithelial Na+ transport by NO. Further studies investigated the involvement of NO in lung disease-associated impairment of transepithelial Na+ transport: The instillation of endotoxin, for example, increased lung NO and cGMP levels and inhibited alveolar fluid clearance in rats (Tsubochi et al., 2003). Furthermore, Pittet et al. (2001) demonstrated an increase in NOS2 after hemorrhagic shock in rats, which was associated with a decrease in alveolar fluid clearance. Similarly, hydrostatic pressure increased NO production and consequently decreased fluid reabsorption in isolated rat and mouse lungs (Kaestle et al., 2007).
Aside from the exogenous administration of NO by NO-liberating molecules, these studies imply that endogenous NO production by NOS also leads to a decrease in pulmonary epithelial Na+ transport (Pittet et al., 2001; Tsubochi et al., 2003; Kaestle et al., 2007). Consistent with these studies, inhibition of NOS2 decreased NO production and increased amiloride-sensitive Na+ currents in a human airway epithelial cell line (H441; Song et al., 2009). These data generally suggest an inhibitory effect of NO on pulmonary epithelial Na+ transport. By contrast, NOS2 knock-out mice exhibited a decreased amiloride-sensitive alveolar fluid clearance (Hardiman et al., 2001), which is likely due to a reduced protein expression of the α and γ subunits of ENaC (Hardiman et al., 2004). Although indirect effects of a long-term NOS inhibition on Na+ transport cannot be excluded (see section about renal physiology), it might be hypothesized that there are different physiological reactions due to acute or long-term changes in pulmonary NO content.
Thus there are various studies available which demonstrate an inhibition of fluid clearance by NO and consequently suggest an impairment of Na+ transport across pulmonary epithelia – especially in the distal lung regions.
Aside from the distal lung epithelia, Na+ transport plays also an important role in the regulation of airway surface liquid and thus airway physiology (Hollenhorst et al., 2011). The administration of NOS2 inhibitors increased amiloride-sensitive nasal potential difference in mice (Kelley and Drumm, 1998; Elmer et al., 1999), indicating that endogenously produced NO keeps Na+ transport across nasal epithelia low. By contrast, Rückes-Nilges et al. (2000) did not detect effects of the NO donors SNP and spermine NONOate on Na+ transport by cultured primary human nasal epithelial cells. The reason for this discrepancy is unknown.
A hyperabsorption of Na+ across the airway epithelia can promote CF-like lung disease (Mall et al., 2004). Interestingly, NOS2 expression is lower in CF compared to non-CF airway epithelia (Kelley and Drumm, 1998; Moeller et al., 2006). Furthermore, the NO concentration in exhaled air is inversely correlated with transepithelial potential difference in patients with CF (Texereau et al., 2005). This might suggest a regulatory mechanism for Na+ hyperabsorption across CF epithelia due to lack of tonic inhibition by endogenously produced NO.
Effects of NO on Na+ transporting molecules in pulmonary epithelia
Consistent with the effects of NO on whole-lung Na+ and fluid clearance, the application of NO-donating molecules decreased amiloride-sensitive Na+ absorption by cultured rat alveolar type 2 cells (Guo et al., 1998) as well as the human airway epithelial cell line H441 (Althaus et al., 2010). In both studies, a cGMP-independent inhibition of ENaC as well as the Na+/K+-ATPase was demonstrated (Guo et al., 1998; Althaus et al., 2010). By contrast, a patch-clamp study on rat alveolar type 2 cells described a cGMP-dependent inhibition of Na+ channel activity due to NO (Jain et al., 1998). The discrepancies between the studies by Jain et al. (1998) and Guo et al. (1998) might be explained by different culture conditions which were demonstrated to affect the phenotype of Na+ channels in alveolar epithelial cells (Jain et al., 2001). Especially the supplement or non-supplement of dexamethasone, in order to induce Na+ transport, or the culture at air/liquid interface, has large effects on transepithelial Na+ transport properties (Althaus et al., 2010). The inhibitory effect of NO on Na+ channel activity was also confirmed in patch-clamp studies on lung slices from rat, although this effect was solely apparent on type 2, but not on type 1 alveolar epithelial cells (Helms et al., 2008). The authors explained this observation by higher levels of reactive oxygen species, such as superoxide anions (), in type 1 cells. might interact with NO and thereby counter inhibitory effects of NO on type 1 cells (Helms et al., 2008).
Nevertheless, although NO-mediated signaling mechanisms vary, the described studies demonstrate a general inhibition of Na+ channels by exogenous NO administration in pulmonary epithelia. By contrast to studies on renal epithelia, potential effects of NO on Na+-coupled transporters in lung epithelia is, to the author’s knowledge, currently unknown.
Pulmonary sources of NO
Direct evidence for endogenous NO synthesis in lungs is the fact that NO can be detected in the exhaled air of humans (Khatri et al., 2001; Texereau et al., 2005). However, the major cellular source of this exhaled NO has not been identified to date.
The expression of NOS has been shown in various pulmonary epithelial cells (Kelley and Drumm, 1998; Ermert et al., 2002; Moeller et al., 2006; Song et al., 2009). In addition, alveolar macrophages express all three NOS isoforms (Ermert et al., 2002). Furthermore, LPS-activated macrophages reduced epithelial amiloride-sensitive Na+ absorption when co-cultured with fetal rat distal lung epithelial cells (Ding et al., 1998). Endothelial cells also contain NOS1 (Ermert et al., 2002; Lührs et al., 2002), NOS3 (Ermert et al., 2002), and can express NOS2 upon stimulation by exposure to endotoxin (Ermert et al., 2002). These data demonstrate that NO might not only serve as an autocrine regulator of Na+ transport in epithelial cells, but may also originate from other cells and tissues, such as macrophages or the pulmonary vasculature.
Taken together, NO is endogenously produced in lung tissues. Although NO-mediated signaling mechanisms vary, the available data suggest an inhibition of Na+ transport across pulmonary epithelia due to impaired activity of Na+ channels and the Na+/K+-ATPase.
Effects of NO on Intestinal Epithelial Na+ Transport
In the intestine, transepithelial Na+ transport drives the absorption of organic substances as well as the absorption of water from the chime. The absorption of amino acids, glucose, or phosphate occurs mainly in the duodenum and jejunum by Na+-coupled symporters, whereas in the ileum, Na+ is taken up by the epithelial cells via NHEs. Na+ absorption in the colon occurs via Na+ selective ion channels (such as ENaC) in most-species in an aldosterone-dependent way.
There are various studies on the effects of NO on electrolyte and water transport across intestinal epithelia and both pro-secretory as well as pro-absorptive actions of NO have been reported (Izzo et al., 1998). However, under physiological conditions, the available studies speak in favor of a NO-dependent pro-absorptive intestinal tone (Izzo et al., 1998). At this point the reader is referred to the excellent review by Izzo et al. (1998).
Although basic effects of NO on electrolyte and fluid transport in intestinal epithelia have been intensively investigated, there are surprisingly few studies on the regulation of distinct Na+ transporting ion channels and transporters by NO.
Consistent with a pro-absorptive role of NO, treatment of rat small intestinal epithelial cells (IEC-18) with L-NAME decreased the activity of the Na+ glucose cotransporter SGLT1 (Coon et al., 2008), suggesting that endogenous NO would simulate SGLT1.
By contrast, the treatment of IEC-18 cells with L-NAME stimulated NHE3 by enhanced protein expression (Coon et al., 2008). The stimulation of NHE3 by L-NAME was also observed on rabbit ileum (Coon et al., 2007). These data suggest that constitutively produced NO inhibits NHE3 in the small intestine. Consistent with these studies, the exposure of Caco-2 cells, a human colon adenocarcinoma cell line, to NO-donating molecules inhibited NHE3 activity via cGMP-dependent mechanisms (Gill et al., 2002).
Furthermore, an increase in Na+/K+-ATPase activity was observed after treatment of IEC-18 cells with L-NAME (Coon et al., 2008), which would suggest an inhibitory action of NO on this enzyme. Furthermore, the NO-donor SNAP decreased Na+/K+-ATPase activity in rat IEC-6 cells (Suzuki et al., 2005).
Those data indicate that, depending on the intestinal segment and distinct Na+ transporting molecules, NO might have net pro- or anti-absorptive effects on electrolyte and Na+ transport across intestinal epithelia. Furthermore it is important to point out that the described studies on Na+ transporting molecules investigated effects of NO on epithelial cells – independently of neuronal regulation. A neuronal input – using NO as a transmitter – might superimpose local effects on Na+ transporting molecules and might also account for different net-effects that are attributed to NO.
NO Regulation of Na+ Transport: Conclusions
There is a convincing body of evidence that speaks in favor of an inhibitory role for NO in the regulation of Na+ transport in renal and pulmonary epithelia. NO is endogenously produced in the kidneys or lungs, affects Na+ transporting molecules via cellular signaling cascades and eventually regulates organ physiology, such as natriuresis/diuresis or Na+-driven alveolar fluid clearance.
By contrast, it is difficult to make a decisive conclusion about a pro- or anti-absorptive role for NO in intestinal epithelia. NO-mediated effects in the intestine seem to be dependent on differences between intestinal segments, species, as well as concentrations of NO (Izzo et al., 1998). Furthermore, NO-mediated signaling mechanisms vary, dependent on the amount of NO, and consequently cGMP, or the availability of other oxidative species and the formation (or quenching) of higher oxidative derivates of NO. The heterogeneity of NO effects in the intestine might therefore not necessarily be contradictory, especially given the fact that the inner milieu of the intestine is more variable (depending on the nutritional status or intestinal flora) when compared to kidneys or lungs.
Carbon Monoxide (CO)
Although generally regarded as a highly toxic gas, carbon monoxide (CO) has been recognized as a physiological cellular signaling mediator. CO is endogenously produced in the human body as a product of heme metabolism and is generated by heme-degrading enzymes referred to as heme oxygenases (Ryter et al., 2006). There are two distinct forms of heme oxygenases: heme oxygenase 1 (Hmox1) an inducible form, and constitutively expressed heme oxygenase 2 (Hmox2; Maines et al., 1986). Originally, CO has been considered simply as a by-product of heme degradation by heme oxygenases, however, in the past decade, evidence accumulated to suggest that CO produced by heme oxygenases serves as a regulated cellular signaling molecule. This has been demonstrated in multiple studies addressing the cellular effects of exogenous administration of physiologically relevant doses of CO. By this means, it was demonstrated that CO has potent cyto- and tissue-protective properties due to its implication in anti-apoptotic, anti-inflammatory, and anti-proliferative signaling mechanisms (Ryter et al., 2006; Motterlini and Otterbein, 2010). In addition, there is recently a growing body of evidence that CO might be a regulator of ion channels/transporters and consequently epithelial ion transport processes.
Effects of CO on Renal Epithelial Na+ Transport
Both heme oxygenase isoforms are expressed in the kidney (Csongradi et al., 2012) and endogenous production of CO has been demonstrated in the kidney (Jackson et al., 2011).
Similar to NO, CO is also an activator of sGC, although with less potency (Ma et al., 2007). Given that NO regulates Na+ transport mainly by sGC/cGMP mediated signaling mechanisms, it is likely that CO also affects Na+ transporting molecules in epithelia. The first hint toward this hypothesis was provided by Nathanson et al. (1995). These authors demonstrated a CO-induced activation of neuronal Na+/K+-ATPase by cGMP-dependent mechanisms (Nathanson et al., 1995). An activation of the Na+/K+-ATPase by CO in epithelia would consequently enhance transepithelial Na+ transport. Consistent with this idea, the inhibition of heme oxygenases by chromium mesoporphyrin (CrMP) and thus endogenous CO production, decreased the absorption of Na+ in the loop of Henle of microperfused rat kidney tubules (Wang et al., 2003). Furthermore, CO was shown to activate ENaC in a M1 mouse kidney CCD cell line (Wang et al., 2009).
By contrast, chronic induction of heme oxygenases by cobalt-protoporphyrin resulted in increased urinary Na+ excretion in cirrhotic rats (Di Pascoli et al., 2011), which would suggest an inhibition of renal epithelial Na+ absorption by CO. Another study has shown that the inhibition of heme oxygenase, and CO production with CrMP leads to a decreased urinary Na+ excretion in rats (Jackson et al., 2011), which is likely due to enhanced Na+ absorption. Those studies would speak in favor of an inhibitory effect of CO on transepithelial Na+ absorption in the kidney.
Interestingly, the studies by Wang et al. (2003) and Jackson et al. (2011) describe oppositional effects of heme oxygenase inhibition on urinary Na+ excretion – despite using the same species, the same inhibitor and without observing effects on glomerular filtration rate. It may be speculated that there are differences between systemic administration of inhibitors (Wang et al., 2003) versus microperfusion studies (Jackson et al., 2011), which would indicate spatiotemporal differences in CO-mediated signaling events. Based on the available studies, whether CO is an activator or inhibitor of transepithelial Na+ absorption in the kidney is yet to be determined.
Effects of CO on Pulmonary Epithelial Na+ Transport
Concerning a putative role of CO on pulmonary Na+ transport, we have previously investigated the effects of CO administration in isolated, ventilated, and perfused rabbit lungs (Althaus et al., 2009). In this model, CO, applied as a gas or by the donor molecule CO-releasing molecule 3 (CORM-3), led to a decreased amiloride-sensitive transepithelial Na+ transport as measured by radioactive 22Na+ clearance and resulted in an increased alveolar lining fluid volume. Thus, CO impaired Na+ transport as well as fluid clearance in rabbit lungs. This effect has been attributed to impaired amiloride-sensitive Na+ transport across pulmonary epithelial cells, as indicated by transepithelial Ussing chamber studies using cultured H441 monolayers as well as primary isolated alveolar type 2 cells from the rat. In contrast to NO which affected both the activity of Na+ channels and the Na+/K+-ATPase (Althaus et al., 2010), CO did not impair Na+/K+-ATPase activity, but did inhibit amiloride-sensitive ENaC (Althaus et al., 2009). This finding is also in contrast to the study by Wang et al. (2009), who observed an increased activity of ENaC due to CO. This discrepancy is interesting, especially since both in M1 cells, and H441 cells, the major Na+ conductance is an ENaC-typical ∼5–10 pS channel (Althaus et al., 2009; Wang et al., 2009). The effect of CO on ENaC in H441 cells likely involves histidine residues since CO-effects can be mimicked and abolished by the histidine-modifying agent diethyl pyrocarbonate (Althaus et al., 2009). As pointed out by Wilkinson and Kemp (2011), there are differences in the histidine contents of human and mouse ENaCs. Those contradictory findings might therefore be explained by taxonomical differences. Furthermore it is important to point out that it is difficult to compare findings from excised patch-clamp experiments (Wang et al., 2009) with intact epithelial monolayers or isolated organ studies (Althaus et al., 2009), as additional regulatory CO-sensitive mediators might be present in intact cells.
These studies indicate that exogenously applied CO affects Na+ transport in the pulmonary epithelium. However, if CO is endogenously produced in the lung and affects Na+ transport is an important question that needs to be addressed. Constitutively expressed Hmox2 is expressed at low levels in lung epithelial cells (Roth et al., 2009). Furthermore, alveolar macrophages express both heme oxygenase isoforms (Maestrelli et al., 2001). In addition, pulmonary endothelial cells express constitutive Hmox2 (Roth et al., 2009). Thus, several cell types in the lung contain the enzyme system required for the generation of CO. Similar to NO, CO can also be detected in the exhaled air of humans (Khatri et al., 2001). Thus, CO is indeed endogenously produced in the lung. However, a putative link between CO production and epithelial Na+ transport needs to be demonstrated.
Effects of CO on Intestinal Epithelial Na+ Transport
In the intestine, CO might also serve as a regulator of epithelial ion transport processes since the expression of heme oxygenases has been demonstrated and the administration of CO can activate a Cl− secretion across colonic epithelia (Pouokam et al., 2011; Steidle and Diener, 2011). However, whether or not CO also influences Na+ absorption in the intestine, as well as other epithelia, is currently unknown.
CO Regulation of Na+ Transport: Conclusions
It was demonstrated that CO is able to affect Na+ transport processes across various epithelia. In the kidneys, both pro- and anti- Na+-absorptive effects of CO have been described. In the lung, exogenous administration of CO decreased Na+ transport and fluid clearance, however, a regulatory contribution of endogenously synthesized CO remains to be demonstrated. Thus, there is promising data available which indicate that CO might be a putative novel regulator of epithelial Na+ transport physiology. However, there are important questions remaining: How is endogenous CO-production regulated? What are the stimulators of CO production? How do concentrations of CO employed in experimental studies reflect endogenously produced levels of CO? These questions need to be answered before a decisive conclusion that CO is a physiological regulator of epithelial Na+ transport physiology can be drawn.
Hydrogen Sulfide (H2S)
Hydrogen sulfide (H2S) is a well known environmental threat with the typical odor of rotten eggs. However, similar to NO and CO, H2S has been suggested to represent the third biologically active gasotransmitter (Wang, 2002). H2S is endogenously produced by mammalian cells from the amino acid L-cysteine by mainly two enzymes: cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE; Stipanuk and Beck, 1982; Wang, 2002). CSE knock-out mice have reduced H2S levels in serum, aorta, and heart and increased systolic blood pressure (Yang et al., 2008), which convincingly showed for the first time a physiological role for H2S. Although H2S has been shown to interact with various ion channels (Tang et al., 2010), putative effects of H2S on Na+ transporting molecules are hardly investigated.
Effects of H2S on Renal Epithelial Na+ Transport
Despite the fact that H2S is produced by CBS/CSE in the kidney (Xu et al., 2009; Aminzadeh and Vaziri, 2012), up to today there are no studies on the impact of H2S on Na+ transport processes across renal epithelia. However, recently it has been shown that H2S can activate AMP-activated protein kinase (AMPK) in rat glomerular epithelial cells (Lee et al., 2012). Since AMPK is an important regulator of the Na+/K+-ATPase and ENaC (Carattino et al., 2005; Woollhead et al., 2007; Albert et al., 2008; Mace et al., 2008), it might be speculated that H2S-mediated AMPK activation affects renal Na+ absorption. This hypothesis remains to be investigated.
Effects of H2S on Pulmonary Epithelial Na+ Transport
A first hint that H2S might affect Na+ transport processes across lung epithelia comes from pathological observations on patients which had prolonged exposure to this gas. One of their symptoms is, amongst others, pulmonary edema (Cordasco and Stone, 1973). Based on the link between alveolar fluid clearance and Na+ transport across the alveolar epithelium (Althaus et al., 2011), it might be speculated that H2S poisoning leads to fluid accumulation in the airspaces due to impairment of transepithelial Na+ and, consequently, water reabsorption. Consistent with this idea, our laboratory has recently shown that the administration of the H2S liberating molecule NaHS decreased amiloride-sensitive Na+ transport across the H441 lung epithelial cell line as well as native tracheal preparations of pigs and mice (Althaus et al., 2012). This was not the result of a direct impairment of Na+ transporting molecules, but rather occurred indirectly via H2S-mediated inhibition of basolateral K+ channels and consequently impairment of Na+/K+-ATPase activity (Althaus et al., 2012). An impaired activity of the Na+/K+-ATPase due to NaHS has also been reported for rat colonic epithelia; although this study did not investigate transepithelial Na+ transport processes (Pouokam and Diener, 2011). In general, an impaired transepithelial Na+ transport by H2S in the lung may provide a molecular explanation for edema development in patients with H2S poisoning. However, whether or not H2S is also an endogenous, physiological regulator of pulmonary epithelial Na+ transport, needs to be addressed. A first step toward the answer to this question is the fact that H2S generating enzymes are expressed in rat lungs (Madden et al., 2012) and H2S production has been shown in lung tissue homogenates of mammals (Olson et al., 2010). If such endogenously produced H2S affects pulmonary Na+ transport is a question that remains to be answered.
Effects of H2S on Intestinal Na+ Transport
In the intestine, production of H2S has also been shown in rat ileum (Zhao et al., 2003). Additionally, the H2S generating enzymes CBS and CSE are expressed in the colon and inhibition of these enzymes affects transepithelial ion transport (Schicho et al., 2006; Hennig and Diener, 2009). Furthermore, the application of NaHS induces a Cl− secretion across colonic epithelia (Schicho et al., 2006; Hennig and Diener, 2009). Putative effects of H2S on intestinal Na+ absorption, however, remain unknown.
H2S Regulation of Na+ Transport: Conclusions
Although H2S is an emerging signaling molecule in organs such as kidneys, lung, or the intestine, its effects on epithelial Na+ transport processes in those organs is largely unknown. Interestingly, a major target for H2S are different types of K+ channels (Telezhkin et al., 2009; Tang et al., 2010). K+ channels play a crucial role in epithelial ion transport physiology since these channels regulate the membrane potential and thus electrochemical gradients which are necessary for ion fluxes. For example, we and others have shown that modulation of K+ channel activity in the basolateral membrane of pulmonary epithelia indirectly affects transepithelial Na+ transport processes (Greenwood et al., 2009; Althaus et al., 2012). Therefore it may be speculated that H2S likely affects ion and Na+ transport processes across various other epithelia by interference with K+ channel activity.
Beside the elucidation of effects of H2S on Na+ transport processes, a major point that needs to be addressed is the question if H2S is a physiological regulator of epithelial transport. Physiological concentrations of H2S are hard to determine and still controversial (Olson, 2011). The future challenges will be the answer to the question what the real physiological concentrations of H2S are and if such concentrations are necessary or sufficient to maintain or affect epithelial Na+ transport processes.
Summary and Perspectives
There are various studies available which demonstrate that the gasotransmitters NO, CO, and H2S affect epithelial Na+ transport processes. However, before one can decisively consider any gasotransmitter as a true physiological regulator of Na+ transport, the following criteria need to be fulfilled:
(1) The transmitter needs to be generated by specific enzymes either directly in the epithelium or in associated tissues, such as endothelium, nerve fibers, or other specific cells.
(2) The production of the transmitter needs to be either tonic or stimulated by specific, physiologically relevant inducers.
(3) The endogenously produced transmitter needs to regulate specific protein targets (Na+ transporting molecules), either directly or by cellular signaling cascades.
Although these criteria seem to be quite straightforward, it is not easy to address them in experimental studies. This is reasoned by the facts that (i) it is difficult to measure physiological concentrations of the transmitters and (ii) consequently it is not easy to decide whether transmitter concentrations, when applied as gas or by donating molecules, are in a physiological range.
So far, NO is the only gasotransmitter which sufficiently fulfills the mentioned criteria. This is evident from studies demonstrating that NO is produced by distinct cell types, either tonic or stimulated, and can regulate specific Na+ transporting molecules in epithelia via cellular signaling mechanisms, such as activation of the sGC/cGMP pathway.
Additionally, there is a growing body of evidence that the gasotransmitters CO and H2S can influence epithelial Na+ transport processes by affecting distinct Na+ transporting molecules in epithelia. However, especially the first two criteria concerning a physiological role of CO and H2S have not been sufficiently addressed. The future challenge will be the investigation of the role of endogenously produced CO/H2S, the identification of stimulators of their synthesis and finally the elucidation of their impact on Na+ transport processes across epithelia.
Taken together, gasotransmitters are emerging as an important class of signaling molecules which might be identified as novel regulators of epithelial Na+ transport processes in the future. The investigation of their physiology and putative dysregulation in pathophysiological situations is an exciting field which eventually may expand our understanding of Na+ transport biology in health and disease.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author would like to thank Wolfgang Clauss and Martin Fronius for outstanding support and the three reviewers for the comments and improvement of the manuscript. The work of the author is supported by the Deutsche Forschungsgemeinschaft, DFG, grant AL1453/1-1.
References
Adachi, Y., Hashimoto, K., Ono, N., Yoshida, M., Suzuki-Kusaba, M., Hisa, H., and Satoh, S. (1997). Renal effects of a nitric oxide donor, NOC 7, in anesthetized rabbits. Eur. J. Pharmacol. 324, 223–226.
Albert, A. P., Woollhead, A. M., Mace, O. J., and Baines, D. L. (2008). AICAR decreases the activity of two distinct amiloride-sensitive Na+-permeable channels in H441 human lung epithelial cell monolayers. Am. J. Physiol. Lung Cell Mol. Physiol. 295, L837–L848.
Althaus, M., Clauss, W. G., and Fronius, M. (2011). Amiloride-sensitive sodium channels and pulmonary edema. Pulm. Med. 8. PMID: 21637371. [Epub ahead of print].
Althaus, M., Fronius, M., Buchäckert, Y., Vadász, I., Clauss, W. G., Seeger, W., Motterlini, R., and Morty, R. E. (2009). Carbon monoxide rapidly impairs alveolar fluid clearance by inhibiting epithelial sodium channels. Am. J. Respir. Cell Mol. Biol. 41, 639–650.
Althaus, M., Pichl, A., Clauss, W. G., Seeger, W., Fronius, M., and Morty, R. E. (2010). Nitric oxide inhibits highly selective sodium channels and the Na+/K+-ATPase in H441 Cells. Am. J. Respir. Cell Mol. Biol. 44, 53–65.
Althaus, M., Urness, K. D., Clauss, W. G., Baines, D. L., and Fronius, M. (2012). The gasotransmitter hydrogen sulphide decreases Na+ transport across pulmonary epithelial cells. Br. J. Pharmacol. doi:10.1111/j.1476-5381.2012.01909.x
Aminzadeh, M. A., and Vaziri, N. D. (2012). Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol. Dial. Transplant. 27, 498–504.
Azad, A. K., Rauh, R., Vermeulen, F., Jaspers, M., Korbmacher, J., Boissier, B., Bassinet, L., Fichou, Y., Des Georges, M., Stanke, F., Boeck, K., de Dupont, L., Balascáková, M., Hjelte, L., Lebecque, P., Radojkovic, D., Castellani, C., Schwartz, M., Stuhrmann, M., Schwarz, M., Skalicka, V., de Monestrol, I., Girodon, E., Férec, C., Claustres, M., Tümmler, B., Cassiman, J. J., Korbmacher, C., and Cuppens, H. (2009). Mutations in the amiloride-sensitive epithelial sodium channel in patients with cystic fibrosis-like disease. Hum. Mutat. 30, 1093–1103.
Bhalla, V., and Hallows, K. R. (2008). Mechanisms of ENaC regulation and clinical implications. J. Am. Soc. Nephrol. 19, 1845–1854.
Cabral, P. D., and Garvin, J. L. (2011). Luminal flow regulates NO and O2(-) along the nephron. Am. J. Physiol. Renal Physiol. 300, F1047–F1053.
Cai, H., Wu, L., Qu, W., Malhotra, D., Xie, Z., Shapiro, J. I., and Liu, J. (2008). Regulation of apical NHE3 trafficking by ouabain-induced activation of the basolateral Na+-K+-ATPase receptor complex. Am. J. Physiol. Cell Physiol. 294, C555–C563.
Carattino, M. D., Edinger, R. S., Grieser, H. J., Wise, R., Neumann, D., Schlattner, U., Johnson, J. P., Kleyman, T. R., and Hallows, K. R. (2005). Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J. Biol. Chem. 280, 17608–17616.
Chang, S. S., Grunder, S., Hanukoglu, A., Rösler, A., Mathew, P. M., Hanukoglu, I., Schild, L., Lu, Y., Shimkets, R. A., Nelson-Williams, C., Rossier, B. C., and Lifton, R. P. (1996). Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat. Genet. 12, 248–253.
Coon, S., Kekuda, R., Saha, P., Talukder, J. R., and Sundaram, U. (2008). Constitutive nitric oxide differentially regulates Na-H and Na-glucose cotransport in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1369–G1375.
Coon, S., Shao, G., Wisel, S., Vulaupalli, R., and Sundaram, U. (2007). Mechanism of regulation of rabbit intestinal villus cell brush border membrane Na/H exchange by nitric oxide. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G475–G481.
Cordasco, E. M., and Stone, F. D. (1973). Pulmonary edema of environmental origin. Chest 64, 182–185.
Csongradi, E., Juncos, L. A., Drummond, H. A., Vera, T., and Stec, D. E. (2012). Role of carbon monoxide in kidney function: is a little carbon monoxide good for the kidney? Curr. Pharm. Biotechnol. PMID: 22201605. [Epub ahead of print].
Di Pascoli, M., Zampieri, F., Quarta, S., Sacerdoti, D., Merkel, C., Gatta, A., and Bolognesi, M. (2011). Heme oxygenase regulates renal arterial resistance and sodium excretion in cirrhotic rats. J. Hepatol. 54, 258–264.
Ding, J. W., Dickie, J., O’Brodovich, H., Shintani, Y., Rafii, B., Hackam, D., Marunaka, Y., and Rotstein, O. D. (1998). Inhibition of amiloride-sensitive sodium-channel activity in distal lung epithelial cells by nitric oxide. Am. J. Physiol. 274, L378–L387.
Eitle, E., Hiranyachattada, S., Wang, H., and Harris, P. J. (1998). Inhibition of proximal tubular fluid absorption by nitric oxide and atrial natriuretic peptide in rat kidney. Am. J. Physiol. 274, C1075–C1080.
Elmer, H. L., Brady, K. G., Drumm, M. L., and Kelley, T. J. (1999). Nitric oxide-mediated regulation of transepithelial sodium and chloride transport in murine nasal epithelium. Am. J. Physiol. 276, L466–L473.
Ermert, M., Ruppert, C., Günther, A., Duncker, H.-R., Seeger, W., and Ermert, L. (2002). Cell-specific nitric oxide synthase-isoenzyme expression and regulation in response to endotoxin in intact rat lungs. Lab. Invest. 82, 425–441.
Garvin, J. L., Herrera, M., and Ortiz, P. A. (2011). Regulation of renal NaCl transport by nitric oxide, endothelin, and ATP: clinical implications. Annu. Rev. Physiol. 73, 359–376.
Garvin, J. L., and Hong, N. J. (1999). Nitric oxide inhibits sodium/hydrogen exchange activity in the thick ascending limb. Am. J. Physiol. 277, F377–F382.
Gill, R. K., Saksena, S., Syed, I. A., Tyagi, S., Alrefai, W. A., Malakooti, J., Ramaswamy, K., and Dudeja, P. K. (2002). Regulation of NHE3 by nitric oxide in Caco-2 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 283, G747–G756.
Grandes, S., Gallego, M. J., Riesco, A., López Farré, A., Millas, I., Casado, S., Hernando, L., and Caramelo, C. (1991). Mechanisms of renal effects of different agents stimulating production of cGMP. Am. J. Physiol. 261, H1109–H1114.
Greenwood, I. A., Yeung, S. Y. M., Hettiarachi, S., Andersson, M., and Baines, D. L. (2009). KCNQ-encoded channels regulate Na+ transport across H441 lung epithelial cells. Pflugers Arch. 457, 785–794.
Guo, Y., DuVall, M. D., Crow, J. P., and Matalon, S. (1998). Nitric oxide inhibits Na+ absorption across cultured alveolar type II monolayers. Am. J. Physiol. 274, L369–L377.
Hardiman, K. M., Lindsey, J. R., and Matalon, S. (2001). Lack of amiloride-sensitive transport across alveolar and respiratory epithelium of iNOS(-/-) mice in vivo. Am. J. Physiol. Lung Cell Mol. Physiol. 281, L722–L731.
Hardiman, K. M., McNicholas-Bevensee, C. M., Fortenberry, J., Myles, C. T., Malik, B., Eaton, D. C., and Matalon, S. (2004). Regulation of amiloride-sensitive Na(+) transport by basal nitric oxide. Am. J. Respir. Cell Mol. Biol. 30, 720–728.
Helms, M. N., Jain, L., Self, J. L., and Eaton, D. C. (2008). Redox regulation of epithelial sodium channels examined in alveolar type 1 and 2 cells patch-clamped in lung slice tissue. J. Biol. Chem. 283, 22875–22883.
Hennig, B., and Diener, M. (2009). Actions of hydrogen sulphide on ion transport across rat distal colon. Br. J. Pharmacol. 158, 1263–1275.
Hess, D. T., Matsumoto, A., Kim, S.-O., Marshall, H. E., and Stamler, J. S. (2005). Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166.
Hollenhorst, M. I., Richter, K., and Fronius, M. (2011). Ion transport by pulmonary epithelia. J. Biomed. Biotechnol. 2011, 174306.
Hummler, E., Barker, P., Gatzy, J., Beermann, F., Verdumo, C., Schmidt, A., Boucher, R., and Rossier, B. C. (1996). Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat. Genet. 12, 325–328.
Ignarro, L. J., Buga, G. M., Wood, K. S., Byrns, R. E., and Chaudhuri, G. (1987). Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 84, 9265–9269.
Izzo, A. A., Mascolo, N., and Capasso, F. (1998). Nitric oxide as a modulator of intestinal water and electrolyte transport. Dig. Dis. Sci. 43, 1605–1620.
Jackson, K. E., Jackson, D. W., Quadri, S., Reitzell, M. J., and Navar, L. G. (2011). Inhibition of heme oxygenase augments tubular sodium reabsorption. Am. J. Physiol. Renal Physiol. 300, F941–F946.
Jain, L., Chen, X. J., Brown, L. A., and Eaton, D. C. (1998). Nitric oxide inhibits lung sodium transport through a cGMP-mediated inhibition of epithelial cation channels. Am. J. Physiol. 274, L475–L484.
Jain, L., Chen, X. J., Ramosevac, S., Brown, L. A., and Eaton, D. C. (2001). Expression of highly selective sodium channels in alveolar type II cells is determined by culture conditions. Am. J. Physiol. Lung Cell Mol. Physiol. 280, L646–L658.
Jin, X.-H., McGrath, H. E., Gildea, J. J., Siragy, H. M., Felder, R. A., and Carey, R. M. (2004). Renal interstitial guanosine cyclic 3′, 5′-monophosphate mediates pressure-natriuresis via protein kinase G. Hypertension 43, 1133–1139.
Kaestle, S. M., Reich, C. A., Yin, N., Habazettl, H., Weimann, J., and Kuebler, W. M. (2007). Nitric oxide-dependent inhibition of alveolar fluid clearance in hydrostatic lung edema. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L859–L869.
Kelley, T. J., and Drumm, M. L. (1998). Inducible nitric oxide synthase expression is reduced in cystic fibrosis murine and human airway epithelial cells. J. Clin. Invest. 102, 1200–1207.
Khatri, S. B., Ozkan, M., McCarthy, K., Laskowski, D., Hammel, J., Dweik, R. A., and Erzurum, S. C. (2001). Alterations in exhaled gas profile during allergen-induced asthmatic response. Am. J. Respir. Crit. Care Med. 164, 1844–1848.
Koefoed-Johnsen, V., and Ussing, H. H. (1958). The nature of the frog skin potential. Acta Physiol. Scand. 42, 298–308.
Kopkan, L., Khan, M. A. H., Lis, A., Awayda, M. S., and Majid, D. S. A. (2009). Cholesterol induces renal vasoconstriction and anti-natriuresis by inhibiting nitric oxide production in anesthetized rats. Am. J. Physiol. Renal Physiol. 297, F1606–F1613.
Lahera, V., Navarro, J., Biondi, M. L., Ruilope, L. M., and Romero, J. C. (1993). Exogenous cGMP prevents decrease in diuresis and natriuresis induced by inhibition of NO synthesis. Am. J. Physiol. 264, F344–F347.
Lahera, V., Salom, M. G., Miranda-Guardiola, F., Moncada, S., and Romero, J. C. (1991). Effects of NG-nitro-L-arginine methyl ester on renal function and blood pressure. Am. J. Physiol. 261, F1033–F1037.
Lee, H. J., Mariappan, M. M., Feliers, D., Cavaglieri, R. C., Sataranatarajan, K., Abboud, H. E., Ghosh Choudhury, G., and Kasinath, B. S. (2012). Hydrogen sulfide inhibits high glucose-induced matrix protein synthesis by activating AMP-activated protein kinase in renal epithelial cells. J. Biol. Chem. 287, 4451–4461.
Liang, M., and Knox, F. G. (1999). Nitric oxide reduces the molecular activity of Na+,K+-ATPase in opossum kidney cells. Kidney Int. 56, 627–634.
Linas, S. L., and Repine, J. E. (1999). Endothelial cells regulate proximal tubule epithelial cell sodium transport. Kidney Int. 55, 1251–1258.
Liu, J., Kesiry, R., Periyasamy, S. M., Malhotra, D., Xie, Z., and Shapiro, J. I. (2004). Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int. 66, 227–241.
Liu, J., Liang, M., Liu, L., Malhotra, D., Xie, Z., and Shapiro, J. I. (2005). Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 67, 1844–1854.
Lu, M., Giebisch, G., and Wang, W. (1997). Nitric oxide-induced hyperpolarization stimulates low-conductance Na+ channel of rat CCD. Am. J. Physiol. 272, F498–F504.
Lührs, H., Papadopoulos, T., Schmidt, H. H., and Menzel, T. (2002). Type I nitric oxide synthase in the human lung is predominantly expressed in capillary endothelial cells. Respir. Physiol. 129, 367–374.
Ma, X., Sayed, N., Beuve, A., and van den Akker, F. (2007). NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 26, 578–588.
Mace, O. J., Woollhead, A. M., and Baines, D. L. (2008). AICAR activates AMPK and alters PIP2 association with the epithelial sodium channel ENaC to inhibit Na+ transport in H441 lung epithelial cells. J. Physiol. (Lond.) 586, 4541–4557.
Madden, J. A., Ahlf, S. B., Dantuma, M. W., Olson, K. R., and Roerig, D. L. (2012). Precursors and inhibitors of hydrogen sulfide synthesis affect acute hypoxic pulmonary vasoconstriction in the intact lung. J. Appl. Physiol. 112, 411–418.
Maestrelli, P., El Messlemani, A. H., Fina, O., de Nowicki, Y., Saetta, M., Mapp, C., and Fabbri, L. M. (2001). Increased expression of heme oxygenase (HO)-1 in alveolar spaces and HO-2 in alveolar walls of smokers. Am. J. Respir. Crit. Care Med. 164, 1508–1513.
Maines, M. D., Trakshel, G. M., and Kutty, R. K. (1986). Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 261, 411–419.
Majid, D. S., Omoro, S. A., Chin, S. Y., and Navar, L. G. (1998). Intrarenal nitric oxide activity and pressure natriuresis in anesthetized dogs. Hypertension 32, 266–272.
Mall, M., Grubb, B. R., Harkema, J. R., O’Neal, W. K., and Boucher, R. C. (2004). Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 10, 487–493.
Moeller, A., Horak, F., Lane, C., Knight, D., Kicic, A., Brennan, S., Franklin, P., Terpolilli, J., Wildhaber, J. H., and Stick, S. M. (2006). Inducible NO synthase expression is low in airway epithelium from young children with cystic fibrosis. Thorax 61, 514–520.
Motterlini, R., and Otterbein, L. E. (2010). The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 9, 728–743.
Nascimento, N. R. F., Kemp, B. A., Howell, N. L., Gildea, J. J., Santos, C. F., Harris, T. E., and Carey, R. M. (2011). Role of SRC family kinase in extracellular renal cyclic guanosine 3′,5′-monophosphate- and pressure-induced natriuresis. Hypertension 58, 107–113.
Nathanson, J. A., Scavone, C., Scanlon, C., and McKee, M. (1995). The cellular Na+ pump as a site of action for carbon monoxide and glutamate: a mechanism for long-term modulation of cellular activity. Neuron 14, 781–794.
Nielsen, V. G., Baird, M. S., Chen, L., and Matalon, S. (2000). DETANONOate, a nitric oxide donor, decreases amiloride-sensitive alveolar fluid clearance in rabbits. Am. J. Respir. Crit. Care Med. 161, 1154–1160.
Olson, K. R. (2011). The therapeutic potential of hydrogen sulfide: separating hype from hope. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R297–R312.
Olson, K. R., Whitfield, N. L., Bearden, S. E., St Leger, J., Nilson, E., Gao, Y., and Madden, J. A. (2010). Hypoxic pulmonary vasodilation: a paradigm shift with a hydrogen sulfide mechanism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R51–R60.
Olver, R. E., Ramsden, C. A., Strang, L. B., and Walters, D. V. (1986). The role of amiloride-blockable sodium transport in adrenaline-induced lung liquid reabsorption in the fetal lamb. J. Physiol. (Lond.) 376, 321–340.
Ortiz, P. A., and Garvin, J. L. (2001). NO Inhibits NaCl absorption by rat thick ascending limb through activation of cGMP-stimulated phosphodiesterase. Hypertension 37, 467–471.
Ortiz, P. A., and Garvin, J. L. (2002). Role of nitric oxide in the regulation of nephron transport. Am. J. Physiol. Renal Physiol. 282, F777–F784.
Ortiz, P. A., Hong, N. J., and Garvin, J. L. (2001). NO decreases thick ascending limb chloride absorption by reducing Na(+)-K(+)-2Cl(-) cotransporter activity. Am. J. Physiol. Renal Physiol. 281, F819–F825.
Ousingsawat, J., Mirza, M., Tian, Y., Roussa, E., Schreiber, R., Cook, D. I., and Kunzelmann, K. (2011). Rotavirus toxin NSP4 induces diarrhea by activation of TMEM16A and inhibition of Na+ absorption. Pflugers Arch. 461, 579–589.
Oweis, S., Wu, L., Kiela, P. R., Zhao, H., Malhotra, D., Ghishan, F. K., Xie, Z., Shapiro, J. I., and Liu, J. (2006). Cardiac glycoside downregulates NHE3 activity and expression in LLC-PK1 cells. Am. J. Physiol. Renal Physiol. 290, F997–F1008.
Perez-Rojas, J. M., Kassem, K. M., Beierwaltes, W. H., Garvin, J. L., and Herrera, M. (2010). Nitric oxide produced by endothelial nitric oxide synthase promotes diuresis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1050–R1055.
Pittet, J. F., Lu, L. N., Morris, D. G., Modelska, K., Welch, W. J., Carey, H. V., Roux, J., and Matthay, M. A. (2001). Reactive nitrogen species inhibit alveolar epithelial fluid transport after hemorrhagic shock in rats. J. Immunol. 166, 6301–6310.
Plato, C. F., Shesely, E. G., and Garvin, J. L. (2000). eNOS mediates L-arginine-induced inhibition of thick ascending limb chloride flux. Hypertension 35, 319–323.
Pouokam, E., and Diener, M. (2011). Mechanisms of actions of hydrogen sulphide on rat distal colonic epithelium. Br. J. Pharmacol. 162, 392–404.
Pouokam, E., Steidle, J., and Diener, M. (2011). Regulation of colonic ion transport by gasotransmitters. Biol. Pharm. Bull. 34, 789–793.
Rauh, R., Diakov, A., Tzschoppe, A., Korbmacher, J., Azad, A. K., Cuppens, H., Cassiman, J.-J., Dötsch, J., Sticht, H., and Korbmacher, C. (2010). A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self inhibition. J. Physiol. (Lond.) 588, 1211–1225.
Roczniak, A., and Burns, K. D. (1996). Nitric oxide stimulates guanylate cyclase and regulates sodium transport in rabbit proximal tubule. Am. J. Physiol. 270, F106–F115.
Roth, M., Rupp, M., Hofmann, S., Mittal, M., Fuchs, B., Sommer, N., Parajuli, N., Quanz, K., Schubert, D., Dony, E., Schermuly, R. T., Ghofrani, H. A., Sausbier, U., Rutschmann, K., Wilhelm, S., Seeger, W., Ruth, P., Grimminger, F., Sausbier, M., and Weissmann, N. (2009). Heme oxygenase-2 and large-conductance Ca2+-activated K+ channels: lung vascular effects of hypoxia. Am. J. Respir. Crit. Care Med. 180, 353–364.
Rückes-Nilges, C., Lindemann, H., Klimek, T., Glanz, H., and Weber, W. M. (2000). Nitric oxide has no beneficial effects on ion transport defects in cystic fibrosis human nasal epithelium. Pflugers Arch. 441, 133–137.
Ryter, S. W., Alam, J., and Choi, A. M. K. (2006). Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 86, 583–650.
Sasaki, S., Siragy, H. M., Gildea, J. J., Felder, R. A., and Carey, R. M. (2004). Production and role of extracellular guanosine cyclic 3′, 5′ monophosphate in sodium uptake in human proximal tubule cells. Hypertension 43, 286–291.
Sato, T., Kamata, Y., Irifune, M., and Nishikawa, T. (1995). Inhibition of purified (Na+,K+)-ATPase activity from porcine cerebral cortex by NO generating drugs. Brain Res. 704, 117–120.
Schicho, R., Krueger, D., Zeller, F., Weyhern, C. W. H., von Frieling, T., Kimura, H., Ishii, I., Giorgio, R., de Campi, B., and Schemann, M. (2006). Hydrogen sulfide is a novel prosecretory neuromodulator in the Guinea-pig and human colon. Gastroenterology 131, 1542–1552.
Schild, L., Canessa, C. M., Shimkets, R. A., Gautschi, I., Lifton, R. P., and Rossier, B. C. (1995). A mutation in the epithelial sodium channel causing Liddle disease increases channel activity in the Xenopus laevis oocyte expression system. Proc. Natl. Acad. Sci. U.S.A. 92, 5699–5703.
Shimkets, R. A., Warnock, D. G., Bositis, C. M., Nelson-Williams, C., Hansson, J. H., Schambelan, M., Gill, J. R., Ulick, S., Milora, R. V., and Findling, J. W. (1994). Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 79, 407–414.
Song, W., Liu, G., Bosworth, C. A., Walker, J. R., Megaw, G. A., Lazrak, A., Abraham, E., Sullender, W. M., and Matalon, S. (2009). Respiratory syncytial virus inhibits lung epithelial Na+ channels by up-regulating inducible nitric-oxide synthase. J. Biol. Chem. 284, 7294–7306.
Steidle, J., and Diener, M. (2011). Effects of carbon monoxide on ion transport across rat distal colon. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G207–G216.
Stipanuk, M. H., and Beck, P. W. (1982). Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 206, 267–277.
Stoos, B. A., Carretero, O. A., and Garvin, J. L. (1994). Endothelial-derived nitric oxide inhibits sodium transport by affecting apical membrane channels in cultured collecting duct cells. J. Am. Soc. Nephrol. 4, 1855–1860.
Stoos, B. A., Garcia, N. H., and Garvin, J. L. (1995). Nitric oxide inhibits sodium reabsorption in the isolated perfused cortical collecting duct. J. Am. Soc. Nephrol. 6, 89–94.
Suzuki, Y., Lu, Q., Xu, D.-Z., Szabó, C., Haskó, G., and Deitch, E. A. (2005). Na+,K+-ATPase activity is inhibited in cultured intestinal epithelial cells by endotoxin or nitric oxide. Int. J. Mol. Med. 15, 871–877.
Tang, G., Wu, L., and Wang, R. (2010). Interaction of hydrogen sulfide with ion channels. Clin. Exp. Pharmacol. Physiol. 37, 753–763.
Telezhkin, V., Brazier, S. P., Cayzac, S., Müller, C. T., Riccardi, D., and Kemp, P. J. (2009). Hydrogen sulfide inhibits human BK(Ca) channels. Adv. Exp. Med. Biol. 648, 65–72.
Texereau, J., Fajac, I., Hubert, D., Coste, J., Dusser, D. J., Bienvenu, T., Dall’Ava-Santucci, J., and Dinh-Xuan, A. T. (2005). Reduced exhaled NO is related to impaired nasal potential difference in patients with cystic fibrosis. Vascul. Pharmacol. 43, 385–389.
Tsubochi, H., Suzuki, S., Kubo, H., Ueno, T., Yoshimura, T., Suzuki, T., Sasano, H., and Kondo, T. (2003). Early changes in alveolar fluid clearance by nitric oxide after endotoxin instillation in rats. Am. J. Respir. Crit. Care Med. 167, 205–210.
Wang, R. (2002). Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 16, 1792–1798.
Wang, S., Publicover, S., and Gu, Y. (2009). An oxygen-sensitive mechanism in regulation of epithelial sodium channel. Proc. Natl. Acad. Sci. U.S.A. 106, 2957–2962.
Wang, T., Sterling, H., Shao, W. A., Yan, Q., Bailey, M. A., Giebisch, G., and Wang, W.-H. (2003). Inhibition of heme oxygenase decreases sodium and fluid absorption in the loop of Henle. Am. J. Physiol. Renal Physiol. 285, F484–F490.
Wilkinson, W. J., and Kemp, P. J. (2011). Carbon monoxide: an emerging regulator of ion channels. J. Physiol. (Lond.) 589, 3055–3062.
Woollhead, A. M., Sivagnanasundaram, J., Kalsi, K. K., Pucovsky, V., Pellatt, L. J., Scott, J. W., Mustard, K. J., Hardie, D. G., and Baines, D. L. (2007). Pharmacological activators of AMP-activated protein kinase have different effects on Na+ transport processes across human lung epithelial cells. Br. J. Pharmacol. 151, 1204–1215.
Wu, X. C., Harris, P. J., and Johns, E. J. (1999). Nitric oxide and renal nerve-mediated proximal tubular reabsorption in normotensive and hypertensive rats. Am. J. Physiol. 277, F560–F566.
Xu, Z., Prathapasinghe, G., Wu, N., Hwang, S.-Y., Siow, Y. L., and O, K. (2009). Ischemia-reperfusion reduces cystathionine-beta-synthase-mediated hydrogen sulfide generation in the kidney. Am. J. Physiol. Renal Physiol. 297, F27–F35.
Yang, G., Wu, L., Jiang, B., Yang, W., Qi, J., Cao, K., Meng, Q., Mustafa, A. K., Mu, W., Zhang, S., Snyder, S. H., and Wang, R. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322, 587–590.
Yu, L., Bao, H. F., Sellf, J. L., Eaton, D. C., and Helmy, M. (2007). Aldosterone-induced increases in superoxide production counters nitric oxide inhibition of epithelial Na channel activity in A6 distal nephron cells. Am. J. Physiol. Renal Physiol. 293, F1666–F1677.
Keywords: Na+ absorption, electrolyte transport, NO, CO, H2S, ENaC, transporter, Na+/K+-ATPase
Citation: Althaus M (2012) Gasotransmitters: novel regulators of epithelial Na+ transport? Front. Physio. 3:83. doi: 10.3389/fphys.2012.00083
Received: 22 February 2012; Paper pending published: 04 March 2012;
Accepted: 20 March 2012; Published online: 09 April 2012.
Edited by:
Wolfgang G. Clauss, Justus Liebig University Giessen, GermanyReviewed by:
Martin Diener, University Giessen, GermanyDeborah Baines, St George’s, University of London, UK
Ritva Tikkanen, Justus Liebig University of Giessen, Germany
Copyright: © 2012 Althaus. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Mike Althaus, Institute of Animal Physiology, Justus Liebig University of Giessen, Heinrich-Buff-Ring 26, 35392 Giessen, Germany. e-mail: mike.althaus@bio.uni-giessen.de