 Chun Gu
Chun Gu Fang Wang1
Fang Wang1- 1State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 2Key Laboratory of Analytical Chemistry for Living Biosystems, Institute of Chemistry Chinese Academy of Science, Beijing, China
- 3Department of Pathology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Lysophosphatidic acid (LPA), one of the simplest phospholipid signaling molecules, participates in formation and disruption of atherosclerotic plaque. Matrix metalloproteinases (MMPs) contribute to atherosclerotic plaque rupture by involving in extracellular matrix (ECM) degradation and then thinning fibrous cap. Our previous study demonstrated that macrophage-derived MMP-9 was associated with coronary plaque instability, but the relationship between LPA and MMP-9 remains unclear. The present work therefore aimed at elucidating association between LPA and MMP-9 and the regulation mechanism of LPA on MMP-9 in macrophages. We found that plasma LPA and MMP-9 levels were correlated positively (r = 0.31, P < 0.05) and both elevated significantly in patients with acute myocardial infarct (AMI). Consistent with peripheral blood levels, histochemical staining indicated that autotaxin (ATX), LPA-producing ectoenzyme, and MMP-9 were expressed frequently in the necrotic core and fibrous cap of human unstable plaques, which might increase the instability of plaque. Experiments in vitro were done with THP-1-derived macrophages and showed that LPA enhanced the expression, secretion and activity of MMP-9 in a time- and dose-dependent manner. Induction of LPA on pro-MMP-9 and active-MMP-9 was confirmed in human peripheral blood monocyte-derived macrophages. PDTC, NF-κB inhibitor, but not inhibitor of AP-1 and PPARγ, effectively prevented LPA-induced MMP-9 expression and NF-κB p65 siRNA decreased MMP-9 transcription, confirming that LPA might induce MMP-9 elevation by activating NF-κB pathway. In addition, knockdown of LPA2 attenuated LPA-induced MMP-9 expression and nucleus p65 levels. These findings revealed that LPA upregulated the expression of MMP-9 through activating NF-κB pathway in the LPA2 dependent manner, hence blocking LPA receptors signaling may provide therapeutic strategy to target plaque destabilization.
Introduction
Acute coronary syndromes (ACS) is a sudden attack without warning, even life-threatening as a result of the atherosclerosis, one of the leading causes of cardiovascular death worldwide. A postmortem study shows that 75% of ACS arises from atherosclerosis plaque rupture (Narula et al., 2013), but the complicated mechanisms underlying plaque destabilization remain fragmentary. Recent evidence suggests that stable atherosclerotic plaques are rich in smooth muscle cells (SMCs) and collagen, whereas unstable plaques are characterized by a large necrotic core covered with a thin fibrous cap and a highly inflammatory cell content (Libby et al., 2011; Silvestre-Roig et al., 2014). Abundant macrophage infiltration and extracellular matrix (ECM) degradation are mainly contributed to necrotic core expansion and fibrous cap thinning, respectively (Moore and Tabas, 2011).
Matrix metalloproteinases (MMPs) are zinc-dependent enzymes and involved in ECM degradation and remodeling, which may hasten plaque disruption (Ketelhuth and Back, 2011; Muller et al., 2014). MMPs are produced by many vascular wall cells, but macrophages are critical in human plaques (Newby, 2008). It has been reported that elevated levels of MMP-1, -3, -8, -9, -12, and -13 are found in macrophage-rich regions of human atherosclerotic plaques (Sukhova et al., 1999; Morgan et al., 2004; Higashikata et al., 2006; Sluijter et al., 2006; Quillard et al., 2011). In particular, the increased content and activity of MMP-9 are associated with plaque stability. ApoE-deficient mice model indicates that the loss of MMP-9 reduces impaired macrophage infiltration and collagen deposition (Luttun et al., 2004), whereas the overexpression of MMP-9 promotes intraplaque hemorrhage and induces acute plaque disruption (de Nooijer et al., 2006; Gough et al., 2006). Our previous study demonstrated macrophage-derived MMP-9 was associated with coronary plaque instability (Fan et al., 2014), and therefore it is of great interest to discuss the regulation mechanism of MMP-9.
Lysophosphatidic acid (LPA), generated by autotaxin (ATX) (Moolenaar and Perrakis, 2011), is a bioactive and endogenous phospholipid signaling factor, regulating cell proliferation, differentiation, migration, survival and apoptosis through the activation of at least six known G protein-coupled receptors (GPCRs), LPA1–LPA6 (Yung et al., 2014). It is found that LPA progressively accumulates in human and mouse atherosclerotic plaques in contrast with normal vessels (Bot et al., 2010; Dohi et al., 2013). Actually, LPA participates in the most processes of atherogenesis and thrombosis, such as promoting adhesion molecule expression in endothelial cells (ECs) (Lee et al., 2004), stimulating SMCs migration (Damirin et al., 2007), inducing foam cell formation (Chang et al., 2008). However, whether LPA plays a role in ECM degradation and what's the downstream signaling pathway in macrophages have not been fully elucidated.
In the present study, we observed the correlation between circulating LPA and MMP-9 levels as well as expression in the advanced atherosclerotic plaques. Furthermore, we also investigated the detailed mechanisms of LPA on MMP-9 in macrophages—via NF-κB signaling in the LPA2 dependent manner, and thus LPA receptors (LPARs) might be potential therapeutic target to prevent plaque rupture.
Materials and Methods
Study Population
Twenty-four patients with unstable angina (UA) and twenty-four patients with non-ST-segment elevation myocardial infarction (NSTEMI) were recruited in Fuwai Hospital (Beijing, China) between March 2013 and January 2014 (Table 1). The diagnoses of UA/NSTEMI were all in line with the standards of the Chinese Medical Society of Cardiology. Twenty-four healthy persons served as a control group with normal echocardiograms (ECGs), negative exercise stress ECGs based on physical examination and laboratory tests and without coronary heart disease history. Patients with inflammatory diseases, acute myocardial injury, severe heart failure, malignant disease, impaired liver function, renal failure, or recent surgery were excluded.
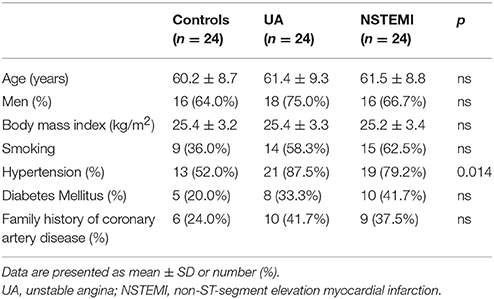
Table 1. Characteristics of patients and controls.
The coronary atherosclerotic plaques were collected from explanted hearts donated by three patients undergoing heart transplantation and stored in liquid nitrogen in Fuwai Hospital. According to histomorphologic features, atherosclerotic plaques were classified as follows: unstable plaques were defined as the lesions with thin fibrous cap (<65 μm) covering large lipid core (>40% plaque volume); stable plaques were described as the lesions with well-matured necrotic core (<40% plaque volume) covered by thick fibrous cap (>65 μm). All specimens were carefully perfusion-fixed in 10% buffered formalin.
The study protocol conformed to the Declaration of Helsinki and was approved by the ethics review board of Fu Wai Hospital and all the patients gave written informed consent.
Hematoxylin-Eosin (H&E) and Immunohistochemistry (IHC) Staining
Formalin-fixed paraffin embedded tissue were cuted into 4-μm-thick sections and immersed in xylene and rehydrated with graded alcohols and subjected to H&E staining before examination by light microscopy.
IHC was performed by the standard protocol. Briefly, antigen retrieval was performed in 0.01 M citrate buffer (pH 6.0), followed by quenching of endogenous peroxidase activity with 3% H2O2 in methanol. Sections were then incubated with MMP-9 (Abcam, Cambridge, UK) and ATX (as described Li and Zhang, 2009) primary antibodies overnight at 4°C. Powervision horseradish peroxidase-IgG was used as secondary antibody. Sections were visualized using diaminobenzidine and counter stained with hematoxylin.
Cell Culture
Human monocytic cell line THP-1 (a gift from Prof. Junjie Zhang, Beijing Normal University, China) and maintained at a density of 106/ml in RPMI 1640 medium (Gibco, CA, USA) containing 10% fetal bovine serum (FBS) and 1% penicillin/Streptomycin, and then incubated at 37°C with 5% CO2. Cells were cultured in six-well plates for 48 h in the presence of phorbol 12-myristate 13-acetate (PMA) (100 ng/mL, Sigma, St Louis, USA), which induced the differentiation into adherent macrophages (Aldo et al., 2013). After serum-starvation for 18–24 h, cells were treated with 0 to 50 μM LPA (oleoyl C 18:1, Avanti Polar Lipids, Alabaster, AL), or pretreated 0.1 μM Actinomycin D (Act.D), 0.1 μM Cycloheximide (CHX), 10 μM GW9662 (PPARγ inhibitor), 10 μM PDTC (NF-κB inhibitor) and 10 μM Tan II A (AP-1 inhibitor) for 1 h.
Isolation of peripheral blood mononuclear cells (PBMCs) and culture of macrophages were performed using classical methods (van Grevenynghe et al., 2003). Briefly, PBMCs were isolated through Ficoll-Paque™ PLUS (GE Healthcare, Piscataway, NJ, USA) gradient centrifugation within 1 h after blood collection. Cells were seeded in culture flasks with RPMI 1640 medium supplemented with 10% FBS and 1% penicillin/Streptomycin for 2 h to adhesion. The adherent monocytes were then added recombinant human granulocyte macrophage colony-stimulating factor (rhGM-CSF) (PeproTech, New Jersey, USA) for 7 days.
Transfection of siRNA For LPA Receptors
Small interfering RNAs (siRNAs) for LPA1, LPA2, and LPA6 were transfected using Lipofectamine™ RNAiMAX according to manufacturer's protocols. The target sequences of these siRNAs were listed as follow:
LPA1 Stealth siRNA duplexes (LPA1-siRNA) targeting sequences: 5′-GAA AUG AGC GCC ACC UUU A-3′and 5′-UAA AGG UGG CGC UCA UUU C -3′; LPA2 Stealth siRNA duplexes (LPA2-siRNA) targeting sequences: 5′-GGU CAA UGC UGC UGU GUA C-3′ and 5′-GUA CAC AGC AGC AUU GAC C-3′; LPA6 Stealth siRNA duplexes (LPA6-siRNA) targeting sequences: 5′-UCA GCA UGG UGU UUG UGC UUG GGU U-3′ and 5′-AAC CCA AGC ACA AAC ACC AUG CUG A-3′. NF-κB p65-siRNA targeting sequences: 5′-GCC CUA UCC CUU UAC GUC A-3′ and 5′-UGA CGU AAA GGG AUA GGG C-3′. The scrambled siRNA controls were used as a negative control.
Quantitative Real Time PCR (qRT-PCR)
Total RNA was isolated from THP-1 derived macrophages using Trizol according to the manufacture's instruction and then quantified by NANODROP 2000 spectrophotometer. cDNA was generated from 2 μg total RNA using M-MLV reverse transcriptase and oligo (dT) 15 primer. qRT -PCR was performed using SYBR GREEN PCR Master Mix in the Applied Biosystems 7300 (Foster city, CS, USA). All gene specific extron primers used in the present study were as follows:
LPA1: 5′-GCT GCC ATC TCT ACT TCC AT-3′ and 5′-CCA TTC TGT GGC AAG ATG CT-3′; LPA2: 5′-CAG CGC ATG GCA GAG CAT GT-3′ and 5′-CCA GGA CAT TGC AGG ACT CAC A-3′; LPA3: 5′-AAC GTG AGC GGA TGT TCA CT-3′ and 5′-CCG CGA TGA CCA GAG AAT TA-3′; LPA4: 5′-TTC GAA CTA ATG TGG AGG AA-3′ and 5′-TGG AAT TGG AAG TCA ATG AA-3′; LPA5: 5′-CAG AGC AAC ACG GAG CAC AG-3′ and 5′-CAC CAG AAT CAT GGC ATG GC-3′; LPA6: 5′-CTT CAC AAC ACG GAA TTG GC-3′ and 5′-AGT TAA CCA CAC GCC AGT GC-3′; MMP-9: 5′-TGG AAA ATT ATT GCG CCT CT-3′ and 5′-CCT CGA TGA GAC CAT CAA CA-3′; GAPDH: 5′-ACT AGG CGC TCA CTG TTC TC-3′ and 5′-GCC CAA TAC GAC CAA ATC CG-3′; NF-κB p65: 5'-GCG AGA GGA GCA CAG ATA CC-3' and 5'-CTG ATA GCC TGC TCC AGG TC-3'. The relative expression of each gene was normalized to GAPDH by subtracting the corresponding GAPDH threshold cycle (Ct) values using comparative 2−ΔΔCt method.
Western Blot Analysis
The treated cells were washed with ice-cold phosphate-buffered saline (PBS) twice and then lysed in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM PMSF and 1% sodium deoxycholate) for 30 min on ice. The BCA assay was used to quantify the extract protein concentration. Equal amounts of protein (20 μg/lane) were separated on 10% SDS-PAGE gels by electrophoresis and then transferred to nitrocellulose membranes using semi-dry electroblotting apparatus. The membranes were blocked for 2 h at room temperature in 5% skim milk and subsequently incubated with primary antibodies overnight at 4°C. In the following day, the members were washed and incubated with secondary antibody for 2 h. Protein bands were visualized by FluorChem M system (Protein Simple, USA) and quantified by densitometry with Quantity One software. The target signals were normalized to the GAPDH levels.
Luciferase Reporter Assay
THP-1 derived macrophages were cultured on 96-well plates (105 cells/well) for 48 h and stimulated with 10 μM LPA for 24 h. Reporter plasmid REPO™NF-κB and REPO™AP-1 (Genomeditech, Shanghai, China) were transfected using Lipofectamine 2000. Luciferase activity, quantified as relative luminescence units (firefly luminescence/Renilla luminescence), was measured with the Dual-Glo® Luciferase Assay System (Promega, Madison, WI) according to the manufacturer's instructions using a microplate reader (Infinite 200, Tecan).
Gelatin Zymography
The MMPs activities were assayed as described (Zhang et al., 2014). Briefly, 2 × 106 cells in a 6-well plate were cultured in serum-free medium and 10 μL supernatants were loaded on 8% SDS-PAGE gels containing 2 mg/ml gelatin under non-denaturing conditions. The gels were washed with 2.5% Triton X-100 and incubated overnight in buffer (150 mM NaCl, 5 mM CaCl2, 50 mM Tris-HCl, pH 7.6) at 37°C. Following staining with Coomassie brilliant blue R250, the gelatinolytic activities were detected as clear bands against a blue background. The scan of the gel was analyzed by densitometry using FluorChem M system (Protein Simple, USA).
Enzyme Linked Immunosorbent Assay (Elisa)
Levels of MMP-9 in plasma or conditioned medium from THP-1 derived macrophages were quantified by the human MMP-9 Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. All plasma samples were diluted 1:100 and the conditioned mediums were diluted 1:10 when testing.
Electrospray Ionization Tandem Mass Spectrometry
Detection of plasma LPA levels were carried out by Prof. Zhenwen Zhao (Institute of Chemistry Chinese Academy of Science, Beijing, China) through high performance-liquid-chromatography electrospray ionization tandem mass spectrometry (HPLCESI-MS/MS) as described (Zhao and Xu, 2009).
Statistical Analysis
All statistical data were presented as mean ± SD and categorical variables were represented as percentages. Comparisons among the groups were performed using the chi-square test or one-way ANOVA. Bivariate correlation analysis was used to analyze the relations of plasma LPA and MMP-9 levels. Two-sided P-values were used and P < 0.05 were considered statistically significant. All the statistical analyses were performed using SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA).
Results
The Plasma LPA and MMP-9 Levels Are Elevated Significantly in Patients with Acute Myocardial Infarction
We measured the plasma levels of LPA and MMP-9 in patients with AMI (n = 24), UA (n = 24) and asymptomatic controls (n = 24) and the data showed that both LPA and MMP-9 with higher levels in plasma in AMI group compared with that in the control group (LPA: 0.66 ± 0.23 vs. 0.42 ± 0.16, P < 0.05; MMP-9: 775.5 ± 242.4 vs. 532.1 ± 204.1, P < 0.05), but no significant difference was found between patients with UA and controls (Figures 1A,B). Pearson correlation analysis showed that there was a positive correlation between plasma LPA and MMP-9 levels (r = 0.31, P < 0.05) (Figure 1C).
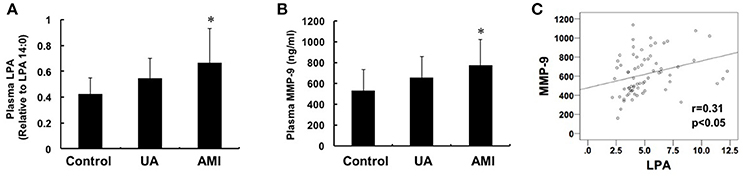
Figure 1. Changes of plasma LPA and MMP-9 levels among control, unstable angina and acute myocardial infarction groups. Plasma LPA (A) and MMP-9 (B) levels were detected by mass spectrometry and ELISA, respectively. The levels of MMP-9 and LPA in the group with AMI were both higher than the control and UA groups (P < 0.05). (C) Pearson correlation analysis showed that there is a positive correlation between LPA and MMP-9 (r = 0.31, P < 0.05). *P < 0.05, compared with control.
The Expression of ATX and MMP-9 Increases in Human Unstable Plaques
The positive correlation between LPA and MMP-9 in the peripheral blood was found, but the presence and localization of them in the local tissue are unclear. ATX, LPA-producing ectoenzyme, were detected because LPA is a bioactive phospholipid molecules and it is difficult to detect in the tissue. As shown in Figures 2A–C, normal artery was characterized by uniform intima-media thickness (IMT) and smooth vascular lumen, stable plaque with large lipid core but thick fibrous cap and the large lipid core and rupture-prone fibrous cap in unstable plaques. The positive staining for MMP-9 was lacked in normal artery (Figure 2D) and mainly located in the shoulder of necrotic core and cap region in stable plaque (Figure 2E). In contrast, unstable plaque showed strong staining for MMP-9 in and surrounding the lesion (Figure 2F). The positive staining for ATX was found in normal artery, stable and unstable plaque and it seemed somewhat a change in distribution. ATX is mainly distributed in the vascular smooth muscle layer and accumulated in the necrotic core and fibrous cap with the increase of plaque instability (Figures 2G–I). Furthermore, distribution patterns of MMP-9 and ATX overlapped in unstable plaque although were different.
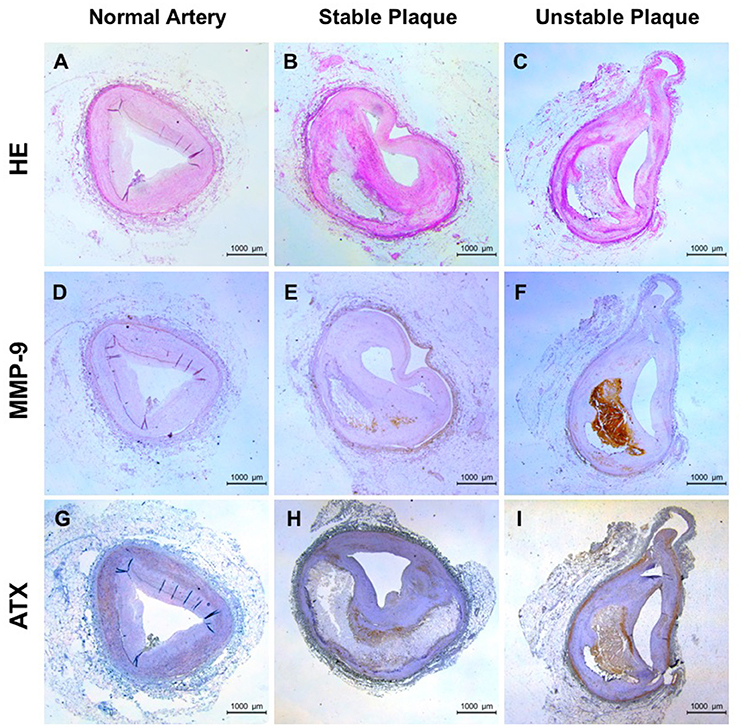
Figure 2. Expression of ATX and MMP-9 in human coronary plaque tissues. (A,D,G) for normal arteries; (B,E,H) for stable plaques; (C,F,I) for unstable plaques; (A–C) HE staining; (D–F) Immunohistochemical staining of MMP-9; (G–I) Immunohistochemical staining of ATX.
LPA Enhances the Expression, Secretion and Activation of MMP-9 in Macrophage
To evaluate the effect of LPA on MMP-9, we first treated THP-1-derived macrophages with different concentrations of LPA (0, 1, 5, 10, 25, 50 μM) for 24 h. The expression and secretion of MMP-9 increased from 1 μM of LPA in dose-dependent manner and reached maximum at 10 μM of LPA (Figures 3A–C). Meanwhile, Figure 3D revealed that the ability of MMP-9 to degrade collagen was enhanced with the strongest at 10 μM of LPA, while MMP-2 made no responses to LPA. As shown in Figures 3E–G, mRNA levels, protein syntheses and secretion of MMP-9 increased time-dependently with 10 μM LPA for 4–48 h. Additionally, the protein levels of pro-MMP-9 and active-MMP-9 were both increased with LPA treatment in human peripheral blood monocyte-derived macrophages (Figure 3H).
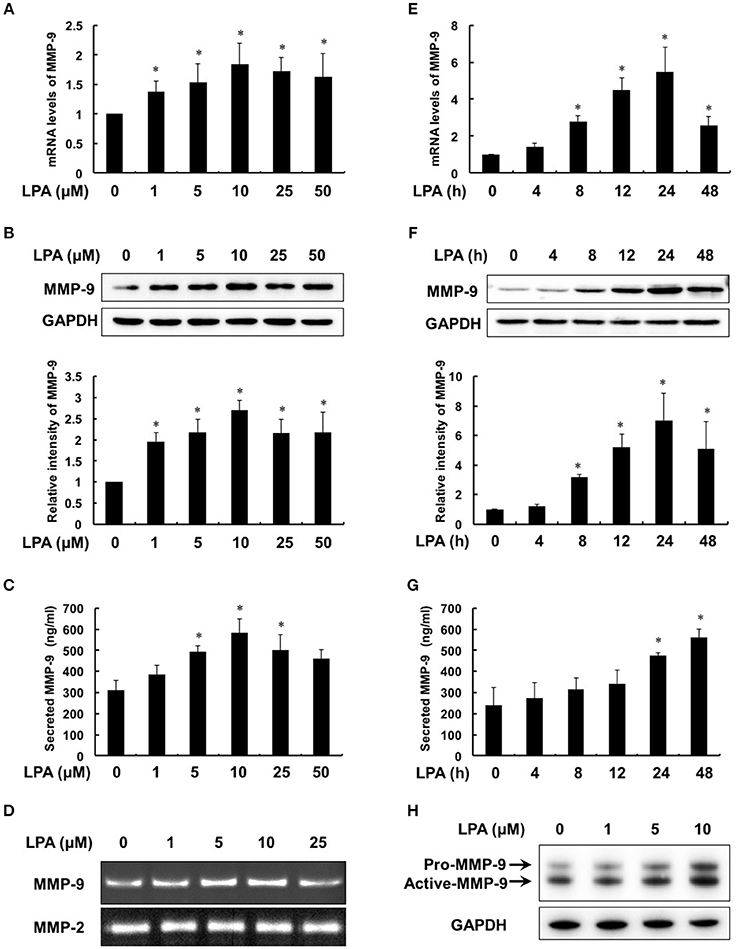
Figure 3. LPA induced the expression, secretion and activity of MMP-9 in a dose- and time- dependent manner. Macrophages were treated with the pointed concentrations of LPA for 24 h or with 10 μM LPA for the pointed time course. The mRNA levels (A,E), protein levels (B,F), secretion (C,G) and activity (D) of MMP-9 were detected by RT-PCR, Western Blot, ELISA and gelatin zymography, respectively. LPA induced MMP-9 expression in human peripheral blood monocyte-derived macrophages (H). Data were expressed as mean ± SD of three independent experiments. *P < 0.05, compared with control.
NF-κB Activation Is Required for LPA-Induced MMP-9 Expression
MMPs are regulated at the levels of transcription, secretion, activation and physiological suppression of tissue inhibitor of matrix metalloproteinases (TIMPs). To determine the regulation at transcriptional or post transcriptional, Act.D and CHX were used to inhibit synthesis of nascent mRNA and protein of MMP-9, respectively. As shown in Figures 4A–C, the expression of MMP-9 failed to increase after pretreatment with Act.D or CHX followed by LPA exposure. It indicated that up-regulation of LPA-induced MMP-9 occurred in the de novo synthesis of mRNA, namely transcription. Thus, we analyzed promoter region of MMP-9 and filtered out three possible transcription factors: PPARγ, NF-κB and AP-1. As shown in Figures 4D,E, after pretreatment with PPARγ inhibitor GW9662 and AP-1 inhibitor, the basic levels of MMP-9 reduced, but LPA-induced MMP-9 still increased, indicating that PPARγ and AP-1 were not involved in the regulating process of LPA on MMP-9. In contrast, LPA-induced MMP-9 expression was markedly attenuated by NF-κB inhibitor PDTC and siRNA for NF-κB p65 (Figures 4E,G,H), and transcriptional activity of NF-κB was up-regulated by LPA as expected (Figure 4F).
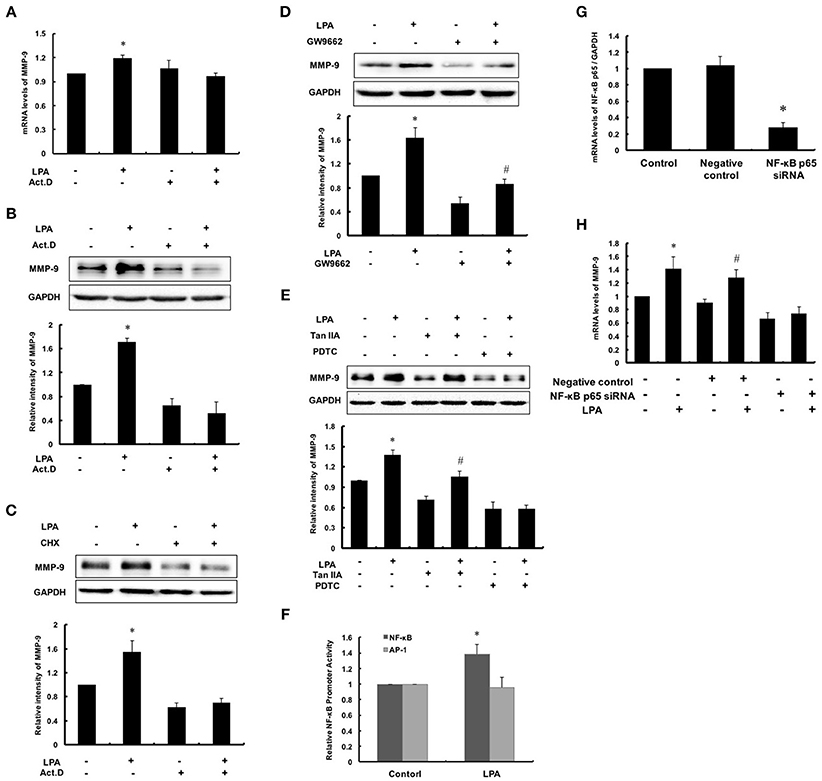
Figure 4. NF-κB activation was required for LPA-induced MMP-9 expression. After pretreating with 0.1 μM Act.D (A,B) and 0.1 μM CHX (C) for 1 h to inhibit mRNA and protein synthesis, and then cells were incubated with 10 μM LPA for 24 h to detect MMP-9 expression. (D,E) Cells were pretreated with 10 μM GW9662 (PPARγ inhibitor), 10 μM PDTC (NF-κB inhibitor) and 10 μM TanIIA (AP-1 inhibitor) respectively, and then incubated with 10 μM LPA for 24 h. The expression of MMP-9 was determined. (F) The NF-κB and AP-1 promoter activity was measured. The mRNA expression of NF-κB p65 (G) and MMP-9 (H) were determined after interference of NF-κB p65-siRNA. Data were expressed as mean ± SD of three independent experiments. *P < 0.05, compared with control; #P < 0.05, compared with inhibitor treatment groups.
LPA Induces MMP-9 Expression through LPA2
LPA exerts various biological effects through different G protein-coupled receptors: LPA1–LPA6, and therefore we investigated which subtype of LPA receptors mediated MMP-9 expression. Since LPA1, LPA2, and LPA6 were highly expressed in THP-1-derived macrophages (Figure 5A), we separately knocked down the three LPA receptors using specific siRNAs and detected the MMP-9 expression with or without LPA treatment. As shown in Figure 5B, LPA1, LPA2, and LPA6 were effectively knocked down and LPA-induced MMP-9 expression and nucleus p65 levels were attenuated significantly by inhibition of LPA2 (Figures 5C–E). These data suggested that LPA2 was involved in LPA-induced upregulation of MMP-9 and activation of NF-κB.
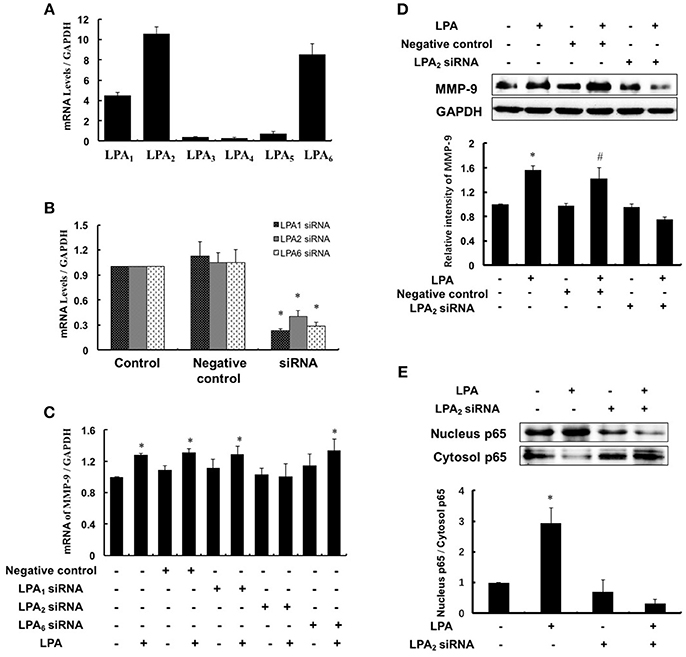
Figure 5. LPA2 mediated LPA-induced MMP-9 expression. (A) The expression of LPAR was detected in THP-1-derived macrophages. The mRNA expression of LPAR (B) and MMP-9 (C) were determined after interference of LPA1-siRNA, LPA2-siRNA and LPA6-siRNA, respectively. The expression of MMP-9 (D), nucleus and cytoplasm p65 (E) were determined after interference of LPA2-siRNA. Data were expressed as mean ± SD of three independent experiments. *P < 0.05, compared with control. #P < 0.05, compared with negative control.
Discussion
In the present study, we found a positive correlation between LPA and MMP-9 concentrations in peripheral blood, which was in accordance with the overlapping distribution of ATX and MMP-9 in the human atherosclerotic plaques. Experiments in vitro clarified that LPA could effectively promote the expression and activity of MMP-9 mainly by binding to G protein-coupled LPA2 receptor that in turn activated NF-κB pathways.
Thinning of the fibrous cap is a crucial step that precedes plaque rupture and consequent secondary thrombotic events. A number of studies have revealed that SMCs contribute to cap thickening by producing elastin, collagen and other matrix components in stable atherosclerotic lesions; while during plaque destabilization, reduced matrix protein deposition resulted in weakening fibrous cap (Libby, 2013). In our study, patients with unstable plaques exhibit significantly elevated LPA/ATX levels in both circulation and vascular tissues, suggesting that LPA may contribute to atherosclerotic plaque instability. This is supported by recent reports demonstrating that LPA contributes to expansion of necrotic core by accumulation of ox-LDL induced macrophages (Schober and Siess, 2012), intraplaque hemorrhage by recruitment of mast cells (Bot et al., 2013), and thrombus formation by induction of platelet aggregation (Pamuklar et al., 2008; Bolen et al., 2011). Macrophages, mainly in necrotic cores (Gautier et al., 2009; Tabas, 2010), are an important source of MMPs for ECM degradation. We found a correlation in plasma and overlapped distribution in macrophage-rich lesions between LPA (ATX) and MMP-9, suggesting a relationship between LPA and MMP-9. Hence, whether LPA could participate in the regulation of the matrix composition through MMP-9 is of high interest. This study validated this hypothesis and established a detailed mechanism underlying LPA on MMP-9 expression in macrophages and provided a new perspective for the progression of atherosclerosis plaque destabilization.
MMP-9 (also known as gelatinase B, 92 kDa collagenase) is one of a family of endopeptidases that cleaves most ECM proteins, particularly collagen types IV and V (Pourmotabbed et al., 1994; Buraczynska et al., 2015). Strong evidence indicates that MMP-9 could contribute to wakening and rupture of atherosclerotic plaques. Consistent with our results, plasma MMP-9 in patients with UA, AMI, and carotid artery stenosis have been found elevated compared to controls (Kai et al., 1998; Ferroni et al., 2003; Renko et al., 2004; Tziakas et al., 2006; Taurino et al., 2007), even similar finding from urine (Fitzsimmons et al., 2007). High levels of MMP-9 are also found in plaque shoulders and regions of foam cells accumulation (Galis et al., 1994). Additionally, genetic studies have demonstrated associations between MMP-9 polymorphism and severity of coronary heart disease (CHD) (Pollanen et al., 2001; Blankenberg et al., 2003). The expression and activity of MMP-9 are regulated on several levels, including transcription, secretion, activation, and inhibition (Vandooren et al., 2013). In our study, Act.D and CHX experiments revealed that LPA-induced MMP-9 expression was up-regulated at the de novo synthesis of mRNA - transcriptional level. A variety of transcription factors for regulating MMP-9 are investigated, such as NF-κB (Dilly et al., 2013; Tseng et al., 2013; Yan et al., 2016), activator protein-1 (AP-1) (Lian et al., 2015; Yang et al., 2016), specificity protein 1 (Sp-1) (Murthy et al., 2012) and peroxisome proliferator-activated receptor-γ (PPARγ) (Lee et al., 2009; Chen et al., 2013). We showed that LPA-mediated MMP-9 expression in macrophages is dependent on NF-κB signaling through inhibitor experiments and promoter analysis.
As we know, six LPA receptor subtypes (LPA1–LPA6) have been discovered so far and mediate the diverse physiological effects of LPA. The expression profiles of LPARs are often different from different cell types. We displayed the distribution of LPA1–6 in THP-1-derived macrophages and found LPA1, LPA2, and LPA6 mRNA were highly expressed. Growing evidence has implicated that LPARs involved in pathophysiologic progression of atherosclerosis. LPA promotes smooth muscle progenitor cell recruitment for neointima formation and releases proinflammatory cytokines from ECs to heighten atherosclerotic plaque burden in an LPA1- and LPA3-receptor dependent manner (Subramanian et al., 2010; Zhou et al., 2011). Zheng et al. (2001) show that LPA stimulates T lymphocytes migration via LPA2. Sumida et al. (2010) indicate that Lpar4−/− mice exhibit embryonic lethality in association with hemorrhage, suggesting that LPA4 may account for post-development blood vessel formation. LPA5 in particular has been reported to mediate LPA signals leading to platelet activation and aggregation (Williams et al., 2009). Almost every subtype of primary or cultured vascular cells exhibits some responses to LPA via LPARs (Smyth et al., 2014). In this study, we highlighted that LPA2 was responsible for LPA-induced NF-κB activation and MMP-9 expression in macrophages, which may provide promising target for therapeutic intervention of atherosclerosis.
Nevertheless, several limitations of the study are worth mentioning. Firstly, the sample size for cardiovascular events (n = 48) and coronary plaques (n = 3) was both small and correlation between LPA and MMP-9 plasma levels might be validated in a larger patient cohort. Secondly, the molecular mechanism by which LPA induced MMP-9 expression was not explored and further in vivo and appropriate animal models remained necessary. Thirdly, we detected LPA distribution in the plaques by ATX antibody staining due to the instability in LPA measurement caused by plaques acquisition and storage. Indeed, LPA levels from heterozygous ATX-null mice were approximately half those from wild-type mice (Tanaka et al., 2006).
In summary, this study demonstrated synchronous increases of LPA and MMP-9 in peripheral blood and overlapping distribution of them in local atherosclerotic plaques. The vitro experiments in macrophages revealed that LPA induced MMP-9 in a time- and dose-dependent manner, mainly by LPA2 to activate NF-κB signaling pathways. Therefore, intervention in LPARs may be an effective therapeutic strategy to target plaque destabilization.
Author Contributions
XC, XfC, FW, and CG conceived and designed the research; CG, HW, and ZZ performed the experiments; CG and FW analyzed the data. CG, XC, and XfC wrote and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The project was supported by the National Natural Science Foundation of China (81371887 and 81470484) and National Key Technology Support Program (2011BAI11B03).
References
Aldo, P. B., Craveiro, V., Guller, S., and Mor, G. (2013). Effect of culture conditions on the phenotype of THP-1 monocyte cell line. Am. J. Reprod Immunol. 70, 80–86. doi: 10.1111/aji.12129
Blankenberg, S., Rupprecht, H. J., Poirier, O., Bickel, C., Smieja, M., Hafner, G., et al. (2003). Plasma concentrations and genetic variation of matrix metalloproteinase 9 and prognosis of patients with cardiovascular disease. Circulation 107, 1579–1585. doi: 10.1161/01.CIR.0000058700.41738.12
Bolen, A. L., Naren, A. P., Yarlagadda, S., Beranova-Giorgianni, S., Chen, L., Norman, D., et al. (2011). The phospholipase A1 activity of lysophospholipase A-I links platelet activation to LPA production during blood coagulation. J. Lipid Res. 52, 958–970. doi: 10.1194/jlr.M013326
Bot, M., Bot, I., Lopez-Vales, R., van de Lest, C. H., Saulnier-Blache, J. S., Helms, J. B., et al. (2010). Atherosclerotic lesion progression changes lysophosphatidic acid homeostasis to favor its accumulation. Am. J. Pathol. 176, 3073–3084. doi: 10.2353/ajpath.2010.090009
Bot, M., de Jager, S. C., MacAleese, L., Lagraauw, H. M., van Berkel, T. J., Quax, P. H., et al. (2013). Lysophosphatidic acid triggers mast cell-driven atherosclerotic plaque destabilization by increasing vascular inflammation. J. Lipid Res. 54, 1265–1274. doi: 10.1194/jlr.M032862
Buraczynska, K., Kurzepa, J., Ksiazek, A., Buraczynska, M., and Rejdak, K. (2015). Matrix Metalloproteinase-9 (MMP-9) gene polymorphism in stroke patients. Neuromolecular Med. 17, 385–390. doi: 10.1007/s12017-015-8367-5
Chang, C. L., Hsu, H. Y., Lin, H. Y., Chiang, W., and Lee, H. (2008). Lysophosphatidic acid-induced oxidized low-density lipoprotein uptake is class A scavenger receptor-dependent in macrophages. Prostaglandins Other Lipid Mediat. 87, 20–25. doi: 10.1016/j.prostaglandins.2008.05.002
Chen, R., Xue, J., and Xie, M. (2013). Osthole regulates TGF-beta1 and MMP-2/9 expressions via activation of PPARalpha/gamma in cultured mouse cardiac fibroblasts stimulated with angiotensin II. J. Pharm. Pharm. Sci. 16, 732–741. doi: 10.18433/J3HK5C
Damirin, A., Tomura, H., Komachi, M., Liu, J. P., Mogi, C., Tobo, M., et al. (2007). Role of lipoprotein-associated lysophospholipids in migratory activity of coronary artery smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 292, H2513–H2522. doi: 10.1152/ajpheart.00865.2006
de Nooijer, R., Verkleij, C. J., von der Thusen, J. H., Jukema, J. W., van der Wall, E. E., van Berkel, T. J., et al. (2006). Lesional overexpression of matrix metalloproteinase-9 promotes intraplaque hemorrhage in advanced lesions but not at earlier stages of atherogenesis. Arterioscler. Thromb. Vasc. Biol. 26, 340–346. doi: 10.1161/01.ATV.0000197795.56960.64
Dilly, A. K., Ekambaram, P., Guo, Y., Cai, Y., Tucker, S. C., Fridman, R., et al. (2013). Platelet-type 12-lipoxygenase induces MMP9 expression and cellular invasion via activation of PI3K/Akt/NF-κB. Int. J. Cancer 133, 1784–1791. doi: 10.1002/ijc.28165
Dohi, T., Miyauchi, K., Ohkawa, R., Nakamura, K., Kurano, M., Kishimoto, T., et al. (2013). Increased lysophosphatidic acid levels in culprit coronary arteries of patients with acute coronary syndrome. Atherosclerosis 229, 192–197. doi: 10.1016/j.atherosclerosis.2013.03.038
Fan, X., Wang, E., Wang, X., Cong, X., and Chen, X. (2014). MicroRNA-21 is a unique signature associated with coronary plaque instability in humans by regulating matrix metalloproteinase-9 via reversion-inducing cysteine-rich protein with Kazal motifs. Exp. Mol. Pathol. 96, 242–249. doi: 10.1016/j.yexmp.2014.02.009
Ferroni, P., Basili, S., Martini, F., Cardarello, C. M., Ceci, F., Di Franco, M., et al. (2003). Serum metalloproteinase 9 levels in patients with coronary artery disease: a novel marker of inflammation. J. Investig. Med. 51, 295–300. doi: 10.1136/jim-51-05-17
Fitzsimmons, P. J., Forough, R., Lawrence, M. E., Gantt, D. S., Rajab, M. H., Kim, H., et al. (2007). Urinary levels of matrix metalloproteinase 9 and 2 and tissue inhibitor of matrix metalloproteinase in patients with coronary artery disease. Atherosclerosis 194, 196–203. doi: 10.1016/j.atherosclerosis.2006.07.027
Galis, Z. S., Sukhova, G. K., Lark, M. W., and Libby, P. (1994). Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Invest. 94, 2493–2503. doi: 10.1172/JCI117619
Gautier, E. L., Huby, T., Witztum, J. L., Ouzilleau, B., Miller, E. R., Saint-Charles, F., et al. (2009). Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation 119, 1795–1804. doi: 10.1161/CIRCULATIONAHA.108.806158
Gough, P. J., Gomez, I. G., Wille, P. T., and Raines, E. W. (2006). Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J. Clin. Invest. 116, 59–69. doi: 10.1172/JCI25074
Higashikata, T., Yamagishi, M., Higashi, T., Nagata, I., Iihara, K., Miyamoto, S., et al. (2006). Altered expression balance of matrix metalloproteinases and their inhibitors in human carotid plaque disruption: results of quantitative tissue analysis using real-time RT-PCR method. Atherosclerosis 185, 165–172. doi: 10.1016/j.atherosclerosis.2005.05.039
Kai, H., Ikeda, H., Yasukawa, H., Kai, M., Seki, Y., Kuwahara, F., et al. (1998). Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J. Am. Coll. Cardiol. 32, 368–372.
Ketelhuth, D. F., and Back, M. (2011). The role of matrix metalloproteinases in atherothrombosis. Curr. Atheroscler. Rep. 13, 162–169. doi: 10.1007/s11883-010-0159-7
Lee, C. S., Kwon, Y. W., Yang, H. M., Kim, S. H., Kim, T. Y., Hur, J., et al. (2009). New mechanism of rosiglitazone to reduce neointimal hyperplasia: activation of glycogen synthase kinase-3beta followed by inhibition of MMP-9. Arterioscler. Thromb. Vasc. Biol. 29, 472–479. doi: 10.1161/ATVBAHA.108.176230
Lee, H., Lin, C. I., Liao, J. J., Lee, Y. W., Yang, H. Y., Lee, C. Y., et al. (2004). Lysophospholipids increase ICAM-1 expression in HUVEC through a Gi- and NF-κB-dependent mechanism. Am. J. Physiol,. Cell Physiol. 287, C1657–C1666. doi: 10.1152/ajpcell.00172.2004
Li, S., and Zhang, J. (2009). Lipopolysaccharide induces autotaxin expression in human monocytic THP-1 cells. Biochem. Biophys. Res. Commun. 378, 264–268. doi: 10.1016/j.bbrc.2008.11.047
Lian, S., Xia, Y., Khoi, P. N., Ung, T. T., Yoon, H. J., Kim, N. H., et al. (2015). Cadmium induces matrix metalloproteinase-9 expression via ROS-dependent EGFR, NF-κB and AP-1 pathways in human endothelial cells. Toxicology 338, 104–116. doi: 10.1016/j.tox.2015.10.008
Libby, P. (2013). Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 368, 2004–2013. doi: 10.1056/NEJMra1216063
Libby, P., Ridker, P. M., and Hansson, G. K. (2011). Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325. doi: 10.1038/nature10146
Luttun, A., Lutgens, E., Manderveld, A., Maris, K., Collen, D., Carmeliet, P., et al. (2004). Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation 109, 1408–1414. doi: 10.1161/01.CIR.0000121728.14930.DE
Moolenaar, W. H., and Perrakis, A. (2011). Insights into autotaxin: how to produce and present a lipid mediator. Nat. Rev. Mol. Cell Biol. 12, 674–679. doi: 10.1038/nrm3188
Moore, K. J., and Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355. doi: 10.1016/j.cell.2011.04.005
Morgan, A. R., Rerkasem, K., Gallagher, P. J., Zhang, B., Morris, G. E., Calder, P. C., et al. (2004). Differences in matrix metalloproteinase-1 and matrix metalloproteinase-12 transcript levels among carotid atherosclerotic plaques with different histopathological characteristics. Stroke 35, 1310–1315. doi: 10.1161/01.STR.0000126822.01756.99
Muller, A., Kramer, S. D., Meletta, R., Beck, K., Selivanova, S. V., Rancic, Z., et al. (2014). Gene expression levels of matrix metalloproteinases in human atherosclerotic plaques and evaluation of radiolabeled inhibitors as imaging agents for plaque vulnerability. Nucl. Med. Biol. 41, 562–569. doi: 10.1016/j.nucmedbio.2014.04.085
Murthy, S., Ryan, A. J., and Carter, A. B. (2012). SP-1 regulation of MMP-9 expression requires Ser586 in the PEST domain. Biochem. J. 445, 229–236. doi: 10.1042/BJ20120053
Narula, J., Nakano, M., Virmani, R., Kolodgie, F. D., Petersen, R., Newcomb, R., et al. (2013). Histopathologic characteristics of atherosclerotic coronary disease and implications of the findings for the invasive and noninvasive detection of vulnerable plaques. J. Am. Coll. Cardiol. 61, 1041–1051. doi: 10.1016/j.jacc.2012.10.054
Newby, A. C. (2008). Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler. Thromb. Vasc. Biol. 28, 2108–2114. doi: 10.1161/ATVBAHA.108.173898
Pamuklar, Z., Lee, J. S., Cheng, H. Y., Panchatcharam, M., Steinhubl, S., Morris, A. J., et al. (2008). Individual heterogeneity in platelet response to lysophosphatidic acid: evidence for a novel inhibitory pathway. Arterioscler. Thromb. Vasc. Biol. 28, 555–561. doi: 10.1161/ATVBAHA.107.151837
Pollanen, P. J., Karhunen, P. J., Mikkelsson, J., Laippala, P., Perola, M., Penttila, A., et al. (2001). Coronary artery complicated lesion area is related to functional polymorphism of matrix metalloproteinase 9 gene: an autopsy study. Arterioscler. Thromb. Vasc. Biol. 21, 1446–1450. doi: 10.1161/hq0901.095545
Pourmotabbed, T., Solomon, T. L., Hasty, K. A., and Mainardi, C. L. (1994). Characteristics of 92 kDa type IV collagenase/gelatinase produced by granulocytic leukemia cells: structure, expression of cDNA in E. coli and enzymic properties. Biochim. Biophys. Acta 1204, 97–107.
Quillard, T., Tesmenitsky, Y., Croce, K., Travers, R., Shvartz, E., Koskinas, K. C., et al. (2011). Selective inhibition of matrix metalloproteinase-13 increases collagen content of established mouse atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 31, 2464–2472. doi: 10.1161/ATVBAHA.111.231563
Renko, J., Kalela, A., Jaakkola, O., Laine, S., Hoyhtya, M., Alho, H., et al. (2004). Serum matrix metalloproteinase-9 is elevated in men with a history of myocardial infarction. Scand. J. Clin. Lab. Invest. 64, 255–261. doi: 10.1080/00365510410006054
Schober, A., and Siess, W. (2012). Lysophosphatidic acid in atherosclerotic diseases. Br. J. Pharmacol. 167, 465–482. doi: 10.1111/j.1476-5381.2012.02021.x
Silvestre-Roig, C., de Winther, M. P., Weber, C., Daemen, M. J., Lutgens, E., and Soehnlein, O. (2014). Atherosclerotic plaque destabilization: mechanisms, models, and therapeutic strategies. Circ. Res. 114, 214–226. doi: 10.1161/CIRCRESAHA.114.302355
Sluijter, J. P., Pulskens, W. P., Schoneveld, A. H., Velema, E., Strijder, C. F., Moll, F., et al. (2006). Matrix metalloproteinase 2 is associated with stable and matrix metalloproteinases 8 and 9 with vulnerable carotid atherosclerotic lesions: a study in human endarterectomy specimen pointing to a role for different extracellular matrix metalloproteinase inducer glycosylation forms. Stroke 37, 235–239. doi: 10.1161/01.STR.0000196986.50059.e0
Smyth, S. S., Mueller, P., Yang, F., Brandon, J. A., and Morris, A. J. (2014). Arguing the case for the autotaxin-lysophosphatidic acid-lipid phosphate phosphatase 3-signaling nexus in the development and complications of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 34, 479–486. doi: 10.1161/ATVBAHA.113.302737
Subramanian, P., Karshovska, E., Reinhard, P., Megens, R. T., Zhou, Z., Akhtar, S., et al. (2010). Lysophosphatidic acid receptors LPA1 and LPA3 promote CXCL12-mediated smooth muscle progenitor cell recruitment in neointima formation. Circ. Res. 107, 96–105. doi: 10.1161/CIRCRESAHA.109.212647
Sukhova, G. K., Schonbeck, U., Rabkin, E., Schoen, F. J., Poole, A. R., Billinghurst, R. C., et al. (1999). Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation 99, 2503–2509.
Sumida, H., Noguchi, K., Kihara, Y., Abe, M., Yanagida, K., Hamano, F., et al. (2010). LPA4 regulates blood and lymphatic vessel formation during mouse embryogenesis. Blood 116, 5060–5070. doi: 10.1182/blood-2010-03-272443
Tabas, I. (2010). Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 10, 36–46. doi: 10.1038/nri2675
Tanaka, M., Okudaira, S., Kishi, Y., Ohkawa, R., Iseki, S., Ota, M., et al. (2006). Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J. Biol. Chem. 281, 25822–25830. doi: 10.1074/jbc.M605142200
Taurino, M., Raffa, S., Mastroddi, M., Visco, V., Rizzo, L., Torrisi, M. R., et al. (2007). Metalloproteinase expression in carotid plaque and its correlation with plasma levels before and after carotid endarterectomy. Vasc. Endovasc. Surg. 41, 516–521. doi: 10.1177/1538574407307405
Tseng, H. C., Lee, I. T., Lin, C. C., Chi, P. L., Cheng, S. E., Shih, R. H., et al. (2013). IL-1β promotes corneal epithelial cell migration by increasing MMP-9 expression through NF-κB- and AP-1-dependent pathways. PLoS ONE 8:e57955. doi: 10.1371/journal.pone.0057955
Tziakas, D. N., Chalikias, G. K., Hatzinikolaou, E. I., Stakos, D. A., Tentes, I. K., Kortsaris, A., et al. (2006). Alteplase treatment affects circulating matrix metalloproteinase concentrations in patients with ST segment elevation acute myocardial infarction. Thromb. Res. 118, 221–227. doi: 10.1016/j.thromres.2005.07.014
Vandooren, J., Van den Steen, P. E., and Opdenakker, G. (2013). Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit. Rev. Biochem. Mol. Biol. 48, 222–272. doi: 10.3109/10409238.2013.770819
van Grevenynghe, J., Rion, S., Le Ferrec, E., Le Vee, M., Amiot, L., Fauchet, R., et al. (2003). Polycyclic aromatic hydrocarbons inhibit differentiation of human monocytes into macrophages. J. Immunol. 170, 2374–2381. doi: 10.4049/jimmunol.170.5.2374
Williams, J. R., Khandoga, A. L., Goyal, P., Fells, J. I., Perygin, D. H., Siess, W., et al. (2009). Unique ligand selectivity of the GPR92/LPA5 lysophosphatidate receptor indicates role in human platelet activation. J. Biol. Chem. 284, 17304–17319. doi: 10.1074/jbc.M109.003194
Yan, W., Fan, W., Chen, C., Wu, Y., Fan, Z., Chen, J., et al. (2016). IL-15 up-regulates the MMP-9 expression levels and induces inflammatory infiltration of macrophages in polymyositis through regulating the NF-κB pathway. Gene 591, 137–147. doi: 10.1016/j.gene.2016.06.055
Yang, C. C., Hsiao, L. D., Yang, C. M., and Lin, C. C. (2016). Thrombin Enhanced Matrix Metalloproteinase-9 Expression and Migration of SK-N-SH Cells via PAR-1, c-Src, PYK2, EGFR, Erk1/2 and AP-1. Mol. Neurobiol. doi: 10.1007/s12035-016-9916-0. [Epub ahead of print].
Yung, Y. C., Stoddard, N. C., and Chun, J. (2014). LPA receptor signaling: pharmacology, physiology, and pathophysiology. J. Lipid Res. 55, 1192–1214. doi: 10.1194/jlr.R046458
Zhang, W., Wang, F., Xu, P., Miao, C., Zeng, X., Cui, X., et al. (2014). Perfluorooctanoic acid stimulates breast cancer cells invasion and up-regulates matrix metalloproteinase-2/-9 expression mediated by activating NF-κB. Toxicol. Lett. 229, 118–125. doi: 10.1016/j.toxlet.2014.06.004
Zhao, Z., and Xu, Y. (2009). Measurement of endogenous lysophosphatidic acid by ESI-MS/MS in plasma samples requires pre-separation of lysophosphatidylcholine. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 877, 3739–3742. doi: 10.1016/j.jchromb.2009.08.032
Zheng, Y., Kong, Y., and Goetzl, E. J. (2001). Lysophosphatidic acid receptor-selective effects on Jurkat T cell migration through a Matrigel model basement membrane. J. Immunol. 166, 2317–2322. doi: 10.4049/jimmunol.166.4.2317
Keywords: lysophosphatidic acid, matrix metalloproteinase-9, macrophages, LPA2, coronary atherosclerotic plaques
Citation: Gu C, Wang F, Zhao Z, Wang H, Cong X and Chen X (2017) Lysophosphatidic Acid Is Associated with Atherosclerotic Plaque Instability by Regulating NF-κB Dependent Matrix Metalloproteinase-9 Expression via LPA2 in Macrophages. Front. Physiol. 8:266. doi: 10.3389/fphys.2017.00266
Received: 26 February 2017; Accepted: 11 April 2017;
Published: 27 April 2017.
Edited by:
Jincai Luo, Peking University, ChinaCopyright © 2017 Gu, Wang, Zhao, Wang, Cong and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Chen, chenxifw@pumc.edu.cn