
- Division of Gastroenterology, Department of Medicine, University of California San Diego, School of Medicine, La Jolla, CA, USA
Toll-like receptors (TLRs) are pattern recognition receptors that distinguish conserved microbial products, also known as pathogen-associated molecular patterns (PAMPs), from host molecules. Liver is the first filter organ between the gastrointestinal tracts and the rest of the body through portal circulation. Thus, the liver is a major organ that must deal with PAMPs and microorganisms translocated from the intestine and to respond to the damage associated molecular patterns (DAMPs) released from injured organs. These PAMPs and DAMPs preferentially activate TLR signaling on various cell types in the liver inducing the production of inflammatory and fibrogenic cytokines that initiate and prolong liver inflammation, thereby leading to fibrosis. We summarize recent findings on the role of TLRs, ligands, and intracellular signaling in the pathophysiology of liver fibrosis due to different etiology, as well as to highlight the potential role of TLR signaling in liver fibrosis associated with hepatitis C infection, non-alcoholic and alcoholic steatoheoatitis, primary biliary cirrhosis, and cystic fibrosis.
Introduction
Hepatic fibrosis is a wound healing response that occurs following chronic liver injuries induced by various causes, such as viral infection, toxin, metabolic disorders, alcohol abuse, cholestasis, and autoimmunity. Cirrhosis, the end stage of liver fibrosis, often causes life-threatening complications, such as portal hypertension, liver failure, and hepatocellular carcinoma (HCC; Bataller and Brenner, 2005; Friedman, 2010).
The following mechanisms are proposed to be critical for pathogenesis of liver fibrosis: (1) acute and chronic hepatocyte damage; (2) recruitment and activation of inflammatory cells including macrophages; (3) release of inflammatory and fibrogenic cytokines; (4) production of reactive oxygen species; (5) activation of hepatic stellate cells (HSCs) and portal fibroblasts that transdifferentiate into collagen producing myofibroblats (Friedman, 2010). Amongst these events, the hepatic innate immune system plays a critical role and is activated in both acute and chronic liver diseases (Seki and Brenner, 2008; Seki and Schnabl, 2012). The healthy intestine prevents the translocation of intestine-derived microbial products by intact barrier system. The small amounts of intestine-derived bacterial products may reach the liver through portal vein. However, liver inflammation usually does not occur in normal healthy liver (Crispe, 2009). When intestinal barriers is disrupted, a large amount of bacteria translocate to the liver, resulting in the activation of hepatic innate immune system. Intestinal microbial products, such as lipopolysaccharide (LPS) and CpG-containing bacterial DNA contain signature motifs called pathogen-associated molecular patterns (PAMP; Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). Toll-like receptors (TLRs) are germline-encoded pattern recognition receptors that are important in the innate immune system involved in initial step of host defense against microorganisms. TLRs are widely expressed on immune cells and non-immune cells to recognize a broad array of microorganism products (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). Accumulated lines of evidence indicate that TLRs are also activated by endogenous ligands, such as high mobility group box 1 (HMGB1), hyaluronan, and heat shock proteins released from damaged tissues, termed damaged associated molecular patterns (DAMPs; Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). In addition, innate immune response contributes not only to liver inflammation, but also to physiological and pathological repair processes including liver fibrosis, regeneration, and carcinogenesis (Seki and Brenner, 2008; Guo and Friedman, 2010; Seki and Schnabl, 2012). This review focuses on the current knowledge of TLRs, their ligands, intracellular signaling and cells expressing TLRs in liver fibrosis and its associated diseases, such as viral hepatitis, non-alcoholic and alcoholic steatohepatitis, primary biliary cirrhosis, and cystic fibrosis (CF).
TLRs and Their Ligands
TLRs are structurally characterized by the presence of a leucine-rich repeat domain in their extracellular domain and a Toll/interleukin (IL)-1 receptor (TIR) domain in their intracellular domain (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). Currently, 10 1nd 12 functional TLRs have been identified in humans and mice, respectively. While TLR1-TLR9 are conserved in both species, mouse TLR10 is not functional due to a retrovirus insertion, and TLR11, TLR12 and TLR13 do not exist in the human genome. TLR1,TLR2, TLR4, TLR5, and TLR6 are expressed on the cell surface whereas intracellular TLRs, TLR3, TLR7/8, TLR9, and TLR11 are located in the endoplasmic reticulum (ER), endosomes, lysosomes, and endolysosomes (Figure 1; Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010).
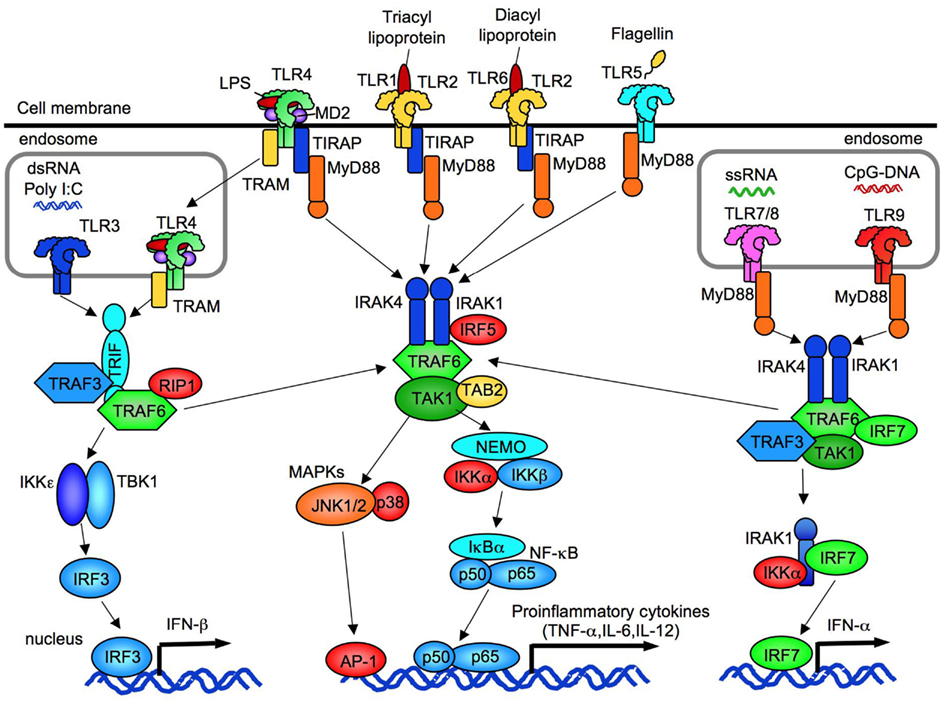
Figure 1. The overview of TLR signaling. TLR1, TLR2, TLR4, TLR5, and TLR6 are expressed on cell membrane. TLR3, TLR7/8, and TLR9 are expressed in endosome. All TLRs, expect for TLR3, activate MyD88-dependent pathway to induce NF-κB and p38/JNK activation. TLR2 and TLR4 signaling require TIRAP and MyD88. TLR3 requires IRIF to activate TBK1/IKKε. Subsequent to TLR4 internalization, TLR4 signaling activates TRAM/TRIF-dependent pathway. TLR3/4-dependent TRIF-dependent signaling induces IRF3 activation and IFN-β production. TLR7/8 and TLR9 induce IFN-α production through IRF7.
The TLR ligands are generally categorized into three types: proteins, nucleic acids, and lipid-based elements. TLR4 recognizes LPS (also known as endotoxin), a Gram-negative bacterial cell wall component. The recognition process is enhanced by LPS-binding protein (LBP) that carries LPS to the CD14 molecule, where it then binds to the MD2–TLR4 complex (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). This complex forms a symmetrical TLR4 homodimer. TLR2 can form a heterodimer with either TLR1 to sense bacterial triacylated lipopeptides and the synthetic ligand Pam3CSK4, or with TLR6 to recognize bacterial diacylated lipopeptides like MALP-2 (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). Because of the sequence similarity with TLR1 or TLR6, TLR10 senses triacylated lipopeptides by heterodimerization with TLR2 (Guan et al., 2010). In addition, the scavenger receptor CD36 enhances recognition capability of TLR2 to their ligands (Yamamoto and Takeda, 2010). Thus, the dimerization of these TLRs increases the specificity and sensitivity of the recognition for a variety of microbial components. TLR5, the receptor for bacterial flagellin, is expressed on dendritic cells (DCs) on the lamina propria to maintain homeostasis of intestinal immunity (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). TLR3 senses synthetic ligand polyriboinosinic polyribocytidylic acid (poly I:C) and double-stranded RNA produced during viral replication in the endolysosome (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). Mouse TLR7 and human TLR7/8 recognize viral single stranded RNA. Synthetic ligands, such as R-848 and imiquimod, can also activate TLR7 and TLR8 signaling pathways (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). TLR9 acts as a receptor for unmethylated CpG islands in bacterial and viral DNA. Synthetic oligonucleotides that contain unmethylated CpG motifs also activate TLR9 (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). The ligands for TLR12 and TLR13 remain unidentified. TLRs also function as sensors for “danger signals.” The endogenous molecules released from damaged cells activate innate immune signaling through TLRs. TLR4 recognizes endogenous ligands, such as HMGB1, hyaluronan, heat shock protein 60, and free fatty acids (C12:0, C14:0, C16:0, and C18:0; Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010).
TLR Adaptor Molecules and Their Dependent Signaling Pathways
Currently, five distinct adaptor molecules [Myeloid differentiation factor 88 (MyD88), TIR domain-containing adaptor-inducing IFN-β (TRIF), TIR domain-containing adaptor protein (TIRAP), TRIF-related adaptor molecule (TRAM), and SARM] have been identified. The interaction between cytoplasmic TIR domain of the TLRs and TIR domain of adaptors are required for the signal-transduction of TLR signaling (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010).
MyD88-Dependent Pathway
MyD88 is a common adaptor molecule that transmits the signals from all members of TLR (except for TLR3) and members of IL-1 receptor family. The signals culminate in the activation of nuclear factor (NF)-κB, MAP kinases and IRF5, and the induction of inflammatory cytokines, such as TNF-α, IL-1β, and IL-6 (Figure 1; Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). MyD88 couples directly to the activated receptor dimers, except in the case of TLR4 and TLR2, which indirectly link with MyD88 via the bridging adaptor TIRAP. MyD88 recruits IL-1 receptor associated kinase (IRAK)-4 and binds through interaction of their death domains. Then, IRAK4 activates IRAK1 and IRAK2. The activated IRAKs dissociate from MyD88, resulting in an interaction with TRAF6, an E3 ubiquitin ligase. TRAF6 and E2 ubiquitin-conjugating enzyme Ubc13 and Uev1A catalyze the synthesis of polyubiquitin chain linked to Lys63 (K63) on TRAF6 itself and IRAK1, which activates a complex of TGF-β activated kinase 1 (TAK1), TAK1-binding protein 2 (TAB2), and TAB3 (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). Subsequently, activated TAK1 complex activates the MAPKs ERK1/2, p38, and JNK, leading to activation of AP-1 (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). Simultaneously, TAK1 activates the IKK complex comprised of NEMO (IKKΓ), IKKα, and IKKβ, which phosphorylates IκBα. The phosphorylated and ubiquitinated IκBα dissociates from NF-κB, resulting in the nuclear translocation of NF-κB; NF-κB binds to the corresponding NF-κB binding elements to induce transcription of inflammatory cytokines, such as IL-6, IL-12p40, and TNF (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). In plasmacytoid DCs, TLR7, and TLR9 signaling forms a complex composed of MyD88, IRAK1, TRAF6, TRAF3, IKK-α, and IRF7, resulting in activation and nuclear translocation of IRF7 to induce type I interferons (Figure 1; Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010).
TRIF-Dependent Pathway
TLR3 and TLR4 utilize TRIF to induce MyD88-independent pathways (Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010). Importantly, CD14-mediated TLR4 internalization and the bridge by TRAM are required for the activation of TLR4-dependent TRIF pathway (Kagan et al., 2008; Zanoni et al., 2011). Subsequently, TRIF binds to TRAF3 and TRAF6. TRIF–TRAF3 transmits the signals to activate IKKε, TBK1, and the transcription factor IRF3, which induces type I interferon production (Figure 1; Hacker et al., 2006). Alternatively, TRIF recruits the adaptor RIP1 to form a multiprotein signaling complex along with TRAF6, TRADD, RIP1, and RIP3 for the activation of TAK1, which in turn activates the NF-κB and MAPK pathways (Hacker et al., 2006). The secretion of IL-1β and IL-18 require a two-step mechanism: the production of proform of IL-1β and IL-18 in cytoplasm, and subsequent maturation by caspase-1 in inflammasome. In Kupffer cells, the proform of IL-1β and IL-18 is produced through the TLR4-MyD88-dependent pathway (Imamura et al., 2009), and the TLR4–TRIF-dependent pathway activates caspase-1 and processes the proform of IL-1β and IL-18 into the matured molecules (Seki et al., 2001; Imamura et al., 2009; Tsutsui et al., 2010).
Negative Regulation of TLR Signaling
TLR signaling is controlled by a number of negative regulators to prevent harmful tissue injury induced by excessive immune responses. These negative regulators include splicing variants for adaptors or their related proteins, ubiquitin ligases, deubiquitinases, transcriptional regulators, and microRNAs (miRNAs), which suppress TLR signaling at distinct levels (Liew et al., 2005; Coll and O’Neill, 2010; Kawai and Akira, 2010; Takeuchi and Akira, 2010; Yamamoto and Takeda, 2010; O’Neill et al., 2011; Figure 2). ST2 (known as IL-33 receptor), single immunoglobulin IL-1R-related molecule (SIGGR/TIR8), and RP105 (a homolog of TLR4, essential for LPS recognition with MD-1 in B cells) negatively regulate TLR signaling at their receptor levels (Liew et al., 2005). A soluble form of TLR9 generated by the cleavage of TLR9 between amino acids 724–735 inhibits TLR9-dependent signaling (Chockalingam et al., 2011). An adaptor molecule, Toll/IL-1R resistance motif containing protein (SARM) localized in mitochondria, inhibits both MyD88 and TRIF-dependent pathways (Belinda et al., 2008). In addition, phosphatase PTP1B can also negatively regulate both MyD88- and TRIF-dependent signaling (Xu et al., 2008a). An isoform of IRAKs, IRAK-M, hampers IRAK activity (Kobayashi et al., 2002). Likewise, a suppressor of cytokine signaling-1 (SOCS-1) can suppress IRAK activity as well as degrade tyrosine-phosphorylated TIRAP to inhibit TLR4 signaling (Nakagawa et al., 2002). SHP-1, a tyrosine phosphatase, inhibits the function of both IRAK1 and IRAK2 (An et al., 2008). A20 acts as an E3 ubiquitin ligase and a deubiquitinase, which prevents NF-κB activation by interfering RIP1 and TRAF6 interaction (Turer et al., 2008). The activation of transcription factor 3 (ATF3) negatively regulates TLR-induced IL-6 and IL-12p40 production (Gilchrist et al., 2006). Zc3h12a, an RNA binding protein with a CCCH-type zinc finger motif, inhibits TLR-mediated inflammatory responses by affecting mRNA stability (Matsushita et al., 2009).
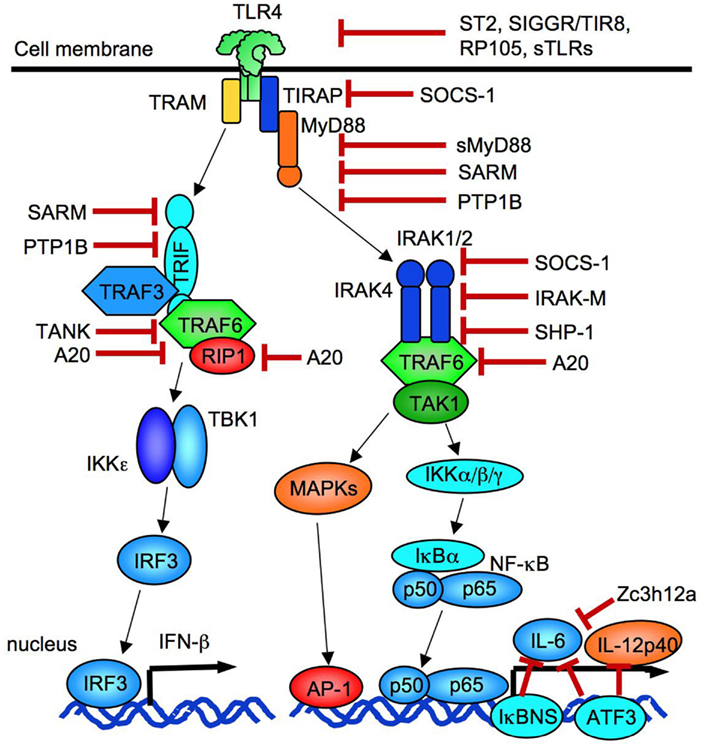
Figure 2. Negative regulators of TLR signaling. TLR signaling is suppressed at levels of receptors by ST2, SIGGR/TIR8, RP105, and soluble forms of TLRs. Adaptor molecules are inhibited by SOCS-1, sMyD88, SARM, and PTP1B. SOCS-1 and IRAK-M suppresses IRAKs. A20 blocks TRAF6 and RIP1. IL-6 transcription is negatively regulated by IκBNS, Zc3h12a, and ATF3. ATF3 also blocks IL-12p40 transcription.
MicroRNAs negatively regulates target gene expression. Some miRNAs are induced by TLR signaling and negatively regulate TLR signaling (Coll and O’Neill, 2010; O’Neill et al., 2011; Figure 3). TLR signaling induces upregulation of miR-155, miR-146, miR-132, miR-21, miR-223, miR-9, and miR-147 (O’Neill et al., 2011). The induction of miR-223 and let-7e suppresses TLR4 expression (O’Neill et al., 2011). The targets of miR-146 are IRAK1, IRAK2 and TRAF6 and IRF5 (Quinn and O’Neill, 2011) while miR-9 targets NFKB1 mRNA (O’Neill et al., 2011). The miR-155 negatively regulates TLR signaling by targeting MyD88, TAB2, IKKε, RIP1, and C/EBPβ (Quinn and O’Neill, 2011). Recent studies demonstrated that both miR-155 and miR-21 cooperatively regulate TLR4 signaling (Figure 4). While miRNA-155 acts as a negative regulator of TLR signaling as mentioned above, TLR4-induced miR-155 can also augment TLR4 signaling via suppression of Src homology 2 domain-containing inositol-5′-phosphatase 1 (SHIP1), a negative regulator of TLR4 signaling. However, in parallel, TLR4 signaling induces miR-21, which targets programmed cell death 4 (PDCD4; Sheedy et al., 2010). Because PDCD4 is a suppressor of IL-10, reduced PDCD4 levels increase IL-10, which further inhibits miR-155. Inhibited miR-155 increases SHIP1 levels to suppress TLR4 signaling (O’Neill et al., 2011; Quinn and O’Neill, 2011). Thus, TLR4 signaling is tightly regulated by miRNAs.
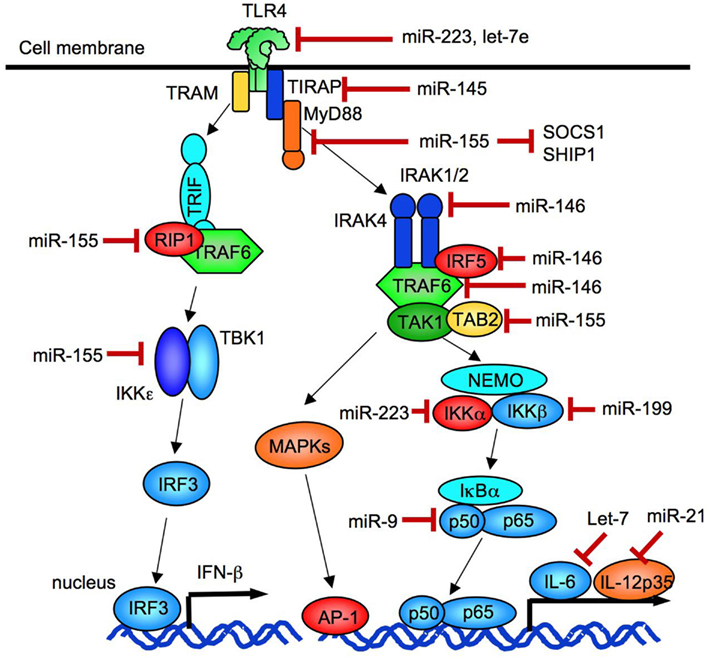
Figure 3. Negative regulation of TLR signaling by miRNA. miR-146 negatively regulates the expression of IRAK1, IRAK2, and TRAF6. miR-21 negatively regulates the expression of a IL-12 p35 subunit. miR-155 inhibits the expression of TAB2, RIP1, IKKε, SOCS-1, and TIRAP. miR-223 suppresses the expression of TLR4 and IKKα, and miR-9 inhibits the expression of an NF-κB p50 subunit.
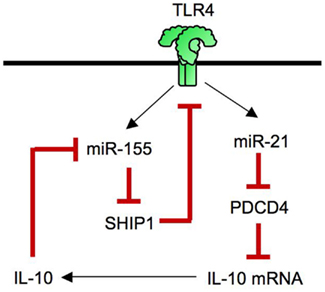
Figure 4. miR-155 and miR-21 tunes TLR4 signaling. TLR4 signaling increases the level of miR-155 that depredates SHIP1, a negative regulator of TLR4 signaling. TLR4 signaling also increases the level of miR-21, which targets PDCD4 mRNA, resulting in increased production of IL-10 as PDCD4 is an inhibitor of IL-10 translation. IL-10 further inhibits miR-155 induction, which in turn, leads to an increase in SHIP1, inhibiting TLR4 signaling.
TLR Expression in the Liver
Toll-like receptor expression in distinct liver cells is shown in Table 1.
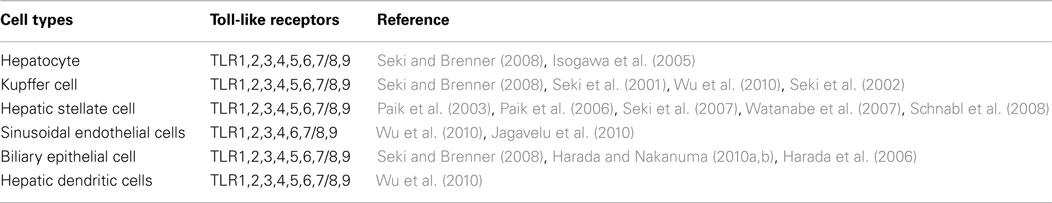
Table 1. Toll-like receptors expression in the liver cell population.
Hepatocytes
Hepatocytes account for 60–80% of the total cell population in the liver. Hepatocytes may uptake and remove endotoxin derived from the intestine through portal circulation (Seki and Brenner, 2008). Primary cultured hepatocytes express mRNA for all TLRs, but the expression levels are very low. While cultured hepatocytes respond to TLR2 and TLR4 ligands, their responses in vivo are quite weak (Isogawa et al., 2005; Seki and Brenner, 2008).
Kupffer Cells (Hepatic Resident Macrophages)
Kupffer cells are the primary cells that encounter the gut-derived toxins such as LPS, but are less responsiveness due to “LPS tolerance” that prevents overt inflammation in the physiological setting (Seki and Brenner, 2008). Kupffer cells respond to all TLR ligands to produce TNF-α, IL-1, IL-6, IL-12, IL-18, and IL-10 (Seki et al., 2001; Wu et al., 2010). IL-12 and IL-18 produced from Kupffer cells synergistically act on hepatic natural killer (NK) cells to produce anti-microbial IFN-Γ (Seki et al., 2002). Kupffer cells also express TLR2, TLR3, and TLR9 and respond to their corresponding ligands.
Hepatic Stellate Cells
HSCs reside in the space of Disse and are the principal cellular source of extracellular matrix proteins, such as collagen type I, III, and IV that are prominent in liver fibrosis (Bataller and Brenner, 2005; Friedman, 2008). HSCs express all TLRs (Paik et al., 2003, 2006; Seki et al., 2007; Watanabe et al., 2007; Wang et al., 2009). In response to LPS, HSCs express inflammatory and fibrogenic features, such as upregulation of chemokines (CCL2, CCL3, and CCL4) and adhesion molecules (VCAM-1, ICAM-1, and E-selectin) as well as downregulation of TGF-β pseudoreceptor, bone morphogenetic protein and activin membrane bound inhibitor (Bambi), to amplify TGF-β signaling (Seki et al., 2007). TLR2 expression is unregulated by LPS or TNF-α stimulation in HSCs (Paik et al., 2006; Seki et al., 2007). TLR9 signaling enhances collagen production, but inhibits the migration of HSCs (Watanabe et al., 2007). A recent study has shown that a soluble TLR4–MD2 fusion protein prevents LPS-induced NF-κB and JNK activation in human HSCs, suggesting possibility of the soluble TLR4–MD2 fusion protein as a new therapeutic tool for liver fibrosis (Schnabl et al., 2008).
Biliary Epithelial Cells
Biliary epithelial cells (BECs) build intrahepatic biliary trees and extrahepatic bile ducts, and participate in the production and transport of bile substances, as well as immune responses to protect host against pathogens through TLRs and expression of anti-microbial peptides. BECs express a variety of TLRs, and at least TLR2 and TLR4 signaling activates NF-κB through MyD88 (Seki and Brenner, 2008; Harada and Nakanuma, 2010a,b). BECs also negatively regulate TLR signaling by induction of IRAK-M to prevent overt inflammation (Harada et al., 2006).
Liver Sinusoidal Endothelial Cells
Liver sinusoidal endothelial cells (LSECs) express and respond to all TLR ligands except for TLR5 ligand. TLR3 and TLR4 ligands induce IL-6 production, and TLR3 ligand produces IFN-β (Wu et al., 2010). LSECs are primarily associated with hepatic immune tolerance to maintain hepatic immune homeostasis. More recent study demonstrated that TLR4 expressed on LSECs mediates angiogenesis that is associated with liver fibrosis (Jagavelu et al., 2010).
Hepatic Dendritic Cells
Dendritic cells are professional antigen-presenting cells that bridge between innate and acquired immunity in the liver. The hepatic plasmacytoid DCs (pDCs) express low levels of MHC class II and costimulatory molecules. Hepatic DCs express all TLRs, however, TLR5 expression is low (Wu et al., 2010). In response to ligands for TLR7 and TLR9, hepatic pDCs produce TNF-α, IL-6, IL-12, and IFN-α (Wu et al., 2010).
TLR4, TLR9, TLR3, and Bacterial Translocation in Liver Fibrosis
Bacterial Translocation and Liver Fibrosis
Previous studies demonstrated that systemic plasma levels of LPS are elevated in experimental liver fibrosis induced by carbon tetrachloride (CCl4), thioacetamide, or bile duct ligation (BDL; Nolan and Leibowitz, 1978; Grinko et al., 1995; Seki et al., 2007). Gut sterilization decreased plasma LPS levels and suppressed the development of liver fibrosis (Rakoff-Nahoum et al., 2004; Seki et al., 2007). This strongly suggests that intestine-derived bacterial components including LPS is involved in promotion of experimental liver fibrosis. The gut leaky caused by the disintegrity of intestinal tight junction, gut mucosal atrophy and edema, systemic inflammation, and oxidative stress, is involved in chronic liver injury (Assimakopoulos et al., 2007a,b). Changes in the composition of intestinal microbiota in patients with liver cirrhosis are also suggested to be associated with the promotion of bacterial translocation. Pathogenic Enterobacteriaceae and Streptococcaceae were increased and beneficial Bifidobacteria and Lachnospiraceae were decreased in feces from patients with liver cirrhosis (Chen et al., 2011; Lu et al., 2011). Animal models of liver fibrosis cause increased levels of Enterobacteriaceae and decreased Gram-positive anaerobic Clostridium groups in the intestine (Zhang et al., 2010a; Gomez-Hurtado et al., 2011). While the altered population of intestinal microbiota may have resulted from hepatic cirrhosis, enhanced bacterial translocation could further amplify liver disease through hepatic expression of TLRs.
TLR4 Signaling in Liver Fibrosis
TLR4 is expressed on both parenchymal and non-parenchymal cell types in the liver, and its signaling is involved in a variety of liver injury induced by viral hepatitis, alcoholic and non-alcoholic steatohepatitis, and cholestatic, autoimmune and drug-induced liver diseases. Several animal studies support the importance of TLR4 in liver fibrosis. Mice mutated in TLR4, CD14, LBP, MyD88, and TRIF have less liver fibrosis induced by bile duct ligation (BDL), or treatment of carbon tetrachloride (CCl4; Isayama et al., 2006; Seki et al., 2007; Jagavelu et al., 2010; Teratani et al., 2012; Zhu et al., 2012). Selective decontamination of gut flora also attenuates liver fibrosis. One study determined that TLR4 on HSCs, but not Kupffer cells and hepatocytes, is crucial for liver fibrosis (Seki et al., 2007). Through our current knowledge, the following mechanisms of TLR4 signaling in promotion of liver fibrosis have been proposed (Figure 5): (1) HSCs respond to even low concentrations of LPS, leading to the activation of TLR4 and downstream signaling (IKK/NF-κB and JNK) to secrete a number of chemokines (CCL2, CCL3, CCL4, and CCL5) and express adhesion molecules (ICAM-1, VCAM-1, and E-selectin; Paik et al., 2003; Seki et al., 2007). These molecules induce the migration of Kupffer cells and the recruitment of extrahepatic monocytes into the liver. Among chemokines, CCL2 and CCL5 were reported to activate HSC directly (Marra et al., 1999; Schwabe et al., 2003). Recent publications confirmed the in vivo roles of CCL5 and chemokine receptors CCR1, CCR2, and CCR5 in liver fibrosis using genetic knockout animals (Seki et al., 2009a,b; Berres et al., 2010). (2) TLR4 signaling mediates the crosstalk with TGF-β signaling in HSCs (Seki et al., 2007). Quiescent HSCs express high levels of Bambi, an endogenous TGF-β receptor inhibitor, to prevent TGF-β signaling. In liver fibrosis, HSC’s Bambi expression is quickly downregulated dependent on TLR4, MyD88, and NF-κB, but not dependent on TRIF (Seki et al., 2007). Bambi downregulation results in the amplification of TGF-β receptor signaling to promote HSC activation. Bambi not only acts as decoy receptor for TGF-β receptors, but also interacts with Smad7, interfering with the complex comprising of type I and type II TGF-β receptors and Smad3, thereby inhibiting TGF-β signaling (Yan et al., 2009). (3) TLR4 signaling inhibits miR-29 expression in HSCs. In human and animal models of liver fibrosis, miR-29 expression is suppressed. Decreased expression of miR-29 suppresses transcription of collagen α1(I)mRNA in HSCs (Roderburg et al., 2011). This suggests that TLR4-mediated miR-29 downregulation is one of the mechanisms by which TLR4 signaling enhances HSC activation and liver fibrosis. (4) TLR4 signaling promotes fibronectin (FN) production in HSCs. HSC-derived FN then induces the migration of LECs and angiogenesis, which promotes liver fibrosis (Zhu et al., 2012).
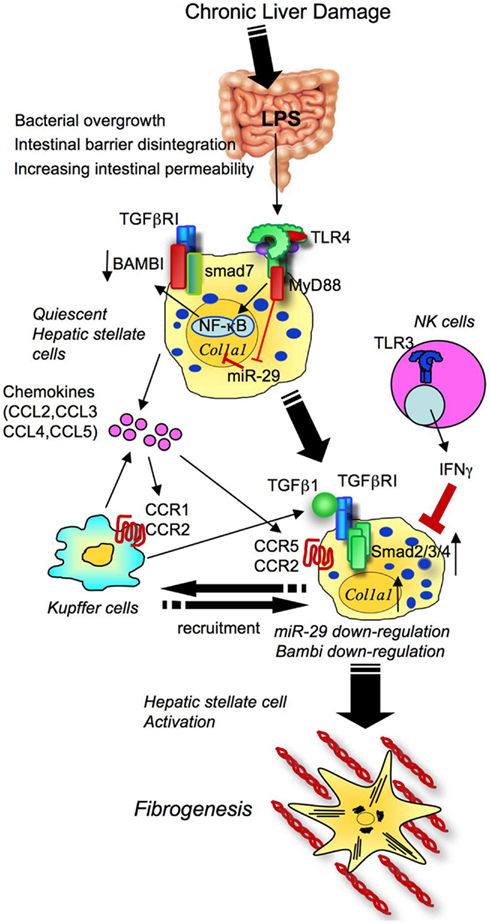
Figure 5. Toll-like receptor signaling drives liver fibrosis through activation of hepatic stellate cells. In chronic liver damage, intestinal permeability is increased due to systemic inflammation, portal hypertension, intestinal dysbiosis, or tight junction disintegrity, leading to bacterial translocation. Translocated LPS stimulates TLR4 on HSCs. Upon activation of TLR4, HSCs produce chemokines to recruit Kupffer cells through CCR1 and CCR2, and Bambi is downregulated through MyD88 and NF-κB. The fully activated TGF-β signaling eventually induces HSC activation and liver fibrosis. TLR4 also inhibits miR-29 to enhance collagen production. CCR5 is important for HSC recruitment. Generally, the NK cells-TLR3-IFNΓ axis suppresses HSC activation.
TLR9 Signaling in Liver Fibrosis
Several clinical and animal studies for liver cirrhosis have demonstrated elevated bacterial DNA levels in blood and ascites (Guarner et al., 1997, 2006; Frances et al., 2004, 2008), which suggests the importance of TLR9 signaling and bacterial DNA in the liver fibrosis, as TLR9 is a receptor for unmethylated CpG-containing DNA derived from bacteria. TLR9 signaling is also activated by denatured endogenous DNA released from dying hepatocytes (Watanabe et al., 2007). Upon activation of TLR9 signaling, HSCs produces CCL2 and collagen, but PDGF-induced HSC migration is inhibited. In mouse models of liver fibrosis induced by BDL and CCl4 treatment, TLR9-deficient mice exhibited a reduction in liver fibrosis (Watanabe et al., 2007; Gabele et al., 2008).
One study has mentioned the critical roles of TLR9 on DCs in liver fibrosis. DCs isolated from fibrotic livers can produce more TNF-α, IL-6, and chemokines than DCs from control livers in response to CpG-DNA. TLR9-mediated, DC-derived TNF-α enhances HSC activation and cytotoxicity of NK cells, which promote liver fibrosis. In fact, depletion of DCs reduces liver fibrosis (Connolly et al., 2009).
A recent report on peripheral blood mononuclear cells (PBMCs) from cirrhotic patients demonstrated less frequency of B cells and hyporesponsiveness of B cells to CD40 and TLR9 ligands as characterized by CD70 upregulation, TNFβ secretion, IgG production, and T-cell allostimulation. The abnormality of B cell function was found in the patients with liver cirrhosis, however, was independent of HCV infection (Doi et al., 2012).
TLR3 and Liver Fibrosis
TLR3 signaling is activated by double-stranded RNA released from viruses and damaged host cells and synthetic poly I:C. Liver fibrosis is highly inhibited in mice treated with poly I:C (Jeong et al., 2006; Radaeva et al., 2006; Yin and Gao, 2010). Poly I:C treatment promotes expression of NK cell activating ligands and TRAIL to enhance the cytotoxicity of NK cells for killing activated HSCs, which eventually suppresses liver fibrosis (Jeong et al., 2006; Radaeva et al., 2006; Yin and Gao, 2010; Figure 5). Interestingly, this effect is observed only in the early stage of liver fibrosis, and not in advanced liver fibrosis (Jeong et al., 2011). Moreover, poly I:C treatment are not able to suppress CCl4-induced liver fibrosis in animals treated with alcohol due to blunted NK cell-mediated HSC killing by ethanol (Jeong et al., 2008). These findings suggest that chronic ethanol consumption inhibits TLR3-dependent NK cell cytotoxicity against HSCs. Thus, the absence of TLR3-mediated NK cell killing is important for amplifying fibrogenic responses in advanced liver fibrosis and alcoholic liver disease (Jeong et al., 2008, 2011).
TLRs in Human Liver Cirrhosis
In PBMC from cirrhotic patients, TLR4 expression was decreased and TLR2 expression was increased (Manigold et al., 2003; Riordan et al., 2003; Tazi et al., 2006). The cirrhotic patients exhibited high levels of serum LPS and soluble CD14 (Manigold et al., 2003; Riordan et al., 2003). Decreased TLR4 expression and hyporesponsiveness to LPS in PBMCs were recovered after antibiotics treatment that targets enteric Gram-negative bacteria (Testro et al., 2010). These findings suggest that decreased TLR4 signaling is a mechanism of systemic LPS hyporesponsiveness and the high incidence of Gram-negative infection in cirrhotic patients. In contrast, hepatic expression of TLR2 and TLR4 was increased or unchanged in patients with cirrhosis (Manigold et al., 2003; Soares et al., 2012).
Evidence from a large patient cohort demonstrated that the TLR4 single nucleotide polymorphisms (SNPs) reduces the risk of liver cirrhosis in patients with chronic hepatitis C (Huang et al., 2007). These investigators continued their research and have reported that TLR4 D299G and T399I SNPs are associated with the reduction of TLR4-mediated signaling and cell death in HSCs (Guo et al., 2009). In contrast, the incidence of bacterial infection is higher in the HCV patients with TLR4 D299G SNP (Guarner-Argente et al., 2010). The TLR7 SNPs have also been studied. The study found that TLR7 c.1-120T > G SNP is more frequent in HCV patients with no or less inflammation and fibrosis than in HCV patients with liver inflammation and fibrosis. The difference was more evident in male patients (Schott et al., 2007).
TLRs in NASH, Alcohol, HBV, and HCV infection, PBC, Cystic Fibrosis
TLRs in NASH Associated Fibrosis
Non-alcoholic steatohepatitis (NASH) is characterized by hepatocyte damage, lipid deposition, ballooning, inflammatory cell infiltration, and fibrosis. NASH may eventually produce liver cirrhosis and HCC. Obesity, insulin resistance, and enteric microbiome are also associated with the promotion of NASH.
In animals, high fat diet (HFD) feeding induces significant obesity, insulin resistance, and hepatic steatosis, but pronounced inflammation and fibrosis are not observed. Methionine-choline-deficient (MCD) diet is preferentially used in the hepatology research field, because MCD diet induces prominent fat accumulation in hepatocytes, ALT level elevation, and inflammatory cell infiltration, but not obesity, insulin resistance, or evident fibrosis. Mice deficient in TLR4 or its coreceptor MD2 are resistant to MCD-induced NASH (Rivera et al., 2007; Csak et al., 2011). In NASH, the importance of intestine-derived bacterial products as a ligand for TLR4 has been proposed. A noteworthy study has reported that an increase in intestinal permeability produced by dextran sulfate sodium (DSS) treatment remarkably worsened HFD-induced fatty liver disease with severe inflammation and fibrosis (Gabele et al., 2011a). In the liver, hepatic TLR4 and TLR9 expression was increased, which suggests that intestinal barrier dysfunction and translocated bacteria products act as a “second hit,” to amplify liver inflammation and fibrosis in NASH. The importance of intestinal microflora in NASH is also supported by another study that showed improvement of NASH induced by MCD diet with probiotic VSL#3 treatment (Velayudham et al., 2009). In contrast to the suggestion that marks translocated bacterial as the inducer of TLR4-mediated NASH, one study suggests HMGB1 released from hepatocytes in response to free fatty acid (FFA) infusion as the endogenous ligand for the TLR4/MyD88 activation and cytokine production that contribute to the early HFD-induced fatty liver disease (Li et al., 2011).
The effect of high cholesterol (HC) diet on liver fibrosis has been demonstrated. Despite the lack of differences in liver injury and liver inflammation between normal chow diet and HC diet fed mice, mice fed with HC diet had aggravated liver fibrosis induced by BDL and CCl4 treatment (Teratani et al., 2012). The study found accumulation of free cholesterol in HSCs induced more TLR4 expression, thereby inhibiting Bambi and sensitization of HSCs to TGFβ for HSC activation (Teratani et al., 2012). This may account for the mechanisms of increased liver fibrosis in some patients with hypercholesterolemia (Teratani et al., 2012).
We have recently reported that TLR9 signaling is critical for the progression of choline-deficient amino acid defined (CDAA) diet-induced steatohepatitis and fibrosis. The CDAA diet produces excellent hepatocyte steatosis, liver damage, inflammatory cell infiltration, and fine fibrosis with increases in body weight and mild insulin resistance, which is more similar to the phenotypes of human NASH than HFD or MCD diet-induced NASH. In addition to LPS, bacterial products such as bacterial DNA, a ligand for TLR9, was present in the blood of mice fed on the CDAA diet for 22 weeks as assessed by PCR for bacterial 16S rRNA (Miura et al., 2010). As a result of NASH, the intestinal wall becomes leaky due to reduced oncotic pressure, oxidant stress, and increased intestinal edema. Bacterial overgrowth and changes in the composition of enteric microbiota are also components associated with bacterial translocation in obese individuals. Upon the transfer of microbiota harvested from obese mice to lean germfree mice on normal chow, the recipient lean mice became obese, suggesting the importance of microbiota in the development of obesity and fatty liver disease (Backhed et al., 2004; Ley et al., 2006; Turnbaugh et al., 2006). The leaky gut and alteration of enteric microbiota enhance translocation of bacterial products, such as LPS and bacterial DNA, from the intestine into the liver. Intestine-derived translocated LPS and bacterial DNA activates hepatic TLR4 and TLR9 signaling that is involved in the pathogenesis of NASH. Consistently, upon CDAA diet feeding, TLR9-deficient mice showed less severe steatohepatitis and liver fibrosis than wild-type mice (Miura et al., 2010). Among inflammatory cytokines increased in steatohepatitis of WT animals, only IL-1β is significantly decreased in TLR9-deficient mice, suggesting the importance of IL-1β in TLR9-mediated steatohepatitis. TLR9 signaling is activated mainly in Kupffer cells to produce IL-1β (Figure 6; Miura et al., 2010). Kupffer cell-derived IL-1β then activates HSCs for the induction of fibrogenic responses and also stimulates hepatocytes to increase fat accumulation with elevated DGAT2 levels. Intriguingly, fat accumulated hepatocytes isolated from CDAA diet fed mice had increase in sensitivity to IL-1β-induced cell death associated with increased expression of proapoptotic Bax and decreased expression of antiapoptotic Bcl-2 (Miura et al., 2010). Then, the study confirmed in vivo roles of IL-1 receptor signaling by using IL-1R-deficient mice. IL-1R-deficient mice had less steatohepatitis and fibrosis than wild-type mice after the CDAA diet feeding (Miura et al., 2010). Mice deficient in MyD88, a common adaptor molecule for TLR and IL-1R signaling, exhibited a marked reduction in steatohepatitis and fibrosis compared to wild-type mice (Miura et al., 2010). These findings indicate that TLR9-mediated IL-1β stimulates hepatocytes and HSCs to promote steatohepatitis and fibrosis.
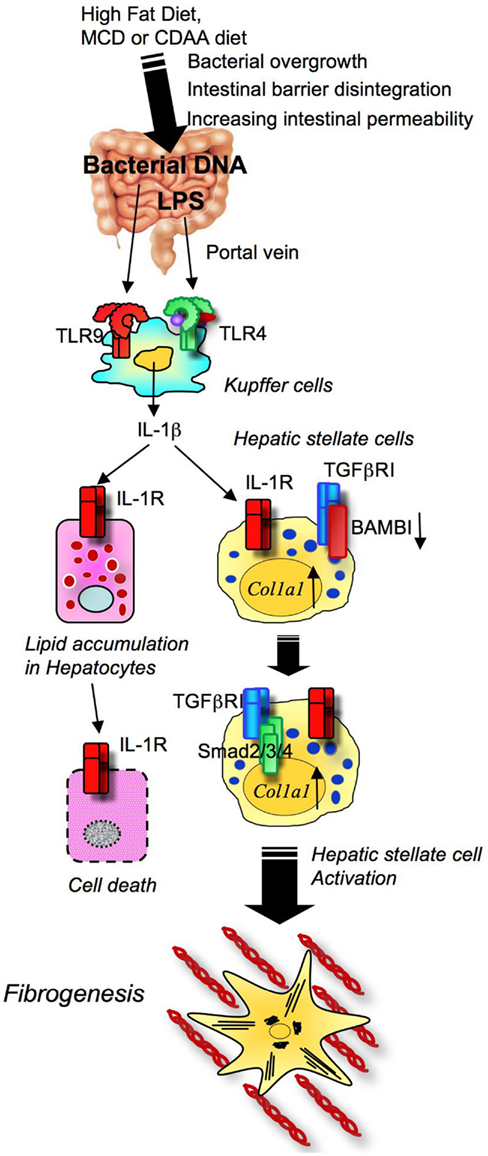
Figure 6. Toll-like receptor signaling and NASH development. High fat diet feeding and obesity conditions affect the composition of intestinal microflora and bacterial overgrowth. In NASH, translocated LPS and CpG-DNA stimulates TLR4 and TLR9 on Kupffer cells to produce IL-1β, which induces hepatocyte steatosis and death, and ultimately activates HSC, resulting in fibrosis.
In contrast to the central roles of TLR4 and TLR9 in NASH as we discussed above, the role of TLR2 has not been discussed well. The two studies showed that TLR2 deficiency does not decrease MCD diet-induced NASH, but increases the sensitivity to TLR4 ligand by increasing expression of TLR4 and CD14. As a result, TLR2-deficient mice have more liver inflammation and damage through TLR4 in response to MCD diet (Szabo et al., 2005; Rivera et al., 2010). These findings suggest the protective roles of TLR2 on NASH pathogenesis.
TLRs in Alcohol Mediated Liver Fibrosis
Because alcohol and its metabolite acetaldehyde are potent hepatotoxins, excessive intake of alcohol induces acute and/or chronic liver damage. The histological findings of alcoholic steatohepatitis include significant lipid deposition in hepatocytes, hepatocyte death, polymononuclear cell infiltration, and fibrosis, which are similar to NASH. Endotoxin levels in portal and peripheral blood are elevated in patients and animals with chronic alcohol abuse. It is postulated that excessive alcohol intake induces bacterial translocation by increasing intestinal permeability; the intestine-derived LPS promotes alcohol-induced steatohepatitis (Bode et al., 1987; Fukui et al., 1991; Uesugi et al., 2001, 2002; Yan et al., 2011). These hypotheses have been proven by several reports demonstrating reduction of alcohol-induced steatohepatitis in mice deficient in TLR4, LBP, and CD14 and mice with gut sterilization by oral administration of antibiotics (Adachi et al., 1995; Uesugi et al., 2001, 2002; Yin et al., 2001). A recent comprehensive analysis for the gut microbiome demonstrated the importance of alteration in microbiota in the development of alcoholic steatohepatitis (Yan et al., 2011).
Previous studies focused on TLR4 signaling in Kupffer cells, but the direct contribution of TLR4 to HSC activation in alcoholic steatohepatitis has yet to be examined (Adachi et al., 1994; Uesugi et al., 2001). Recently, we investigated the relative roles of TLR4 between Kupffer cells and HSCs in alcoholic steatohepatitis (Inokuchi et al., 2011). In this study, we found TLR4 to be important in both Kupffer cells (BM-derived cells) and HSCs (endogenous liver cells) in alcohol-induced hepatocyte injury, lipid accumulation, inflammatory cell infiltration, and HSC activation (Figure 7; Inokuchi et al., 2011). In contrast to the impact of the TLR4/MyD88 pathway on NASH, the TLR4–TRIF–IRF3 pathway is responsible for alcoholic steatohepatitis (Hritz et al., 2008a,b; Petrasek et al., 2011). Moreover, anti-inflammatory adiponectin and HO-1 have an inhibitory effect on the MyD88-independent TRIF-dependent pathway in chronic alcohol feeding (Mandal et al., 2010). Intriguingly, while the TLR4–TRIF–IRF3 pathway in Kupffer cells promotes alcoholic steatohepatitis, this pathway on hepatocytes has a protective role on alcohol-induced liver damage (Petrasek et al., 2011).
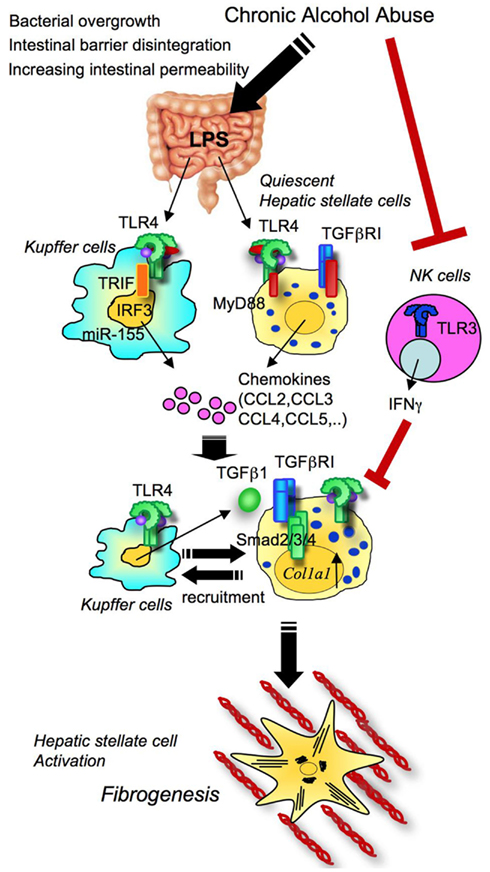
Figure 7. TLR4 signaling in alcoholic steatohepatitis and fibrosis. Excessive alcohol abuse induces changes in the composition of intestinal microbiota and bacterial overgrowth. With tight junction disintegrity, intestinal permeability increases, causing the translocation of gut microflora-derived LPS into the liver through the portal vein. Translocated LPS activates TLR4 on both Kupffer cells and HSCs. TLR4 signaling induces the production of chemokines that promote migration of Kupffer cells and HSCs. The TLR4–TRIF–IRF3 pathway and miR-155 control TLR4 activation in Kupffer cells. These events induce liver inflammation, hepatocyte steatosis, and fibrosis.
There is a strong epidemiological link between alcohol consumption, obesity, and insulin resistance on the progression of steatohepatitis (Naveau et al., 1997). An animal study demonstrated a synergistic effect between alcohol and HFD on the development of hepatic fibrosis, potentially through TLR4 signaling (Gabele et al., 2011b; Xu et al., 2011).
The miR-155 has contradictory effects on TLR4 signaling. On one hand, miR-155 inhibits TLR signaling by targeting MyD88 and TAB2 (Figure 3). On the other hand, miR-155 augments TLR4 signaling by suppression of SHIP (Quinn and O’Neill, 2011; Figure 4). miR-155 expression is upregulated in hepatic macrophages by chronic alcohol treatment, and is further elevated by LPS treatment. The increased miR-155 enhances alcohol- and TLR4-induced TNF-α production through stabilization of mRNA (Bala et al., 2011).
HCV Infection, TLRs, and Fibrosis
HCV infection as a major inducer of liver fibrosis activates innate immune receptors including TLRs and RNA helicases, such as RIG-I (Dolganiuc et al., 2004; Saito et al., 2008). In PBMCs from patients with chronic hepatitis, expressions of TLR2, TLR3, TLR4, TLR6, TLR7, and TLR8 were increased (He et al., 2006; Shehata et al., 2006; Sato et al., 2007). On the contrary, TLR9 expression was suppressed in PBMCs from HCV patients and negatively correlated with their serum viral copies (Zhou et al., 2009). When PBMCs from HCV patients and control subjects were treated with HCV core protein, TLR2 expression increased, while expressions of TLR4 and TLR7 decreased (Sato et al., 2007).
HCV core and NS3 proteins activate TLR2/TLR1 and TLR2/TLR6 in human PBMCs (Dolganiuc et al., 2004; Chang et al., 2007; Figure 8). Similarly, in human HSCs, HCV core protein activates TLR2 signaling to induce expressions of fibrogenic genes and matrix metalloproteinases, suggesting the direct contribution of HCV to fibrogenic responses through TLRs (Coenen et al., 2011). HCV NS5A protein induces TLR4 expression on B cells and hepatocytes to amplify the sensitivity to LPS stimulation (Machida et al., 2006). In the cultured hepatoma cell line system, TLR3 recognizes HCV infection to activate NF-κB and produce inflammatory cytokines and chemokines. Because UV-inactivated cells do not induce this response, TLR3 senses products generated through HCV replication (Li et al., 2012). HCV also activates TLR7 signaling in pDCs to induce type I interferon responses. To trigger this response, the direct interaction between pDCs and HCV infected cells is required (Takahashi et al., 2010). The HCV-specific G/U fragment as a PAMP activates TLR7 on human PBMCs (Zhang et al., 2009).
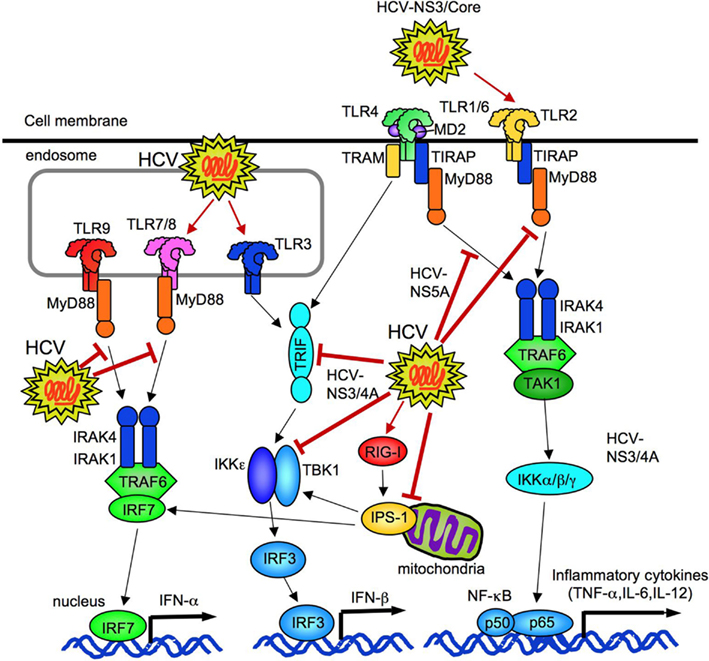
Figure 8. HCV regulates innate immune responses. HCV can activate TLR2, TLR3, TLR7, and cytosolic RIG-I. HCV (NS3/4A and NS3) inhibits TRIF and TBK1 to suppress IFN-β production. HCV (NS5A) binds to MyD88 to prevent TLR2, 4, 7, and 9 signaling. HCV (NS3/4A) cleaves of IPS-1 at C508, resulting in prevention of IRF3-mediated IFN-β production.
HCV composed proteins not only stimulate innate immune signaling, but also suppress the signaling. HCV NS3/4a proteins prevent TLR3-mediated TRIF-dependent IRF3 activation and IFN-β production by degrading TRIF and interfering the interaction between TBK1 and IRF3 (Li et al., 2005; Otsuka et al., 2005; Figure 8). Another study reported that NS5A blocks the recruitment of IRAK1 to MyD88, resulting in an inhibition of inflammatory cytokine production in response to the ligands for TLR2, TLR4, TLR7, and TLR9 (Abe et al., 2007; Figure 8). Experiments using cell culture-produced HCV virus (HCVcc) demonstrated HCVcc to inhibit TLR9-mediated IFN-α production in PBMCs and pDCs, but does not impair DC maturation and production of IL-12, IL-6, IL-10, IFN-Γ, and TNF-α in response to TLR3 and TLR4 ligands (Shiina and Rehermann, 2008).
Infection of HCV to hepatocytes initially activates RIG-I, IPS-1, and IRF3 to induce IFN-β production, but the subsequent cleavage of IPS-1 at C508 by NS3/4A dampens the RIG-I–IPS-1–IRF3 pathway to prevent HCV clearance (Foy et al., 2005; Meylan et al., 2005; Loo et al., 2006). HCV envelope proteins also downregulate the expression of TLR3 and RIG-I (Eksioglu et al., 2011). Thus, HCV possesses unique ability to escape from host immune response and maintain its ability to infect.
As mentioned above, there is a strong correlation between TLR4 SNPs and the risk of liver fibrosis in HCV patients (Huang et al., 2007). Currently, TLR agonists are considered as a potential therapeutic strategy for HCV infection. TLR3, TLR4, TLR7, TLR8, and TLR9 agonists potently induce type I interferon response and cytokine production that promote HCV eradication (Thomas et al., 2007).
TLRs and HBV Infection
HBV infection usually causes acute hepatitis, and 90% of patients will recover, 10% of patients sustain their infection and develop chronic hepatitis that progresses to liver cirrhosis, a major risk factor for the cause of HCC. The gene expression of TLR1, TLR2, TLR4, TLR6, TLR7, and TLR9 was suppressed in PBMCs from patients with chronic hepatitis B compared to healthy subject, and the expression of TLR2 and TLR4 are upregulated in peripheral PBMCs from hepatitis B patients with liver cirrhosis (Chen et al., 2008; Xu et al., 2008b; Lian et al., 2009; Vincent et al., 2011). A negative correlation between TLR2 expression, and T-cell number or HBV viral load in cirrhotic patients was also reported (Chen et al., 2008; Lian et al., 2009). The response of PBMCs from chronic hepatitis B patients to TLR2 and TLR4 ligands is impaired (Chen et al., 2008).
Immunosuppressive effects of HBV infection have been shown by several reports. In cirrhotic hepatitis B patients, TLR4 expression correlates with the number of regulatory T (Tregs) cells, and increased TLR2 and TLR4 expressions are associated with the immunotolerance of chronic HBV infection through modulation of Treg functions (Lian et al., 2009; Zhang et al., 2010b). Alternatively, HBeAg interacts with TRAM, TIRAP, and TLR2, which inhibit NF-κB and Interferon-β promoter activity (Lang et al., 2011). The HBV particle also inhibits TLR9 signaling by inhibiting MyD88 and IRAK4 to impair IFN-α production (Vincent et al., 2011).
TLR signaling also induces antiviral effects on HBV. Ligands for TLR3, TLR4, TLR5, TLR7, and TLR9 inhibit HBV replication in HBV transgenic mice (Isogawa et al., 2005). Mediators secreted from non-parenchymal cells including Kupffer cells and LSECs in response to ligands for TLR3 and TLR4 inhibit HBV replication in HBV Met cells (Wu et al., 2007). These mediators are produced through TRIF-dependent, but not MyD88-dependent signaling (Wu et al., 2007). Another study shows that TLR2 ligand also inhibits HBV replication (Thompson et al., 2009), and the agonists for TLR7 and TLR9 assist the immunogenicity of HBV vaccination. These findings indicate that TLR agonists are potential therapies for HBV as well as a booster for its vaccination (Weeratna et al., 2005; Ma et al., 2007).
TLRs in Primary Biliary Cirrhosis
Primary biliary cirrhosis (PBC) is an autoimmune disease characterized by chronic inflammation and destruction of small bile ducts, which causes hepatocyte injury, progressive inflammation and fibrosis and ultimately cirrhosis. Because of the anatomically direct connection between biliary tract and intestinal lumen, intestinal-derived bacterial products may contribute to disease progression of PBC (Harada and Nakanuma, 2010a,b). PBMCs in PBC patients produce higher amount of inflammatory cytokines in response to the ligands for TLR2, TLR3, TLR4, TLR5, and TLR9 than PBMCs in healthy subjects (Mao et al., 2005). In PBC patients, B cells produce more intracellular immunoglobulin M and antimitochondrial antibodies in response to CpG-B due to high expression of TLR9 (Kikuchi et al., 2005a; Moritoki et al., 2007). Furthermore, B cells with TLR9 SNPs produce higher amount of intracellular immunoglobulin M (Kikuchi et al., 2005b). In the livers of PBC patients, TLR3 is highly expressed on macrophages surrounding portal tract and hepatocytes. Type I interferon is also expressed on these cells in the PBC livers (Takii et al., 2005). Liver macrophages produce IFN-α through TLR3 signaling. This IFN-α synergistically enhances LPS-induced NK cell cytotoxicity to autologous BECs. Furthermore, the cytotoxicity of liver NK cells in PBC patients is more profound than cells in control subjects when incubated with poly I:C and LPS-primed liver macrophages (Shimoda et al., 2011). Mice immunized with 2-octynoic acid developed autoimmune cholangitis with positive for antimitochondrial antibodies, similar to the features of human PBC livers, but did not induce liver fibrosis. Poly I:C treatment exacerbated CD8 T cell infiltration, inflammatory cytokine expression and evident liver fibrosis in this PBC mice model, which further supports the impact of TLR3 signaling in PBC (Ambrosini et al., 2011).
TLRs in Cystic Fibrosis
Cystic fibrosis is an inherited disease that affects lungs, pancreas, liver and intestines, and is associated with the mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. However, only 30% of CF-patients develop liver disease and CFTR-deficient mice do not develop liver disease spontaneously. Thus, additional factors are required for CF liver disease progression (Fiorotto et al., 2011). One study examined the effect of bacterial translocation in CF liver disease by treating CFTR-deficient mice with DSS. DSS-treated mice developed higher blood LPS levels in both WT and CFTR-deficient mice, but only CFTR-deficient mice exhibited ALT elevation and ductular reaction, which were suppressed by antibiotics treatment, suggesting intestine-derived LPS to be important in CF disease progression (Fiorotto et al., 2011). BECs express high levels of CFTR. In response to LPS, CFTR-deficient BECs produce more inflammatory cytokines than wild-type cells. These findings suggest that translocated intestine-derived LPS is a factor to develop CF liver disease in individuals with mutated CFTR (Fiorotto et al., 2011).
TLRs as Potential Targets for the Therapy of Liver Fibrosis
Pharmacological Inhibition of TLR4 Signaling
Several TLR4 inhibitors have been developed and are potential drugs for diseases that are related to TLR4 activation. Lipid A antagonists, such as lipid IVa and E5564, can inhibit LPS-mediated inflammation by blocking TLR4 signaling (Saitoh et al., 2004; Kitazawa et al., 2010; Barochia et al., 2011). Preclinical studies have demonstrated the inhibitory effects of E5564 in vitro and in vivo on animal models of sepsis and LPS-induced acute liver injury, and its efficacy on clinical usage are under investigation (Kitazawa et al., 2010; Barochia et al., 2011). CRX-526, a lipid A-mimetic TLR4 antagonist, inhibits LPS-induced TLR4 signaling and cytokine production in vitro and in vivo (Fort et al., 2005). CRX-526 treatment suppressed a mouse model of colitis induced by DSS and genetic mouse colitis model (Fort et al., 2005). TAK-242, a small molecule inhibitor for TLR4 signaling, inhibits LPS-induced inflammatory cytokine production by binding to intracellular domain of TLR4. TAK-242 interferes the interaction between intracellular domain of TLR4 and TIRAP or TRAM, which blocks both MyD88- and TRIF-dependent pathways (Matsunaga et al., 2011). TAK-242 prevented LPS-mediated sepsis, peritonitis and acute kidney injury (Sha et al., 2007; Fenhammar et al., 2011; Goldfarb et al., 2011). A soluble TLR4–MD2 fusion protein binds to free LPS and inhibits LPS-induced NF-κB and JNK activation in HSCs (Schnabl et al., 2008). These TLR4 inhibitors have not been tested yet for liver fibrosis. In the basis of the importance of TLR4 in liver fibrosis, alcoholic steatohepatitis and obesity-related fatty liver disease, pharmacological inhibition of TLR4 will be innovative therapeutic approaches for the treatment of these diseases.
Targeting Intestinal Microbiota as TLR Ligands
As discussed above, changes in the composition of gut microflora and bacterial translocation are suggested to be important for the pathogenesis of liver cirrhosis. Modulation of intestinal microbiota is an alternative approach for the therapy of liver cirrhosis. Oral administration of antibiotics is an approach to reduce TLR ligands by decontaminating Gram-negative bacteria, a source of LPS, in intestine. Selective decontamination of intestine improved liver fibrosis with reduction of bacterial translocation in mice (Seki et al., 2007; Zhu et al., 2012). Gut sterilization by oral administration of antibiotics also inhibited alcohol-induced steatohepatitis in rats (Adachi et al., 1995). However, long-term antibiotics treatment may cause unfavorable alteration of microbiome, and is not a perfect approach for the treatment of liver fibrosis.
A second approach to the reduction of TLR ligands is the use of probiotics to complement beneficial microbiome in the intestine. There are several studies demonstrating beneficial effects of probiotics. Probiotics protected patients with hepatic cirrhosis from bacterial translocation and bacterial peritonitis (Adawi et al., 1997; Rayes et al., 2002, 2005; Liu et al., 2004; Ewaschuk et al., 2007; Osman et al., 2007), and prevented NASH in patients and animals (Li et al., 2003; Loguercio et al., 2005; Velayudham et al., 2009). This approach also remarkably inhibited alcohol-induced steatohepatitis (Nanji et al., 1994; Ewaschuk et al., 2007; Osman et al., 2007).
Another approach for regulation of enteric microflora is to use prebiotics to maintain beneficial microbiome in intestine. Administration of prebiotic fructo-oligosaccharides (FOS), indigestive short-chain saccharides, stimulates probiotic bacteria Lactobacilli and Bifdobacteria, suppressing intestinal bacterial overgrowth and alcohol-induced steatohepatitis (Yan et al., 2011). Thus, pre- or probiotics are attractive agents to improve intestinal dysbiosis, bacterial overgrowth, and liver fibrosis induced by different etiology, such as viral hepatitis, non-alcoholic fatty liver disease, and alcoholic steatohepatitis.
Conclusion
Animals evolutionally acquired innate immune signaling cascade to protect their organs from pathogenic microorganisms (bacteria, viruses, fungi, and parasites) as their first line of host defense. Mammalian homologs of Drosophila Toll, TLRs, were identified in the late 1990s as essential signal sensors that recognizes specific molecular patterns derived from microorganisms. The TLR signaling induces the production of inflammatory mediators and anti-microbial peptides to eradicate invading microorganisms from the host as well as to bridge the acquired immunity to amplify immune responses. The innate immune signaling also contributes to tissue homeostasis, such as intestinal microflora, proliferation, and apoptosis of intestinal epithelial cells, and liver regeneration after the loss of liver mass. However, abnormal activation of innate immune signaling may also cause sepsis, chronic inflammation, autoimmune diseases, tissue and organ injuries, fibrosis and carcinogenesis that are unfavorable to the host. It appears that these pattern recognition receptors function in turning “on” the diseases rather than protectively turning “off” the diseases in some conditions. As we mentioned earlier in this review, the liver is susceptible to the exposure of intestine-derived microbial products, such as LPS and CpG-DNA, because of a specific anatomical connection between the intestinal lumen and the liver through portal vein and bile ducts. The hepatic innate immune signaling in response to intestine-derived products may affect the development of liver inflammation. Damaged hepatocytes may also release endogenous TLR ligands, such as HMGB1, to stimulate TLR signaling and further promote liver disease. Furthermore, hepatic inflammation may induce systemic inflammation followed by the destruction of intestinal barrier and subsequent bacterial translocation. These events are able to induce a second activation of TLR signaling in the liver. Thus, to develop new effective therapy for liver disease including liver fibrosis should target either the initial or the secondary responses caused by TLR signaling. Besides blocking TLR signaling, activation of TLR signaling by agonists may also become an attractive approach for some liver diseases, such as hepatitis B and C infection, liver fibrosis, and cancer. Liver is composed of hepatocytes, Kupffer cells, recruited BM-derived monocytes, DCs, endothelial cells, and HSCs. Among these cells, some cells cause harmful responses by activating TLR signaling, while TLR signaling in other type of cells may act as a beneficial player. Therefore, cell specific approach should be important for developing new therapy targeting TLRs. Future studies are required for discovering further mechanisms on TLR-mediated liver fibrosis and new clinical relevance of TLRs as biomarkers and therapeutic targets for liver fibrosis/cirrhosis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support: This study is supported by the pilot grants from the UCSD Digestive Diseases Research Development Center (DK080506), the research grants from ABMRF, NIH grant R01AA02172 and R01DK085252. We thank Ms. Isabelle Jingyi Song (Department of Medicine at UC San Diego) for editing the manuscript.
Abbreviations
ATF, activating transcription factor; Bambi, bone morphogenetic protein and activin membrane bound inhibitor; BDL, bile duct ligation; BEC, biliary epithelial cell; CCl4, carbon tetrachloride; CDAA, choline-deficient amino acid defined; CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; DAMP, damage associated molecular pattern; DC, dendritic cell; DSS, dextran sulfate sodium; FN, fibronectin; HC, high cholesterol; HCVcc, cell culture-produced HCV virus; HFD, high fat diet; HMGB, high mobility group box; HSC, hepatic stellate cell; IL, interleukin; IRAK, IL-1 receptor associated kinase; LBP, LPS-binding protein; LPS, lipopolysaccharide; LSEC, liver sinusoidal endothelial cell; MCD, methionine-choline-deficient; miRNA, microRNA; MyD88, myeloid differentiation factor 88; NASH, non-alcoholic steatohepatitis; NF, nuclear factor; NK, natural killer; PAMP, pathogen-associated molecular pattern; PBC, primary biliary cirrhosis; PBMC, peripheral blood mononuclear cell; pDC, plasmacytoid DC; PDCD, programmed cell death; SARM, toll/IL-1R resistance motif containing protein; SHIP1, Src homology 2 domain-containing inositol-5′-phosphatase 1; SIGGR, single immunoglobulin IL-1R-related molecule; SNP, single nucleotide polymorphism; SOCS, suppressor of cytokine signaling; TAK1, TGF-β activated kinase 1; TAB, TAK1-binding protein; TIR, toll/IL-1 receptor; TIRAP, TIR domain-containing adaptor protein; TLR, toll-like receptor.
References
Abe, T., Kaname, Y., Hamamoto, I., Tsuda, Y., Wen, X., Taguwa, S., Moriishi, K., Takeuchi, O., Kawai, T., Kanto, T., Hayashi, N., Akira, S., and Matsuura, Y. (2007). Hepatitis C virus nonstructural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J. Virol. 81, 8953–8966.
Adachi, Y., Bradford, B. U., Gao, W., Bojes, H. K., and Thurman, R. G. (1994). Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology 20, 453–460.
Adachi, Y., Moore, L. E., Bradford, B. U., Gao, W., and Thurman, R. G. (1995). Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108, 218–224.
Adawi, D., Kasravi, F. B., Molin, G., and Jeppsson, B. (1997). Effect of Lactobacillus supplementation with and without arginine on liver damage and bacterial translocation in an acute liver injury model in the rat. Hepatology 25, 642–647.
Ambrosini, Y. M., Yang, G. X., Zhang, W., Tsuda, M., Shu, S., Tsuneyama, K., Leung, P. S., Ansari, A. A., Coppel, R. L., and Gershwin, M. E. (2011). The multi-hit hypothesis of primary biliary cirrhosis: polyinosinic-polycytidylic acid (poly I:C) and murine autoimmune cholangitis. Clin. Exp. Immunol. 166, 110–120.
An, H., Hou, J., Zhou, J., Zhao, W., Xu, H., Zheng, Y., Yu, Y., Liu, S., and Cao, X. (2008). Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1. Nat. Immunol. 9, 542–550.
Assimakopoulos, S. F., Scopa, C. D., and Vagianos, C. E. (2007a). Pathophysiology of increased intestinal permeability in obstructive jaundice. World J. Gastroenterol. 13, 6458–6464.
Assimakopoulos, S. F., Maroulis, I., Patsoukis, N., Vagenas, K., Scopa, C. D., Georgiou, C. D., and Vagianos, C. E. (2007b). Effect of antioxidant treatments on the gut-liver axis oxidative status and function in bile duct-ligated rats. World J. Surg. 31, 2023–2032.
Backhed, F., Ding, H., Wang, T., Hooper, L. V., Koh, G. Y., Nagy, A., Semenkovich, C. F., and Gordon, J. I. (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U.S.A. 101, 15718–15723.
Bala, S., Marcos, M., Kodys, K., Csak, T., Catalano, D., Mandrekar, P., and Szabo, G. (2011). Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA, half-life in alcoholic liver disease. J. Biol. Chem. 286, 1436–1444.
Barochia, A., Solomon, S., Cui, X., Natanson, C., and Eichacker, P. Q. (2011). Eritoran tetrasodium (E5564) treatment for sepsis: review of preclinical and clinical studies. Expert Opin. Drug Metab. Toxicol. 7, 479–494.
Belinda, L. W., Wei, W. X., Hanh, B. T., Lei, L. X., Bow, H., and Ling, D. J. (2008). SARM: a novel toll-like receptor adaptor, is functionally conserved from arthropod to human. Mol. Immunol. 45, 1732–1742.
Berres, M. L., Koenen, R. R., Rueland, A., Zaldivar, M. M., Heinrichs, D., Sahin, H., Schmitz, P., Streetz, K. L., Berg, T., Gassler, N., Weiskirchen, R., Proudfoot, A., Weber, C., Trautwein, C., and Wasmuth, H. E. (2010). Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J. Clin. Invest. 120, 4129–4140.
Bode, C., Kugler, V., and Bode, J. C. (1987). Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J. Hepatol. 4, 8–14.
Chang, S., Dolganiuc, A., and Szabo, G. (2007). Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 82, 479–487.
Chen, Y., Yang, F., Lu, H., Wang, B., Lei, D., Wang, Y., Zhu, B., and Li, L. (2011). Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 54, 562–572.
Chen, Z., Cheng, Y., Xu, Y., Liao, J., Zhang, X., Hu, Y., Zhang, Q., Wang, J., Zhang, Z., Shen, F., and Yuan, Z. (2008). Expression profiles and function of toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin. Immunol. 128, 400–408.
Chockalingam, A., Cameron, J. L., Brooks, J. C., and Leifer, C. A. (2011). Negative regulation of signaling by a soluble form of toll-like receptor 9. Eur. J. Immunol. 41, 2176–2184.
Coenen, M., Nischalke, H. D., Kramer, B., Langhans, B., Glassner, A., Schulte, D., Korner, C., Sauerbruch, T., Nattermann, J., and Spengler, U. (2011). Hepatitis C virus core protein induces fibrogenic actions of hepatic stellate cells via toll-like receptor 2. Lab. Invest. 91, 1375–1382.
Coll, R. C., and O’Neill, L. A. (2010). New insights into the regulation of signalling by toll-like receptors and nod-like receptors. J. Innate Immun. 2, 406–421.
Connolly, M. K., Bedrosian, A. S., Mallen-St Clair, J., Mitchell, A. P., Ibrahim, J., Stroud, A., Pachter, H. L., Bar-Sagi, D., Frey, A. B., and Miller, G. (2009). In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J. Clin. Invest. 119, 3213–3225.
Csak, T., Velayudham, A., Hritz, I., Petrasek, J., Levin, I., Lippai, D., Catalano, D., Mandrekar, P., Dolganiuc, A., Kurt-Jones, E., and Szabo, G. (2011). Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G433–G441.
Doi, H., Iyer, T. K., Carpenter, E., Li, H., Chang, K. M., Vonderheide, R. H., and Kaplan, D. E. (2012). Dysfunctional B-cell activation in cirrhosis due to hepatitis C infection associated with disappearance of CD27+ B-cell population. Hepatology 55, 709–719.
Dolganiuc, A., Oak, S., Kodys, K., Golenbock, D. T., Finberg, R. W., Kurt-Jones, E., and Szabo, G. (2004). Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 127, 1513–1524.
Eksioglu, E. A., Zhu, H., Bayouth, L., Bess, J., Liu, H. Y., Nelson, D. R., and Liu, C. (2011). Characterization of HCV interactions with toll-like receptors and RIG-I in liver cells. PLoS ONE 6, e21186. doi:10.1371/journal.pone.0021186
Ewaschuk, J., Endersby, R., Thiel, D., Diaz, H., Backer, J., Ma, M., Churchill, T., and Madsen, K. (2007). Probiotic bacteria prevent hepatic damage and maintain colonic barrier function in a mouse model of sepsis. Hepatology 46, 841–850.
Fenhammar, J., Rundgren, M., Forestier, J., Kalman, S., Eriksson, S., and Frithiof, R. (2011). Toll-like receptor 4 inhibitor TA, K-242 attenuates acute kidney injury in endotoxemic sheep. Anesthesiology 114, 1130–1137.
Fiorotto, R., Scirpo, R., Trauner, M., Fabris, L., Hoque, R., Spirli, C., and Strazzabosco, M. (2011). Loss of CFTR, affects biliary epithelium innate immunity and causes TLR4-NF-kappaB-mediated inflammatory response in mice. Gastroenterology 141, 1498–1508; 1508.e1-5.
Fort, M. M., Mozaffarian, A., Stover, A. G., Correia Jda, S., Johnson, D. A., Crane, R. T., Ulevitch, R. J., Persing, D. H., Bielefeldt-Ohmann, H., Probst, P., Jeffery, E., Fling, S. P., and Hershberg, R. M. (2005). A synthetic TL, R4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J. Immunol. 174, 6416–6423.
Foy, E., Li, K., Sumpter, R. Jr., Loo, Y. M., Johnson, C. L., Wang, C., Fish, P. M., Yoneyama, M., Fujita, T., Lemon, S. M., and Gale, M. Jr. (2005). Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 2986–2991.
Frances, R., Benlloch, S., Zapater, P., Gonzalez, J. M., Lozano, B., Munoz, C., Pascual, S., Casellas, J. A., Uceda, F., Palazon, J. M., Carnicer, F., Perez-Mateo, M., and Such, J. (2004). A sequential study of serum bacterial DNA in patients with advanced cirrhosis and ascites. Hepatology 39, 484–491.
Frances, R., Zapater, P., Gonzalez-Navajas, C., Munoz, R., Cano, R., Moreu, S., Pascual, S., Bellot, P., Perez-Mateo, M., and Such, J. (2008). Bacterial DNA in patients with cirrhosis and noninfected ascites mimics the soluble immune response established in patients with spontaneous bacterial peritonitis. Hepatology 47, 978–985.
Friedman, S. L. (2010). Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 7, 425–436.
Fukui, H., Brauner, B., Bode, J. C., and Bode, C. (1991). Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J. Hepatol. 12, 162–169.
Gabele, E., Dostert, K., Hofmann, C., Wiest, R., Scholmerich, J., Hellerbrand, C., and Obermeier, F. (2011a). DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 55, 1391–1399.
Gabele, E., Dostert, K., Dorn, C., Patsenker, E., Stickel, F., and Hellerbrand, C. (2011b). A new model of interactive effects of alcohol and high-fat diet on hepatic fibrosis. Alcohol. Clin. Exp. Res. 35, 1361–1367.
Gabele, E., Muhlbauer, M., Dorn, C., Weiss, T. S., Froh, M., Schnabl, B., Wiest, R., Scholmerich, J., Obermeier, F., and Hellerbrand, C. (2008). Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 376, 271–276.
Gilchrist, M., Thorsson, V., Li, B., Rust, A. G., Korb, M., Roach, J. C., Kennedy, K., Hai, T., Bolouri, H., and Aderem, A. (2006). Systems biology approaches identify ATF3 as a negative regulator of toll-like receptor 4. Nature 441, 173–178.
Goldfarb, R. D., Ortegel, J. W., Parrillo, J. E., Zanotti-Cavazzoni, S., Casey, L. C., and Dellinger, R. P. (2011). TAKEDA-143242 increased survival via reduced cytokines in porcine peritonitis. J. Surg. Res. 166, e165–e173.
Gomez-Hurtado, I., Santacruz, A., Peiro, G., Zapater, P., Gutierrez, A., Perez-Mateo, M., Sanz, Y., and Frances, R. (2011). Gut microbiota dysbiosis is associated with inflammation and bacterial translocation in mice with CCl(4)-induced fibrosis. PLoS ONE 6, e23037. doi:10.1371/journal.pone.0023037
Grinko, I., Geerts, A., and Wisse, E. (1995). Experimental biliary fibrosis correlates with increased numbers of fat-storing and Kupffer cells, and portal endotoxemia. J. Hepatol. 23, 449–458.
Guan, Y., Ranoa, D. R., Jiang, S., Mutha, S. K., Li, X., Baudry, J., and Tapping, R. I. (2010). Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J. Immunol. 184, 5094–5103.
Guarner, C., Gonzalez-Navajas, J. M., Sanchez, E., Soriando, G., Frances, R., Chiva, M., Zapater, P., Benlloch, S., Munoz, C., Pascual, S., Balanzo, J., Perez-Mateo, M., and Such, J. (2006). The detection of bacterial DNA in blood of rats with CCl4-induced cirrhosis with ascites represents episodes of bacterial translocation. Hepatology 44, 633–639.
Guarner, C., Runyon, B. A., Young, S., Heck, M., and Sheikh, M. Y. (1997). Intestinal bacterial overgrowth and bacterial translocation in cirrhotic rats with ascites. J. Hepatol. 26, 1372–1378.
Guarner-Argente, C., Sanchez, E., Vidal, S., Roman, E., Concepcion, M., Poca, M., Sanchez, D., Juarez, C., Soriano, G., and Guarner, C. (2010). Toll-like receptor 4 D299G polymorphism and the incidence of infections in cirrhotic patients. Aliment. Pharmacol. Ther. 31, 1192–1199.
Guo, J., and Friedman, S. L. (2010). Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair 3, 21.
Guo, J., Loke, J., Zheng, F., Hong, F., Yea, S., Fugita, M., Tarocchi, M., Abar, O. T., Huang, H., Sninsky, J. J., and Friedman, S. L. (2009). Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of toll-like receptor 4 to hepatic stellate cell responses. Hepatology 49, 960–968.
Hacker, H., Redecke, V., Blagoev, B., Kratchmarova, I., Hsu, L. C., Wang, G. G., Kamps, M. P., Raz, E., Wagner, H., Hacker, G., Mann, M., and Karin, M. (2006). Specificity in toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439, 204–207.
Harada, K., Isse, K., Sato, Y., Ozaki, S., and Nakanuma, Y. (2006). Endotoxin tolerance in human intrahepatic biliary epithelial cells is induced by upregulation of IRAK-M. Liver Int. 26, 935–942.
Harada, K., and Nakanuma, Y. (2010a). Biliary innate immunity: function and modulation. Mediators Inflamm. 2010, 9.
Harada, K., and Nakanuma, Y. (2010b). Biliary innate immunity in the pathogenesis of biliary diseases. Inflamm. Allergy Drug Targets 9, 83–90.
He, Q., Graham, C. S., Durante Mangoni, E., and Koziel, M. J. (2006). Differential expression of toll-like receptor mRNA, in treatment non-responders and sustained virologic responders at baseline in patients with chronic hepatitis C. Liver Int. 26, 1100–1110.
Hritz, I., Mandrekar, P., Velayudham, A., Catalano, D., Dolganiuc, A., Kodys, K., Kurt-Jones, E., and Szabo, G. (2008a). The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 48, 1224–1231.
Hritz, I., Velayudham, A., Dolganiuc, A., Kodys, K., Mandrekar, P., Kurt-Jones, E., and Szabo, G. (2008b). Bone marrow-derived immune cells mediate sensitization to liver injury in a myeloid differentiation factor 88-dependent fashion. Hepatology 48, 1342–1347.
Huang, H., Shiffman, M. L., Friedman, S., Venkatesh, R., Bzowej, N., Abar, O. T., Rowland, C. M., Catanese, J. J., Leong, D. U., Sninsky, J. J., Layden, T. J., Wright, T. L., White, T., and Cheung, R. C. (2007). A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology 46, 297–306.
Imamura, M., Tsutsui, H., Yasuda, K., Uchiyama, R., Yumikura-Futatsugi, S., Mitani, K., Hayashi, S., Akira, S., Taniguchi, S., Van Rooijen, N., Tschopp, J., Yamamoto, T., Fujimoto, J., and Nakanishi, K. (2009). Contribution of TIR domain-containing adapter inducing IFN-beta-mediated IL-18 release to LPS-induced liver injury in mice. J. Hepatol. 51, 333–341.
Inokuchi, S., Tsukamoto, H., Park, E. J., Liu, Z.-X., Brenner, D. A., and Seki, E. (2011). Toll-like receptor 4 mediates alcohol-induced steatohepatitis through bone marrow-derived and endogenous liver cells in mice. Alcohol. Clin. Exp. Res. 35, 1509–1518.
Isayama, F., Hines, I. N., Kremer, M., Milton, R. J., Byrd, C. L., Perry, A. W., McKim, S. E., Parsons, C., Rippe, R. A., and Wheeler, M. D. (2006). LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G1318–G1328.
Isogawa, M., Robek, M. D., Furuichi, Y., and Chisari, F. V. (2005). Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 79, 7269–7272.
Jagavelu, K., Routray, C., Shergill, U., O’Hara, S. P., Faubion, W., and Shah, V. H. (2010). Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology 52, 590–601.
Jeong, W. I., Park, O., and Gao, B. (2008). Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology 134, 248–258.
Jeong, W. I., Park, O., Radaeva, S., and Gao, B. (2006). STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 44, 1441–1451.
Jeong, W.-I., Park, O., Suh, Y.-G., Byun, J.-S., Park, S.-Y., Choi, E., Kim, J.-K., Ko, H., Wang, H., Miller, A. M., and Gao, B. (2011). Suppression of innate immunity (natural killer cell/interferon-Γ) in the advanced stages of liver fibrosis in mice. Hepatology 53, 1342–1351.
Kagan, J. C., Su, T., Horng, T., Chow, A., Akira, S., and Medzhitov, R. (2008). TRAM, couples endocytosis of toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 9, 361–368.
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat. Immunol. 11, 373–384.
Kikuchi, K., Lian, Z. X., Yang, G. X., Ansari, A. A., Ikehara, S., Kaplan, M., Miyakawa, H., Coppel, R. L., and Gershwin, M. E. (2005a). Bacterial CpG, induces hyper-IgM, production in CD, 27(+) memory B cells in primary biliary cirrhosis. Gastroenterology 128, 304–312.
Kikuchi, K., Lian, Z. X., Kimura, Y., Selmi, C., Yang, G. X., Gordon, S. C., Invernizzi, P., Podda, M., Coppel, R. L., Ansari, A. A., Ikehara, S., Miyakawa, H., and Gershwin, M. E. (2005b). Genetic polymorphisms of toll-like receptor 9 influence the immune response to CpG, and contribute to hyper-IgM, in primary biliary cirrhosis. J. Autoimmun. 24, 347–352.
Kitazawa, T., Tsujimoto, T., Kawaratani, H., and Fukui, H. (2010). Salvage effect of E5564, toll-like receptor 4 antagonist on d-galactosamine and lipopolysaccharide-induced acute liver failure in rats. J. Gastroenterol. Hepatol. 25, 1009–1012.
Kobayashi, K., Hernandez, L. D., Galan, J. E., Janeway, C. A. Jr., Medzhitov, R., and Flavell, R. A. (2002). IRAK-M is a negative regulator of toll-like receptor signaling. Cell 110, 191–202.
Lang, T., Lo, C., Skinner, N., Locarnini, S., Visvanathan, K., and Mansell, A. (2011). The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J. Hepatol. 55, 762–769.
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023.
Li, K., Foy, E., Ferreon, J. C., Nakamura, M., Ferreon, A. C., Ikeda, M., Ray, S. C., Gale, M. Jr., and Lemon, S. M. (2005). Immune evasion by hepatitis C virus NS, 3/4A protease-mediated cleavage of the toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U.S.A. 102, 2992–2997.
Li, L., Chen, L., Hu, L., Liu, Y., Sun, H. Y., Tang, J., Hou, Y. J., Chang, Y. X., Tu, Q. Q., Feng, G. S., Shen, F., Wu, M. C., and Wang, H. Y. (2011). Nuclear factor high-mobility group box1 mediating the activation of toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 54, 1620–1630.
Li, K., Li, N. L., Wei, D., Pfeffer, S. R., Fan, M., and Pfeffer, L. M. (2012). Activation of chemokine and inflammatory cytokine response in HCV-infected hepatocytes depends on TLR3 sensing of HCV dsRNA intermediates. Hepatology 55, 666–675.
Li, Z., Yang, S., Lin, H., Huang, J., Watkins, P. A., Moser, A. B., Desimone, C., Song, X. Y., and Diehl, A. M. (2003). Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 37, 343–350.
Lian, J. Q., Wang, X. Q., Zhang, Y., Huang, C. X., and Bai, X. F. (2009). Correlation of circulating TLR2/4 expression with CD, 3+/4+/8+. T cells and Treg cells in HB, V-related liver cirrhosis. Viral Immunol. 22, 301–308.
Liew, F. Y., Xu, D., Brint, E. K., and O’Neill, L. A. (2005). Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 5, 446–458.
Liu, Q., Duan, Z. P., Ha, D. K., Bengmark, S., Kurtovic, J., and Riordan, S. M. (2004). Synbiotic modulation of gut flora: effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology 39, 1441–1449.
Loguercio, C., Federico, A., Tuccillo, C., Terracciano, F., D’Auria, M. V., De Simone, C., and Del Vecchio Blanco, C. (2005). Beneficial effects of a probiotic VS, L#3 on parameters of liver dysfunction in chronic liver diseases. J. Clin. Gastroenterol. 39, 540–543.
Loo, Y. M., Owen, D. M., Li, K., Erickson, A. K., Johnson, C. L., Fish, P. M., Carney, D. S., Wang, T., Ishida, H., Yoneyama, M., Fujita, T., Saito, T., Lee, W. M., Hagedorn, C. H., Lau, D. T., Weinman, S. A., Lemon, S. M., and Gale, M. Jr. (2006). Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. U.S.A. 103, 6001–6006.
Lu, H., Wu, Z., Xu, W., Yang, J., Chen, Y., and Li, L. (2011). Intestinal microbiota was assessed in cirrhotic patients with hepatitis B virus infection. Intestinal microbiota of HBV cirrhotic patients. Microb. Ecol. 61, 693–703.
Ma, R., Du, J. L., Huang, J., and Wu, C. Y. (2007). Additive effects of CpG ODN and R-848 as adjuvants on augmenting immune responses to HBsAg vaccination. Biochem. Biophys. Res. Commun. 361, 537–542.
Machida, K., Cheng, K. T., Sung, V. M., Levine, A. M., Foung, S., and Lai, M. M. (2006). Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J. Virol. 80, 866–874.
Mandal, P., Roychowdhury, S., Park, P. H., Pratt, B. T., Roger, T., and Nagy, L. E. (2010). Adiponectin and heme oxygenase-1 suppress TLR4/MyD, 88-independent signaling in rat Kupffer cells and in mice after chronic ethanol exposure. J. Immunol. 185, 4928–4937.
Manigold, T., Bocker, U., Hanck, C., Gundt, J., Traber, P., Antoni, C., and Rossol, S. (2003). Differential expression of toll-like receptors 2 and 4 in patients with liver cirrhosis. Eur. J. Gastroenterol. Hepatol. 15, 275–282.
Mao, T. K., Lian, Z. X., Selmi, C., Ichiki, Y., Ashwood, P., Ansari, A. A., Coppel, R. L., Shimoda, S., Ishibashi, H., and Gershwin, M. E. (2005). Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology 42, 802–808.
Marra, F., Romanelli, R. G., Giannini, C., Failli, P., Pastacaldi, S., Arrighi, M. C., Pinzani, M., Laffi, G., Montalto, P., and Gentilini, P. (1999). Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology 29, 140–148.
Matsunaga, N., Tsuchimori, N., Matsumoto, T., and Ii, M. (2011). TAK-242 (resatorvid), a small-molecule inhibitor of toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 79, 34–41.
Matsushita, K., Takeuchi, O., Standley, D. M., Kumagai, Y., Kawagoe, T., Miyake, T., Satoh, T., Kato, H., Tsujimura, T., Nakamura, H., and Akira, S. (2009). Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA, decay. Nature 458, 1185–1190.
Meylan, E., Curran, J., Hofmann, K., Moradpour, D., Binder, M., Bartenschlager, R., and Tschopp, J. (2005). Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172.
Miura, K., Kodama, Y., Inokuchi, S., Schnabl, B., Aoyama, T., Ohnishi, H., Olefsky, J. M., Brenner, D. A., and Seki, E. (2010). Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139, 323–34 e7.
Moritoki, Y., Lian, Z. X., Wulff, H., Yang, G. X., Chuang, Y. H., Lan, R. Y., Ueno, Y., Ansari, A. A., Coppel, R. L., Mackay, I. R., and Gershwin, M. E. (2007). AM, A production in primary biliary cirrhosis is promoted by the TLR9 ligand CpG, and suppressed by potassium channel blockers. Hepatology 45, 314–322.
Nakagawa, R., Naka, T., Tsutsui, H., Fujimoto, M., Kimura, A., Abe, T., Seki, E., Sato, S., Takeuchi, O., Takeda, K., Akira, S., Yamanishi, K., Kawase, I., Nakanishi, K., and Kishimoto, T. (2002). SOCS-1 participates in negative regulation of LPS responses. Immunity 17, 677–687.
Nanji, A. A., Khettry, U., and Sadrzadeh, S. M. (1994). Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver (disease). Proc. Soc. Exp. Biol. Med. 205, 243–247.
Naveau, S., Giraud, V., Borotto, E., Aubert, A., Capron, F., and Chaput, J. C. (1997). Excess weight risk factor for alcoholic liver disease. Hepatology 25, 108–111.
Nolan, J. P., and Leibowitz, A. I. (1978). Endotoxin and the liver. III Modification of acute carbon tetrachloride injury by polymyxin b – an antiendotoxin. Gastroenterology 75, 445–449.
O’Neill, L. A., Sheedy, F. J., and McCoy, C. E. (2011). MicroRNAs: the fine-tuners of toll-like receptor signalling. Nat. Rev. Immunol. 11, 163–175.
Osman, N., Adawi, D., Ahrne, S., Jeppsson, B., and Molin, G. (2007). Endotoxin- and D-galactosamine-induced liver injury improved by the administration of Lactobacillus, Bifidobacterium and blueberry. Dig. Liver Dis. 39, 849–856.
Otsuka, M., Kato, N., Moriyama, M., Taniguchi, H., Wang, Y., Dharel, N., Kawabe, T., and Omata, M. (2005). Interaction between the HCV NS3 protein and the host TBK1 protein leads to inhibition of cellular antiviral responses. Hepatology 41, 1004–1012.
Paik, Y. H., Lee, K. S., Lee, H. J., Yang, K. M., Lee, S. J., Lee, D. K., Han, K. H., Chon, C. Y., Lee, S. I., Moon, Y. M., and Brenner, D. A. (2006). Hepatic stellate cells primed with cytokines upregulate inflammation in response to peptidoglycan or lipoteichoic acid. Lab. Invest. 86, 676–686.
Paik, Y. H., Schwabe, R. F., Bataller, R., Russo, M. P., Jobin, C., and Brenner, D. A. (2003). Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 37, 1043–1055.
Petrasek, J., Dolganiuc, A., Csak, T., Nath, B., Hritz, I., Kodys, K., Catalano, D., Kurt-Jones, E., Mandrekar, P., and Szabo, G. (2011). Interferon regulatory factor 3 and type I interferons are protective in alcoholic liver injury in mice by way of crosstalk of parenchymal and myeloid cells. Hepatology 53, 649–660.
Quinn, S. R., and O’Neill, L. A. (2011). A trio of microRNAs that control toll-like receptor signalling. Int. Immunol. 23, 421–425.
Radaeva, S., Sun, R., Jaruga, B., Nguyen, V. T., Tian, Z., and Gao, B. (2006). Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 130, 435–452.
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S., and Medzhitov, R. (2004). Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241.
Rayes, N., Seehofer, D., Hansen, S., Boucsein, K., Muller, A. R., Serke, S., Bengmark, S., and Neuhaus, P. (2002). Early enteral supply of lactobacillus and fiber versus selective bowel decontamination: a controlled trial in liver transplant recipients. Transplantation 74, 123–127.
Rayes, N., Seehofer, D., Theruvath, T., Schiller, R. A., Langrehr, J. M., Jonas, S., Bengmark, S., and Neuhaus, P. (2005). Supply of pre- and probiotics reduces bacterial infection rates after liver transplantation – a randomized, double-blind trial. Am. J. Transplant. 5, 125–130.
Riordan, S. M., Skinner, N., Nagree, A., McCallum, H., McIver, C. J., Kurtovic, J., Hamilton, J. A., Bengmark, S., Williams, R., and Visvanathan, K. (2003). Peripheral blood mononuclear cell expression of toll-like receptors and relation to cytokine levels in cirrhosis. Hepatology 37, 1154–1164.
Rivera, C. A., Adegboyega, P., van Rooijen, N., Tagalicud, A., Allman, M., and Wallace, M. (2007). Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 47, 571–579.
Rivera, C. A., Gaskin, L., Allman, M., Pang, J., Brady, K., Adegboyega, P., and Pruitt, K. (2010). Toll-like receptor-2 deficiency enhances non-alcoholic steatohepatitis. BMC Gastroenterol. 10, 52. doi:10.1186/1471-230X-10-52
Roderburg, C., Urban, G. W., Bettermann, K., Vucur, M., Zimmermann, H., Schmidt, S., Janssen, J., Koppe, C., Knolle, P., Castoldi, M., Tacke, F., Trautwein, C., and Luedde, T. (2011). MicroRNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53, 209–218.
Saito, T., Owen, D. M., Jiang, F., Marcotrigiano, J., and Gale, M. Jr. (2008). Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454, 523–527.
Saitoh, S., Akashi, S., Yamada, T., Tanimura, N., Kobayashi, M., Konno, K., Matsumoto, F., Fukase, K., Kusumoto, S., Nagai, Y., Kusumoto, Y., Kosugi, A., and Miyake, K. (2004). Lipid A antagonist, lipid IVa, is distinct from lipid A in interaction with toll-like receptor 4 (TLR4)-MD-2 and ligand-induced TLR4 oligomerization. Int. Immunol. 16, 961–969.
Sato, K., Ishikawa, T., Okumura, A., Yamauchi, T., Sato, S., Ayada, M., Matsumoto, E., Hotta, N., Oohashi, T., Fukuzawa, Y., and Kakumu, S. (2007). Expression of toll-like receptors in chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 22, 1627–1632.
Schnabl, B., Brandl, K., Fink, M., Gross, P., Taura, K., Gabele, E., Hellerbrand, C., and Falk, W. (2008). A TLR4/MD2 fusion protein inhibits LPS-induced pro-inflammatory signaling in hepatic stellate cells. Biochem. Biophys. Res. Commun. 375, 210–214.
Schott, E., Witt, H., Neumann, K., Taube, S., Oh, D. Y., Schreier, E., Vierich, S., Puhl, G., Bergk, A., Halangk, J., Weich, V., Wiedenmann, B., and Berg, T. (2007). A toll-like receptor 7 single nucleotide polymorphism protects from advanced inflammation and fibrosis in male patients with chronic HCV-infection. J. Hepatol. 47, 203–211.
Schwabe, R. F., Bataller, R., and Brenner, D. A. (2003). Human hepatic stellate cells express CCR5 and RANTES, to induce proliferation and migration. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G949–G958.
Seki, E., and Brenner, D. A. (2008). Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48, 322–335.
Seki, E., de Minicis, S., Inokuchi, S., Taura, K., Miyai, K., van Rooijen, N., Schwabe, R. F., and Brenner, D. A. (2009a). CCR2 promotes hepatic fibrosis in mice. Hepatology 50, 185–197.
Seki, E., De Minicis, S., Gwak, G. Y., Kluwe, J., Inokuchi, S., Bursill, C. A., Llovet, J. M., Brenner, D. A., and Schwabe, R. F. (2009b). CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Invest. 119, 1858–1870.
Seki, E., De Minicis, S., Osterreicher, C. H., Kluwe, J., Osawa, Y., Brenner, D. A., and Schwabe, R. F. (2007). TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 13, 1324–1332.
Seki, E., and Schnabl, B. (2012). Role of innate immunity and the microbiota in liver fibrosis – crosstalk between the liver and gut. J. Physiol. (Lond.) 590, 447–458.
Seki, E., Tsutsui, H., Nakano, H., Tsuji, N., Hoshino, K., Adachi, O., Adachi, K., Futatsugi, S., Kuida, K., Takeuchi, O., Okamura, H., Fujimoto, J., Akira, S., and Nakanishi, K. (2001). Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J. Immunol. 166, 2651–2657.
Seki, E., Tsutsui, H., Tsuji, N. M., Hayashi, N., Adachi, K., Nakano, H., Futatsugi-Yumikura, S., Takeuchi, O., Hoshino, K., Akira, S., Fujimoto, J., and Nakanishi, K. (2002). Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 169, 3863–3868.
Sha, T., Sunamoto, M., Kitazaki, T., Sato, J., Ii, M., and Iizawa, Y. (2007). Therapeutic effects of TAK-242, a novel selective toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur. J. Pharmacol. 571, 231–239.
Sheedy, F. J., Palsson-McDermott, E., Hennessy, E. J., Martin, C., O’Leary, J. J., Ruan, Q., Johnson, D. S., Chen, Y., and O’Neill, L. A. (2010). Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA, miR-21. Nat. Immunol. 11, 141–147.
Shehata, M. A., Abou El-Enein, A., and El-Sharnouby, G. A. (2006). Significance of toll-like receptors 2 and 4 mRNA, expression in chronic hepatitis C virus infection. Egypt J. Immunol. 13, 141–152.
Shiina, M., and Rehermann, B. (2008). Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology 47, 385–395.
Shimoda, S., Harada, K., Niiro, H., Shirabe, K., Taketomi, A., Maehara, Y., Tsuneyama, K., Nakanuma, Y., Leung, P., Ansari, A. A., Gershwin, M. E., and Akashi, K. (2011). Interaction between toll-like receptors and natural killer cells in the destruction of bile ducts in primary biliary cirrhosis. Hepatology 53, 1270–1281.
Soares, J. B., Pimentel-Nunes, P., Afonso, L., Rolanda, C., Lopes, P., Roncon-Albuquerque, R. Jr., Goncalves, N., Boal-Carvalho, I., Pardal, F., Lopes, S., Macedo, G., Lara-Santos, L., Henrique, R., Moreira-Dias, L., Goncalves, R., Dinis-Ribeiro, M., and Leite-Moreira, A. F. (2012). Increased hepatic expression of TLR2 and TLR4 in the hepatic inflammation-fibrosis-carcinoma sequence. Innate Immun. (in press).
Szabo, G., Velayudham, A., Romics, L. Jr., and Mandrekar, P. (2005). Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: the role of toll-like receptors 2 and 4. Alcohol. Clin. Exp. Res. 29, 140S–145S.
Takahashi, K., Asabe, S., Wieland, S., Garaigorta, U., Gastaminza, P., Isogawa, M., and Chisari, F. V. (2010). Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. U.S.A. 107, 7431–7436.
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820.
Takii, Y., Nakamura, M., Ito, M., Yokoyama, T., Komori, A., Shimizu-Yoshida, Y., Nakao, R., Kusumoto, K., Nagaoka, S., Yano, K., Abiru, S., Ueki, T., Matsumoto, T., Daikoku, M., Taniguchi, K., Fujioka, H., Migita, K., Yatsuhashi, H., Nakashima, M., Harada, M., and Ishibashi, H. (2005). Enhanced expression of type I interferon and toll-like receptor-3 in primary biliary cirrhosis. Lab. Invest. 85, 908–920.
Tazi, K. A., Quioc, J. J., Saada, V., Bezeaud, A., Lebrec, D., and Moreau, R. (2006). Upregulation of TNF-alpha production signaling pathways in monocytes from patients with advanced cirrhosis: possible role of Akt and IRAK-M. J. Hepatol. 45, 280–289.
Teratani, T., Tomita, K., Suzuki, T., Oshikawa, T., Yokoyama, H., Shimamura, K., Tominaga, S., Hiroi, S., Irie, R., Okada, Y., Kurihara, C., Ebinuma, H., Saito, H., Hokari, R., Sugiyama, K., Kanai, T., Miura, S., and Hibi, T. (2012). A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 142, 152–164.
Testro, A. G., Gow, P. J., Angus, P. W., Wongseelashote, S., Skinner, N., Markovska, V., and Visvanathan, K. (2010). Effects of antibiotics on expression and function of toll-like receptors 2 and 4 on mononuclear cells in patients with advanced cirrhosis. J. Hepatol. 52, 199–205.
Thomas, A., Laxton, C., Rodman, J., Myangar, N., Horscroft, N., and Parkinson, T. (2007). Investigating toll-like receptor agonists for potential to treat hepatitis C virus infection. Antimicrob. Agents Chemother. 51, 2969–2978.
Thompson, A. J., Colledge, D., Rodgers, S., Wilson, R., Revill, P., Desmond, P., Mansell, A., Visvanathan, K., and Locarnini, S. (2009). Stimulation of the interleukin-1 receptor and toll-like receptor 2 inhibits hepatitis B virus replication in hepatoma cell lines in vitro. Antivir. Ther. (Lond.) 14, 797–808.
Tsutsui, H., Imamura, M., Fujimoto, J., and Nakanishi, K. (2010). The TLR4/TRIF-mediated activation of NLRP3 inflammasome underlies endotoxin-induced liver injury in mice. Gastroenterol. Res. Pract. 2010, 641–865.
Turer, E. E., Tavares, R. M., Mortier, E., Hitotsumatsu, O., Advincula, R., Lee, B., Shifrin, N., Malynn, B. A., and Ma, A. (2008). Homeostatic MyD, 88-dependent signals cause lethal inflammation in the absence of A20. J. Exp. Med. 205, 451–464.
Turnbaugh, P. J., Ley, R. E., Mahowald, M. A., Magrini, V., Mardis, E. R., and Gordon, J. I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031.
Uesugi, T., Froh, M., Arteel, G. E., Bradford, B. U., and Thurman, R. G. (2001). Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 34, 101–108.
Uesugi, T., Froh, M., Arteel, G. E., Bradford, B. U., Wheeler, M. D., Gabele, E., Isayama, F., and Thurman, R. G. (2002). Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J. Immunol. 168, 2963–2969.
Velayudham, A., Dolganiuc, A., Ellis, M., Petrasek, J., Kodys, K., Mandrekar, P., and Szabo, G. (2009). VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology 49, 989–997.
Vincent, I. E., Zannetti, C., Lucifora, J., Norder, H., Protzer, U., Hainaut, P., Zoulim, F., Tommasino, M., Trepo, C., Hasan, U., and Chemin, I. (2011). Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS ONE 6, e26315. doi:10.1371/journal.pone.0026315
Wang, B., Trippler, M., Pei, R., Lu, M., Broering, R., Gerken, G., and Schlaak, J. F. (2009). Toll-like receptor activated human and murine hepatic stellate cells are potent regulators of hepatitis C virus replication. J. Hepatol. 51, 1037–1045.
Watanabe, A., Hashmi, A., Gomes, D. A., Town, T., Badou, A., Flavell, R. A., and Mehal, W. Z. (2007). Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 46, 1509–1518.
Weeratna, R. D., Makinen, S. R., McCluskie, M. J., and Davis, H. L. (2005). TLR agonists as vaccine adjuvants: comparison of CpG ODN and resiquimod (R-848). Vaccine 23, 5263–5270.
Wu, J., Lu, M., Meng, Z., Trippler, M., Broering, R., Szczeponek, A., Krux, F., Dittmer, U., Roggendorf, M., Gerken, G., and Schlaak, J. F. (2007). Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology 46, 1769–1778.
Wu, J., Meng, Z., Jiang, M., Zhang, E., Trippler, M., Broering, R., Bucchi, A., Krux, F., Dittmer, U., Yang, D., Roggendorf, M., Gerken, G., Lu, M., and Schlaak, J. F. (2010). Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology 129, 363–374.
Xu, H., An, H., Hou, J., Han, C., Wang, P., Yu, Y., and Cao, X. (2008a). Phosphatase PTP1B negatively regulates MyD88- and TRIF-dependent proinflammatory cytokine and type I interferon production in TLR-triggered macrophages. Mol. Immunol. 45, 3545–3552.
Xu, N., Yao, H. P., Sun, Z., and Chen, Z. (2008b). Toll-like receptor 7 and 9 expression in peripheral blood mononuclear cells from patients with chronic hepatitis B and related hepatocellular carcinoma. Acta Pharmacol. Sin. 29, 239–244.
Xu, J., Lai, K. K., Verlinsky, A., Lugea, A., French, S. W., Cooper, M. P., Ji, C., and Tsukamoto, H. (2011). Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J. Hepatol. 55, 673–682.
Yamamoto, M., and Takeda, K. (2010). Current views of toll-like receptor signaling pathways. Gastroenterol. Res. Pract. 2010, 240–365.
Yan, A. W., Fouts, D. E., Brandl, J., Stärkel, P., Torralba, M., Schott, E., Tsukamoto, H., Nelson, K. E., Brenner, D. A., and Schnabl, B. (2011). Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 53, 96–105.
Yan, X., Lin, Z., Chen, F., Zhao, X., Chen, H., Ning, Y., and Chen, Y. G. (2009). Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-beta signaling. J. Biol. Chem. 284, 30097–30104.
Yin, M., Bradford, B. U., Wheeler, M. D., Uesugi, T., Froh, M., Goyert, S. M., and Thurman, R. G. (2001). Reduced early alcohol-induced liver injury in CD14-deficient mice. J. Immunol. 166, 4737–4742.
Yin, S., and Gao, B. (2010). Toll-like receptor 3 in liver diseases. Gastroenterol. Res. Pract. 2010, 6.
Zanoni, I., Ostuni, R., Marek, L. R., Barresi, S., Barbalat, R., Barton, G. M., Granucci, F., and Kagan, J. C. (2011). CD14 controls the LPS-induced endocytosis of toll-like receptor 4. Cell 147, 868–880.
Zhang, W., Gu, Y., Chen, Y., Deng, H., Chen, L., Chen, S., Zhang, G., and Gao, Z. (2010a). Intestinal flora imbalance results in altered bacterial translocation and liver function in rats with experimental cirrhosis. Eur. J. Gastroenterol. Hepatol. 22, 1481–1486.
Zhang, Y., Lian, J. Q., Huang, C. X., Wang, J. P., Wei, X., Nan, X. P., Yu, H. T., Jiang, L. L., Wang, X. Q., Zhuang, Y., Li, X. H., Li, Y., Wang, P. Z., Robek, M. D., and Bai, X. F. (2010b). Overexpression of toll-like receptor 2/4 on monocytes modulates the activities of CD, 4(+)CD, 25(+) regulatory T cells in chronic hepatitis B virus infection. Virology 397, 34–42.
Zhang, Y. L., Guo, Y. J., Bin, L., and Sun, S. H. (2009). Hepatitis C virus single-stranded RNA induces innate immunity via toll-like receptor 7. J. Hepatol. 51, 29–38.
Zhou, J., Huang, Y., Tian, D., Xu, D., Chen, M., and Wu, H. (2009). Expression of toll-like receptor 9 in peripheral blood mononuclear cells from patients with different hepatitis B and C viral loads. J. Huazhong Univ. Sci. Technol. Med. Sci. 29, 313–317.
Keywords: toll-like receptors, non-alcoholic steatohepatitis, alcoholic liver disease, hepatitis C, primary biliary cirrhosis, cystic fibrosis
Citation: Yang L and Seki E (2012) Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms. Front. Physio. 3:138. doi: 10.3389/fphys.2012.00138
Received: 01 January 2012; Paper pending published: 28 January 2012;
Accepted: 24 April 2012; Published online: 22 May 2012.
Edited by:
Honglei Weng, University of Heidelberg, GermanyReviewed by:
Hiroko Tsutsui, Hyogo College of Medicine, JapanJinsheng Guo, Fu Dan University, Zhongshan Hospital, China
Copyright: © 2012 Yang and Seki. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ekihiro Seki, Division of Gastroenterology, Department of Medicine, University of California San Diego, School of Medicine, 9500 Gilman Drive MC 0702, Leichtag Biomedical Research Building Room 118B, La Jolla, CA 92093-0702, USA. e-mail: ekseki@ucsd.edu