 S. D. L. Postnikoff T. A. A. Harkness*
S. D. L. Postnikoff T. A. A. Harkness*- Department of Anatomy and Cell Biology, University of Saskatchewan, Saskatoon, SK, Canada
The longevity of an organism depends on the health of its cells. Throughout life cells are exposed to numerous intrinsic and extrinsic stresses, such as free radicals, generated through mitochondrial electron transport, and ultraviolet irradiation. The cell has evolved numerous mechanisms to scavenge free radicals and repair damage induced by these insults. One mechanism employed by the yeast Saccharomyces cerevisiae to combat stress utilizes the Anaphase Promoting Complex (APC), an essential multi-subunit ubiquitin-protein ligase structurally and functionally conserved from yeast to humans that controls progression through mitosis and G1. We have observed that yeast cells expressing compromised APC subunits are sensitive to multiple stresses and have shorter replicative and chronological lifespans. In a pathway that runs parallel to that regulated by the APC, members of the Forkhead box (Fox) transcription factor family also regulate stress responses. The yeast Fox orthologs Fkh1 and Fkh2 appear to drive the transcription of stress response factors and slow early G1 progression, while the APC seems to regulate chromatin structure, chromosome segregation, and resetting of the transcriptome in early G1. In contrast, under non-stress conditions, the Fkhs play a complex role in cell-cycle progression, partially through activation of the APC. Direct and indirect interactions between the APC and the yeast Fkhs appear to be pivotal for lifespan determination. Here we explore the potential for these interactions to be evolutionarily conserved as a mechanism to balance cell-cycle regulation with stress responses.
Introduction
Throughout history humanity has sought to understand the reasons for aging and dying. Relatively recently, genetic and biochemical studies have offered some insight into these complex processes. The health of an organism is directly related to the health of its cellular constituents, with genomic instability being a dominant force that leads to either senescence of stem cells or uncontrolled growth and tumor formation. Advances in our understanding of these processes have been made through the identification of gene products that can increase or decrease cellular health span, influencing the incidence of tissue degeneration and age-related diseases, such as Alzheimer’s, diabetes, and cancer (reviewed in Kloet and Burgering, 2011; Stünkel and Campbell, 2011; Ziv and Hu, 2011; Jia et al., 2012; Salminen and Kaarniranta, 2012). Genetic screens in the nematode Caenorhabditis elegans identified members of the insulin-signaling pathway as regulators of the aging process. Specifically, decreased activity of the PI3K/AKT pathway, a prominent pathway overactive in many cancer cells, increases longevity in a variety of model organisms (Kloet and Burgering, 2011; Speakman and Mitchell, 2011).
Regulation of growth in conjunction with stress resistance and genomic stability was found to rely on the worm Daf-16, a stress response transcription factor featuring the forkhead box (Fox) DNA-binding domain (Kenyon et al., 1993; Lin et al., 2001; Libina et al., 2003; Murphy et al., 2003). Fox-containing proteins have been identified from yeast to worms and insects to humans (Baldauf, 1999; Mazet et al., 2003). Fox family members regulate diverse biological processes, such as metabolism, embryonic development, differentiation, cell migration, invasion, cell-cycle progression, apoptosis, autophagy, immunity, DNA-damage repair, and toxin scavenging (Tuteja and Kaestner, 2007). A large number of Fox genes have been identified in higher eukaryotic systems (Kaufmann and Knöchel, 1996; Murakami et al., 2010), which makes it very difficult to gain insight into any one Fox protein or subfamily. The brewing yeast Saccharomyces cerevisiae contains only four Fox proteins (Fkh1, Fkh2, Fhl1, and Hcm1; Murakami et al., 2010), presenting an opportunity to learn in greater depth how individual Fox proteins are regulated and what they control. It has become apparent that unraveling the regulation, targets, and evolution of the Fox family of transcription factors is crucial for understanding cancer biology and aging, as this group contains suspected oncogenes, as well as bona fide tumor suppressors and longevity determining factors. Emerging evidence highlights the importance of these factors in cell-cycle regulation and stress responses, through the regulation of the evolutionary conserved Cyclins, Cyclin Dependent Kinase inhibitors (CDKIs), and the Anaphase Promoting Complex (APC), a ubiquitin-protein ligase. Here we explore the evolutionary conservation of these mechanisms from yeast to humans.
The Evolutionarily Conserved Fox Factors
Fox transcription factors have been identified in a wide range of species from yeast to humans based on the common 110-amino-acid winged-helix DNA-binding domain, known as the forkhead box (Kaufmann et al., 1995; Kaufmann and Knöchel, 1996). Due to the highly conserved nature of the Fox DNA-binding domain, all Fox proteins bind to the consensus core nucleotide sequence A/CAAC/TA (Lalmansingh et al., 2012). Although Fox genes have been found in animals, as well as yeast and other fungi, plants do not encode these transcription factors. This suggests the proto-Fox gene originated in the animal/fungal ancestor after the evolutionary split of autotrophs and heterotrophs (Baldauf, 1999). Phylogenetic and comparative analyses have identified over 100 Fox genes in humans that can be subgrouped into 19 subclasses (A to S) with further subdivision based on the relationship between vertebrate and invertebrate genes (Kaufmann and Knöchel, 1996; Hannenhalli and Kaestner, 2009; Murakami et al., 2010). Analyses of fungal Fox genes found they were equally related to all animal Fox genes, suggesting only one proto-Fox gene was present at the divergence of animals and fungus (Baldauf, 1999).
A fascinating aspect of Fox gene function is their involvement in many developmental defects and cancer (Lehmann et al., 2003; Myatt and Lam, 2007). This review will focus on two closely related subclasses: the aging and tumor suppressor FoxO and the oncogenic FoxM.
The FoxO subclass is highly conserved throughout animals and is primarily responsible for regulation of G2-M and G1-S cell-cycle checkpoints, as well as for the expression of stress response, DNA repair, and apoptotic genes (Brunet et al., 1999; Medema et al., 2000; Dijkers et al., 2002; Kops et al., 2002; Nemoto and Finkel, 2002; Tran et al., 2002). The FoxO subclass is of special interest as its members seem intrinsic to tumor suppression and lifespan extension. Intense investigation of post-translational regulation of the FoxOs is underway, wherein phosphorylation, acetylation, and ubiquitination (both poly and mono) are known to influence FoxO nuclear shuttling, DNA-binding ability, transcriptional activity, and protein stability (Calnan and Brunet, 2008; Boccitto and Kalb, 2011; Daitoku et al., 2011; Huang and Tindall, 2011; Tzivion et al., 2011; Zhao et al., 2011). Under non-stress conditions, phosphorylation of FoxOs by growth factor (insulin/insulin-like growth factor, TOR2C and Mitogen activated kinase) pathways results in cytosolic (rather than nuclear) localization and proteasome-dependent degradation via ubiquitination by the Skp/Cullin/F-box ubiquitin-protein ligase (E3) complex SCFSkp2. Conversely, stress-induced phosphorylation signals (oxidative stress activated c-Jun N-terminal kinase and MST1, as well as starvation response AMPK) result in nuclear localization and the transcription of specific factors. Furthermore, FoxO acetylation leads to dissociation from DNA, while simultaneously stabilizing FoxO proteins by blocking ubiquitination and subsequent degradation.
FoxM1, the single member of the FoxM subfamily, is found only in proliferating cells, and is also involved in cell-cycle regulation, aging, and cancer (Korver et al., 1997; Mazet et al., 2003; Laoukili et al., 2007; Tang et al., 2008; Pandit et al., 2009; Petrovic et al., 2010; Wang et al., 2010). Cells deficient in FoxM1 show delays in G2/M and G1/S progression, as well as defects in chromosome segregation and cytokinesis (Laoukili et al., 2005; Wang et al., 2005, 2008; Ustiyan et al., 2009). Increased FoxM1 protein has been found in numerous types of cancer and may be involved in early stages of tumorigenesis (Wilson et al., 2011). Like FoxO proteins, FoxM1 is also involved in DNA repair, implicating a need for FoxM1 in genomic stability and survival. Phylogenetic analyses suggest that the FoxM subclass is an offshoot of the FoxO family that split early in vertebrate evolution as no known structural ortholog has been found in modern chordate or invertebrate species (Mazet et al., 2003).
A possible direct connection between FoxO3a and FoxM1 was suggested by microarray analyses of FoxO3a overexpressing cells, which identified FoxM1 as a gene differentially repressed when Fox3a levels were increased (Delpuech et al., 2007). Of the 151 differentially expressed genes (>2-fold up or down), 59 (39.1%) were downregulated. A large percentage of the cell-cycle regulated genes were downregulated, consistent with a role for FoxO3a in cell-cycle inhibition. FoxM1 was previously identified as a Myc target gene containing E-boxes (CACTGT) within the FoxM1 promoter at −1244 and −1091 (Figure 1; Fernandez et al., 2003). Myc forms a heterodimer with Max, which binds the E-box to activate gene expression. Contrary to this, Max can also dimerize with Mxi1 to bind E-boxes, resulting in transcriptional repression (Delpuech et al., 2007). Increased FoxO3a expression upregulated members of the Mad/Mxi family, which was predicted to antagonize Myc function. It was concluded that downregulation of FoxM1 following FoxO3a induction may be an indirect effect of Myc inhibition via up-regulation of the Myc antagonists Mxi1 and Mad. It was also observed that increased FoxO3a expression decreased Myc protein levels, providing an additional mechanism for FoxM1 repression. However, it should be noted that FoxM1 contains a TAAACA Fox binding site at position −88 within the FoxM1 promoter (Figure 1), indicating that FoxO3a may indeed bind and repress the FoxM1 promoter, perhaps first requiring the displacement of Myc.
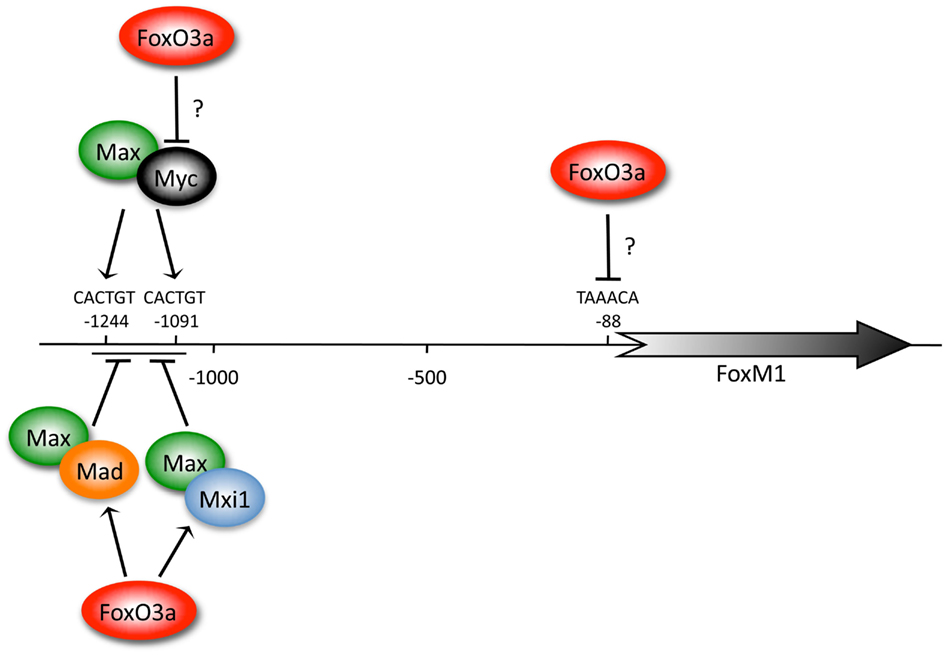
Figure 1. FoxM1 expression is repressed by FoxO3a. FoxO3a may repress FoxM1 expression in one of three ways, which may not be mutually exclusive. First, FoxO3a may bind to a Fox consensus site at position −88 of the FoxM1 promoter. This could lead to FoxM1 repression. Second, expression of the Myc antagonists Mad and Mxi1 are driven by FoxO3a. Mad and Mxi1 compete with Myc to dimerize with Max. The Max/Myc dimer binds to E-boxes (CACTGT) located within the FoxM1 promoter to drive FoxM1 expression, while Mad/Max and Mxi1/Max dimers bind the same E-boxes, but repress expression. Thus, increased expression of Mxi1 and Mad by FoxO3a could inhibit FoxM1 expression by blocking Myc/Max dimerization. Third, Myc protein levels decrease when FoxO3a expression is increased, perhaps through a post-translational mechanism, providing another method to potentially repress FoxM1 expression following FoxO3a activation. This figure is based on work from Delpuech et al. (2007) and Fernandez et al. (2003).
The reciprocal interaction of FoxO3a and FoxM1 is critical to stave off cancer (Wilson et al., 2011). Previous work indicated that repression of FoxM1, via FoxO3a, was necessary to suppress the growth of breast cancer cells treated with the EGRF inhibitor Gefitinib (McGovern et al., 2009). It was observed that Gefitinib induced the expression of FoxO3a in association with the repression of FoxM1. Silencing of FoxO3a increased FoxM1 expression in response to Gefitinib, which was associated with increased cell proliferation and reduced cell death. Similarly, increased FoxM1 expression reduced Gefitinib induced cell-cycle arrest.
FoxO3a and FoxM1 also appear to oppose one another in ERα-positive breast cancer cells. FoxO3a was identified as a binding partner for ERα and ERβ, which was important for the suppression of estrogen-dependent breast cancer (Zou et al., 2008). This interaction resulted in the repression of ERα-regulated gene expression (Figure 2). Conversely, FoxM1 was shown to drive the expression of ERα via “promoter A” of the two-set ERα promoter (Figure 2), and that silencing of FoxM1 completely blocked ERα expression (Madureira et al., 2006). It was also observed that FoxO3a could promote weak expression of ERα through ERα “promoter B.” However, both FoxM1 and FoxO3a could bind to both ERα promoter sets. Lastly, although FoxM1 and FoxO3a could be co-immunoprecipitated the relevance of this observation was not pursued. The compiled evidence suggests that FoxM1 and FoxO3a may form a complex that cooperatively regulates ERα expression. However, these interactions could also be interpreted to imply that FoxO3a can bind FoxM1 at promoters to inhibit FoxM1 activity. Much more work is required to elucidate the mechanisms regulating ERα expression and breast cancer progression.
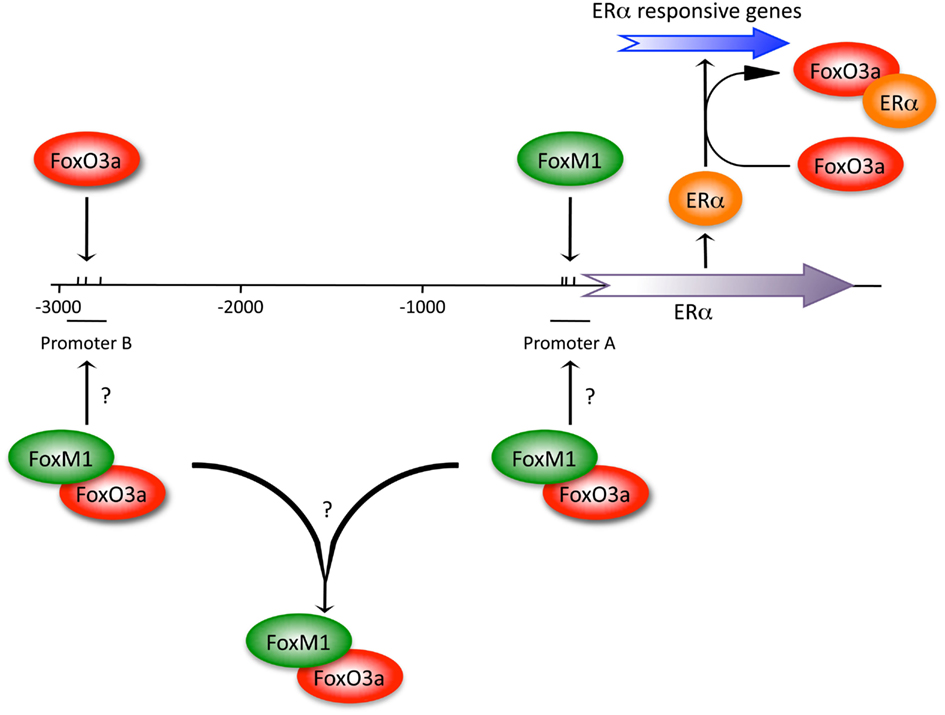
Figure 2. A complex interaction between FoxM1 and FoxO3a controls ERα expression. The ERα promoter contains two clusters of Fox binding sites, one immediately upstream of the ERα start site, “Promoter A” and a second site at approximately position −3000, “Promoter B.” ChIP studies showed that FoxM1 and FoxO3a could bind both promoters. Based on RNAi experiments, FoxM1 and FoxO3a were both capable of driving ERα expression, although through Promoters A and B, respectively, with FoxM1 playing a dominant role. A second level of complexity involves a protein/protein interaction between FoxO3a and ERα that blocks ERα from promoting the transcription of ERα responsive genes. A potential FoxO3a/FoxM1 physical interaction provides a third possible layer of complexity. The relevance of the FoxO3a/FoxM1 interaction remains unknown. This figure is based on work by Zou et al. (2008) and Madureira et al. (2006).
Yeast Fox Proteins
The budding yeast S. cerevisiae contains four Fox genes: FKH1, FKH2, HCM1, and FHL1. FHL1 regulates ribosome biogenesis (Rudra et al., 2005), and HCM1 regulates progression through G2, preparing the cell for mitosis (Pramila et al., 2006). Although both may play a role in lifespan determination, their individual deletion did not influence yeast replicative lifespan (RLS; a measure of how many daughter cells a single mother can produce; Kennedy et al., 1994; Wei et al., 2008). Here we focus on FKH1 and FKH2 as they show conserved function with human FoxM/FoxO genes (Murakami et al., 2010; Postnikoff et al., 2012). Genetic redundancy is suggested for these two factors as the combined deletion of both FKH1 and FKH2 is necessary to alter growth, stress response, longevity, cell morphology, and gene transcription phenotypes (Hollenhorst et al., 2000; Zhu et al., 2000; Shapira et al., 2004; Sherriff et al., 2007; Voth et al., 2007; Postnikoff et al., 2012). Evolutionary conservation for FKH1 and FKH2 with higher Fox genes is suggested by their similar involvement in ROS induced cell-cycle arrest and resistance to oxidative stress during stationary phase (Shapira et al., 2004; Postnikoff et al., 2012), as well as in cell-cycle regulation through both G1 and G2/M gene clusters (Zhu et al., 2000). Finally, we have recently found that deletion of both FKH1 and FKH2 reduces lifespan in a manner that inhibits lifespan extension due to caloric restriction, while over-expression of one or both genes extends lifespan (Postnikoff et al., 2012), hallmarks of human FoxO genes.
Fkh1 and Fkh2 are capable of binding the Forkhead box consensus site (TAAACA) first identified for Xenopus XFD1, 2, and 3 (Kaufmann et al., 1995). Under normal laboratory conditions Fkh2 primarily occupies known binding sites of the Clb2 gene cluster. Fkh2 forms a complex with the MADS-box transcription factor Mcm1, while Fkh1 does not (Hollenhorst et al., 2001). Cell-cycle specific activation of this complex is initiated by the expression and binding of the co-activator Ndd1 to Fkh2, which switches the function of Fkh2 from repressor to activator (Loy et al., 1999; Koranda et al., 2000). Fkh1 may function as a co-regulator of this process, or may function as a primary regulator under alternate growth conditions. In FKH2 deletion strains, cell-cycle specific expression of Fkh2 targets is not disrupted, as it is in fkh1Δ fkh2Δ double mutant strains (Zhu et al., 2000; Hollenhorst et al., 2001), suggesting Fkh1 can function at the same loci without Fkh2/Ndd1 occupancy (Reynolds et al., 2003). However the mechanism by which Fkh1 regulates these genes in a periodic cell-cycle dependent manner, in the absence of Fkh2 and Ndd1, requires further investigation.
Common Biology of the Fkhs and the Foxs
Mitotic progression genes are common targets of both yeast and human Fox proteins. As described earlier, the yeast Fkh1 and Fkh2 regulate clusters of genes required for cell-cycle progression, such as the CLN2 and CLB2 gene clusters (Zhu et al., 2000), which include targets (Iqg1, Cdc20, the B type cyclins Clb1, Clb2, Clb5, and the yeast the polo-like kinase Cdc5) and regulators (Clb2, Cdc20, and Cdc5) of the APC (Ko et al., 2007; Sari et al., 2007; Qiao et al., 2010). The APC is a highly conserved ubiquitin-protein ligase (E3) that primarily controls progression through mitosis and G1. The observation that the Fkh proteins control transcription of many genes required for APC function suggests that the APC may be a critical downstream target of the Fkhs (discussed in more detail below). The Fkh proteins also control the transcription of the Histone gene cluster (Zhu et al., 2000). Interestingly, the yeast APC is required for histone protein expression and post-translational modification, which may be a shared feature with the Fkh proteins, as deletion of FKH1 and FKH2 in a mutant apc5CA background further impairs histone protein levels (Turner et al., 2010; Postnikoff et al., 2012). In mammals, FoxM1 primarily regulates G2/M genes, such as B type cyclins, the polo-like kinase (PLK-1), Aurora B, Skp2, INCENP, Nek2, and the centromere specific proteins CENP-A, CENP-B, and CENP-F (Laoukili et al., 2005; Wang et al., 2005). Like in yeast, many of the FoxM1 controlled genes are either required for APC function (PLK-1, B type cyclins), or are targeted for degradation by the APC (Aurora B, Skp2, Nek2, PLK-1, and B type cyclins; Pfleger and Kirschner, 2000; Qiao et al., 2010). An additional level of striking conservation between mammalian and yeast cells is the positive feedback loop that initiates an irreversible commitment to mitosis, wherein Cdk-cyclin B complexes and polo-like kinases phosphorylate FoxM1 and the Fkhs to increase their transcriptional activity (Murakami et al., 2010).
Many of the FoxM1 targets are required for genomic stability (O’Brien et al., 2007; van der Waal et al., 2012). For example, silencing of CENP-F (activated by FoxM1) resulted in the loss of the mitotic checkpoint proteins Mad1 and BubR1, which are required to block APC activity until appropriate (Laoukili et al., 2005). The subsequent inappropriate activation of the APC in the presence of DNA damage could result in potential mitotic catastrophe (D’Arcy et al., 2010; Lara-Gonzalez et al., 2011). Likewise, the Chromosomal Passenger Complex, composed of Aurora B kinase, INCENP, Survivin, and Borealin, regulates the mitotic checkpoint to ensure accurate segregation of mitotic chromosomes (van der Waal et al., 2012). Thus, FoxM1 expression at least in part increases mitotic checkpoint control and APC function, ensuring proper transit through mitosis. Considering that improved mitotic checkpoints should result in increased genomic stability, not in cancer development, this apparent paradox may reflect a mechanism whereby increased FoxM1 expression in cancer cells allows the defective cell to bypass cellular mechanisms aimed at destroying it. FoxM1 itself is a target of the APCCdh1 G1 specific complex (Laoukili et al., 2008; Park et al., 2008). This is an interesting finding since FoxM1 joins the growing list of APC activators that are later targeted by the APC for degradation to complete a negative feedback loop. It is thought that in order to shut down the expression of mitosis-specific genes, FoxM1 must be degraded as cells exit mitosis. Our unpublished data suggests that the targeting of Fox proteins by the APC is indeed a conserved process, as the yeast Fkh1 is targeted by the APC during G1 for degradation (Malo, Postnikoff, and Harkness, unpublished).
Stability of the FoxO and FoxM proteins is controlled antagonistically by the SCF and APC ubiquitin-protein ligase/E3 complexes, respectively. The APC targets FoxM1 for degradation, and indirectly stabilizes FoxO during G1 (Laoukili et al., 2008; Park et al., 2008). The SCFSkp2 complex targets phosphorylated FoxO proteins for degradation (Huang et al., 2005; Huang and Tindall, 2011). The APC potentially controls this process through targeted degradation of the F-box protein Skp2 (van Leuken et al., 2008); degradation of Skp2 by the APCCdh1 blocks SCFSkp2 function, thereby delaying FoxO degradation and allowing increased FoxO tumor suppressor activity. There appears to be a battle over the control of Skp2 stability in mammalian cells. AKT phosphorylates FoxO proteins, leading to their SCFSkp2-dependent ubiquitination and degradation. AKT also phosphorylates Skp2 (Gao et al., 2009; Lin et al., 2009). AKT phosphorylation of Skp2 is believed to signal cytosolic localization of Skp2, thereby protecting Skp2 from APC-dependent ubiquitination and degradation. However, this mechanism may be cell type dependent, as another study was not able to reproduce these results (Bashir et al., 2010). Nonetheless, the APC and AKT may be competing for Skp2’s attention, with the winner perhaps deciding between cell health and death.
Opposing FoxO3a and FoxM1 function exists at the transcriptional target level as well. As already mentioned, FoxM1 is repressed by FoxO3a (Figure 1). However, FoxO3a also represses the expression of many cell-cycle specific genes, consistent with its role in blocking cell-cycle progression (Delpuech et al., 2007). The G2/M genes activated by FoxM1, such as UBE2C, NEK2, CENP-F, and Aurora, were repressed by FoxO3a (Laoukili et al., 2005; Delpuech et al., 2007). FoxO3a was more likely to activate the expression of genes required for apoptosis/stress response, transcription, and signaling (Delpuech et al., 2007). This pattern was also observed when asynchronous fkh1Δ fkh2Δ cells were used for microarray analyses, with downregulation of genes involved in cell stress response (RNR1, ALK1, IRC8, PHO5/11/12, DSE1/2, CRG1; Zhu et al., 2000). Importantly, these genes all contain the Fox TAAACA binding site within 1 kb of the start site. Thus, the yeast Fkh1 and Fkh2 stress response transcription factors appear to possess activities associated with both FoxM1 and FoxO3a.
Another conserved role for these proteins is in the regulation of cell-cycle arrest and check point genes through two different mechanisms: the induction of cell-cycle arresting agents, such as CDKIs, and the repression of cell-cycle regulators, such as cyclins and polo-like kinases. The FoxOs regulate G1/G0 cell-cycle arrest by induction of the CDKIs p27Kip1, p21Cip1, p15Ink4b, p19INK4d, and p19Arf, which inhibit the formation of S-phase entry cyclin-CDK complexes (Medema et al., 2000; Seoane et al., 2004; Gomis et al., 2006; Bouchard et al., 2007; Miyamoto et al., 2007; Katayama et al., 2008). In addition, the FoxOs are involved in increasing the level of expression of the quiescent cyclin G2 while repressing that of cell-cycle entry cyclin D family members (Ramaswamy et al., 2002; Schmidt et al., 2002; Martínez-Gac et al., 2004). Furthermore, the FoxOs prevent the transcriptional activation of cell-cycle entry proteins through the up-regulation of the retinoblastoma protein family member p130 (Kops et al., 2002). Similarly, the FoxOs may be involved in check point arrest through the regulation of GADD45α, a component of the G2 checkpoint and DNA-damage repair systems (Tran et al., 2002; Laoukili et al., 2005).
The yeast Fkhs appear to function in a similar manner to the FoxOs through complimentary mechanisms. As mentioned earlier, Fkh2 in vivo is dominant over Fkh1 at promoter-binding sites where it acts as a transcriptional inhibitor until it binds the co-activator Ndd1 (Loy et al., 1999; Koranda et al., 2000; Hollenhorst et al., 2001). Thus, Fkh2 acts to block the expression of G2/M progression genes until the appropriate growth signals regulating the binding of Ndd1 to Fkh2 are present. At this point there is a switch to mitotic progression via the up-regulation of Clb2 and Cdc5, which further activates the Fkh2/Ndd1 complex (Reynolds et al., 2003). The expression of other genes, such as the rest of the Clb2 gene cluster is similarly regulated: this includes the APC subunits/activators and the G1 transcription factors Swi5 and Ace2, as well as the Histone gene cluster (Zhu et al., 2000). Swi5 and Ace2 are responsible for the expression of M/G1 progression genes as well as the yeast CDKI Sic1. In late mitosis the proteasomal degradation of Ndd1 switches Fkh2 to a transcriptional repressor of the Clb2 gene cluster. Interestingly, the Fkhs also bind to and repress many Swi5/Ace2 targeted promoters, preventing early G1 progression. As it appears Fkh1 is functionally redundant with Fkh2 (Hollenhorst et al., 2000; Zhu et al., 2000; Shapira et al., 2004; Sherriff et al., 2007; Voth et al., 2007; Postnikoff et al., 2012), it can be confidently postulated that the Fkhs follow the same pattern of cell-cycle regulation as the human Fox proteins, switching from cell-cycle repression in G2, to activation in M, followed by early G1 repression.
The Role of Fox Proteins in Lifespan Determination
The FoxO family of proteins have been reproducibly found to extend lifespan when expression is increased in many model systems studied, including flies, worms, and yeast (Libina et al., 2003; Giannakou et al., 2007; Postnikoff et al., 2012). The C. elegans FoxO ortholog DAF-16 was found to serve as a direct downstream target of the worm insulin-signaling pathway (Kenyon et al., 1993). Worm mutants encoding defective daf-2 insulin receptor genes exhibited a twofold lifespan extension, which was abolished when daf-16 was mutated. Since the first studies of DAF-16 in worms, inactivation of the insulin-signaling pathway from flies and worms to mice has resulted in extended lifespan (Kenyon et al., 1993; Carter et al., 2002; Blüher et al., 2003; Holzenberger et al., 2003; Rincon et al., 2004). For the most part, lifespan extension through reduced insulin-signaling depends on the evolutionarily conserved FoxO factors. Mutation to the FoxO stress response factors reduces lifespan and stress response in many model systems (Lin et al., 2001; Greer and Brunet, 2008; Moskalev et al., 2011; Yamamoto and Tatar, 2011; Postnikoff et al., 2012). While independent mutation to either yeast FKH1 or FKH2 has no effect on replicative or chronological lifespan (CLS; Wei et al., 2008; Postnikoff et al., 2012), a combined deletion of both FKH1 and FKH2 in the same cell dramatically impaired CLS and these cells could not respond to severe caloric restriction induced by maintenance in water (Postnikoff et al., 2012). As opposed to RLS, CLS measures how long stationary phase cells can remain metabolically active (Fabrizio et al., 2001; Longo and Fabrizio, 2012). The requirement for Fkh1 or Fkh2 for maintenance of stationary phase metabolic activity indicates that the yeast Fkh1 and Fkh2 proteins are fully active in non-dividing cells. This was supported by experiments demonstrating that fkh1Δ fkh2Δ cells are far more sensitive to oxidative stress when in stationary phase than when rapidly dividing (Postnikoff et al., 2012). Since yeast cells do not naturally respond to insulin, this indicates that yeast respond to nutrients directly through evolutionarily conserved insulin-like signaling mechanisms. In this respect, insulin may act as a “middle-man” in multi-cellular organisms, indicating to cells that express an insulin receptor that nutrients are available.
A series of studies focused on long-lived human populations have convincingly shown that long-lived individuals express altered insulin-signaling. A study of female human centenarians revealed that these individuals carry a heterozygous mutation in the IGF1 receptor that was over-representative compared to controls (Suh et al., 2008). Transformed lymphocytes generated from these individuals revealed reduced insulin-signaling. Other long-lived human cohorts were found to carry an altered FoxO3a allele that was not generally found in the population (Willcox et al., 2008; Chung et al., 2010; Ziv and Hu, 2011). Taken together, the findings in model systems ranging from yeast to mice clearly show that increased lifespan as a result of diminished insulin-signaling is a trait likely conserved in humans.
FoxM1 also appears to play a critical role in cell survival with respect to cancer cells. FoxM1 is highly expressed in many cancer cells and most weakly expressed in prematurely aging fibroblasts, such as those isolated from individuals suffering from Progeria, a rapid aging phenotype (Laoukili et al., 2007; Zeng et al., 2009; Anders et al., 2011). From these observations, it is clear that FoxM1 expression is associated with the proliferative capacity of the cell, consistent with its role in primarily driving the expression of G2/M specific genes (Laoukili et al., 2005), with associated phenotypic expression of mitotic defects and chromosome aberrations when defective (Wonsey and Follettie, 2005). As such, FoxM1 appears to be tightly linked with inducing the expression of genes required for G2 and mitotic progression, very similar to the yeast Fkh1 and Fkh2 Fox proteins. FoxM1 seems to counter senescence, but primarily to maintain the life of cancer cells.
Downstream Fox Targets Required for Increased Lifespan
A major quest over the past decade has been to identify downstream targets of the FoxO proteins specifically required for lifespan extension in model systems. The primary model used for these screens to identify FoxO lifespan targets has been the nematode C. elegans, largely due to the emergence of RNAi libraries (Murphy et al., 2003; Murphy, 2006; Oh et al., 2006; Szewczyk et al., 2006; Pinkston-Gosse and Kenyon, 2007; Shmookler et al., 2009; Schuster et al., 2010). An early study performed microarray on a series of long- and short-lived worm mutants over a lengthy time course spanning their overall development (Murphy et al., 2003). The 60 experiments were combined and mined for differentially expressed genes. Genes were found that grouped into distinct clusters representing signaling, stress response, and antimicrobial responses. Many of these genes were then silenced by RNAi in order to determine lifespan. Although many of the silenced genes conferred lifespans that altered from wild type controls, none of the lifespan differences approached those observed with daf-2 mutants, leading to the conclusion that multiple effector genes involved in longevity determination are regulated by the insulin-signaling pathway. Using chromatin immunoprecipitation (ChIP) 103 specific promoters were identified as bound by DAF-16 (Oh et al., 2006). Again using RNAi, few of these genes were seen to be involved in lifespan in isolation, although several were controlled by insulin-signaling. This study suggested that either (i) multiple proteins are involved in DAF-16 functions, or (ii) DAF-16 may have non-productive promoter interactions. A rigorous examination of microarray data from long-lived worms, flies, and mice that were impaired in insulin-signaling showed a striking lack of convergence between the differentially expressed genes (McElwee et al., 2007). Nonetheless, several functional categories were found to be conserved, such as protein biosynthesis, sugar catabolism, energy generation, and cellular detoxification. A novel approach to identifying DAF-16 targets involved fusing DAF-16 with a bacterial DNA adenine methyltransferase (DAM), which methylates adenine residues within the sequence GATC anywhere within 2 kb of its binding site (Schuster et al., 2010). An original list of 907 promoters was whittled down to 65 DAF-16 targets. This list was largely enriched in genes involved in signaling, and not in genes required for somatic maintenance processes, suggesting that DAF-16 is more likely activating signaling processes that lead to cellular maintenance and protection, rather than the individual genes themselves. Although dramatic gains have been made in identifying DAF-16 targets required for longevity determination, surprisingly little ground has been made toward defining networks leading from DAF-16 that result in definitive increased lifespan.
The Anaphase Promoting Complex, an Emerging Fox Specific Target Bridging the Longevity Gap
Emerging evidence suggests that the APC may be an important downstream target necessary for many Fox/Fkh functions (Laoukili et al., 2008; Park et al., 2008; Postnikoff et al., 2012). The APC is a highly conserved multi-subunit ubiquitin-protein ligase (E3) that primarily targets proteins that inhibit chromosome segregation and mitotic exit for ubiquitin- and proteasome-dependent degradation (Qiao et al., 2010; McLean et al., 2011). Initiation of and progression through mitosis is mediated by the APCCdc20 complex, while exit from mitosis and maintenance of G1 is controlled by the APCCdh1 complex, which targets components that drive mitosis, such as Clb2, Cdc5, and Cdc20, for degradation. Cdc20 is activated by the polo-like kinase Cdc5, as well as by the Cdc28/Clb2 Cdk complex prior to chromosome segregation. APCCdc20 targets spindle checkpoint proteins, such as the securin Pds1 in yeast, for proteasomal degradation (Hilioti et al., 2001; Wang et al., 2001). A specific example of such regulation involves the mouse spindle checkpoint protein BubR1. The spindle checkpoint functions in normal mitotic progression to inhibit APCCdc20 as a counter to polo-like kinase activation, ensuring that chromosomes are properly attached to the mitotic spindle, thereby preventing changes in chromosome numbers (D’Arcy et al., 2010; Lara-Gonzalez et al., 2011). Mice deficient in BubR1, either through mutation or as a natural property of aging, show signs of early aging such as kyphosis, cataracts, cardiovascular disease, muscle wasting, and susceptibility to carcinogens (Baker et al., 2004, 2005; Kim and Kao, 2005). Proper regulation of APCCdc20 may increase chromosome segregation fidelity, reducing non-disjunction events, and potentially increasing cellular healthspan. Other non-mitotic functions have now been ascribed to the APC, including maintaining neuronal development and genomic stability, as well as regulating chromatin metabolism and enhancing longevity in yeast and higher eukaryotic organisms (Harkness et al., 2002, 2004, 2005; Baker et al., 2004; Arnason et al., 2005; Turnell et al., 2005; Li et al., 2008; Turner et al., 2010; Eguren et al., 2011; Islam et al., 2011; Puram and Bonni, 2011; Postnikoff et al., 2012). As diverse as APC functions seem to be, regulatory mechanisms controlling APC output remain largely unknown.
Our recent work demonstrates that the APC may serve as a downstream Fkh1/Fkh2 target that regulates Fox-dependent longevity (Postnikoff et al., 2012). The APC and the Fkhs interact genetically and functionally to ensure normal yeast lifespan, and to respond to severe caloric restriction and stress in non-dividing cells. As mentioned above, increased expression of FKH1 or FKH2 could increase both RLS and CLS in yeast (Postnikoff et al., 2012). As expected from redundant factors, deletion of both FKH1 and FKH2, rather than either one alone, was required to reduce yeast CLS. However, disruption of both FKH1 and FKH2 in cells harboring a temperature sensitive allele of the gene encoding the APC subunit APC5, exhibited a CLS that was the same as fkh1Δ fkh2Δ cells, indicating that fkh1Δ fkh2Δ CLS is epistatic to apc5CA CLS under normal culture conditions. This indicates that under non-stress conditions Fkh1/2 acts directly upstream of the APC. We believe that this is through the direct transcriptional regulation of APC activators and APC substrates (Zhu et al., 2000).
The apc5CA mutant had a lifespan shorter than fkh1Δ fkh2Δ cells (Postnikoff et al., 2012). Unlike an earlier C. elegans study where RNAi of many DAF-16 targets (determined by microarray analyses) did not generate lifespan alterations as impressive as daf-2 or daf-16 mutants (Murphy et al., 2003), our data indicates that the APC may indeed be capable of mediating the lifespan effects of Fkh1/2 (Postnikoff et al., 2012). However, under stress conditions, such as maintenance of stationary phase cells in water, or exposure to either oxidative or heat stress, apc5CA fkh1Δ fkh2Δ cells grew much slower, were far more sensitive to stress and had a dramatically reduced CLS. While fkh1Δ fkh2Δ cells do not show the normal increased CLS in water, the triple mutant exhibited a dramatically shortened lifespan. This indicated that the Fkhs and the APC work together in a redundant manner to respond to stress and to ensure prolonged longevity. This is likely coordinated through the transcriptional up-regulation of stress response genes by Fkh1/2 in tandem with the chromatin assembly and histone modification functions of the APC (Harkness et al., 2002, 2005; Arnason et al., 2005; Turner et al., 2010; Islam et al., 2011). Together, the APC and the Fkhs drive response to stress and protect the genome from environmental stressors.
Conclusion
A pattern is emerging where pro-mitotic processes act as cell-cycle inhibitors to slow progression through early G1, allowing for preparation of S phase by resetting the transcriptome, repairing cellular damage, or remaining in a non-dividing state until conditions are right for the next cell cycle. Stress and starvation may act to prolong the function of this mechanism, allowing more time for stress response and cell repair. However, growth factors and energy sources inactivate these processes, favoring rapid growth over maintenance of youth. In mammals this process is regulated, at least in part, by the Fox class of transcription factors and the APC. Specifically, FoxM1 and APCCdc20 function together to maintain genomic stability by regulating separation of sister chromosomes and chromatin structure, while the FoxOs and APCCdh1 regulate cellular repair and maintenance, as well as the removal of built up pro-mitotic signals resetting the daughter cells. In yeast, new evidence suggests that this process is co-regulated by Fkh1 and Fkh2, which redundantly function in both mitotic progression and G1 maintenance, acting as both FoxO3a and FoxM1. This suggests a common role for evolutionary conserved proto-Fox proteins that regulates orthologous processes, such as cell-cycle progression and stress response. With this understanding, the resourcefulness of yeast biology and genetics, in addition to the ease of environmental control, could be utilized for untangling the mechanisms of Fox function, especially with regards to cancer and lifespan.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anders, L., Ke, N., Hydbring, P., Choi, Y. J., Widlund, H. R., Chick, J. M., Zhai, H., Vidal, M., Gygi, S. P., Braun, P., and Sicinski, P. (2011). A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 20, 620–634.
Arnason, T. G., Pisclevich, M. G., Dash, M. D., Davies, G. F., and Harkness, T. A. (2005). Novel interaction between Apc5p and Rsp5p in an intracellular signaling pathway in Saccharomyces cerevisiae. Eukaryot. Cell 4, 134–146.
Baker, D. J., Chen, J., and van Deursen, J. M. (2005). The mitotic checkpoint in cancer and aging: what have mice taught us? Curr. Opin. Cell Biol. 17, 583–589.
Baker, D. J., Jeganathan, K. B., Cameron, J. D., Thompson, M., Juneja, S., Kopecka, A., Kumar, R., Jenkins, R. B., de Groen, P. C., Roche, P., and van Deursen, J. M. (2004). BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 36, 744–749.
Baldauf, S. L. (1999). A search for the origins of animals and fungi: comparing and combining molecular data. Am. Nat. 154, S178–S188.
Bashir, T., Pagan, J. K., Busino, L., and Pagano, M. (2010). Phosphorylation of Ser72 is dispensable for Skp2 assembly into an active SCF ubiquitin ligase and its subcellular localization. Cell Cycle 9, 971–974.
Blüher, M., Kahn, B. B., and Kahn, C. R. (2003). Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 299, 572–574.
Boccitto, M., and Kalb, R. G. (2011). Regulation of Foxo-dependent transcription by post-translational modifications. Curr. Drug Targets 12, 1303–1310.
Bouchard, C., Lee, S., Paulus-Hock, V., Loddenkemper, C., Eilers, M., and Schmitt, C. A. (2007). FoxO transcription factors suppress Myc-driven lymphomagenesis via direct activation of Arf. Genes Dev. 21, 2775–2787.
Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., Anderson, M. J., Arden, K. C., Blenis, J., and Greenberg, M. E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 96, 857–868.
Carter, C. S., Ramsey, M. M., and Sonntag, W. E. (2002). A critical analysis of the role of growth hormone and IGF-1 in aging and lifespan. Trends Genet. 18, 295–301.
Chung, W.-H., Dao, R.-L., Chen, L.-K., and Hung, S.-I. (2010). The role of genetic variants in human longevity. Ageing Res. Rev. 9(Suppl. 1), S67–S78.
Daitoku, H., Sakamaki, J., and Fukamizu, A. (2011). Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochim. Biophys. Acta 1813, 1954–1960.
D’Arcy, S., Davies, O. R., Blundell, T. L., and Bolanos-Garcia, V. M. (2010). Defining the molecular basis of BubR1 kinetochore interactions and APC/C-CDC20 inhibition. J. Biol. Chem. 285, 14764–14776.
Delpuech, O., Griffiths, B., East, P., Essafi, A., Lam, E. W., Burgering, B., Downward, J., and Schulze, A. (2007). Induction of Mxi1-SR alpha by FOXO3a contributes to repression of Myc-dependent gene expression. Mol. Cell. Biol. 27, 4917–4930.
Dijkers, P. F., Birkenkamp, K. U., Lam, E. W., Thomas, N. S., Lammers, J. W., Koenderman, L., and Coffer, P. J. (2002). FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J. Cell Biol. 156, 531–542.
Eguren, M., Manchado, E., and Malumbres, M. (2011). Non-mitotic functions of the anaphase-promoting complex. Semin. Cell Dev. Biol. 22, 572–578.
Fabrizio, P., Pozza, F., Pletcher, S. D., Gendron, C. M., and Longo, V. D. (2001). Regulation of longevity and stress resistance by Sch9 in yeast. Science 292, 288–290.
Fernandez, P. C., Frank, S. R., Wang, L., Schroeder, M., Liu, S., Greene, J., Cocito, A., and Amati, B. (2003). Genomic targets of the human c-Myc protein. Genes Dev. 17, 1115–1129.
Gao, D., Inuzuka, H., Tseng, A., Chin, R. Y., Toker, A., and Wei, W. (2009). Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat. Cell Biol. 11, 397–408.
Giannakou, M. E., Goss, M., Jacobson, J., Vinti, G., Leevers, S. J., and Partridge, L. (2007). Dynamics of the action of dFOXO on adult mortality in Drosophila. Aging Cell 6, 429–438.
Gomis, R. R., Alarcón, C., Nadal, C., Van Poznak, C., and Massagué, J. (2006). C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell 10, 203–214.
Greer, E. L., and Brunet, A. (2008). FOXO transcription factors in ageing and cancer. Acta Physiol. (Oxf.) 192, 19–28.
Hannenhalli, S., and Kaestner, K. H. (2009). The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 10, 233–240.
Harkness, T. A., Arnason, T. G., Legrand, C., Pisclevich, M. G., Davies, G. F., and Turner, E. L. (2005). Contribution of CAF-I to anaphase-promoting-complex-mediated mitotic chromatin assembly in Saccharomyces cerevisiae. Eukaryot. Cell 4, 673–684.
Harkness, T. A., Davies, G. F., Ramaswamy, V., and Arnason, T. G. (2002). The ubiquitin-dependent targeting pathway in Saccharomyces cerevisiae plays a critical role in multiple chromatin assembly regulatory steps. Genetics 162, 615–632.
Harkness, T. A., Shea, K. A., Legrand, C., Brahmania, M., and Davies, G. F. (2004). A functional analysis reveals dependence on the anaphase-promoting complex for prolonged life span in yeast. Genetics 168, 759–774.
Hilioti, Z., Chung, Y.-S., Mochizuki, Y., Hardy, C. F. J., and Cohen-Fix, O. (2001). The anaphase inhibitor Pds1 binds to the APC/C-associated protein Cdc20 in a destruction box-dependent manner. Curr. Biol. 11, 1347–1352.
Hollenhorst, P. C., Bose, M. E., Mielke, M. R., Müller, U., and Fox, C. A. (2000). Forkhead genes in transcriptional silencing, cell morphology and the cell cycle. Overlapping and distinct functions for FKH1 and FKH2 in Saccharomyces cerevisiae. Genetics 154, 1533–1548.
Hollenhorst, P. C., Pietz, G., and Fox, C. A. (2001). Mechanisms controlling differential promoter-occupancy by the yeast forkhead proteins Fkh1p and Fkh2p: implications for regulating the cell cycle and differentiation. Genes Dev. 15, 2445–2456.
Holzenberger, M., Dupont, J., Ducos, B., Leneuve, P., Géloën, A., Even, P. C., Cervera, P., and Le Bouc, Y. (2003). IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421, 182–187.
Huang, H., Regan, K. M., Wang, F., Wang, D., Smith, D. I., van Deursen, J. M., and Tindall, D. J. (2005). Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc. Natl. Acad. Sci. U.S.A. 102, 1649–1654.
Huang, H., and Tindall, D. J. (2011). Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim. Biophys. Acta 1813, 1961–1964.
Islam, A., Turner, E. L., Menzel, J., Malo, M. E., and Harkness, T. A. (2011). Antagonistic Gcn5-Hda1 interactions revealed by mutations to the anaphase promoting complex in yeast. Cell Div. 6, 13.
Jia, G., Su, L., Singhal, S., and Liu, X. (2012). Emerging roles of SIRT6 on telomere maintenance, DNA repair, metabolism and mammalian aging. Mol. Cell. Biochem. 364, 345–350.
Katayama, K., Nakamura, A., Sugimoto, Y., Tsuruo, T., and Fujita, N. (2008). FOXO transcription factor-dependent p15(INK4b) and p19(INK4d) expression. Oncogene 27, 1677–1686.
Kaufmann, E., Müller, D., and Knöchel, W. (1995). DNA recognition site analysis of Xenopus winged helix proteins. J. Mol. Biol. 248, 239–254.
Kennedy, B. K., Austriaco, N. R., and Guarente, L. (1994). Daughter cells of Saccharomyces cerevisiae from old mothers display a reduced life span. J. Cell Biol. 127 (Pt 2), 1985–1993.
Kenyon, C., Chang, J., Gensch, E., Rudner, A., and Tabtiang, R. (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464.
Kim, M., and Kao, G. D. (2005). Newly identified roles for an old guardian: profound deficiency of the mitotic spindle checkpoint protein BubR1 leads to early aging and infertility. Cancer Biol. Ther. 4, 164–165.
Kloet, D. E., and Burgering, B. M. (2011). The PKB/FOXO switch in aging and cancer. Biochim. Biophys. Acta 1813, 1926–1937.
Ko, N., Nishihama, R., Tully, G. H., Ostapenko, D., Solomon, M. J., Morgan, D. O., and Pringle, J. R. (2007). Identification of yeast IQGAP (Iqg1p) as an anaphase-promoting-complex substrate and its role in actomyosin-ring-independent cytokinesis. Mol. Biol. Cell 18, 5139–5153.
Kops, G. J., Medema, R. H., Glassford, J., Essers, M. A., Dijkers, P. F., Coffer, P. J., Lam, E. W., and Burgering, B. M. (2002). Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol. Cell. Biol. 22, 2025–2036.
Koranda, M., Schleiffer, A., Endler, L., and Ammerer, G. (2000). Forkhead-like transcription factors recruit Ndd1 to the chromatin of G2/M-specific promoters. Nature 406, 94–98.
Korver, W., Roose, J., Wilson, A., and Clevers, H. (1997). The winged-helix transcription factor Trident is expressed in actively dividing lymphocytes. Immunobiology 198, 157–161.
Lalmansingh, A. S., Karmakar, S., Jin, Y., and Nagaich, A. K. (2012). Multiple modes of chromatin remodeling by forkhead box proteins. Biochim. Biophys. Acta. PMID: 22406422. [Epub ahead of print].
Laoukili, J., Alvarez-Fernandez, M., Stahl, M., and Medema, R. H. (2008). FoxM1 is degraded at mitotic exit in a Cdh1-dependent manner. Cell Cycle 7, 2720–2726.
Laoukili, J., Kooistra, M. R., Brás, A., Kauw, J., Kerkhoven, R. M., Morrison, A., Clevers, H., and Medema, R. H. (2005). FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 7, 126–136.
Laoukili, J., Stahl, M., and Medema, R. H. (2007). FoxM1: at the crossroads of ageing and cancer. Biochim. Biophys. Acta 1775, 92–102.
Lara-Gonzalez, P., Scott, M. I. F., Diez, M., Sen, O., and Taylor, S. S. (2011). BubR1 blocks substrate recruitment to the APC/C in a KEN-box-dependent manner. J. Cell. Sci. 124, 4332–4345.
Lehmann, O. J., Sowden, J. C., Carlsson, P., Jordan, T., and Bhattacharya, S. S. (2003). Fox’s in development and disease. Trends Genet. 19, 339–344.
Li, M., Shin, Y. H., Hou, L., Huang, X., Wei, Z., Klann, E., and Zhang, P. (2008). The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nat. Cell Biol. 10, 1083–1089.
Libina, N., Berman, J. R., and Kenyon, C. (2003). Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115, 489–502.
Lin, H. K., Wang, G., Chen, Z., Teruya-Feldstein, J., Liu, Y., Chan, C. H., Yang, W. L., Erdjument-Bromage, H., Nakayama, K. I., Nimer, S., Tempst, P., and Pandolfi, P. P. (2009). Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat. Cell Biol. 11, 420–432.
Lin, K., Hsin, H., Libina, N., and Kenyon, C. (2001). Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 28, 139–145.
Longo, V. D., and Fabrizio, P. (2012). Chronological aging in Saccharomyces cerevisiae. Subcell. Biochem. 57, 101–121.
Loy, C. J., Lydall, D., and Surana, U. (1999). NDD1, a high-dosage suppressor of cdc28-1N, is essential for expression of a subset of late-S-phase-specific genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 19, 3312–3327.
Madureira, P. A., Varshochi, R., Constantinidou, D., Francis, R. E., Coombes, R. C., Yao, K. M., and Lam, E. W. (2006). The forkhead box M1 protein regulates the transcription of the estrogen receptor alpha in breast cancer cells. J. Biol. Chem. 281, 25167–25176.
Martínez-Gac, L., Marqués, M., García, Z., Campanero, M. R., and Carrera, A. C. (2004). Control of cyclin G2 mRNA expression by forkhead transcription factors: novel mechanism for cell cycle control by phosphoinositide 3-kinase and forkhead. Mol. Cell. Biol. 24, 2181–2189.
Mazet, F., Yu, J. K., Liberles, D. A., Holland, L. Z., and Shimeld, S. M. (2003). Phylogenetic relationships of the fox (forkhead) gene family in the bilateria. Gene 316, 79–89.
McElwee, J. J., Schuster, E., Blanc, E., Piper, M. D., Thomas, J. H., Patel, D. S., Selman, C., Withers, D. J., Thornton, J. M., Partridge, L., and Gems, D. (2007). Evolutionary conservation of regulated longevity assurance mechanisms. Genome Biol. 8, R132.
McGovern, U. B., Francis, R. E., Peck, B., Guest, S. K., Wang, J., Myatt, S. S., Krol, J., Kwok, J. M., Polychronis, A., Coombes, R. C., and Lam, E. W. (2009). Gefitinib (iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol. Cancer Ther. 8, 582–591.
McLean, J. R., Chaix, D., Ohi, M. D., and Gould, K. L. (2011). State of the APC/C: organization, function, and structure. Crit. Rev. Biochem. Mol. Biol. 46, 118–136.
Medema, R. H., Kops, G. J., Bos, J. L., and Burgering, B. M. (2000). AFX-like forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404, 782–787.
Miyamoto, K., Araki, K. Y., Naka, K., Arai, F., Takubo, K., Yamazaki, S., Matsuoka, S., Miyamoto, T., Ito, K., Ohmura, M., Chen, C., Hosokawa, K., Nakauchi, H., Nakayama, K., Nakayama, K. I., Harada, M., Motoyama, N., Suda, T., and Hirao, A. (2007). Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 1, 101–112.
Moskalev, A. A., Plyusnina, E. N., and Shaposhnikov, M. V. (2011). Radiation hormesis and radioadaptive response in Drosophila melanogaster flies with different genetic backgrounds: the role of cellular stress-resistance mechanisms. Biogerontology 12, 253–263.
Murakami, H., Aiba, H., Nakanishi, M., and Murakami-Tonami, Y. (2010). Regulation of yeast forkhead transcription factors and FoxM1 by cyclin-dependent and polo-like kinases. Cell Cycle 9, 3233–3242.
Murphy, C. T. (2006). The search for DAF-16/FOXO transcriptional targets: approaches and discoveries. Exp. Gerontol. 41, 910–921.
Murphy, C. T., McCarroll, S. A., Bargmann, C. I., Fraser, A., Kamath, R. S., Ahringer, J., Li, H., and Kenyon, C. (2003). Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283.
Myatt, S. S., and Lam, E. W. (2007). The emerging roles of forkhead box (fox) proteins in cancer. Nat. Rev. Cancer 7, 847–859.
Nemoto, S., and Finkel, T. (2002). Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 295, 2450–2452.
O’Brien, S. L., Fagan, A., Fox, E. J., Millikan, R. C., Culhane, A. C., Brennan, D. J., McCann, A. H., Hegarty, S., Moyna, S., Duffy, M. J., Higgins, D. G., Jirström, K., Landberg, G., and Gallagher, W. M. (2007). CENP-F expression is associated with poor prognosis and chromosomal instability in patients with primary breast cancer. Int. J. Cancer 120, 1434–1443.
Oh, S. W., Mukhopadhyay, A., Dixit, B. L., Raha, T., Green, M. R., and Tissenbaum, H. A. (2006). Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat. Genet. 38, 251–257.
Pandit, B., Halasi, M., and Gartel, A. L. (2009). p53 Negatively regulates expression of FoxM1. Cell Cycle 8, 3425–3427.
Park, H. J., Costa, R. H., Lau, L. F., Tyner, A. L., and Raychaudhuri, P. (2008). Anaphase-promoting complex/cyclosome-CDH1-mediated proteolysis of the forkhead box M1 transcription factor is critical for regulated entry into S phase. Mol. Cell. Biol. 28, 5162–5171.
Petrovic, V., Costa, R. H., Lau, L. F., Raychaudhuri, P., and Tyner, A. (2010). Negative regulation of the oncogenic transcription factor FoxM1 by thiazolidinediones and mithramycin. Cancer Biol. Ther. 9, 1008–1016.
Pfleger, C. M., and Kirschner, M. W. (2000). The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14, 655–665.
Pinkston-Gosse, J., and Kenyon, C. (2007). DAF-16/FOXO targets genes that regulate tumor growth in Caenorhabditis elegans. Nat. Genet. 39, 1403–1409.
Postnikoff, S. D., Malo, M. E., Wong, B., and Harkness, T. A. (2012). The yeast forkhead transcription factors fkh1 and fkh2 regulate lifespan and stress response together with the anaphase-promoting complex. PLoS Genet. 8, e1002583. doi:10.1371/journal.pgen.1002583
Pramila, T., Wu, W., Miles, S., Noble, W. S., and Breeden, L. L. (2006). The forkhead transcription factor Hcm1 regulates chromosome segregation genes and fills the S-phase gap in the transcriptional circuitry of the cell cycle. Genes Dev. 20, 2266–2278.
Puram, S. V., and Bonni, A. (2011). Novel functions for the anaphase-promoting complex in neurobiology. Semin. Cell Dev. Biol. 22, 586–594.
Qiao, X., Zhang, L., Gamper, A. M., Fujita, T., and Wan, Y. (2010). APC/C-Cdh1: from cell cycle to cellular differentiation and genomic integrity. Cell Cycle 9, 3904–3912.
Ramaswamy, S., Nakamura, N., Sansal, I., Bergeron, L., and Sellers, W. R. (2002). A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell 2, 81–91.
Reynolds, D., Shi, B. J., McLean, C., Katsis, F., Kemp, B., and Dalton, S. (2003). Recruitment of Thr 319-phosphorylated Ndd1p to the FHA domain of Fkh2p requires Clb kinase activity: a mechanism for CLB cluster gene activation. Genes Dev. 17, 1789–1802.
Rincon, M., Muzumdar, R., Atzmon, G., and Barzilai, N. (2004). The paradox of the insulin/IGF-1 signaling pathway in longevity. Mech. Ageing Dev. 125, 397–403.
Rudra, D., Zhao, Y., and Warner, J. R. (2005). Central role of Ifh1p-Fhl1p interaction in the synthesis of yeast ribosomal proteins. EMBO J. 24, 533–542.
Salminen, A., and Kaarniranta, K. (2012). AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 11, 230–241.
Sari, F., Braus, G. H., and Irniger, S. (2007). A process independent of the anaphase-promoting complex contributes to instability of the yeast S phase cyclin Clb5. J. Biol. Chem. 282, 26614–26622.
Schmidt, M., Fernandez de Mattos, S., van der Horst, A., Klompmaker, R., Kops, G. J., Lam, E. W., Burgering, B. M., and Medema, R. H. (2002). Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell. Biol. 22, 7842–7852.
Schuster, E., McElwee, J. J., Tullet, J. M., Doonan, R., Matthijssens, F., Reece-Hoyes, J. S., Hope, I. A., Vanfleteren, J. R., Thornton, J. M., and Gems, D. (2010). DamID in C. elegans reveals longevity-associated targets of DAF-16/FoxO. Mol. Syst. Biol. 6.
Seoane, J., Le, H. V., Shen, L., Anderson, S. A., and Massagué, J. (2004). Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117, 211–223.
Shapira, M., Segal, E., and Botstein, D. (2004). Disruption of yeast forkhead-associated cell cycle transcription by oxidative stress. Mol. Biol. Cell 15, 5659–5669.
Sherriff, J. A., Kent, N. A., and Mellor, J. (2007). The Isw2 chromatin-remodeling ATPase cooperates with the Fkh2 transcription factor to repress transcription of the B-type cyclin gene CLB2. Mol. Cell. Biol. 27, 2848–2860.
Shmookler, R. J., Bharill, P., Tazearslan, C., and Ayyadevara, S. (2009). Extreme-longevity mutations orchestrate silencing of multiple signaling pathways. Biochim. Biophys. Acta 1790, 1075–1083.
Stünkel, W., and Campbell, R. M. (2011). Sirtuin 1 (SIRT1): the misunderstood HDAC. J. Biomol. Screen 16, 1153–1169.
Suh, Y., Atzmon, G., Cho, M. O., Hwang, D., Liu, B., Leahy, D. J., Barzilai, N., and Cohen, P. (2008). Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl. Acad. Sci. U.S.A. 105, 3438–3442.
Szewczyk, N. J., Udranszky, I. A., Kozak, E., Sunga, J., Kim, S. K., Jacobson, L. A., and Conley, C. A. (2006). Delayed development and lifespan extension as features of metabolic lifestyle alteration in C. elegans under dietary restriction. J. Exp. Biol. 209, 4129–4139.
Tang, S. Y., Jiao, Y., and Li, L. Q. (2008). Significance of forkhead Box m1b (Foxm1b) gene in cell proliferation and carcinogenesis. Ai Zheng 27, 894–896.
Tran, H., Brunet, A., Grenier, J. M., Datta, S. R., Fornace, A. J., DiStefano, P. S., Chiang, L. W., and Greenberg, M. E. (2002). DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 296, 530–534.
Turnell, A. S., Stewart, G. S., Grand, R. J., Rookes, S. M., Martin, A., Yamano, H., Elledge, S. J., and Gallimore, P. H. (2005). The APC/C and CBP/p300 cooperate to regulate transcription and cell-cycle progression. Nature 438, 690–695.
Turner, E. L., Malo, M. E., Pisclevich, M. G., Dash, M. D., Davies, G. F., Arnason, T. G., and Harkness, T. A. (2010). The Saccharomyces cerevisiae anaphase-promoting complex interacts with multiple histone-modifying enzymes to regulate cell cycle progression. Eukaryot. Cell 9, 1418–1431.
Tzivion, G., Dobson, M., and Ramakrishnan, G. (2011). FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 1813, 1938–1945.
Ustiyan, V., Wang, I. C., Ren, X., Zhang, Y., Snyder, J., Xu, Y., Wert, S. E., Lessard, J. L., Kalin, T. V., and Kalinichenko, V. V. (2009). Forkhead box M1 transcriptional factor is required for smooth muscle cells during embryonic development of blood vessels and esophagus. Dev. Biol. 336, 266–279.
van der Waal, M. S., Hengeveld, R. C., van der Horst, A., and Lens, S. M. (2012). Cell division control by the chromosomal passenger complex. Exp. Cell Res. PMID: 22472345. [Epub ahead of print]..
van Leuken, R., Clijsters, L., and Wolthuis, R. (2008). To cell cycle, swing the APC/C. Biochim. Biophys. Acta 1786, 49–59.
Voth, W. P., Yu, Y., Takahata, S., Kretschmann, K. L., Lieb, J. D., Parker, R. L., Milash, B., and Stillman, D. J. (2007). Forkhead proteins control the outcome of transcription factor binding by antiactivation. EMBO J. 26, 4324–4334.
Wang, H., Liu, D., Wang, Y., Qin, J., and Elledge, S. J. (2001). Pds1 phosphorylation in response to DNA damage is essential for its DNA damage checkpoint function. Genes Dev. 15, 1361–1372.
Wang, I. C., Chen, Y. J., Hughes, D., Petrovic, V., Major, M. L., Park, H. J., Tan, Y., Ackerson, T., and Costa, R. H. (2005). Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol. Cell. Biol. 25, 10875–10894.
Wang, I. C., Chen, Y. J., Hughes, D. E., Ackerson, T., Major, M. L., Kalinichenko, V. V., Costa, R. H., Raychaudhuri, P., Tyner, A. L., and Lau, L. F. (2008). FoxM1 regulates transcription of JNK1 to promote the G1/S transition and tumor cell invasiveness. J. Biol. Chem. 283, 20770–20778.
Wang, Z., Ahmad, A., Li, Y., Banerjee, S., Kong, D., and Sarkar, F. H. (2010). Forkhead box M1 transcription factor: a novel target for cancer therapy. Cancer Treat. Rev. 36, 151–156.
Wei, M., Fabrizio, P., Hu, J., Ge, H., Cheng, C., Li, L., and Longo, V. D. (2008). Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 4, e13. doi:10.1371/journal.pgen.0040013
Willcox, B. J., Donlon, T. A., He, Q., Chen, R., Grove, J. S., Yano, K., Masaki, K. H., Willcox, D. C., Rodriguez, B., and Curb, J. D. (2008). FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. U.S.A. 105, 13987–13992.
Wilson, M. S., Brosens, J. J., Schwenen, H. D., and Lam, E. W. (2011). FOXO and FOXM1 in cancer: the FOXO-FOXM1 axis shapes the outcome of cancer chemotherapy. Curr. Drug Targets 12, 1256–1266.
Wonsey, D. R., and Follettie, M. T. (2005). Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 65, 5181–5189.
Yamamoto, R., and Tatar, M. (2011). Insulin receptor substrate chico acts with the transcription factor FOXO to extend Drosophila lifespan. Aging Cell 10, 729–732.
Zeng, J., Wang, L., Li, Q., Li, W., Björkholm, M., Jia, J., and Xu, D. (2009). FoxM1 is up-regulated in gastric cancer and its inhibition leads to cellular senescence, partially dependent on p27 kip1. J. Pathol. 218, 419–427.
Zhao, Y., Wang, Y., and Zhu, W. G. (2011). Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell Biol. 3, 276–282.
Zhu, G., Spellman, P. T., Volpe, T., Brown, P. O., Botstein, D., Davis, T. N., and Futcher, B. (2000). Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature 406, 90–94.
Ziv, E., and Hu, D. (2011). Genetic variation in insulin/IGF-1 signaling pathways and longevity. Ageing Res. Rev. 10, 201–204.
Keywords: FoxO3a, FoxM1, Fkh1, Fkh2, Anaphase Promoting Complex
Citation: Postnikoff SDL and Harkness TAA (2012) Mechanistic insights into aging, cell-cycle progression, and stress response. Front. Physio. 3:183. doi: 10.3389/fphys.2012.00183
Received: 15 April 2012; Paper pending published: 03 May 2012;
Accepted: 17 May 2012; Published online: 04 June 2012.
Edited by:
Vladimir Titorenko, Concordia University, CanadaReviewed by:
Yasser Mohamed El-Wazir, Suez Canal University, EgyptGema Frühbeck, University of Navarra, Spain
Copyright: © 2012 Postnikoff and Harkness. This is an openaccess article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: T. A. A. Harkness, Department of Anatomy and Cell Biology, University of Saskatchewan, B313 Health Science Building, 107 Wiggins Road, Saskatoon, SK, Canada S7N 5E5. e-mail: troy.harkness@usask.ca