 Viktória Jeney
Viktória Jeney György Balla
György Balla József Balla
József Balla- 1Department of Medicine, University of Debrecen, Debrecen, Hungary
- 2MTA-DE Vascular Biology, Thrombosis and Hemostasis Research Group, Hungarian Academy of Sciences, Debrecen, Hungary
- 3Department of Pediatrics, University of Debrecen, Debrecen, Hungary
For decades plaque neovascularization was considered as an innocent feature of advanced atherosclerotic lesions, but nowadays growing evidence suggest that this process triggers plaque progression and vulnerability. Neovascularization is induced mostly by hypoxia, but the involvement of oxidative stress is also established. Because of inappropriate angiogenesis, neovessels are leaky and prone to rupture, leading to the extravasation of red blood cells (RBCs) within the plaque. RBCs, in the highly oxidative environment of the atherosclerotic lesions, tend to lyse quickly. Both RBC membrane and the released hemoglobin (Hb) possess atherogenic activities. Cholesterol content of RBC membrane contributes to lipid deposition and lipid core expansion upon intraplaque hemorrhage. Cell-free Hb is prone to oxidation, and the oxidation products possess pro-oxidant and pro-inflammatory activities. Defense and adaptation mechanisms evolved to cope with the deleterious effects of cell free Hb and heme. These rely on plasma proteins haptoglobin (Hp) and hemopexin (Hx) with the ability to scavenge and eliminate free Hb and heme form the circulation. The protective strategy is completed with the cellular heme oxygenase-1/ferritin system that becomes activated when Hp and Hx fail to control free Hb and heme-mediated stress. These protective molecules have pharmacological potential in diverse pathologies including atherosclerosis.
Introduction
Complications of cardiovascular disease, and in particularly luminal thrombosis triggered by rupture of atherosclerotic lesions, are the leading cause of mortality and morbidity worldwide. Not all the plaques are prone to rupture, only the vulnerable ones, characterized by thin fibrous cap. Recently, plaque neovascularization and intraplaque hemorrhage (IPH) have been linked to plaque progression and vulnerability and these processes gained substantial interest (reviewed in Moreno et al., 2012).
In this review we briefly summarize what is known regarding the triggers of neovascularization and IPH. We overview the fate of red blood cells (RBCs) in the highly oxidative environment of the atherosclerotic plaque, and discuss the defense and adaptation mechanisms which have evolved to control the deleterious effects of cell free Hb.
Neovascularization in Atherosclerotic Lesions
Oxygen and nutrients are diffused from the vessel lumen into the intimal and medial cells of healthy vessels, while the outer layers of media and the adventitia are nurtured by the capillary network of vasa vasorum (Moreno et al., 2006, 2012). Neovascularization, that is the growth of capillary-like microvessels into the thickened media and intima, has long been considered as a prominent feature of late-stage atherosclerotic plaques (O'Brien et al., 1994). Nowadays growing evidence support that in fact neovascularization is present in early atherosclerotic lesions (Jeziorska and Woolley, 1999) particularly when the thickness of the tunica intima exceeds the maximum oxygen diffusion distance that is ~200–250 μm (Geiringer, 1951; Torres Filho et al., 1994; Moulton et al., 1999).
Hypoxia as a trigger of plaque neovascularization
Hypoxia, a condition when oxygen tension drop below its normal level in the particular tissue (20–100 mmHg), is a long-recognized stimulus for angiogenesis (Knighton et al., 1983). Using oxygen microelectrodes or specific hypoxia markers, hypoxia of the mid-region of the atherosclerotic plaques was demonstrated in various animal models (Jurrus and Weiss, 1977; Zemplenyi et al., 1989; Crawford and Blankenhorn, 1991; Bjornheden et al., 1999). In humans, the presence of hypoxic milieu in advanced atherosclerotic lesions of carotid arteries was also shown (Sluimer et al., 2008). As a consequence of hypoxia, switch from aerobic to anaerobic metabolism, characterized by glucose and ATP depletion and lactate accumulation, occurs in both human and experimental atheroma (Levin et al., 2003; Leppanen et al., 2006). Recently it has been shown that both sustained and intermittent hypoxia accelerates the progression of atherosclerosis in apolipoprotein E (apoE) deficient mice (Nakano et al., 2005; Jun et al., 2010).
Hypoxia-inducible factor-1 (HIF-1) pathway is the major mediator of the biological effects of hypoxia (Wang and Semenza, 1995). HIF-1 is active exclusively as a heterodimer of HIF-1α and HIF-1β subunits. While HIF-1β is stable, the level of HIF-1α is regulated by oxygen (Wang et al., 1995). Under normoxia, HIF-1α subunits are hydroxylated by the Fe2+-dependent prolyl hydroxylases (PHD) followed by ubiquitination and subsequent degradation by the proteasome (Maxwell et al., 1999; Ivan et al., 2001). In contrast, under hypoxia PHDs are inactive and HIF-1α subunits are no longer degraded. This allows the formation of the active HIF-1 heterodimer, which then translocate into the nucleus, binds to the hypoxic response elements and initiates transcription of target genes (Wenger et al., 2005). These genes are involved in the adaptation of the organism to hypoxic condition, such as vascular endothelial growth factor (VEGF) that has a pivotal role in angiogenesis (Forsythe et al., 1996).
Expression of HIF-1α is increased in deep and less-vascularized layers of human carotid and femoral endarterectomy specimens (Vink et al., 2007; Higashida et al., 2008). Increased HIF-1 alpha expression is associated with elevated level of VEGF suggesting that HIF-1 pathway is active and most probably play a role in neoangiogenesis in these hypoxic regions of the atherosclerotic plaques (Vink et al., 2007; Higashida et al., 2008; Gao et al., 2012).
Inflammation and ROS as triggers of plaque neovascularization
Although hypoxia is by far the most studied angiogenic factor, recent discoveries highlighted the role of reactive oxygen species (ROS) that are implicated in both physiological and pathological angiogenesis under normoxic conditions (reviewed in Kim and Byzova, 2014). ROS activates the HIF-1/VEGF pathway that serves as the major underlying mechanism of ROS-mediated angiogenesis. Additionally, recent discoveries highlighted the role of toll-like receptors (TLR) behind angiogenic activity of ROS. The activation of various TLR receptors (TLR2, TLR3, TLR4, TLR2/6) can lead to angiogenesis in both HIF-1/VEGF-dependent and HIF-1/VEGF-independent manners (Leibovich et al., 2002; Pollet et al., 2003; Grote et al., 2010; Paone et al., 2010; Spirig et al., 2010) (reviewed in Bordon, 2010). For example activation of TLR4 by lipopolysaccharide activates the HIF-1 pathway (Vink et al., 2007), whereas activation of TLR2 by its novel endogenous ligand, ω-(2-carboxyethyl) pyrrole, leads to an angiogenic response that is independent of VEGF (West et al., 2010).
Besides its direct angiogenic potential, ROS have been implicated in the generation of lipid oxidation products with proangiogenic activities, such as oxidized phospholipids that can be found in large amounts in atherosclerotic lesions (Bochkov et al., 2006; West et al., 2010; Hutter et al., 2013).
Physiological and pathological angiogenesis
Angiogenesis in general is fundamental for development and repair. It was proposed that physiologic angiogenesis can serve as a defense mechanism in atherosclerosis to compensate tissue hypoxia and restore homeostasis in the vessel wall (Moreno et al., 2006). Theoretically neovessels could provide channels for immune cells and bone marrow-derived progenitors to resolve inflammation and facilitate repair of the diseased vessel, respectively. It was also postulated that physiological angiogenesis contributes to the elimination of accumulated lipids from the intima (Moreno et al., 2006). Regardless of these potential beneficial effects, growing body of evidence suggest that plaque neovascularization correlates with the progression of atherosclerosis and neovessel density was found to be an independent risk factor for aortic plaque rupture (McCarthy et al., 1999; Moreno et al., 2004). Many studies revealed that inhibition of angiogenesis with different approaches reduces plaque growth (Moulton et al., 1999, 2003; Luttun et al., 2002; Petrovan et al., 2007; Drinane et al., 2009; Bot et al., 2011), whereas stimulation of angiogenesis with VEGF or nicotine results in elevated lesion progression in experimental atherosclerosis (Celletti et al., 2001; Heeschen et al., 2001). The observed disadvantageous effects of plaque neovascularization might be explained by pathological angiogenesis that proceeds in an uncontrolled manner, and results the formation of an abnormal neovessel structure.
Neovessles can originate from three sources. Sprouting of the adventitial vasa vasorum in response to angoigenic stimuli is the most widely accepted mechanism of neovessel formation. Besides vasa vasorum, luminal endothelial cells, or recruitment and differentiation of vascular progenitor cells inside the plaque can be involved in the formation of neovessels (reviewed in Galis and Lessner, 2009). Regardless of their origin, plaque neovessels differ both anatomically and in their response to different stimuli from the normal vessels (Ritman and Lerman, 2007). Neovessels are dysmorphic and characterized by discontinuous basement membrane and a relatively low number of tight junctions between endothelial cells (Heistad et al., 1981; Dunmore et al., 2007; Sluimer et al., 2009). Moreover these premature vessels are relatively poor in smooth muscle cells or pericytes (Kolodgie et al., 2003). Consequently, neovessels are leaky and unable to control intraluminal pressure therefore they are prone to rupture (Zhang et al., 1993; Sluimer et al., 2009).
Intraplaque Hemorrhage
Continuous leakage or rupture of immature neovessels leads to extravasation of RBCs within plaques which process is defined as IPH. IPH is present in about 40% of high-risk plaques (Kockx et al., 2003). Recently IPH has been linked to plaque progression and vulnerability and nowadays is considered as a critical event in triggering atherosclerosis-associated acute clinical symptoms (Michel et al., 2011). Different theories evolved about the molecular mechanisms via which IPH contribute to plaque progression.
RBC membrane-derived cholesterol as a trigger for lipid core expansion and inflammation
The casual relationship between elevated cholesterol level and atherosclerosis is known for more than 60 years. Early atherosclerotic lesions are characterized by subendothelial accumulation of cholesterol-laden macrophages called foam-cells. During plaque progression foam cells dye and release free cholesterol that deposits inside the plaque forming the necrotic core a characteristic feature of more advanced lesions (Lusis, 2000). For decades low-density lipoprotein (LDL) was considered as the main source of atherosclerotic plaque lipid content, and lowering circulating LDL-cholesterol level is still a major approach for anti-atherosclerotic therapies (Sahebkar and Watts, 2013).
Recently it has been shown that in human atherosclerotic lesions cholesterol crystals are co-localized with glyophorin A, a characteristic protein of RBC membrane, suggesting that cholesterol content of RBC membrane contributes to lipid deposition and lipid core expansion upon IPH (Kolodgie et al., 2003, 2007). In fact, RBC membrane is particularly abundant in cholesterol (Yeagle, 1985). RBCs are not able to synthetize lipids, but there is an active exchange between RBC membrane lipids and plasma lipoproteins. Therefore lipid composition of RBC membrane reflects plasma lipoprotein levels. For example it has been shown that familial hypercholesterolemia is associated with elevated RBC membrane-associated cholesterol (Koter et al., 2002) and that high-fat diet increase membrane lipid content of RBCs in experimental animal models (Bhandaru et al., 1982; Ivanov et al., 1991; Tziakas et al., 2013). Accordingly, lipid lowering strategies such as statin treatment and life-style changes have been shown to positively modulate RBC lipid composition which might contribute to the atheroprotective effects of these approaches (Tynan et al., 1995; Koter et al., 2002; Caspar-Bauguil et al., 2010; Tziakas et al., 2013).
The direct evidence that RBC contribute to lesion progression is provided by the experiment of Kolodgie et al. in which they injected packed RBCs directly into quiescent atherosclerotic lesions in rabbit aortas. RBC injection triggered the enlargement of necrotic core and formation of free cholesterol crystals along with excessive macrophage infiltration (Kolodgie et al., 2003).
Inflammation has a fundamental role in mediating all stages of atherosclerosis (Libby, 2002). Discoveries of the last 20 years made us to understand that besides pathogen-associated molecular patterns (PAMPs) several endogenous molecules, called danger- or damage-associated molecular patterns (DAMPs) can activate cellular receptors leading to downstream inflammation (Matzinger, 1994, 2002). Rajamaki et al. showed that cholesterol crystals serve as DAMPs and cause the activation of the NLRP3 [nucleotide-binding domain leucine-rich repeat containing (NLR) family, pyrin domain containing 3] inflammasome in macrophages (Rajamaki et al., 2010). Activation of NLRP3 inflammasome by cholesterol crystals leads to the activation of cytoplasmic caspase-1 that promotes maturation and secretion of the proinflammatory cytokine IL-1β (Rajamaki et al., 2010) and thus link altered cholesterol metabolism and inflammation in atherosclerotic lesions.
RBC lysis, Hb release and Hb oxidation upon IPH
While compartmentalized in RBCs oxidation of Hb is controlled by a highly effective antioxidant defense system including enzymatic (Cu/Zn superoxide dismutase, catalase, glutathione peroxidase, and peroxiredoxins) and non-enzymatic (glutathione) scavengers (Siems et al., 2000; Jeney et al., 2013). Upon IPH RBCs enter to the highly oxidative milieu of atherosclerotic lesion, the “death zone” that contains cytotoxic products of lipid peroxidation such as lipid hydroperoxides, aldehydes, and carbonyls (Li et al., 2006). The high occurrence of IPH prompted us to study the interaction of RBC and atheroma lipids. We revealed that these reactive lipids, extracted from human atheroma trigger the lysis of RBCs (Figure 1) (Nagy et al., 2010). Oxidized LDL and cumene hydroperoxide mimic the effect of plaque lipid extract on RBC lysis (Nagy et al., 2010). Moreover, enzymatic conversion of lipid-hydroperoxides to lipid-alcohols by glutathione/glutathione peroxidase causes significant inhibition of RBC lysis triggered by oxLDL and plaque lipids highlighting the critical role of lipid-hydroperoxides in RBC lysis (Nagy et al., 2010).
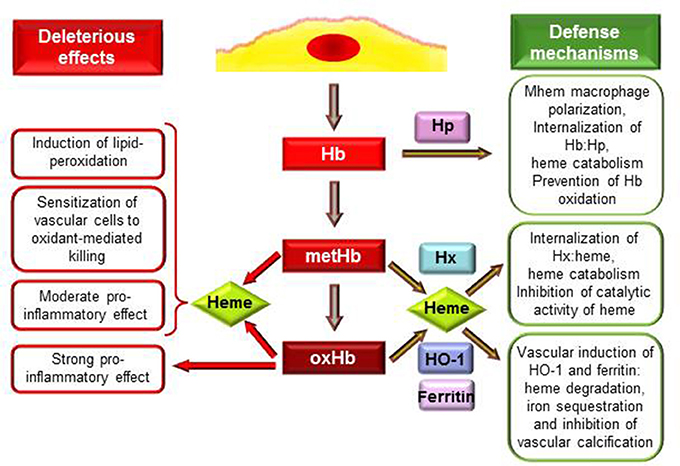
Figure 1. Deleterious effects and defense mechanisms triggered by extracellular hemoglobin and its oxidation products upon intraplaque hemorrhage. Interactions between RBCs and plaque lipids lead to lysis of erythrocytes and release of Hb. Extracellular Hb by reacting with plaque lipids undergo rapid oxidation to metHb and oxHb. MetHb and oxHb release their heme moieties that possess pro-oxidant and pro-inflammatory properties. OxHb is a strong pro-inflammatory agonist which effect is independent of heme release. As an atheroprotective mechanism in response to Hb stress Mhem macrophages polarization occurs. Hp binding Hb but not oxHb attenuates Hb oxidation and their uptake by Mhem macrophages results in heme catabolism. Heme liberated from MetHb and oxHb can be captured by hemopexin (Hx). Hx inhibits catalytic activity of heme, and after internalization of Hx:heme complex heme is catabolized. Induction of heme oxygenase-1 (HO-1) and ferritin in vascular cells in response to such and insult (Hb, oxHb, heme, lipid oxidation) provides inhibition of oxidant damage and inflammation. HO-1 degrades heme into biliverdin, CO and iron which is sequestered by ferritin. Products of HO-1 mediated heme degradation: CO and biliverdin—that is readily converted to bilirubin—possess different antioxidant and anti-inflammatory properties. Additionally, ferritin inhibits vascular calcification.
Hb once outside the protective environment of RBC is prone to oxidation (Figure 1). Auto-oxidation of Hb occurs resulting in metHb generation meanwhile superoxide anions are formed (Table 1, equation 1). Peroxides, such as H2O2 can trigger a two-electron oxidation of Hb leading to the formation of ferryl (Fe4+ = O−2) Hb (Table 1, equation 2), whereas the reaction of metHb with H2O2 yields ferrylHb radical (Hb·+(Fe4+ = O−2)) in which the unpaired electron is associated with the globin or the porphyrin ring (Table 1, equation 3) (Harel and Kanner, 1988; Patel et al., 1996; Alayash et al., 2001; Jia et al., 2007).
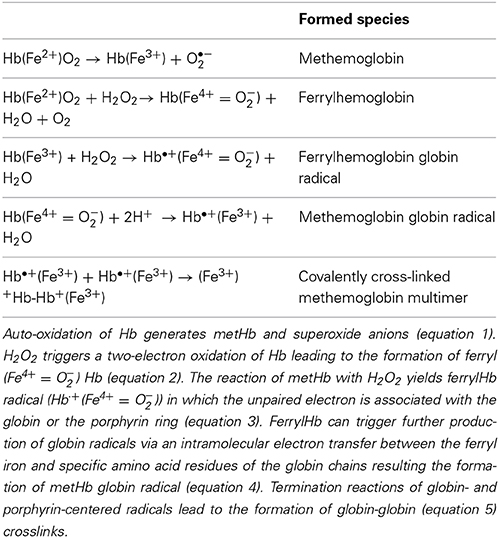
Table 1. Routes of hemoglobin oxidation.
The generated high-valence iron compounds are highly reactive intermediates that can decay by several routes (Reeder et al., 2008). FerrylHb can trigger further production of globin radicals via an intramolecular electron transfer between the ferryl iron and specific amino acid residues such as αTyr-24, αTyr-42, αHis-20, βTyr-35, βTyr-130, and βCys-93 of the globin chains resulting the formation of metHb globin radical (Table 1, equation 4) (Ramirez et al., 2003; Deterding et al., 2004; Jeney et al., 2013). Termination reactions of globin- and porphyrin-centered radicals lead to the formation of globin-globin (Table 1, equation 5) or porphyrin-globin crosslinks. The common feature of these structurally heterogeneous molecules is the modification of the globin chain. The nomenclature of these molecules is not concise in these days. Nevertheless, along this review in order to distinguish from metHb and ferrylHb—in which only the oxidation state of heme iron is altered but no globin modification is present—we will refer to those globin-modified molecules as oxidatively modified Hb (oxHb).
Studying the interaction of Hb and atheroma lipids, we observed a severe oxidation of Hb leading to the generation of metHb and oxHb (Figure 1). Moreover, we revealed significant accumulation of metHb and oxHb within human complicated atherosclerotic lesions—covalently cross-linked globin-globin multimers, and dityrosine formation occurs upon IPH—suggesting that the above-mentioned reactions take place in such lesions (Nagy et al., 2010; Jeney et al., 2013). We suggested that oxidation of Hb in the atherosclerotic plaque might be triggered by reactive lipid mediators (Figure 1). Besides atheroma lipids oxLDL was also shown to cause oxidation of cell-free Hb, producing metHb as well as ferrylHb and oxHb (Tynan et al., 1995; Nagy et al., 2005; Potor et al., 2013). Oxidation of Hb provoked by reactive lipid mediators can be inhibited by the heme scavenging Hx and by the elimination of lipid hydroperoxides, suggesting that interactions between the heme moiety and the hydroperoxide group drive the oxidation (Jeney et al., 2013).
Extracellular Hb, oxidized Hb species and heme as triggers of lipid peroxidation and endothelial damage
Oxidative modification of LDL and endothelial damage are key elements of atherogenesis. More than 20 years ago Balla et al. showed that heme, the prosthetic group of Hb, is a very efficient trigger of LDL oxidation in vitro and suggested that it might be a physiological mediator of LDL oxidation in vivo (Balla et al., 1991a). We also showed that heme greatly amplifies oxidant-mediated endothelial damage (Balla et al., 1990, 1991b). Several lines of evidence support, that these heme-triggered events have etiopathogenic roles in diverse vascular pathologies, including atherosclerosis (Balla et al., 2007). Deficiency of the heme-catabolizing enzyme, heme oxygenase-1 (HO-1), in humans was found to be associated with elevated plasma heme levels, extensive LDL oxidation, severe endothelial damage and accelerated atherosclerosis (Yachie et al., 1999; Jeney et al., 2002; Kawashima et al., 2002; Radhakrishnan et al., 2011). The role of HO-1 in atherogenesis was also examined in animal models. It has been shown that overexpression of HO-1 in apoE deficient mice inhibit lesion formation (Juan et al., 2001), whereas HO-1 deficiency is associated with accelerated atherosclerosis in apoE deficient mice (Yet et al., 2003). In heme-mediated LDL oxidation a unique oxidation product, 5-hydroxy-2-amino valeric acid (HAVA) is formed (Julius and Pietzsch, 2005). HAVA is a hallmark of heme-mediated LDL oxidation, because other known triggers of LDL oxidation, such as HOCl, H2O2 alone or in combination with Cu2+ or Fe2+ induce no or minor HAVA formation (Julius and Pietzsch, 2005). HAVA levels in LDL was found to be elevated in patients with impaired glucose tolerance and with diabetes mellitus suggesting that heme-mediated LDL oxidation occurs in these patients (Julius and Pietzsch, 2005).
Not only free heme, but metHb and oxHb trigger LDL oxidation and sensitize endothelial cells to oxidant-mediated killing (Balla et al., 1993; Paone et al., 2010; Potor et al., 2013). These Hb species readily release heme moiety (Bunn and Jandl, 1968), which step is of crucial importance in mediating their effect (Figure 1). This notion is supported by the observation that restriction of heme release using different approaches, such as binding of Hb to Hp or strengthening the globin-heme binding, inhibits the deleterious effects of these Hb species (Balla et al., 1993; Jeney et al., 2002; Nagy et al., 2010; Potor et al., 2013).
Extracellular Hb, oxidized Hb species and heme as modulators of inflammation
Inflammation is an important etiopathogenic component of atherogenesis, and several evidence suggest that cell free Hb, oxidized Hb species and heme possess specific immunomodulatory activities (Figure 1). Hemolytic or hemorrhagic episodes are often associated with inflammation even in the absence of infectious agents (Arruda et al., 2005; Gram et al., 2013). Vascular endothelium, that provides a barrier between blood and tissue has a critical role in the inflammatory response mainly by inducing the leukocyte adhesion cascade to facilitate transmigration of inflammatory cells to the inflamed tissue. Endothelial cells when exposed to heme or oxHb up-regulate the expression of adhesion molecules: intracellular adhesion molecule-1 (Icam-1), vascular cell adhesion molecule-1 (Vcam-1) and E selectin (Wagener et al., 1997; Silva et al., 2009). Comparing to heme, oxHb is a more robust inducer of this inflammatory response, as one-tenth of oxHb provoke the same response as heme. Also, the mechanism of oxHb-triggered inflammatory response seems to be different from the one that heme initiates. OxHb mediated inflammatory response is independent of heme release, which notion is supported by the observation that metHb that can release heme moiety similarly to oxHb has no pro-inflammatory properties (Figure 1) (Silva et al., 2009). Additionally, endothelial cells exposed to oxHb show rearrangement of the actin cytoskeleton leading to disruption of the endothelial cell monolayer, intercellular gap formation and increased permeability of the monolayer, which did not occur upon heme exposure (Silva et al., 2009). Both heme and oxHb have been shown to induce inflammation in mice, with the notion that oxHb seems to be a 10-times more potent agonist than heme (Wagener et al., 2001; Silva et al., 2009). Both heme and oxHb are chemotactic for neutrophils when injected into the peritoneal cavity of mice, but again, oxHb is a much stronger chemotactic agent compared to heme (Porto et al., 2007; Silva et al., 2009). Importantly, heme and oxHb-mediated inflammatory responses do not share a common signaling pathway, as heme mediated response has been shown to be TLR4-depenedent (Figueiredo et al., 2007; Belcher et al., 2014), whereas oxHb acts on a TLR4-independent manner (Silva et al., 2009).
Macrophages are considered to be the major immune cell type involved in atherogenesis. These macrophages originate from blood monocytes which are attracted to the subendothelial space. The plaque microenvironment dictates the differentiation of these cells functionally diverse phenotypes. Besides the most extensively studied M1 and M2 subtypes, several other macrophage populations have been identified in atherosclerotic plaques (reviewed in Leitinger and Schulman, 2013; Vinchi et al., 2014). Boyle et al. recently identified a novel hemorrhage-associated macrophage phenotype (Mhem, HA-mac) in human hemorrhaged atherosclerotic plaques (Boyle et al., 2009). It has been demonstrated that polarization of these Mhem macrophages is driven by Hb bound to its endogenous scavenger Hp (Boyle et al., 2009; Finn et al., 2012). The major function of Mhem macrophages is the safe elimination of cell free Hb from the plaque, therefore they highly express CD163, the receptor for uptake of Hb:Hp complex and HO-1, the rate limiting enzyme of heme catabolism (Boyle et al., 2009, 2012). Moreover, Mhem differentiation prevents foam cell formation via decreased lipid uptake and increased cholesterol efflux (Finn et al., 2012). All of these properties can contribute to the atheroprotective nature of these Mhem macrophages (Figure 1).
Defense and Adaptation Mechanisms
Extracellular Hb and heme are harmful therefore efficient mechanisms have evolved to control their deleterious effects (Figure 1). The plasma acute phase proteins Hp and Hx are in the first line of defense upon intravascular hemolysis. The protective strategy is completed with the HO-1/ferritin system that could serve as the last line of defense and become activated when the Hp and Hx cannot control free Hb and heme mediated stress (Figure 1). The pharmacological potential of these molecules emerged recently to neutralize the adverse effects of Hb and heme in diverse pathologies (Durante, 2010; Schaer et al., 2013a).
Control of free Hb by Hp
Hp is present in plasma in high amounts (0.41–1.65 mg/ml) with the exclusive recognized function of capturing and chaperoning cell free Hb to macrophages for degradation (Figure 1) (reviewed in Alayash, 2011). Hp binding accelerates the elimination of circulating Hb through the CD163 macrophage scavenger receptor-mediated endocytosis (Kristiansen et al., 2001). The Hp:Hb complex is highly stable and protects Hb from H2O2-induced oxidation (Miller et al., 1997; Buehler et al., 2009; Pimenova et al., 2010; Banerjee et al., 2012; Potor et al., 2013; Schaer et al., 2013b). Recent resolution of the crystal structure of the porcine Hp:Hb complex revealed that Hb residues known to be prone to oxidative modifications are buried in the Hp:Hb interface thereby explaining the protective effect of Hp against H2O2-induced oxidation (Andersen et al., 2012). Hp binding not just provide structural stabilization of Hb but also inhibits heme transfer from Hb toward LDL or vascular endothelial cells (Balla et al., 1993; Nagy et al., 2010; Schaer et al., 2013b).
In humans there are two alleles for the Hp gene resulting 3 different genotypes Hp1-1, Hp2-1 and Hp2-2 (reviewed in Goldenstein et al., 2012) accompanied by structurally different proteins. This molecular heterogeneity of Hp was found to be associated with cardiovascular diseases. Many clinical observations revealed that the Hp2-2 genotype is a risk factor for cardiovascular complications in diverse patient populations (reviewed in Costacou and Levy, 2012), however the attempt to understand the underlying mechanisms lead to controversial results. It has been demonstrated that Hp1-1 is more efficient in blocking heme transfer from Hb to LDL or endothelial cells than Hp2-2 (Melamed-Frank et al., 2001; Bamm et al., 2004) but recently it was reported that the two proteins are equally efficient (Lipiski et al., 2013). Furthermore, Hp2-2:Hb complex was found to be associated with higher functional affinity for the macrophage scavenger receptor CD163 than the Hp1-1:Hb complex (Kristiansen et al., 2001), though other group observed the opposite (Asleh et al., 2003).
Nevertheless, the protective effect and the therapeutic potential of Hp in various hemolytic models has been reported (reviewed in Schaer et al., 2013a), but whether Hb scavenging by Hp acts in an atheroprotective manner remained to be elucidated.
Control of free hem by Hx
Upon excessive hemolysis Hp is consumed, causing accumulation and oxidation of cell-free Hb that eventually lead to the release of heme. Hx is an acute-phase plasma protein that binds heme with the highest affinity of any known heme-binding proteins (Hrkal et al., 1974). Hx-heme complexes are internalized via the scavenger receptor LDL receptor-related protein 1/CD91 (Hvidberg et al., 2005) mainly by hepatocytes and macrophages (Figure 1) (Herz and Strickland, 2001). Following endocytosis heme is degraded by HO-1 and iron is stored by ferritin (Alam and Smith, 1989). Although it is well-established that Hx binding inhibits the catalytic activity of heme in oxidative reactions including LDL oxidation (Figure 1) (Gutteridge and Smith, 1988; Vincent et al., 1988; Balla et al., 1991a), its role in atherogenesis remained to be elucidated.
Larsen et al. showed that Hx can be used therapeutically to attenuate heme-mediated tissue damage upon severe sepsis in mice (Larsen et al., 2010). Following this study the protective nature of Hx has been shown in different hemolytic mice models, in which administration of Hx improved cardiac function of those mice (Vinchi et al., 2013). These studies raise the possibility of Hx-based therapeutics in the treatment of diverse pathologies in which heme-mediated tissue damage play an etiopathogenetic role.
The HO-1 ferritin system
Cells exposed to free heme or heme-releasing Hb species, i.e., metHb and oxHb up-regulate HO-1 and ferritin (Figure 1) (Balla et al., 1992a,b; Nath et al., 1992; Potor et al., 2013). These proteins provide cellular and tissue protection in diverse pathologies. The mechanism of cytoprotection by HO-1 was recently reviewed (Gozzelino et al., 2010), and it relies on the ability of HO-1 to degrade heme into biliverdin—that is promptly converted into biliverdin—carbon-monoxide (CO) and iron (Tenhunen et al., 1968). The subsequent upregulation of ferritin is essential to obtain the protective effect, as it can store the released iron in a catalitically inactive form (Balla et al., 1992b). Additionally, the side-products of heme degradation—bilirubin and CO—exert various antioxidant and anti-inflammatory properties (Gozzelino et al., 2010).
Many lines of evidences support the atheroprotective nature of HO-1 in humans and in experimental atherosclerosis (reviewed in Vinchi et al., 2014). Recently it has been shown that HO-1 not simply slow down, but reverse plaque progression from a vulnerable plaque to a more stable phenotype (Cheng et al., 2009).
Autopsy examinations of human atherosclerotic lesions revealed that HO-1 expression correlates with plaque instability and the level of pro-inflammatory markers. This might be explained by the stress responsive nature of HO-1. Initial induction of HO-1 expression may act as a compensatory atheroprotective mechanism, whereas at later stages HO-1 expression reflects oxidative stress, inflammation and tissue damage.
Upregulation of HO-1 in atherosclerotic lesions generally appears to coincide with ferritin induction in vitro (Juckett et al., 1995; Pang et al., 1996). The increased expression of ferritin also reflects cellular response to heme or heme-iron generated lipid peroxidation products (Agarwal et al., 1996; Hill-Kapturczak et al., 2003). Such induction correlates with the oxidative insult imposed by reactive oxygen and iron. The mechanism by which ferritin provides cytoprotection relies on the ferroxidase activity of H-ferritin subunit (Balla et al., 1992b). Beyond cytoprotection, ferritin serves as a regulator for cell proliferation, inflammation and vascular calcification (Figure 1) (reviewed in Crawford and Blankenhorn, 1991; Zarjou et al., 2009).
Impaired defense mechanisms in the atherosclerotic lesion
The elimination of cell-free Hb and heme by Hp and Hx is well characterized in hemolytic pathologies where Hb is released into the circulation. But our knowledge is quite limited when Hb is released from RBC outside of the circulatory system. Hp and Hx are plasma proteins and their penetration into the deeper compartments of atherosclerosic plaque might be limited. This could be particularly true for Hp2-2 that is a large molecule thereby its restricted diffusion may explain the apparent association of the Hp2-2 genotype with more severe symptoms in different pathologies.
Following IPH oxidation of Hb occurs, leading to the formation of structurally altered (e.g., covalently cross-linked) Hb species. It was hypothesized that these structural changes might be associated with the impairment of the endogenous scavenging pathways. Recent studies have revealed that elimination of oxidized Hb species via both high-affinity and low-affinity pathways are severely compromised (Schaer et al., 2006; Vallelian et al., 2008).
Impaired defense mechanisms following IPH might limit the clearance of extracellular Hb and heme from the atherosclerotic plaque thereby this could be a new etiopathogenic factor to address in details in the future.
Control of free heme in extravascular sites by α-1 microglobulin (A1M)
A1M is a small glycoprotein that is found ubiquitously in all tissues. Recently it has been demonstrated that A1M can bind small molecules in its hydrophobic pocket, scavenge free radicals and possesses reductase activity. Based on these features A1M plays a crucial role in tissue housekeeping (reviewed in Akerstrom and Gram, 2014). Importantly, heme is a major ligand for A1M that can bind heme with high affinity and degrade it (Allhorn et al., 2002). The protective effect of A1M against Hb/heme-mediated oxidative stress has been shown in different in vitro models (Olsson et al., 2008, 2011). Moreover, recently it was demonstrated that A1M infusion attenuates Hb-induced kidney damage in rats (Sverrisson et al., 2014). Based on these properties, we can assume that A1M plays a beneficial role upon IPH by neutralizing and eliminating radicals, oxidants and free heme, but this hypothesis and the potential therapeutic potential of A1M needs to be tested in the future.
Conclusions
In the last decade, our understanding of atherosclerotic plaque progression and vulnerability underwent a fundamental revision, and neovascularization accompanied by IPH shifted from being an innocent bystander to a pathogenic event that plays a critical role in atherogenesis.
Extravasation of RBCs into the plaque is of crucial importance in triggering IPH-associated reactions. RBC membrane lipids contribute to plaque expansion, whereas cell-free Hb and its oxidation products are strong pro-oxidants and pro-inflammatory agonists targeting cell types with major roles in atherogenesis, such as vascular endothelial cells and macrophages. Systemic and cellular defense strategies to cope with extracellular Hb and its oxidation products might not be efficient or sufficient enough to control the deleterious effects of these molecules deep inside the atherosclerotic plaque, the “death zone.”
Comprehensive understanding the role of neovascularization, IPH and Hb release and oxidation on atherogenesis may lead to the development of novel therapeutics intended to interrupt these pathological events.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The research group is supported by the Hungarian Academy of Sciences (11003). This work was supported by Hungarian Government grants, OTKA-K83478 and K-112333 (József Balla), OTKA- PD83435 (Viktória Jeney), European Reintegration Grant FP7-PEOPLE-2010-268332 (Viktória Jeney), and by the TÁMOP-4.2.4.A/2-11/1-2012-0001 projects. The project is co-financed by the European Union and the European Social Fund.
References
Agarwal, A., Balla, J., Balla, G., Croatt, A. J., Vercellotti, G. M., and Nath, K. A. (1996). Renal tubular epithelial cells mimic endothelial cells upon exposure to oxidized LDL. Am. J. Physiol. 271, F814–F823.
Akerstrom, B., and Gram, M. (2014). A1M, an extravascular tissue cleaning and housekeeping protein. Free Radic. Biol. Med. 74C, 274–282. doi: 10.1016/j.freeradbiomed.2014.06.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alam, J., and Smith, A. (1989). Receptor-mediated transport of heme by hemopexin regulates gene expression in mammalian cells. J. Biol. Chem. 264, 17637–17640.
Alayash, A. I. (2011). Haptoglobin: Old protein with new functions. Clin. Chim. Acta 412, 493–498. doi: 10.1016/j.cca.2010.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alayash, A. I., Patel, R. P., and Cashon, R. E. (2001). Redox reactions of hemoglobin and myoglobin: biological and toxicological implications. Antioxid. Redox Signal. 3, 313–327. doi: 10.1089/152308601300185250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Allhorn, M., Berggard, T., Nordberg, J., Olsson, M. L., and Akerstrom, B. (2002). Processing of the lipocalin alpha(1)-microglobulin by hemoglobin induces heme-binding and heme-degradation properties. Blood 99, 1894–1901. doi: 10.1182/blood.V99.6.1894
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andersen, C. B., Torvund-Jensen, M., Nielsen, M. J., de Oliveira, C. L., Hersleth, H. P., Andersen, N. H., et al. (2012). Structure of the haptoglobin-haemoglobin complex. Nature 489, 456–459. doi: 10.1038/nature11369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arruda, M. A., Graca-Souza, A. V., and Barja-Fidalgo, C. (2005). Heme and innate immunity: new insights for an old molecule. Mem. Inst. Oswaldo Cruz 100, 799–803. doi: 10.1590/S0074-02762005000700022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Asleh, R., Marsh, S., Shilkrut, M., Binah, O., Guetta, J., Lejbkowicz, F., et al. (2003). Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ. Res. 92, 1193–1200. doi: 10.1161/01.RES.0000076889.23082.F1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Balla, G., Jacob, H. S., Balla, J., Rosenberg, M., Nath, K., Apple, F., et al. (1992b). Ferritin: a cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 267, 18148–18153.
Balla, G., Jacob, H. S., Eaton, J. W., Belcher, J. D., and Vercellotti, G. M. (1991a). Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. 11, 1700–1711.
Balla, G., Vercellotti, G., Eaton, J. W., and Jacob, H. S. (1990). Heme uptake by endothelium synergizes polymorphonuclear granulocyte-mediated damage. Trans. Assoc. Am. Physicians 103, 174–179.
Balla, G., Vercellotti, G. M., Muller-Eberhard, U., Eaton, J., and Jacob, H. S. (1991b). Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab. Invest. 64, 648–655.
Balla, J., Jacob, H. S., Balla, G., Nath, K., Eaton, J. W., and Vercellotti, G. M. (1993). Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. U.S.A. 90, 9285–9289. doi: 10.1073/pnas.90.20.9285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Balla, J., Jacob, H. S., Balla, G., Nath, K., and Vercellotti, G. M. (1992a). Endothelial cell heme oxygenase and ferritin induction by heme proteins: a possible mechanism limiting shock damage. Trans. Assoc. Am. Physicians 105, 1–6.
Balla, J., Vercellotti, G. M., Jeney, V., Yachie, A., Varga, Z., Jacob, H. S., et al. (2007). Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid. Redox Signal. 9, 2119–2137. doi: 10.1089/ars.2007.1787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bamm, V. V., Tsemakhovich, V. A., Shaklai, M., and Shaklai, N. (2004). Haptoglobin phenotypes differ in their ability to inhibit heme transfer from hemoglobin to LDL. Biochemistry 43, 3899–3906. doi: 10.1021/bi0362626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Banerjee, S., Jia, Y., Siburt, C. J., Abraham, B., Wood, F., Bonaventura, C., et al. (2012). Haptoglobin alters oxygenation and oxidation of hemoglobin and decreases propagation of peroxide-induced oxidative reactions. Free Radic. Biol. Med. 53, 1317–1326. doi: 10.1016/j.freeradbiomed.2012.07.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belcher, J. D., Chen, C., Nguyen, J., Milbauer, L., Abdulla, F., Alayash, A. I., et al. (2014). Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123, 377–390. doi: 10.1182/blood-2013-04-495887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhandaru, R., Srinivasan, S. R., Radhakrisnamurthy, B., and Berenson, G. S. (1982). Effects of diabetes and high fat-high cholesterol diet on plasma lipid levels and on erythrocyte membrane composition. Atherosclerosis 42, 263–272. doi: 10.1016/0021-9150(82)90156-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bjornheden, T., Levin, M., Evaldsson, M., and Wiklund, O. (1999). Evidence of hypoxic areas within the arterial wall in vivo. Arterioscler. Thromb. Vasc. Biol. 19, 870–876. doi: 10.1161/01.ATV.19.4.870
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bochkov, V. N., Philippova, M., Oskolkova, O., Kadl, A., Furnkranz, A., Karabeg, E., et al. (2006). Oxidized phospholipids stimulate angiogenesis via autocrine mechanisms, implicating a novel role for lipid oxidation in the evolution of atherosclerotic lesions. Circ. Res. 99, 900–908. doi: 10.1161/01.RES.0000245485.04489.ee
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bordon, Y. (2010). A new vein of TLR biology. Nat. Rev. Immunol. 10:748. doi: 10.1038/nri2875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bot, I., Jukema, J. W., Lankhuizen, I. M., van Berkel, T. J., and Biessen, E. A. (2011). Atorvastatin inhibits plaque development and adventitial neovascularization in ApoE deficient mice independent of plasma cholesterol levels. Atherosclerosis 214, 295–300. doi: 10.1016/j.atherosclerosis.2010.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boyle, J. J., Harrington, H. A., Piper, E., Elderfield, K., Stark, J., Landis, R. C., et al. (2009). Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 174, 1097–1108. doi: 10.2353/ajpath.2009.080431
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boyle, J. J., Johns, M., Kampfer, T., Nguyen, A. T., Game, L., Schaer, D. J., et al. (2012). Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ. Res. 110, 20–33. doi: 10.1161/CIRCRESAHA.111.247577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buehler, P. W., Abraham, B., Vallelian, F., Linnemayr, C., Pereira, C. P., Cipollo, J. F., et al. (2009). Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood 113, 2578–2586. doi: 10.1182/blood-2008-08-174466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bunn, H. F., and Jandl, J. H. (1968). Exchange of heme among hemoglobins and between hemoglobin and albumin. J. Biol. Chem. 243, 465–475.
Caspar-Bauguil, S., Garcia, J., Galinier, A., Periquet, B., Ferrieres, J., Allenbach, S., et al. (2010). Positive impact of long-term lifestyle change on erythrocyte fatty acid profile after acute coronary syndromes. Arch. Cardiovasc. Dis. 103, 106–114. doi: 10.1016/j.acvd.2009.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Celletti, F. L., Waugh, J. M., Amabile, P. G., Brendolan, A., Hilfiker, P. R., and Dake, M. D. (2001). Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat. Med. 7, 425–429. doi: 10.1038/86490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheng, C., Noordeloos, A. M., Jeney, V., Soares, M. P., Moll, F., Pasterkamp, G., et al. (2009). Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation 119, 3017–3027. doi: 10.1161/CIRCULATIONAHA.108.808618
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Costacou, T., and Levy, A. P. (2012). Haptoglobin genotype and its role in diabetic cardiovascular disease. J. Cardiovasc. Transl. Res. 5, 423–435. doi: 10.1007/s12265-012-9361-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crawford, D. W., and Blankenhorn, D. H. (1991). Arterial wall oxygenation, oxyradicals, and atherosclerosis. Atherosclerosis 89, 97–108. doi: 10.1016/0021-9150(91)90049-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deterding, L. J., Ramirez, D. C., Dubin, J. R., Mason, R. P., and Tomer, K. B. (2004). Identification of free radicals on hemoglobin from its self-peroxidation using mass spectrometry and immuno-spin trapping: observation of a histidinyl radical. J. Biol. Chem. 279, 11600–11607. doi: 10.1074/jbc.M310704200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drinane, M., Mollmark, J., Zagorchev, L., Moodie, K., Sun, B., Hall, A., et al. (2009). The antiangiogenic activity of rPAI-1(23) inhibits vasa vasorum and growth of atherosclerotic plaque. Circ. Res. 104, 337–345. doi: 10.1161/CIRCRESAHA.108.184622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dunmore, B. J., McCarthy, M. J., Naylor, A. R., and Brindle, N. P. (2007). Carotid plaque instability and ischemic symptoms are linked to immaturity of microvessels within plaques. J. Vasc. Surg. 45, 155–159. doi: 10.1016/j.jvs.2006.08.072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Durante, W. (2010). Targeting heme oxygenase-1 in vascular disease. Curr. Drug Targets 11, 1504–1516. doi: 10.2174/1389450111009011504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Figueiredo, R. T., Fernandez, P. L., Mourao-Sa, D. S., Porto, B. N., Dutra, F. F., Alves, L. S., et al. (2007). Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 282, 20221–20229. doi: 10.1074/jbc.M610737200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Finn, A. V., Nakano, M., Polavarapu, R., Karmali, V., Saeed, O., Zhao, X., et al. (2012). Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 59, 166–177. doi: 10.1016/j.jacc.2011.10.852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Forsythe, J. A., Jiang, B. H., Iyer, N. V., Agani, F., Leung, S. W., Koos, R. D., et al. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613.
Galis, Z. S., and Lessner, S. M. (2009). Will the real plaque vasculature please stand up? Why we need to distinguish the vasa plaquorum from the vasa vasorum. Trends Cardiovasc. Med. 19, 87–94. doi: 10.1016/j.tcm.2009.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, L., Chen, Q., Zhou, X., and Fan, L. (2012). The role of hypoxia-inducible factor 1 in atherosclerosis. J. Clin. Pathol. 65, 872–876. doi: 10.1136/jclinpath-2012-200828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geiringer, E. (1951). Intimal vascularization and atherosclerosis. J. Pathol. Bacteriol. 63, 201–211. doi: 10.1002/path.1700630204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goldenstein, H., Levy, N. S., and Levy, A. P. (2012). Haptoglobin genotype and its role in determining heme-iron mediated vascular disease. Pharmacol. Res. 66, 1–6. doi: 10.1016/j.phrs.2012.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gozzelino, R., Jeney, V., and Soares, M. P. (2010). Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 50, 323–354. doi: 10.1146/annurev.pharmtox.010909.105600
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gram, M., Sveinsdottir, S., Ruscher, K., Hansson, S. R., Cinthio, M., Akerstrom, B., et al. (2013). Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J. Neuroinflammation 10:100. doi: 10.1186/1742-2094-10-100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grote, K., Schuett, H., Salguero, G., Grothusen, C., Jagielska, J., Drexler, H., et al. (2010). Toll-like receptor 2/6 stimulation promotes angiogenesis via GM-CSF as a potential strategy for immune defense and tissue regeneration. Blood 115, 2543–2552. doi: 10.1182/blood-2009-05-224402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gutteridge, J. M., and Smith, A. (1988). Antioxidant protection by haemopexin of haem-stimulated lipid peroxidation. Biochem. J. 256, 861–865.
Harel, S., and Kanner, J. (1988). The generation of ferryl or hydroxyl radicals during interaction of haemproteins with hydrogen peroxide. Free Radic. Res. Commun. 5, 21–33. doi: 10.3109/10715768809068555
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heeschen, C., Jang, J. J., Weis, M., Pathak, A., Kaji, S., Hu, R. S., et al. (2001). Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat. Med. 7, 833–839. doi: 10.1038/89961
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heistad, D. D., Armstrong, M. L., and Marcus, M. L. (1981). Hyperemia of the aortic wall in atherosclerotic monkeys. Circ. Res. 48, 669–675. doi: 10.1161/01.RES.48.5.669
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herz, J., and Strickland, D. K. (2001). LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779–784. doi: 10.1172/JCI200113992
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Higashida, T., Kanno, H., Nakano, M., Funakoshi, K., and Yamamoto, I. (2008). Expression of hypoxia-inducible angiogenic proteins (hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and E26 transformation-specific-1) and plaque hemorrhage in human carotid atherosclerosis. J. Neurosurg. 109, 83–91. doi: 10.3171/JNS/2008/109/7/0083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hill-Kapturczak, N., Voakes, C., Garcia, J., Visner, G., Nick, H. S., and Agarwal, A. (2003). A cis-acting region regulates oxidized lipid-mediated induction of the human heme oxygenase-1 gene in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 23, 1416–1422. doi: 10.1161/01.ATV.0000081656.76378.A7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hrkal, Z., Vodrazka, Z., and Kalousek, I. (1974). Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. Eur. J. Biochem. 43, 73–78. doi: 10.1111/j.1432-1033.1974.tb03386.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hutter, R., Speidl, W. S., Valdiviezo, C., Sauter, B., Corti, R., Fuster, V., et al. (2013). Macrophages transmit potent proangiogenic effects of oxLDL in vitro and in vivo involving HIF-1alpha activation: a novel aspect of angiogenesis in atherosclerosis. J. Cardiovasc. Transl. Res. 6, 558–569. doi: 10.1007/s12265-013-9469-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hvidberg, V., Maniecki, M. B., Jacobsen, C., Hojrup, P., Moller, H. J., and Moestrup, S. K. (2005). Identification of the receptor scavenging hemopexin-heme complexes. Blood 106, 2572–2579. doi: 10.1182/blood-2005-03-1185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., et al. (2001). HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468. doi: 10.1126/science.1059817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ivanov, A. S., Torkhovskaya, T. I., Khalilov, E. M., and Archakov, A. I. (1991). Diet-induced hypercholesterolaemia in the rabbit. Biomed. Sci. 2, 285–288.
Jeney, V., Balla, J., Yachie, A., Varga, Z., Vercellotti, G. M., Eaton, J. W., et al. (2002). Pro-oxidant and cytotoxic effects of circulating heme. Blood 100, 879–887. doi: 10.1182/blood.V100.3.879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeney, V., Eaton, J. W., Balla, G., and Balla, J. (2013). Natural history of the bruise: formation, elimination, and biological effects of oxidized hemoglobin. Oxid. Med. Cell. Longev. 2013:703571. doi: 10.1155/2013/703571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeziorska, M., and Woolley, D. E. (1999). Neovascularization in early atherosclerotic lesions of human carotid arteries: its potential contribution to plaque development. Hum. Pathol. 30, 919–925. doi: 10.1016/S0046-8177(99)90245-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jia, Y., Buehler, P. W., Boykins, R. A., Venable, R. M., and Alayash, A. I. (2007). Structural basis of peroxide-mediated changes in human hemoglobin: a novel oxidative pathway. J. Biol. Chem. 282, 4894–4907. doi: 10.1074/jbc.M609955200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Juan, S. H., Lee, T. S., Tseng, K. W., Liou, J. Y., Shyue, S. K., Wu, K. K., et al. (2001). Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation 104, 1519–1525. doi: 10.1161/hc3801.095663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Juckett, M. B., Balla, J., Balla, G., Jessurun, J., Jacob, H. S., and Vercellotti, G. M. (1995). Ferritin protects endothelial cells from oxidized low density lipoprotein in vitro. Am. J. Pathol. 147, 782–789.
Julius, U., and Pietzsch, J. (2005). Glucose-induced enhancement of hemin-catalyzed LDL oxidation in vitro and in vivo. Antioxid. Redox Signal. 7, 1507–1512. doi: 10.1089/ars.2005.7.1507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jun, J., Reinke, C., Bedja, D., Berkowitz, D., Bevans-Fonti, S., Li, J., et al. (2010). Effect of intermittent hypoxia on atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 209, 381–386. doi: 10.1016/j.atherosclerosis.2009.10.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jurrus, E. R., and Weiss, H. S. (1977). In vitro tissue oxygen tensions in the rabbit aortic arch. Atherosclerosis 28, 223–232. doi: 10.1016/0021-9150(77)90172-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kawashima, A., Oda, Y., Yachie, A., Koizumi, S., and Nakanishi, I. (2002). Heme oxygenase-1 deficiency: the first autopsy case. Hum. Pathol. 33, 125–130. doi: 10.1053/hupa.2002.30217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, Y. W., and Byzova, T. V. (2014). Oxidative stress in angiogenesis and vascular disease. Blood 123, 625–631. doi: 10.1182/blood-2013-09-512749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Knighton, D. R., Hunt, T. K., Scheuenstuhl, H., Halliday, B. J., Werb, Z., and Banda, M. J. (1983). Oxygen tension regulates the expression of angiogenesis factor by macrophages. Science 221, 1283–1285. doi: 10.1126/science.6612342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kockx, M. M., Cromheeke, K. M., Knaapen, M. W., Bosmans, J. M., De Meyer, G. R., Herman, A. G., et al. (2003). Phagocytosis and macrophage activation associated with hemorrhagic microvessels in human atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 23, 440–446. doi: 10.1161/01.ATV.0000057807.28754.7F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kolodgie, F. D., Burke, A. P., Nakazawa, G., Cheng, Q., Xu, X., and Virmani, R. (2007). Free cholesterol in atherosclerotic plaques: where does it come from? Curr. Opin. Lipidol. 18, 500–507. doi: 10.1097/MOL.0b013e3282efa35b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kolodgie, F. D., Gold, H. K., Burke, A. P., Fowler, D. R., Kruth, H. S., Weber, D. K., et al. (2003). Intraplaque hemorrhage and progression of coronary atheroma. N. Engl. J. Med. 349, 2316–2325. doi: 10.1056/NEJMoa035655
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koter, M., Broncel, M., Chojnowska-Jezierska, J., Klikczynska, K., and Franiak, I. (2002). The effect of atorvastatin on erythrocyte membranes and serum lipids in patients with type-2 hypercholesterolemia. Eur. J. Clin. Pharmacol. 58, 501–506. doi: 10.1007/s00228-002-0507-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kristiansen, M., Graversen, J. H., Jacobsen, C., Sonne, O., Hoffman, H. J., Law, S. K., et al. (2001). Identification of the haemoglobin scavenger receptor. Nature 409, 198–201. doi: 10.1038/35051594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larsen, R., Gozzelino, R., Jeney, V., Tokaji, L., Bozza, F. A., Japiassu, A. M., et al. (2010). A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2:51ra71. doi: 10.1126/scitranslmed.3001118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leibovich, S. J., Chen, J. F., Pinhal-Enfield, G., Belem, P. C., Elson, G., Rosania, A., et al. (2002). Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A(2A) receptor agonists and endotoxin. Am. J. Pathol. 160, 2231–2244. doi: 10.1016/S0002-9440(10)61170-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leitinger, N., and Schulman, I. G. (2013). Phenotypic polarization of macrophages in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 33, 1120–1126. doi: 10.1161/ATVBAHA.112.300173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leppanen, O., Bjornheden, T., Evaldsson, M., Boren, J., Wiklund, O., and Levin, M. (2006). ATP depletion in macrophages in the core of advanced rabbit atherosclerotic plaques in vivo. Atherosclerosis 188, 323–330. doi: 10.1016/j.atherosclerosis.2005.11.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levin, M., Leppanen, O., Evaldsson, M., Wiklund, O., Bondjers, G., and Bjornheden, T. (2003). Mapping of ATP, glucose, glycogen, and lactate concentrations within the arterial wall. Arterioscler. Thromb. Vasc. Biol. 23, 1801–1807. doi: 10.1161/01.ATV.0000092872.54026.8D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, W., Ostblom, M., Xu, L. H., Hellsten, A., Leanderson, P., Liedberg, B., et al. (2006). Cytocidal effects of atheromatous plaque components: the death zone revisited. FASEB J. 20, 2281–2290. doi: 10.1096/fj.06-6114com
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Libby, P. (2002). Inflammation in atherosclerosis. Nature 420, 868–874. doi: 10.1038/nature01323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lipiski, M., Deuel, J., Baek, J. H., Engelsberger, W. R., Buehler, P. W., and Schaer, D. J. (2013). Human phenotype specific haptoglobin therapeutics are both effective in vitro and in vivo to attenuate hemoglobin toxicity in guinea pigs. Antioxid. Redox Signal. doi: 10.1089/ars.2012.5089
Luttun, A., Tjwa, M., Moons, L., Wu, Y., Angelillo-Scherrer, A., Liao, F., et al. (2002). Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat. Med. 8, 831–840. doi: 10.1038/nm731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matzinger, P. (1994). Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12, 991–1045. doi: 10.1146/annurev.iy.12.040194.005015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matzinger, P. (2002). The danger model: a renewed sense of self. Science 296, 301–305. doi: 10.1126/science.1071059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maxwell, P. H., Wiesener, M. S., Chang, G. W., Clifford, S. C., Vaux, E. C., Cockman, M. E., et al. (1999). The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275. doi: 10.1038/20459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCarthy, M. J., Loftus, I. M., Thompson, M. M., Jones, L., London, N. J., Bell, P. R., et al. (1999). Angiogenesis and the atherosclerotic carotid plaque: an association between symptomatology and plaque morphology. J. Vasc. Surg. 30, 261–268. doi: 10.1016/S0741-5214(99)70136-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Melamed-Frank, M., Lache, O., Enav, B. I., Szafranek, T., Levy, N. S., Ricklis, R. M., et al. (2001). Structure-function analysis of the antioxidant properties of haptoglobin. Blood 98, 3693–3698. doi: 10.1182/blood.V98.13.3693
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michel, J. B., Virmani, R., Arbustini, E., and Pasterkamp, G. (2011). Intraplaque haemorrhages as the trigger of plaque vulnerability. Eur. Heart J. 32, 1977–1985, 1985a, 1985b, 1985c. doi: 10.1093/eurheartj/ehr054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, Y. I., Altamentova, S. M., and Shaklai, N. (1997). Oxidation of low-density lipoprotein by hemoglobin stems from a heme-initiated globin radical: antioxidant role of haptoglobin. Biochemistry 36, 12189–12198. doi: 10.1021/bi970258a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moreno, P. R., Purushothaman, K. R., Fuster, V., Echeverri, D., Truszczynska, H., Sharma, S. K., et al. (2004). Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: implications for plaque vulnerability. Circulation 110, 2032–2038. doi: 10.1161/01.CIR.0000143233.87854.23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moreno, P. R., Purushothaman, K. R., Sirol, M., Levy, A. P., and Fuster, V. (2006). Neovascularization in human atherosclerosis. Circulation 113, 2245–2252. doi: 10.1161/CIRCULATIONAHA.105.578955
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moreno, P. R., Purushothaman, M., and Purushothaman, K. R. (2012). Plaque neovascularization: defense mechanisms, betrayal, or a war in progress. Ann. N.Y. Acad. Sci. 1254, 7–17. doi: 10.1111/j.1749-6632.2012.06497.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moulton, K. S., Heller, E., Konerding, M. A., Flynn, E., Palinski, W., and Folkman, J. (1999). Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 99, 1726–1732. doi: 10.1161/01.CIR.99.13.1726
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moulton, K. S., Vakili, K., Zurakowski, D., Soliman, M., Butterfield, C., Sylvin, E., et al. (2003). Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 100, 4736–4741. doi: 10.1073/pnas.0730843100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagy, E., Eaton, J. W., Jeney, V., Soares, M. P., Varga, Z., Galajda, Z., et al. (2010). Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 30, 1347–1353. doi: 10.1161/ATVBAHA.110.206433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagy, E., Jeney, V., Yachie, A., Szabo, R. P., Wagner, O., Vercellotti, G. M., et al. (2005). Oxidation of hemoglobin by lipid hydroperoxide associated with low-density lipoprotein (LDL) and increased cytotoxic effect by LDL oxidation in heme oxygenase-1 (HO-1) deficiency. Cell. Mol. Biol. (Noisy-le-grand). 51, 377–385. doi: 10.1170/T641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakano, D., Hayashi, T., Tazawa, N., Yamashita, C., Inamoto, S., Okuda, N., et al. (2005). Chronic hypoxia accelerates the progression of atherosclerosis in apolipoprotein E-knockout mice. Hypertens. Res. 28, 837–845. doi: 10.1291/hypres.28.837
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nath, K. A., Balla, G., Vercellotti, G. M., Balla, J., Jacob, H. S., Levitt, M. D., et al. (1992). Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J. Clin. Invest. 90, 267–270. doi: 10.1172/JCI115847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O'Brien, E. R., Garvin, M. R., Dev, R., Stewart, D. K., Hinohara, T., Simpson, J. B., et al. (1994). Angiogenesis in human coronary atherosclerotic plaques. Am. J. Pathol. 145, 883–894.
Olsson, M. G., Allhorn, M., Larsson, J., Cederlund, M., Lundqvist, K., Schmidtchen, A., et al. (2011). Up-regulation of A1M/alpha1-microglobulin in skin by heme and reactive oxygen species gives protection from oxidative damage. PLoS ONE 6:e27505. doi: 10.1371/journal.pone.0027505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Olsson, M. G., Olofsson, T., Tapper, H., and Akerstrom, B. (2008). The lipocalin alpha1-microglobulin protects erythroid K562 cells against oxidative damage induced by heme and reactive oxygen species. Free Radic. Res. 42, 725–736. doi: 10.1080/10715760802337265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pang, J. H., Jiang, M. J., Chen, Y. L., Wang, F. W., Wang, D. L., Chu, S. H., et al. (1996). Increased ferritin gene expression in atherosclerotic lesions. J. Clin. Invest. 97, 2204–2212. doi: 10.1172/JCI118661
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paone, A., Galli, R., Gabellini, C., Lukashev, D., Starace, D., Gorlach, A., et al. (2010). Toll-like receptor 3 regulates angiogenesis and apoptosis in prostate cancer cell lines through hypoxia-inducible factor 1 alpha. Neoplasia 12, 539–549. doi: 10.1593/neo.92106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patel, R. P., Svistunenko, D. A., Darley-Usmar, V. M., Symons, M. C., and Wilson, M. T. (1996). Redox cycling of human methaemoglobin by H2O2 yields persistent ferryl iron and protein based radicals. Free Radic. Res. 25, 117–123. doi: 10.3109/10715769609149916
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Petrovan, R. J., Kaplan, C. D., Reisfeld, R. A., and Curtiss, L. K. (2007). DNA vaccination against VEGF receptor 2 reduces atherosclerosis in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 27, 1095–1100. doi: 10.1161/ATVBAHA.106.139246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pimenova, T., Pereira, C. P., Gehrig, P., Buehler, P. W., Schaer, D. J., and Zenobi, R. (2010). Quantitative mass spectrometry defines an oxidative hotspot in hemoglobin that is specifically protected by haptoglobin. J. Proteome Res. 9, 4061–4070. doi: 10.1021/pr100252e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pollet, I., Opina, C. J., Zimmerman, C., Leong, K. G., Wong, F., and Karsan, A. (2003). Bacterial lipopolysaccharide directly induces angiogenesis through TRAF6-mediated activation of NF-kappaB and c-Jun N-terminal kinase. Blood 102, 1740–1742. doi: 10.1182/blood-2003-01-0288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Porto, B. N., Alves, L. S., Fernandez, P. L., Dutra, T. P., Figueiredo, R. T., Graca-Souza, A. V., et al. (2007). Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J. Biol. Chem. 282, 24430–24436. doi: 10.1074/jbc.M703570200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Potor, L., Banyai, E., Becs, G., Soares, M. P., Balla, G., Balla, J., et al. (2013). Atherogenesis may involve the prooxidant and proinflammatory effects of ferryl hemoglobin. Oxid. Med. Cell. Longev. 2013:676425. doi: 10.1155/2013/676425
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Radhakrishnan, N., Yadav, S. P., Sachdeva, A., Pruthi, P. K., Sawhney, S., Piplani, T., et al. (2011). Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 33, 74–78. doi: 10.1097/MPH.0b013e3181fd2aae
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rajamaki, K., Lappalainen, J., Oorni, K., Valimaki, E., Matikainen, S., Kovanen, P. T., et al. (2010). Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS ONE 5:e11765. doi: 10.1371/journal.pone.0011765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramirez, D. C., Chen, Y. R., and Mason, R. P. (2003). Immunochemical detection of hemoglobin-derived radicals formed by reaction with hydrogen peroxide: involvement of a protein-tyrosyl radical. Free Radic. Biol. Med. 34, 830–839. doi: 10.1016/S0891-5849(02)01437-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reeder, B. J., Cutruzzola, F., Bigotti, M. G., Hider, R. C., and Wilson, M. T. (2008). Tyrosine as a redox-active center in electron transfer to ferryl heme in globins. Free Radic. Biol. Med. 44, 274–283. doi: 10.1016/j.freeradbiomed.2007.06.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ritman, E. L., and Lerman, A. (2007). The dynamic vasa vasorum. Cardiovasc. Res. 75, 649–658. doi: 10.1016/j.cardiores.2007.06.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sahebkar, A., and Watts, G. F. (2013). New LDL-cholesterol lowering therapies: pharmacology, clinical trials, and relevance to acute coronary syndromes. Clin. Ther. 35, 1082–1098. doi: 10.1016/j.clinthera.2013.06.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, C. A., Deuel, J. W., Bittermann, A. G., Rubio, I. G., Schoedon, G., Spahn, D. R., et al. (2013b). Mechanisms of haptoglobin protection against hemoglobin peroxidation triggered endothelial damage. Cell Death Differ. 20, 1569–1579. doi: 10.1038/cdd.2013.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, D. J., Buehler, P. W., Alayash, A. I., Belcher, J. D., and Vercellotti, G. M. (2013a). Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121, 1276–1284. doi: 10.1182/blood-2012-11-451229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, D. J., Schaer, C. A., Buehler, P. W., Boykins, R. A., Schoedon, G., Alayash, A. I., et al. (2006). CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood 107, 373–380. doi: 10.1182/blood-2005-03-1014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Siems, W. G., Sommerburg, O., and Grune, T. (2000). Erythrocyte free radical and energy metabolism. Clin. Nephrol. 53, S9–S17.
Silva, G., Jeney, V., Chora, A., Larsen, R., Balla, J., and Soares, M. P. (2009). Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J. Biol. Chem. 284, 29582–29595. doi: 10.1074/jbc.M109.045344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sluimer, J. C., Gasc, J. M., van Wanroij, J. L., Kisters, N., Groeneweg, M., Sollewijn Gelpke, M. D., et al. (2008). Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J. Am. Coll. Cardiol. 51, 1258–1265. doi: 10.1016/j.jacc.2007.12.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sluimer, J. C., Kolodgie, F. D., Bijnens, A. P., Maxfield, K., Pacheco, E., Kutys, B., et al. (2009). Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J. Am. Coll. Cardiol. 53, 1517–1527. doi: 10.1016/j.jacc.2008.12.056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spirig, R., Djafarzadeh, S., Regueira, T., Shaw, S. G., von Garnier, C., Takala, J., et al. (2010). Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1alpha and dendritic cell maturation under normoxic conditions. PLoS ONE 5:e0010983. doi: 10.1371/journal.pone.0010983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sverrisson, K., Axelsson, J., Rippe, A., Gram, M., Akerstrom, B., Hansson, S. R., et al. (2014). Extracellular fetal hemoglobin induces increases in glomerular permeability: inhibition with alpha1-microglobulin and tempol. Am. J. Physiol. Renal Physiol. 306, F442–F448. doi: 10.1152/ajprenal.00502.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tenhunen, R., Marver, H. S., and Schmid, R. (1968). The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. U.S.A. 61, 748–755. doi: 10.1073/pnas.61.2.748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Torres Filho, I. P., Leunig, M., Yuan, F., Intaglietta, M., and Jain, R. K. (1994). Noninvasive measurement of microvascular and interstitial oxygen profiles in a human tumor in SCID mice. Proc. Natl. Acad. Sci. U.S.A. 91, 2081–2085. doi: 10.1073/pnas.91.6.2081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tynan, M. B., Nicholls, D. P., Maguire, S. M., Steele, I. C., McMaster, C., Moore, R., et al. (1995). Erythrocyte membrane fatty acid composition as a marker of dietary compliance in hyperlipidaemic subjects. Atherosclerosis 117, 245–252. doi: 10.1016/0021-9150(95)05578-K
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tziakas, D., Chalikias, G., Kapelouzou, A., Tentes, I., Schafer, K., Karayannakos, P., et al. (2013). Erythrocyte membrane cholesterol and lipid core growth in a rabbit model of atherosclerosis: modulatory effects of rosuvastatin. Int. J. Cardiol. 170, 173–181. doi: 10.1016/j.ijcard.2013.10.070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vallelian, F., Pimenova, T., Pereira, C. P., Abraham, B., Mikolajczyk, M. G., Schoedon, G., et al. (2008). The reaction of hydrogen peroxide with hemoglobin induces extensive alpha-globin crosslinking and impairs the interaction of hemoglobin with endogenous scavenger pathways. Free Radic. Biol. Med. 45, 1150–1158. doi: 10.1016/j.freeradbiomed.2008.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vincent, S. H., Grady, R. W., Shaklai, N., Snider, J. M., and Muller-Eberhard, U. (1988). The influence of heme-binding proteins in heme-catalyzed oxidations. Arch. Biochem. Biophys. 265, 539–550. doi: 10.1016/0003-9861(88)90159-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vinchi, F., De Franceschi, L., Ghigo, A., Townes, T., Cimino, J., Silengo, L., et al. (2013). Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 127, 1317–1329. doi: 10.1161/CIRCULATIONAHA.112.130179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vinchi, F., Muckenthaler, M. U., Da Silva, M. C., Balla, G., Balla, J., and Jeney, V. (2014). Atherogenesis and iron: from epidemiology to cellular level. Front. Pharmacol. 5:94. doi: 10.3389/fphar.2014.00094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vink, A., Schoneveld, A. H., Lamers, D., Houben, A. J., van der Groep, P., van Diest, P. J., et al. (2007). HIF-1 alpha expression is associated with an atheromatous inflammatory plaque phenotype and upregulated in activated macrophages. Atherosclerosis 195, e69–e75. doi: 10.1016/j.atherosclerosis.2007.05.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagener, F. A., Eggert, A., Boerman, O. C., Oyen, W. J., Verhofstad, A., Abraham, N. G., et al. (2001). Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 98, 1802–1811. doi: 10.1182/blood.V98.6.1802
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagener, F. A., Feldman, E., de Witte, T., and Abraham, N. G. (1997). Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc. Soc. Exp. Biol. Med. 216, 456–463. doi: 10.3181/00379727-216-44197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, G. L., Jiang, B. H., Rue, E. A., and Semenza, G. L. (1995). Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U.S.A. 92, 5510–5514. doi: 10.1073/pnas.92.12.5510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, G. L., and Semenza, G. L. (1995). Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 270, 1230–1237. doi: 10.1074/jbc.270.3.1230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wenger, R. H., Stiehl, D. P., and Camenisch, G. (2005). Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005:re12. doi: 10.1126/stke.3062005re12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
West, X. Z., Malinin, N. L., Merkulova, A. A., Tischenko, M., Kerr, B. A., Borden, E. C., et al. (2010). Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467, 972–976. doi: 10.1038/nature09421
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yachie, A., Niida, Y., Wada, T., Igarashi, N., Kaneda, H., Toma, T., et al. (1999). Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Invest. 103, 129–135. doi: 10.1172/JCI4165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yeagle, P. L. (1985). Cholesterol and the cell membrane. Biochim. Biophys. Acta 822, 267–287. doi: 10.1016/0304-4157(85)90011-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yet, S. F., Layne, M. D., Liu, X., Chen, Y. H., Ith, B., Sibinga, N. E., et al. (2003). Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 17, 1759–1761. doi: 10.1096/fj.03-0187fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zarjou, A., Jeney, V., Arosio, P., Poli, M., Antal-Szalmas, P., Agarwal, A., et al. (2009). Ferritin prevents calcification and osteoblastic differentiation of vascular smooth muscle cells. J. Am. Soc. Nephrol. 20, 1254–1263. doi: 10.1681/ASN.2008070788
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zemplenyi, T., Crawford, D. W., and Cole, M. A. (1989). Adaptation to arterial wall hypoxia demonstrated in vivo with oxygen microcathodes. Atherosclerosis 76, 173–179. doi: 10.1016/0021-9150(89)90101-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: atherosclerosis, intraplaque hemorrhage, red blood cell lysis, hemoglobin oxidation, haptoglobin, hemopexin, heme-oxygenase, ferritin
Citation: Jeney V, Balla G and Balla J (2014) Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front. Physiol. 5:379. doi: 10.3389/fphys.2014.00379
Received: 31 July 2014; Accepted: 13 September 2014;
Published online: 02 October 2014.
Edited by:
Magnus Gram, Lund University, SwedenReviewed by:
Wang Min, Yale University, USAMagnus Gram, Lund University, Sweden
Dominik J. Schaer, University of Zurich, Switzerland
Copyright © 2014 Jeney, Balla and Balla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: József Balla, Department of Medicine, University of Debrecen, Pf. 19, Nagyerdei krt. 98, 4012 Debrecen, Hungary e-mail: balla@internal.med.unideb.hu