
- 1Laboratory of Molecular Biology, Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Kyoto, Japan
- 2Advanced Biomedical Engineering Research Unit, Kyoto University, Kyoto, Japan
- 3CREST, Japan Science and Technology Agency, Chiyoda-ku, Tokyo, Japan
- 4Laboratory of Environmental Systems Biology, Department of Technology and Ecology, Hall of Global Environmental Studies, Kyoto University, Kyoto, Japan
The transient receptor potential (trp) gene superfamily encodes cation channels that act as multimodal sensors for a wide variety of stimuli from outside and inside the cell. Upon sensing, they transduce electrical and Ca2+ signals via their cation channel activities. These functional features of TRP channels allow the body to react and adapt to different forms of environmental changes. Indeed, members of one class of TRP channels have emerged as sensors of gaseous messenger molecules that control various cellular processes. Nitric oxide (NO), a vasoactive gaseous molecule, regulates TRP channels directly via cysteine (Cys) S-nitrosylation or indirectly via cyclic GMP (cGMP)/protein kinase G (PKG)-dependent phosphorylation. Recent studies have revealed that changes in the availability of molecular oxygen (O2) also control the activation of TRP channels. Anoxia induced by O2-glucose deprivation and severe hypoxia (1% O2) activates TRPM7 and TRPC6, respectively, whereas TRPA1 has recently been identified as a novel sensor of hyperoxia and mild hypoxia (15% O2) in vagal and sensory neurons. TRPA1 also detects other gaseous molecules such as hydrogen sulfide (H2S) and carbon dioxide (CO2). In this review, we focus on how signaling by gaseous molecules is sensed and integrated by TRP channels.
Introduction
Transient receptor potential (TRP) proteins are the product of trp genes, which were first discovered in Drosophila melanogaster, but later found to have homologs in other species. These gene products form cation channels that detect and transduce cellular stimuli into electrical signals (via changes in membrane potential) or chemical signals [via changes in intracellular Ca2+ concentration ([Ca2+]i)] (Montell et al., 2002; Clapham, 2003; Voets et al., 2005). TRP proteins are putative six-transmembrane domain polypeptide subunits that assemble into tetramers to form channels (Figure 1A). In mammalian systems, TRP channels comprise six related protein subfamilies: TRPC, TRPV, TRPM, TRPA, TRPP, and TRPML (Clapham et al., 2005) (Figure 1B). The TRPC homologs form receptor-activated Ca2+-permeable cation channels (RACCs) that, when activated by receptor stimulation, induce phospholipase C (PLC) to hydrolyze phosphatidylinositol-4,5-biphosphate (PIP2) into inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) (Zhu et al., 1996; Vazquez et al., 2004). Store-operated channels (SOCs), which are activated by IP3-induced Ca2+ release and depletion of endoplasmic reticulum (ER) Ca2+ stores, can also be categorized as RACCs. In contrast, TRPV Ca2+-permeable channels can be functionally defined as thermosensors (Caterina et al., 1997; Clapham, 2003; Patapoutian et al., 2003; Clapham et al., 2005). TRPV1, originally identified as the receptor for the vanilloid compound capsaicin, is responsive to heat (>43°C); proton (H+) concentration (pH < 5.6); the intrinsic ligand, anandamide; and receptor-driven PLC activity (Clapham et al., 2005). High temperature also activates TRPV2 (>52°C), TRPV3 (>31°C or >39°C), and TRPV4 (>27°C). TRPV5 and TRPV6 comprise a different subfamily because they are activated by [Ca2+]i (Clapham, 2003; Clapham et al., 2005). The TRPM subfamily is named after melastatin (TRPM1), a tumor suppressor protein isolated in a screen for genes whose level of expression is inversely correlated with the severity of metastatic potential of a melanoma cell line (Duncan et al., 1998), and contains eight mammalian members. TRPM8 channels, in contrast to TRPV channels, are activated by low temperatures (<25°C) and menthol (McKemy et al., 2002; Peier et al., 2002). The sole member of the TRPA subfamily, TRPA1, has a large N-terminal domain with 17 predicted ankyrin repeat (AnkR) domains (Gaudet, 2008). Pungent compounds, such as the allyl isothiocyanate found in mustard oil, trigger TRPA1 activation (Jordt et al., 2004). TRPA1 has been shown to respond to noxious cold (<17°C), although its activation by cold remains controversial (Story et al., 2003; Bandell et al., 2004; Jordt et al., 2004; Nagata et al., 2005; Bautista et al., 2006; Kwan et al., 2006; Sawada et al., 2007; Karashima et al., 2009). Thus, TRP channels serve as sensors for a variety of environmental factors. With the exception of TRPM4 and TRPM5 (Nilius, 2007), all of these TRP channels have some Ca2+ influx activity that is a component of their regulation of diverse cellular processes.
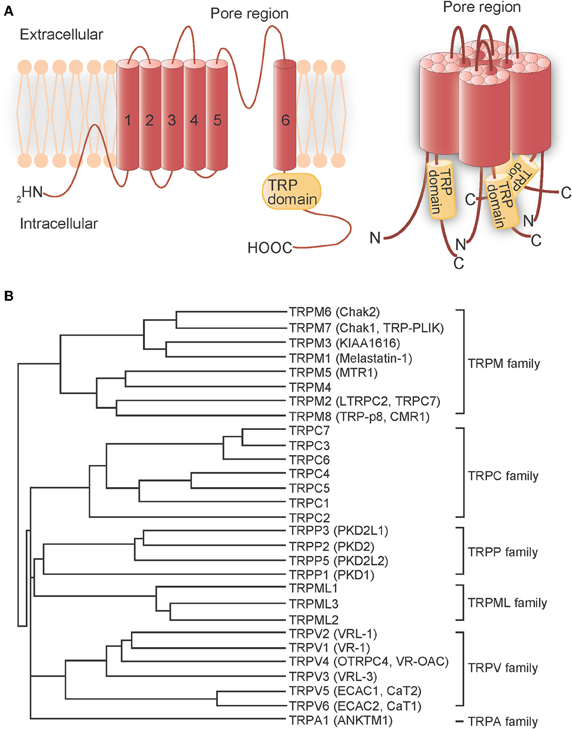
Figure 1. Transmembrane topology and phylogenetic tree of mammalian TRP channels. (A) Transmembrane topology (left) and the quartenary structure of TRP channels (right). The TRP protein has six putative transmembrane domains, a pore region between the fifth and sixth transmembrane domains and a TRP domain in the C-terminal region. The TRP protein assembles into homo-tetramers or hetero-tetramers to form channels. (B) Phylogenetic tree of mammalian TRP channels based on their homology.
Gaseous molecules, such as oxygen (O2), nitric oxide (NO), carbon monoxide (CO), hydrogen sulfide (H2S), and carbon dioxide (CO2), have been shown to play important roles in biological signal transduction. These molecules share several unique physicochemical properties and exert their biological activities through mechanisms distinct from those of other signaling molecules as summarized by Suematsu (2003). Firstly, these molecules are highly membrane-permeable and can readily convey signals in an autocrine, paracrine and/or juxtacrine manner. Secondly, they exert their biological actions via a variety of interactions with macromolecules: covalent binding of gases to prosthetic metal complexes in receptor proteins; non-covalent binding to the regulatory subunits of proteins; and space occupancy in and around the protein structure that impedes the access of other gases to the functionally critical protein motifs. Recent evidence suggest that NO, CO, H2S, and CO2 function as signaling molecules that also play critical roles in mediating the biological effects of changes in O2 availability (Semenza and Prabhakar, 2012).
Among the gaseous signaling molecules, NO is the most extensively studied. Its biological significance and the systems by which it is generated were first revealed in the 1980s (Furchgott and Zawadzki, 1980; Palmer et al., 1988; Sakuma et al., 1988) and it is now known to regulate a variety of biological events, including vascular relaxation and neurotransmission. Conversely, excessive generation of NO and NO-derived reactive nitrogen species (RNS) has been implicated in a number of pathological conditions (Reiter, 2006). Cyclic GMP (cGMP) is the canonical mediator of NO signaling. However, the importance of a cGMP-independent signaling pathway involving protein S-nitrosylation is becoming increasingly recognized (Jaffrey et al., 2001; Hess et al., 2005). S-nitrosylation of cysteine (Cys) is readily reversible with high spatial and temporal specificity. The NADH-dependent oxidoreductase, S-nitrosoglutathione reductase, specifically catalyzes the denitrosylation of S-nitrosoglutathione, by which protein S-nitrosylation is regulated in the cellular equilibrium between S-nitrosylated proteins and S-nitrosoglutathione. Thioredoxin also mediates direct denitrosylation of multiple S-nitrosylated proteins. Thus, the temporal and spatial regulation of S-nitrosylation and denitrosylation confers specificity to NO-based cellular signaling (Benhar et al., 2009).
Ca2+ and NO signals are precisely coordinated with each other and converge at two main points (Milbourne and Bygrave, 1995). Firstly, in order to be activated, constitutive NO synthase (NOS) must bind calmodulin (CaM), an event controlled by the level of [Ca2+]i (Nathan, 1992). An increase in [Ca2+]i has been shown to activate constitutive NOS, especially in endothelial and neuronal cells, and this results in NO production (Moncada et al., 1997). However, it is still unclear whether activation of endothelial NOS (eNOS) requires specific modes of upstream Ca2+-mobilization, or RACC subtypes formed by particular TRPCs (Hutcheson and Griffith, 1997; Lantoine et al., 1998; Lin et al., 2000; Koyama et al., 2002; Yao and Garland, 2005). In contrast, it is well characterized that Ca2+ influx through N-methyl-D-aspartate (NMDA) receptors activates neuronal NOS (nNOS) (Dawson et al., 1991) to elicit S-nitrosylation of many synaptic proteins (Jaffrey et al., 2001). The second point of convergence is that NO can directly affect intracellular Ca2+ levels by acting on cell surface receptors to promote Ca2+ mobilization (Milbourne and Bygrave, 1995). This feedback regulation of Ca2+ signaling by NO remains controversial in non-excitable cells (Khan and Hare, 2003), where NO has been reported to regulate Ca2+ mobilization pathways (including RACCs) both positively (Volk et al., 1997; Chen et al., 2000; Li et al., 2003) and negatively (Kwan et al., 2000; Dedkova and Blatter, 2002). In contrast, the regulation of Ca2+ signaling by NO is better defined in neurons, where NO nitrosylates NR1 and NR2 subunits of NMDA receptors at specific Cys/thiol groups and decreases Ca2+ entry via this ionotropic receptor (Choi et al., 2000; Jaffrey et al., 2001; Lipton et al., 2002). This negative feedback mechanism is important in precluding over-activation of NMDA receptors. However, NMDA receptors have a very restricted tissue distribution and specific physiological functions, whereas NO signaling via protein S-nitrosylation and Ca2+ signaling are much more generalized and have a wide array of functions. Therefore, to understand better the molecular mechanisms that link these two key signals, S-nitrosylation targets must be distinguished from the more ubiquitous Ca2+-mobilizing ion channels. As described below, this ultimately provides insights into the activation gating that underlies sensor function of TRP channels. In this review, we focus on how the gaseous signaling is sensed and integrated by TRP channels (Figure 2).
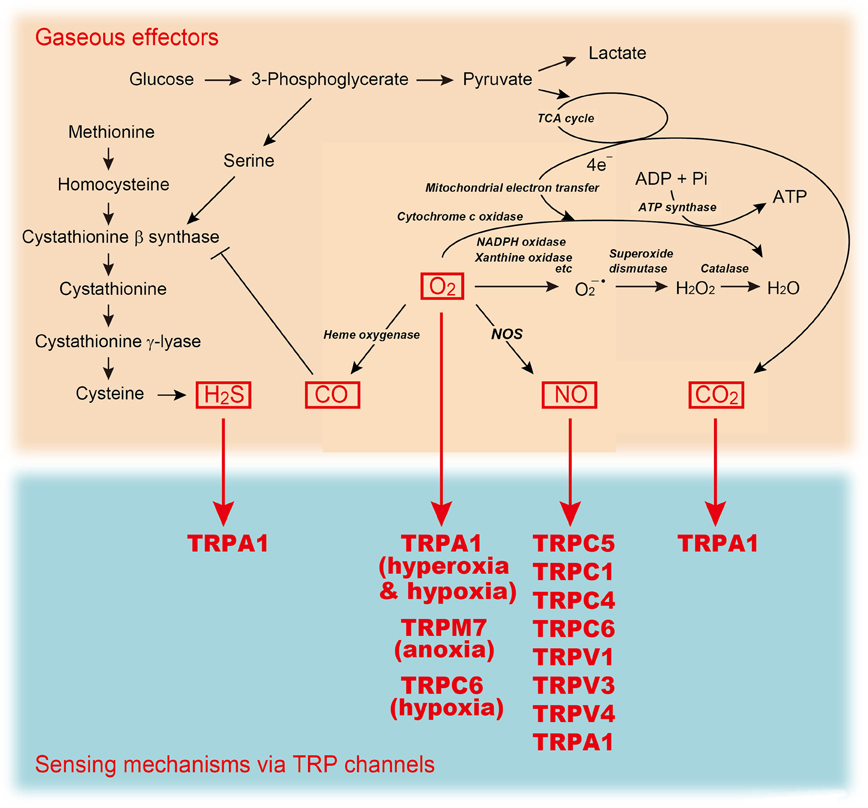
Figure 2. A sensing mechanism for gaseous molecules linked to metabolic pathways. TRP channels mediate sensing mechanism for H2S, O2 (anoxia, hypoxia, or hyperoxia), NO, and CO2.
Correlation Between RACCs and NO Signals in Native Tissue Preparations
Tight correlation between native RACCs and NO signaling has been presumed in many types of cells (Xu et al., 1994; Volk et al., 1997; Ma et al., 1999; Chen et al., 2000; van Rossum et al., 2000; Thyagarajan et al., 2001; Li et al., 2003). In pancreatic acini isolated from rat, agonist-mediated Ca2+ release from internal stores activates a cellular pool of NOS to generate cGMP, which then modulates Ca2+ entry through the plasma membrane (Xu et al., 1994). This mechanism might be responsible for the capacitative nature of Ca2+ entry. Interestingly, different concentrations of an NO donor sodium nitroprusside (SNP) revealed that cGMP has a dual effect on Ca2+ entry. Increasing cGMP levels by up to 10-fold above that in control cells treated with N-nitro-L-arginine, a specific inhibitor of NOS, is associated with activation of Ca2+ entry. Further increases in cGMP to levels up to 80-fold above control inhibit Ca2+ entry in a concentration-dependent manner. The biphasic effect of cGMP provides the cells with a negative feedback mechanism and inhibits Ca2+ entry during periods of high [Ca2+]i, allowing oscillatory behavior in Ca2+ entry. In vascular endothelial cells isolated from bovine aorta, exogenous NO gas potentiates Ca2+ influx, which markedly increases the sustained phase of [Ca2+]i elevation and possibly NO production (Chen et al., 2000). In DDT1MF-2 smooth muscle cell line derived from hamster vas deferens and DC-3F Chinese hamster lung fibroblast cell line, Ca2+ entry activated by the lipophilic NO donor, GEA3162 [5-amino-3-(3,4-dichlorophenyl)1,2,3,4-oxatriazolium] (25 μM), or the alkylator, N-ethylmaleimide (10 μM), is strongly activated by transient external Ca2+ removal that depletes internal Ca2+ stores, closely resembling activation of SOC activity in the same cells (Ma et al., 1999). In cortical astrocytes isolated from mice, physiological concentrations of a natural neuromessenger, ATP (10 μM), induces Ca2+-dependent NO production. By promoting Ca2+ influx, NO may facilitate the refilling of internal stores that have become partially depleted as a result of Ca2+ release during neurotransmitter-induced Ca2+ signaling (Li et al., 2003).
Suppressive effects of NO on Ca2+ entry have also been demonstrated (Kwan et al., 2000; Dedkova and Blatter, 2002). In vascular endothelial cells, the membrane permeant cGMP analogue 8-Br-cGMP (>300 μM) attenuates SOC activity via a protein kinase G (PKG)-dependent mechanism. These results suggest a role of cGMP and PKG in the regulation of Ca2+ entry in vascular endothelial cells (Kwan et al., 2000; Dedkova and Blatter, 2002). Contradictory effects of NO on Ca2+ influx may depend on the difference of the type of NO donors used for experiments and their concentrations. More importantly, the contradictory effects are at least in part attributable to diversity in the NO susceptibility of Ca2+ entry channels and signaling molecules that regulate Ca2+ influx. Thus, different ensembles of NO-regulated mechanisms may lead to a wide variety of net Ca2+ entry responses in the presence of NO.
Regulation of TRP Channels by NO Via Cys S-Nitrosylation
Some TRP channels are potently regulated by Cys modifications, including Cys S-nitrosylation by NO in heterologous systems and bovine aortic endothelial cells (Yoshida et al., 2006). By performing labeling and functional assays with Cys mutants, it has been shown that Cys553 and nearby Cys558 on the N-terminal side of the putative pore-forming region between the fifth and sixth transmembrane domains S5 and S6 are essential for mouse TRPC5 activation in response to an NO donor, S-nitroso-N-acetyl-DL-penicillamine (SNAP) (300 μM) (Figure 3). In Drosophila melanogaster Shaker voltage-gated K+ channels, the activation gate formed by S6 residues near the intracellular entrance of the pore cavity has been identified (del Camino and Yellen, 2001). Considering the longer S5–S6 linkers of TRPC5, the TRPC5 S5–S6 linker with modified Cys553 and Cys558 may be invaginated toward the cytoplasm to reach the S6 activation gate (Figure 3). The corresponding Cys sites of TRPC1, TRPC4, TRPV1, TRPV3, and TRPV4 are potential targets of nitrosylation leading to channel activation in heterologous expression systems. Although the differences in maximal [Ca2+]i responses [Δ ratio (340/380)] to SNAP, in control cells and cells heterologously expressing either TRPC1 or TRPC4β (a splice isoform of TRPC4) are not statistically significant, a larger fraction (7–9%) of the TRP-expressing cells shows a Δ ratio (340/380) >0.5 when compared to control cells (2–5%). Cells heterologously co-expressing TRPC4β and TRPC5 give responses to SNAP comparable to those in cells heterologously expressing TRPC5 alone, whereas cells heterologously co-expressing TRPC1 and TRPC5 give slightly suppressed yet robust responses. Co-immunoprecipitation of TRPC5 with TRPC1 or TRPC4β suggests that heteromultimeric TRPC5/TRPC1 and TRPC5/TRPC4β channels also have NO sensitivity. The thermosensor TRP channels TRPV1, TRPV3, and TRPV4 also show SNAP (300 μM)-induced activation, as predicted from conserved Cys residues in the corresponding regions of these homologs. Indeed, substitutions of two conserved Cys residues in TRPV1 lead to significantly suppressed responses to SNAP and to Cys S-nitrosylation. Notably, the sensitivity of TRPV1 to H+ and heat is enhanced by SNAP but abolished by the mutations, despite normal surface expression and intact control H+ and heat responses. Thus, channel activation regulated by nitrosylation is conserved among a number of TRP channels belonging to different subfamilies.
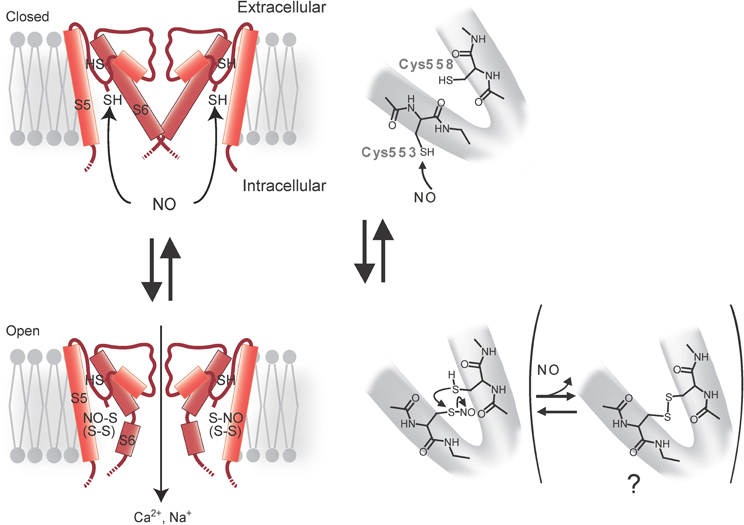
Figure 3. Model for activation of TRPC5 channel by NO. Cys553 is nitrosylated by NO, which triggers TRPC5 channel opening. The free sulfhydryl group of Cys558 nucleophilically attacks nitrosylated Cys553 to form a disulfide bond that stabilizes the open state.
SNAP-activated TRPC5 channels are not entirely inactivated by ascorbate, which reduces S-nitrosothiols to thiols but not disulfides. However, dithiothreitol (DTT), which reduces both S-nitrosothiols and disulfides to thiols, fully suppressed SNAP-activated TRPC5 channel activity, suggesting that both nitrosylation and disulfide bond formation are involved in SNAP-induced TRPC5 activation. In an S-nitrosylation assay (Jaffrey et al., 2001), S-nitrosylation is abolished by mutation of Cys553 in TRPC5 but not by mutation of Cys558. As proposed for the acid–base catalysis of hemoglobin nitrosylation in proteins with high NO sensitivity, basic and acidic amino acids surrounding S-nitrosylated Cys may enhance the nucleophilicity of the sulfhydryl (SH) group, and therefore the S-nitrosylation of this group of proteins (Hess et al., 2005). Charged residues flanking Cys553 and Cys558 may confer modification susceptibility to NO in TRPC5. It is possible that the TRPC5 channel is opened via S-nitrosylation of Cys553 and a subsequent nucleophilic attack of nitrosylated Cys553 by the free SH group of Cys558 to form a disulfide bond that stabilizes the open state (Figure 3). The NO sensitivity of TRPC5 channels has been disputed by several groups (Xu et al., 2008; Wong et al., 2010). NO sensitivity of TRPC5 may depend on culturing conditions, the way drugs are administered, cell density during measurements, levels of antioxidants or other experimental conditions, molecular and cellular states that may affect the modification state of TRPC5 proteins.
Previous reports have provided important information with respect to the formation in endothelial cells of a TRPC5 “channelsome,” a molecular assembly centered upon a channel. As mentioned above, TRPC1 forms heterotetrameric channels with TRPC5 (Strübing et al., 2001) and a protein complex with caveolin-1 in caveolae/lipid raft domains (Lockwich et al., 2000; Bergdahl et al., 2003), which regulate the plasma membrane trafficking of TRPC1 (Brazer et al., 2003). It is therefore possible that TRPC5 forms indirect protein complexes with caveolin-1 via TRPC1. Indeed, we have found an interaction of TRPC5 with caveolin-1 and eNOS in co-immunoprecipitation experiments as well as by colocalization of TRPC5 with caveolin-1 in heterologous systems and bovine aortic endothelial cells (Mori et al., unpublished data). Among numerous caveolin-associated proteins linked to signaling cascades (Couet et al., 1997; García-Cardeña et al., 1997; Sato et al., 2004; Quest et al., 2008), three isoforms of NOS, including eNOS, have been identified (Kone et al., 2003). The inhibitory association of caveolin is disrupted by the binding of Ca2+-CaM to eNOS, leading to eNOS activation (Ju et al., 1997; Michel et al., 1997a,b; Rizzo et al., 1998; Bernatchez et al., 2005). In addition, eNOS is activated by different kinases, including Akt, protein kinase A (PKA), and protein kinase C (PKC) (García-Cardeña et al., 1996; Fulton et al., 1999; Michell et al., 2001; Boo and Jo, 2003; Heijnen et al., 2004). Based on data from our and other laboratories, we can propose a plausible model to describe the role of the TRPC5 channelsome in regulating receptor-activated NO production in vascular endothelial cells (Figure 4). TRPC5 proteins form complexes with vasodilator receptors, G-proteins, PLCβ s and eNOS, and are anchored in caveolae by the scaffolding protein, caveolin-1. Upon vasodilator receptor stimulation, TRPC5 is activated by the PLCβ cascade to induce Ca2+ influx, which elevates the [Ca2+]i to form Ca2+-CaM. This then releases eNOS from the inhibitory control of caveolin-1 and leads to an initial NO production that activates TRPC5 channels. Ca2+ influx via NO-activated TRPC5 channels then induces secondary activation of eNOS to amplify the production of NO, resulting in a positive feedback cycle of receptor-activated Ca2+ and NO signaling. This model has been neatly summarized in a short review by Stamler and colleagues (Foster et al., 2006), based largely on our own data (Yoshida et al., 2006).
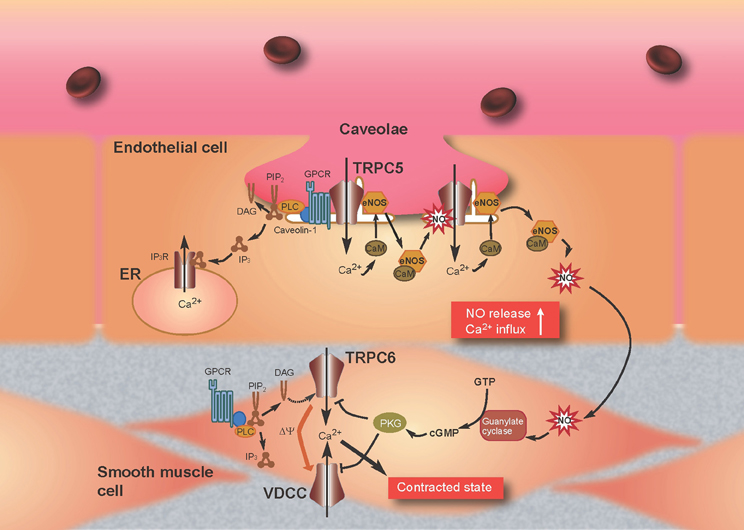
Figure 4. Model for TRPC5-mediated feedback of Ca2+ and NO signaling in endothelial cells and attenuation of Ca2+ entry through TRPC6 by NO in smooth muscle cells. Stimulation of G protein-coupled receptors (GPCRs) (such as the ATP-activated P2Y receptor) induces Ca2+ influx and activation of eNOS as a consequence of binding of Ca2+-CaM and release of eNOS from caveolin-1. TRPC5 undergoes eNOS-dependent S-nitrosylation after GPCR stimulation, resulting in amplified Ca2+ entry and secondary activation of eNOS to amplify production of NO. NO diffuses out of endothelial cells into adjacent smooth muscle cells and stimulates the guanylate cyclase, which leads to the activation of PKG in smooth muscle cells. In the most prevailing hypothesis, the magnitude of continuous Ca2+ influx through VDCC, which critically determines the contractile status of vascular smooth muscle cells, decreases and increases by membrane hyperpolarization and depolarization, respectively. TRPC6 likely functions as a depolarization (Δψ)-inducing channel or a direct Ca2+-entry pathway, activated in response to receptor stimulation. The NO/cGMP/PKG pathway suppresses TRPC6 and VDCC activity to induce relaxation of smooth muscle.
Recent reports have shown that SNAP (30 μM) and another NO donor (6)-(E)-Ethyl-2-[(E)-hydroxyimino]-5-nitro-3-hexeneamide (NOR3) (300 μM) activate human TRPA1 in heterologous systems and mouse TRPA1 in dissociated sensory neurons (Sawada et al., 2008; Takahashi et al., 2008a; Miyamoto et al., 2009). Functional characterization of site-directed Cys mutants of TRPA1 has demonstrated that Cys421, Cys641, and Cys665 are responsible for human TRPA1 activation by NO (Takahashi et al., 2008a). Cys421, Cys641, and Cys665 are located within the eleventh AnkR domain, seventeenth AnkR domain, and the N-terminal cytoplasmic region between seventeenth AnkR domain and S1, respectively (Gaudet, 2008).
Injection of NOR3 (1.5 mM) in the hind paw after PLC and PKA pathway activation (which sensitizes nociceptors, including TRPV1 and TRPA1), causes nociceptive behavior (Miyamoto et al., 2009). Interestingly, a decrease in nociception is observed in mice lacking both TRPV1 and TRPA1, but not in individual knockout animals (Miyamoto et al., 2009) despite in vitro results showing that the NO donor can activate both ion channels. Note that due to solubility issues of NO donors, limitations exist in using NO donors at relatively high concentrations for behavioral experiments (<10-fold the in vitro EC50). For capsaicin, injections of capsaicin at >1000-times the in vitro EC50 are typically used to observe acute pain behavior. It is therefore not surprising that significant acute nocifensive behavior was not observed in terms of NO donors. Another potential explanation for the lack of a phenotype in individual knockout mice is due to functional compensation by each other. Indeed, TRPV1 and TRPA1 have overlapping expression in a subset of DRG neurons (Story et al., 2003) and thereby could potentially interact via intracellular signaling in vivo. Recently, icilin, an agonist of TRPA1 and TRPM8, has been implicated as a trigger for shaking and hyperthermia which require NO production and NMDA receptor activation (Ding et al., 2008; Werkheiser et al., 2009). In this context, it would be interesting to explore the mechanism of the concerted regulation of Ca2+ and NO signaling by the TRP channels, NMDA receptors and nNOS in neurons.
Regulation of TRP Channels Via the NO/cGMP/PKG Pathway
One of the principal consequences of activation of the NO/cGMP/PKG cascade in blood vessels is vasorelaxation, which is mediated by phosphorylation of proteins that regulate intracellular Ca2+ levels and the sensitivity of the contractile machinery. Several distinct mechanisms have been proposed for the reduction in [Ca2+]i caused by PKG (Lincoln et al., 2001; Feil et al., 2003). Increased activity of the BKCa channel following PKG activation has been reported to induce membrane hyperpolarization, thereby decreasing the rate of Ca2+ entry into vascular smooth muscle cells (VSMCs) through voltage-dependent Ca2+ channels (VDCCs). Phosphorylation of the inositol 1,4,5-trisphosphate receptor (IP3R)-associated protein, IRAG (IP3R-associated PKG substrate), is thought to inhibit agonist-induced Ca2+ release from internal stores. The possibility has also been suggested that phosphorylation of phospholamban by PKG increases the activity of sarcoplasmic Ca2+-ATPase, facilitating the active transport of Ca2+ into internal stores and thereby decreasing [Ca2+]i. In many different types of blood vessel, activation of PKG has been found to inhibit vasoconstrictor-induced Ca2+ influx through plasmalemmal pathways other than those employing VDCCs (Karaki et al., 1997). However, little information exists regarding the molecular components of such Ca2+ influx pathways and, accordingly, it remains unclear how phosphorylation via PKG activation reduces the rate of Ca2+ influx.
TRPC6 is a Ca2+-permeable channel regulated negatively by PKG-mediated phosphorylation in the NO/cGMP signaling pathway in heterologous systems and the rat vascular myocytes A7r5 (Takahashi et al., 2008b). Macroscopic and single-channel current recordings using patch clamp techniques have demonstrated that SNAP (100 μM)-induced inhibition of receptor-activated TRPC6 currents is abolished by pharmacological blockade of cGMP/PKG signaling with 1H-[1,2,4]oxadiazolo [4,3-a]quinoxalin-1-one (ODQ), 2,3,9,10,11,12-hexahydro-10R-methoxy-2,9-dimethyl-1-oxo-9S,12R-epoxy-1H-diindolo[1, 2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester (KT5823) or membrane permeable PKG inhibitory peptide (DT3). It is also ablated by site-directed alanine mutation of a PKG phosphorylation site [threonine (Thr) 69] within the N-terminal cytoplasmic region of TRPC6. The critical involvement of Thr69 in PKG phosphorylation is confirmed by 32P-incorporation assays of wild-type and alanine-substitution mutant TRPC6 proteins. Similar NO/cGMP/PKG pathway-mediated negative regulation is also observed for TRPC6-like currents recorded in A7r5 VSMCs. Indeed, vasopressin-evoked membrane depolarization of these cells, which is expected secondarily to activate VDCCs, is significantly slowed and attenuated after application of SNAP (100 μM). The TRPC6 protein is abundantly expressed in various types of VSMCs and has been shown to be a constituent of vasoconstrictor-activated cation channels, which increase Ca2+ entry into VSMCs via direct Ca2+ permeation or secondary activation of a VDCC and/or Na+-Ca2+ exchanger (Inoue et al., 2006; Dietrich et al., 2007; Poburko et al., 2007). Thus, it is highly possible that, in a direct or indirect manner (i.e., via changes in membrane potential or an increase in intracellular Na+ concentration), PKG-mediated mechanisms may work as a universal negative feedback to regulate neurohormonal Ca2+ mobilization across the VSMC membrane (Figure 4). This mechanism may be physiologically important in vascular tissues where NO is constantly released from vascular endothelial cells or nitrergic nerves.
O2 Sensing Mechanisms
Of the gaseous molecules, O2 is the most well-known to physicians, scientists and laymen alike as an essential physical requirement. O2 functions primarily as a terminal acceptor of electrons on mitochondrial electron transport. Most of the O2 consumed in this process is reduced to generate water through the actions of cytochrome oxidase. The remainder is used to generate compounds that exert potent biological actions, including prostaglandins, reactive oxygen species (ROS) and gaseous molecules such as NO and CO (Suematsu et al., 2003). In the last decade, O2 itself has been increasingly recognized as an important signal molecule that mediates many physiological and pathophysiological processes including proliferation of stem cells, ischemia injury and tumor progression (Csete, 2006; Swartz et al., 2007). Thus, O2 is not only required for cellular respiration, but also serves as a signaling molecule, and as the essential substrate for the formation of other signaling molecules (Figure 2). However, O2 also exerts toxicity causing aging, respiratory disorders and eventually death in a high O2 (hyperoxic) environment. Because of the ambivalent physiological nature of O2, aerobic life forms must adapt to hyperoxia and low O2 environment (hypoxia) by sensing O2 availability and transmitting this information to effector systems.
Cellular responses to changes in O2 availability can be acute or chronic (López-Barneo et al., 2001). Acute responses rely mainly on O2-regulated ion channels, which mediate adaptive changes in cell excitability, contractility, and secretory activity (Gonzalez et al., 1994; Neubauer and Sunderram, 2004; Weir et al., 2005). Chronic responses depend on the modulation of transcription factors such as hypoxia-inducible factor (HIF), which determines the expression of numerous genes encoding growth factors, enzymes, and transporters (Semenza and Wang, 1992; Schofield and Ratcliffe, 2004; Webb et al., 2009). O2-regulated ion channels and transcription factors are part of a widely operating signaling system that helps to provide an appropriate amount of O2 to the tissues and to protect the cells against toxicity damage due to excess or deficient O2.
In mammals, the carotid bodies, located near the carotid artery bifurcations, and brainstem catecholaminergic neurons rapidly detect changes in partial O2 pressure (PO2) in arterial blood (Gonzalez et al., 1994; Neubauer and Sunderram, 2004). It is known that BKCa, TASK, and KV K+ channels are involved in the mechanism of arterial O2 sensing in the carotid bodies (Gonzalez et al., 1994; Weir et al., 2005). Hypoxia inhibits K+ channels through several mechanisms, such as CO production by hemeoxygenase and intracellular ATP depletion that depolarizes glomus cells. This inhibition leads to activation of VDCC, exocytosis, and the excitation of carotid sinus nerves. In contrast, hyperoxia attenuates depolarization and inhibits exocytosis (Gonzalez et al., 1994; Weir et al., 2005). The chemosensory inputs of the carotid sinus nerve are carried toward the medullary centers that regulate the ventilatory pattern. The local O2 tension is also rapidly detected by other tissues including vagal and sensory neurons in the airway and lungs, chromaffin cells of the fetal adrenal medulla, smooth muscle cells of the pulmonary resistance arteries, cerebral neurons, fetoplacental arteries, systemic arteries, and the doctus arteriosus. However, detection of hypoxia by these tissues remains to be fully characterized. Recently, a class of TRP channels has been demonstrated to act as cell sensors for changes in O2 availability (Aarts et al., 2003; Weissmann et al., 2006; Takahashi et al., 2011).
Anoxia-Sensing Mediated by TRPM7 Channels in the Brain
Excitotoxicity in brain ischemia triggers neuronal death and neurological disability, and yet these are not prevented by antiexcitotoxic therapy (AET) in humans. The failure of AET in the face of a clear role for excitotoxicity in acute neurological disorders is paradoxical (Birmingham, 2002; Ikonomidou and Turski, 2002). In addressing this problem, Aarts et al. have revealed that the TRPM7 channel, termed chanzyme because it possess a channel and α-kinase domain (Runnels et al., 2001; Nadler et al., 2001), is activated by oxygen-glucose deprivation (OGD) through the production of ROS and RNS, permitting Ca2+ uptake that further stimulates ROS and TRPM7 activation in heterologous systems and cortical neurons isolated form rats (Aarts et al., 2003). Suppressing TRPM7 expression in rat cortical neurons prevents anoxic neuronal death even in the absence of AET, indicating that TRPM7 is an essential mediator of anoxic death. It is possible that patients enrolled in failed trials studying the use of AET for stroke or traumatic brain injury are selected to have severe injuries (Morris et al., 1999), or that these disorders in humans, by their nature, induce severe ischemia. Therefore, future treatment of such disorders may also need to inhibit TRPM7. Indeed, it has been shown that suppression of hippocampal TRPM7 by intrahippocampal injections of viral vectors bearing shRNA specific for TRPM7 makes neurons resistant to ischemic death after brain ischemia and preserves neuronal morphology and function in rats (Sun et al., 2009). TRPM7 suppression also prevents ischemia-induced deficits in long-term potentiation and preserves performance in fear-associated and spatial-navigational memory tasks. Thus, regional suppression of TRPM7 is feasible, well tolerated and inhibits delayed neuronal death in vivo in an animal model.
Hypoxia-Sensing by TRPC6 Channels in Pulmonary Smooth Muscle Cells
Regional alveolar hypoxia causes local vasoconstriction in the lungs, shifting blood flow from hypoxic to normoxic areas, thereby maintaining gas exchange. This mechanism is known as hypoxic pulmonary vasoconstriction (HPV) (Jeffery and Wanstall, 2001; Weissmann et al., 2001; Schermuly et al., 2005). Disturbances in HPV can cause life-threatening hypoxemia, whereas chronic hypoxia triggers vascular remodeling in the lungs and pulmonary hypertension (Sartori et al., 1999). In studying signaling cascades of this vitally important mechanism, Weissmann et al. have shown that severe hypoxia (1% O2)-induced cation influx and currents in smooth muscle cells are largely absent in precapillary pulmonary arteries of Trpc6 knockout mice, although recombinant TRPC6 expressed heterologously cannot be activated by hypoxia (Weissmann et al., 2006). Hypoxia-induced TRPC6 activation in smooth muscle cells is mediated by DAG accumulation probably by activated phospholipases. TRPC6 appears to be a key regulator of acute HPV, because this regulatory mechanism is absent in Trpc6 knockout mice, whereas the pulmonary vasoconstrictor response to the thromboxane mimetic, U46619, is unchanged. Accordingly, induction of regional hypoventilation results in severe arterial hypoxemia in Trpc6 knockout mice, but not in wild type mice. Notably, chronic hypoxia-induced pulmonary hypertension is independent of TRPC6 activity. Thus, TRPC6 plays a unique and indispensable role in acute HPV. Manipulation of TRPC6 function may thus offer a therapeutic strategy for the control of pulmonary hemodynamics and gas exchange.
O2-Sensing by TRPA1 Channel in Vagal and Sensory Neurons
Sensory and vagal afferent neurons, which project nerve endings throughout the body, are thought to detect hypoxia in organs such as the airway, lungs and heart after ischemia, and other conditions of low O2 supply (Howe et al., 1981; De Sanctis et al., 1991; Longhurst et al., 2001; Gruss et al., 2006). However, the characteristics and mechanisms of hypoxia detection by sensory and vagal neurons have yet to be fully defined (Longhurst et al., 2001). In terms of hyperoxia, Caenorhabditis elegans has been reported to be adept at avoiding hyperoxia due to detection mechanisms in sensory neurons (Gray et al., 2004). Furthermore, insects breathe discontinuously to avoid O2 toxicity during hyperoxia (Hetz and Bradley, 2005). However, the physiological relevance of hyperoxia detection through sensory systems is less clear in vertebrates, and the delineation of these neuronal hyperoxia-sensing molecular processes in vertebrates remains an exciting area of research.
Systematic evaluation of TRP channels using reactive disulfides with different redox potentials has revealed that TRPA1 can sense O2 (Takahashi et al., 2011). O2 sensing by TRPA1 is based upon disparate processes: proline (Pro) hydroxylation by Pro hydroxylases (PHDs), and direct oxidation of Cys residues. In normoxia, PHDs hydroxylate conserved Pro394 within the tenth AnkR domain of TRPA1, inhibiting its activity. In hypoxia, the decrease in O2 concentration diminishes PHD activity, relieving TRPA1 from the inhibitory action of Pro hydroxylation and leading to its activation. This recovery of TRPA1 activity can be achieved by insertion of fresh, unmodified TRPA1 proteins into the plasma membrane or by dehydroxylation of modified proteins through an unidentified molecular mechanism. In hyperoxia, O2 activates TPRA1 by oxidizing Cys633, Cys856 or both. Cys633 and Cys856 are located within the seventeenth AnkR domain and the intracellular linker region between S4 and S5, respectively. TRPA1 can assume at least two oxidized forms during hyperoxia: a relatively unstable oxidized state (state 1) readily reversed by glutathione, and a relatively stable oxidized state (state 2). SH groups on the key Cys residues (Cys633 and Cys856) may be modified to sulfenic acid (S-OH) in state 1, and form disulfide bonds (S-S) in state 2. This oxidation mechanism over-rides the inhibition by Pro hydroxylation to activate TRPA1. In mice, disruption of the Trpa1 gene abolishes hyperoxia- and mild hypoxia (15% O2)-induced cationic currents in vagal and sensory neurons and thereby impedes enhancement of in vivo vagal discharges induced by hyperoxia and hypoxia. The results suggest a new O2-sensing mechanism mediated by TRPA1 (Figure 5).

Figure 5. Model for O2-sensing of TRPA1 channel. PHDs hydroxylate specific Pro residue on the N-terminal AnkR domain of TRPA1 protein in normoxia, whereas a decrease in O2 concentrations diminishes PHD activity and relieves TRPA1 from the hydroxylation, leading to its activation in hypoxia. The relief can be achieved by insertion of unmodified TRPA1 proteins into the plasma membrane or by dehydroxylation through an unidentified molecular mechanism. In hyperoxia, O2 oxidizes specific Cys residues, thereby activating TRPA1. TRPA1 may at least take two oxidized state upon hyperoxia: a relatively unstable oxidized state (state 1) readily reversed by glutathione and a relatively stable oxidized state (state 2). Sulfhydryl group(s) of the key Cys residues may be modified to sulfenic acid in the former state of TRPA1, whereas that in the latter state of TRPA1 may form a disulfide bond(s). These oxidation mechanisms over-ride the inhibition by Pro hydroxylation to activate TRPA1.
The vagal nerve conveys sensory information about the state of the organs to the central nervous system, in addition to providing output to the various organs in the body. Enhanced discharges in vagal afferents induce respiratory, cardiac, and vascular responses (Meller and Gebhart, 1992; Longhurst et al., 2001; Kubin et al., 2006). Chemicals encountered in the airway are detected by airway vagal C fibers (Kubin et al., 2006). Recently, TRPA1 has been shown to sense environmental irritants, thus initiating defensive reflexes such as coughing and respiratory depression in the C fibers (Bessac and Jordt, 2008; Bessac et al., 2008; Nassenstein et al., 2008). Notably, TRPA1 activation shows an inverted bell-shaped O2-dependency curve with a minimum at the PO2 of 137 mmHg, which is slightly below the atmospheric PO2 (159 mmHg) (Takahashi et al., 2011). Considering that tracheal PO2 (149 mmHg) is comparable to atmospheric PO2 (Cottrell, 1988), it is highly possible that TRPA1 expressed in the trachea is slightly but significantly activated to act as a frontline defense against mild hyperoxia in the atmosphere.
Weather records (McElroy, 2002) suggest that atmospheric PO2 at sea level ranges between approximately 137 and 170 mmHg. Because our O2 dependence data reveal that minimal TRPA1 activity at a PO2 of 137 mmHg is approximately 30% of the maximum activity at a PO2 of 170 mmHg, so-called normoxia can be hyperoxic in the context of mammalian TRPA1 channels. This is reminiscent of O2 avoidance in Caenorhabditis elegans (Gray et al., 2004) and insects (Hetz and Bradley, 2005).
Sensing of Other Gaseous Molecules by TRP Channels
H2S has emerged as a new gaseous modulator of various biological functions including nociception (Szabó, 2007; Gadalla and Snyder, 2010). In rodents, topical application of an H2S donor, NaHS (1 nmol) produces pain responses through potentiation of T-type Ca2+ channels probably in the primary afferents (Kawabata et al., 2007; Matsunami et al., 2009). Endogenous H2S is also reported to contribute to pain transmission in rat models of formalin-induced inflammation (Lee et al., 2008) and irritable bowel syndrome (Xu et al., 2009).
NaHS (1 mM) activates capsaicin-sensitive sensory neurons in isolated rat urinary bladder (Patacchini et al., 2005). Although the mechanisms underlying the action of H2S are yet to be clarified, the pharmacological profile of H2S hints at the involvement of TRPA1. Indeed, Streng et al. have shown that NaHS (1 mM) causes activation of human and mouse TRPA1 in heterologous expression systems, suggesting that TRPA1 is indeed a molecular target for H2S in the bladder (Streng et al., 2008). In this context, it is interesting to note that bladder inflammation can be triggered by TRPA1 activation (Cox, 1979; McMahon and Abel, 1987) and that potential pathogens, such as Escherichia coli, can produce H2S (Berglin and Carlsson, 1985). Recently, Miyamoto et al. have shown that NaHS (1 μM)-evoked increases in [Ca2+]i are inhibited by removal of external Ca2+ and by a TRPA1-specific inhibitor, HC-030031, suggesting that H2S stimulates sensory neurons via activation of TRPA1 (Miyamoto et al., 2011). Furthermore, the hyperalgesia/allodynia induced in mice by intraplantar administration of NaHS (100 pmol) is significantly suppressed by pre-administration of a TRPA1-specific blocker AP18 and by silencing TRPA1 channels in sensory neurons (Okubo et al., 2012). Thus, H2S-induced mechanical hyperalgesia and allodynia require activation of TRPA1 channels.
In humans, gaseous CO2 produces a pungent sensation, as noted by the Scottish philosopher Alexander Bain more than 100 years ago (Cain and Murphy, 1980). A variety of sensory structures and receptors mediate the responses to CO2 in different organisms (Luo et al., 2009). For example, in flies, gaseous CO2 is detected by gustatory receptors on the antenna, whereas dissolved CO2 is detected on the proboscis: CO2 is either aversive or attractive depending on the sensory structure activated (Suh et al., 2004; Fischler et al., 2007; Jones et al., 2007; Kwon et al., 2007). In mice, ingested CO2 is sensed by taste receptors in the mouth (Chandrashekar et al., 2009), and blood CO2 is detected in the brainstem by K+ channels (Trapp et al., 2008) and in the amygdala by acid-sensing ion channel (ASIC) 1a (Ziemann et al., 2009). Atmospheric levels of CO2 are detected by guanylyl cyclase D, which is expressed in a subset of olfactory sensory neurons in mice (Luo et al., 2009). However, this latter system is missing in humans and most other primates (Young et al., 2007). It is widely believed that the noxious sensation of CO2 is due to activation of the trigeminal nerve fibers that innervate the nasal and oral cavities (Silver and Moulton, 1982; Steen et al., 1992). Recently, Wang et al. have shown that CO2 specifically activates a subpopulation of trigeminal neurons expressing a functional Trpa1 gene (Wang et al., 2010). CO2-induced activation of TRPA1 is downstream of intracellular acidification, consistent with our observation that TRPA1 is activated by H+ (Takahashi et al., 2008a). Thus, TRPA1 makes an important contribution to nociceptive responses to CO2.
Conclusions
TRP channels respond to multiple activation triggers and therefore serve as polymodal signal detectors. An important aspect of this multimodal activation of TRP channels is their role in signal integration and amplification. When a TRP channel is activated by downstream or upstream constituents (molecules/proteins/enzymes in a specific signaling cascade), in addition to the primary activation trigger immediately upstream, the TRP channel plays an important role in forming positive feedback or feed-forward loops in the cascade. For example, in vascular endothelial cells, Ca2+ influx via NO-activated TRPC5 channels can amplify production of NO by eNOS, resulting in the enhancement of NO production in nearby endothelial cells and NO-dependent relaxation of smooth muscle cells. In smooth muscle cells, NO exerts inhibitory effects on TRPC6 channel activity through PKG-mediated phosphorylation. Together, these mechanisms are capable of ensuring the fidelity of cellular responses and minimizing variation in their magnitude, synchronizing the responses of neighboring cells that comprise functional domains within vascular tissues to reduce blood pressure and maintain local blood flow.
The accumulating evidence summarized above strongly suggests that TRPV1 and TRPA1 are sensors that transduce gaseous signals into electrical signals in sensory and vagal neurons. The chemosensory inputs to these neurons are propagated toward the central nervous system to induce pain sensation or to change ventilatory patterns. However, the roles of Ca2+ influx via TRPV1 and TRPA1 in controlling Ca2+ signaling pathways in neurons remain elusive. Considering that the PHD-HIF pathway is central to chronic hypoxia responses that increase red blood cell mass and stimulate new blood vessel growth (Semenza and Wang, 1992; Webb et al., 2009), it would be interesting to examine the effect of Ca2+ influx through O2-sensitive TRPA1 channels on the functional regulation of PHD and HIF. Studies of TRP channels have been extended dramatically from the simple functional description of single molecules to the holistic analysis and integration of the molecular systems controlled by TRP channels.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Emiko Mori and Shaun Johnson for providing materials in constructing Figure 4.
References
Aarts, M., Iihara, K., Wei, W. L., Xiong, Z. G., Arundine, M., Cerwinski, W., MacDonald, J. F., and Tymianski, M. (2003). A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877.
Bandell, M., Story, G. M., Hwang, S. W., Viswanath, V., Eid, S. R., Petrus, M. J., Earley, T. J., and Patapoutian, A. (2004). Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41, 849–857.
Bautista, D. M., Jordt, S. E., Nikai, T., Tsuruda, P. R., Read, A. J., Poblete, J., Yamoah, E. N., Basbaum, A. I., and Julius, D. (2006). TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124, 1269–1282.
Benhar, M., Forrester, M. T., and Stamler, J. S. (2009). Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 10, 721–732.
Bergdahl, A., Gomez, M. F., Dreja, K., Xu, S. Z., Adner, M., Beech, D. J., Broman, J., Hellstrand, P., and Swärd, K. (2003). Cholesterol depletion impairs vascular reactivity to endothelin-1 by reducing store-operated Ca2+ entry dependent on TRPC1. Circ. Res. 93, 839–847.
Berglin, E. H., and Carlsson, J. (1985). Potentiation by sulfide of hydrogen peroxide-induced killing of Escherichia coli. Infect. Immun. 49, 538–543.
Bernatchez, P. N., Bauer, P. M., Yu, J., Prendergast, J. S., He, P., and Sessa, W. C. (2005). Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc. Natl. Acad. Sci. U.S.A. 102, 761–766.
Bessac, B. F., and Jordt, S. E. (2008). Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology (Bethesda) 23, 360–370.
Bessac, B. F., Sivula, M., von Hehn, C. A., Escalera, J., Cohn, L., and Jordt, S. E. (2008). TRPA1 is a major oxidant sensor in murine airway sensory neurons. J. Clin. Invest. 118, 1899–1910.
Boo, Y. C., and Jo, H. (2003). Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am. J. Physiol. Cell Physiol. 285, C499–C508.
Brazer, S. C., Singh, B. B., Liu, X., Swaim, W., and Ambudkar, I. S. (2003). Caveolin-1 contributes to assembly of store-operated Ca2+ influx channels by regulating plasma membrane localization of TRPC1. J. Biol. Chem. 278, 27208–27215.
Cain, W. S., and Murphy, C. L. (1980). Interaction between chemoreceptive modalities of odour and irritation. Nature 284, 255–257.
Caterina, M. J., Schumacher, M. A., Tominaga, M., Rosen, T. A., Levine, J. D., and Julius, D. (1997). The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824.
Chandrashekar, J., Yarmolinsky, D., von Buchholtz, L., Oka, Y., Sly, W., Ryba, N. J., and Zuker, C. S. (2009). The taste of carbonation. Science 326, 443–445.
Chen, J., Wang, Y., Nakajima, T., Iwasawa, K., Hikiji, H., Sunamoto, M., Choi, D. K., Yoshida, Y., Sakaki, Y., and Toyo-oka, T. (2000). Autocrine action and its underlying mechanism of nitric oxide on intracellular Ca2+ homeostasis in vascular endothelial cells. J. Biol. Chem. 275, 28739–28749.
Choi, Y. B., Tenneti, L., Le, D. A., Ortiz, J., Bai, G., Chen, H. S., and Lipton, S. A. (2000). Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat. Neurosci. 3, 15–21.
Clapham, D. E., Julius, D., Montell, C., and Schultz, G. (2005). Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol. Rev. 57, 427–450.
Couet, J., Li, S., Okamoto, T., Ikezu, T., and Lisanti, M. P. (1997). Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J. Biol. Chem. 272, 6525–6533.
Cox, P. J. (1979). Cyclophosphamide cystitis–identification of acrolein as the causative agent. Biochem. Pharmacol. 28, 2045–2049.
Dawson, V. L., Dawson, T. M., London, E. D., Bredt, D. S., and Snyder, S. H. (1991). Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. U.S.A. 88, 6368–6371.
Dedkova, E. N., and Blatter, L. A. (2002). Nitric oxide inhibits capacitative Ca2+ entry and enhances endoplasmic reticulum Ca2+ uptake in bovine vascular endothelial cells. J. Physiol. 539, 77–91.
del Camino, D., and Yellen, G. (2001). Tight steric closure at the intracellular activation gate of a voltage-gated K+ channel. Neuron 32, 649–656.
De Sanctis, G. T., Green, F. H., and Remmers, J. E. (1991). Ventilatory responses to hypoxia and hypercapnia in awake rats pretreated with capsaicin. J. Appl. Physiol. 70, 1168–1174.
Dietrich, A., Kalwa, H., Fuchs, B., Grimminger, F., Weissmann, N., and Gudermann, T. (2007). In vivo TRPC functions in the cardiopulmonary vasculature. Cell Calcium 42, 233–244.
Ding, Z., Gomez, T., Werkheiser, J. L., Cowan, A., and Rawls, S. M. (2008). Icilin induces a hyperthermia in rats that is dependent on nitric oxide production and NMDA receptor activation. Eur. J. Pharmacol. 578, 201–208.
Duncan, L. M., Deeds, J., Hunter, J., Shao, J., Holmgren, L. M., Woolf, E. A., Tepper, R. I., and Shyjan, A. W. (1998). Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res. 58, 1515–1520.
Feil, R., Lohmann, S. M., de Jonge, H., Walter, U., and Hofmann, F. (2003). Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ. Res. 93, 907–916.
Fischler, W., Kong, P., Marella, S., and Scott, K. (2007). The detection of carbonation by the Drosophila gustatory system. Nature 448, 1054–1057.
Foster, M. W., Hess, D. T., and Stamler, J. S. (2006). S-nitrosylation TRiPs a calcium switch. Nat. Chem. Biol. 2, 570–571.
Fulton, D., Gratton, J. P., McCabe, T. J., Fontana, J., Fujio, Y., Walsh, K., Franke, T. F., Papapetropoulos, A., and Sessa, W. C. (1999). Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399, 597–601.
Furchgott, R. F., and Zawadzki, J. V. (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288, 373–376.
Gadalla, M. M., and Snyder, S. H. (2010). Hydrogen sulfide as a gasotransmitter. J. Neurochem. 113, 14–26.
García-Cardeña, G., Fan, R., Stern, D. F., Liu, J., and Sessa, W. C. (1996). Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. J. Biol. Chem. 271, 27237–27240.
García-Cardeña, G., Martasek, P., Masters, B. S., Skidd, P. M., Couet, J., Li, S., Lisanti, M. P., and Sessa, W. C. (1997). Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J. Biol. Chem. 272, 25437–25440.
Gaudet, R. (2008). A primer on ankyrin repeat function in TRP channels and beyond. Mol. BioSyst. 4, 372–379.
Gonzalez, C., Almaraz, L., Obeso, A., and Rigual, R. (1994). Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol. Rev. 74, 829–898.
Gray, J. M., Karow, D. S., Lu, H., Chang, A. J., Chang, J. S., Ellis, R. E., Marletta, M. A., and Bargmann, C. I. (2004). Oxygen sensation and social feeding mediated by a C. elegans guanylate cyclase homologue. Nature 430, 317–322.
Gruss, M., Ettorre, G., Stehr, A. J., Henrich, M., Hempelmann, G., and Scholz, A. (2006). Moderate hypoxia influences excitability and blocks dendrotoxin sensitive K+ currents in rat primary sensory neurones. Mol. Pain 2, 12.
Heijnen, H. F., Waaijenborg, S., Crapo, J. D., Bowler, R. P., Akkerman, J. W., and Slot, J. W. (2004). Colocalization of eNOS and the catalytic subunit of PKA in endothelial cell junctions: a clue for regulated NO production. J. Histochem. Cytochem. 52, 1277–1285.
Hess, D. T., Matsumoto, A., Kim, S. O., Marshall, H. E., and Stamler, J. S. (2005). Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166.
Hetz, S. K., and Bradley, T. J. (2005). Insects breathe discontinuously to avoid oxygen toxicity. Nature 433, 516–519.
Howe, A., Pack, R. J., and Wise, J. C. (1981). Arterial chemoreceptor-like activity in the abdominal vagus of the rat. J. Physiol. 320, 309–318.
Hutcheson, I. R., and Griffith, T. M. (1997). Central role of intracellular calcium stores in acute flow- and agonist-evoked endothelial nitric oxide release. Br. J. Pharmacol. 122, 117–125.
Ikonomidou, C., and Turski, L. (2002). Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 1, 383–386.
Inoue, R., Jensen, L. J., Shi, J., Morita, H., Nishida, M., Honda, A., and Ito, Y. (2006). Transient receptor potential channels in cardiovascular function and disease. Circ. Res. 99, 119–131.
Jaffrey, S. R., Erdjument-Bromage, H., Ferris, C. D., Tempst, P., and Snyder, S. H. (2001). Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3, 193–197.
Jeffery, T. K., and Wanstall, J. C. (2001). Pulmonary vascular remodeling: a target for therapeutic intervention in pulmonary hypertension. Pharmacol. Ther. 92, 1–20.
Jones, W. D., Cayirlioglu, P., Kadow, I. G., and Vosshall, L. B. (2007). Two chemosensory receptors together mediate carbon dioxide detection in Drosophila. Nature 445, 86–90.
Jordt, S. E., Bautista, D. M., Chuang, H. H., McKemy, D. D., Zygmunt, P. M., Höqestätt, E. D., Meng, I. D., and Julius, D. (2004). Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427, 260–265.
Ju, H., Zou, R., Venema, V. J., and Venema, R. C. (1997). Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J. Biol. Chem. 272, 18522–18525.
Karaki, H., Ozaki, H., Hori, M., Mitsui-Saito, M., Amano, K., Harada, K., Miyamoto, S., Nakazawa, H., Won, K. J., and Sato, K. (1997). Calcium movements, distribution, and functions in smooth muscle. Pharmacol. Rev. 49, 157–230.
Karashima, Y., Talavera, K., Everaerts, W., Janssens, A., Kwan, K. Y., Vennekens, R., Nilius, B., and Voets, T. (2009). TRPA1 acts as a cold sensor in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 1273–1278.
Kawabata, A., Ishiki, T., Nagasawa, K., Yoshida, S., Maeda, Y., Takahashi, T., Sekiguchi, F., Wada, T., Ichida, S., and Nishikawa, H. (2007). Hydrogen sulfide as a novel nociceptive messenger. Pain 132, 74–81.
Khan, S. A., and Hare, J. M. (2003). The role of nitric oxide in the physiological regulation of Ca2+ cycling. Curr. Opin. Drug Discov. Devel. 6, 658–666.
Kone, B. C., Kuncewicz, T., Zhang, W., and Yu, Z. Y. (2003). Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am. J. Physiol. Renal Physiol. 285, F178–F190.
Koyama, T., Kimura, C., Park, S. J., Oike, M., and Ito, Y. (2002). Functional implications of Ca2+ mobilizing properties for nitric oxide production in aortic endo-thelium. Life Sci. 72, 511 –520.
Kubin, L., Alheid, G. F., Zuperku, E. J., and McCrimmon, D. R. (2006). Central pathways of pulmonary and lower airway vagal afferents. J. Appl. Physiol. 101, 618–627.
Kwan, H. Y., Huang, Y., and Yao, X. (2000). Store-operated calcium entry in vascular endothelial cells is inhibited by cGMP via a protein kinase G-dependent mechanism. J. Biol. Chem. 275, 6758–6763.
Kwan, K. Y., Allchorne, A. J., Vollrath, M. A., Christensen, A. P., Zhang, D. S., Woolf, C. J., and Corey, D. P. (2006). TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron 50, 277–289.
Kwon, J. Y., Dahanukar, A., Weiss, L. A., and Carlson, J. R. (2007). The molecular basis of CO2 reception in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 104, 3574–3578.
Lantoine, F., Iouzalen, L., Devynck, M. A., Millanvoye-Van Brussel, E., and David-Dufilho, M. (1998). Nitric oxide production in human endothelial cells stimulated by histamine requires Ca2+ influx. Biochem. J. 330, 695–699.
Lee, A. T., Shah, J. J., Li, L., Cheng, Y., Moore, P. K., and Khanna, S. (2008). A nociceptive-intensity-dependent role for hydrogen sulphide in the formalin model of persistent inflammatory pain. Neuroscience 152, 89–96.
Li, N., Sul, J. Y., and Haydon, P. G. (2003). A calcium-induced calcium influx factor, nitric oxide, modulates the refilling of calcium stores in astrocytes. J. Neurosci. 23, 10302–10310.
Lin, S., Fagan, K. A., Li, K. X., Shaul, P. W., Cooper, D. M., and Rodman, D. M. (2000). Sustained endothelial nitric-oxide synthase activation requires capacitative Ca2+ entry. J. Biol. Chem. 275, 17979–17985.
Lincoln, T. M., Dey, N., and Sellak, H. (2001). Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J. Appl. Physiol. 91, 1421–1430.
Lipton, S. A., Choi, Y. B., Takahashi, H., Zhang, D., Li, W., Godzik, A., and Bankston, L. A. (2002). Cysteine regulation of protein function–as exemplified by NMDA-receptor modulation. Trends Neurosci. 25, 474–480.
Lockwich, T. P., Liu, X., Singh, B. B., Jadlowiec, J., Weiland, S., and Ambudkar, I. S. (2000). Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J. Biol. Chem. 275, 11934–11942.
Longhurst, J. C., Tjen-A-Looi, S. C., and Fu, L. W. (2001). Cardiac sympathetic afferent activation provoked by myocardial ischemia and reperfusion. Mechanisms and reflexes. Ann. N.Y. Acad. Sci. 940, 74–95.
López-Barneo, J., Pardal, R., and Ortega-Sáenz, P. (2001). Cellular mechanism of oxygen sensing. Annu. Rev. Physiol. 63, 259–287.
Luo, M., Sun, L., and Hu, J. (2009). Neural detection of gases–carbon dioxide, oxygen–in vertebrates and invertebrates. Curr. Opin. Neurobiol. 19, 354–361.
Ma, H. T., Favre, C. J., Patterson, R. L., Stone, M. R., and Gill, D. L. (1999). Ca2+ entry activated by S-nitrosylation. Relatioship to store-operated Ca2+ entry. J. Biol. Chem. 274, 35318–35324.
Matsunami, M., Tarui, T., Mitani, K., Nagasawa, K., Fukushima, O., Okubo, K., Yoshida, S., Takemura, M., and Kawabata, A. (2009). Luminal hydrogen sulfide plays a pronociceptive role in mouse colon. Gut 58, 751–761.
McElroy, M. B. (2002). The Atmospheric Environment: Effects of Human Activity. New Jersey, NY: Princeton University Press.
McKemy, D. D., Neuhausser, W. M., and Julius, D. (2002). Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416, 52–58.
McMahon, S. B., and Abel, C. (1987). A model for the study of visceral pain states: chronic inflammation of the chronic decerebrate rat urinary bladder by irritant chemicals. Pain 28, 109–127.
Meller, S. T., and Gebhart, G. F. (1992). A critical review of the afferent pathways and the potential chemical mediators involved in cardiac pain. Neuroscience 48, 501–524.
Michel, J. B., Feron, O., Sacks, D., and Michel, T. (1997a). Reciprocal regulation of endothelial nitricoxide synthase by Ca2+-calmodulin and caveolin. J. Biol. Chem. 272, 15583–15586.
Michel, J. B., Feron, O., Sase, K., Prabhakar, P., and Michel, T. (1997b). Caveolin versus calmodulin. Counterbalancing allosteric modulators of endothelial nitric oxide synthase. J. Biol. Chem. 272, 25907–25912.
Michell, B. J., Chen, Z. p., Tiganis, T., Stapleton, D., Katsis, F., Power, D. A., Sim, A. T., and Kemp, B. E. (2001). Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J. Biol. Chem. 276, 17625–17628.
Milbourne, E. A., and Bygrave, F. L. (1995). Do nitric oxide and cGMP play a role in calcium cycling? Cell Calcium 18, 207–213.
Miyamoto, R., Otsuguro, K., and Ito, S. (2011). Time- and concentration-dependent activation of TRPA1 by hydrogen sulfide in rat DRG neurons. Neurosci. Lett. 499, 137–142.
Miyamoto, T., Dubin, A. E., Petrus, M. J., and Patapoutian, A. (2009). TRPV1 and TRPA1 mediate peripheral nitric oxide-induced nociception in mice. PLoS ONE 4:e7596. doi: 10.1371/journal.pone.0007596
Moncada, S., Higgs, A., and Furchgott, R. (1997). International Union of Pharmacology Nomenclature in Nitric Oxide Research. Pharmacol. Rev. 49, 137–142.
Montell, C., Birnbaumer, L., and Flockerzi, V. (2002). The TRP channels, a remarkably functional family. Cell 108, 595–598.
Morris, G. F., Bullock, R., Marshall, S. B., Marmarou, A., Maas, A., and Marshall, L. F. (1999). Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The selfotel investigators. J. Neurosurg. 91, 737–743.
Nadler, M. J., Hermosura, M. C., Inabe, K., Perraud, A. L., Zhu, Q., Stokes, A. J., Kurosaki, T., Kinet, J. P., Penner, R., Scharenberg, A. M., and Fleig, A. (2001). LTRPC7 is a Mg·ATP-regulated divalent cation channel required for cell viability. Nature 411, 590–595.
Nagata, K., Duggan, A., Kumar, G., and García-Añoveros, J. (2005). Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J. Neurosci. 25, 4052–4061.
Nassenstein, C., Kwong, K., Taylor-Clark, T., Kollarik, M., Macglashan, D. M., Braun, A., and Undem, B. J. (2008). Expression and function of the ion channel TRPA1 in vagal afferent nerves innervating mouse lungs. J. Physiol. 586, 1595–1604.
Neubauer, J. A., and Sunderram, J. (2004). Oxygen-sensing neurons in the central nervous system. J. Appl. Physiol. 96, 367–374.
Okubo, K., Matsumura, M., Kawaishi, Y., Aoki, Y., Matsunami, M., Okawa, Y., Sekiguchi, F., and Kawabata, A. (2012). Hydrogen sulfide-induced mechanical hyperalgesia and allodynia require activation of both CaV 3.2 and TRPA1 channels in mice. Br. J. Pharmacol. 166, 1738–1743.
Palmer, R. M., Ashton, D. S., and Moncada, S. (1988). Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 333, 664–666.
Patacchini, R., Santicioli, P., Giuliani, S., and Maggi, C. A. (2005). Pharmacological investigation of hydrogen sulfide (H2S) contractile activity in rat detrusor muscle. Eur. J. Pharmacol. 509, 171–177.
Patapoutian, A., Peier, A. M., Story, G. M., and Viswanath, V. (2003). ThermoTRP channels and beyond: mechanisms of temperature sensation. Nat. Rev. Neurosci. 4, 529–539.
Peier, A. M., Moqrich, A., Hergarden, A. C., Reeve, A. J., Andersson, D. A., Story, G. M., Earley, T. J., Dragoni, I., McIntyre, P., Bevan, S., and Patapoutian, A. (2002). A TRP channel that senses cold stimuli and menthol. Cell 108, 705–715.
Pubmed Abstract | Pubmed Full Text
| CrossRef Full TextPoburko, D., Liao, C. H., Lemos, V. S., Lin, E., Maruyama, Y., Cole, W. C., and van Breemen, C. (2007). Transient receptor potential channel 6-mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger-mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ. Res. 101, 1030–1038.
Quest, A. F., Gutierrez-Pajares, J. L., and Torres, V. A. (2008). Caveolin-1, an ambiguous partner in cell signaling and cancer. J. Cell. Mol. Med. 12, 1130–1150.
Rizzo, V., McIntosh, D. P., Oh, P., and Schnitzer, J. E. (1998). In situ flow activates endothelial nitric oxide synthase in luminal caveolae of endothelium with rapid caveolin dissociation and calmodulin association. J. Biol. Chem. 273, 34724–34729.
Runnels, L. W., Yue, L., and Clapham, D. E. (2001). TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 291, 1043–1047.
Sakuma, I., Stuehr, D. J., Gross, S. S., Nathan, C., and Levi, R. (1988). Identification of arginine as a precursor of endothelium-derived relaxing factor. Proc. Natl. Acad. Sci. U.S.A. 85, 8664–8667.
Sartori, C., Allemann, Y., Trueb, L., Delabays, A., Nicod, P., and Scherrer, U. (1999). Augmented vasoreactivity in adult life associated with perinatal vascular insult. Lancet 353, 2205–2207.
Sato, Y., Sagami, I., and Shimizu, T. (2004). Identification of caveolin-1-interacting sites in neuronal nitric-oxide synthase. Molecular mechanism for inhibition of NO formation. J. Biol. Chem. 279, 8827–8836.
Sawada, Y., Hosokawa, H., Hori, A., Matsumura, K., and Kobayashi, S. (2007). Cold sensitivity of recombinant TRPA1 channels. Brain Res. 1160, 39–46.
Sawada, Y., Hosokawa, H., Matsumura, K., and Kobayashi, S. (2008). Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur. J. Neurosci. 27, 1131–1142.
Schermuly, R. T., Dony, E., Ghofrani, H. A., Pullamsetti, S., Savai, R., Roth, M., Sydykov, A., Lai, Y. J., Weissmann, N., Seeger, W., and Grimminger, F. (2005). Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Invest. 115, 2811–2821.
Schofield, C. J., and Ratcliffe, P. J. (2004). Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343–354.
Semenza, G. L., and Prabhakar, N. R. (2012). Gas biology: small molecular medicine. J. Mol. Med. 90, 213–215.
Semenza, G. L., and Wang, G. L. (1992). A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 12, 5447–5454.
Silver, W. L., and Moulton, D. G. (1982). Chemosensitivity of rat nasal trigeminal receptors. Physiol. Behav. 28, 927–931.
Steen, K. H., Reeh, P. W., Anton, F., and Handwerker, H. O. (1992). Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro. J. Neurosci. 12, 86–95.
Story, G. M., Peier, A. M., Reeve, A. J., Eid, S. R., Mosbacher, J., Hricik, T. R., Earley, T. J., Hergarden, A. C., Andersson, D. A., Hwang, S. W., McIntyre, P., Jegla, T., Bevan, S., and Patapoutian, A. (2003). ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112, 819–829.
Streng, T., Axelsson, H. E., Hedlund, P., Andersson, D. A., Jordt, S. E., Bevan, S., Andersson, K. E., Högestätt, E. D., and Zygmunt, P. M. (2008). Distribution and function of the hydrogen sulfide-sensitive TRPA1 ion channel in rat urinary bladder. Eur. Urol. 53, 391–400.
Strübing, C., Krapivinsky, G., Krapivinsky, L., and Clapham, D. E. (2001). TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 29, 645–655.
Suematsu, M., Suganuma, K., and Kashiwagi, S. (2003). Mechanistic probing of gaseous signal transduction in microcirculation. Antioxid. Redox Signal. 5, 485–492.
Suh, G. S., Wong, A. M., Hergarden, A. C., Wang, J. W., Simon, A. F., Benzer, S., Axel, R., and Anderson, D. J. (2004). A single population of olfactory sensory neurons mediates an innate avoidance behaviour in Drosophila. Nature 431, 854–859.
Sun, H. S., Jackson, M. F., Martin, L. J., Jansen, K., Teves, L., Cui, H., Kiyonaka, S., Mori, Y., Jones, M., Forder, J. P., Golde, T. E., Orser, B. A., Macdonald, J. F., and Tymianski, M. (2009). Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat. Neurosci. 12, 1300–1307.
Swartz, H. M., Khan, N., and Khramtsov, V. V. (2007). Use of electron paramagnetic resonance spectroscopy to evaluate the redox state in vivo. Antioxid. Redox Signal. 9, 1757–1771.
Szabó, C. (2007). Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 6, 917–935.
Takahashi, N., Kuwaki, T., Kiyonaka, S., Numata, T., Kozai, D., Mizuno, Y., Yamamoto, S., Naito, S., Knevels, E., Carmeliet, P., Oga, T., Kaneko, S., Suga, S., Nokami, T., Yoshida, J., and Mori, Y. (2011). TRPA1 underlies a sensing mechanism for O2. Nat. Chem. Biol. 7, 701–711.
Takahashi, N., Mizuno, Y., Kozai, D., Yamamoto, S., Kiyonaka, S., Shibata, T., Uchida, K., and Mori, Y. (2008a). Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels (Austin) 2, 287–298.
Takahashi, S., Lin, H., Geshi, N., Mori, Y., Kawarabayashi, Y., Takami, N., Mori, M. X., Honda, A., and Inoue, R. (2008b). Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J. Physiol. 586, 4209–4223.
Thyagarajan, B., Poteser, M., Romanin, C., Kahr, H., Zhu, M. X., and Groschner, K. (2001). Expression of Trp3 determines sensitivity of capacitative Ca2+ entry to nitric oxide and mitochondrial Ca2+ handling: evidence for a role of Trp3 as a subunit of capacitative Ca2+ entry channels. J. Biol. Chem. 276, 48149–48158.
Trapp, S., Aller, M. I., Wisden, W., and Gourine, A. V. (2008). A role for TASK-1 (KCNK3) channels in the chemosensory control of breathing. J. Neurosci. 28, 8844–8850.
van Rossum, D. B., Patterson, R. L., Ma, H. T., and Gill, D. L. (2000). Ca2+ entry mediated by store depletion, S-nitrosylation, and TRP3 channels. Comparison of coupling and function. J. Biol. Chem. 275, 28562–28568.
Vazquez, G., Wedel, B. J., Aziz, O., Trebak, M., and Putney, J. W. Jr. (2004). The mammalian TRPC cation channels. Biochim. Biophys. Acta 1742, 21–36.
Voets, T., Talavera, K., Owsianik, G., and Nilius, B. (2005). Sensing with TRP channels. Nat. Chem. Biol. 1, 85–92.
Volk, T., Mäding, K., Hensel, M., and Kox, W. J. (1997). Nitric oxide induces transient Ca2+ changes in endothelial cells independent of cGMP. J. Cell. Physiol. 172, 296–305.
Wang, Y. Y., Chang, R. B., and Liman, E. R. (2010). TRPA1 is a component of the nociceptive response to CO2. J. Neurosci. 30, 12958–12963.
Webb, J. D., Coleman, M. L., and Pugh, C. W. (2009). Hypoxia, hypoxia-inducible factors (HIF), HIF hydroxylases and oxygen sensing. Cell. Mol. Life Sci. 66, 3539–3554.
Weir, E. K., López-Barneo, J., Buckler, K. J., and Archer, S. L. (2005). Acute oxygen-sensing mechanisms. N. Engl. J. Med. 353, 2042–2055.
Weissmann, N., Dietrich, A., Fuchs, B., Kalwa, H., Ay, M., Dumitrascu, R., Olschewski, A., Storch, U., Mederos y Schnitzler, M., Ghofrani, H. A., Schermuly, R. T., Pinkenburg, O., Seeger, W., Grimminger, F., and Gudermann, T. (2006). Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc. Natl. Acad. Sci. U.S.A. 103, 19093–19098.
Weissmann, N., Winterhalder, S., Nollen, M., Voswinckel, R., Quanz, K., Ghofrani, H. A., Schermuly, R. T., Seeger, W., and Grimminger, F. (2001). NO and reactive oxygen species are involved in biphasic hypoxic vasoconstriction of isolated rabbit lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L638–L645.
Werkheiser, J., Cowan, A., Gomez, T., Henry, C., Parekh, S., Chau, S., Baron, D. A., and Rawls, S. M. (2009). Icilin-induced wet-dog shakes in rats are dependent on NMDA receptor activation and nitric oxide production. Pharmacol. Biochem. Behav. 92, 543–548.
Wong, C. O., Sukumar, P., Beech, D. J., and Yao, X. (2010). Nitric oxide lacks direct effect on TRPC5 channels but suppresses endogenous TRPC5-containing channels in endothelial cells. Pflügers Arch. 460, 121–130.
Xu, G. Y., Winston, J. H., Shenoy, M., Zhou, S., Chen, J. D., and Pasricha, P. J. (2009). The endogenous hydrogen sulfide producing enzyme cystathionine-β synthase contributes to visceral hypersensitivity in a rat model of irritable bowel syndrome. Mol. Pain 5, 44.
Xu, S. Z., Sukumar, P., Zeng, F., Li, J., Jairaman, A., English, A., Naylor, J., Ciurtin, C., Majeed, Y., Milligan, C. J., Bahnasi, Y. M., Al-Shawaf, E., Porter, K. E., Jiang, L. H., Emery, P., Sivaprasadarao, A., and Beech, D. J. (2008). TRPC channel activation by extracellular thioredoxin. Nature 451, 69–72.
Xu, X., Star, R. A., Tortorici, G., and Muallem, S. (1994). Depletion of intracellular Ca2+ stores activates nitric-oxide synthase to generate cGMP and regulate Ca2+ influx. J. Biol. Chem. 269, 12645–12653.
Yao, X., and Garland, C. J. (2005). Recent developments in vascular endothelial cell transient receptor potential channels. Circ. Res. 97, 853–863.
Yoshida, T., Inoue, R., Morii, T., Takahashi, N., Yamamoto, S., Hara, Y., Tominaga, M., Shimizu, S., Sato, Y., and Mori, Y. (2006). Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2, 596–607.
Young, J. M., Waters, H., Dong, C., Fülle, H. J., and Liman, E. R. (2007). Degeneration of the olfactory guanylyl cyclase D gene during primate evolution. PLoS ONE 2:e884. doi: 10.1371/journal.pone.0000884
Zhu, X., Jiang, M., Peyton, M., Boulay, G., Hurst, R., Stefani, E., and Birnbaumer, L. (1996). trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 85, 661–671.
Keywords: TRP channels, gasotransmitter, nitric oxide, oxygen, TRPC5, TRPC6, TRPV1, TRPA1
Citation: Takahashi N, Kozai D and Mori Y (2012) TRP channels: sensors and transducers of gasotransmitter signals. Front. Physio. 3:324. doi: 10.3389/fphys.2012.00324
Received: 12 May 2012; Paper pending published: 06 June 2012;
Accepted: 24 July 2012; Published online: 09 August 2012.
Edited by:
Mike Althaus, Justus-Liebig University of Giessen, GermanyReviewed by:
Scott Earley, Colorado State University, USAJohn Q. Wang, University of Missouri-Kansas City School of Medicine, USA
Christina Nassenstein, Justus-Liebig-University Giessen, Germany
Copyright © 2012 Takahashi, Kozai and Mori. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Yasuo Mori, Laboratory of Molecular Biology, Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, and Laboratory of Environmental Systems Biology, Department of Technology and Ecology, Hall of Global Environmental Studies, Kyoto University, Kyoto 615-8510, Japan. e-mail: mori@sbchem.kyoto-u.ac.jp