 Beibei Luo1Feng Huang2Yanli Liu1Yiying Liang1Zhe Wei1Honghong Ke1Zhiyu Zeng1*Weiqiang Huang1*
Beibei Luo1Feng Huang2Yanli Liu1Yiying Liang1Zhe Wei1Honghong Ke1Zhiyu Zeng1*Weiqiang Huang1* Yan He1*
Yan He1*- 1Department of Geriatric Cardiology, The First Affiliated Hospital of Guangxi Medical University, Nanning, China
- 2Department of Cardiology, Institute of Cardiovascular Diseases, The First Affiliated Hospital of Guangxi Medical University, Nanning, China
Diabetic cardiomyopathy (DCM), a common consequence of longstanding diabetes mellitus, is initiated by death of cardiomyocyte. Hyperglycemia-induced reactive oxygen species (ROS) overproduction is a major contributor of the chronic low-grade inflammation that characterizes as the DCM. ROS may promote the activation of nucleotide-binding oligomerization domain like receptor (NLR) pyrin domain containing 3 (NLRP3) inflammasome, a novel regulator of inflammation and cell death, by nuclear factor-kB (NF-κB) and thioredoxin interacting/inhibiting protein (TXNIP). NLRP3 inflammasome regulates the death of cardiomyocyte and activation of fibroblast in DCM, which is involved in the structural and functional disorder of DCM. However, comprehensive understanding of molecular mechanisms linking NLRP3 inflammasome and disorder of cardiomyocyte and fibroblast in DCM is lacking. Here, we review the molecular mechanism(s) of NLRP3 inflammasome activation in response to hyperglycemia in DCM.
Introduction
Diabetic cardiomyopathy (DCM), one of the severe complication of diabetes mellitus, is the leading cause of death in diabetes patients (Shaw et al., 2010). DCM is characterized by structural and functional disorders, including myocardial cell death, myocardial fibroblast activation, left ventricular dysfunction, and metabolic deregulation (Westermeier et al., 2015). Among them, the death of cardiomyocyte is thought to be the basic change of DCM, which initiates cardiac remodeling and results in left ventricular dysfunction (Kuethe et al., 2007; Liu et al., 2013).
From the standpoints of cellular death and inflammation, a newly described inflammatory mechanism fundamental to innate immune system is proposed to contribute to DCM (Luo et al., 2014a,b). In particular, the nucleotide-binding oligomerization domain like receptor (NLR) pyrin domain containing 3 (NLRP3) inflammasome, expressed abundantly in cardiomyotytes, may play important roles in the process of myocardial cell death.
A growing list of molecules has been examined as possible molecular markers for heart failure (HF) and cardiovascular disease (CVD), exemplified by the myocyte injury markers troponins I and T, the myocyte stress markers Brain natriuretic peptide (BNP) and ST2/IL-33, and the inflammation markers C-reactive protein (CRP) and certain cytokines (TNF, IL-1β, and IL-18; Braunwald, 2008). Troponin I and T are used to diagnose acute myocardial infarction and prognose thrombotic ACS. BNP and NT-pro-BNP are used to predict death or re-hospitalization for HF in a number of conditions and distinguish acute from chronic HF. ST2/IL-33 also can be used to predict cardiac damage and disease progression. CRP measurement is a useful tool for determining prognosis in HF and myocarditis patients. According to the “cytokine hypothesis,” cytokines play important roles in the pathogenesis of HF and CVD and are valuable to indicate inflammation and disease progression.
Increasing evidence reveals that NLRP3 inflammasome can be used as a promising molecular marker for future development of effective and targeted therapies with lower toxicity in DCM and other CVD. We found that the protective effect of the anti-inflammation drug rosuvastatin (RSV) was associated with its ability to inhibit the activation of NLRP3 inflammasome via TXNIP and suppress the phosphorylation of MAPK signal pathways in DCM (Luo et al., 2014a). The synthetized NLRP3 inflammasome inhibitor INF4E was also found to protect against the IR-induced myocardial injury by inhibiting NLRP3 inflammasome, leading to the activation of the prosurvival RISK pathway and improvement in mitochondrial function (Mastrocola et al., 2016b). Since previous anti-inflammatory drug therapy can lead to compromised host defense or secondary inflammatory processes due to the compensatory responses, targeting specific inflammatory pathways (such as NLRP3 inflammasome) may provide a more precise approach to reducing deleterious inflammation without affecting other innate host defense (Butts et al., 2015). Moreover, blocking early inflammatory mediators such as TXNIP (as well as NLRP3 inflammasome components) that significantly induced by hyperglycemia will represent the excellent targets to prevent diabetes and its complications DCM (Singh, 2014). In this review, we will summarize the available evidence, including the regulatory mechanism and biological function of NLRP3 inflammasome in DCM, which will elucidate the significance of NLRP3 inflamamsome as the molecular marker of DCM.
NLRP3 Inflammasome Biology
NLRP3 inflammasome consists of NLRP3, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and pro-caspase-1 (Shao et al., 2015). NLRP3 contains three domains: a C-terminal leucine-rich repeats domain (LRRs), a central nucleotide binding and oligomerization domain (NACHT), and an N-terminal pyrin domain (PYD). ASC is composed of a C-terminal caspase recruitment domain (CARD) and an N-terminal PYD. Pro-caspase-1 is consists of a CARD, a p20 and a p10 domain (Jo et al., 2016). Once activated, the PYD of NLRP3 could bind to that of ASC, and subsequently the CARD of ASC recruits and interacts with pro-caspase-1. These interactions form the NLRP3 inflammasome and promote the autocatalytic cleavage of pro-caspase-1, which results in the production of activated caspase-1 (Lamkanfi and Dixit, 2014).
The activated caspase-1 has two major functions: the regulated function that processes pro-IL-18 and pro-IL-1β into their mature forms (IL-18 and IL-1β) and the novel function that induces pyroptosis. Accumulated evidence indicated that IL-18 and IL-1β were important proinflammatory cytokines in the development of CVD by suppressing cardiac contractility, promoting myocardial hypertrophy, and inducing cardiomyocyte apoptosis (Loppnow et al., 2001; Apostolakis et al., 2008; Qamar and Rader, 2012). Pyroptosis, an inflammatory form of programmed cell death, was dependent on caspase-1 activity (Guarda and So, 2010). The morphology of pyroptosis partially overlaps with both apoptosis and necrosis (Coll et al., 2011). Similar to apoptotic cells, pyroptotic cells incur DNA damage, TUNEL staining positive, annexin V staining positive (Miao et al., 2011). As in necrosis, pyroptosis triggers pore formation in the cell membrane, release of pro-inflammatory cytosolic content, and cell lysis. Pyroptosis has been found in response to infection with several bacteria and viruses (Allen et al., 2009; Bergsbaken et al., 2009). However, aberrant or excessive activation of the NLRP3 inflammasome contribute to development of autoimmune and even metabolic diseases, such as type 2 diabetes (Donath and Shoelson, 2011), atherosclerotic disease (Rajamaki et al., 2016; Wang et al., 2017), obesity (Ahmad et al., 2016), gouty Arthritis (Liu Y. F. et al., 2016; Table 1). Recently, we and other researchers reported that pyroptosis were involved in non-infectious disease, such as atherosclerosis and DCM (Luo et al., 2014b; Wree et al., 2014).

Table 1. NLRP3 inflammasome and metabolic disorders.
The Activation of NLRP3 Inflammasome in DCM
A diverse set of pathogen- and host-derived ligands can activate the NLRP3 inflammasome. These include pathogen-associated molecular patterns (PAMPs), bacterial pore-forming toxins, hemozoin, silica, asbestos, UV light, ATP, glucose, MSU, calcium pyrophosphate dehydrate, amyloid β, hyaluronan, alum, danger-associated molecular patterns (DAMPs), and environmental stimuli (Schroder et al., 2010). The stimuli of NLRP3 inflammasome were classically seen to converge on three distinct signal pathways: (1) NLRP3 senses potassium efflux and loss of membrane integrity through purinergic receptor P2X7 and pannexin-1, respectively; (2) crystalline or particulate-accelerated lysosomal destabilization promote cathepsin B to direct NLRP3 ligands; (3) DAMP/PAMP-induced reactive oxygen species (ROS) trigger the activation of NLRP3 inflammasome (Zhou et al., 2010). Recent researches have indicated various other activating mechanisms of NLRP3 inflammasome. Among these, mitochondrial dysfunction was thought to be the pivotal step for the NLRP3 inflammasome activation induced by NLRP3 agonists. Mitochondria can promote NLRP3 inflammasome activation through mitochondrial-derived signals, such as mROS, mtDNA, and cadiolipin (Zhou et al., 2010; Nakahira et al., 2011; Iyer et al., 2013). Besides mitochondria, ER-stress, guanylate-binding protein 5 (GBP5), and double-stranded RNA-dependent protein kinase (PKR) were also shown to be correlated with NLRP3 inflammasome activation (Lerner et al., 2012; Lu et al., 2012; Shenoy et al., 2012).
During NLRP3 inflammasome activation, the agonist can induce an initial priming step (the first signal) that promotes NLRP3 expression, followed by the structural modulation step (the second signal) that induces NLRP3 inflammasome assembly. Nuclear factor-kB (NF-κB), which controls the transcriptional induction of NLRP3, was shown to provide the first signal in the activation of NLRP3 inflammasome (Corsini et al., 2013; Liao et al., 2013; Luo et al., 2014b). Thioredoxin interacting/inhibiting protein (TXNIP), deubiquitination, and oxidized mitochondrial DNA were suggested to provide the second signal by binding with NLRP3 directly and modulating its oligomerization (Zhou et al., 2010; Luo et al., 2014b). Selected DAMPs associated with atherosclerosis are found to provide both signals for inflammasome activation (Robbins et al., 2014).
Type 1 diabetes, resulting from the autoimmune destruction of insulin-producing β cells in the pancreas, is characterized by low level of insulin and hyperglycemia (Nakayama et al., 2015). Most diabetes patients have type 2 diabetes, which is characterized by hyperglycemia, hyperlipidemia, and insulin resistance (Pandey et al., 2015). Several studies have indicated that NLRP3 inflammasome expression was increased in circumstance of glycotoxicity and lipotoxicity (Zhou et al., 2010; Vandanmagsar et al., 2011). Furthermore, NLRP3 inflammsome could be activated by saturated fatty acid, ceramides, modified LDL, and hyperglycemia in obesity and type 2 diabetes (Wen et al., 2011; Jin and Flavell, 2013). During the process of DCM, DAMP -induced ROS generation is the most well-studied pathway of NLRP3 inflammasome activation. Our previous studies have addressed the role of NLRP3 inflammasome in DCM using the rat model of T2DM (Luo et al., 2014a,b). High glucose-mediated ROS generation could upregulate NF-κB phosphorylation and TXNIP expression, which account for NLRP3 priming and the secondary step of activation (Bryant and Fitzgerald, 2009; Franchi et al., 2012; Kumar et al., 2013). NF-κB has been shown to increase the expression of NLRP3, pro-caspase-18 and pro-IL-1β, which facilitating the activation of NLRP3 inflammasome (Donath et al., 2010; Qiao et al., 2012; Boaru et al., 2015). TXNIP has been reported to be another essential link between ROS and NLRP3 inflammasome by priming the expression of NLRP3 inflammasome or modulating the structure of NLRP3 directly (Martinon, 2010).
Lipotoxicity occurring in type 2 diabetes plays a non-negligible role on inflammasome assembly. Free fatty acid might induce the activation of NLRP3 inflammasome by ROS production and ER stress (Legrand-Poels et al., 2014). And intramyocellular lipid accumulation in cardiomyocytes has been frequently reported in diet-induced diabetes (Wolf et al., 2016; Zlobine et al., 2016). However, there was no evidence that intramyocellular lipid could promote the activation of intercellular NLRP3 inflammasome directly.
NLRP3 Inflammasome-Mediated Inflammation in DCM
Glucose has been reported to be one of the effective activators of NLRP3 inflammasome (Shi H. et al., 2015; Zu et al., 2015). Recent data also suggest that NLRP3 is responsible for the cardiac inflammation of glycotoxicity during the process of T2DM and DCM (Vandanmagsar et al., 2011; Luo et al., 2014b). IL-1β and IL-18 are the main effectors of ROS-mediated NLRP3 inflammasome activation in DCM. IL-1β and IL-18 have a central role in the cardiomyocyte apoptosis and fibroblast activation of DCM, which is thought to be the initiator of the structural disorder (Santiago et al., 2014; Somanna et al., 2015). NLRP3 inflammasome-mediated pyroptosis in DCM.
Pyroptosis is a caspase-1- or caspase-11-dependent cell death process, which is characterized by plasma membrane pore formation, cell swelling, cell osmotic lysis, and release of proinflammastory intracellular contents (de Zoete et al., 2014; Shalini et al., 2015). Pyroptosis was firstly reported in the research of the infection with Shigell flexneri and Salmonell typhimurium, which was considered to be key responses of immune response to pathogens (Zychlinsky et al., 1992; Monack et al., 1996). Subsequent studies discovered that pyroptosis also occurred in cells induced by non-infectious stimuli (Jang et al., 2015; Lebeaupin et al., 2015; Lopez-Pastrana et al., 2015). Within the type 2 diabetes rat model and H9c2 cardiomyoctye cell line, our previous work showed the important characteristics of pyroptosis in myocardium and cardiomyocyte, including activated caspase-1, cytoplasmic swelling, and nucleus DNA damage. NLRP3-siRNA lentivirus treatment, followed by reduced activation of caspase-1, abrogated the pyroptosis both in the myocardium of DCM and the high glucose-treated cardiomyocyte. NLRP3 inflammasome turned out to be a pivotal regulator of caspase-1-depended pyroptosis in the pathogenesis of DCM.
Not much research has been done to explore the key molecules involved in caspase-1-induced pyroptosis. Recent studies identified that the protein gasdermin D (GSDMD), encoded by a gene named Gsdmd, was required and sufficient for pyroptosis in mouse and human cells (He et al., 2015; Kayagaki et al., 2015; Shi J. et al., 2015; Shi et al., 2017). Mechanistically, caspase-1 and other inflammatory caspases (caspase-4/5/11) cleave GSDMD into two fragments, and the resulting amino-terminal fragment promotes pyroptosis by its pore-forming activity to rupture the membrane (Ding et al., 2016; Liu X. et al., 2016). Given the similar pore-forming activity in different gasdermins, the concept of pyroptosis is thus redefined as gasdermin-mediated programmed necrotic cell death (Shi et al., 2017). Other pyroptosis-inducing caspase-1 substrates still await to be discovered.
NLRP3 Inflammasome-Promoted Fibrosis in DCM
Our previous work showed that NLRP3-siRNA lentivirus treatment reduced the aberrant expression of collage I and III in myocardium of DCM rat model (Luo et al., 2014b). Other research revealed that NLRP3 inflammasome suppressed the cAMP expression of cardiac fibroblast in mice with sepsis, which inhibited the cardiac contraction (Zhang et al., 2014). With TGF-β stimulation, NLRP3 inflammasome increased in cardiac fibroblast, which facilitated the myofibroblast and receptor associated Smad (R-Smad) activation. The activated R-Smad could associate with co-Smad to form the transcription complex that promotes the expression of profibrotic gene (Bracey et al., 2014). Whether the R-Smad is the pivotal link between NLRP3 inflammasome and high glucose-induced cardiac fibroblast activation remains unknown.
Conclusions
NLRP3 inflammasome is a multiprotein signaling complex of the innate immune system essential for controlling the inflammatory response and coordinating antimicrobial host defense. Hyperglycemia-induced ROS overproduction is a major contributor of chronic low-grade inflammation. Therefore, NLRP3-dependent pyroptosis and maturation of pro-inflammatory effectors (IL-1β/IL-18) induced by ROS could contribute to development of autoimmune and even metabolic diseases, such as DCM.
Identification of NLRP3 inflammasome platforms is the main breakthrough on DCM research, as it is in the area of HF (Butts et al., 2015) and other cardiometabolic diseases (Janket et al., 2015; Mastrocola et al., 2016a). ROS-dependent NF-κB and TXNIP appear to regulate both priming and post-translational steps in the activation of NLRP3 inflammasome. After NLRP3 inflammasome activation, caspase-1 promotes a novel programmed cell death process, named pyroptosis, in cardiomyocyte of DCM. Pro-inflammatory cytokines IL-1β and IL-18 are direct substrates of caspase-1. Caspase-1 is activated by the NLRP3 inflammasome, in which a central platform, consisted of NLRP3, ASC1, and caspase1, recognizes an unknown signal or ligand. Active caspase-1 then cleaves and maturates of these cytokines and triggers pyroptosis. Together with this observation, caspase-1-mediated IL-1β and IL-18 activation initiate apoptosis of cardiomyocyte and activation of cardiofibroblast. On the other hand, NLRP3 inflammasome can induce fibrosis in DCM (Figure 1). Further investigations on the mechanism underlying caspase-1-regulated pyroptosis and IL-18/IL-1β-regulated fibroblast disorder are required to elucidate the function of NLRP3 inflammasome in DCM.
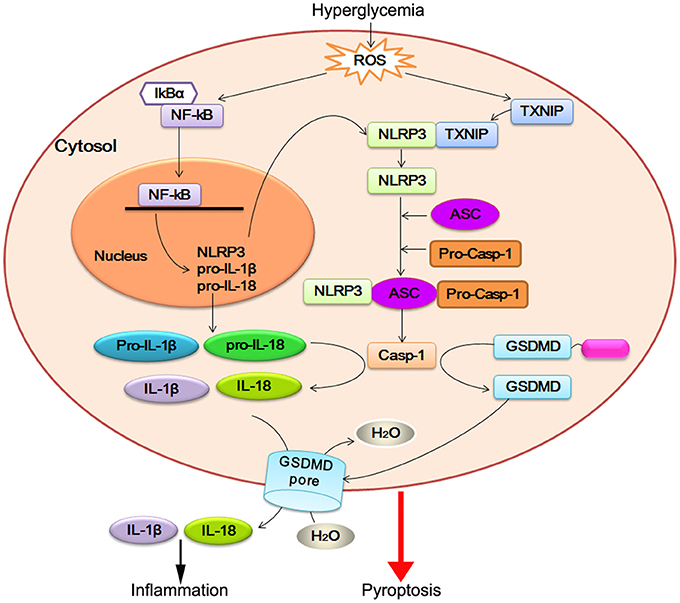
Figure 1. NLRP3 inflammasome activation in DCM. Hyperglycemia-induced reactive oxygen species (ROS) leads to nuclear factor-kB (NF-κB) and TXNIP overexpression. NF-κB increases the expression of NLRP3, pro-IL-18, and pro-IL-1β. TXNIP modulates the biological structure of NLRP3 leading to NLRP3 inflammasome assembly and pro-caspase-1 (pro-casp-1) autocleavage. Active caspase-1 (Casp-1) promotes pro-IL-18 and pro-IL-1β maturation, which facilitate inflammatory reaction. On the other hand, active caspase-1 cleaves GSDMD within the linker between its N-terminal (blue) and C-terminal (magenta). The released GSDMD-N domain oligomerizes to generate membrane pores, which disrupts the osmotic potential and leads to cell swelling and eventual lysis.
As in DCM, NLRP3 inflammasome also plays an important role in myocardial ischemia/reperfusion (I/R) injury after high-fat high-fructose (HFHF) diet, which is a common type of cardiometabolic disease (Mastrocola et al., 2016a). The HFHF diet mouse showed excessive intake of fatty acids and sugars in cardiomyocyte, which induced the overexpression of ROS and then triggered the activation of NLRP3 inflamamsome. The upregulation of NLRP3 inflammasome enhanced the susceptibility to (I/R) injury by promoting the mitochondrial disorder via IL-1β and IL-18. On the other hand, the protective pathways of Reperfusion Injury Salvage Kinases (RISK) pathway (including AKT, ERK, and GSK-3β) and hypoxia inducible factor-2α (HIF-2α) was inhibited by fatty acids and sugar. The impairment of RISK/HIF-2α pathway can worsen the mitochondrial oxidative stress regulated by NLRP3 inflammasome, which increase the susceptibility to I/R injury in HFHF diet mouse (Figure 2).
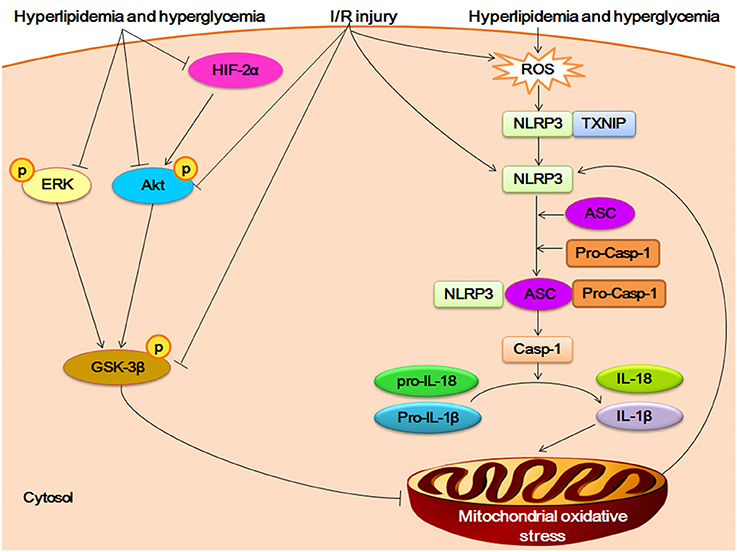
Figure 2. Crosstalk between NLRP3 inflammasome and RISK/HIF-2α in myocardial I/R injury. The HFHF diet induced-hyperlipidemia and hyperglycemia promote overexpression of ROS and then triggers the assembly of NLRP3 inflamamsome. The activation of NLRP3 inflammasome increase the secretion of IL-1β and IL-18. The proinflammatory cytokines accelerate the oxidative stress of mitochondrial, which in turn promotes the activation of NlRP3 inflammasome. Hyperlipidemia and hyperglycemia inhibit the phosphorylation of Reperfusion Injury Salvage Kinases (RISK) pathway (including AKT, ERK, and GSK-3β), and negatively impacts hypoxia inducible factor-2α (HIF-2α), which can worsen the mitochondrial oxidative unbalance. The I/R challenge can suppress the phosphorylation of AKT and GSK-3β, and induce the expression of ROS and activation of NLRP3.
As mentioned above, molecular markers are important tools for clinical management in CVD, facilitating in early detection of disease, diagnostic, monitor of disease state, assessment of disease risk, determination of therapy, and evaluation of therapeutic activity (Braunwald, 2008). Inflammation associated markers (CRP, cytokines and the NLRP3 inflammasome) are important in that they are also risk factors directly involved in the pathogenesis of CVD. Of these, the NLRP3 inflammasome is unique in that it provides a mechanistic explanation of cytokine activation which leads to disease progression (Luo et al., 2014a; Mastrocola et al., 2016b). Therefore, understanding the molecular mechanism of the NLRP3 inflammasome activation and targeting specifically the NLRP3 inflammasome and its interacting counterparts (e.g., TXNIP) will be of great value in clinical management of DCM and other CVD (Singh, 2014; Butts et al., 2015).
Author Contributions
BL and ZZ wrote the manuscript. FH, YaL, and YiL revised the language of the manuscript. ZW and HK prepared the figure and figure legend. BL, WH, and YH conceived the review.
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 81600185), the Province Natural Science Foundation of Guangxi (grant no. 2016GXNSFBA380019), and the Province Young Teacher Foundation of Guangxi (grant no. KY2016YB078).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Zhenhua Ming for discussion and critical reading of the article.
References
Ahmad, F., Chung, Y. W., Tang, Y., Hockman, S. C., Liu, S., Khan, Y., et al. (2016). Phosphodiesterase 3B (PDE3B) regulates NLRP3 inflammasome in adipose tissue. Sci. Rep. 6:28056. doi: 10.1038/srep28056
Allen, I. C., Scull, M. A., Moore, C. B., Holl, E. K., McElvania-TeKippe, E., Taxman, D. J., et al. (2009). The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565. doi: 10.1016/j.immuni.2009.02.005
Apostolakis, S., Vogiatzi, K., Krambovitis, E., and Spandidos, D. A. (2008). IL-1 cytokines in cardiovascular disease: diagnostic, prognostic and therapeutic implications. Cardiovasc. Hematol. Agents Med. Chem. 6, 150–158. doi: 10.2174/187152508783955006
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109. doi: 10.1038/nrmicro2070
Boaru, S. G., Borkham-Kamphorst, E., Van de Leur, E., Lehnen, E., Liedtke, C., and Weiskirchen, R. (2015). NLRP3 inflammasome expression is driven by NF-κB in cultured hepatocytes. Biochem. Biophys. Res. Commun. 458, 700–706. doi: 10.1016/j.bbrc.2015.02.029
Bracey, N. A., Gershkovich, B., Chun, J., Vilaysane, A., Meijndert, H. C., Wright, J. R. Jr., et al. (2014). Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 289, 19571–19584. doi: 10.1074/jbc.M114.550624
Braunwald, E. (2008). Biomarkers in heart failure. N. Engl. J. Med. 358, 2148–2159. doi: 10.1056/NEJMra0800239
Bryant, C., and Fitzgerald, K. A. (2009). Molecular mechanisms involved in inflammasome activation. Trends Cell Biol. 19, 455–464. doi: 10.1016/j.tcb.2009.06.002
Butts, B., Gary, R. A., Dunbar, S. B., and Butler, J. (2015). The importance of NLRP3 inflammasome in heart failure. J. Card. Fail. 21, 586–593. doi: 10.1016/j.cardfail.2015.04.014
Coll, N. S., Epple, P., and Dangl, J. L. (2011). Programmed cell death in the plant immune system. Cell Death Differ. 18, 1247–1256. doi: 10.1038/cdd.2011.37
Corsini, E., Galbiati, V., Nikitovic, D., and Tsatsakis, A. M. (2013). Role of oxidative stress in chemical allergens induced skin cells activation. Food Chem. Toxicol. 61, 74–81. doi: 10.1016/j.fct.2013.02.038
de Zoete, M. R., Palm, N. W., Zhu, S., and Flavell, R. A. (2014). Inflammasomes. Cold Spring Harb. Perspect. Biol. 6:a016287. doi: 10.1101/cshperspect.a016287
Ding, J., Wang, K., Liu, W., She, Y., Sun, Q., Shi, J., et al. (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. doi: 10.1038/nature18590
Donath, M. Y., and Shoelson, S. E. (2011). Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11, 98–107. doi: 10.1038/nri2925
Donath, M. Y., Boni-Schnetzler, M., Ellingsgaard, H., Halban, P. A., and Ehses, J. A. (2010). Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol. Metab. 21, 261–267. doi: 10.1016/j.tem.2009.12.010
Franchi, L., Munoz-Planillo, R., and Nunez, G. (2012). Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332. doi: 10.1038/ni.2231
Guarda, G., and So, A. (2010). Regulation of inflammasome activity. Immunology 130, 329–336. doi: 10.1111/j.1365-2567.2010.03283.x
He, W. T., Wan, H., Hu, L., Chen, P., Wang, X., Huang, Z., et al. (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 25, 1285–1298. doi: 10.1038/cr.2015.139
Iyer, S. S., He, Q., Janczy, J. R., Elliott, E. I., Zhong, Z., Olivier, A. K., et al. (2013). Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323. doi: 10.1016/j.immuni.2013.08.001
Jang, Y., Lee, A. Y., Jeong, S. H., Park, K. H., Paik, M. K., Cho, N. J., et al. (2015). Chlorpyrifos induces NLRP3 inflammasome and pyroptosis/apoptosis via mitochondrial oxidative stress in human keratinocyte HaCaT cells. Toxicology 338, 37–46. doi: 10.1016/j.tox.2015.09.006
Janket, S. J., Javaheri, H., Ackerson, L. K., Ayilavarapu, S., and Meurman, J. H. (2015). Oral infections, metabolic inflammation, genetics, and cardiometabolic diseases. J. Dent. Res. 94(9 Suppl.), 119S–127S. doi: 10.1177/0022034515580795
Jin, C., and Flavell, R. A. (2013). Innate sensors of pathogen and stress: linking inflammation to obesity. J. Allergy Clin. Immunol. 132, 287–294. doi: 10.1016/j.jaci.2013.06.022
Jo, E. K., Kim, J. K., Shin, D. M., and Sasakawa, C. (2016). Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 13, 148–159. doi: 10.1038/cmi.2015.95
Kayagaki, N., Stowe, I. B., Lee, B. L., O'Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. doi: 10.1038/nature15541
Kuethe, F., Sigusch, H. H., Bornstein, S. R., Hilbig, K., Kamvissi, V., and Figulla, H. R. (2007). Apoptosis in patients with dilated cardiomyopathy and diabetes: a feature of diabetic cardiomyopathy? Horm. Metab. Res. 39, 672–676. doi: 10.1055/s-2007-985823
Kumar, S., Prasad, S., and Sitasawad, S. L. (2013). Multiple antioxidants improve cardiac complications and inhibit cardiac cell death in streptozotocin-induced diabetic rats. PLoS ONE 8:e67009. doi: 10.1371/journal.pone.0067009
Lamkanfi, M., and Dixit, V. M. (2014). Mechanisms and functions of inflammasomes. Cell 157, 1013–1022. doi: 10.1016/j.cell.2014.04.007
Lebeaupin, C., Proics, E., de Bieville, C. H., Rousseau, D., Bonnafous, S., Patouraux, S., et al. (2015). ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 6:e1879. doi: 10.1038/cddis.2015.248
Legrand-Poels, S., Esser, N., L'Homme, L., Scheen, A., Paquot, N., and Piette, J. (2014). Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem. Pharmacol. 92, 131–141. doi: 10.1016/j.bcp.2014.08.013
Lerner, A. G., Upton, J. P., Praveen, P. V., Ghosh, R., Nakagawa, Y., Igbaria, A., et al. (2012). IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264. doi: 10.1016/j.cmet.2012.07.007
Liao, P. C., Chao, L. K., Chou, J. C., Dong, W. C., Lin, C. N., Lin, C. Y., et al. (2013). Lipopolysaccharide/adenosine triphosphate-mediated signal transduction in the regulation of NLRP3 protein expression and caspase-1-mediated interleukin-1β secretion. Inflamm. Res. 62, 89–96. doi: 10.1007/s00011-012-0555-2
Liu, L., Ding, W. Y., Zhao, J., Wang, Z. H., Zhong, M., Zhang, W., et al. (2013). Activin receptor-like kinase 7 mediates high glucose-induced H9c2 cardiomyoblast apoptosis through activation of Smad2/3. Int. J. Biochem. Cell Biol. 45, 2027–2035. doi: 10.1016/j.biocel.2013.06.018
Liu, X., Zhang, Z., Ruan, J., Pan, Y., Magupalli, V. G., Wu, H., et al. (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. doi: 10.1038/nature18629
Liu, Y. F., Wen, C. Y., Chen, Z., Wang, Y., Huang, Y., and Tu, S. H. (2016). Effects of berberine on NLRP3 and IL-1β expressions in monocytic THP-1 cells with monosodium urate crystals-induced inflammation. Biomed Res. Int. 2016:2503703. doi: 10.1155/2016/2503703
Lopez-Pastrana, J., Ferrer, L. M., Li, Y. F., Xiong, X., Xi, H., Cueto, R., et al. (2015). Inhibition of caspase-1 activation in endothelial cells improves angiogenesis: a novel therapeutic potential for ischemia. J. Biol. Chem. 290, 17485–17494. doi: 10.1074/jbc.M115.641191
Loppnow, H., Westphal, E., Buchhorn, R., Wessel, A., and Werdan, K. (2001). Interleukin-1 and related proteins in cardiovascular disease in adults and children. Shock 16(Suppl. 1), 3–9. doi: 10.1097/00024382-200116001-00002
Lu, B., Nakamura, T., Inouye, K., Li, J., Tang, Y., Lundback, P., et al. (2012). Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674. doi: 10.1038/nature11290
Luo, B., Li, B., Wang, W., Liu, X., Liu, X., Xia, Y., et al. (2014a). Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc. Drugs Ther. 28, 33–43. doi: 10.1007/s10557-013-6498-1
Luo, B., Li, B., Wang, W., Liu, X., Xia, Y., Zhang, C., et al. (2014b). NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 9:e104771. doi: 10.1371/journal.pone.0104771
Martinon, F. (2010). Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 40, 616–619. doi: 10.1002/eji.200940168
Mastrocola, R., Collino, M., Penna, C., Nigro, D., Chiazza, F., Fracasso, V., et al. (2016a). Maladaptive modulations of NLRP3 inflammasome and cardioprotective pathways are involved in diet-induced exacerbation of myocardial ischemia/reperfusion injury in mice. Oxid. Med. Cell. Longev. 2016:3480637. doi: 10.1155/2016/3480637
Mastrocola, R., Penna, C., Tullio, F., Femmino, S., Nigro, D., Chiazza, F., et al. (2016b). Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways. Oxid. Med. Cell. Longev. 2016:5271251. doi: 10.1155/2016/5271251
Miao, E. A., Rajan, J. V., and Aderem, A. (2011). Caspase-1-induced pyroptotic cell death. Immunol. Rev. 243, 206–214. doi: 10.1111/j.1600-065X.2011.01044.x
Monack, D. M., Raupach, B., Hromockyj, A. E., and Falkow, S. (1996). Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc. Natl. Acad. Sci. U.S.A. 93, 9833–9838. doi: 10.1073/pnas.93.18.9833
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
Nakayama, M., Simmons, K. M., and Michels, A. W. (2015). Molecular interactions governing autoantigen presentation in type 1 diabetes. Curr. Diab. Rep. 15, 113. doi: 10.1007/s11892-015-0689-z
Pandey, A., Chawla, S., and Guchhait, P. (2015). Type-2 diabetes: current understanding and future perspectives. IUBMB Life 67, 506–513. doi: 10.1002/iub.1396
Qamar, A., and Rader, D. J. (2012). Effect of interleukin 1β inhibition in cardiovascular disease. Curr. Opin. Lipidol. 23, 548–553. doi: 10.1097/MOL.0b013e328359b0a6
Qiao, Y., Wang, P., Qi, J., Zhang, L., and Gao, C. (2012). TLR-induced NF-κB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 586, 1022–1026. doi: 10.1016/j.febslet.2012.02.045
Rajamaki, K., Mayranpaa, M. I., Risco, A., Tuimala, J., Nurmi, K., Cuenda, A., et al. (2016). p38delta MAPK: a novel regulator of NLRP3 inflammasome activation with increased expression in coronary atherogenesis. Arterioscler. Thromb. Vasc. Biol. 36, 1937–1946. doi: 10.1161/ATVBAHA.115.307312
Robbins, G. R., Wen, H., and Ting, J. P. (2014). Inflammasomes and metabolic disorders: old genes in modern diseases. Mol. Cell 54, 297–308. doi: 10.1016/j.molcel.2014.03.029
Santiago, J. J., McNaughton, L. J., Koleini, N., Ma, X., Bestvater, B., Nickel, B. E., et al. (2014). High molecular weight fibroblast growth factor-2 in the human heart is a potential target for prevention of cardiac remodeling. PLoS ONE 9:e97281. doi: 10.1371/journal.pone.0097281
Schroder, K., Zhou, R., and Tschopp, J. (2010). The NLRP3 inflammasome: a sensor for metabolic danger? Science 327, 296–300. doi: 10.1126/science.1184003
Shalini, S., Dorstyn, L., Dawar, S., and Kumar, S. (2015). Old, new and emerging functions of caspases. Cell Death Differ. 22, 526–539. doi: 10.1038/cdd.2014.216
Shao, B. Z., Xu, Z. Q., Han, B. Z., Su, D. F., and Liu, C. (2015). NLRP3 inflammasome and its inhibitors: a review. Front. Pharmacol. 6:262. doi: 10.3389/fphar.2015.00262
Shaw, J. E., Sicree, R. A., and Zimmet, P. Z. (2010). Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 87, 4–14. doi: 10.1016/j.diabres.2009.10.007
Shenoy, A. R., Wellington, D. A., Kumar, P., Kassa, H., Booth, C. J., Cresswell, P., et al. (2012). GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336, 481–485. doi: 10.1126/science.1217141
Shi, H., Zhang, Z., Wang, X., Li, R., Hou, W., Bi, W., et al. (2015). Inhibition of autophagy induces IL-1β release from ARPE-19 cells via ROS mediated NLRP3 inflammasome activation under high glucose stress. Biochem. Biophys. Res. Commun. 463, 1071–1076. doi: 10.1016/j.bbrc.2015.06.060
Shi, J., Gao, W., and Shao, F. (2017). Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 42, 245–254. doi: 10.1016/j.tibs.2016.10.004
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Singh, L. P. (2014). The NLRP3 inflammasome and diabetic cardiomyopathy: editorial to: “Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model” by Beibei Luo et al. Cardiovasc. Drugs Ther. 28, 5–6. doi: 10.1007/s10557-013-6501-x
Somanna, N. K., Yariswamy, M., Garagliano, J. M., Siebenlist, U., Mummidi, S., Valente, A. J., et al. (2015). Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2. Cell. Signal. 27, 1928–1938. doi: 10.1016/j.cellsig.2015.07.001
Vandanmagsar, B., Youm, Y. H., Ravussin, A., Galgani, J. E., Stadler, K., Mynatt, R. L., et al. (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188. doi: 10.1038/nm.2279
Wang, R., Wang, Y., Mu, N., Lou, X., Li, W., Chen, Y., et al. (2017). Activation of NLRP3 inflammasomes contributes to hyperhomocysteinemia-aggravated inflammation and atherosclerosis in apoE-deficient mice. Lab. Invest. doi: 10.1038/labinvest.2017.30. [Epub ahead of print].
Wen, H., Gris, D., Lei, Y., Jha, S., Zhang, L., Huang, M. T., et al. (2011). Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415. doi: 10.1038/ni.2022
Westermeier, F., Navarro-Marquez, M., Lopez-Crisosto, C., Bravo-Sagua, R., Quiroga, C., Bustamante, M., et al. (2015). Defective insulin signaling and mitochondrial dynamics in diabetic cardiomyopathy. Biochim. Biophys. Acta 1853, 1113–1118. doi: 10.1016/j.bbamcr.2015.02.005
Wolf, P., Winhofer, Y., Krssak, M., Smajis, S., Harreiter, J., Kosi-Trebotic, L., et al. (2016). Suppression of plasma free fatty acids reduces myocardial lipid content and systolic function in type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 26, 387–392. doi: 10.1016/j.numecd.2016.03.012
Wree, A., Eguchi, A., McGeough, M. D., Pena, C. A., Johnson, C. D., Canbay, A., et al. (2014). NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 59, 898–910. doi: 10.1002/hep.26592
Zhang, W., Xu, X., Kao, R., Mele, T., Kvietys, P., Martin, C. M., et al. (2014). Cardiac fibroblasts contribute to myocardial dysfunction in mice with sepsis: the role of NLRP3 inflammasome activation. PLoS ONE 9:e107639. doi: 10.1371/journal.pone.0107639
Zhou, R., Tardivel, A., Thorens, B., Choi, I., and Tschopp, J. (2010). Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140. doi: 10.1038/ni.1831
Zlobine, I., Gopal, K., and Ussher, J. R. (2016). Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochim. Biophys. Acta 1861, 1555–1568. doi: 10.1016/j.bbalip.2016.02.011
Zu, Y., Wan, L. J., Cui, S. Y., Gong, Y. P., and Li, C. L. (2015). The mitochondrial Na+/Ca2+ exchanger may reduce high glucose-induced oxidative stress and nucleotide-binding oligomerization domain receptor 3 inflammasome activation in endothelial cells. J. Geriatr. Cardiol. 12, 270–278. doi: 10.11909/j.issn.1671-5411.2015.03.003
Keywords: NLRP3 inflammasome, thioredoxin interacting/inhibiting protein, pyroptosis, inflammation, diabetic cardiomyopathy
Citation: Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H, Zeng Z, Huang W and He Y (2017) NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Front. Physiol. 8:519. doi: 10.3389/fphys.2017.00519
Received: 03 January 2017; Accepted: 06 July 2017;
Published: 25 July 2017.
Edited by:
Xiaogang Wu, Institute for Systems Biology, United StatesReviewed by:
Fan Zhang, Icahn School of Medicine at Mount Sinai, United StatesSyed Aun Muhammad, Bahauddin Zakariya University, Pakistan
Copyright © 2017 Luo, Huang, Liu, Liang, Wei, Ke, Zeng, Huang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyu Zeng, zhiyuzengedu@sina.com
Weiqiang Huang, weiqianghuangedu@sina.com
Yan He, yanheedu@126.com