 Emanuele Berardi
Emanuele Berardi Daniela Annibali
Daniela Annibali Marco Cassano
Marco Cassano Stefania Crippa
Stefania Crippa Maurilio Sampaolesi
Maurilio Sampaolesi- 1Translational Cardiomyology Laboratory, Department of Development and Reproduction, KUL University of Leuven, Leuven, Belgium
- 2Interuniversity Institute of Myology, Italy
- 3Laboratory of Cell Metabolism and Proliferation, Vesalius Research Center, Vlaamse Institute voor Biotechnologie, Leuven, Belgium
- 4School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland
- 5Department of Medicine, University of Lausanne Medical School, Lausanne, Switzerland
- 6Division of Human Anatomy, Department of Public Health, Experimental and Forensic Medicine, University of Pavia, Pavia, Italy
Despite the advances achieved in understanding the molecular biology of muscle cells in the past decades, there is still need for effective treatments of muscular degeneration caused by muscular dystrophies and for counteracting the muscle wasting caused by cachexia or sarcopenia. The corticosteroid medications currently in use for dystrophic patients merely help to control the inflammatory state and only slightly delay the progression of the disease. Unfortunately, walkers and wheel chairs are the only options for such patients to maintain independence and walking capabilities until the respiratory muscles become weak and the mechanical ventilation is needed. On the other hand, myostatin inhibition, IL-6 antagonism and synthetic ghrelin administration are examples of promising treatments in cachexia animal models. In both dystrophies and cachectic syndrome the muscular degeneration is extremely relevant and the translational therapeutic attempts to find a possible cure are well defined. In particular, molecular-based therapies are common options to be explored in order to exploit beneficial treatments for cachexia, while gene/cell therapies are mostly used in the attempt to induce a substantial improvement of the dystrophic muscular phenotype. This review focuses on the description of the use of molecular administrations and gene/stem cell therapy to treat muscular degenerations. It reviews previous trials using cell delivery protocols in mice and patients starting with the use of donor myoblasts, outlining the likely causes for their poor results and briefly focusing on satellite cell studies that raise new hope. Then it proceeds to describe recently identified stem/progenitor cells, including pluripotent stem cells and in relationship to their ability to home within a dystrophic muscle and to differentiate into skeletal muscle cells. Different known features of various stem cells are compared in this perspective, and the few available examples of their use in animal models of muscular degeneration are reported. Since non coding RNAs, including microRNAs (miRNAs), are emerging as prominent players in the regulation of stem cell fates we also provides an outline of the role of microRNAs in the control of myogenic commitment. Finally, based on our current knowledge and the rapid advance in stem cell biology, a prediction of clinical translation for cell therapy protocols combined with molecular treatments is discussed.
Introduction
Muscular dystrophies are heterogeneous genetic diseases caused by progressive degeneration of skeletal muscle fibers (Emery, 2002). Mutations in genes encoding for crucial skeletal muscle proteins located either at the plasma membrane (i.e., dystrophin-glycoprotein complex) or, less frequently, within internal cellular membranes are responsible for those disorders. The lack of those proteins increases the probability of damage during contraction and eventually leads to fiber degeneration (Blake et al., 2002; Gumerson and Michele, 2011). Despite the extensive literature reported on this topic, the molecular mechanisms responsible for the progressive muscular degeneration are not yet understood in detail. Physiologically, muscular fiber degeneration is counterbalanced by the regeneration of new fibers formed at the expense of resident myogenic cells and usually each degeneration process is followed by a new regenerative cycle. Skeletal muscle regeneration is mainly sustained by satellite cells (Mauro, 1961), local myogenic progenitors localized underneath the basal lamina of muscle fibers (Tedesco et al., 2010).
When it is damaged, a muscle undergoes a remodeling process and the resident myogenic cells differentiate into myofibroblasts to produce extracellular matrix (ECM), which is required for the adequate tissue repair. Following repeated cycles of degeneration/regeneration, such myofibroblasts accumulate in the muscle producing large amounts of ECM proteins and thus ultimately leading to fibrosis. However, after repeated injuries, the satellite cells in the muscles become exhausted, losing their regenerative capacity. In this view, the genetic manipulation of satellite cells could potentially guarantee an improved muscle regeneration and function. In this review we provide information about the different sources of myogenic stem cells, highlighting their common features and characteristics as well as their controversies in the therapeutic approaches. Advantage and disadvantage for autologous and heterologous cell therapy will be discussed, considering the different sources of myogenic stem cells.
Alterations in skeletal muscle homeostasis can result in either atrophy or hypo-metabolism. Etiologically, the molecular determinants responsible for such metabolic changes are known as common players in different muscular wasting diseases. In this view, they represent promising therapeutic targets common to the wide range of the known muscular diseases that could determine a strong impact in terms of prognosis, clinical setting and management. Pharmacotherapy still represents the most common strategy adopted to counteract muscle wasting for a large spectrum of muscular diseases such as cancer mediated cachexia, Rheumatoid Arthritis (RA) and sarcopenia, while muscular dystrophies can also be potentially treated by a multi-therapeutic approach based on gene/cell therapies combined with molecular treatments.
Pathophysiology and Clinical Relevance of Muscle Wasting
The state of progressive loss of muscular and fat mass known as cachexia syndrome is a condition associated with several chronic diseases such as AIDS, cancer, chronic obstructive lung disease, multiple sclerosis, congestive heart failure, sepsis, diabetes, RA and tuberculosis (Laviano et al., 2003; Fearon et al., 2011). According to its multifactorial and complex nature, as well as to both the pathophysiologic and epidemiologic features of its primary-related disease, such syndrome depicts a worse global epidemiologic scenario if compared with the other musculoskeletal disorders. The dramatic effects that cachexia have on the prognosis are well-known in clinical management of cancer diseases. According with the Global Burden Diseases (GBD) estimations, up to 50% of the oncologic patients suffer from cachexia and up to 80% of them show clear signs of cachexia in the late stages of cancer progression (Laviano et al., 2003; Fearon et al., 2011; Suzuki et al., 2013). Moreover, cancer-related cachexia counteracts the efficacy of radio- and chemotherapeutic treatments by increasing their side effects and decreasing patient's quality of life (Tisdale, 2002). Such complications are directly reliable for a high percentage of mortality in cancer patients, about 20–40%, accounting for more than 2 million of global premature deaths for year (Bruera, 1997).
Among the wide range of the muscular diseases that affect musculoskeletal system by hampering respiratory and locomotive functions, RA, dystrophies and cachexia syndrome represent the most common and are considered as a serious problem for human health. In 2010 GBD estimates showed that musculoskeletal disorders accounted for more than 150,000 deaths, with an increment of 121% between 1990 and 2010 (Lim et al., 2012).
The progressive skeletal muscle weakness and wasting are the main prognostic features exhibited by the heterogeneous musculoskeletal disorders (Leung and Wagner, 2013) (Figure 1). Although musculoskeletal diseases and cachexia have different origins (due to genetic alterations the first and to complications of several chronic diseases the latter), body weight loss, muscle atrophy, fatigue, weakness and loss of appetite are common clinical features observed in both. Nevertheless, while many autoimmune diseases ultimately result in a cachectic state of the patients, they are often associated with unintentional weight loss. RA is an autoimmune disease where the energetic balance is normal and eventually fat mass is increased. Thus, RA is a unique example of autoimmune disease in which cachexia is not associated with a general body-wide wasting and depends exclusively on the reduction in the muscle mass that might be responsible in lowering the average survival of the patients. Therefore, muscle wasting is the key player responsible for the induction of muscle atrophy in musculoskeletal disorders, which is triggered by catabolic events occurring into the affected skeletal muscle tissue (Figure 1). At the molecular level, this is due to an unbalance between protein anabolism and catabolism in favor of proteolysis of some crucial proteins occurring into the muscle fibers, mediated by the expression of muscle-specific ubiquitin ligase (E3 protein) atrogin1/MAFbx and MuRF1 (Bodine et al., 2001; Gomes et al., 2001).
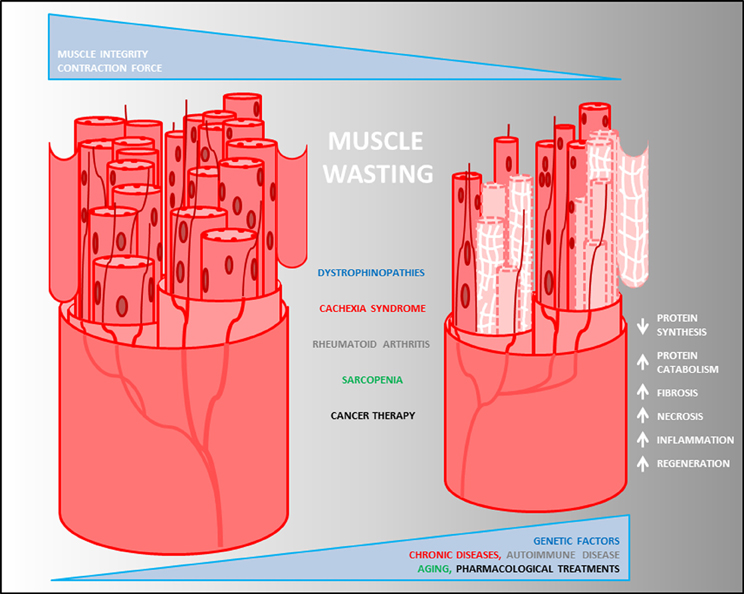
Figure 1. Pathological heterogeneity of muscle wasting. Descriptive model of muscle degeneration in chronic diseases. Loss of muscle mass, decrease of fiber size and myonuclear content, reduction of contraction force and increase of fibrosis (white net) are common pathophysiological features of muscle degeneration mediated by changes into the biological process (white arrows) triggered by muscle diseases.
Beyond the basic action of mechanic contraction, the skeletal muscle is a tissue involved in many other metabolic activities such as glucose, glycogen and lipid metabolism (Jensen and Richter, 2012), as well as endocrine (Pedersen and Febbraio, 2008) and immunogenic activities (Nielsen and Pedersen, 2008). Such biological heterogeneity reflects the histological diversity observed into the skeletal muscle tissue and, in turn, highlights multifaceted possibilities for the therapeutic interventions. It has been recently demonstrated that the microenvironment outside the myofibers can actively participate to the cancer-mediated muscle wasting. This happens when circulating tumor factors induce muscle damage by activation of both satellite and non-satellite muscle progenitor cells, and such process is followed by inhibition of their myogenic differentiation due to a persistent expression of Pax7 (He et al., 2013). On the other hand, the metabolic complexity of the skeletal muscle also renders it susceptible to environmental stimuli. Epidemiological studies show indeed the potential role of environmental and lifestyle factors (i.e., physical activity, diet and sun exposure) on the increasing susceptibility of the insurgence of sarcopenia (Scott et al., 2011). Overall, studies focused on the investigation of the general molecular mechanisms responsible for muscle wasting identified some potential therapeutic targets involved in the main catabolic pathways and that could be inhibited by pharmacological and by gene- or cell-therapy based approaches. Specifically, we will discuss pharmacological strategies aimed to counteract the effects of pro-inflammatory stimuli (i.e., TNF-α, IL-6) in cachexia, sarcopenia and RA, as well vector-based micro-dystrophin transfer, oligonucleotide-induced exon-skipping and cell therapy strategy based on the use of healthy myogenic cell precursors [i.e., satellite cells, side population (SP), fibro-adipogenic progenitors (FAPs), mesoangioblasts, ES, and iPS cells] in dystrophinopathies (Figure 2).
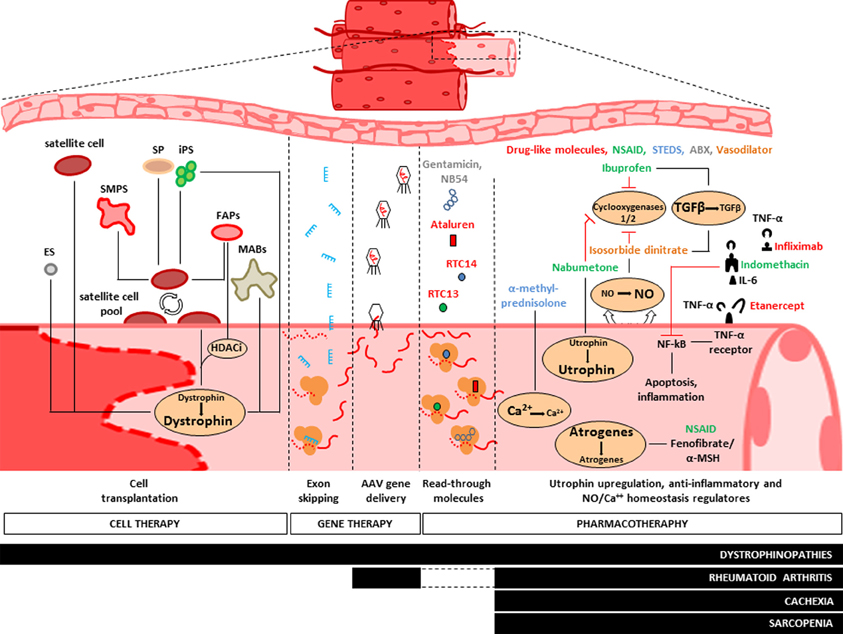
Figure 2. Treatments of muscle diseases. Representative scheme of the main therapeutic approaches adopted to counteract muscle wasting. Pharmacotherapy aims to maintain muscle integrity by neutralization of ubiquitin-proteasome pathway (UPP), provoked by circulating pro-inflammatory stimuli (i.e., TNF-α and Il-6). Administration of non-steroidal anti-inflammatory drugs (NSAID, green), fenofibrates and steroids (STEDS, blue) reduces the overall expressions of atrogenes. It also stimulates utrophin expression and regulates the cytosolic homeostasis of NO and Ca++ elements, while drug-like molecules (red) and antibiotics (green) provide the “read-through” strategy to obtain semi-functional dystrophin protein. To date, NSAID and STEDS are the most diffused drugs to treat dystrophinopathies, cachexia syndrome, rheumatoid arthritis and sarcopenia. Gene therapy is experimentally adopted for dystrophinopathies treatments. Such method is based on the use of Adeno-associated viruses (AAV) and lentiviral vectors to mediate the delivery of micro-dystrophin or mini-utrophin and by use of exon skipping strategy to increase the endogenous expression of dystrophin (see text). Skeletal Myogenic Precursors (SMPS), Side Population (SP), Fibro-Adipogenic Progenitors (FAPs) and Mesoangioblasts (MABs) are potential candidates for cell therapeutic approaches of dystrophinopathies. MABs were recently enrolled in PhaseI/II clinical trial either for their ability to repopulate the endogenous pool of satellite cells and for their myogenic differentiation capability to produce dystrophin.
Pharmacological Approach
Lack or alteration of structural proteins into the musculoskeletal system causes chronic inflammation. Although there are no specific cures for muscle wasting mediated by the different forms of muscular dystrophies and cachexia, pharmacotherapy has been the first historical clinical approach used to modulate the progression of such diseases (Abdel-Hamid and Clemens, 2012) by counteracting chronic inflammation (Figure 2). Because dystrophin plays a crucial role in preserving the integrity of the muscular membrane by permitting the anchorage of the dystrophin-associated protein complex, lack or genetic mutations of dystrophin result in a chronic influx of calcium into the myofibers, causing cellular death and inflammatory responses. In addition, fibrosis can occur to replace the damaged muscular fibers, causing muscle weakness (Figure 1). Pilot studies performed in patients affected by Duchenne/Becker and Limb-Girdle muscular dystrophies (DMD, BMD, and LGMD respectively) based on the administration of non-steroidal anti-inflammatory drugs, such as ibuprofen and nabumetone, or on the use of isosorbide dinitrate, a NO donor vasodilator, showed an improvement of the general pathophysiologic conditions (Figure 2). This effect was mediated by a deregulation of circulating level of TGF-β (D'Angelo et al., 2012), a known mediator of fibrosis in dystrophinopathies (Goldstein and Mcnally, 2010). Corticosteroids have been proposed as a pharmacological therapy for dystrophinopathies, in order to counteract muscle necrosis, inflammation and to reduce the muscle membrane susceptibility to damage (Abdel-Hamid and Clemens, 2012). In particular, prednisone (Griggs et al., 1991, 1993; Bonifati et al., 2000) and deflazacort (Bonifati et al., 2000) induce improvement and a long-term stabilization of the muscle strength (Bonifati et al., 2000), as well as a substantial reduction of weakness progression in DMD patients (Moxley et al., 2005). Moreover, since the elevation of cytosolic calcium concentration can trigger apoptotic and/or necrosis events in the dystrophic muscles, such physiological alteration represents another important glucocorticoid-based therapeutic target. Recently, studies in preclinical models proposed α-methylprednisolone, administrated either alone (Ruegg et al., 2002) or in combination with taurine (an aminoacid with antioxidant properties), as a candidate for pharmacological regulation of the cytosolic calcium flux in dystrophic muscles (Cozzoli et al., 2011).
The pharmacological efforts aiming to counteract muscle degeneration in dystrophinopathies mainly point to stabilize the muscular membrane integrity. This is the case of drugs designed to increase the expression level of native utrophin, as a mechanisms used to compensate for the dystrophin lack (Tinsley et al., 1998; Gilbert et al., 1999). Nabumetone is a novel promising small molecule with anti-inflammatory properties (COX1 and 2 inhibitor) that in vitro can activate the promoter of the A isoform of utrophin (Moorwood et al., 2011). The administration of aminoglycosides antibiotics (i.e., Gentamicin, NB54) (Barton-Davis et al., 1999; Politano et al., 2003; Nudelman et al., 2009) and read-through compounds such as RTC13, RTC14 (Kayali et al., 2012), or ataluren (PTC124) (Hamed, 2006; Finkel, 2010) has been proposed as a new strategy to induce ribosomal read-through of premature termination mutations, to obtain a full-length dystrophin protein in patients with DMD and Becker Muscular Dystrophy (BMD) (Figure 2). Various pro-inflammatory stimuli are involved in cancer mediated muscle wasting (Todorov et al., 1996; Suzuki et al., 2013), RA (Gomez-Sanmiguel et al., 2013) and sarcopenia (Malafarina et al., 2012). In this case the pharmacological approaches used so far aim to counteract the biological activity of secreted pro-inflammatory mediators, such as interleukins (Il-1β, IL-6), interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α) (Todorov et al., 1996) and proteolysis inducing factor (PIF) (Todorov et al., 1999). Unfortunately, anti-cytokine therapy aimed to block TNF-α by administration of Infliximab (monoclonal TNF antibody) or Etanercept (soluble TNF-α receptor) in cancer patients showed only poor ameliorative effects on cachexia pathophysiology (Gueta et al., 2010; Wu et al., 2013), whereas in patients with RA mediated cachexia, Etanercept was shown to reduced mortality (Morgan et al., 2014) and ameliorate the muscular function (Marcora et al., 2006). Indomethacin showed anti-cachectic effects in muscles from tumor bearing mice by inducing reduction in the levels of NF-kappaB, TNF-α and IL-6 (Zhou et al., 2003). Notably, dithiocarbamate inhibits IL-6 synthesis (Nai et al., 2007). Other treatments proposed in in vivo models in order to counteract oxidative and inflammatory burden in cancer-mediated muscle wasting are based on administration of glycine (Ham et al., 2013), simvastatin (Palus et al., 2013), eicosapentaenoic acid (Vaughan et al., 2012) and use of proteasome inhibitors to block the ubiquitin-proteasome pathway (Zhang et al., 2013). Such treatments efficiently counteract the expression of genes associated with the muscle protein breakdown observed in cancer cachexia (i.e., Atrogin-1 and MuRF-1) On the contrary, fenofibrate, a PPARα agonist (Castillero et al., 2011), and α-Melanocyte-stimulating hormone (α-MSH) (Gomez-Sanmiguel et al., 2013) ameliorate the pathophysiology of muscles in an adjuvant-induced arthritis rat model by preventing the overexpression of Atrogin-1, MuRF-1, and myostatin observed in RA (Castillero et al., 2011; Gomez-Sanmiguel et al., 2013). Pharmacological treatments used to counteract the progressive loss of skeletal muscle mass observed in sarcopenia are based on the administration of ghrelin, testosterone, Growth Hormone (GH), myostatin inhibitors and supplementation of vitamin D (Malafarina et al., 2012). Therapeutically, despite the efforts spent so far for sarcopenia treatment, only few results have been achieved in terms of increased muscle mass and strength, and decrease of muscle catabolism. Because vitamin D levels decrease with elderly, promising results were obtained in dietary supplementation of vitamin D in aged people, specially in muscle functional improvement (Malafarina et al., 2012).
Gene Therapy
Gene replacement strategy was historically conceived to counteract the lack of dystrophin that affects DMD and BDM patients. Transgenic mice (mdx), dogs with X-linked muscular dystrophy (GRMD), and non-human primates (cynomolgus macaques) are examples of animal models extensively used to test novel methods for dystrophin gene delivery. Adeno-associated viruses (AAV) and lentivirus based vectors mediate efficient delivery of micro-dystrophin or mini-utrophin (Cerletti et al., 2003) and provide an alternative option for dystrophin-deficient mdx mouse (Gregorevic et al., 2004, 2006; Yoshimura et al., 2004; Liu et al., 2005; Rodino-Klapac et al., 2007), in non-human primate animal models (Rodino-Klapac et al., 2007) and Golden Retriever Muscular Dystrophy (GRMD) dogs (Cerletti et al., 2003; Sampaolesi et al., 2006; Koo et al., 2011). Nevertheless, all the dystrophin delivery methods proposed so far showed poor restoration of dystrophin within a small area of the skeletal muscle tissue targeted and only a partial improvement of the contractile properties (Rodino-Klapac et al., 2013) (Figure 2). Since deletions of single or multiple exons in the dystrophin gene are the most pathogenic mutations in DMD and BDM, antisense-mediated exon skipping (Douglas and Wood, 2013) represents a promising additional strategy adopted to increase dystrophin expression in DMD and BDM models, by restoring the genetic reading frame. Notably, this can be obtained either by single- (Van Deutekom et al., 2007; Jorgensen et al., 2009; Kinali et al., 2009) or multi-exon skipping approaches (Aartsma-Rus et al., 2006; McClorey et al., 2006; Goyenvalle et al., 2012). So far, many antisense oligonucleotide, such as morpholino oligomers (PMOs) and 2′O-methylphosphorothioate oligoribonucleotides (2′OMe), have been synthetized and successfully tested both in vitro and in vivo (Benedetti et al., 2013). They act by targeting of specific exons allowing their skipping during the splicing of dystrophin mRNA (Figure 2). In 2007 van Deutekom and colleagues tested the ability of PRO051oligonucleotide to restore dystrophin into the tibialis anterior of 4 DMD patients. In 2009 Kinali and colleagues treated the extensor digitorum brevis of 7 DMD patients with morpholino splice-switching oligonucleotide (AVI-4658) (Kinali et al., 2009). These trials provided evidences for local restoration of dystrophin in the treated muscles and for the safety of the protocols adopted (Van Deutekom et al., 2007).
However, because myoastin negatively affects skeletal muscle growth, AAV-mediated gene delivery of myostatin inhibitors (i.e., MRPO) has been proposed as a therapeutic strategy to maintain muscle mass (Morine et al., 2010) and improve the contraction force (Qiao et al., 2008) in both mdx mice (Qiao et al., 2008; Morine et al., 2010) and dogs (Qiao et al., 2009). Noteworthy, gene therapy AAV-mediated approaches were also used to restore structural, such as sarcoglycans (Sampaolesi et al., 2003), and non-structural proteins (Goonasekera et al., 2011). In fact, it is known that cytosolic alteration of Ca2+ flux observed in muscular dystrophies leads to sarcolemmal instability. This could be reduced by overexpressing the sarcoplasmic reticulum Ca2+ ATPase 1 (SERCA1) in both mdx and δ-sarcoglycan-null (Sgcd−/−) mice (Goonasekera et al., 2011). Preclinical studies about the therapeutic applications of AAV-based gene delivery strategies were also performed to treat RA (Dai and Rabie, 2007). In particular, because the synovial lining is poorly transduced, subsynovial muscle tissues have been predominantly transfected in various RA models to investigate the effects of anti-inflammatory mediators such as IL-4 (Cottard et al., 2000) and IL-10 (Apparailly et al., 2002) either in mice (Cottard et al., 2000; Apparailly et al., 2002) as well as in human and murine synovial cell lines (Katakura et al., 2004).
Cell Therapy
As already previously mentioned, satellite cells are quiescent unipotent stem cells, located underneath the basal lamina of adult skeletal muscle fibers (Mauro, 1961). They are formed during the second wave of embryonic myogenesis after which they exit the cell cycle, contributing significantly to the first post-natal muscle growth (Gros et al., 2005; Kassar-Duchossoy et al., 2005; Relaix et al., 2005). In case of muscle damages, satellite cells can re-enter the cell cycle resulting in an increasing number of myogenic progenitors able to fuse and form new muscle fibers (Huard et al., 2002; Jarvinen et al., 2005; Tedesco et al., 2010; Wang and Rudnicki, 2012). Given their natural commitment, it has been easy to consider satellite cells as the leading candidate for muscle regeneration in dystrophic mice (Partridge et al., 1989). In the case of transplantation of individual fibers into the tibialis anterior of irradiated mdx mice (specific treatment used to remove the existing population of satellite cells), satellite cells of the fiber donor expand, repopulating the endogenous pool, and differentiate into functional myofibers. Pax7+/CD34+/GFP+ satellite cells, isolated from the diaphragm of Pax3::GFP mice, proved a good cellular model for the treatment of irradiated mdx muscles, resulting in restoration of the expression of dystrophin in many skeletal fibers and reconstitution of the pool of resident satellites cells (Montarras et al., 2005) (Figure 2). It has been shown by in vivo imaging that a single CD34+/integrinα+ satellite cell can replenish the resident satellite pool that, in the presence of further damage, can quickly re-enter a new wave of proliferation, generating new myofibers (Sacco et al., 2008). Fifty years of studies about the role of stem cells in skeletal muscle regeneration have identified a complex pattern of expression for surface markers within the endogenous pool of satellite cells (Rinaldi and Perlingeiro, 2013). Specific subpopulations of satellite cells positive for CD34 (Beauchamp et al., 2000), M-cadherin (Irintchev et al., 1994), α7β 1-integrin (Burkin and Kaufman, 1999; Gnocchi et al., 2009), syndecan-3/4 (Cornelison et al., 2004), the chemokine receptor CXCR4 (Ratajczak et al., 2003) barx2 (Meech et al., 2012) and caveolin-1 (Volonte et al., 2005) have been identified and investigated for their in vitro, as well as in vivo, therapeutic potential.
The intra-muscular injections, however, have revealed some significant problems. One of the major limitations on the use of satellite cells in therapy is the cell heterogeneity. The variable rate of satellite cell homing after transplantation of a single myofiber could be due to the cell heterogeneity and their functional niche of origin. Moreover, satellite cells show limited migratory ability, reduced myogenic capacity when expanded in vitro, while the host immune rejection still represents a major issue when satellite cells are used for transplantation (Mouly et al., 2005; Kuang and Rudnicki, 2008). Nevertheless, many others methodological issues concerning intramuscular cell transplantation of myogenic stem cells in the treatment of dystrophinopathies have been partially solved by a further basic knowledge of the skeletal muscle stem cell biology, achieved in the recent years by both clinical and pre-clinical data (Hill et al., 2006; Skuk and Tremblay, 2011; Brack and Rando, 2012).
Recently, interesting results have been obtained by the prospective isolation of skeletal myogenic precursors (SMPS) (Figure 2), a distinct population of the satellite cells pool, characterized by the expression of Cxcr4 and β 1integrin and the absence of CD45, Sca1 and Mac1(Sherwood et al., 2004). Once injected into immunodeficient mdx muscles, SMPS contribute to the muscle regeneration (up to 94%) and to the satellite cell pool. SMPS need to be injected freshly isolated, in order to have beneficial effects. Although their migratory capacity remains limited to the areas surrounding the site of injection, the contractile force of limb muscles was significantly higher in the treated mice compared to controls (Cerletti et al., 2008).
A subpopulation of progenitors associated with skeletal muscles is a so-called SP. SP cells are defined as myogenic cells SCA1+/ CD45+ (Polesskaya et al., 2003; Seale et al., 2004), unable to retain the intercalating Hoechst 33342 dyes (Gussoni et al., 1999; Polesskaya et al., 2003; Montanaro et al., 2004). When co-cultured with myoblasts, they can fuse to form myotubes in vitro and, if injected into the femoral artery in mice mdx5cv mice, can contribute up to 5–8% of the regenerated fibers (Perez et al., 2009). Remarkable results were obtained with the isolation of a rare subset (0.25%) of SP cells, identified as SCA1+/ABCG2+/Syndecan4+/Pax7+ cells (Tanaka et al., 2009). Once injected, those cells in muscle treated with 1.2% BaCl2 regenerate up to 30% of the fibers and, surprisingly, reconstitute up to 75% of the endogenous satellite pool (Tanaka et al., 2009). However, the muscle damage induced by BaCl2 is not a commonly accepted model of regeneration and this must be taken into account when interpreting these results. Another class of myogenic precursor was isolated from the population of endothelial cells in human muscles, through the prospective isolation of cells by FACS CD56+/CD34+/CD144+ (Okada et al., 2008). These myoendothelial progenitors, after injection into injured muscle of SCID mice can be grafted into existing muscle fibers and form neofibres (Tamaki et al., 2002). A population of muscle-interstitial cells, referred to as FAPs, was recently identified. FAPs have been shown to mediate the beneficial effects of histone deacetylase inhibitors (HDACi) in mdx mice (Mozzetta et al., 2013). HDACi are known as promoters of endogenous regeneration and functional recovery of dystrophic muscles in the mdx mouse. They act by increasing the fiber size and reducing both the fibrosis and the fat deposition (Minetti et al., 2006). FAPs isolated from young dystrophic subjects show a minimal myogenic commitment that can be implemented by HDACi at the expense of their fibro-adipogenic potential. In addition, HDACi enhance FAPs ability to promote differentiation of adjacent satellite cells (Figure 2).
In recent years, a new class of stem cells associated with vasculatures and termed mesoangioblasts (MABs) have been studied as potential therapeutic protocols to threat dystrophic muscles (Figure 2). MABs were originally isolated from the dorsal aorta of the embryo (E9.5) (Minasi et al., 2002) and then from adult skeletal muscles in mice, dogs, and humans (Tonlorenzi et al., 2007; Quattrocelli et al., 2012). MABs are positive for several markers, including CD34, SMA, Pdgfrα, Pdgfrβ, Ng2, and AP, supporting the hypothesis that they belong to a subgroup of pericytes. MABs are multipotent as highlighted by their myogenic, osteogenic, chondrogenic, and adipogenic differentiation potential observed in quail-chick chimeras and in vitro and in vivo experiments in mouse model. After intra-arterial injection in dystrophic muscles of Scga-null mice or GRMD dogs, MABs are able to regenerate (up to 50%) muscle architecture, with functionality (Sampaolesi et al., 2006). Similarly, promised results were obtained with the transplantation of human MABs in immunodeficient mdx mice (Sampaolesi et al., 2003). Building up on those promising preclinical studies a Phase I/II clinical trial of donor mesoangioblasts transplantation from HLA-identical donors in 5 DMD patients is nearing completion (EudraCT Number: 2011-000176-33). However, due the ethical rules, several limitations are present in this first attempt of stem cell systemic delivery in DMD patients. First, the age of the patients and progression of the disease were advanced in the enrolled patients. Second, cell dose was quite low, from 1/5 to 1/10 of that administered to the GRMD dogs. Third, injections were limited into the femoral arteries, confining the cell treatment mainly to the muscles downstream the femoral artery. Taken into account those limitations, this trial will give important information about the safety of systemic delivery of adult stem cells in DMD patients. This trial will also hopefully answer some questions regarding the capability of donor stem cells to migrate towards regenerating muscles and undergo myogenic differentiation, by producing dystrophin.
In the last few years there is a growing interest about the therapeutic prospective offered by pluripotent stem cells. Embryonic Stem (ES) cells have been isolated from the inner cell mass of the blastocyst in mouse (Evans and Kaufman, 1981; Martin, 1981) and in human (Thomson et al., 1998), showing pluripotent features. Mouse and human ES cells can efficiently differentiate into the three germ layers, mesoderm, ectoderm and endoderm and a consistent number of studies showed their myogenic differentiation potential in vitro and in vivo (Bhagavati and Xu, 2005; Barberi et al., 2007; Filareto et al., 2012). The therapeutic use of ES cells has been strongly debated in the scientific community, mainly because of limitations linked to immune rejection and ethical concerns. However, part of these obstacles has been overcome by a pioneering study of Yamanaka in 2006 (Takahashi and Yamanaka, 2006) showing that induced pluripotent stem (iPS) cells can be generated from somatic cells. In addition, a very recent paper published in Nature showed that is possible to generate pluripotent stem cells by STAP, stimulus-triggered acquisition of pluripotency (Obokata et al., 2014). Basically, pluripotent conversion can be achieved by brief exposition of low-passage source cells to acidic conditions (pH 5.7). Since STAP reprogramming takes a very short period, only few days unlike transgene- or chemical-induced iPS cell conversion, it could be clinically relevant for tailoring cell therapy approaches.
To date, the use of iPS cells to potentially correct the dystrophic phenotype has been reported in several studies on murine (Mizuno et al., 2010; Darabi et al., 2011; Quattrocelli et al., 2011; Filareto et al., 2013) and human iPS cells (Darabi et al., 2012). Preclinical evidences show that myoblasts and mesenchymal cells derived from human ES and iPS can efficiently fuse with mature muscle fibers (Awaya et al., 2012; Goudenege et al., 2012) and improve the performances of engrafted muscles (Darabi et al., 2012) (Figure 2). After introduction of factor-based reprogramming, generation of iPS cells is feasible from any kind of cell population. However, iPS cells own an epigenetic memory, which results in a biased cell-differentiation towards the cell lineage of its source. This is probably due to the conservation of epigenetic marks, like CpG island (CGI) methylation and histone modifications, after reprogramming (Kim et al., 2010; Polo et al., 2010). Also iPS cells generated from murine skeletal MABs upon teratoma analysis differentiated with a significantly greater efficiency towards skeletal myocytes compared to fibroblast-reprogrammed iPS cells (Quattrocelli et al., 2011). If the myogenic memory will be confirmed in human iPS cells, this phenomenon could have an unpredicted impact for future translational studies.
Overall, these evidences suggest that, although all the efforts made so far on the use of pluripotent stem cells have been focalized on dystrophinopathies, the therapeutic use of iPS and ES cells could be an extraordinary potential clinical tool useful in the treatment of any skeletal muscle degenerations.
New Challenge: microRNAs and their Therapeutic Potential
In the early '90 a 22 nt non-coding transcript RNA, lin-4, was identified in C. elegans. It represses the expression of lin-14, a nuclear protein necessary for the larval development (Lee et al., 1993; Wightman et al., 1993). This discovery was the trigger for thousands of subsequent publications regarding the identification of micro inhibitory RNAs (miRNAs) in early embryogenesis (Berardi et al., 2012) and in cardiac and skeletal myogenesis (Ge and Chen, 2011; Crippa et al., 2012). miRNAs target sites in the 3'UTR of the mRNA leading to inhibition of mRNA translation and/or enhanced mRNA degradation, thus resulting in the decrease of protein expression levels (Djuranovic et al., 2011). miRNAs are generally transcribed by RNA polymerase II, as a primary miRNA (pri-miRNA)s then the RNase III enzyme Drosha removes hairpins from pri-miRNAs generating pre-miRNAs. In the cytoplasm, the pre-miRNA is further cleaved by the RNase III enzyme Dicer to generate mature miRNAs.
Highly expressed miRNAs in skeletal muscle tissue are termed myomiRs, which include miR-1, miR-133a, miR133-b, miR-206, miR-208, miR208b, miR486, and miR-499 (Van Rooij et al., 2008). They can be responsible for metabolic changes in skeletal muscle tissue (Figure 3). For example, muscular hypertrophy in Texel sheep is caused by a point mutation in the 3'UTR of myostatin RNA messengers, which creates a target site for miR-1 and miR-206 (Clop et al., 2006). Those miRNAs are abundant in the skeletal muscle tissue and can negatively affect myostatin expression, one of the most effective repressor of muscle growth. However, since another possible target for miR-206 is utrophin, a valuable substitute of dystrophin, several researchers believe that miR-206 can be responsible to sustain the dystrophic phenotype (Rosenberg et al., 2006). Transgenic mice for gain and loss of function studies are necessary to shed light in those controversies.
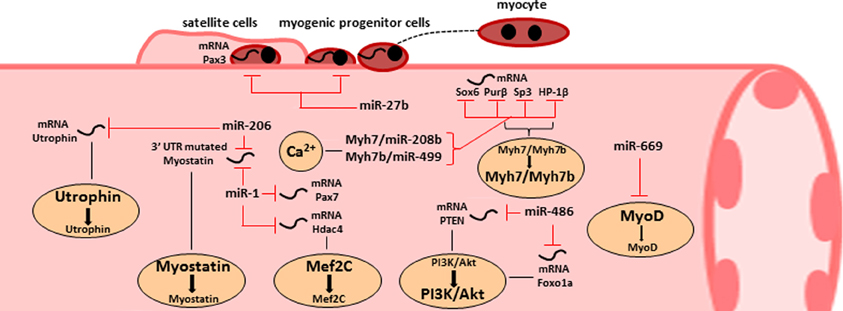
Figure 3. The roles of miRNA in skeletal muscle homeostasis and dysfunction. Representative scheme of the main miRNAs involved in skeletal muscle functions. Specific miRNAs are critical for satellite cell activation (miR-27b) or for skeletal muscle differentiation (miR-1) and can be induced for therapeutic approaches. Other miRNAs are involved in muscle metabolism and they are able to modulate the AKT/PI3K pathway. Two miRNA families are peculiar for their opposite dual biological functions: miR-669 can inhibit MyoD causing a benefit in cardiac progenitors but in the same time reduced myogenic potential in skeletal muscle progenitors; miR-206 can induce hypertrophy by targeting mutated 3'UTR of myostatin messengers and can sustain dystrophic phenotype by inhibiting utrophin expression.
miRNA are also critic regulators for muscle wasting. In fact, possible targets for miR-486 are PTEN (phosphatase and tensin homolog) and FoxO1A, elements of the PI3/Akt pathway (Small et al., 2010) involved in muscle wasting and apoptosis. The activity of miR-486 results in muscle hypertrophy, since it leads to the activation of mTOR via PTEN inhibiton, and to the reduction of the ubiquitin ligase expression, which sustains atrophy in skeletal muscle tissue. The expression profiles of miR-486 strongly support its role in muscle homeostasis, since it is decreased in denervated muscles and almost absent in Duchenne patients (Eisenberg et al., 2007).
miR-1 and miR-133 modulate skeletal and cardiac muscle growth and differentiation. miR-1 promotes skeletal muscle differentiation by targeting the histone deacetylase 4 (HDAC4), that in turn represses Mef2C, an essential muscle transcription factor. On the contrary, miR-133 stimulates myoblast proliferation by targeting SRF (Chen et al., 2006), while miR-206 promotes myoblast differentiation targeting the mRNA of PolA1 (Kim et al., 2006), a DNA polymerase subunit. MyoD and myogenin can regulate miR-206 expression by binding specific elements in the enhancer region upstream miR-206 gene. Overexpression of miR-27b causes premature differentiation of muscle satellite cells: once myoblasts exit the cell cycle, miR-27 indeed targets the 3'UTR of Pax3 (Crist et al., 2009).
The fine-tuned expression of miR-499 and miR-208b plays a role in the control of skeletal muscle performance. In response to calcium signaling, miR-208b and miR-499 indeed reinforce slow fiber conversion by inducing the expression of β-MHC and Myh7b (Van Rooij et al., 2009). They were considered initially as MyomiRs since they are encoded by introns of their host myosin genes Myh7 and Myh7b. These two intronic miRNAs target the transcriptional repressors of slow myofiber genes, including Sox6, Purβ, Sp3, and HP-1β, (Van Rooij et al., 2009). Recently, we have also identified a microRNA family, called miR-669, involved in the muscle lineage switch (Crippa et al., 2011) and used them as therapeutic molecules for long term treatments (Quattrocelli et al., 2013) in an animal model of limb girdle muscular dystrophy type 2E. Up to now, crucial information concerning the effects of miR669 in human setting for cardiac and skeletal muscle differentiation are still missing.
In summary, the possibility to increase the pool of myogenic stem cells, induce hypertrophy or reduce atrophic cell signaling by the perturbation of miRNA expression profiles offers a new opportunity to re-establish the skeletal muscle homeostasis lost in the wide range of muscolo-skeletal disorders (Figure 3).
Concluding Remarks
In conclusion, stem cell research will ride the third millennium as highlighted by the Nobel Prize in Physiology or Medicine 2012 jointly conferred to John Gurdon and Shinya Yamanaka for their discoveries on cell reprogramming which paved the way for new therapeutic horizons. We accumulated evidences that stem cell therapy with donor adult cells has produced dramatic amelioration in dystrophic mice and dogs. However, their finite lifespan and replication capacity, limit their therapeutic potential. Several types or subtypes of resident stem cells have been isolated and characterized from adult skeletal muscles. Further translational studies are still necessary to get molecular insights on how to improve the myogenic potential of each cell type. Moreover, it will be relevant to reveal the molecular and epigenetic signatures of myogenic progenitors to identify all molecules involved in the crosstalk among the different pools. In addition, several articles have documented a subset of miRNAs that regulate myogenic cell proliferation, differentiation, and contractility. The possibility to improve myogenic commitment of stem cells by targeting the expression of specific miRNAs is now implicated in several preclinical studies (Crippa et al., 2012). New insights into additional mechanism of post-transcriptional regulation mediated by lncRNAs are desirable since they have an impact on the distribution of miRNA molecules on their targets (Twayana et al., 2013). In the following years miRNA technologies combined to stem cell treatments will test novel therapeutic strategies for skeletal muscle disorders.
Muscle progenitors may be generated from patient iPS cells, genetically modified, systemically injected, then recruited to and integrated in the areas of damage. This would circumvent problems related to allogeneic transplantation and difficulty in obtaining autologous stem cells. The novel reprogramming methods (Obokata et al., 2014) do not require nuclear transfer or genetic manipulation and thus they are more suitable for translational studies with clinical implications. It is interesting that such a great potential has not been explored yet in cachexia and sarcopenia, where it could be employed avoiding genetic manipulation. However, the enormous research impetus on regenerative medicine and stem cell-based therapy could strongly influence the future scientific directions. Emerging literature supports the hypothesis that downregulating myonuclear apoptosis might preserve muscle mass and function in the elderly. In principle, employing pharmacological or genetic interventions to target muscle protein turnover, autophagy and myogenic stem cell function may provide a more thorough protection against muscle aging and atrophy. These multi-therapeutic approaches will face several challenges, including the clear determination of feasible therapeutic windows for each specific intervention, especially if systemic delivery is employed. Nevertheless, pursuing this path is certainly worth in order to relieve the individual and societal burden associated with muscular degeneration.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Translational Cardiomyology laboratory is supported by CARE-MIFP7, Association française contre les myopathies (AFM), CARIPLO FOUNDATION, Fonds Wetenschappelijk Onderzoek (FWO), Geconcerteerde Onderzoeksacties (GOA), Interuniversity Attraction Poles (IUAP), and Onderzoekstoelagen (OT) grants. Emanuele Berardi is a postdoctoral fellow supported by FWO and Maurilio Sampaolesi is recipient of an Excellentiefinanciering KUL Project grant. The authors would like to thanks also Paolo Luban and Rondoufonds voor Duchenne Onderzoek for kind donations. We appreciated Jan Deprest, Paul Holvoet, Danny Huylebroeck, Arnold Luttun, Frank Luyten, Karin Sipido, Catherine Verfaillie, An Zwijsen for critical discussion. The authors would like to thank Christina Vochten and Vicky Raets for professional secretarial service.
References
Aartsma-Rus, A., Kaman, W. E., Weij, R., Den Dunnen, J. T., Van Ommen, G. J., and Van Deutekom, J. C. (2006). Exploring the frontiers of therapeutic exon skipping for Duchenne muscular dystrophy by double targeting within one or multiple exons. Mol. Ther. 14, 401–407. doi: 10.1016/j.ymthe.2006.02.022
Abdel-Hamid, H., and Clemens, P. R. (2012). Pharmacological therapies for muscular dystrophies. Curr. Opin. Neurol. 25, 604–608. doi: 10.1097/Wco.0b013e328357f44c
Apparailly, F., Millet, V., Noel, D., Jacquet, C., Sany, J., and Jorgensen, C. (2002). Tetracycline-inducible interleukin-10 gene transfer mediated by an adeno-associated virus: application to experimental arthritis. Hum. Gene Ther. 13, 1179–1188. doi: 10.1089/104303402320138961
Awaya, T., Kato, T., Mizuno, Y., Chang, H., Niwa, A., Umeda, K., et al. (2012). Selective development of myogenic mesenchymal cells from human embryonic and induced pluripotent stem cells. PLoS ONE 7:e51638. doi: 10.1371/journal.pone.0051638
Barberi, T., Bradbury, M., Dincer, Z., Panagiotakos, G., Socci, N. D., and Studer, L. (2007). Derivation of engraftable skeletal myoblasts from human embryonic stem cells. Nat. Med. 13, 642–648. doi: 10.1038/nm1533
Barton-Davis, E. R., Cordier, L., Shoturma, D. I., Leland, S. E., and Sweeney, H. L. (1999). Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Invest. 104, 375–381. doi: 10.1172/JCI7866
Beauchamp, J. R., Heslop, L., Yu, D. S., Tajbakhsh, S., Kelly, R. G., Wernig, A., et al. (2000). Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J. Cell Biol. 151, 1221–1234. doi: 10.1083/jcb.151.6.1221
Benedetti, S., Hoshiya, H., and Tedesco, F. S. (2013). Repair or replace? Exploiting novel gene and cell therapy strategies for muscular dystrophies. FEBS J. 280, 4263–4280. doi: 10.1111/febs.12178
Berardi, E., Pues, M., Thorrez, L., and Sampaolesi, M. (2012). miRNAs in ESC differentiation. Am. J. Physiol. Heart Circ. Physiol. 303, H931–H939. doi: 10.1152/ajpheart.00338.2012
Bhagavati, S., and Xu, W. (2005). Generation of skeletal muscle from transplanted embryonic stem cells in dystrophic mice. Biochem. Biophys. Res. Commun. 333, 644–649. doi: 10.1016/j.bbrc.2005.05.135
Blake, D. J., Weir, A., Newey, S. E., and Davies, K. E. (2002). Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 82, 291–329. doi: 10.1152/physrev.00028.2001
Bodine, S. C., Latres, E., Baumhueter, S., Lai, V. K. M., Nunez, L., Clarke, B. A., et al. (2001). Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708. doi: 10.1126/science.1065874
Bonifati, M. D., Ruzza, G., Bonometto, P., Berardinelli, A., Gorni, K., Orcesi, S., et al. (2000). A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve 23, 1344–1347. doi: 10.1002/1097-4598(200009)23:9<1344::AID-MUS4>3.0.CO;2-F
Brack, A. S., and Rando, T. A. (2012). Tissue-specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell 10, 504–514. doi: 10.1016/j.stem.2012.04.001
Bruera, E. (1997). ABC of palliative care. Anorexia, cachexia, and nutrition. BMJ 315, 1219–1222. doi: 10.1136/bmj.315.7117.1219
Burkin, D. J., and Kaufman, S. J. (1999). The alpha7beta1 integrin in muscle development and disease. Cell Tissue Res. 296, 183–190. doi: 10.1007/s004410051279
Castillero, E., Nieto-Bona, M. P., Fernandez-Galaz, C., Martin, A. I., Lopez-Menduina, M., et al. (2011). Fenofibrate, a PPAR{alpha} agonist, decreases atrogenes and myostatin expression and improves arthritis-induced skeletal muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 300, E790–E799. doi: 10.1152/ajpendo.00590.2010
Cerletti, M., Jurga, S., Witczak, C. A., Hirshman, M. F., Shadrach, J. L., Goodyear, L. J., et al. (2008). Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell 134, 37–47. doi: 10.1016/j.cell.2008.05.049
Cerletti, M., Negri, T., Cozzi, F., Colpo, R., Andreetta, F., Croci, D., et al. (2003). Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther. 10, 750–757. doi: 10.1038/sj.gt.3301941
Chen, J. F., Mandel, E. M., Thomson, J. M., Wu, Q. L., Callis, T. E., Hammond, S. M., et al. (2006). The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 38, 228–233. doi: 10.1038/Ng1725
Clop, A., Marcq, F., Takeda, H., Pirottin, D., Tordoir, X., Bibe, B., et al. (2006). A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 38, 813–818. doi: 10.1038/ng1810
Cornelison, D. D., Wilcox-Adelman, S. A., Goetinck, P. F., Rauvala, H., Rapraeger, A. C., and Olwin, B. B. (2004). Essential and separable roles for Syndecan-3 and Syndecan-4 in skeletal muscle development and regeneration. Genes Dev. 18, 2231–2236. doi: 10.1101/gad.1214204
Cottard, V., Mulleman, D., Bouille, P., Mezzina, M., Boissier, M. C., and Bessis, N. (2000). Adeno-associated virus-mediated delivery of IL-4 prevents collagen-induced arthritis. Gene Ther. 7, 1930–1939. doi: 10.1038/sj.gt.3301324
Cozzoli, A., Rolland, J. F., Capogrosso, R. F., Sblendorio, V. T., Longo, V., Simonetti, S., et al. (2011). Evaluation of potential synergistic action of a combined treatment with alpha-methyl-prednisolone and taurine on the mdx mouse model of Duchenne muscular dystrophy. Neuropathol. Appl. Neurobiol. 37, 243–256. doi: 10.1111/j.1365-2990.2010.01106.x
Crippa, S., Cassano, M., Messina, G., Galli, D., Galvez, B. G., Curk, T., et al. (2011). miR669a and miR669q prevent skeletal muscle differentiation in postnatal cardiac progenitors. J. Cell Biol. 193, 1197–1212. doi: 10.1083/jcb.201011099
Crippa, S., Cassano, M., and Sampaolesi, M. (2012). Role of miRNAs in muscle stem cell biology: proliferation, differentiation and death. Curr. Pharm. Des. 18, 1718–1729. doi: 10.2174/138161212799859620
Crist, C. G., Montarras, D., Pallafacchina, G., Rocancourt, D., Cumano, A., Conway, S. J., et al. (2009). Muscle stem cell behavior is modified by microRNA-27 regulation of Pax3 expression. Proc. Natl. Acad. Sci. U.S.A. 106, 13383–13387. doi: 10.1073/pnas.0900210106
Dai, J., and Rabie, A. B. M. (2007). The use of recombinant adeno-associated virus for skeletal gene therapy. Orthod. Craniofac. Res. 10, 1–14. doi: 10.1111/j.1601-6343.2007.00381.x
D'Angelo, M. G., Gandossini, S., Martinelli Boneschi, F., Sciorati, C., Bonato, S., Brighina, E., et al. (2012). Nitric oxide donor and non steroidal anti inflammatory drugs as a therapy for muscular dystrophies: evidence from a safety study with pilot efficacy measures in adult dystrophic patients. Pharmacol. Res 65, 472–479. doi: 10.1016/j.phrs.2012.01.006
Darabi, R., Arpke, R. W., Irion, S., Dimos, J. T., Grskovic, M., Kyba, M., et al. (2012). Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 10, 610–619. doi: 10.1016/j.stem.2012.02.015
Darabi, R., Pan, W., Bosnakovski, D., Baik, J., Kyba, M., and Perlingeiro, R. C. (2011). Functional myogenic engraftment from mouse iPS cells. Stem Cell Rev. 7, 948–957. doi: 10.1007/s12015-011-9258-2
Djuranovic, S., Nahvi, A., and Green, R. (2011). A parsimonious model for gene regulation by miRNAs. Science 331, 550–553. doi: 10.1126/science.1191138
Douglas, A. G., and Wood, M. J. (2013). Splicing therapy for neuromuscular disease. Mol. Cell. Neurosci. 56, 169–185. doi: 10.1016/j.mcn.2013.04.005
Eisenberg, I., Eran, A., Nishino, I., Moggio, M., Lamperti, C., Amato, A. A., et al. (2007). Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. U.S.A. 104, 17016–17021. doi: 10.1073/pnas.0708115104
Emery, A. E. H. (2002). The muscular dystrophies. Lancet 359, 687–695. doi: 10.1016/S0140-6736(02)07815-7
Evans, M. J., and Kaufman, M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156. doi: 10.1038/292154a0
Fearon, K., Strasser, F., Anker, S. D., Bosaeus, I., Bruera, E., Fainsinger, R. L., et al. (2011). Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495. doi: 10.1016/S1470-2045(10)70218-7
Filareto, A., Darabi, R., and Perlingeiro, R. C. (2012). Engraftment of ES-derived myogenic progenitors in a severe mouse model of muscular dystrophy. J. Stem Cell Res. Ther. 10:S10-001. doi: 10.4172/2157-7633.S10-001
Filareto, A., Parker, S., Darabi, R., Borges, L., Iacovino, M., Schaaf, T., et al. (2013). An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat. Commun. 4, 1549. doi: 10.1038/ncomms2550
Finkel, R. S. (2010). Read-through strategies for suppression of nonsense mutations in Duchenne/Becker muscular dystrophy: aminoglycosides and ataluren (PTC124). J. Child Neurol. 25, 1158–1164. doi: 10.1177/0883073810371129
Ge, Y., and Chen, J. (2011). MicroRNAs in skeletal myogenesis. Cell Cycle 10, 441–448. doi: 10.4161/cc.10.3.14710
Gilbert, R., Nalbantoglu, J., Petrof, B. J., Ebihara, S., Guibinga, G. H., Tinsley, J. M., et al. (1999). Adenovirus-mediated utrophin gene transfer mitigates the dystrophic phenotype of mdx mouse muscles. Hum. Gene Ther. 10, 1299–1310. doi: 10.1089/10430349950017987
Gnocchi, V. F., White, R. B., Ono, Y., Ellis, J. A., and Zammit, P. S. (2009). Further characterisation of the molecular signature of quiescent and activated mouse muscle satellite cells. PLoS ONE 4:E5205. doi: 10.1371/Journal.Pone.0005205
Goldstein, J. A., and Mcnally, E. M. (2010). Mechanisms of muscle weakness in muscular dystrophy. J. Gen. Physiol. 136, 29–34. doi: 10.1085/jgp.201010436
Gomes, M. D., Lecker, S. H., Jagoe, R. T., Navon, A., and Goldberg, A. L. (2001). Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. U.S.A. 98, 14440–14445. doi: 10.1073/pnas.251541198
Gomez-Sanmiguel, A. B., Martin, A. I., Nieto-Bona, M. P., Fernandez-Galaz, C., Lopez-Menduina, M., Villanua, M. A., et al. (2013). Systemic alpha-melanocyte-stimulating hormone administration decreases arthritis-induced anorexia and muscle wasting. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R877–R886. doi: 10.1152/ajpregu.00447.2012
Goonasekera, S. A., Lam, C. K., Millay, D. P., Sargent, M. A., Hajjar, R. J., Kranias, E. G., et al. (2011). Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Invest. 121, 1044–1052. doi: 10.1172/JCI43844
Goudenege, S., Lebel, C., Huot, N. B., Dufour, C., Fujii, I., Gekas, J., et al. (2012). Myoblasts derived from normal hESCs and dystrophic hiPSCs efficiently fuse with existing muscle fibers following transplantation. Mol. Ther. 20, 2153–2167. doi: 10.1038/mt.2012.188
Goyenvalle, A., Wright, J., Babbs, A., Wilkins, V., Garcia, L., and Davies, K. E. (2012). Engineering multiple U7snRNA constructs to induce single and multiexon-skipping for Duchenne muscular dystrophy. Mol. Ther. 20, 1212–1221. doi: 10.1038/mt.2012.26
Gregorevic, P., Allen, J. M., Minami, E., Blankinship, M. J., Haraguchi, M., Meuse, L., et al. (2006). rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 12, 787–789. doi: 10.1038/nm1439
Gregorevic, P., Blankinship, M. J., Allen, J. M., Crawford, R. W., Meuse, L., Miller, D. G., et al. (2004). Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 10, 828–834. doi: 10.1038/nm1085
Griggs, R. C., Moxley, R. T. 3rd. Mendell, J. R., Fenichel, G. M., Brooke, M. H., Pestronk, A., et al. (1991). Prednisone in Duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical Investigation of Duchenne Dystrophy Group. Arch. Neurol. 48, 383–388. doi: 10.1001/archneur.1991.00530160047012
Griggs, R. C., Moxley, R. T. 3rd. Mendell, J. R., Fenichel, G. M., Brooke, M. H., Pestronk, A., et al. (1993). Duchenne dystrophy: randomized, controlled trial of prednisone (18 months) and azathioprine (12 months). Neurology 43, 520–527. doi: 10.1212/WNL.43.3_Part_1.520
Gros, J., Manceau, M., Thome, V., and Marcelle, C. (2005). A common somitic origin for embryonic muscle progenitors and satellite cells. Nature 435, 954–958. doi: 10.1038/Nature03572
Gueta, I., Altman, A., and Shoenfeld, Y. (2010). [The effect of blocking TNF-alpha in patients with cancer-related cachexia and anorexia]. Harefuah 149, 512–514, 550, 551.
Gumerson, J. D., and Michele, D. E. (2011). The dystrophin-glycoprotein complex in the prevention of muscle damage. J. Biomed. Biotechnol. 2011:210797. doi: 10.1155/2011/210797
Gussoni, E., Soneoka, Y., Strickland, C. D., Buzney, E. A., Khan, M. K., Flint, A. F., et al. (1999). Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature 401, 390–394. doi: 10.1038/43922
Ham, D. J., Murphy, K. T., Chee, A., Lynch, G. S., and Koopman, R. (2013). Glycine administration attenuates skeletal muscle wasting in a mouse model of cancer cachexia. Clin. Nutr. doi: 10.1016/j.clnu.2013.06.013. [Epub ahead of print].
Hamed, S. A. (2006). Drug evaluation: PTC-124–a potential treatment of cystic fibrosis and Duchenne muscular dystrophy. IDrugs 9, 783–789.
He, W. A., Berardi, E., Cardillo, V. M., Acharyya, S., Aulino, P., Thomas-Ahner, J., et al. (2013). NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Invest. 123, 4821–4835. doi: 10.1172/JCI68523
Hill, E., Boontheekul, T., and Mooney, D. J. (2006). Designing scaffolds to enhance transplanted myoblast survival and migration. Tissue Eng. 12, 1295–1304. doi: 10.1089/ten.2006.12.1295
Huard, J., Li, Y., and Fu, F. H. (2002). Current concepts review - Muscle injuries and repair: current trends in research. J. Bone Joint Surg. Am. 84A, 822–832.
Irintchev, A., Zeschnigk, M., Starzinski-Powitz, A., and Wernig, A. (1994). Expression pattern of M-cadherin in normal, denervated, and regenerating mouse muscles. Dev. Dyn. 199, 326–337. doi: 10.1002/aja.1001990407
Jarvinen, T. A. H. J., Jarvinen, T. L. N., Kaariainen, M., Kalimo, A., and Jarvinen, M. (2005). Muscle injuries - Biology and treatment. Am. J. Sports Med. 33, 745–764. doi: 10.1177/0363546505274714
Jensen, T. E., and Richter, E. A. (2012). Regulation of glucose and glycogen metabolism during and after exercise. J. Physiol. 590, 1069–1076. doi: 10.1113/jphysiol.2011.224972
Jorgensen, L. H., Larochelle, N., Orlopp, K., Dunant, P., Dudley, R. W., Stucka, R., et al. (2009). Efficient and fast functional screening of microdystrophin constructs in vivo and in vitro for therapy of duchenne muscular dystrophy. Hum. Gene Ther. 20, 641–650. doi: 10.1089/hum.2008.162
Kassar-Duchossoy, L., Giacone, E., Gayraud-Morel, B., Jory, A., Gomes, D., and Tajbakhsh, S. (2005). Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes Dev. 19, 1426–1431. doi: 10.1101/Gad.345505
Katakura, S., Jennings, K., Watanabe, S., Adachi, E., Thornton, S., Gao, G. P., et al. (2004). Recombinant adeno-associated virus preferentially transduces human, compared to mouse, synovium: implications for arthritis therapy. Mod. Rheumatol. 14, 18–24. doi: 10.1007/s10165-003-0260-7
Kayali, R., Ku, J. M., Khitrov, G., Jung, M. E., Prikhodko, O., and Bertoni, C. (2012). Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum. Mol. Genet. 21, 4007–4020. doi: 10.1093/hmg/dds223
Kim, H. K., Lee, Y. S., Sivaprasad, U., Malhotra, A., and Dutta, A. (2006). Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 174, 677–687. doi: 10.1083/jcb.200603008
Kim, K., Doi, A., Wen, B., Ng, K., Zhao, R., Cahan, P., et al. (2010). Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290. doi: 10.1038/nature09342
Kinali, M., Arechavala-Gomeza, V., Feng, L., Cirak, S., Hunt, D., Adkin, C., et al. (2009). Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 8, 918–928. doi: 10.1016/S1474-4422(09)70211-X
Koo, T., Okada, T., Athanasopoulos, T., Foster, H., Takeda, S., and Dickson, G. (2011). Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J. gene Med. 13, 497–506. doi: 10.1002/jgm.1602
Kuang, S., and Rudnicki, M. A. (2008). The emerging biology of satellite cells and their therapeutic potential. Trends Mol. Med. 14, 82–91. doi: 10.1016/j.molmed.2007.12.004
Laviano, A., Meguid, M. M., and Rossi-Fanelli, F. (2003). Cancer anorexia: clinical implications, pathogenesis, and therapeutic strategies. Lancet Oncol. 4, 686–694. doi: 10.1016/S1470-2045(03)01247-6
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The c-elegans heterochronic gene lin-4 encodes small rnas with antisense complementarity to lin-14. Cell 75, 843–854. doi: 10.1016/0092-8674(93)90529-Y
Leung, D. G., and Wagner, K. R. (2013). Therapeutic advances in muscular dystrophy. Ann. Neurol. 74, 404–411. doi: 10.1002/Ana.23989
Lim, S. S., Vos, T., Flaxman, A. D., Danaei, G., Shibuya, K., Adair-Rohani, H., et al. (2012). A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2224–2260. doi: 10.1016/S0140-6736(12)61766-8
Liu, M., Yue, Y., Harper, S. Q., Grange, R. W., Chamberlain, J. S., and Duan, D. (2005). Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol. Ther. 11, 245–256. doi: 10.1016/j.ymthe.2004.09.013
Malafarina, V., Uriz-Otano, F., Iniesta, R., and Gil-Guerrero, L. (2012). Sarcopenia in the elderly: diagnosis, physiopathology and treatment. Maturitas 71, 109–114. doi: 10.1016/j.maturitas.2011.11.012
Marcora, S. M., Chester, K. R., Mittal, G., Lemmey, A. B., and Maddison, P. J. (2006). Randomized phase 2 trial of anti-tumor necrosis factor therapy for cachexia in patients with early rheumatoid arthritis. Am. J. Clin. Nutr. 84, 1463–1472.
Martin, G. R. (1981). Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. U.S.A. 78, 7634–7638. doi: 10.1073/pnas.78.12.7634
Mauro, A. (1961). Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9, 493–495. doi: 10.1083/jcb.9.2.493
McClorey, G., Moulton, H. M., Iversen, P. L., Fletcher, S., and Wilton, S. D. (2006). Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 13, 1373–1381. doi: 10.1038/sj.gt.3302800
Meech, R., Gonzalez, K. N., Barro, M., Gromova, A., Zhuang, L., Hulin, J. A., et al. (2012). Barx2 is expressed in satellite cells and is required for normal muscle growth and regeneration. Stem Cells 30, 253–265. doi: 10.1002/stem.777
Minasi, M. G., Riminucci, M., De Angelis, L., Borello, U., Berarducci, B., Innocenzi, A., et al. (2002). The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 129, 2773–2784.
Minetti, G. C., Colussi, C., Adami, R., Serra, C., Mozzetta, C., Parente, V., et al. (2006). Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 12, 1147–1150. doi: 10.1038/Nm1479
Mizuno, Y., Chang, H., Umeda, K., Niwa, A., Iwasa, T., Awaya, T., et al. (2010). Generation of skeletal muscle stem/progenitor cells from murine induced pluripotent stem cells. FASEB J. 24, 2245–2253. doi: 10.1096/fj.09-137174
Montanaro, F., Liadaki, K., Schienda, J., Flint, A., Gussoni, E., and Kunkel, L. M. (2004). Demystifying SP cell purification: viability, yield, and phenotype are defined by isolation parameters. Exp. Cell Res. 298, 144–154. doi: 10.1016/j.yexcr.2004.04.010
Montarras, D., Morgan, J., Collins, C., Relaix, F., Zaffran, S., Cumano, A., et al. (2005). Direct isolation of satellite cells for skeletal muscle regeneration. Science 309, 2064–2067. doi: 10.1126/science.1114758
Moorwood, C., Lozynska, O., Suri, N., Napper, A. D., Diamond, S. L., and Khurana, T. S. (2011). Drug discovery for Duchenne muscular dystrophy via utrophin promoter activation screening. PLoS ONE 6:e26169. doi: 10.1371/journal.pone.0026169
Morgan, C. L., Emery, P., Porter, D., Reynolds, A., Young, A., Boyd, H., et al. (2014). Treatment of rheumatoid arthritis with etanercept with reference to disease-modifying anti-rheumatic drugs: long-term safety and survival using prospective, observational data. Rheumatology 53, 186–194. doi: 10.1093/rheumatology/ket333
Morine, K. J., Bish, L. T., Pendrak, K., Sleeper, M. M., Barton, E. R., and Sweeney, H. L. (2010). Systemic myostatin inhibition via liver-targeted gene transfer in normal and dystrophic mice. PLoS ONE 5:e9176. doi: 10.1371/journal.pone.0009176
Mouly, V., Aamiri, A., Perie, S., Mamchaoui, K., Barani, A., Bigot, A., et al. (2005). Myoblast transfer therapy: is there any light at the end of the tunnel? Acta Myol. 24, 128–133.
Moxley, R. T. 3rd. Ashwal, S., Pandya, S., Connolly, A., Florence, J., Mathews, K., et al. (2005). Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 64, 13–20. doi: 10.1212/01.WNL.0000148485.00049.B7
Mozzetta, C., Consalvi, S., Saccone, V., Tierney, M., Diamantini, A., Mitchell, K. J., et al. (2013). Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Mol. Med. 5, 626–639. doi: 10.1002/emmm.201202096
Nai, Y. J., Jiang, Z. W., Wang, Z. M., Li, N., and Li, J. S. (2007). Prevention of cancer cachexia by pyrrolidine dithiocarbamate (PDTC) in colon 26 tumor-bearing mice. JPEN 31, 18–25. doi: 10.1177/014860710703100118
Nielsen, S., and Pedersen, B. K. (2008). Skeletal muscle as an immunogenic organ. Curr. Opin. Pharmacol. 8, 346–351. doi: 10.1016/j.coph.2008.02.005
Nudelman, I., Rebibo-Sabbah, A., Cherniavsky, M., Belakhov, V., Hainrichson, M., Chen, F., et al. (2009). Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J. Med. Chem. 52, 2836–2845. doi: 10.1021/jm801640k
Obokata, H., Wakayama, T., Sasai, Y., Kojima, K., Vacanti, M. P., Niwa, H., et al. (2014). Stimulus-triggered fate conversion of somatic cells into pluripotency. Nature 505, 641–647. doi: 10.1038/nature12968
Okada, M., Payne, T. R., Zheng, B., Oshima, H., Momoi, N., Tobita, K., et al. (2008). Myogenic endothelial cells purified from human skeletal muscle improve cardiac function after transplantation into infarcted myocardium. J. Am. Coll. Cardiol. 52, 1869–1880. doi: 10.1016/j.jacc.2008.07.064
Palus, S., Von Haehling, S., Flach, V. C., Tschirner, A., Doehner, W., Anker, S. D., et al. (2013). Simvastatin reduces wasting and improves cardiac function as well as outcome in experimental cancer cachexia. Int. J. Cardiol. 168, 3412–3418. doi: 10.1016/j.ijcard.2013.04.150
Partridge, T. A., Morgan, J. E., Coulton, G. R., Hoffman, E. P., and Kunkel, L. M. (1989). Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature 337, 176–179. doi: 10.1038/337176a0
Pedersen, B. K., and Febbraio, M. A. (2008). Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol. Rev. 88, 1379–1406. doi: 10.1152/physrev.90100.2007
Perez, A. L., Bachrach, E., Illigens, B. M. W., Jun, S. J., Bagden, E., Steffen, L., et al. (2009). Cxcr4 enhances engraftment of muscle progenitor cells. Muscle Nerve 40, 562–572. doi: 10.1002/Mus.21317
Polesskaya, A., Seale, P., and Rudnicki, M. A. (2003). Wnt signaling induces the myogenic specification of resident CD45(+) adult stem cells during muscle regeneration. Cell 113, 841–852. doi: 10.1016/S0092-8674(03)00437-9
Politano, L., Nigro, G., Nigro, V., Piluso, G., Papparella, S., Paciello, O., et al. (2003). Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 22, 15–21.
Polo, J. M., Liu, S., Figueroa, M. E., Kulalert, W., Eminli, S., Tan, K. Y., et al. (2010). Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 28, 848–855. doi: 10.1038/nbt.1667
Qiao, C., Li, J., Jiang, J., Zhu, X., Wang, B., and Xiao, X. (2008). Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum. Gene Ther. 19, 241–254. doi: 10.1089/hum.2007.159
Qiao, C., Li, J., Zheng, H., Bogan, J., Yuan, Z., Zhang, C., et al. (2009). Hydrodynamic limb vein injection of adeno-associated virus serotype 8 vector carrying canine myostatin propeptide gene into normal dogs enhances muscle growth. Hum. Gene Ther. 20, 1–10. doi: 10.1089/hum.2008.135
Quattrocelli, M., Crippa, S., Montecchiani, C., Camps, J., Cornaglia, A. I., Boldrin, L., et al. (2013). Long-term miR-669a therapy alleviates chronic dilated cardiomyopathy in dystrophic mice. J. Am. Heart Assoc. 2:e000284. doi: 10.1161/JAHA.113.000284
Quattrocelli, M., Palazzolo, G., Floris, G., Schöffski, P., Anastasia, L., Orlacchio, A., et al. (2011). Intrinsic cell memory reinforces myogenic commitment of pericyte-derived iPSCs. J. Pathol. 223, 593–603. doi: 10.1002/path.2845
Quattrocelli, M., Palazzolo, G., Perini, I., Crippa, S., Cassano, M., and Sampaolesi, M. (2012). Mouse and human mesoangioblasts: isolation and characterization from adult skeletal muscles. Methods Mol. Biol. 798, 65–76. doi: 10.1007/978-1-61779-343-1_4
Ratajczak, M. Z., Majka, M., Kucia, M., Drukala, J., Pietrzkowski, Z., Peiper, S., et al. (2003). Expression of functional CXCR4 by muscle satellite cells and secretion of SDF-1 by muscle-derived fibroblasts is associated with the presence of both muscle progenitors in bone marrow and hematopoietic stem/progenitor cells in muscles. Stem Cells 21, 363–371. doi: 10.1634/stemcells.21-3-363
Relaix, F., Rocancourt, D., Mansouri, A., and Buckingham, M. (2005). A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 435, 948–953. doi: 10.1038/Nature03594
Rinaldi, F., and Perlingeiro, R. C. (2013). Stem cells for skeletal muscle regeneration: therapeutic potential and roadblocks. Transl. Res. doi: 10.1016/j.trsl.2013.11.006. [Epub ahead of print].
Rodino-Klapac, L. R., Janssen, P. M., Montgomery, C. L., Coley, B. D., Chicoine, L. G., Clark, K. R., et al. (2007). A translational approach for limb vascular delivery of the micro-dystrophin gene without high volume or high pressure for treatment of Duchenne muscular dystrophy. J. Trans. Med. 5:45. doi: 10.1186/1479-5876-5-45
Rodino-Klapac, L. R., Janssen, P. M., Shontz, K. M., Canan, B., Montgomery, C. L., Griffin, D., et al. (2013). Micro-dystrophin and follistatin co-delivery restores muscle function in aged DMD model. Hum. Mol. Genet. 22, 4929–4937. doi: 10.1093/hmg/ddt342
Rosenberg, M. I., Georges, S. A., Asawachaicharn, A., Analau, E., and Tapscott, S. J. (2006). MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J. Cell Biol. 175, 77–85. doi: 10.1083/jcb.200603039
Ruegg, U. T., Nicolas-Metral, V., Challet, C., Bernard-Helary, K., Dorchies, O. M., Wagner, S., et al. (2002). Pharmacological control of cellular calcium handling in dystrophic skeletal muscle. Neuromuscul. Disord. 12(Suppl. 1), S155–S161. doi: 10.1016/S0960-8966(02)00095-0
Sacco, A., Doyonnas, R., Kraft, P., Vitorovic, S., and Blau, H. M. (2008). Self-renewal and expansion of single transplanted muscle stem cells. Nature 456, 502–506. doi: 10.1038/Nature07384
Sampaolesi, M., Blot, S., D'Antona, G., Granger, N., Tonlorenzi, R., Innocenzi, A., et al. (2006). Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 444, 574–579. doi: 10.1038/nature05282
Sampaolesi, M., Torrente, Y., Innocenzi, A., Tonlorenzi, R., D'Antona, G., Pellegrino, M. A., et al. (2003). Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science 301, 487–492. doi: 10.1126/science.1082254
Scott, D., Blizzard, L., Fell, J., and Jones, G. (2011). The epidemiology of sarcopenia in community living older adults: what role does lifestyle play? J. Cachexia Sarcopenia Muscle 2, 125–134. doi: 10.1007/s13539-011-0036-4
Seale, P., Ishibashi, J., Scime, A., and Rudnicki, M. A. (2004). Pax7 is necessary and sufficient for the myogenic specification of CD45(+): Sca1(+) stem cells from injured muscle. PLoS Biol. 2:e130. doi: 10.1371/journal.pbio.0020130
Sherwood, R. I., Christensen, J. L., Conboy, I. M., Conboy, M. J., Rando, T. A., Weissman, I. L., et al. (2004). Isolation of adult mouse myogenic progenitors: Functional heterogeneity of cells within and engrafting skeletal muscle. Cell 119, 543–554. doi: 10.1016/j.cell.2004.10.021
Skuk, D., and Tremblay, J. P. (2011). Intramuscular cell transplantation as a potential treatment of myopathies: clinical and preclinical relevant data. Exp. Opin. Biol. Ther. 11, 359–374. doi: 10.1517/14712598.2011.548800
Small, E. M., O'Rourke, J. R., Moresi, V., Sutherland, L. B., McAnally, J., Gerard, R. D., et al. (2010). Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc. Natl. Acad. Sci. U.S.A. 107, 4218–4223. doi: 10.1073/pnas.1000300107
Suzuki, H., Asakawa, A., Amitani, H., Nakamura, N., and Inui, A. (2013). Cancer cachexia–pathophysiology and management. J. Gastroenterol. 48, 574–594. doi: 10.1007/s00535-013-0787-0
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Tamaki, T., Akatsuka, A., Ando, K., Nakamura, Y., Matsuzawa, H., Hotta, T., et al. (2002). Identification of myogenic-endothelial progenitor cells in the interstitial spaces of skeletal muscle. J. Cell Biol. 157, 571–577. doi: 10.1083/jcb.200112106
Tanaka, K. K., Hall, J. K., Troy, A. A., Cornelison, D. D. W., Majka, S. M., and Olwin, B. B. (2009). Syndecan-4-expressing muscle progenitor cells in the SP engraft as satellite cells during muscle regeneration. Cell Stem Cell 4, 217–225. doi: 10.1016/j.stem.2009.01.016
Tedesco, F. S., Dellavalle, A., Diaz-Manera, J., Messina, G., and Cossu, G. (2010). Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J. Clin. Invest. 120, 11–19. doi: 10.1172/JCI40373
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. doi: 10.1126/science.282.5391.1145
Tinsley, J., Deconinck, N., Fisher, R., Kahn, D., Phelps, S., Gillis, J. M., et al. (1998). Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 4, 1441–1444. doi: 10.1038/4033
Tisdale, M. J. (2002). Cachexia in cancer patients. Nat. Rev. Cancer 2, 862–871. doi: 10.1038/nrc927
Todorov, P., Cariuk, P., McDevitt, T., Coles, B., Fearon, K., and Tisdale, M. (1996). Characterization of a cancer cachectic factor. Nature 379, 739–742. doi: 10.1038/379739a0
Todorov, P. T., Field, W. N., and Tisdale, M. J. (1999). Role of a proteolysis-inducing factor (PIF) in cachexia induced by a human melanoma (G361). Br. J. Cancer 80, 1734–1737. doi: 10.1038/sj.bjc.6690590
Tonlorenzi, R., Dellavalle, A., Schnapp, E., Cossu, G., and Sampaolesi, M. (2007). Isolation and characterization of mesoangioblasts from mouse, dog, and human tissues. Curr. Protoc. Stem Cell Biol. Chapter 2: Unit 2B.1. doi: 10.1002/9780470151808.sc02b01s3
Twayana, S., Legnini, I., Cesana, M., Cacchiarelli, D., Morlando, M., and Bozzoni, I. (2013). Biogenesis and function of non-coding RNAs in muscle differentiation and in Duchenne muscular dystrophy. Biochem. Soc. Trans. 41, 844–849. doi: 10.1042/BST20120353
Van Deutekom, J. C., Janson, A. A., Ginjaar, I. B., Frankhuizen, W. S., Aartsma-Rus, A., Bremmer-Bout, M., et al. (2007). Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 357, 2677–2686. doi: 10.1056/NEJMoa073108
Van Rooij, E., Liu, N., and Olson, E. N. (2008). MicroRNAs flex their muscles. Trends Genet. 24, 159–166. doi: 10.1016/j.tig.2008.01.007
Van Rooij, E., Quiat, D., Johnson, B. A., Sutherland, L. B., Qi, X. X., Richardson, J. A., et al. (2009). A Family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 17, 662–673. doi: 10.1016/j.devcel.2009.10.013
Vaughan, V. C., Sullivan-Gunn, M., Hinch, E., Martin, P., and Lewandowski, P. A. (2012). Eicosapentaenoic acid and oxypurinol in the treatment of muscle wasting in a mouse model of cancer cachexia. PLoS ONE 7:e45900. doi: 10.1371/journal.pone.0045900
Volonte, D., Liu, Y., and Galbiati, F. (2005). The modulation of caveolin-1 expression controls satellite cell activation during muscle repair. FASEB J. 19, 237–239. doi: 10.1096/fj.04-2215fje
Wang, Y. X., and Rudnicki, M. A. (2012). Satellite cells, the engines of muscle repair. Nature Rev. Mol. Cell Biol. 13, 127–133. doi: 10.1038/nrm3265
Wightman, B., Ha, I., and Ruvkun, G. (1993). Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862. doi: 10.1016/0092-8674(93)90530-4
Wu, C., Fernandez, S. A., Criswell, T., Chidiac, T. A., Guttridge, D., Villalona-Calero, M., et al. (2013). Disrupting cytokine signaling in pancreatic cancer: a phase I/II study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas 42, 813–818. doi: 10.1097/MPA.0b013e318279b87f
Yoshimura, M., Sakamoto, M., Ikemoto, M., Mochizuki, Y., Yuasa, K., Miyagoe-Suzuki, Y., et al. (2004). AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol. Ther. 10, 821–828. doi: 10.1016/j.ymthe.2004.07.025
Zhang, L., Tang, H., Kou, Y., Li, R., Zheng, Y., Wang, Q., et al. (2013). MG132-mediated inhibition of the ubiquitin-proteasome pathway ameliorates cancer cachexia. J. Cancer Res. Clin. Oncol. 139, 1105–1115. doi: 10.1007/s00432-013-1412-6
Keywords: muscle degeneration, molecular treatments, stem cells, gene and cell therapies, cachexia
Citation: Berardi E, Annibali D, Cassano M, Crippa S and Sampaolesi M (2014) Molecular and cell-based therapies for muscle degenerations: a road under construction. Front. Physiol. 5:119. doi: 10.3389/fphys.2014.00119
Received: 25 November 2013; Accepted: 12 March 2014;
Published online: 08 April 2014.
Edited by:
Carlos Hermano J. Pinheiro, University of São Paulo, BrazilReviewed by:
Alessandra Sacco, Sanford-Burnham Medical Research Institute, USARadbod Darabi, University of Texas Medical Health Center at Houston, USA
Copyright © 2014 Berardi, Annibali, Cassano, Crippa and Sampaolesi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurilio Sampaolesi, Translational Cardiomyology Laboratory, Department of Development and Reproduction, KUL University of Leuven, Stem Cell Research Institute, 49 Herestraat B-3000 Leuven, Belgium e-mail: maurilio.sampaolesi@med.kuleuven.be