 Sabrina Stücker
Sabrina Stücker Nico Kresin1,2
Nico Kresin1,2 Lucie Carrier
Lucie Carrier Felix W. Friedrich
Felix W. Friedrich- 1Department of Experimental Pharmacology and Toxicology, Cardiovascular Research Center, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2German Centre for Cardiovascular Research (DZHK), Hamburg, Germany
Background: Hypertrophic cardiomyopathy (HCM) patients often present with diastolic dysfunction and a normal to supranormal systolic function. To counteract this hypercontractility, guideline therapies advocate treatment with beta-adrenoceptor and Ca2+ channel blockers. One well established pathomechanism for the hypercontractile phenotype frequently observed in HCM patients and several HCM mouse models is an increased myofilament Ca2+ sensitivity. Nebivolol, a commonly used beta-adrenoceptor antagonist, has been reported to lower maximal force development and myofilament Ca2+ sensitivity in rabbit and human heart tissues. The aim of this study was to evaluate the effect of nebivolol in cardiac muscle strips of an established HCM Mybpc3 mouse model. Furthermore, we investigated actions of nebivolol and epigallocatechin-gallate, which has been shown to desensitize myofilaments for Ca2+ in mouse and human HCM models, in cardiac strips of HCM patients with a mutation in the most frequently mutated HCM gene MYBPC3.
Methods and Results: Nebivolol effects were tested on contractile parameters and force-Ca2+ relationship of skinned ventricular muscle strips isolated from Mybpc3-targeted knock-in (KI), wild-type (WT) mice and cardiac strips of three HCM patients with MYBPC3 mutations. At baseline, KI strips showed no difference in maximal force development compared to WT mouse heart strips. Neither 1 nor 10 μM nebivolol had an effect on maximal force development in both genotypes. 10 μM nebivolol induced myofilament Ca2+ desensitization in WT strips and to a greater extent in KI strips. Neither 1 nor 10 μM nebivolol had an effect on Ca2+ sensitivity in cardiac muscle strips of three HCM patients with MYBPC3 mutations, whereas epigallocatechin-gallate induced a right shift in the force-Ca2+ curve.
Conclusion: Nebivolol induced a myofilament Ca2+ desensitization in both WT and KI strips, which was more pronounced in KI muscle strips. In human cardiac muscle strips of three HCM patients nebivolol had no effect on myofilament Ca2+ sensitivity.
Introduction
Hypertrophic cardiomyopathy (HCM) is the most frequent cardiac genetic disease primarily caused by mutations in sarcomeric protein genes (Friedrich and Carrier, 2012; Maron et al., 2014; Ho et al., 2015). The most commonly mutated genes are MYBPC3 (encoding cardiac myosin-binding protein C) and MYH7 (encoding β-myosin-heavy chain) (Walsh et al., 2017). HCM is principally characterized by asymmetric left ventricular hypertrophy, diastolic dysfunction and myocardial disarray (Elliott et al., 2008). Current pharmacological treatment of HCM mainly relies on beta-adrenoceptor (AR) and Ca2+ channel blockers, which improve clinical symptoms, partially prevent arrhythmias and improve diastolic dysfunction by prolonging left ventricular (LV) filling time and reducing outflow tract obstruction (Maron et al., 2003; Gersh et al., 2011; Spoladore et al., 2012; Hamada et al., 2014; Tardiff et al., 2015). Increased Ca2+ sensitivity seems to be a common factor in HCM as seen in animal HCM models (Tardiff et al., 1999; Cazorla et al., 2006; Pohlmann et al., 2007; Vignier et al., 2009; Fraysse et al., 2012; Barefield et al., 2014; Wijnker et al., 2016), and human HCM samples (Jacques et al., 2008; van Dijk et al., 2009, 2012). The increased Ca2+ response may contribute to diastolic dysfunction and arrhythmias (Morimoto et al., 1998; Baudenbacher et al., 2008). Even though the mechanisms accountable for increased myofilament Ca2+ sensitivity remain unclear, targeting this pathomechanism by interventions decreasing myofilament Ca2+ sensitivity may be an attractive alternative for the treatment of HCM and improvement in symptoms (Jagatheesan et al., 2007; Alves et al., 2014; Tardiff et al., 2015). Among beta-AR blockers that are commonly used in the treatment of cardiovascular diseases, nebivolol has been reported to lower maximal force development and to desensitize rabbit and human cardiac myofilaments to Ca2+ (Zeitz et al., 2000; Janssen et al., 2001). However, the effects of nebivolol were never evaluated in HCM models with increased myofilament Ca2+ sensitivity. An established HCM mouse model carrying the human c.772G>A MYBPC3 mutation is the Mybpc3 KI mouse model (Vignier et al., 2009). This mutation was frequently found in unrelated HCM patients in Tuscany and is associated with a bad prognosis (Richard et al., 2003; Girolami et al., 2006; Ho et al., 2015). At the homozygous state, this mouse model exhibits HCM-like features such as left ventricular hypertrophy, diastolic dysfunction and increased myofilament Ca2+ sensitivity (Vignier et al., 2009; Fraysse et al., 2012). We recently showed that epigallocatechin-3-gallate (EGCg), a major component of green tea, hastened relaxation and Ca2+ transient in KI cardiomyocytes and decreased Ca2+ sensitivity of KI myofilaments (Friedrich et al., 2016). In this study, we investigated nebivolol effects on myofilament Ca2+ sensitivity in Mybpc3 KI cardiac muscle strips. We furthermore assessed nebivolol and EGCg effects in cardiac strips of three HCM patients with MYBPC3 mutations.
Materials and Methods
Human Samples
Human myocardial samples were obtained from three HCM patients carrying heterozygous MYBPC3 mutations (c.1960C>T, c.2308G>A, c.2234A>G) who underwent septal myectomy due to outflow tract obstruction. The material was taken with written informed consent of the donor and with written approval of the local ethical boards. The study has been carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki).
Animals
The Mybpc3 KI cardiomyopathy mouse model was generated by the targeted insertion of a G>A transition on the last nucleotide of exon 6 (Vignier et al., 2009; Fraysse et al., 2012; Schlossarek et al., 2012, 2014; Gedicke-Hornung et al., 2013; Mearini et al., 2013, 2014; Stohr et al., 2013; Friedrich et al., 2014; Najafi et al., 2015; Thottakara et al., 2015; Flenner et al., 2016, 2017). Mice were maintained on the C57 background. As controls, Mybpc3 WT mice of the same background were used. The study was exerted in accordance with the recommendations of the guide for the care and use of laboratory animals published by the NIH (Publication No. 85–23, revised 2011 published by National Research Council) and comply with the ARRIVE guidelines (http://www.nc3rs.org.uk/arrive-animal-research-reporting-vivo-experiments). All experimental procedures were in accordance with the German Law for the Protection of Animals and the protocol was approved by the Ministry of Science and Public Health of the City State of Hamburg, Germany (Org 653).
Skinned Ventricular Trabeculae Force Measurements
For the determination of force-Ca2+ relationships, trabeculae were prepared from ventricular endocardial surface of WT and KI mice or human myocardium of a septal myectomy (Flenner et al., 2016; Friedrich et al., 2016). Dimensions of strips were 2.91 ± 0.14 mm in length, 0.36 ± 0.01 mm in width and 0.11 ± 0.01 mm2 in cross-sectional area (CSA), calculated by 2πr2 assuming a circular shape, nWT = 17, nKI = 18, nhuman = 57. Strips were permeabilized in relaxing solution (pCa 9) in EGTA-buffer (5.89 mM Na2ATP, 14.5 mM CrP, 6.48 mM MgCl2, 40.76 mM Kprop, 100 mM BES and 7 mM EGTA, pH 7.1) (Kooij et al., 2010; Stoehr et al., 2014) containing 1% Triton X-100 at 4°C for 18 h. The next day strips were either directly used for measurements or stored at −20°C in a 50% glycerol/relaxing solution containing protease inhibitors (EDTA-free, complete tablets, mini, Roche). The Ca2+-sensitivity of permeabilized cardiac strips was evaluated using a fiber test system (1400A; Aurora Scientific) by mounting them between a force transducer and a length controller. Strips were stretched above slack length until they developed force in activating solution (pCa 4.5) at 15°C. For contraction-relaxation cycles strips were kept in pCa 9 to achieve full relaxation. Then they were moved to pCa 4.5 until maximal force development was reached. Maximal force was related to cross-sectional area (mN/mm2). For force-Ca2+-curves they were exposed to increasing Ca2+ concentrations from pCa 9 to pCa 4.5 in EGTA-buffer. Force development was measured in each pCa solution. Measurements were repeated in the presence of 1 or 10 μM nebivolol (nebivolol hydrochloride, Sigma Life Sciences) or 30 μM epigallocatechin-gallate (Sigma Life Sciences) after 5 min preincubation in relaxing solution (Flenner et al., 2016; Friedrich et al., 2016). In every second measurement, nebivolol was tested first and a control measurement was performed 5 min after nebivolol washout to exclude time-dependent force rundown. Each strip was measured in a pairwise manner (paired analysis baseline vs. intervention) serving as its own control. Data were analyzed using the Hill equation (Hill et al., 1980), with pCa50 as the free Ca2+ concentration which yields 50% of the maximal force and nH representing the Hill coefficient. The pCa50 represents the measure of myofilament Ca2+ sensitivity.
Statistical Analysis
Data were expressed as mean ± SEM. Comparisons were performed by paired or unpaired Student's t-test and with one-way ANOVA, followed by Bonferroni's post-test as indicated in the figure legends. Concentration response curves were fitted to the data points and force-pCa relationship comparison was done by using extra sum-of-squares F-test (GraphPad, Prism 6). A value of P < 0.05 was considered statistically significant.
Results
Nebivolol (1 and 10 μM) Has No Effect on Maximal Force Development in Permeabilized Cardiac Strips of Mybpc3 WT and KI Mice
The hypercontractile phenotype observed in HCM patients could be attributed to an increased myofilament Ca2+ sensitivity. Since nebivolol has been reported to lower maximal force development and to desensitize rabbit and human cardiac myofilaments (Zeitz et al., 2000; Janssen et al., 2001), we investigated nebivolol effects on myofilament Ca2+ sensitivity in cardiac muscle strips of Mybpc3 KI mice with an increased myofilament Ca2+ sensitivity (Vignier et al., 2009; Fraysse et al., 2012; Flenner et al., 2016; Friedrich et al., 2016). There are conflicting reports concerning the effects of nebivolol on maximal force development and myofilament Ca2+ sensitivity in cardiac muscle strips. Whereas Zeitz et al. reported that 1 μM nebivolol lowered maximal force development and myofilament Ca2+ sensitivity in skinned trabeculae (Zeitz et al., 2000), Bundkirchen and colleagues did not observe such an effect at 10 μM (Bundkirchen et al., 2001). We therefore used 1 and 10 μM for our experiments. To investigate nebivolol effects on force development we measured contraction-relaxation cycles in skinned myofilaments (Figure 1A). Analysis showed that maximal force development in baseline conditions did not significantly differ between WT and KI muscle strips (Figure 1B). Neither 1 nor 10 μM nebivolol had an effect on maximal force development (Figure 1B).
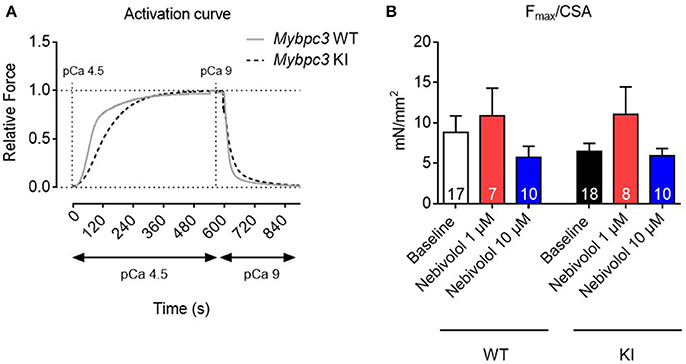
Figure 1. Contractile parameters of permeabilized cardiac muscle strips of Mybpc3 WT and KI mice with and without nebivolol treatment. (A) Representative normalized activation curves of Mybpc3 WT (gray) and KI (black striped) mouse strips. (B) Quantification of maximal force development related to cross sectional area (CSA) in pCa 4.5 ± nebivolol 1 and 10 μM; number of strips is indicated in the bars.
Nebivolol Decreases Myofilament Ca2+ Sensitivity to a Greater Extent in KI than in WT Skinned Ventricular Trabeculae
Nebivolol has been reported to decrease Ca2+ sensitivity in rabbit and human cardiac myofilaments (Zeitz et al., 2000; Janssen et al., 2001). To assess whether this is also the case in myofilaments of Mybpc3 KI mouse hearts, we measured force-pCa relationships in skinned ventricular trabeculae from WT and KI mice. In analogy to the experiments on maximal force development, we performed force-pCa relationships in the absence and presence of 1 and 10 μM nebivolol, respectively. As observed before (Fraysse et al., 2012; Flenner et al., 2016; Friedrich et al., 2016), skinned KI trabeculae showed a higher pCa50 than WT trabeculae in baseline conditions, representing higher myofilament Ca2+ sensitivity (Figures 2A,B). In WT strips, only incubation with 10 μM (by extra sum-of-squares F-test) shifted the force-Ca2+ relationship to the right resulting in a lower pCa50, whereas in KI strips both 1 and 10 μM lowered pCa50 (Figures 2A,B) indicating myofilament Ca2+ desensitization. This effect was concentration-dependent since incubation with 10 μM nebivolol induced a stronger shift (Δ pCa50) to the right (Figure 2C). The nHill coefficient (Hill slope) as a an index for myofilament co-operativity did not differ between the genotypes neither with nor without nebivolol (Figure 2D).
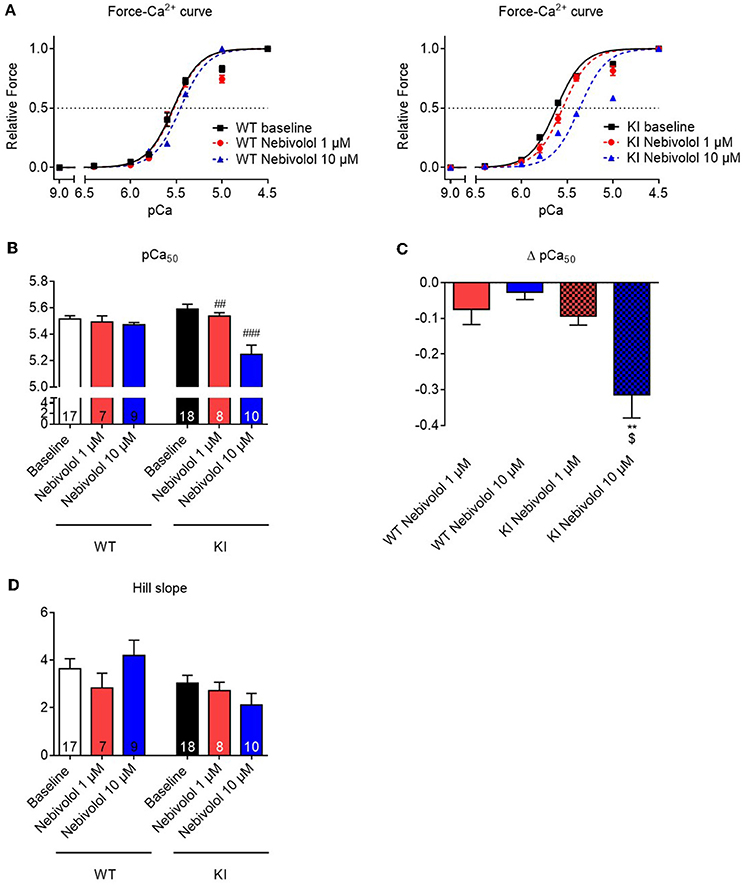
Figure 2. Force-Ca2+ relationship of permeabilized cardiac muscle strips of Mybpc3 WT and KI mice with and without nebivolol treatment. (A) Force-Ca2+ relationship in WT (left) and KI (right) strips ±nebivolol 1 and 10 μM. (B) pCa50 representing the negative logarithm of the calcium concentration needed for half-maximal activation ±nebivolol 1 and 10 μM. (C) Delta of pCa50 ± nebivolol 1 and 10 μM. (D) nHill coefficient (Hill slope) ± nebivolol 1 and 10 μM. **P < 0.01 vs. WT in the same condition and $p < 0.05 vs. KI 1 μM, unpaired Student's t-test; ##P < 0.01 and ###P < 0.001 vs. baseline, paired Student's t-test, concentration response curves were fitted to the data points and curve comparison was done by using extra sum-of-squares F-test; number of strips is indicated in the bars.
Nebivolol Does Not Impact on Maximal Force Development or Myofilament Ca2+ Sensitivity in Muscle Strips Derived from Cardiac Tissue of HCM Patients with MYBPC3 Mutations
Since nebivolol has been reported to desensitize human myofilaments for Ca2+ and since both 1 and 10 μM nebivolol had induced a right-ward shift of the force-pCa curves in Mybpc3 KI cardiac muscle strips we sought to investigate whether it would also affect myofilament Ca2+ sensitivity in human HCM tissue. Similar to the experiments performed with mouse cardiac strips we investigated the effects of 1 and 10 μM nebivolol on contraction-relaxation cycles in muscle strips of three HCM patients carrying different MYBPC3 mutations. Neither 1 nor 10 μM nebivolol had an influence on Fmax (Figure 3A). Furthermore, no shift in Ca2+ sensitivity nor change in Hill slope was observed for the Ca2+-dependent force development (Figures 3B–D). In contrast and as reported before by us in Mybpc3 WT and KI strips (Friedrich et al., 2016), incubation with a positive control compound (30 μM epigallocatechin-gallate; EGCg) shifted the force-Ca2+-relationship to the right in strips from the same three HCM patients (Figures 3E,F).
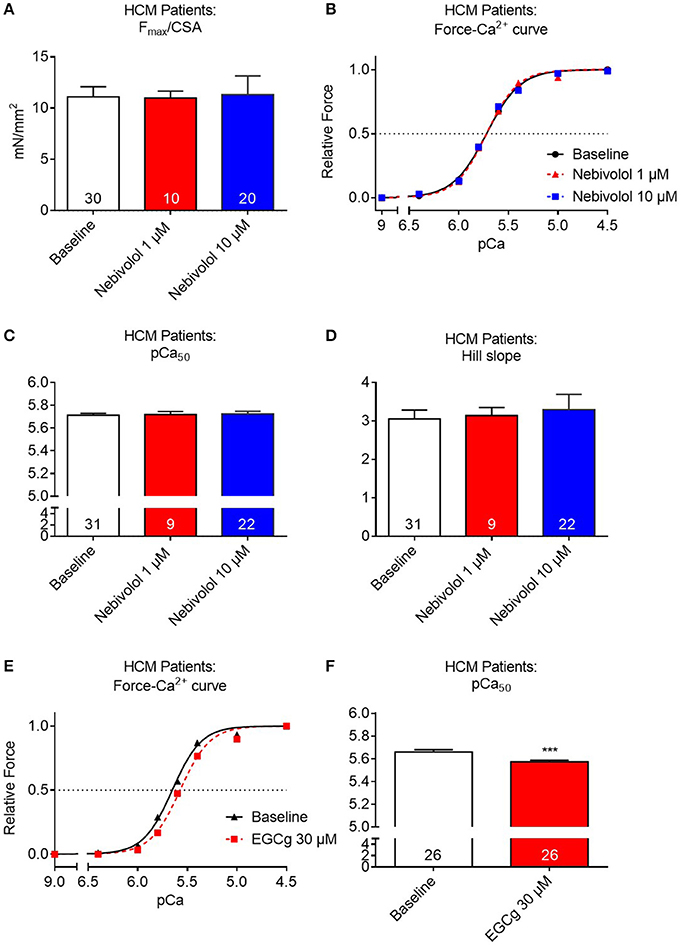
Figure 3. Contractile parameters of permeabilized cardiac muscle strips of three human HCM patients carrying different Mybpc3 mutations in the absence or presence of nebivolol. (A) Quantification of maximal force development related to cross sectional area (CSA) in pCa 4.5 ±nebivolol 1 and 10 μM. (B) Force-Ca2+ relationship ±nebivolol 1 and 10 μM. (C) pCa50 representing the negative logarithm of the calcium concentration needed for half-maximal activation ±nebivolol 1 and 10 μM. (D) nHill coefficient (Hill slope) ±nebivolol 1 and 10 μM. (E) Force-Ca2+ relationship ±EGCg 30 μM. (F) pCa50 ±EGCg 30 μM. ***P < 0.001 vs. baseline, paired Student's t-test. Concentration response curves were fitted to the data points and curve comparison was done by using extra sum-of-squares F-test; number of strips is indicated in the bars.
Discussion
HCM patients often present with a normal to supranormal systolic function and diastolic dysfunction. To counteract this hypercontractility, guideline therapies advocate treatment with beta-AR and Ca2+ channel blockers. One well established pathomechanism for the hypercontractile phenotype frequently observed in HCM patients and several HCM mouse models is an increased myofilament Ca2+ sensitivity (Morimoto et al., 1998; Robinson et al., 2007; Huke and Knollmann, 2010; Kimura, 2010; Fraysse et al., 2012; Moore et al., 2012; van Dijk et al., 2012; Barefield et al., 2014; Elliott et al., 2014; Flenner et al., 2016; Friedrich et al., 2016). Nebivolol, a commonly used beta-AR antagonist, has been reported to lower maximal force development and myofilament Ca2+ sensitivity in rabbit and human heart tissues (Zeitz et al., 2000; Janssen et al., 2001). Given the hypercontractile phenotype mentioned above, these pleiotropic actions predestine it for HCM treatment. The aim of this study was to evaluate whether nebivolol would exert similar effects in permeabilized myofilaments of an Mybpc3 HCM mouse model and of HCM patients with mutations in the most frequently mutated gene MYBPC3. The main findings of this study were: (1) At baseline, permeabilized left ventricular trabeculae isolated from Mybpc3 KI mouse hearts showed no difference in maximal force development compared to WT mouse heart strips. (2) Neither 1 nor 10 μM nebivolol had an effect on maximal force development in both genotypes. (3) 10 μM nebivolol induced myofilament Ca2+ desensitization in both WT and KI strips and this effect was more pronounced in KI muscle strips, respectively. (4) Nebivolol had no effect on Ca2+ sensitivity in cardiac muscle strips of three HCM patients with MYBPC3 mutations, whereas 30 μM of EGCg induced a right shift in the force-Ca2+ curve.
In mice, nebivolol did not influence maximal force development. On the other hand, it affected myofilament Ca2+ sensitivity in mouse strips. The mechanism behind this is unknown so far. In analogy to the mouse results, 1 and 10 μM nebivolol had no effect on maximal force development in human tissues. In contrast to the observations made in mouse strips, it did not impact on myofilament Ca2+ sensitivity.
The reason why nebivolol exerted a myofilament Ca2+ desensitizing effect in KI strips at both 1 and 10 μM, whereas in WT strips only 10 μM had an effect and no effect at all in human HCM tissues remains unclear. As mentioned before, discrepancies concerning the effects of nebivolol on maximal force development and myofilament Ca2+ sensitivity in cardiac muscle strips have been previously described. Whereas Zeitz et al. reported that 1 μM nebivolol lowered maximal force development and myofilament Ca2+ sensitivity in skinned rabbit and human trabeculae (Zeitz et al., 2000), Bundkirchen and colleagues did not observe such an effect at 10 μM in human tissue (Bundkirchen et al., 2001). The findings are contradictory but could be explained by either differences (i) between species (rabbit vs. mouse vs. human), (ii) in experimental setups, (iii) the functional status of the tissues (non-failing vs. failing), or (iv) a combination of it. Interspecies- or setup-dependent differences have been reported in permeabilized strip experiments with other drug interventions (Lues et al., 1988; Edes et al., 1995). Zeitz et al. used non-failing cardiac tissue from rabbit and explanted human tissue from end-stage failing myocardium from patients undergoing heart transplantation and saw an effect in tissues of both species. In another study the same group did not observe any effect on maximal force development in human explanted heart muscle preparations (Janssen et al., 2001). We observed no effect on maximal force development in neither mouse nor human tissue, but on myofilament Ca2+ sensitivity in mouse tissue, which was more pronounced in the KI strips. Similar to this study we observed in a previous study with skinned trabeculae that EGCg, another compound with Ca2+-desensitizing properties had a more profound effect on strips of the KI than the WT genotype (Friedrich et al., 2016). This is also compatible with results of a study in which the Ca2+-desensitizing effect of ranolazine was only present in KI, but not in WT muscle strips (Flenner et al., 2016). The reason for the difference between KI and WT is unclear but could be related to the higher baseline myofilament Ca2+ sensitivity in KI or to the proposed antioxidative activity of ranolazine, which might be important in a potentially hyperoxidized KI tissues (Lovelock et al., 2012; Flenner et al., 2016). In analogy to the study of Bundkirchen et al., in which nebivolol had no effect in explanted left ventricular tissue of patients with dilated cardiomyopathy, we did not observe any effect on Ca2+ sensitivity at 1 or 10 μM in the HCM samples. In contrast, 30 μM of the positive control compound EGCg, which has been suggested to alter the interaction between cardiac troponin C and I and therefore the sensitivity of the myofilaments to Ca2+ (Liou et al., 2008; Robertson et al., 2009), induced a rightward shift in the force-Ca2+ curve in human HCM strips. EGCg has been shown to lower the myofilament Ca2+ sensitivity in a transgenic HCM mouse model expressing a human cardiac troponin T mutant (Tadano et al., 2010) and in HCM-associated human cardiac troponin I and T mutants (Tadano et al., 2010; Warren et al., 2015; Messer et al., 2016). Similarly, we reported that 30 μM EGCg decreased Ca2+ sensitivity in our Mybpc3 KI mouse model that carries a frequent Mybpc3 HCM mutation (Friedrich et al., 2016). EGCg action on myofilament Ca2+ sensitivity in cardiac muscle strips of patients carrying a heterozygous MYBPC3 mutation indicates that the human strips can be desensitized for Ca2+.
Yet the precise mechanism of Ca2+-desensitization of nebivolol in mouse heart tissue remains unaddressed. Nebivolol is a third-generation beta-AR antagonist that exhibits vasodilating properties, most likely due to stimulation of nitric oxide synthase (Cockcroft et al., 1995). Since it was shown to attenuate hydroxyl radical-induced myocardial damage which has been associated with altered intracellular calcium handling and calcium overload of the myocytes (Josephson et al., 1991; Janssen et al., 1999; Piccini et al., 2012), it was proposed that nebivolol has direct free-radical scavenging properties (Janssen et al., 1999). Whether such an indirect effect is the main reason for a decrease in myofilament Ca2+ sensitivity or another direct mechanism on the moyfilaments, such as binding to the C-terminal region of cardiac troponin C altering the interaction between cTnC and cTnI as in the case of the positive control compound EGCg (Liou et al., 2008; Robertson et al., 2009), exists, remains to be shown.
Clinically, beta-AR-antagonists are the mainstay of HCM therapy (Elliott et al., 2014). They are thought to potentially improve diastolic filling by a negative chronotropic effect. Some studies support the use of beta-AR-antagonists in HF patients with preserved ejection fraction (EF) but impaired relaxation similar to diastolic dysfunction seen in HCM patients (Lund et al., 2014). Recent data suggest that the effect of nebivolol is similar in HF patients with reduced and preserved EF (van Veldhuisen et al., 2009). This initiated the design of a still ongoing trial (https://clinicaltrials.gov/ct2/show/NCT02619526) investigating the effects of nebivolol and carvedilol on diastolic function of the left ventricle in older HF patients with preserved EF (Park and Park, 2016).
Limitations of the Study
(1) The Mybpc3 KI model shows many HCM characteristics only at the homozygous state. Moreover, Mybpc3 KI mice present a reduced EF. These two points are in contrast to the more common findings in HCM patients who present left ventricular hypertrophy, interstitial fibrosis and diastolic dysfunction with heterozygous mutation states and normal or even supra-normal EF. (2) Our study does not explain the precise mechanism of Ca2+-desensitization of nebivolol in mouse heart tissue. (3) The Ca2+ desensitizing effect of nebivolol in mouse tissues occurred at concentrations which were above the plasma concentrations (0.8–3.7 nM) observed in humans (Stoschitzky et al., 2004; Prisant, 2008). (4) Even though we did not observe any nebivolol effect in the human strips in this study, this observation cannot be generalized to all HCM patients since the number of tissues was low and they were derived from HCM patients carrying only mutations in MYBPC3.
Conclusion
Nebivolol had no effect on maximal force, but induced a myofilament Ca2+ desensitization in both WT and KI mouse cardiac muscle strips, which was more pronounced in KI muscle strips. In human cardiac muscle strips, nebivolol had no effect on force development and myofilament Ca2+ sensitivity. Further studies should investigate the exact target and mechanism for Ca2+ desensitization in mouse cardiac tissues in order to be able to develop modified compounds with even more potency and specificity for use in human tissue.
Author Contributions
SS and NK: Isolation and treatment of cardiac muscle strips and execution of experiments. LC: Analysis and interpretation of data, and correction of the manuscript. FF: Conception and design of research, execution of experiments, analysis and interpretation of data, figure preparation, and drafting of the manuscript. All authors critically discussed the results, and reviewed and approved the manuscript before submission.
Funding
This work was supported by the DZHK (German Centre for Cardiovascular Research).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patient for donating the cardiac tissue for scientific research. We thank Julia Münch and Monica Patten (University Heart Center Hamburg, Hamburg, Germany) for patients' recruitment, Elisabeth Krämer, Giulia Mearini, and Frederik Flenner (UKE-Pharmacology, Hamburg, Germany) for help in preservation of human septal myectomies and database maintenance.
References
Alves, M. L., Dias, F. A., Gaffin, R. D., Simon, J. N., Montminy, E. M., Biesiadecki, B. J., et al. (2014). Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins. Circ Cardiovasc Genet 7, 132–143. doi: 10.1161/CIRCGENETICS.113.000324
Barefield, D., Kumar, M., De Tombe, P. P., and Sadayappan, S. (2014). Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 306, H807–H815. doi: 10.1152/ajpheart.00913.2013
Baudenbacher, F., Schober, T., Pinto, J. R., Sidorov, V. Y., Hilliard, F., Solaro, R. J., et al. (2008). Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Invest. 118, 3893–3903. doi: 10.1172/JCI36642
Bundkirchen, A., Brixius, K., Bolck, B., Mehlhorn, U., Bloch, W., and Schwinger, R. H. (2001). Nebivolol, carvedilol and metoprolol do not influence cardiac Ca2+ sensitivity. Eur. J. Pharmacol. 422, 175–180. doi: 10.1016/S0014-2999(01)01082-2
Cazorla, O., Szilagyi, S., Vignier, N., Salazar, G., Kramer, E., Vassort, G., et al. (2006). Length and protein kinase A modulations of myocytes in cardiac myosin binding protein C-deficient mice. Cardiovasc. Res. 69, 370–380. doi: 10.1016/j.cardiores.2005.11.009
Cockcroft, J. R., Chowienczyk, P. J., Brett, S. E., Chen, C. P., Dupont, A. G., Van Nueten, L., et al. (1995). Nebivolol vasodilates human forearm vasculature: evidence for an L-arginine/NO-dependent mechanism. J. Pharmacol. Exp. Ther. 274, 1067–1071.
Edes, I., Kiss, E., Kitada, Y., Powers, F. M., Papp, J. G., Kranias, E. G., et al. (1995). Effects of Levosimendan, a cardiotonic agent targeted to troponin C, on cardiac function and on phosphorylation and Ca2+ sensitivity of cardiac myofibrils and sarcoplasmic reticulum in guinea pig heart. Circ. Res. 77, 107–113. doi: 10.1161/01.RES.77.1.107
Elliott, P., Andersson, B., Arbustini, E., Bilinska, Z., Cecchi, F., Charron, P., et al. (2008). Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 29, 270–276. doi: 10.1093/eurheartj/ehm342
Elliott, P. M., Anastasakis, A., Borger, M. A., Borggrefe, M., Cecchi, F., Charron, P., et al. (2014). 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 35, 2733–2779. doi: 10.1093/eurheartj/ehu284
Flenner, F., Friedrich, F. W., Ungeheuer, N., Christ, T., Geertz, B., Reischmann, S., et al. (2016). Ranolazine antagonizes catecholamine-induced dysfunction in isolated cardiomyocytes, but lacks long-term therapeutic effects in vivo in a mouse model of hypertrophic cardiomyopathy. Cardiovasc. Res. 109, 90–102. doi: 10.1093/cvr/cvv247
Flenner, F., Geertz, B., Reischmann-Dusener, S., Weinberger, F., Eschenhagen, T., Carrier, L., et al. (2017). Diltiazem prevents stress-induced contractile deficits in cardiomyocytes, but does not reverse the cardiomyopathy phenotype in Mybpc3-knock-in mice. J. Physiol. 595, 3987–3999. doi: 10.1113/JP273769
Fraysse, B., Weinberger, F., Bardswell, S. C., Cuello, F., Vignier, N., Geertz, B., et al. (2012). Increased myofilament Ca2+ sensitivity and diastolic dysfunction as early consequences of Mybpc3 mutation in heterozygous knock-in mice. J. Mol. Cell. Cardiol. 52, 1299–1307. doi: 10.1016/j.yjmcc.2012.03.009
Friedrich, F. W., and Carrier, L. (2012). Genetics of hypertrophic and dilated cardiomyopathy. Curr. Pharm. Biotechnol. 13, 2467–2476. doi: 10.2174/1389201011208062467
Friedrich, F. W., Flenner, F., Nasib, M., Eschenhagen, T., and Carrier, L. (2016). Epigallocatechin-3-gallate accelerates relaxation and Ca2+ transient decay and desensitizes myofilaments in healthy and mybpc3-targeted knock-in cardiomyopathic mice. Front. Physiol. 7:607. doi: 10.3389/fphys.2016.00607
Friedrich, F. W., Reischmann, S., Schwalm, A., Unger, A., Ramanujam, D., Munch, J., et al. (2014). FHL2 expression and variants in hypertrophic cardiomyopathy. Basic Res. Cardiol. 109:451. doi: 10.1007/s00395-014-0451-8
Gedicke-Hornung, C., Behrens-Gawlik, V., Reischmann, S., Geertz, B., Stimpel, D., Weinberger, F., et al. (2013). Rescue of cardiomyopathy through U7snRNA-mediated exon skipping in Mybpc3-targeted knock-in mice. EMBO Mol. Med. 5, 1128–1145. doi: 10.1002/emmm.201202168
Gersh, B. J., Maron, B. J., Bonow, R. O., Dearani, J. A., Fifer, M. A., Link, M. S., et al. (2011). 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Thorac. Cardiovasc. Surg. 142, e153–e203. doi: 10.1016/j.jtcvs.2011.10.020
Girolami, F., Olivotto, I., Passerini, I., Zachara, E., Nistri, S., Re, F., et al. (2006). A molecular screening strategy based on beta-myosin heavy chain, cardiac myosin binding protein C and troponin T genes in Italian patients with hypertrophic cardiomyopathy. J. Cardiov. Med. 7, 601–607. doi: 10.2459/01.JCM.0000237908.26377.d6
Hamada, M., Ikeda, S., and Shigematsu, Y. (2014). Advances in medical treatment of hypertrophic cardiomyopathy. J. Cardiol. 64, 1–10. doi: 10.1016/j.jjcc.2014.02.022
Hill, T. L., Eisenberg, E., and Greene, L. (1980). Theoretical model for the cooperative equilibrium binding of myosin subfragment 1 to the actin-troponin-tropomyosin complex. Proc. Natl. Acad. Sci. U.S.A. 77, 3186–3190. doi: 10.1073/pnas.77.6.3186
Ho, C. Y., Charron, P., Richard, P., Girolami, F., Van Spaendonck-Zwarts, K. Y., and Pinto, Y. (2015). Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc. Res. 105, 397–408. doi: 10.1093/cvr/cvv025
Huke, S., and Knollmann, B. C. (2010). Increased myofilament Ca2+-sensitivity and arrhythmia susceptibility. J. Mol. Cell. Cardiol. 48, 824–833. doi: 10.1016/j.yjmcc.2010.01.011
Jacques, A., Hoskins, A. C., Kentish, J. C., and Marston, S. B. (2008). From genotype to phenotype: a longitudinal study of a patient with hypertrophic cardiomyopathy due to a mutation in the MYBPC3 gene. J. Muscle Res. Cell Motil. 29, 239–246. doi: 10.1007/s10974-009-9174-0
Jagatheesan, G., Rajan, S., Petrashevskaya, N., Schwartz, A., Boivin, G., Arteaga, G. M., et al. (2007). Rescue of tropomyosin-induced familial hypertrophic cardiomyopathy mice by transgenesis. Am. J. Physiol. Heart Circ. Physiol. 293, H949–H958. doi: 10.1152/ajpheart.01341.2006
Janssen, P. M., Zeitz, O., and Hasenfuss, G. (1999). Transient and sustained impacts of hydroxyl radicals on sarcoplasmic reticulum function: protective effects of nebivolol. Eur. J. Pharmacol. 366, 223–232. doi: 10.1016/S0014-2999(98)00907-8
Janssen, P. M., Zeitz, O., Rahman, A., and Hasenfuss, G. (2001). Protective role of nebivolol in hydroxyl radical induced injury. J. Cardiovasc. Pharmacol. 38(Suppl. 3), S17–S23. doi: 10.1097/00005344-200112003-00004
Josephson, R. A., Silverman, H. S., Lakatta, E. G., Stern, M. D., and Zweier, J. L. (1991). Study of the mechanisms of hydrogen peroxide and hydroxyl free radical-induced cellular injury and calcium overload in cardiac myocytes. J. Biol. Chem. 266, 2354–2361.
Kimura, A. (2010). Molecular basis of hereditary cardiomyopathy: abnormalities in calcium sensitivity, stretch response, stress response and beyond. J. Hum. Genet. 55, 81–90. doi: 10.1038/jhg.2009.138
Kooij, V., Boontje, N., Zaremba, R., Jaquet, K., Dos Remedios, C., Stienen, G. J., et al. (2010). Protein kinase C alpha and epsilon phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic Res. Cardiol. 105, 289–300. doi: 10.1007/s00395-009-0053-z
Liou, Y. M., Kuo, S. C., and Hsieh, S. R. (2008). Differential effects of a green tea-derived polyphenol (–)-epigallocatechin-3-gallate on the acidosis-induced decrease in the Ca2+ sensitivity of cardiac and skeletal muscle. Pflugers Arch. 456, 787–800. doi: 10.1007/s00424-008-0456-y
Lovelock, J. D., Monasky, M. M., Jeong, E. M., Lardin, H. A., Liu, H., Patel, B. G., et al. (2012). Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circ. Res. 110, 841–850. doi: 10.1161/CIRCRESAHA.111.258251
Lues, I., Siegel, R., and Harting, J. (1988). Effect of isomazole on the responsiveness to calcium of the contractile elements in skinned cardiac muscle fibres of various species. Eur. J. Pharmacol. 146, 145–153. doi: 10.1016/0014-2999(88)90496-7
Lund, L. H., Benson, L., Dahlstrom, U., Edner, M., and Friberg, L. (2014). Association between use of beta-blockers and outcomes in patients with heart failure and preserved ejection fraction. JAMA 312, 2008–2018. doi: 10.1001/jama.2014.15241
Maron, B. J., McKenna, W. J., Danielson, G. K., Kappenberger, L. J., Kuhn, H. J., Seidman, C. E., et al. (2003). American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur. Heart. J. 24, 1965–1991. doi: 10.1016/S0195-668X(03)00479-2
Maron, B. J., Ommen, S. R., Semsarian, C., Spirito, P., Olivotto, I., and Maron, M. S. (2014). Hypertrophic cardiomyopathy: present and future, with translation into contemporary cardiovascular medicine. J. Am. Coll. Cardiol. 64, 83–99. doi: 10.1016/j.jacc.2014.05.003
Mearini, G., Stimpel, D., Geertz, B., Weinberger, F., Kramer, E., Schlossarek, S., et al. (2014). Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat. Commun. 5:5515. doi: 10.1038/ncomms6515
Mearini, G., Stimpel, D., Kramer, E., Geertz, B., Braren, I., Gedicke-Hornung, C., et al. (2013). Repair of Mybpc3 mRNA by 5′-trans-splicing in a Mouse Model of Hypertrophic Cardiomyopathy. Mol. Ther. Nucleic Acids 2:e102. doi: 10.1038/mtna.2013.31
Messer, A. E., Bayliss, C. R., El-Mezgueldi, M., Redwood, C. S., Ward, D. G., Leung, M. C., et al. (2016). Mutations in troponin T associated with Hypertrophic Cardiomyopathy increase Ca2+-sensitivity and suppress the modulation of Ca2+-sensitivity by troponin I phosphorylation. Arch. Biochem. Biophys. 601, 113–120. doi: 10.1016/j.abb.2016.03.027
Moore, J. R., Leinwand, L., and Warshaw, D. M. (2012). Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 111, 375–385. doi: 10.1161/CIRCRESAHA.110.223842
Morimoto, S., Yanaga, F., Minakami, R., and Ohtsuki, I. (1998). Ca2+-sensitizing effects of the mutations at Ile-79 and Arg-92 of troponin T in hypertrophic cardiomyopathy. Am. J. Physiol. 275, C200–C207.
Najafi, A., Schlossarek, S., Van Deel, E. D., van Den Heuvel, N., Guclu, A., Goebel, M., et al. (2015). Sexual dimorphic response to exercise in hypertrophic cardiomyopathy-associated MYBPC3-targeted knock-in mice. Pflugers Arch. 467, 1303–1317. doi: 10.1007/s00424-014-1570-7
Park, K., and Park, T. H. (2016). Comparative effects of nebivolol and carvedilol on left ventricular diastolic function in older heart failure patients with preserved ejection fraction: study protocol for a randomized controlled trial. Trials 17:530. doi: 10.1186/s13063-016-1656-y
Piccini, J. P., Sinner, M. F., Greiner, M. A., Hammill, B. G., Fontes, J. D., Daubert, J. P., et al. (2012). Outcomes of Medicare beneficiaries undergoing catheter ablation for atrial fibrillation. Circulation 126, 2200–2207. doi: 10.1161/CIRCULATIONAHA.112.109330
Pohlmann, L., Kroger, I., Vignier, N., Schlossarek, S., Kramer, E., Coirault, C., et al. (2007). Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ. Res. 101, 928–938. doi: 10.1161/CIRCRESAHA.107.158774
Prisant, L. M. (2008). Nebivolol: pharmacologic profile of an ultraselective, vasodilatory beta1-blocker. J. Clin. Pharmacol. 48, 225–239. doi: 10.1177/0091270007310378
Richard, P., Charron, P., Carrier, L., Ledeuil, C., Cheav, T., Pichereau, C., et al. (2003). Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107, 2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54
Robertson, I. M., Li, M. X., and Sykes, B. D. (2009). Solution structure of human cardiac troponin C in complex with the green tea polyphenol, (–)-epigallocatechin 3-gallate. J. Biol. Chem. 284, 23012–23023. doi: 10.1074/jbc.M109.021352
Robinson, P., Griffiths, P. J., Watkins, H., and Redwood, C. S. (2007). Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 101, 1266–1273. doi: 10.1161/CIRCRESAHA.107.156380
Schlossarek, S., Englmann, D. R., Sultan, K. R., Sauer, M., Eschenhagen, T., and Carrier, L. (2012). Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res. Cardiol. 107:235. doi: 10.1007/s00395-011-0235-3
Schlossarek, S., Singh, S. R., Geertz, B., Schulz, H., Reischmann, S., Hubner, N., et al. (2014). Proteasome inhibition slightly improves cardiac function in mice with hypertrophic cardiomyopathy. Front. Physiol. 5:484. doi: 10.3389/fphys.2014.00484
Spoladore, R., Maron, M. S., D'amato, R., Camici, P. G., and Olivotto, I. (2012). Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur. Heart J. 33, 1724–1733. doi: 10.1093/eurheartj/ehs150
Stoehr, A., Neuber, C., Baldauf, C., Vollert, I., Friedrich, F. W., Flenner, F., et al. (2014). Automated analysis of contractile force and Ca2+ transients in engineered heart tissue. Am. J. Physiol. Heart Circ. Physiol. 306, H1353–H1363. doi: 10.1152/ajpheart.00705.2013
Stohr, A., Friedrich, F. W., Flenner, F., Geertz, B., Eder, A., Schaaf, S., et al. (2013). Contractile abnormalities and altered drug response in engineered heart tissue from Mybpc3-targeted knock-in mice. J. Mol. Cell. Cardiol. 63, 189–198. doi: 10.1016/j.yjmcc.2013.07.011
Stoschitzky, K., Stoschitzky, G., Klein, W., Muller, F., Buhring, K., Lamprecht, G., et al. (2004). Different effects of exercise on plasma concentrations of nebivolol, bisoprolol and carvedilol. Cardiovasc. Drugs Ther. 18, 135–138. doi: 10.1023/B:CARD.0000029031.87129.05
Tadano, N., Du, C. K., Yumoto, F., Morimoto, S., Ohta, M., Xie, M. F., et al. (2010). Biological actions of green tea catechins on cardiac troponin C. Br. J. Pharmacol. 161, 1034–1043. doi: 10.1111/j.1476-5381.2010.00942.x
Tardiff, J. C., Carrier, L., Bers, D. M., Poggesi, C., Ferrantini, C., Coppini, R., et al. (2015). Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc. Res. 105, 457–470. doi: 10.1093/cvr/cvv023
Tardiff, J. C., Hewett, T. E., Palmer, B. M., Olsson, C., Factor, S. M., Moore, R. L., et al. (1999). Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J. Clin. Invest. 104, 469–481. doi: 10.1172/JCI6067
Thottakara, T., Friedrich, F. W., Reischmann, S., Braumann, S., Schlossarek, S., Kramer, E., et al. (2015). The E3 ubiquitin ligase Asb2beta is downregulated in a mouse model of hypertrophic cardiomyopathy and targets desmin for proteasomal degradation. J. Mol. Cell. Cardiol. 87, 214–224. doi: 10.1016/j.yjmcc.2015.08.020
van Dijk, S. J., Dooijes, D., Dos Remedios, C., Michels, M., Lamers, J. M., Winegrad, S., et al. (2009). Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 119, 1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672
van Dijk, S. J., Paalberends, E. R., Najafi, A., Michels, M., Sadayappan, S., Carrier, L., et al. (2012). Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ. Heart Fail. 5, 36–46. doi: 10.1161/CIRCHEARTFAILURE.111.963702
van Veldhuisen, D. J., Cohen-Solal, A., Bohm, M., Anker, S. D., Babalis, D., Roughton, M., et al. (2009). Beta-blockade with nebivolol in elderly heart failure patients with impaired and preserved left ventricular ejection fraction: data From SENIORS (Study of Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors With Heart Failure). J. Am. Coll. Cardiol. 53, 2150–2158. doi: 10.1016/j.jacc.2009.02.046
Vignier, N., Schlossarek, S., Fraysse, B., Mearini, G., Kramer, E., Pointu, H., et al. (2009). Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ. Res. 105, 239–248. doi: 10.1161/CIRCRESAHA.109.201251
Walsh, R., Thomson, K. L., Ware, J. S., Funke, B. H., Woodley, J., Mcguire, K. J., et al. (2017). Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 19, 192–203. doi: 10.1038/gim.2016.90
Warren, C. M., Karam, C. N., Wolska, B. M., Kobayashi, T., De Tombe, P. P., Arteaga, G. M., et al. (2015). Green tea catechin normalizes the enhanced Ca2+ sensitivity of myofilaments regulated by a hypertrophic cardiomyopathy-associated mutation in human cardiac troponin I (K206I). Circ. Cardiovasc. Genet. 8, 765–773. doi: 10.1161/CIRCGENETICS.115.001234
Wijnker, P. J., Friedrich, F. W., Dutsch, A., Reischmann, S., Eder, A., Mannhardt, I., et al. (2016). Comparison of the effects of a truncating and a missense MYBPC3 mutation on contractile parameters of engineered heart tissue. J. Mol. Cell. Cardiol. 97, 82–92. doi: 10.1016/j.yjmcc.2016.03.003
Keywords: nebivolol, myofilament, Ca2+ sensitivity, hypertrophic cardiomyopathy, Mybpc3, mouse, human, epigallocatechin-3-gallate
Citation: Stücker S, Kresin N, Carrier L and Friedrich FW (2017) Nebivolol Desensitizes Myofilaments of a Hypertrophic Cardiomyopathy Mouse Model. Front. Physiol. 8:558. doi: 10.3389/fphys.2017.00558
Received: 04 May 2017; Accepted: 17 July 2017;
Published: 02 August 2017.
Edited by:
P. Bryant Chase, Florida State University, United StatesReviewed by:
Paul M. L. Janssen, Ohio State University Columbus, United StatesAdriano Martins, Maimonides Medical Center, United States
Copyright © 2017 Stücker, Kresin, Carrier and Friedrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felix W. Friedrich, f.friedrich@uke.de