 Youn Jung Kim1Jenny Kang2
Youn Jung Kim1Jenny Kang2 Figen Seymen3
Figen Seymen3 Mine Koruyucu3Koray Gencay3Teo Jeon Shin2Hong-Keun Hyun2Zang Hee Lee4
Mine Koruyucu3Koray Gencay3Teo Jeon Shin2Hong-Keun Hyun2Zang Hee Lee4 Jan C.-C. Hu5James P. Simmer5
Jan C.-C. Hu5James P. Simmer5 Jung-Wook Kim1,2*
Jung-Wook Kim1,2*- 1Department of Molecular Genetics and Dental Research Institute, School of Dentistry, Seoul National University, Seoul, Korea
- 2Department of Pediatric Dentistry and Dental Research Institute, School of Dentistry, Seoul National University, Seoul, Korea
- 3Faculty of Dentistry, Department of Pedodontics, Istanbul University, Istanbul, Turkey
- 4Department of Cell and Developmental Biology and Dental Research Institute, School of Dentistry, Seoul National University, Seoul, Korea
- 5Department of Biologic and Materials Sciences, University of Michigan School of Dentistry, Ann Arbor, MI, USA
Amelogenesis imperfecta is a group of rare inherited disorders that affect tooth enamel formation, quantitatively and/or qualitatively. The aim of this study was to identify the genetic etiologies of two families presenting with hypomaturation amelogenesis imperfecta. DNA was isolated from peripheral blood samples obtained from participating family members. Whole exome sequencing was performed using DNA samples from the two probands. Sequencing data was aligned to the NCBI human reference genome (NCBI build 37.2, hg19) and sequence variations were annotated with the dbSNP build 138. Mutations in MMP20 were identified in both probands. A homozygous missense mutation (c.678T>A; p.His226Gln) was identified in the consanguineous Family 1. Compound heterozygous MMP20 mutations (c.540T>A, p.Tyr180* and c.389C>T, p.Thr130Ile) were identified in the non-consanguineous Family 2. Affected persons in Family 1 showed hypomaturation AI with dark brown discoloration, which is similar to the clinical phenotype in a previous report with the same mutation. However, the dentition of the Family 2 proband exhibited slight yellowish discoloration with reduced transparency. Functional analysis showed that the p.Thr130Ile mutant protein had reduced activity of MMP20, while there was no functional MMP20 in the Family 1 proband. These results expand the mutational spectrum of the MMP20 and broaden our understanding of genotype-phenotype correlations in amelogenesis imperfecta.
Introduction
Non-syndromic amelogenesis imperfecta (AI), hereditary enamel defects, can be divided into 3 major categories based on the quantity and quality of the enamel (Witkop, 1988). In hypoplastic AI, the enamel is thin with interdental spacing and the affected individuals are often sensitive to thermal changes and possess an increased tendency of anterior open bite (Ravassipour et al., 2005). In hypocalcification AI, the affected enamel is extremely soft with normal thickness prior to tooth eruption, which may be lost rapidly after eruption leaving the remaining enamel rough, discolored, and thin. Hypomaturation AI is caused by failures during the maturation stage of amelogenesis. The resulting phenotype is characteristically (dark) brown or yellowish discolored less mineralized enamel with normal thickness. But because the enamel is not matured well, prolonged attrition can result in excessive enamel wear facets or localized enamel fractures (Wright et al., 2011). However, definitive characterization of the phenotype may be challenging in some cases. Therefore, a broader classification scheme with two categories has been used: hypoplastic AI and hypomineralized AI. The hypomineralized AI includes hypocalcification AI and hypomaturation AI (Prasad et al., 2016).
To date, mutations in more than 17 genes are involved in non-syndromic AI. Hypoplastic AI can be caused by mutations in AMELX (MIM: 300391), ENAM (MIM: 606585), AMBN (MIM: 601259), LAMB3 (MIM: 150310), LAMA3 (MIM: 600805), COL17A1 (MIM: 113811), ITGB6 (MIM: 147558), and ACPT (MIM: 606362) (Lagerstrom et al., 1991; McGrath et al., 1996; Rajpar et al., 2001; Yuen et al., 2012; Kim et al., 2013; Wang et al., 2014; Poulter et al., 2014a,b,c; Seymen et al., 2016). Autosomal dominant hypocalcification AI is caused by mutations in FAM83H (MIM: 611927) (Kim et al., 2008). Some AMELX mutations can cause hypomaturation AI with enamel hypoplasia (Hart et al., 2000). Recessive mutations in SLC24A4 (MIM: 609840), WDR72 (MIM: 613214), MMP20 (MIM: 604629), KLK4 (MIM: 603767), and GPR68 (MIN: 601404) cause hypomaturation AI (Wright et al., 2003; Hart et al., 2004; Kim et al., 2005; El-Sayed et al., 2009; Parry et al., 2013, 2016). Clinical phenotype caused by autosomal recessive mutations of C4orf26 (MIM: 614829) and autosomal dominant mutation of AMTN (MIM: 610912) were reported as hypomineralization AI (Parry et al., 2012; Smith et al., 2016).
Two proteinases secreted by ameloblasts during mammalian enamel formation are matrix metalloproteinase 20 (MMP20, enamelysin) and kallikrein 4 (KLK4) (Hu et al., 2002). MMP20 is the early protease expressed by ameloblasts throughout the secretory stage and early maturation stage of amelogenesis. KLK4 is the late protease expressed by ameloblasts from the transition stage to the maturation stage. Lack of proteinase function in the maturing enamel matrix prevents proper degradation and removal of the enamel matrix proteins resulting in enamel hypomaturation AI.
Here we report the identification of MMP20 mutations in two Turkish families with hypomaturation AI by whole exome sequencing and the mutational effect on the protein secretion and proteolytic activity.
Materials and Methods
Identification and Enrollment of AI Families
Clinical and radiographic examinations of the probands and their available family members were performed, and blood samples were collected with the understanding and written consent of each participant according to the Declaration of Helsinki. Affected individuals were healthy, except hypomaturation enamel defects. The study protocol was independently reviewed and approved by the Institution Review Board at the Seoul National University Dental Hospital, the University of Istanbul and the University of Michigan.
DNA Isolation and Whole-Exome Sequencing
Genomic DNA was isolated from peripheral whole blood. The purity and concentration of the DNA were quantified by spectrophotometry measurement and the OD260/OD280 ratio obtained. Whole-exome sequencing was performed with the DNA sample of the probands using Illumina HiSeq 2000 platform. The NimbleGen (Roche Diagnostics, Indianapolis, IL, USA) exome capture reagent was used for exome capturing.
Autozygosity Mapping
The affected individuals in family 1 (IV:3 and IV:4) were genotyped with the Affymetrix Genome-Wide Human SNP array 6.0 (DNALINK INC., Seoul, Korea). The annotated SNP files were analyzed with HomozygosityMapper (http://www.homozygositymapper.org/) (Seelow et al., 2009) to identify the shared regions of homozygosity in the affected individuals.
Segregation Analysis by Polymerase Chain Reaction (PCR)
The sequence variations in the MMP20 gene and segregation within each family was confirmed by Sanger sequencing with primers and conditions described previously (Kim et al., 2005). PCR amplifications were done with the HiPi DNA polymerase premix (Elpis Biotech, Daejeon, Korea), and DNA sequencing was performed at a DNA sequencing center (Macrogen, Seoul, Korea).
Cloning and Mutagenesis of the MMP20 cDNA
Human MMP20 cDNA, previously cloned into the pcDNA3.1 vector, was used to introduce the identified mutations using PCR mutagenesis (sense: 5′-TACCGTTGCTGCTCAAGAATTTGGCCATGC, antisense: 5′-GCATGGCCAAATTCTTGAGCAGCAACGGTA for the p.His226Gln and sense: 5′-GAATATCTAAATACATACCTTCCATGAGTT, antisense: 5′-AACTCATGGAAGGTATGTATTTAGATATTC for the p.Thr130Ile) (Lee et al., 2010). Sequences of normal and mutant MMP20 pcDNA3.1 vectors were confirmed by direct plasmid sequencing.
Transfection
HEK293T cells were grown and maintained in DMEM supplemented with 10% FBS and antibiotics in a 5% CO2 atmosphere at 37°C. Cells at ~2 × 105 quantity were seeded in each well of the 6-well culture dish. Each plasmid construct at 2 ug quantity was transiently transfected into HEK293T cells with Genjet in vitro DNA transfection reagent (SigmaGen Laboratories, Ijamsville, MD, USA). The culture medium of each well was harvested after 30 h of incubation and concentrated using Amicon ultra-4 centrifugal filter units (Millipore, Bedford, MA, USA).
Zymography
Four ml of conditioned medium from the culture was collected and concentrated to 50 ul. The concentrated media of 20 ul was mixed with 4 ul of 5x non-reducing buffer, then loaded onto the 11% SDS-polyacrylamide gel with β-casein (Sigma-Aldrich, St. Louis, MO, USA) as a substrate. The zymogram was developed, stained with Coomassie brilliant blue R-250 staining solution (Bio-rad, Hercules, CA, USA) for 20 min, and visualized after incubation in a destaining solution (10% MeOH, 10% acetic acid) for 3 h.
Western Blot
Concentrated media and cell lysates were run on the 11% SDS-polyacrylamide gel and subjected to Western blotting. Specifically, 50 ug cell lysate from each sample and 10 ul of concentrated media were used. After gel transfer to the PVDF membrane and blocking, MMP20 was detected by incubating the membrane with primary antibody overnight at 4°C and with secondary antibody for 2 h at room temperature. The primary antibodies used were a rabbit polyclonal anti-MMP20 antibody (ab39038, abcam plc., Cambridge, UK) and a mouse monoclonal anti-ACTB antibody (A2228, Sigma-Aldrich, St. Louis, MO, USA); both of which were diluted in 1:10,000.
Results
Clinical Phenotype
The proband of Family 1 (IV:4) was an 11-year-old girl from a consanguineous marriage of first cousins (Figure 1A). Her prenatal and perinatal history was uneventful and her parents reported no other medical problem. Her teeth exhibited generalized brown discoloration with exogenous black pigmentation mainly on occlusal surface of the posterior teeth (Figures 1B–D). Maxillary left central incisor was lost due to trauma. The radiopacity of enamel did not contrast well with dentin in the panoramic radiograph, consistent with hypomineralization (Figure 1E). Her 24-year-old brother (IV:3) was also affected and almost all of his teeth have been reconstructed with full-coverage prosthodontics. His remaining natural teeth exhibited dark brown discoloration with exogenous pigmentation (Figure S1).
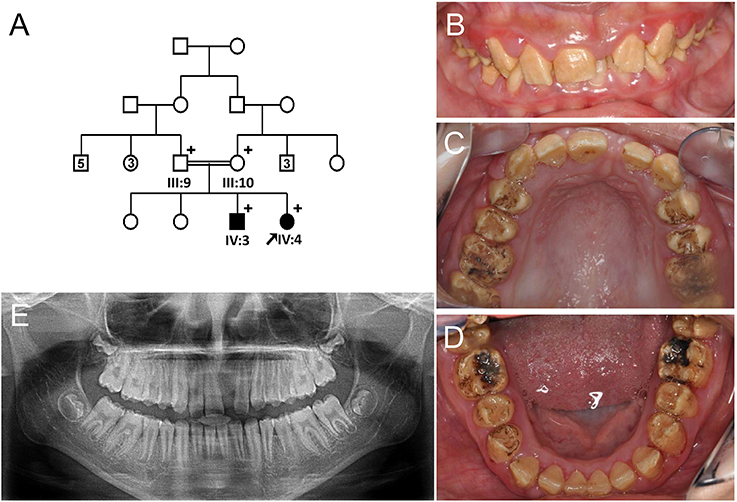
Figure 1. Pedigree and clinical phenotype of the proband of Family 1. (A) Pedigree of family 1. (B) Frontal view of the proband (IV:4) at age 11. (C) Maxillary occlusal view. (D) Mandibular occlusal view. (E) Panoramic radiograph of the proband at age 11. Numbers in the subject symbol indicate the number of siblings. Plus symbols indicate individuals who participated in this study.
The proband of the family 2 (III:1) was a 10-year-old girl from a non-consanguineous family (Figure 2A). Her past medical history was unremarkable. Her anterior permanent teeth were not severely discolored, but slightly yellow and less transparent than normal teeth (Figures 2B,C). Her right second premolar was congenitally absent based on the panoramic radiograph (Figure 2D).
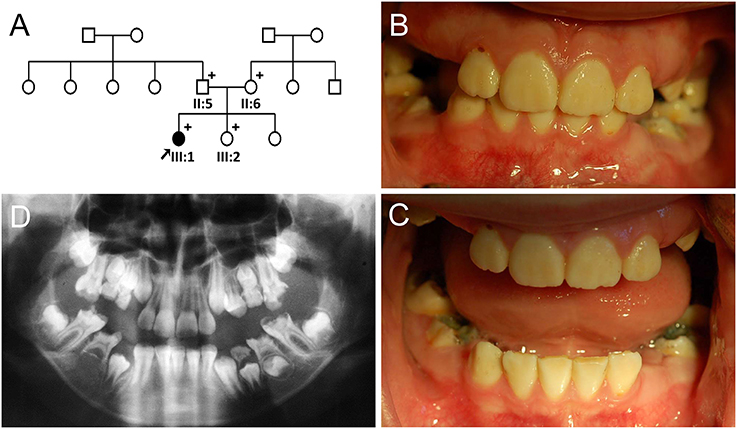
Figure 2. Pedigree and clinical phenotype of the proband of Family 2. (A) Pedigree of family 2. (B) Frontal view of the proband (III:1) at age 10. (C) Frontal view with bite open. (D) Panoramic radiograph of the proband at age 10. Plus symbols indicate individuals who participated in this study.
Mutational Analysis
Sequencing reads were aligned to the UCSC human reference genome (hg19) with Burrow-Wheeler Aligner, and the sequence variations were annotated by referencing dbSNP build 138, which preceded variant calling with SAMtools and GATK (Table S1). Annotated variants were filtered with the criteria of minor allele frequency of 0.01.
Autozygosity mapping of the family 1 revealed 3 shared regions of loss of heterozygosity: chr4:65,904,881–82,427,846, chr11:83,358,629–113,318,007, and chr21:11,039,570–17,728,224 (Figure S2). The exome data in the shared regions of the proband in family 1 revealed a homozygous variant in exon 5 of the MMP20 gene (NM_004771.3: c.678T>A). This transversion of thymine to adenine changed histidine to glutamine at amino acid position 226 (p.His226Gln). There was no other variation in the known AI-causing genes and the MMP20 mutation (c.678T>A, p.His226Gln) was previously reported as an AI-causing mutation (Ozdemir et al., 2005; Wright et al., 2011).
Whole exome sequencing of the Family 2 proband revealed compound heterozygous MMP20 mutations (c.389C>T and c.540T>A). There was no other variation in the known AI-causing genes. A cytosine to thymine transition in exon 3 changed threonine to isoleucine at amino acid position 130 (p.Thr130Ile). This variation was listed in the Exome Aggregation Consortium (ExAC) database (rs61730849) with an allele frequency of 0.00165 (200/121176). But the frequency was relatively high (0.0294) in a certain subset of small population (ss86247256, AGI_ASP_population; Coriell Apparently Healthy Collection). In addition, it was previously reported as a disease-causing mutation (Gasse et al., 2013). The other variation, a transversion of thymine to adenine in exon 4, would introduce a premature stop codon (p.Tyr180*) and the mutant transcript would be degraded by the nonsense-mediated decay system. This variant was not listed in any database.
Segregation within the families by Sanger sequencing confirmed that the nonsense mutation (c.540T>A, p.Tyr180*) was transmitted paternally and the missense mutation (c.389C>T, p.Thr130Ile) was transmitted maternally to the proband (Figure 3A, Figure S3). These amino acids at the mutation sites (Thr130, Tyr180, and His226) are strictly conserved among eutherian mammal orthologs (Figure 3B).
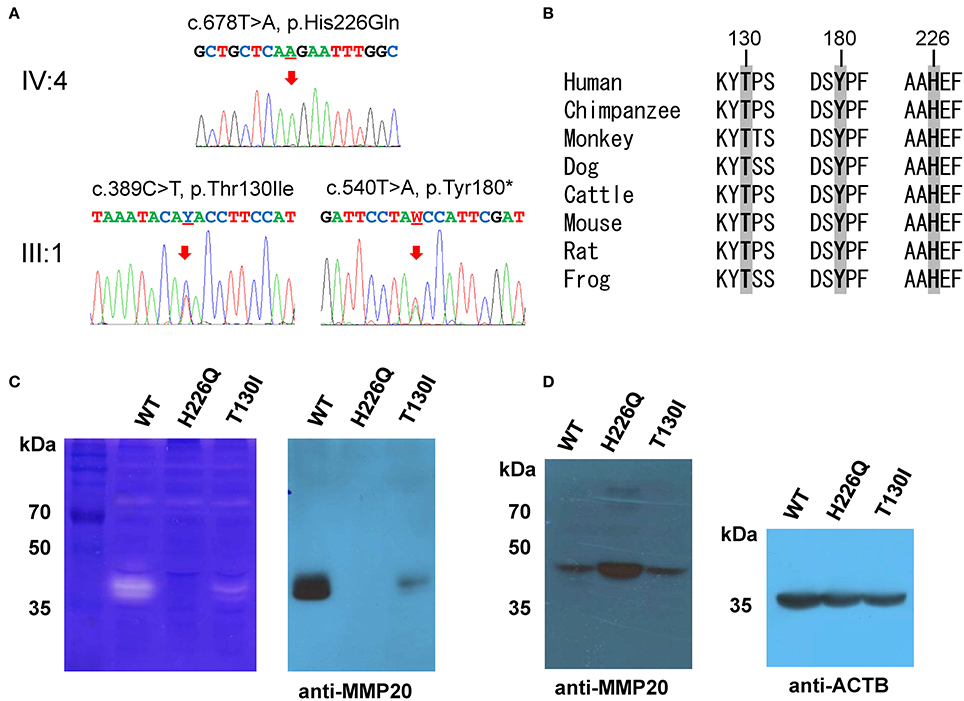
Figure 3. Orthologs alignment, sequencing chromatograms and in vitro translation. (A) Sanger sequencing chromatograms of the probands (IV:4 of family 1 and III:1 of family 2). The mutated nucleotide is indicated by a red arrow and underlined (Y; C or T and W; A or T). (B) Sequence alignment of vertebrate orthologs. Amino acids affected by the mutations are indicated with bold character and gray highlight. Numbers above the amino acids are based on the human MMP20 sequence. (C) Casein zymography indicated that the p.Thr130Ile mutant protein retains proteolytic function, but the p.His226Gln mutant protein has no proteolytic activity. Western blot of the conditioned media revealed that the secretion of the p.Thr130Ile mutant protein into the culture media was greatly reduced, but the p.His226Gln mutant protein cannot be secreted at all. (D) Western blot of the cell lysate demonstrated that the p.His226Gln mutant protein remained in the cell. (ACTB: beta actin).
Western blotting and zymography determining the function of the MMP20 mutants demonstrated that the p.Thr130Ile mutant protein was secreted at a reduced amount and had proteolytic activity. Western blot of cell lysate revealed that the p.His226Gln mutant protein was retained in the cell and likely not able to be secreted (Figures 3C,D).
Discussion
MMP20 is one of 23 human matrix metalloproteinases. It processes structural enamel matrix proteins into functional fragments in the secretory stage and facilitates the removal of those proteins during the maturation stage. MMP20 gene is located in a cluster with 7 other MMPs on chromosomal location 11q22.3. MMP20 encodes a 483-amino-acid protein, which has a signal peptide (Met1 to Ala22), a prodomain (Ala23 to Asn107), a catalytic domain (Tyr108 to Gly271), a linker (Pro272 to Leu295) and a hemopexin domain (Cys296 to Cys483) (Llano et al., 1997).
The homozygous missense mutation (c.678T>A, p.His226Gln) identified in Family 1 was previously reported (Ozdemir et al., 2005; Wright et al., 2011). His226 is one of the three histidine residues involved in the coordination of zinc ion at the active site (Llano et al., 1997). This study showed that the p.His226Gln mutant protein cannot be secreted into the developing extracellular matrix, probably due to a structural change in the core area of the protein.
The Family 2 proband had a paternal nonsense mutation (c.540T>A, p.Tyr180*) and a maternal missense mutation (c.389C>T, p.Thr130Ile) (Gasse et al., 2013). This novel nonsense mutation would introduce a premature stop codon in exon 4, so the mutant mRNA transcript would be degraded by the nonsense-mediated decay system. This study showed that the p.Thr130Ile mutant protein could be secreted into the developing enamel matrix and retained proteolytic function.
The functional analysis suggested that the Family 2 proband would have reduced MMP20 activity, while there's no functional MMP20 in the Family 1 proband. This reduced functional activity of MMP20 potentially explains the difference in clinical phenotype between the probands of these two families. Among nine mutations in MMP20 gene reported to date (Kim et al., 2005; Ozdemir et al., 2005; Papagerakis et al., 2008; Lee et al., 2010; Wright et al., 2011; Gasse et al., 2013; Wang et al., 2013; Seymen et al., 2015), mutations presumed to have retained functional activity would likely present less severe discoloration compared to nullifying mutations (Table 1). The degree of discoloration could be an indicator of the enamel porosity and reflects an altered level of maturation. Therefore, such clinical feature reflecting enamel quality should be considered by clinicians when devising a treatment plan for the patient.

Table 1. Disease-causing mutations of the MMP20 gene.
As mutations of the MMP20 gene are characterized, their functional impact investigated, and clinical features of the affected individuals documented, it will enhance our ability to establish genotype and phenotype correlation and provide the needed evidence to improve clinical diagnosis and management of patients with AI.
Author Contributions
Study design: FS, JH, JS, and JWK. Data collection: MK, KG, TS, HH, and ZL. Data analysis: YK, MK, KG, TS, HH, and JWK. Drafting manuscript: YK, JH, JS, and JWK. Revising manuscript content: JK, JH, JS, and JWK. Approving final version of manuscript: YK, JK, FS, MK, KG, TS, HH, ZL, JH, JS, and JWK. JWK takes responsibility for the integrity of the data analysis.
Funding
This work was supported by grants from the National Research Foundation of Korea (NRF) grant funded by the Korea government (2014R1A2A1A11049931) and the National Institute for Dental and Craniofacial Research (DE015846).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors sincerely thank all the family members for their participation in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphys.2017.00229/full#supplementary-material
References
Chan, H. C., Estrella, N. M., Milkovich, R. N., Kim, J. W., Simmer, J. P., and Hu, J. C. (2011). Target gene analyses of 39 amelogenesis imperfecta kindreds. Eur. J. Oral Sci. 119(Suppl 1), 311–323. doi: 10.1111/j.1600-0722.2011.00857.x
El-Sayed, W., Parry, D. A., Shore, R. C., Ahmed, M., Jafri, H., Rashid, Y., et al. (2009). Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am. J. Hum. Genet. 85, 699–705. doi: 10.1016/j.ajhg.2009.09.014
Gasse, B., Karayigit, E., Mathieu, E., Jung, S., Garret, A., Huckert, M., et al. (2013). Homozygous and compound heterozygous MMP20 mutations in amelogenesis imperfecta. J. Dent. Res. 92, 598–603. doi: 10.1177/0022034513488393
Hart, P. S., Hart, T. C., Michalec, M. D., Ryu, O. H., Simmons, D., Hong, S., et al. (2004). Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J. Med. Genet. 41, 545–549. doi: 10.1136/jmg.2003.017657
Hart, S., Hart, T., Gibson, C., and Wright, J. T. (2000). Mutational analysis of X-linked amelogenesis imperfecta in multiple families. Arch. Oral Biol. 45, 79–86. doi: 10.1016/S0003-9969(99)00106-5
Hu, J. C. C., Sun, X., Liu, S., Zhang, C., Bartlett, J. D., and Simmer, J. P. (2002). Enamelysin and kallikrein-4 expression in developing mouse molars. Eur. J. Oral Sci. 110, 307–315. doi: 10.1034/j.1600-0722.2002.21301.x
Kim, J. W., Lee, S. K., Lee, Z. H., Park, J. C., Lee, K. E., Lee, M. H., et al. (2008). FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am. J. Hum. Genet. 82, 489–494. doi: 10.1016/j.ajhg.2007.09.020
Kim, J. W., Seymen, F., Lee, K. E., Ko, J., Yildirim, M., Tuna, E. B., et al. (2013). LAMB3 Mutations Causing Autosomal-dominant Amelogenesis Imperfecta. J. Dent. Res. 92, 899–904. doi: 10.1177/0022034513502054
Kim, J. W., Simmer, J. P., Hart, T. C., Hart, P. S., Ramaswami, M. D., Bartlett, J. D., et al. (2005). MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J. Med. Genet. 42, 271–275. doi: 10.1136/jmg.2004.024505
Lagerstrom, M., Dahl, N., Nakahori, Y., Nakagome, Y., Backman, B., Landegren, U., et al. (1991). A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1). Genomics 10, 971–975. doi: 10.1016/0888-7543(91)90187-J
Lee, S. K., Seymen, F., Kang, H. Y., Lee, K. E., Gencay, K., Tuna, B., et al. (2010). MMP20 hemopexin domain mutation in amelogenesis imperfecta. J. Dent. Res. 89, 46–50. doi: 10.1177/0022034509352844
Llano, E., Pendas, A. M., Knauper, V., Sorsa, T., Salo, T., Salido, E., et al. (1997). Identification and structural and functional characterization of human enamelysin (MMP-20). Biochemistry 36, 15101–15108. doi: 10.1021/bi972120y
McGrath, J. A., Gatalica, B., Li, K., Dunnill, M. G., McMillan, J. R., Christiano, A. M., et al. (1996). Compound heterozygosity for a dominant glycine substitution and a recessive internal duplication mutation in the type XVII collagen gene results in junctional epidermolysis bullosa and abnormal dentition. Am. J. Pathol. 148, 1787–1796.
Ozdemir, D., Hart, P. S., Ryu, O. H., Choi, S. J., Ozdemir-Karatas, M., Firatli, E., et al. (2005). MMP20 active-site mutation in hypomaturation amelogenesis imperfecta. J. Dent. Res. 84, 1031–1035. doi: 10.1177/154405910508401112
Papagerakis, P., Lin, H. K., Lee, K. Y., Hu, Y., Simmer, J. P., Bartlett, J. D., et al. (2008). Premature stop codon in MMP20 causing amelogenesis imperfecta. J. Dent. Res. 87, 56–59. doi: 10.1177/154405910808700109
Parry, D. A., Brookes, S. J., Logan, C. V., Poulter, J. A., El-Sayed, W., Al-Bahlani, S., et al. (2012). Mutations in C4orf26, encoding a peptide with in vitro hydroxyapatite crystal nucleation and growth activity, cause amelogenesis imperfecta. Am. J. Hum. Genet. 91, 565–571. doi: 10.1016/j.ajhg.2012.07.020
Parry, D. A., Poulter, J. A., Logan, C. V., Brookes, S. J., Jafri, H., Ferguson, C. H., et al. (2013). Identification of mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta. Am. J. Hum. Genet. 92, 307–312. doi: 10.1016/j.ajhg.2013.01.003
Parry, D. A., Smith, C. E., El-Sayed, W., Poulter, J. A., Shore, R. C., Logan, C. V., et al. (2016). Mutations in the pH-Sensing G-protein-Coupled Receptor GPR68 Cause Amelogenesis Imperfecta. Am. J. Hum. Genet. 99, 984–990. doi: 10.1016/j.ajhg.2016.08.020
Poulter, J. A., Brookes, S. J., Shore, R. C., Smith, C. E., Abi Farraj, L., Kirkham, J., et al. (2014a). A missense mutation in ITGB6 causes pitted hypomineralized amelogenesis imperfecta. Hum. Mol. Genet. 23, 2189–2197. doi: 10.1093/hmg/ddt616
Poulter, J. A., El-Sayed, W., Shore, R. C., Kirkham, J., Inglehearn, C. F., and Mighell, A. J. (2014b). Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. Eur. J. Hum. Genet. 22, 132–135. doi: 10.1038/ejhg.2013.76
Poulter, J. A., Murillo, G., Brookes, S. J., Smith, C. E., Parry, D. A., Silva, S., et al. (2014c). Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Hum. Mol. Genet. 23, 5317–5324. doi: 10.1093/hmg/ddu247
Prasad, M. K., Laouina, S., El Alloussi, M., Dollfus, H., and Bloch-Zupan, A. (2016). Amelogenesis Imperfecta: 1 Family, 2 Phenotypes, and 2 Mutated Genes. J. Dent. Res. 95, 1457–1463. doi: 10.1177/0022034516663200
Rajpar, M. H., Harley, K., Laing, C., Davies, R. M., and Dixon, M. J. (2001). Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum. Mol. Genet. 10, 1673–1677. doi: 10.1093/hmg/10.16.1673
Ravassipour, D. B., Powell, C. M., Phillips, C. L., Hart, P. S., Hart, T. C., Boyd, C., et al. (2005). Variation in dental and skeletal open bite malocclusion in humans with amelogenesis imperfecta. Arch. Oral Biol. 50, 611–623. doi: 10.1016/j.archoralbio.2004.12.003
Seelow, D., Schuelke, M., Hildebrandt, F., and Nurnberg, P. (2009). HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res. 37, W593–W599. doi: 10.1093/nar/gkp369
Seymen, F., Kim, Y. J., Lee, Y. J., Kang, J., Kim, T. H., Choi, H., et al. (2016). Recessive mutations in ACPT, encoding testicular acid phosphatase, cause hypoplastic amelogenesis imperfecta. Am. J. Hum. Genet. 99, 1199–1205. doi: 10.1016/j.ajhg.2016.09.018
Seymen, F., Park, J. C., Lee, K. E., Lee, H. K., Lee, D. S., Koruyucu, M., et al. (2015). Novel MMP20 and KLK4 mutations in amelogenesis imperfecta. J. Dent. Res. 94, 1063–1069. doi: 10.1177/0022034515590569
Smith, C. E., Murillo, G., Brookes, S. J., Poulter, J. A., Silva, S., Kirkham, J., et al. (2016). Deletion of amelotin exons 3-6 is associated with amelogenesis imperfecta. Hum. Mol. Genet. 25, 3578–3587. doi: 10.1093/hmg/ddw203
Wang, S. K., Choi, M., Richardson, A. S., Reid, B. M., Lin, B. P., Wang, S. J., et al. (2014). ITGB6 loss-of-function mutations cause autosomal recessive amelogenesis imperfecta. Hum. Mol. Genet. 23, 2157–2163. doi: 10.1093/hmg/ddt611
Wang, S. K., Hu, Y., Simmer, J. P., Seymen, F., Estrella, N. M., Pal, S., et al. (2013). Novel KLK4 and MMP20 mutations discovered by whole-exome sequencing. J. Dent. Res. 92, 266–271. doi: 10.1177/0022034513475626
Witkop, C. J. Jr. (1988). Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J. Oral Pathol. 17, 547–553. doi: 10.1111/j.1600-0714.1988.tb01332.x
Wright, J. T., Hart, P. S., Aldred, M. J., Seow, K., Crawford, P. J., Hong, S. P., et al. (2003). Relationship of phenotype and genotype in X-linked amelogenesis imperfecta. Connect Tissue Res. 44(Suppl. 1), 72–78. doi: 10.1080/03008200390152124
Wright, J. T., Torain, M., Long, K., Seow, K., Crawford, P., Aldred, M. J., et al. (2011). Amelogenesis imperfecta: genotype-phenotype studies in 71 families. Cells Tissues Organs 194, 279–283. doi: 10.1159/000324339
Keywords: amelogenesis imperfecta, enamelysin, proteinase, enamel, matrix, hypomaturation
Citation: Kim YJ, Kang J, Seymen F, Koruyucu M, Gencay K, Shin TJ, Hyun H-K, Lee ZH, Hu JCC, Simmer JP and Kim J-W (2017) Analyses of MMP20 Missense Mutations in Two Families with Hypomaturation Amelogenesis Imperfecta. Front. Physiol. 8:229. doi: 10.3389/fphys.2017.00229
Received: 10 February 2017; Accepted: 31 March 2017;
Published: 20 April 2017.
Edited by:
Ariane Berdal, UMRS 1138 INSERM University Paris-Diderot Team POM, FranceReviewed by:
Flavia Martinez De Carvalho, Oswaldo Cruz Foundation, BrazilRafaela Scariot De Moraes, Universidade Positivo, Brazil
Agnes Bloch-Zupan, University of Strasbourg, France
Copyright © 2017 Kim, Kang, Seymen, Koruyucu, Gencay, Shin, Hyun, Lee, Hu, Simmer and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jung-Wook Kim, pedoman@snu.ac.kr