 Claire E. Olson
Claire E. Olson Steven B. Roberts
Steven B. Roberts- School of Aquatic and Fishery Sciences, University of Washington, Seattle, WA, USA
DNA methylation patterns and functions are variable across invertebrate taxa. In order to provide a better understanding of DNA methylation in the Pacific oyster (Crassostrea gigas), we characterized the genome-wide DNA methylation profile in male gamete cells using whole-genome bisulfite sequencing. RNA-Seq analysis was performed to examine the relationship between DNA methylation and transcript expression. Methylation status of over 7.6 million CpG dinucleotides was described with a majority of methylated regions occurring among intragenic regions. Overall, 15% of the CpG dinucleotides were determined to be methylated and the mitochondrial genome lacked DNA methylation. Integrative analysis of DNA methylation and RNA-Seq data revealed a positive association between methylation status, both in gene bodies and putative promoter regions, and expression. This study provides a comprehensive characterization of the distribution of DNA methylation in the oyster male gamete tissue and suggests that DNA methylation is involved in gene regulatory activity.
Introduction
DNA methylation is an important epigenetic process that varies in genomic distribution and biological function across taxa. DNA methylation involves the addition of a methyl group to a cytosine pyrimidine ring and most often occurs as part of C-G nucleotide pairs, frequently referred to as CpG dinucleotides. Mammals exhibit a pattern commonly referred to as global methylation, in which 70–80% of CpG dinucleotides are methylated (Bird, 1980). In contrast, invertebrates display relatively low levels of DNA methylation, from almost no methylation in Drosophila melanogaster (Gowher et al., 2000) to intermediate levels in the sea urchin Echinus esculentus (Bird et al., 1979). In mammals, a primary function of DNA methylation is to suppress gene expression through increased promoter DNA methylation (Bell and Felsenfeld, 2000). However, the function of DNA methylation in invertebrates is variable and likely differs among invertebrate taxa. The roles of methylation include regulation of transcriptional activity (Suzuki and Bird, 2008), alternatively exon splicing (Lyko et al., 2010), and developmental activity (Riviere et al., 2013).
While in general there is a limited amount of comprehensive information regarding DNA methylation in non-mammalian taxa, some recent studies have focused on DNA methylation in the Pacific oyster. The Pacific oyster is an excellent model for studying epigenetic modifications because its life history characteristics make it an important aquaculture species (Glude and Chew, 1982) and genomic resources for this species have recently become available (Zhang et al., 2012). Gavery and Roberts (2010) first reported the presence of DNA methylation in the Pacific oyster. In the same study, in silico analysis revealed a significant correlation between gene function and methylation level (Gavery and Roberts, 2010). The relationship between gene methylation and gene function was experimentally corroborated with high-throughput sequencing and it has been proposed that limited methylation in select genes may contribute to increased phenotypic plasticity in highly fluctuating environments (Roberts and Gavery, 2012). More recently, methylation enrichment and bisulfite sequencing were used to describe high-resolution DNA methylation patterns in pooled oyster gill tissue (Gavery and Roberts, 2013). A characterization of DNA methylation during oyster larval development has also been performed, revealing that DNA methylation varies through early development and treatment with 5-Aza-cytidine, a DNA methyltransferase (DNMT) inhibitor, leads to developmental alterations (Riviere et al., 2013). In the same study, researchers found an inverse correlation between methylation proximal to the transcription start site and expression of hox genes (Riviere et al., 2013). In addition to studies that investigate putative function of DNA methylation, other research has begun to evaluate relationships between epigenetic and genetic variations in C. gigas mass selection procedures (Jiang et al., 2013).
While a better understanding of DNA methylation is emerging for this species, there are still several questions that remain. Importantly we still do not fully understand the relationship between DNA methylation and gene expression, nor DNA methylation patterns in a single cell type. Examining a single cell type is important as methylation levels and patterns may differ between multiple cell types and life history stages, and our research attempted to limit this potential variability. Spermatozoa are an ideal resource for studying a single cell type and also provide the secondary benefit of understanding more about oyster spermatogenesis. The oyster male gonad consists of numerous gonadal tubules that grow during tissue development (Franco et al., 2008) and evolve according to four successive reproductive stages annually (Berthelin et al., 2000). These gonadal stages include undifferentiated (stage 0), mitosis of spermatogonia and differentiation of germ cells (stage 1), visible spermatogenesis (stage 2), and mature gametes (stage 3) (Franco et al., 2008). This is the first time DNA methylation has been characterized in Pacific oyster gametes, however spermatozoa methylation has been previously examined in other marine invertebrates. For example, spermatozoa DNA methylation has been described in both the marine annelid worm Chaetopterus variopedatus (del Gaudio et al., 1997) and Ciona intestinalis (Suzuki et al., 2013).
This research represents the first high-resolution characterization of DNA methylation patterns from a single cell type in a mollusc, including an examination of the relationship between gene expression and promoter region methylation. Our results demonstrate that DNA methylation is predominant in intragenic regions (exons and introns) and that there is a positive relationship between methylation and gene expression in C. gigas. Furthermore, we were surprised to find similar patterns of tissue-specific methylation in male gametes as has been previously described in oyster gill tissue, thus suggesting that overall methylation levels do not dramatically vary between tissue types, and specifically between gametic and somatic cells.
Methods
Bisulfite Treated DNA Sequencing (BS-Seq)
A single male adult oyster was collected from Thorndyke Bay, WA and thermally conditioned and fed for 6 weeks in the laboratory. Male gamete tissue was scored with a razor blade, gametes rinsed with sterile seawater, centrifuged, and immediately placed on dry ice then stored at −80°C until further processing. Genomic DNA was extracted using DNAzol according to the manufacturer's protocol (Molecular Research Center, Inc. Cincinnati, OH). High molecular weight genomic DNA (6 ug) was used to prepare a library for whole-genome bisulfite sequencing. Lambda DNA (Promega Co. Madison, WI) was added to the sample prior to fragmentation and library construction to serve as a measure of bisulfite conversion efficiency. Extracted DNA was fragmented to an average length of 250 bp using a Covaris S2 (Covaris Inc. Woburn, MA) and fragment size was confirmed by gel electrophoresis. The library was constructed using the Paired-End DNA Sample Prep Kit (Illumina, San Diego, CA) with standard protocols. DNA was treated with sodium bisulfite using the EpiTect Bisulfite Kit (Qiagen, Valencia, CA) and 72 bp paired-end sequencing was performed on the Illumina HiSeq 2000 system. Library construction and sequencing was performed by the High Throughput Genomics Center (htSEQ, Seattle, WA).
DNA sequence reads were mapped to all genomic scaffolds from the Crassostrea gigas draft genome (Fang et al., 2012). Sequences were mapped using Bisulfite Sequencing Mapping Program BSMAP v2.73 (Xi and Li, 2009). Resulting data from mapping bisulfite treated reads was analyzed with methratio, a Python script that accompanies BSMAP to calculate and extract methylation ratios. Parameters for methratio included reporting loci with zero methylation ratios (-z), combining CpG methylation ratios on both strands (-g) and only using unique mappings (-u). The same mapping procedure was also performed with the Crassostrea gigas mitochondrial genome (Accession # AF177226). The resulting methratio outputs were uploaded to SQLShare (Howe et al., 2011) and queried to examine distribution of methylation. Methylation characteristics were initially calculated for all cytosines.
Genomic Features and CpG Dinucleotide Methylation
Methylation of CpG dinucleotides was characterized in relation to genomic features. A CpG locus was considered methylated if it had at least 5× coverage and at least half the reads remained unconverted after bisulfite treatment. Methylation ratios were calculated for individual loci as well as for full-length genes and intragenic regions (introns and exons). Methylation on a per gene basis was determined by obtaining the number of methylated cytosines divided by the total number of CpG dinucleotides per region. Genome feature tracks were generated in order to characterize the distribution of methylation in the male gamete tissue. All CpG dinucleotides were identified using the EMBOSS tool fuzznuc (Rice et al., 2000). Methylated CpGs (5× coverage, ≥50% unconverted), sparsely methylated CpGs (5× coverage, 0–50% unconverted) and unmethylated CpG loci (5x coverage, 0% unconverted) within genomic regions were determined using Bedtools (i.e., intersectBED) (Quinlan and Hall, 2010). Methylation was examined within exons and introns (Fang et al., 2012), promoter regions (characterized as 1 kb regions upstream from transcription start sites), and putative transposable elements identified using RepeatMasker and the Transposable Element Protein Database (Smit et al., 1996–2010).
Transcriptome Sequencing
Total RNA was isolated using TRI reagent (Molecular Research Center) from the same oyster gamete tissue used for bisulfite sequencing. RNA was enriched for mRNA using Sera-Mag oligo dT beads (Thermo Scientific). A shotgun library was constructed from double stranded cDNA for paired end sequencing by end-polishing, A-tailing and ligation of sequencing adaptors. Sequencing and library preparation were performed on the Illumina HiSeq 2000 platform at the Northwest Genomics Center at the University of Washington (Seattle, WA). RNA-Seq analysis was performed using CLC Genomics Workbench version 6.5 (CLC Bio, Aarhus, Denmark) with high-throughput reads (50 bp paired end) mapped back to the oyster transcriptome (Fang et al., 2012). Initially, sequences were trimmed based on quality scores of 0.05 (Phred; Ewing and Green, 1998; Ewing et al., 1998), and the number of ambiguous nucleotides (>2 on ends). Sequences smaller than 20 bp were also removed. For RNA-Seq analysis, expression values for each gene (28,027) were measured as RPKM (reads per kilobase of exon model per million mapped reads) (Mortazavi et al., 2008) with an unspecific match limit of 10 and maximum number of 2 mismatches.
A Chi-squared test was performed to determine if the degree of gene methylation with respect to gene expression levels (RPKM) was different from what would be expected from a random distribution of methylation levels in promoter regions (p-value <0.05 was considered significant). For promoter region analysis, these regions were determined to be the 1 kb regions upstream from transcription start sites that did not overlap with neighboring genes. In addition, only promoter regions with at least 10 CpG dinucleotides were considered. Oyster genes were classified as either heavily methylated (methylation ratio ≥0.5), sparsely methylated (methylation ratio 0–0.5) or unmethylated (methylation ratio = 0).
Results
Bisulfite Treated DNA Sequencing (BS-Seq)
Bisulfite treated DNA sequence reads (171.5 million) were produced and are available (NCBI Sequence Read Archive: accession number SRX386228). A total of 90 million paired end reads (53% of total reads) and 32 million single end reads (9.6%) mapped to the Crassostrea gigas genome. Sodium bisulfite conversion efficiency was estimated to be 99.72% based on analysis of lambda phage DNA. All cytosine dinucleotide motifs were examined and a majority of methylated cytosines were reported in CpG dinucleotides. We found that 15% of the CpG dinucleotides were methylated while the next highest motif (CpA) methylated at 0.14%, which falls within the sodium bisulfite conversion efficiency margin of error (0.28%).
DNA Methylation and Genomic Features
The bisulfite sequencing effort provided ≥1x coverage for 8.52 million of the 9.98 million CpGs (85%) in the oyster nuclear genome. Using a 5x coverage threshold, which corresponds to 7.64 million CpGs (77%), the majority of CpGs were not methylated (Figure 1).
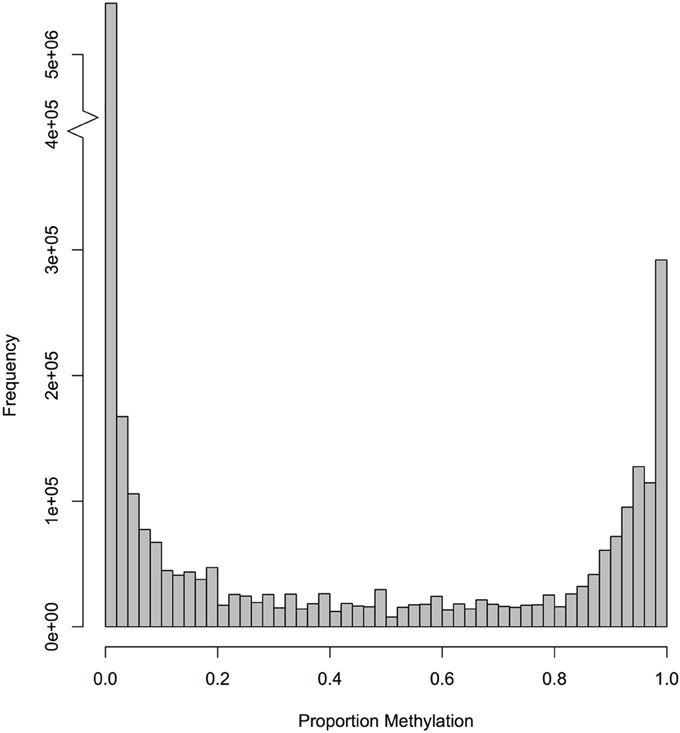
Figure 1. Frequency of CpG methylation ratios in C. gigas male gamete tissue. A total of 7,642,816 CpG dinucleotides with at least 5× coverage were examined.
The proportion of CpG methylation occurring in specific regions of the oyster genomic landscape were characterized. Methylation occurs predominantly in intragenic regions, with 74% of methylated CpGs found in exons and introns. A total of 30% of CpGs in exons were methylated and 18% of CpGs in introns were methylated (Figure 2). These are particularly high levels of methylation when compared to methylation levels of other oyster genomic regions, wherein between 4 and 7% of CpGs were methylated.
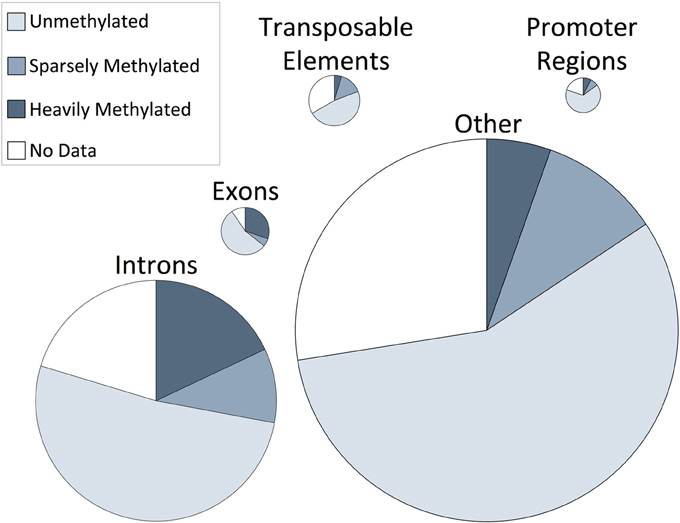
Figure 2. Genome-wide distribution of CpG methylation in C. gigas male gamete tissue within genomic regions. The proportion of CpG unmethylated (ratio = 0), sparsely methylated (ratio between 0 and 0.5), heavily methylated (ratio ≥0.5), or CpG dinucleotides with less than 5× coverage (“No data”) are shown as percentage contributions to specific oyster genomic regions. Genomic regions are scaled according to their relative CpG contribution to the genome.
The oyster mitochondrial genome is predominantly unmethylated. With an average coverage of 39.76-fold, of the 2518 cytosines with at least 5x coverage, 2316 cytosines were converted upon bisulfite treatment and no cytosines were considered methylated.
Whole Transcriptome Sequencing Expression Patterns
After quality trimming, 50.3 million reads (paired end 50 bp) remained (NCBI Sequence Read Archive: accession number SRX390346). Expression (RPKM) was detected in a majority of the genes (17,093 genes or 63%). Median expression level was 0.749 and expression level ranged from 0 to 35637.3 RPKM. The relationship between gene methylation and expression levels was examined by determining methylation on a per gene basis.
A minimum of one methylated CpG dinucleotide with ≥ 5× coverage was observed for every 14,517 genes in the C. gigas genome, or 53% of genes. The proportion of methylated CpGs was characterized with respect to RNA-Seq data on expression levels for full-length genes. Specifically, within genes and putative promoter regions we found a greater proportion of fully methylated CpGs for genes that have elevated expression levels (>1 RPKM) (Figure 3). The observed distributions of methylated CpGs within genes (X2 = 5493.85, df = 2, p < 0.0001) and promoter regions (X2 = 1765.56, df = 2, p < 0.0001) were significantly different than what would be expected if methylated CpGs were randomly distributed among genes and promoter regions.
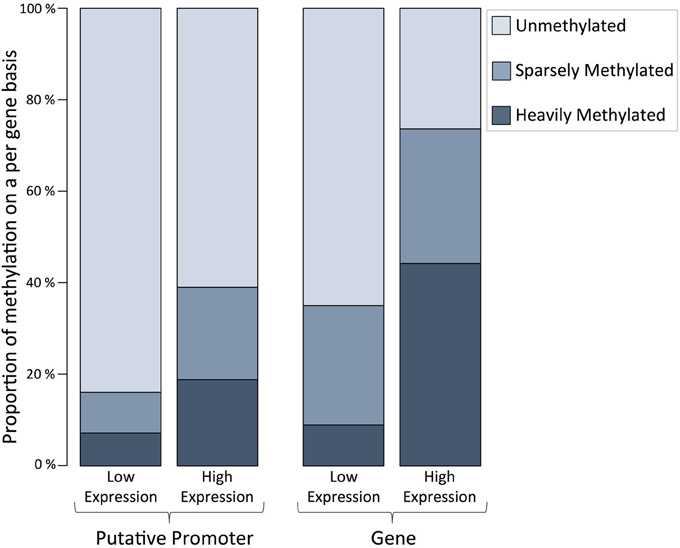
Figure 3. Proportion of methylation on a per gene basis for putative promoter regions and gene bodies (exon and introns) for high and low expression levels indicate that DNA methylation is positively correlated to gene expression in C. gigas male gamete tissue. Two classifications shown include genes with of low expression (Promoter regions = 13,919 and genes = 14,377, RPKM ≤1, including no expression) and high expression (Promoter regions = 12,477 and genes = 12,877; RPKM > 1). The proportion of corresponding genes that are unmethylated (methylation ratio = 0), sparsely methylated (methylation ratio 0—0.5), and heavily methylated (methylation ratio ≥ 0.5) are shown.
Additionally, our transcriptome data was compared to a comprehensive microarray experiment focused on gametogenesis in C. gigas (Dheilly et al., 2012). Elevated gene expression was observed in genes identified by Dheilly et al. (2012) to increase in expression over spermatogenesis (clusters 5 and 6) (Olson and Roberts, 2014). Based on this analysis, we suspect our sample was likely from a fully mature male gonad.
Discussion
Genome-wide shotgun bisulfite sequencing was conducted on gametes from a male oyster to illustrate the role of DNA methylation in a single cell type. While reduced representation CpG DNA methylation has been previously quantified for this species, this study represents the first examination of other dinucleotide motifs. Although CpG DNA methylation is ubiquitous across organisms, types and levels of methylation vary considerably among invertebrates. For example, Caenorhabditis elegans essentially lacks DNA methylation in its genome where neither 5-methylcytosine nor DNA methyltransferase are present (Bird, 2002). The Drosophila melanogaster genome contains extremely low 5-methylcytosine levels (Gowher et al., 2000; Lyko et al., 2000) and mostly in the CpT dinucleotide context rather than CpG (Lyko et al., 2000). In this study, DNA methylation was only observed in CpG motifs.
This is the first characterization of DNA methylation in bivalve mitochondrial DNA. A lack of methylation in oyster mitochondrial DNA in our study is consistent with previous invertebrate research, which found that genes encoding mitochondrial DNA were unmethylated in the brain tissue of the honey bee Apis mellifera (Lyko et al., 2010). Recent research has also demonstrated the absence of CpG methylation in the mitochondrial genome of the sea squirt Ciona intestinalis (Suzuki et al., 2013). Studies on the mammalian system have found similar results, confirming a lack of methylation among CpGs in human mitochondrial DNA (Hong et al., 2013).
Overall, approximately 15% of the CpGs are methylated in the oyster male gamete nuclear genome. This is the same estimated proportion from analysis of DNA methylation in oyster gill tissue (Gavery and Roberts, 2013). Similarly, (Suzuki et al., 2013) found identical gene groups to be methylated in tissues from C. intestinalis sperm and muscle cells. In their study 23% of the genome was determined to be methylated (Suzuki et al., 2013). The degree of methylation we found in oyster male gamete tissue falls within those previously described for other molluscs. For instance, recent research examining DNA methylation in multiple tissues of the mollusc Chlamys farreri found methylation levels in the genome to be around 21% using a methylation-sensitive amplification polymorphism approach (Sun et al., 2014). Studies of DNA methylation in the foot tissue of the gastropod mollusc Biomphalaria glabrata found approximately 2% of the CpG dinucleotides to be methylated (Fneich et al., 2013). Our findings, in agreement with previous studies in C. gigas, corroborates that overall genome methylation in C. gigas is at an intermediate level and suggests that DNA methylation does not significantly vary among tissue type.
DNA methylation is predominantly found in exons and introns in oyster male gametes (Figure 2). These findings are consistent with previous work characterizing DNA methylation in Crassostrea gigas gill tissue (Gavery and Roberts, 2013). In several animal and plant genomes, transcribed regions of genes, including intragenic regions, have higher levels of DNA methylation than neighboring regions (Suzuki et al., 2007; Zilberman et al., 2007). DNA methylation in insects, however, appears to be primarily confined to exons (Lyko et al., 2000). Recent work examining DNA methylation in C. intestinalis sperm found that methylated domains of the genome are primarily contained within transcription units and promoter regions, with intergenic regions completely unmethylated (Suzuki et al., 2013). Together these studies demonstrate that the distribution of DNA methylation in invertebrate genomes is diverse and that most invertebrate genomes exhibit interspersed regions of methylated and unmethylated DNA. Although we have a relatively comprehensive profile of where DNA methylation occurs across the oyster genome, there is no definitive evidence of a link between DNA methylation and function. However, based on RNA-Seq data there is likely an association between methylation status and gene expression in C. gigas.
Recently, Riviere et al. (2013) proposed that proximal promoter and first exon methylation in C. gigas larvae have similar functions than those in mammalian systems, where increased methylation in promoter regions corresponds to decreased gene expression. Specifically, Riviere et al. (2013) reported a negative correlation between DNA methylation and expression of some homeobox gene orthologs during early oyster development. Our results indicate the opposite- a positive correlation between DNA methylation and gene expression (Figure 3), in which genes with high expression levels had high methylation levels. This is similar to what has been reported for oyster gill tissue (Gavery and Roberts, 2013). Riviere et al. (2013) suggested that a lack of DNA methylation influences gene expression specifically by controlling the transcription level of homeobox orthologs in the proximal promoter and first exon. Similarly, the presence of promoter methylation has specifically been associated with transcriptional silencing among many organisms (Suzuki and Bird, 2008). Here we found a significant difference in promoter methylation in high versus low gene expression. It is likely that the relationship of DNA methylation and gene expression is complex and dependent on several factors (Gavery and Roberts, 2014). For example, it has been shown that methylation pattern is dependent on gene function (Roberts and Gavery, 2012) and the current study focused on the entire transcriptome where Riviere et al. (2013) specifically looked at homeobox orthologs. The differences we observed between DNA methylation and expression than was previously reported (Riviere et al., 2013) could also be attributable to differences in cell type or a differing role of methylation throughout C. gigas life history stages. It should also be noted both studies have limitations as they rely on accurate genome annotation, and, as recently reported (Elsik et al., 2014), this can significantly alter research findings.
Here we provide the first genome-wide characterization of DNA methylation in a bivalve mollusc that focused on a single-cell type. We found that DNA methylation levels in male gametes are similar to those previously reported in gill tissue (Gavery and Roberts, 2013). In examining the relationship between DNA methylation and transcript expression, we observed a pattern suggesting that promoter regions with a higher proportion of CpG methylation are associated with highly expressed genes. Further explorations into the roles of miRNA and histone modifications will help to elucidate our understanding of epigenetic regulatory functions in oysters.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bell, A. C., and Felsenfeld, G. (2000). Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485. doi: 10.1038/35013100
Berthelin, C., Kellner, K., and Mathieu, M. (2000). Storage metabolism in the Pacific oyster (Crassostrea gigas) in relation to summer mortalities and reproductive cycle (West Coast of France). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 125, 359–369. doi: 10.1016/S0305-0491(99)00187-X
Bird, A. (1980). DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 8, 1499–1504. doi: 10.1093/nar/8.7.1499
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. doi: 10.1101/gad.947102
Bird, A. P., Taggart, M. H., and Smith, B. A. (1979). Methylated and unmethylated DNA compartments in the sea urchin genome. Cell 17, 889–901. doi: 10.1016/0092-8674(79)90329-5
del Gaudio, R., Di Giamio, R., and Geraci, G. (1997). Genome methylation of the marine annelid worm Chaetopterus variopedatus: methylation of a CpG in an expressed H1 histone gene. FEBS Lett. 417, 48–52. doi: 10.1016/S0014-5793(97)01262-3
Dheilly, N. M., Lelong, C., Huvet, A., Kellner, K., Dubos, M., Riviere, G., et al. (2012). Gametogenesis in the pacific oyster Crassostrea gigas: a microarrays-based analysis identifies sex and stage specific genes. PLoS ONE 7:e36353. doi: 10.1371/journal.pone.0036353
Elsik, C. G., Worley, K. C., Bennett, A. K., Beye, M., Camara, F., Childers, C. P., et al. (2014). Finding the missing honey bee genes: lessons learned from a genome upgrade. BMC Genomics 15:86. doi: 10.1186/1471-2164-15-86
Ewing, B., and Green, P. (1998). Base-calling of automated sequencer traces using phred. II. error probabilities. Genome Res. 8, 186–194. doi: 10.1101/gr.8.3.175
Ewing, B., Hillier, L., Wendl, M. C., and Green, P. (1998). Base-calling of automated sequencer traces using phred. I. accuracy assessment. Genome Res. 8, 175–185. doi: 10.1101/gr.8.3.175
Fang, X., Li, L., Luo, R., Xu, F., Wang, X., Zhu, Y., et al. (2012). Genomic data from the Pacific oyster (Crassostrea gigas). GigaScience. doi: 10.5524/100030
Fneich, S., Dheilly, N., Adema, C., Rognon, A., Reichelt, M., Bulla, J., et al. (2013). 5-methyl-cytosine and 5-hydroxy-methyl-cytosine in the genome of Biomphalaria glabrata, a snail intermediate host of Schistosoma mansoni. Parasit. Vectors 6:167. doi: 10.1186/1756-3305-6-167
Franco, A., Heude-Berthelin, C., Goux, D., Sourdaine, P., and Mathieu, M. (2008). Fine structure of the early stages of spermatogenesis in the Pacific oyster, Crassostrea gigas (Mollusca, Bivalvia). Tissue Cell 40, 251–260. doi: 10.1016/j.tice.2007.12.006
Gavery, M. R., and Roberts, S. B. (2010). DNA methylation patterns provide insight into epigenetic regulation in the Pacific Oyster (Crassostrea gigas). BMC Genomics 11:483. doi: 10.1186/1471-2164-11-483
Gavery, M. R., and Roberts, S. B. (2013). Predominant intragenic methylation is associated with gene expression characteristics in a bivalve mollusc. PeerJ 1:e215. doi: 10.7717/peerj.215
Gavery, M. R., and Roberts, S. B. (2014). A context dependent role for DNA methylation in bivalves. Brief. Funct. Genomics 13, 1–6. doi: 10.1093/bfgp/elt054
Glude, J. B., and Chew, K. K. (1982). Shellfish Aquaculture in the Pacific Northwest. Alaska Sea Grant Report, 82-2. Anchorage: University of Alaska.
Gowher, H., Leismann, O., and Jeltsch, A. (2000). DNA of Drosophila melanogaster contains 5-methylcytosine. Eur. Mol. Biol. Organ. J. 19, 6918–6923. doi: 10.1093/emboj/19.24.6918
Hong, E. E., Okitsu, C. Y., Smith, A. D., and Hsieh, C. L. (2013). Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol. Cell. Biol. 33, 2683–2690. doi: 10.1128/MCB.00220-13
Howe, B., Cole, G., Souroush, E., Koutris, P., Key, A., Khoussainova, N., et al. (2011). Database-as-a-service for long-tail science. Sci. Stat. Database Manag. 6809, 480–489. doi: 10.1007/978-3-642-22351-8_31
Jiang, Q., Li, Q., Yu, H., and Kong, L. F. (2013). Genetic and epigenetic variation in mass selection populations of Pacific oyster Crassostrea gigas. Genes Genomics 35, 641–647. doi: 10.1007/s13258-013-0114-4
Lyko, F., Foret, S., Kucharski, R., Wolf, S., Falckenhayn, C., and Maleszka, R. (2010). The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS ONE 8:1000506. doi: 10.1371/journal.pbio.1000506
Lyko, F., Whittaker, A. J., Orr-Weaver, T. L., and Jaenisch, R. (2000). The putative Drosophila methyltransferase gene dDnmt2 is contained in a transposon-like element and is expressed specifically in ovaries. Mech. Dev. 95, 215–217. doi: 10.1016/S0925-4773(00)00325-7
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Olson, C., and Roberts, S. (2014). Crassostrea gigas male gonad transcriptome data comparison. Figshare. doi: 10.6084/m9.figshare.1004464
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rice, P., Longden, I., and Bleasby, A. (2000). EMBOSS: the european molecular biology open software suite. Trends Genetics 16, 276–272. doi: 10.1016/S0168-9525(00)02024-2
Riviere, G., Wu, G. C., Fellous, A., Goux, D., Sourdaine, P., and Favrel, P. (2013). DNA methylation is crucial for the early development in the oyster C. gigas. Mar. Biotechnol. 15, 739–753. doi: 10.1007/s10126-013-9523-2
Roberts, S. B., and Gavery, M. R. (2012). Is there a relationship between DNA methylation and phenotypic plasticity in invertebrates? Front. Physiol. 2:116. doi: 10.3389/fphys.2011.00116
Smit, A. F. A., Hubley, R., and Green, P. (1996–2010). RepeatMasker Open-3.0. Available online at http://www.repeatmasker.org
Sun, Y., Hou, R., Fu, X., Sun, C., Wang, S., Wang, C., et al. (2014). Genome-wide analysis of DNA methylation in five tissues of zhikong scallop, Chlamys farreri. PLoS ONE 9:e86232. doi: 10.1371/journal.pone.0086232
Suzuki, M. M., and Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genetics 9, 465–476. doi: 10.1038/nrg2341
Suzuki, M. M., Kerr, A. R., De Sousa, D., and Bird, A. (2007). CpG methylation is targeted to transcription units in an invertebrate genome. Genome Res. 17, 625–631. doi: 10.1101/gr.6163007
Suzuki, M. M., Yoshinari, A., Obara, M., Takuno, S., Shigenobu, S., Sasakura, Y., et al. (2013). Identical sets of methylated and nonmethylated genes in Ciona intestinalis sperm and muscle cells. Epigenetics Chromatin 6:38. doi: 10.1186/1756-8935-6-38
Xi, Y., and Li, W. (2009). BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10:232. doi: 10.1186/1471-2105-10-232
Zhang, G., Fang, X., Guo, X., Li, L., Luo, R., Xu, F., et al. (2012). The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54. doi: 10.1038/nature11413
Keywords: epigenetics, DNA methylation, oyster, bivalves, gene expression
Citation: Olson CE and Roberts SB (2014) Genome-wide profiling of DNA methylation and gene expression in Crassostrea gigas male gametes. Front. Physiol. 5:224. doi: 10.3389/fphys.2014.00224
Received: 10 February 2014; Accepted: 28 May 2014;
Published online: 17 June 2014.
Edited by:
Elzbieta M. Pyza, Jagiellonian University, PolandReviewed by:
Pierre Boudry, Ifremer, FranceGiovanna Benvenuto, Stazione Zoologica Anton Dohrn, Italy
Copyright © 2014 Olson and Roberts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven B. Roberts, School of Aquatic and Fishery Sciences, 1122 Boat Street, Box 355020, Seattle, WA 98105-5020, USA e-mail: sr320@u.washington.edu