 Igor A. Yakovlev
Igor A. Yakovlev Carl G. Fossdal
Carl G. Fossdal- Norwegian Institute of Bioeconomy Research, Ås, Norway
Epigenetic memory in Norway spruce affects the timing of bud burst and bud set, vitally important adaptive traits for this long-lived forest species. Epigenetic memory is established in response to the temperature conditions during embryogenesis. Somatic embryogenesis at different epitype inducing (EpI) temperatures closely mimics the natural processes of epigenetic memory formation in seeds, giving rise to epigenetically different clonal plants in a reproducible and predictable manner, with respect to altered bud phenology. MicroRNAs (miRNAs) and other small non-coding RNAs (sRNAs) play an essential role in the regulation of plant gene expression and may affect this epigenetic mechanism. We used NGS sequencing and computational in silico methods to identify and profile conserved and novel miRNAs among small RNAs in embryogenic tissues of Norway spruce at three EpI temperatures (18, 23 and 28°C). We detected three predominant classes of sRNAs related to a length of 24 nt, followed by a 21–22 nt class and a third 31 nt class of sRNAs. More than 2100 different miRNAs within the prevailing length 21–22 nt were identified. Profiling these putative miRNAs allowed identification of 1053 highly expressed miRNAs, including 523 conserved and 530 novels. 654 of these miRNAs were found to be differentially expressed (DEM) depending on EpI temperature. For most DEMs, we defined their putative mRNA targets. The targets represented mostly by transcripts of multiple-repeats proteins, like TIR, NBS-LRR, PPR and TPR repeat, Clathrin/VPS proteins, Myb-like, AP2, etc. Notably, 124 DE miRNAs targeted 203 differentially expressed epigenetic regulators. Developing Norway spruce embryos possess a more complex sRNA structure than that reported for somatic tissues. A variety of the predicted miRNAs showed distinct EpI temperature dependent expression patterns. These putative EpI miRNAs target spruce genes with a wide range of functions, including genes known to be involved in epigenetic regulation, which in turn could provide a feedback process leading to the formation of epigenetic marks. We suggest that TIR, NBS and LRR domain containing proteins could fulfill more general functions for signal transduction from external environmental stimuli and conversion them into molecular response. Fine-tuning of the miRNA production likely participates in both developmental regulation and epigenetic memory formation in Norway spruce.
Introduction
Adaptation to the changing environments is vitally important for long-lived plant species like forest trees. Epigenetic modifications and specifically epigenetic memory could be important mechanisms for diversifying environmental responses and widening the total plasticity of populations. The epigenetic memory of a plant is defined by the reproducible set of modifications of DNA and chromatin (without alteration of the DNA sequence) induced by external stimuli, which alter gene expression and therefore the properties and behavior of the plant. Memorizing of specific responses, laid down by epigenetic mechanisms, could provide significant strategic benefits to those plants, since the most successful response could be tuned or reenacted in response to a modified environmental condition and this would be retained in future cell lineages, and potentially inherited and altered by selection in future generations (Bräutigam et al., 2013; Baulcombe and Dean, 2014; Iwasaki and Paszkowski, 2014; Kinoshita and Seki, 2014). Much remains to be known about the enigmatic repertoire of epigenetic mechanisms that operate in forest trees but earlier studies firmly confirmed the presence of epitype inducing (EpI) temperature-dependent plant phenotypes (Yakovlev et al., 2012; Liu et al., 2015) and significant transcriptomic changes in such epitypes (Yakovlev et al., 2016).
Both long non-coding RNAs (lncRNAs) and small RNAs (sRNAs) such as short non-coding RNAs are known to be core components of signaling networks involved in epigenetic modification, transcription regulation and participate in transgenerational epigenetic inheritance in plants and animals (Hauser et al., 2011; Heard and Martienssen, 2014). Epigenetic regulation can be mediated through a dynamic interplay between sRNAs, DNA methylation, histone modifications, histone variants, and chromatin architecture, which together modulate transcriptional silencing, activation and the accessibility of DNA in variety of ways (Heo and Sung, 2011; Simon and Meyers, 2011; Lee, 2012; Mirouze, 2012; Bond and Baulcombe, 2014).
MicroRNAs (miRNAs) are specific class of sRNA regulators, with having roles in phenotypic plasticity, plant development and as well as metabolism, all of which act through posttranscriptional regulation of gene expression. miRNAs are widely recognized as having a 20–24 nt length, and characteristically originate from a hairpin-folded single-stranded RNA precursor structure (Meyers et al., 2008). MicroRNA precursors are transcribed from specific miRNA genes (MIR), and are processed predominantly by a multi-functional DICER-LIKE1 (DCL1). The miRNAs in turn negatively regulate gene expression by forming miRNA-inducing silencing complex (miRISC) in association with the ARGONAUTE (AGO) proteins (Bartel, 2004). miRNAs have potential to regulate virtually all cellular mechanisms (Sun, 2012; Wu, 2013), and they do so by restricting translation or cleaving multiple target transcripts. In some instances, they have already been identified as key players in producing rapid adaptation to changing environmental conditions (Sunkar et al., 2012; Ferdous et al., 2015; Nguyen et al., 2015). As miRNAs target transcripts through the imperfect match of sequence composition between miRNA and target, the consequence of gene regulation by miRNAs is that a few miRNAs can specifically change the expression pattern, or fine tune, many specific genes simultaneously. The consequence of gene regulation by miRNAs is similar to that by transcription factors (TFs) (Morris and Mattick, 2014). Moreover, a regulatory cascade essential for appropriate execution of several biological events is triggered through the combinatorial network action of both miRNAs and TFs (Sunkar, 2010; Arora et al., 2013). Besides TFs being among miRNA targets there are known epigenetic regulators (Pikaard and Mittelsten Scheid, 2014), and these in turn, regulate the expression of the individual miRNAs (Gruber and Zavolan, 2013; Song et al., 2015). More specifically, miRNAs are shown to be directly involved in epigenetic regulation and memorizing the responses to different types of stress both in plants and animals (Khraiwesh et al., 2012; Osella et al., 2014; Stief et al., 2014; Hilker et al., 2016). Thus miRNAs have the ability to regulate many target genes, initiate transcriptional and silencing cascades, provide feedback loops, and split transcriptional regulation off into a separate dedicated parallel tracks including those already known to be in the epigenetic regulatory pathway itself.
Conserved and novel miRNAs were identified in angiosperm and gymnosperm species (Montes et al., 2014), including multiple conifers—pines (Lu et al., 2007; Oh et al., 2008; Wan et al., 2012b; Quinn et al., 2014), spruces (Yakovlev et al., 2010; Nystedt et al., 2013), and larches (Zhang et al., 2012, 2013), but their diversity, abundance and regulatory functions are still largely to be studied. Norway spruce is a suitable Pinaceae member to conduct experimental studies for epigenetic modification in gymnosperms since a variety of resources exist, including the possibilities for in vitro propagation of plant material (Kvaalen and Johnsen, 2008) and draft genome (Nystedt et al., 2013). Furthermore, epigenetic memory in Norway spruce affects vitally important adaptive traits such as the timing of bud burst and bud set, adaptive traits crucial for this species environmental success. Epigenetic alteration of these traits are presumed to be established or modified in response to the EpI temperature conditions prevailing during early seed formation, based phenotypic and molecular responses (Kvaalen and Johnsen, 2008; Johnsen et al., 2009). Moreover, Picea abies somatic embryogenesis (SE) is an ideal experimental system for studying this phenomenon since these responses are recapitulated through in vitro SE. SE at different temperatures closely mimic the processes of epigenetic memory formation, naturally occurring in zygotic seeds, and give rise to epigenetically different plants (epitypes), which have a clonal character, with a reproducible and predictable temperature-dependent altered bud phenology.
Some progress has already been achieved toward understanding molecular mechanisms underlying the epigenetic memory formation. A significant number of epigenetic regulators, including sRNA biogenesis pathways, are differentially expressed at different EpI conditions, supporting that methylation of DNA, histones modifications and sRNAs are pivotal for the establishment of the epigenetic memory (Yakovlev et al., 2014, 2016). We found several miRNAs differentially expressed in buds of different epitypes and suggesting their involvement in the epigenetic memory (Yakovlev et al., 2010), and this stimulated the need for a much deeper study of the various miRNA during SE in spruce, the life stage at which the epigenetic memory is laid down. The main goal of the current study was to further characterize and identify the extensive miRNA fraction in the small non-coding part of Norway spruce transcriptome. We aimed to identify the key miRNAs regulating differentially expressed genes (DEGs) and those especially related to epigenetic regulators that could potentially establish the epigenetic memory process during embryogenesis, by examining their expression profiles during SE at contrasting EpI temperature conditions. To our knowledge, this is also the first genome-wide in silico characterization of miRNAs and study of their transcript profiles during SE in spruce. We show an extensive number of miRNAs that can target epigenetic regulators including those modifying DNA and histone methylation, and sRNA pathways genes, supporting the notion that these predicted miRNAs and their target genes could be among central players in epigenetic memory formation.
Methods
Plant Material and RNA Extraction
Somatic embryos, and their induction and growth, used in this analysis are already previously described (Kvaalen and Johnsen, 2008; Yakovlev et al., 2014, 2016). Embryogenic samples were those obtained from two seeds (genotypes) originating from a controlled cross of a defined female (♀#2650) and male (♂#2707) of Norway spruce parents, with those crosses being performed either in outdoor conditions; a cold originated genotype, denoted as A2C, or in greenhouse conditions as a warm originated genotype, denoted as B10W. With the current analysis, nine samples were collected from each of the two different genotypes, representing three maturation stages and three different temperatures in which the epitypes form, providing 18 samples in total. Twenty to Thirty milligram of embryo containing callus or 2–5 embryos were collected per sample, combined and immediately snap-frozen and stored in liquid nitrogen until RNA extraction. Embryo tissues were ground in 1.5 ml Eppendorf tubes by pestle and the total RNA extracted using an Epicentre MasterPure™ Plant RNA Purification Kit (Epicentre, Madison, WI, USA, #MCR85102) according to the manufacture's instruction. Total RNA preparations were stored at −80°C and the integrity and quantity of total RNA was assessed by Agilent 2100 Bioanalyzer with RNA 6000 Nano Kit and also the Small RNA kit (Agilent, Santa Clara, CA, USA #5067-1511 and #5067-1548 respectively).
Small RNA Library Construction, Sequencing and Bioinformatics
The 18 small RNA libraries were each constructed from 1 μg of total RNA, using the Ion Total RNA-Seq Kit v2 for Small RNA Libraries (#4476289), with the enrichment steps as outlined in the Ion RNA-Seq Library Preparation guide (#4476286 revision E). Quality and quantity of amplified libraries were analyzed with the Agilent Technologies 2100 Bioanalyzer with Agilent High Sensitivity DNA Kit (Agilent, Santa Clara, CA, USA, #5067-4626). Template-positive Ion Sphere™ Particles (ISPs) containing clonally amplified DNA were prepared with the Ion OneTouch™ 2 Instrument using the Ion PGM Template OT2 200 Kit (#4480974) according manufacturer instructions. Sequencing was performed using Ion Personal Genome Machine® (PGM™) Sequencer (Thermo Fisher Scientific Inc.) and each library was sequenced individually on 316v2 chips using the Ion PGM™ sequencing 200 Kit v2 (#4482006). Data was initially processed on Ion Torrent Server using Torrent Suite software (v.4.2) and fastq formatted files were analyzed using CLC Genomics Workbench software (V 8.+) (QIAGEN, Aarhus A/S, Denmark).
In Silico Identification of Conserved and Novel Mirnas and miRNA Genes in Spruce
All the single-read and sRNA sequences beyond 19–29 bp from the 18 libraries were filtered out to remove rare and too short or too long reads. To search for conserved miRNAs, the filtered sRNA sequences were compared to known miRNAs in the miRBase v21.0 (Kozomara and Griffiths-Jones, 2011) restricted to all tree species in addition to miRNAs from the genomes of Arabidopsis, Vitis, and Physcomitrella allowing up to 2 nucleotides mismatch using the sRNA module of CLC genomics workbench software (v.8). To identify miRNA gene loci and novel miRNAs, we mapped all the filtered sRNA reads to the gene models encompassing high-, medium- and low-confidence as defined in the Picea abies genome v1 (http://congenie.org/) (Nystedt et al., 2013). A gene model was considered a putative miRNA gene loci when at least 100 reads of distinct sRNA tags mapped to the loci or gene model with a minimum of 0–2 mismatches. Gene sequences having 80–300 nt flanking the candidate miRNA sequence were manually scrutinized based on the criteria for miRNA definition described by Meyers et al. (2008). In addition, secondary structures of putative miRNA genes were predicted using different folding algorithms by the CLC genomics workbench software. When the stem-loop hairpin for the putative MIR was confirmed, then the existence of putative novel miRNAs was estimated. sRNA(s) with higher frequency was/were considered as guide miRNAs, sRNA(s) on the opposite strand of the loop was/were considered as star-miRNAs (*miRNA). We allowed shifting of star sequences relative to miRNA guide sequences for 1–6 bp.
During analysis, we established that some miRNA candidates (guide and star sequences) were determined to have considerably long hairpin structures, so we extended our search for gene models within the fragments for up to 1 kb, using the same procedure for miRNA detection as those used in the shorter fragments.
Expression Analysis of Predicted MiRNAs and in Silico Identification of their Targets
Expression analysis was performed using RNA-Seq tool of the CLC Genomic Workbench v8 with defined lists of miRNAs for annotation of the sRNA reads. Prediction of miRNA targets was carried out by searching for complementary regions between the identified miRNAs in this study and by using all the Picea abies gene models v1 as the transcript sequence input using online web server psRobot—Plant Small RNA Analysis Toolbox (Wu et al., 2012), and the psRNATarget—Plant Small RNA Target Analysis Server (Dai and Zhao, 2011).
To substantiate putative targets and to refine potential miRNA-mRNA target pairs, we additionally analyzed correlations between transcript amounts of miRNA and their defined targets at three different EpI temperatures. mRNA transcript amounts were taken based on our previous study (Yakovlev et al., 2016).
qRT-PCR for miRNAs
To validate sequencing data we quantified transcript levels for 10 selected conserved and novel miRNAs with quantitative real-time RT-PCR. For analysis we used the same small RNA extracts which were used for sequencing. cDNAs were synthesized from 600 ng of the small RNA extracts with the Mir-X™ miRNA First-Strand Synthesis kit (Clontech, #638315) following manufacturer recommendations. Real-time RT-PCR amplification was performed using Mir-X™ miRNA qRT-PCR SYBR® Kit (Clontech, #638314) in a 25 μl reaction volume, using 2 μl of a diluted cDNA solution described above as template and 200 nM of each primer. qPCRs were performed on a ViiA™ 7 Real-Time PCR System (Applied Biosystems, USA) following the manufacturer's instructions. After PCR, dissociation curves were carried out to verify the specificity of the amplification. There were three biological replicates for each sample. All expression levels were normalized to geometric mean of three selected ribosomal and transfer RNA genes (Pa4.5S, Pa5S and PatRNA-R), showing most stable expression profiles as describe previously (Yakovlev et al., 2010). Forward primers were designed based on mature miRNA sequence. If Tm of mature miRNA was less than 60°C, it had been adjusted by adding G's to the 5′ end of the miRNA sequence. The list of studied miRNAs and their primer sequences are shown in Table S10. The 3′ primer for qPCR was the mRQ 3′ Primer supplied with the kit.
Data acquisition and analysis were done using ViiA™ 7-system SDS software for absolute quantification and MS Excel software.
Data Submission
Unique transcripts from 18 libraries sequenced using Ion Torrent PGM™ Sequencer were deposited to the SRA (Short Read Archive, NCBI) and got the following accession: submission ID SUB1781210; BioProject ID PRJNA339513 and accession IDs: SAMN05592191–SAMN05592208.
Results
Small RNA Library Sequencing
In total, we sequenced 18 small RNA (sRNA) libraries, representing three stages of in vitro spruce embryo development and three different EpI temperature treatments. This produced nearly 50 million reads in the length range from 7 to 50 nt (Table S1). Three clear read length peaks were found in the embryonic sRNA pool after the trimming—and these corresponded with the lengths of 24–23, 21–20, and 31–32 nt (Figure 1A). The 31–32 nt group of sRNAs consist of ~14,5 thousands sRNAs among more than 3 million reads. To reduce complexity and focus attention on the canonical miRNA population, we filtered all reads to 19–27 nt and removed all single reads to avoid sequencing and stochastic errors. In total, over 13 million reads were retained for further analysis with two clear peak classes—prevailing with length of 24–23 nt and then of 21–20 nt, in both genotypes A2K and B10W (Figure 1B).
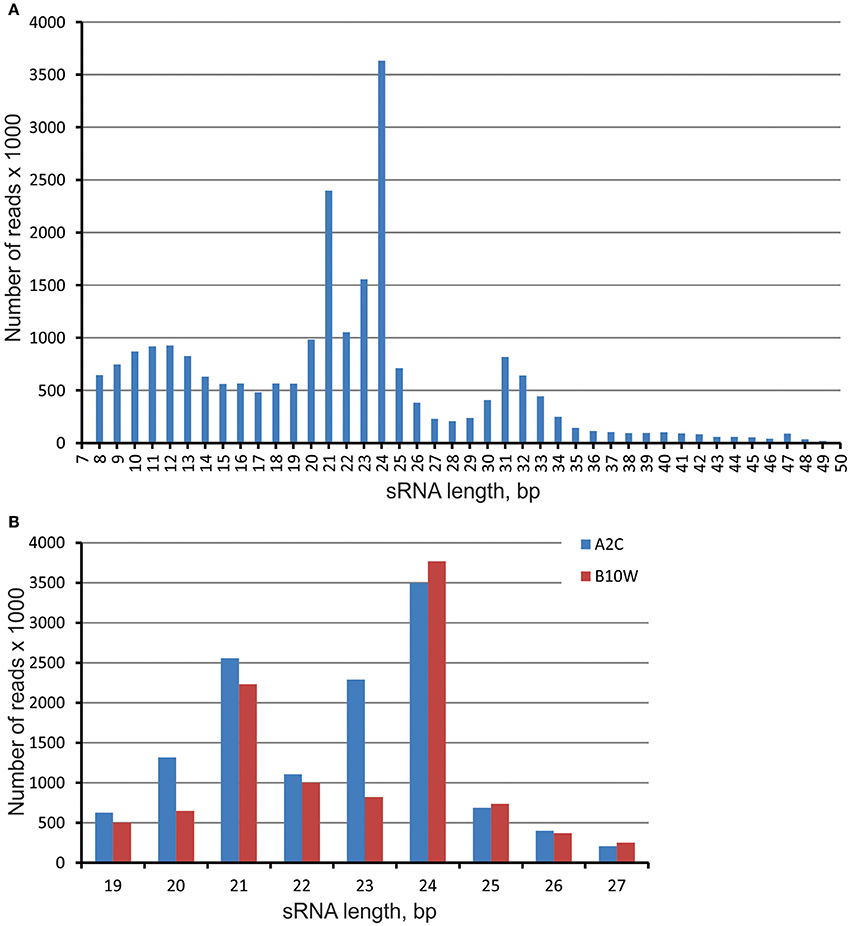
Figure 1. Length distribution of sRNA in libraries prepared from the embryos of two genotypes of the full-sib family of Norway spruce from cross ♀#2650 × ♂#2707 produced in outdoor conditions—A2C and produced in greenhouse conditions—B10W. (A) averaged sRNA length distribution in all libraries; (B) sRNA length distribution in miRNA length range—19–27 bp in two genotypes.
In Silico Identification of Conserved and Novel miRNAs in Spruce
Using CLC Genomics Workbench a sRNA analysis was conducted. Search and annotation of conserved miRNAs was based on the miRBase v21 database using a criteria allowing up to two mismatches. A total of 636 conserved miRNAs were defined and these belonged to 51 miRNA families. These could originate from 99 defined precursors. Not all precursors for the conserved miRNA members were found, but at least one precursor was identified for the majority of miRNA families and we considered that sufficiently supported the internal origin of the defined class of conserved miRNAs (Table S2).
Additionally we defined 1316 novel miRNAs that had no homology to miRBase v21 annotations. They could belong to 630 families and could originate from 740 predicted precursors (Table S3).
The majority of identified miRNAs in spruce embryos were 21 nt (41%) and 22 nt (34%) in length, all other length classes count less than 10% (Figure 2) (Figure S1). More often the miRNAs at the initial positions contain uridine (U–37%) and adenine (A–25%) and less C and G (16 and 22%% correspondingly).
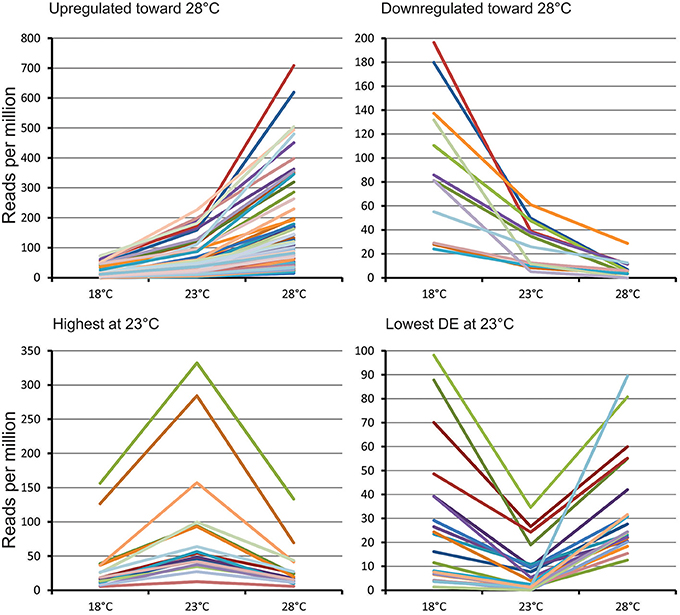
Figure 2. Main expression profiles of predicted differentially expressed miRNAs in Norway spruce embryos cultivating at three different EpI temperatures.
For nearly all conserved miRNA families we found large numbers of miRNA members (isomiRs). In average, there were 15 isomiRs per family, but varied from 1 to 102. The largest quantity of isomiRs was found for the highly conserved family miRNA166, with 102 miRNAs. More than 40 miRNAs were found across the identified miRNA families—of miR156, miR159, miR946, miR950, miR951, miR1311 and miR3701. Eleven families had more than 10 but less than 40 member miRNAs and these included miR319, miR390, miR396, miR397, miR482, miR947, miR1312, miR1316, miR3705, miR3710, and miR3712. The remaining 39 families had less than 10 isomiRs (Table 1).
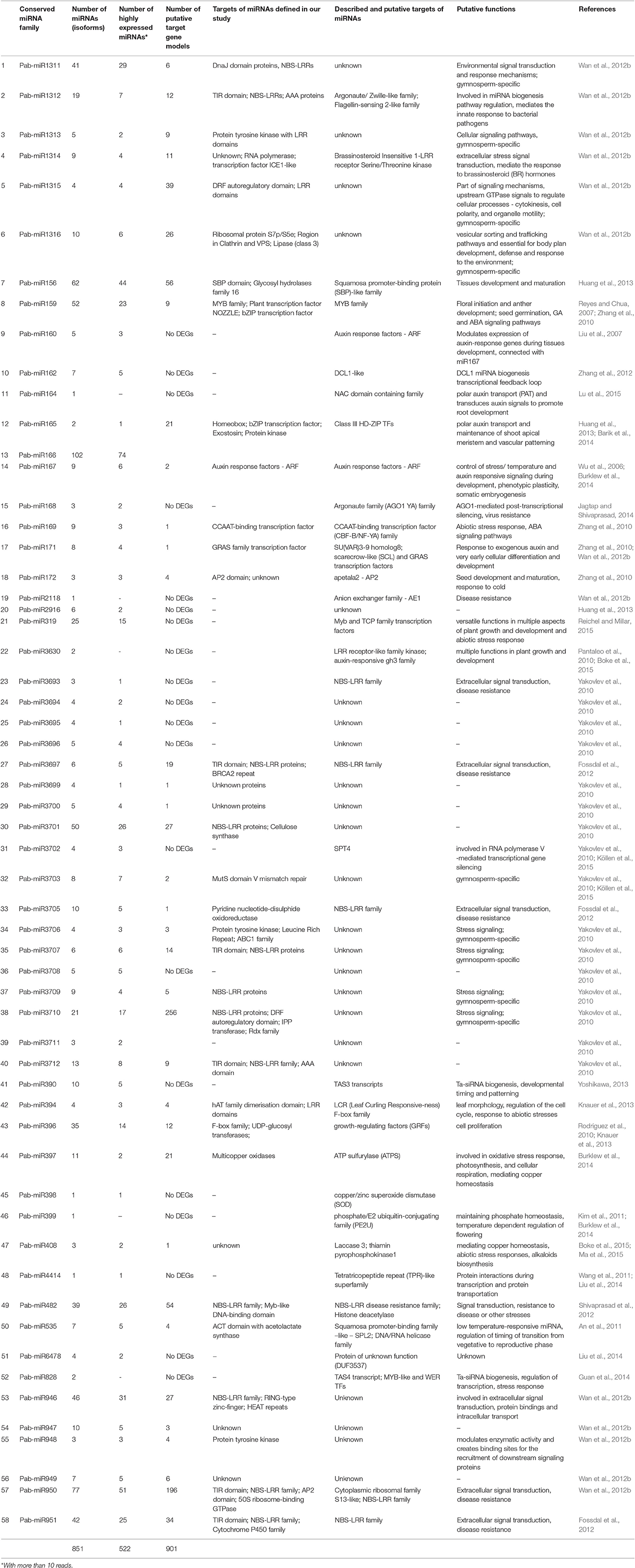
Table 1. Family description, targets and putative functions of predicted conserved miRNAs families defined in within the sRNAs of Norway spruce embryos in comparison with previously published data.
Quantification of Transcripts and Identification of Differentially Expressed miRNA Profiles
For expression analysis, we used all miRNAs with average read counts greater or equal to 10 in at least one of the sequenced libraries. From the 2267 miRNAs we defined in the transcriptome of Norway spruce embryos, 1115 miRNAs were further used for differential expression analysis.
Differentially expressed miRNAs (DEMs) were identified through pair-wise comparison of libraries by setting the threshold |log2 RPKM ratio| to more than 1 and p-value < 0.05. Temperature responsive miRNAs displaying more than two fold difference between EpI temperatures were considered as differentially expressed and these were further examined. In total, we detected 676 DEMs while the remaining 439 miRNAs, did not show any differences in transcript numbers at different EpI temperatures. Most of the defined miRNAs were present in all treatments. Among the DEMs, only one miRNA—Pab-miRn931 was expressed solely at 28°C and 15 miRNAs were expressed at two temperatures and were absent at third particular temperature.
Based on their transcript profiles at the three different EpI temperatures, 654 DEMs could be grouped into 12 clusters. Main transcription profiles shown on Figure 1 and detailed description of clusters presented on Table S4. The first two clusters included 159 miRNAs significantly upregulated at 28°C and decreasing in abundance with decreasing the temperature. The most abundant here were conserved miRNAs from miR156, miR159, miR166, miR167, miR396, miR1311, miR3701, and miR951, as well as 26 novel miRNAs. Two other clusters (5–6) contained 50 miRNAs significantly upregulated at 18°C and decreasing in abundance with increasing temperature. Most of miRNAs in this cluster were novel, and only conserved miRNAs from miR950 and miR482 families were identified based on sequence similarities. Two clusters (10–11) encompass 22 miRNAs found most abundant at 23°C (Figure 1). Two conserved miRNAs from miR319 and miR3701 families were found here, yet all the other discovered miRNAs were found to be novel. Other clusters consisted of various other miRNAs having similar transcript profiles, the largest of which were clusters 3 and 4. Cluster 3 encompassed 101 miRNAs that were highly expressed at 28°C, yet were of equally low expression at 18° and 23°C, while cluster 4 had an opposing profile, and this encompassed 151 miRNAs showing equal high expression at 28° and 23°C, while showing low expression at 18°C. Both clusters include different miRNAs from the families of miR156, miR159, miR166, miR167, miR396, miR946, miR1311, miR1312, miR3701 miR951 as well as many novel miRNAs (Table S4).
Additionally, we specifically analyzed the changes in sequence and abundance of DEMs from conserved families at different EpI temperatures. Within each family, we found wide range of modifications, including nucleotide substitutions, 5′ and 3′ uridylation and adenylation, trimming and tailing. In addition, we found quite variable transcription patterns for different family members, sometimes opposite. However, we did not find any EpI temperature specific isomiRs presented only at one specific culturing temperature and did not find any clear influence of EpI temperature on modification type. Some examples of miRNA diversity within families, their expression patterns and their corresponding stem-loop RNA secondary structure of hairpin-forming precursors presented at Figure S2.
In Silico Prediction of Targets of Conserved and Novel Norway Spruce miRNAs
For the assignment of functional roles to the whole set of defined miRNAs, the target gene transcripts were predicted by the online web server psRobot—Plant Small RNA Analysis Toolbox (Wu et al., 2012) and the psRNATarget—Plant Small RNA Target Analysis Server (Dai and Zhao, 2011). In the first instance, we searched for the respective target genes for all miRNAs as defined in Norway spruce v1 coding sequences (Nystedt et al., 2013) irrespective of their transcript profiles. This resulted in 2050 miRNAs being identified as the cognate miRNAs to 6058 annotated gene models from around 1414 gene families with diverse biological functions and 4701 gene models without matches to the database. The largest number of gene models, which could be regulated by miRNAs, were in gene families containing following Pfam domains: Leucine Rich Repeat (LRR) protein genes, protein kinase domain, pentatricopeptide (PPR) repeat, NB-ARC (nucleotide-binding adaptor R-gene shared) domain, ATPase family associated with various cellular activities (AAA), Toll-Interleukin receptor (TIR) domain, Clathrin heavy chains/VPS (vacuolar protein sorting-associated), tetratricopeptide (TPR), Myb-like DNA-binding domain, mTERF (Mitochondrial transcription termination factor), Multicopper oxidase, AP2 domain, Cytochrome P450, F-box domain and many others (Table S5). TIR and NBS-LRR comprise one of the largest groups of genes in spruce. We found more than 1900 gene models containing different LRR domains and more than 740 gene models containing NB-ARC domain and close to 370 models containing TIR domain. In total, 2594 for the TIR or NB-ARC LRR gene models were found.
Differential Expression of miRNAs and their Predicted Targets
Afterwards we selected putative targets among DEGs for the 522 conserved and 593 novel DEMs under the inductive conditions required of epitype differentiation. We additionally analyzed correlations between miRNA and their target transcription patterns at different EpI temperatures to refine potential cognate miRNA-mRNA target pairs. All target pairs with correlation below −0.6 were considered as prospective miRNA regulated gene models, especially as correlations between target transcript and miRNAs at all temperatures could help build a robust definition functional pairs, providing further insight into temperature-dependent processes leading to the formation of epigenetic memory in developing embryos (Tables S6–S8).
In total, we defined 1921 miRNA—mRNAs (DEM–DEG) pairs, consist of 470 miRNAs and 1139 target genes, incl. 930 annotated gene models from around 212 gene families with diverse biological functions and 209 gene models without match at the NCBI databases. Similar to the whole set of miRNAs, the largest number of DEM/DEG pairs were found in gene families coding for tandem repeat domain (TRD) containing proteins. Among the gene families are the 166 LRR gene models, which could be targeted by 278 miRNAs; 90 NB-ARC—by 169 miRNAs, and 52 TIRs—by 138 miRNAs. TIR, NBS-LRR proteins could be targeted by both conserved (miR482, miR946, miR950, miR1311-1316, miR3710, etc.), and novel miRNAs, like Pab-miRnY45_str, Pab-miRn00543, Pab-miRn00468, Pab-miRn00930, Pab-miRn00202_3p, Pab-miRn00386, Pab-miRn00492, Pab-miRn00930, Pab-miRn01804_5p, Pab-miRnB5, etc. Clathrin and vacuolar protein sorting (VPS) domain proteins often contain PPR and TPR repeat domains, and these could be the targets for regulation by miRNAs based on duplex sequence similarity. 30 Clathrin/VPS and 128 PPR/TPR gene models could be targeted by 202 miRNAs, including Pab-miRn00676, which could regulate the translation of 43 genes (Table 2; Table S6).

Table 2. Enrichment of Pfam domains based on the preliminary functional characterization of most abundant differentially expressed gene families, which could be regulated by highly differentially expressed miRNAs.
DEMs were also showed to target several transcription factors such as Squamosa promoter-binding (SPB) protein (target of miR156), plant transcription factor NOZZLE-like (miR159), Myb-like (miR159, miR482), Homeobox domain bZIP transcription factor (miR166), HD-ZIP protein (miR165/miR166), CCAAT-binding transcription factor (miR169) and AP2-like transcription factors (miR172, miR950), involved in developmental timing and transition from juvenility. Besides transcription factors, other targets included F-box protein (miR396), laccase (miR397), plant U-box (PUB) proteins (miR946).
We found that 317 DEGs could be regulated by several miRNAs and 290 DEMs could regulate more than one gene (Figures S3.1–20). For example, six DEMs- Pab-miR950.59, Pab-miR950.67, Pab-miR950.68, Pab-miR950.69, Pab-miR950.70 and Pab-miR950.72 from the miR950 family could target the same gene model MA_10433003g0010, coding for multi-domain TIR-NBS P-loop containing Nucleoside Triphosphate Hydrolase domain and involved in signal transduction and response to different external stimuli (Figure S3.1). Pab-miRn0282.1, Pab-miRn0054.4_5p, Pab-miR950.59, and Pab-miR950.72 could target the same gene model MA_55143g0010 at four different target sites (Figure S3.2). MA_55143g0010 is coding for multi-domain TIR-NBS-LRR protein, containing Helix-hairpin-helix motif and probably in addition to signal sensing, monitoring and transduction functions could directly bind to DNA and could fulfill “reader” functions.
Similarly, the novel Pab-miRn0539_5p was found to potentially regulate 12 different gene transcripts of NBS-LRR and ATPases; while conserved Pab-miR1315.1 could regulate 9 genes, mostly LRRs and cytoskeleton remodeling proteins participating in regulation of cellular processes such as cytokinesis, cell polarity, and organelle motility. In most cases, such redundant targeting occurs in gene families of repeat proteins, such as multiple multi-domain TIR-NBS-LRR proteins, proteins kinases, HEAT-repeat proteins and tetra- (TPR) or pentatricopeptide (PPR) repeat proteins (Figures S3.4,5).
In Silico Prediction of Differentially Expressed miRNAs Targeting Epigenetic Regulators
To evaluate miRNAs regulating epigenetic regulators and the potential for feedback loops within the sRNA biogenesis pathways we focused on the putative miRNA targets encoded within genes of the known epigenetic pathways described earlier (Yakovlev et al., 2016). All miRNAs and their predicted targets were analyzed, irrespective of their expression levels and distributing the target gene models by the type of epigenetic modification, they could be related to (Table 3, Table S9).
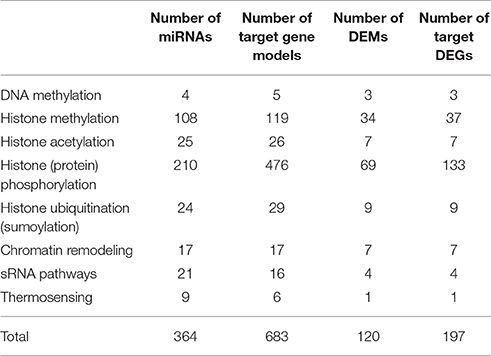
Table 3. Predicted miRNAs targeting epigenetic regulator with the distribution of the target gene models by the type of epigenetic modification.
In total, we found 84 conserved miRNAs from 32 families and 280 novel miRNAs that could target and post-transcriptionally regulate 683 gene models spanning all pathways of epigenetic regulation. Among them, 22 conserved miRNAs from 12 families and 98 novel miRNAs, with 197 of predicted targets, showed opposite transcription patterns and considered as involved into post-transcriptional silencing of epigenetic regulators during epitype formation in Norway spruce embryos (Table 3). As the miRNAs targets, we found several gene families, coding for genes involved in epigenetic regulation, like WD domain, G-beta repeat; SNF2 family N-terminal domain; DEAD/DEAH box helicase; BRCA1 C Terminus (BRCT) domain; KH (K Homology) RNA-binding domain; PHD-finger; Core histone H2A/H2B/H3/H4; SET domain; BTB/POZ (BR-C, ttk and bab/Pox virus and Zinc finger) domain; E1-E2 ATPase; ThiF family of Ubiquitin-activating enzyme (E1 enzyme) and some others.
We found only 4 miRNAs targeting the genes involved in DNA methylation, including methylation marks setting, erasing or reading (Table S9-1) and 3 miRNA-target pairs showed opposite transcription profiles, considering repression of target transcripts by high levels of miRNA transcripts and vice versa. 108 miRNAs were found to target 119 epigenetic regulators related to histone methylation (Table S9-2). Among them, 34 miRNA targeting 37 gene models, showed opposing transcriptional profiles. At least three SET-domain and one polycomb-like protein genes look to be regulated by conserved Pab-miR156.61 and three novel miRs—Pab-miRn0254.1_5p, Pab-miRn0305.2_3p, and Pab-miRn0252.1_3p correspondingly. In addition, large number of histone methylation readers could be regulated by miRNAs.
Phosphorylation was also an over-represented processes which appears to be highly regulated by miRNAs. Protein kinases (PK) were abundantly found among the gene families and had the highest number of total miRNA targets. We found 210 miRNAs, which could target 476 PK gene models, from which 69 miRNAs had opposite transcription profiles with 133 targets. Here we note, the conserved miRNAs from families' miR162, miR390, miR948, miR1313 and miR 3706 and abundant number of novel miRNAs, including Pab-miRn0117.1_3p; Pab-miRn0117.3_3p; Pab-miRn0165_3p; Pab-miRn0286.2_5p; Pab-miRn0301; Pab-miRn0367.1; Pab-miRn0441; Pab-miRn0592; Pab-miR1313.2 and others. All of them could target multiple gene models (Table S9-4).
Gene models related to histone acetylation were also revealed to be miRNA targets, but only 25 miRNAs were predicted to target such genes, and seven of these miRNA-target pairs had negatively correlated transcription profiles, indication miRNA regulatory effect (Table S9-3). Similarly, genes involved in ubiquitination/de-ubiquitination were infrequent amongst the miRNA targets. In this case, we found 24 miRNA targeting 29 gene models and 9 miRNA-target pairs with opposing transcriptional profiles (Table S9-5). Only 17 miRNAs were found to target chromatin remodelers, and these occurred mainly from the SNF2 family and in one case from a SWI gene family. In the latter case, 7 miRNA-target pairs had opposing transcription profiles, characteristic of a miRNA-target pair, and in each case these miRNAs were all of novel sequence composition (Table S9-6).
Conserved miRNAs were found to target genes participating in miRNA and other sRNAs biogenesis pathways. Several conserved miRNAs from miR156, miR162 and miR482 could target DCL1-like gene transcripts but transcription patterns did not confirm any regulatory effect. While, novels Pab-miRn miRn0030.3 shown to regulate ARGONAUTE7 (AGO7), Pab-miRn0009.3_3p—3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG1) and Pab-miRn0305.2_3p; Pab-miR482.29 shown to regulate different SUPPRESSOR OF GENE SILENCING 3 (SGS3) genes (Table S9-7).
Another important and noteworthy observation was that a few genes were related to those already identified to be involved in thermosensing. Six genes were found to be potentially regulated by 9 miRNAs. We found just one pair Pab-miRn0407—Calcium-activated BK potassium channel (MA_10433576g0020) with opposite transcript profiles. Most of other genes belong to the Ankyrin repeat family and Myb-like DNA-binding domain, which could be targeted by numerous miRNAs, so it is difficult to establish clear transcription patterns using whole embryo samples (Table S9-8).
Verification of miRNA-Seq Expressions by qRT-PCR
To validate the miRNA-Seq expression data, a subset of 10 specific DEMs (Table S1) was selected for verification by qRT-PCR. The selected miRNAs (5 conserved and 5 novel) showed a distinct differential profiles during embryo development at different epitype-inducing temperatures. For all of the 10 studied miRNAs the qRT-PCR outcomes closely correlated with the transcript abundance estimated by miRNA-Seq (Figure S4).
Discussion
Different EpI temperatures of SE result in epigenetically different plants (epitypes) with altered bud phenology observed phenotypically later in the sporophytes annual growth cycle, in a predictable and reproducible manner (Kvaalen and Johnsen, 2008). During early development, complex gene expression, together with epigenetic changes, control and determine the embryonic cell fates (de Vega-Bartol et al., 2013; De-la-Peña et al., 2015). Some of these processes are determined by epigenetic modifications and networks of gene expression are directed and mediated by non-coding RNAs (Simon and Meyers, 2011; Lee, 2012; Mirouze, 2012). To focus on the genetic pathways initiating and establishing the epigenetic memory response, we obtained sRNAs using deep miRNA sequencing on embryos from contrasting EpI temperatures. Our sRNA analysis revealed over 3000 miRNA candidates from somatic embryonic tissues, including those of the 24-nt miRNA class. This result reconfirms their earlier found presence (Nystedt et al., 2013), in other gymnosperm species (Wan et al., 2012a,b; Wang et al., 2015), and firmly reject the earlier hypothesis that the 24 nt miRNA and siRNA class are missing and the notion that the DCL4-mediated miRNA biogenesis is absent within gymnosperm plants. We found a more complex structure of sRNA pool in embryonic tissues, compared to developed plant tissues, with the presence of additional longer fraction of sRNAs of 31–32 nt length. These sRNAs were not reported earlier in any plant species, but a same size class were described in animals germ line cells as PIWI-related sRNA (Le Thomas et al., 2014). Appearance of such longer fraction was very recently described during callus formation in tobacco plants Lunerova, (personal communication). Therefore, we could speculate that appearance of longer sRNA fraction could be related to pluripotent state of cells, presented in callus or developing SE. This longer sRNA fraction was not the topic of our present study and should be pursued further in a separate study. Aside from this, our in silico analysis uncovers numerous predicted novel miRNA families and identifies miRNAs with potential involvement in epigenetic regulation and thermosensing based on anti-correlated expression patterns of miRNA—target pairs.
Whilst this sRNA sequencing effort and in silico analysis is not an exhaustive search or analysis of the complex spruce sRNA population as we were limited only to one tissue type, it is the most exhaustive examination of 24–23 nt and 21–20 nt small RNAs in spruce to date. We limited our miRNA candidates' pool to predicted novel and conserved miRNAs with more than 10 reads in any libraries. In total, we identified 1115 miRNAs; nearly half of which (593) were putatively novel miRNAs and 522 conserved miRNAs. This conserved pool included 21 spruce specific miRNAs defined in previous studies (Yakovlev et al., 2010).
Analysis of the conserved miRNAs pool revealed a high number of miRNAs isoforms (isomiRs) nearly for all of the 58 conserved miRNA families. Various mechanisms are associated with the diversification of miRNA sequences, including imprecise DCL processing or post-transcriptional modifications, like trimming and tailing (Li et al., 2014). As we did not find specific genomic fragments with precursors for the most of isomiRs, most probably, they were originate from the common precursors and modified post-transcriptionally. Existence of isomiRs could be also explained by the possibility of miRNAs to originate from several MIRs. In this case, any SNP changes in miRNA genes would cause the creation of specific isomiRs. Generation of isomiRs from the same miRNA locus may extend its functional influence. As miRNA isoforms vary in size and sequence from the canonical miRNA, alternative targets may be regulated and differential AGO loading could arise, resulting in diverse regulatory outcomes (Neilsen et al., 2012; Ameres and Zamore, 2013; Li et al., 2014). Large amount of isoforms for conserved miRNAs are present in non-model species (e.g., Mica et al., 2009; Lin and Lai, 2013; Liu et al., 2014), but these isomiRs are not well characterized and, in most cases, their origins and functions if any remains unknown (Neilsen et al., 2012). It has previously been demonstrated that temperature treatments altered the expression of a specific subset of mature miRNAs and displayed differential expression of numbers of miRNA isoforms (Baev et al., 2014).
This is the first report of *miRNAs in gymnosperms. Even if our predictions detect the presence of both guide and *miRNA, we have not equivalently identified specific*miRNAs since such identification should be supported with more exact experimental knowledge of each miRNAs origin precursor. For most putative miRNA we found several potential precursors and we cannot at present define which exact precursor of origin or which arm of these precursors is the guiding (functional) mature miRNA and which is the passenger (non-functional) ones. We have defined miRNAs closer to 5′ end as -5p and putative *miRNA on the opposite strand of the predicted precursor (closer to 3′ end) as -3p. In many cases both predicted miRNAs (guide and *miRNA) from both strands were expressed and occasionally in opposite manner. Our sRNAs originate from whole embryos containing various tissues and cell types, and since different tissues and cell types may preferentially express either the -5p or the -3p variant (or vice versa), it is impossible in the present material to define with sufficient certainty which variant is main miRNA and which is star. Future cell type specific studies should be performed to clear up this matter.
Our target prediction revealed a large range of gene families with diverse biological functions. Usually, miRNAs regulate posttranslational repression of mRNAs via two different mechanisms: the miRNAs induce mRNA translational repression and the miRNAs induce mRNA cleavage under the help of ARGONAUTE protein (Bartel, 2009). Due to lack of necessity of full complementarity between miRNA and its target, it is difficult to establish clear relations between miRNA transcript profiles and their putative targets and transcript profiles as in many cases same gene transcript could be regulated by several miRNAs and, opposite, same miRNA could target several gene transcripts. High redundancy of miRNA—mRNA interplay and the multiplicity of miRNA genes and miRNA binding sites in the UTR of target genes may play a synergistic or additive function in the regulation of such targets (Bartel, 2009). Hence, the role of miRNA in gene expression would most likely be that of a fine-tuning process rather than an ON/OFF switch. One gene may be targeted by up and downregulated miRNA at the same time in order to attain the optimum concentration required for a specific function (Herranz and Cohen, 2010). This is the case for the mediation of cell fate decisions, where miRNAs act in synergy with other transcription regulators to establish gene regulatory networks (Herranz and Cohen, 2010).
We want to highlight the predictive nature of our in silico predictions using psRobot and psRNATarget analysis server and that future experimental validation studies are needed to confirm or refute our predictions. Notwithstanding, in most cases when we found inverse relations between miRNAs transcript levels and transcript levels of their predicted targets we presently considered them as a likely functional miRNA target pairs and have predicted their putative functional importance.
The highest number of predicted target gene models, which could be regulated by miRNAs, were found among multiple repeats containing proteins gene families, like: Leucine Rich Repeat (LRR) protein genes, protein kinase domain, NB-ARC (nucleotide-binding adaptor R-gene shared) domain, ATPase family associated with various cellular activities (AAA), Toll-Interleukin receptor (TIR) domain, Clathrin heavy chains/VPS (vacuolar protein sorting-associated), tetra- (TPR) and pentatricopeptide (PPR) repeat protein genes and some others. TIR-, NBS-LRR genes are considered as one of the first lines of defense against pathogen infection (Dangl and Jones, 2001; Meyers et al., 2005). However, our in vitro culture was axenic, free from any pathogens, and should not initiate any defense-related responses. Even so, we could see a large amount of differentially expressed LRR-containing genes showing clear dependence on epitype inducing temperature, and this imply their involvement in processes far removed from pathogen-mediated interactions. We consider that TIR, NBS, and LRR domain containing proteins may fulfill more general role in signals transduction from external environment (both biotic and abiotic) and conversion into molecular responses of diverse nature. TPR proteins can promote the formation of highly specific multiprotein complexes and can support the binding of different ligands (Zeytuni and Zarivach, 2012). A typical PPR protein could binds one or several organellar transcripts, and influences their expression by altering RNA sequence, turnover, processing, or translation (Barkan and Small, 2014). Clathrin and vacuolar protein sorting (VPS) domain proteins are another large group of protein involved in the vesicular sorting and trafficking pathways and essential for body plan development, defense and response to the environment (Chen et al., 2011). They often contain penta- and tetratricopeptide repeat domains, which could be the targets for regulation by miRNAs. In response to temperature differences, these may help fine tune intracellular traffic or the delivery of signaling molecules, but it is hard to envisage otherwise how they may directly contribute to temperature-dependent formation of epigenetic memory in the spruce embryos.
More specifically, we were looking for the genes involved in epigenetic regulation. It is shown, that a significant part of sRNA can serve as a pointer and participate in chromatin modification of promoters or DNA methylation, preventing, or activating the transcription of the individual is often remotely located sRNA coding genes or clusters (Mirouze, 2012). It is remarkably, that from the more than 700 gene models of putative epigenetic regulators described in developing embryos (Yakovlev et al., 2016), more than half are predicted targets by miRNAs. Moreover, we found that in EpI temperature dependent manner 197 DEGs of epigenetic regulators could be post-transcriptionally regulated by 120 miRNAs, including 22 conserved miRNAs from 12 families. miRNAs were mostly involved in regulation of genes related to methylation modifications, both in DNA and histones. In addition, several miRNAs were shown to target sRNA biogenesis pathway's gene models, confirming the existence of tight regulatory feedback loops within the miRNA and siRNA pathways in both gymnosperms and angiosperms (Henderson and Jacobsen, 2007; Niu et al., 2015). The opposite may occur, that miRNAs expression could be regulated by specific genes in response to changes in an extracellular microenvironment and considered as one of the major mechanisms for epigenetic modifications of the cell. It was shown that ion channels/transporters could transduce extracellular signals into miRNA transcript level changes, which, in turn, regulating target genes, and proposed potential link between cells and their microenvironment through ion channels/transporters (Jiang et al., 2012).
Most of miRNAs targeting epigenetic regulators predicted here were novel, so their possible existence also in other plant species should be pursued to verify their general importance. However, the fact that we find miRNA directly targeting all types of epigenetic modifiers indicated that miRNAs are central players involved in formation of epigenetic memory or at least in regulating the expression of the epigenetic machinery. In light of their important functions in the epigenetic memory formation, future validation work on these miRNAs and their targets is required.
Conclusion
In this in silico analysis, we defined a predicted repertoire of conserved and novel miRNAs that could play crucial roles in regulating embryo development and epigenetic memory formation in Norway spruce. We showed that developing Norway spruce embryos possess a more complex sRNA structure than reported for somatic tissues. A variety of the predicted miRNAs showed distinct EpI temperature dependent expression patterns. These putative EpI miRNAs target spruce genes with a wide range of functions, including genes known to be involved in epigenetic regulation, which in turn could provide a feedback process leading to the formation of epigenetic marks. We suggest that TIR, NBS, and LRR domain containing proteins could fulfill more general functions for signal transduction from external environmental stimuli and conversion them into molecular response. Fine-tuning of the miRNA production likely participates in both developmental regulation and epigenetic memory formation in Norway spruce. This study also provides important information for comparative studies of miRNAs with other plant species and their predicted involvement in epigenetic regulation.
Author Contributions
IY and CF conceived and designed research. IY conducted experiments. IY analyzed data and wrote the manuscript together with CF. Both authors read and approved the final manuscript.
Funding
This work was supported by the Norwegian Research Council (FRIBIO Grant#191455/V40 and FRIMEDBIO Grant#240766/F20), the Norwegian Forest and Landscape Institute (SGB #135117-1) and the EU FP7 project ProCoGen.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Ksenia Zakharova (St.-Petersburg State University, Russia) for assistance during library preparation and sequencing; Adam Vivian-Smith (NIBIO, Norway) for advice and technical help during sequencing, for commenting on the manuscript; Thomas Solvin (NIBIO, Norway) for help in miRNA cataloging and YeonKyeong Lee (Norwegian University of Life Sciences) for assistance in growing of embryogenic cultures.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphys.2017.00674/full#supplementary-material
Abbreviations
DEG, differentially expressed gene; DEM, differentially expressed defined miRNA; SE, somatic embryogenesis; sRNA, small non-coding RNA; miRNA, microRNA; isomiRs, isoform microRNAs; EpI, epitype inducing.
References
Ameres, S. L., and Zamore, P. D. (2013). Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 14, 475–488. doi: 10.1038/nrm3611
An, F.-M., Hsiao, S.-R., and Chan, M.-T. (2011). Sequencing-based approaches reveal low ambient temperature-responsive and tissue-specific microRNAs in Phalaenopsis Orchid. PLoS ONE 6:e18937. doi: 10.1371/journal.pone.0018937
Arora, S., Rana, R., Chhabra, A., Jaiswal, A., and Rani, V. (2013). miRNA–transcription factor interactions: a combinatorial regulation of gene expression. Mol. Genet. Genomics 288, 77–87. doi: 10.1007/s00438-013-0734-z
Baev, V., Milev, I., Naydenov, M., Vachev, T., Apostolova, E., Mehterov, N., et al. (2014). Insight into small RNA abundance and expression in high- and low-temperature stress response using deep sequencing in Arabidopsis. Plant Physiol. Biochem. 84, 105–114. doi: 10.1016/j.plaphy.2014.09.007
Barik, S., SarkarDas, S., Singh, A., Gautam, V., Kumar, P., Majee, M., et al. (2014). Phylogenetic analysis reveals conservation and diversification of micro RNA166 genes among diverse plant species. Genomics 103, 114–121. doi: 10.1016/j.ygeno.2013.11.004
Barkan, A., and Small, I. (2014). Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 65, 415–442. doi: 10.1146/annurev-arplant-050213-040159
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi: 10.1016/j.cell.2009.01.002
Baulcombe, D. C., and Dean, C. (2014). Epigenetic regulation in plant responses to the environment. Cold Spring Harb. Perspect. Biol. 6:a019471. doi: 10.1101/cshperspect.a019471
Boke, H., Ozhuner, E., Turktas, M., Parmaksiz, I., Ozcan, S., and Unver, T. (2015). Regulation of the alkaloid biosynthesis by miRNA in opium poppy. Plant Biotechnol. J. 13, 409–420. doi: 10.1111/pbi.12346
Bond, D. M., and Baulcombe, D. C. (2014). Small RNAs and heritable epigenetic variation in plants. Trends Cell Biol. 24, 100–107. doi: 10.1016/j.tcb.2013.08.001
Bräutigam, K., Vining, K. J., Lafon-Placette, C., Fossdal, C. G., Mirouze, M., Marcos, J. G., et al. (2013). Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol. Evol. 3, 399–415. doi: 10.1002/ece3.461
Burklew, C., Xie, F., Ashlock, J., and Zhang, B. (2014). Expression of microRNAs and their targets regulates floral development in tobacco (Nicotiana tabacum). Funct. Integr. Genomics 14, 299–306. doi: 10.1007/s10142-014-0359-2
Chen, X., Irani, N. G., and Friml, J. (2011). Clathrin-mediated endocytosis: the gateway into plant cells. Curr. Opin. Plant Biol. 14, 674–682. doi: 10.1016/j.pbi.2011.08.006
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39(suppl_2), W155–W159. doi: 10.1093/nar/gkr319
Dangl, J. L., and Jones, J. D. G. (2001). Plant pathogens and integrated defence responses to infection. Nature 411, 826–833. doi: 10.1038/35081161
de Vega-Bartol, J., Simoes, M., Lorenz, W., Rodrigues, A., Alba, R., Dean, J., et al. (2013). Transcriptomic analysis highlights epigenetic and transcriptional regulation during zygotic embryo development of Pinus pinaster. BMC Plant Biol. 13:123. doi: 10.1186/1471-2229-13-123
De-la-Peña, C., Nic-Can, G. I., Galaz-Ávalos, R. M., Avilez-Montalvo, R., and Loyola-Vargas, V. M. (2015). The role of chromatin modifications in somatic embryogenesis in plants. Front. Plant Sci. 6:635. doi: 10.3389/fpls.2015.00635
Ferdous, J., Hussain, S. S., and Shi, B.-J. (2015). Role of microRNAs in plant drought tolerance. Plant Biotechnol. J. 13, 293–305. doi: 10.1111/pbi.12318
Fossdal, C., Yaqoob, N., Krokene, P., Kvaalen, H., Solheim, H., and Yakovlev, I. (2012). Local and systemic changes in expression of resistance genes, NB-LRR genes and their putative microRNAs in norway spruce after wounding and inoculation with the pathogen Ceratocystis polonica. BMC Plant Biol. 12:105. doi: 10.1186/1471-2229-12-105
Gruber, A. J., and Zavolan, M. (2013). Modulation of epigenetic regulators and cell fate decisions by miRNAs. Epigenomics 5, 671–683. doi: 10.2217/epi.13.65
Guan, X., Pang, M., Nah, G., Shi, X., Ye, W., Stelly, D. M., et al. (2014). miR828 and miR858 regulate homoeologous MYB2 gene functions in Arabidopsis trichome and cotton fibre development. Nat. Commun. 5:3050. doi: 10.1038/ncomms4050
Hauser, M.-T., Aufsatz, W., Jonak, C., and Luschnig, C. (2011). Transgenerational epigenetic inheritance in plants. Biochim. Biophy. Acta 1809, 459–468. doi: 10.1016/j.bbagrm.2011.03.007
Heard, E., and Martienssen, R A., (2014). Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157, 95–109. doi: 10.1016/j.cell.2014.02.045
Henderson, I. R., and Jacobsen, S. E. (2007). Epigenetic inheritance in plants. Nature 447, 418–424. doi: 10.1038/nature05917
Heo, J. B., and Sung, S. (2011). Vernalization-mediated epigenetic silencing by a long intronic noncoding, RNA. Science 331, 76–79. doi: 10.1126/science.1197349
Herranz, H., and Cohen, S. M. (2010). MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev. 24, 1339–1344. doi: 10.1101/gad.1937010
Hilker, M., Schwachtje, J., Baier, M., Balazadeh, S., Bäurle, I., Geiselhardt, S., et al. (2016). Priming and memory of stress responses in organisms lacking a nervous system. Biol. Rev. 91, 1118–1133. doi: 10.1111/brv.12215
Huang, D., Koh, C., Feurtado, J. A., Tsang, E., and Cutler, A. (2013). MicroRNAs and their putative targets in Brassica napus seed maturation. BMC Genomics 14:140. doi: 10.1186/1471-2164-14-140
Iwasaki, M., and Paszkowski, J. (2014). Epigenetic memory in plants. EMBO J. 33, 1987–1998. doi: 10.15252/embj.201488883
Jagtap, S., and Shivaprasad, P. (2014). Diversity, expression and mRNA targeting abilities of Argonaute-targeting miRNAs among selected vascular plants. BMC Genomics 15:1049. doi: 10.1186/1471-2164-15-1049
Jiang, X., Zhang, J., and Chan, H. (2012). Ion channels/transporters as epigenetic regulators? —a microRNA perspective. Sci. China Life Sci. 55, 753–760. doi: 10.1007/s11427-012-4369-9
Johnsen, Ø., Kvaalen, H., Yakovlev, I. A., Dæhlen, O. G., Fossdal, C. G., and Skrøppa, T. (2009). “An epigenetic memory from time of embryo development affects climatic adaptation in Norway spruce,” in Plant Cold Hardiness. From the Laboratory to the Field, eds L. V. Gusta, M. E. Wisniewski and K. K. Tanino. (Wallingford, CT: CABI), 99–107.
Khraiwesh, B., Zhu, J.-K., and Zhu, J. (2012). Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophy. Acta 1819, 137–148. doi: 10.1016/j.bbagrm.2011.05.001
Kim, W., Ahn, H. J., Chiou, T.-J., and Ahn, J. H. (2011). The Role of the miR399-PHO2 module in the regulation of flowering time in response to different ambient temperatures in Arabidopsis thaliana. Mol. Cells 32, 83–88. doi: 10.1007/s10059-011-1043-1
Kinoshita, T., and Seki, M. (2014). Epigenetic memory for stress response and adaptation in plants. Plant Cell Physiol. 55, 1859–1863. doi: 10.1093/pcp/pcu125
Knauer, S., Holt, A. L., Rubio-Somoza, I., Tucker, E. J., Hinze, A., Pisch, M., et al. (2013). A protodermal miR394 signal defines a region of stem cell competence in the Arabidopsis shoot meristem. Dev. Cell 24, 125–132. doi: 10.1016/j.devcel.2012.12.009
Köllen, K., Dietz, L., Bies-Etheve, N., Lagrange, T., Grasser, M., and Grasser, K. D. (2015). The zinc-finger protein SPT4 interacts with SPT5L/KTF1 and modulates transcriptional silencing in Arabidopsis. FEBS Lett. 589, 3254–3257. doi: 10.1016/j.febslet.2015.09.017
Kozomara, A., and Griffiths-Jones, S. (2011). miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39(Suppl. 1), D152–D157. doi: 10.1093/nar/gkq1027
Kvaalen, H., and Johnsen, O. (2008). Timing of bud set in Picea abies is regulated by a memory of temperature during zygotic and somatic embryogenesis. N. Phytol. 177, 49–59. doi: 10.1111/j.1469-8137.2007.02222.x
Le Thomas, A., Toth, K., and Aravin, A. (2014). To be or not to be a piRNA: genomic origin and processing of piRNAs. Genome Biol. 15:204. doi: 10.1186/gb4154
Lee, J. T. (2012). Epigenetic regulation by long noncoding RNAs. Science 338, 1435–1439. doi: 10.1126/science.1231776
Li, J., Reichel, M., Li, Y., and Millar, A. A. (2014). The functional scope of plant microRNA-mediated silencing. Trends Plant Sci. 19, 750–756. doi: 10.1016/j.tplants.2014.08.006
Lin, Y., and Lai, Z. (2013). Comparative analysis reveals dynamic changes in miRNAs and their targets and expression during somatic embryogenesis in longan (Dimocarpus longan Lour.). PLoS ONE 8:e60337. doi: 10.1371/journal.pone.0060337
Liu, J., Feng, L., Li, J., and He, Z. (2015). Genetic and epigenetic control of plant heat responses. Front. Plant Sci. 6:267. doi: 10.3389/fpls.2015.00267
Liu, P.-P., Montgomery, T. A., Fahlgren, N., Kasschau, K. D., Nonogaki, H., and Carrington, J. C. (2007). Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 52, 133–146. doi: 10.1111/j.1365-313X.2007.03218.x
Liu, W., Yu, W., Hou, L., Wang, X., Zheng, F., Wang, W., et al. (2014). Analysis of miRNAs and their targets during adventitious shoot organogenesis of Acacia crassicarpa. PLoS ONE 9:e93438. doi: 10.1371/journal.pone.0093438
Lu, S., Sun, Y.-H., Amerson, H., and Chiang, V. L. (2007). MicroRNAs in loblolly pine (Pinus taeda L.) and their association with fusiform rust gall development. Plant J. 51, 1077–1098. doi: 10.1111/j.1365-313X.2007.03208.x
Lu, Y.-B., Qi, Y.-P., Yang, L.-T., Guo, P., Li, Y., and Chen, L.-S. (2015). Boron-deficiency-responsive microRNAs and their targets in Citrus sinensis leaves. BMC Plant Biol. 15:271. doi: 10.1186/s12870-015-0642-y
Ma, C., Burd, S., and Lers, A. (2015). miR408 is involved in abiotic stress responses in Arabidopsis. Plant J. 84, 169–187. doi: 10.1111/tpj.12999
Meyers, B. C., Axtell, M. J., Bartel, B., Bartel, D. P., Baulcombe, D., Bowman, J. L., et al. (2008). Criteria for annotation of plant microRNAs. Plant Cell Tpc. 108:064311. doi: 10.1105/tpc.108.064311
Meyers, B. C., Kaushik, S., and Nandety, R. S. (2005). Evolving disease resistance genes. Curr. Opin. Plant Biol. 8, 129–134. doi: 10.1016/j.pbi.2005.01.002
Mica, E., Piccolo, V., Delledonne, M., Ferrarini, A., Pezzotti, M., Casati, C., et al. (2009). High throughput approaches reveal splicing of primary microRNA transcripts and tissue specific expression of mature microRNAs in Vitis vinifera. BMC Genomics 10:558. doi: 10.1186/1471-2164-10-558
Mirouze, M. (2012). The small RNA-based odyssey of epigenetic information in plants: from cells to species. DNA Cell Biol. 31, 1650–1656. doi: 10.1089/dna.2012.1681
Montes, R. A. C., de Fátima Rosas-Cárdenas, F., De Paoli, E., Accerbi, M., Rymarquis, L. A., Mahalingam, G., et al. (2014). Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat. Commun. 5:3722. doi: 10.1038/ncomms4722
Morris, K. V., and Mattick, J. S. (2014). The rise of regulatory RNA. Nat. Rev. Genet. 15, 423–437. doi: 10.1038/nrg3722
Neilsen, C. T., Goodall, G. J., and Bracken, C. P. (2012). IsomiRs – the overlooked repertoire in the dynamic microRNAome. Trends Genet. 28, 544–549. doi: 10.1016/j.tig.2012.07.005
Nguyen, G., Rothstein, S., Spangenberg, G., and Kant, S. (2015). Role of MicroRNAs involved in plant response to nitrogen and phosphorous limiting conditions. Front. Plant Sci. 6:629. doi: 10.3389/fpls.2015.00629
Niu, S.-H., Liu, C., Yuan, H.-W., Li, P., Li, Y., and Li, W. (2015). Identification and expression profiles of sRNAs and their biogenesis and action-related genes in male and female cones of Pinus tabuliformis. BMC Genomics 16:693. doi: 10.1186/s12864-015-1885-6
Nystedt, B., Street, N. R., Wetterbom, A., Zuccolo, A., Lin, Y.-C., Scofield, D. G., et al. (2013). The Norway spruce genome sequence and conifer genome evolution. Nature 497, 579–584. doi: 10.1038/nature12211
Oh, T. J., Wartell, R. M., Cairney, J., and Pullman, G. S. (2008). Evidence for stage-specific modulation of specific microRNAs (miRNAs) and miRNA processing components in zygotic embryo and female gametophyte of loblolly pine (Pinus taeda). N. Phytol. 179, 67–80. doi: 10.1111/j.1469-8137.2008.02448.x
Osella, M., Riba, A., Testori, A., Corà, D., and Caselle, M. (2014). Interplay of microRNA and epigenetic regulation in the human regulatory network. Front. Genet. 5:345. doi: 10.3389/fgene.2014.00345
Pantaleo, V., Szittya, G., Moxon, S., Miozzi, L., Moulton, V., Dalmay, T., et al. (2010). Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 62, 960–976. doi: 10.1111/j.0960-7412.2010.04208.x
Pikaard, C. S., and Mittelsten Scheid, O. (2014). Epigenetic regulation in plants. Cold Spring Harb. Perspect. Biol. 6:a019315. doi: 10.1101/cshperspect.a019315
Quinn, C., Iriyama, R., and Fernando, D. (2014). Expression patterns of conserved microRNAs in the male gametophyte of loblolly pine (Pinus taeda). Plant Reprod. 27, 69–78. doi: 10.1007/s00497-014-0241-3
Reichel, M., and Millar, A. A. (2015). Specificity of plant microRNA target MIMICs: cross-targeting of miR159 and miR319. J. Plant Physiol. 180, 45–48. doi: 10.1016/j.jplph.2015.03.010
Reyes, J. L., and Chua, N.-H. (2007). ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 49, 592–606.
Rodriguez, R. E., Mecchia, M. A., Debernardi, J. M., Schommer, C., Weigel, D., and Palatnik, J. F. (2010). Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development 137, 103–112. doi: 10.1242/dev.043067
Shivaprasad, P. V., Chen, H.-M., Patel, K., Bond, D. M., Santos, B. A. C. M., and Baulcombe, D. C. (2012). A microRNA superfamily regulates nucleotide binding site–leucine-rich repeats and other mRNAs. Plant Cell 24, 859–874. doi: 10.1105/tpc.111.095380
Simon, S. A., and Meyers, B. C. (2011). Small RNA-mediated epigenetic modifications in plants. Curr. Opin. Plant Biol. 14, 148–155. doi: 10.1016/j.pbi.2010.11.007
Song, Y., Tian, M., Ci, D., and Zhang, D. (2015). Methylation of microRNA genes regulates gene expression in bisexual flower development in andromonoecious poplar. J. Exp. Bot. 66, 1891–1905. doi: 10.1093/jxb/eru531
Stief, A., Brzezinka, K., Lämke, J., and Bäurle, I. (2014). Epigenetic responses to heat stress at different time scales and the involvement of small RNAs. Plant Signal. Behav. 9:e970430. doi: 10.4161/15592316.2014.970430
Sun, G. (2012). MicroRNAs and their diverse functions in plants. Plant Mol. Biol. 80, 17–36. doi: 10.1007/s11103-011-9817-6
Sunkar, R. (2010). MicroRNAs with macro-effects on plant stress responses. Semin. Cell Dev. Biol. 21, 805–811. doi: 10.1016/j.semcdb.2010.04.001
Sunkar, R., Li, Y., and Jagadeeswaran, G. (2012). Functions of microRNAs in plant stress. Trends Plant Sci. 17, 196–203. doi: 10.1016/j.tplants.2012.01.010
Wan, L.-C., Wang, F., Guo, X., Lu, S., Qiu, Z., Zhao, Y., et al. (2012a). Identification and characterization of small non-coding RNAs from Chinese fir by high throughput sequencing. BMC Plant Biol. 12:146. doi: 10.1186/1471-2229-12-146
Wan, L.-C., Zhang, H., Lu, S., Zhang, L., Qiu, Z., Zhao, Y., et al. (2012b). Transcriptome-wide identification and characterization of miRNAs from Pinus densata. BMC Genomics 13:132. doi: 10.1186/1471-2164-13-132
Wang, L., Zhao, J., Zhang, M., Li, W., Luo, K., Lu, Z., et al. (2015). Identification and characterization of microRNA expression in Ginkgo biloba L. leaves. Tree Genet. Genomes 11, 1–13. doi: 10.1007/s11295-015-0897-3
Wang, T., Chen, L., Zhao, M., Tian, Q., and Zhang, W.-H. (2011). Identification of drought-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. BMC Genomics 12:367. doi: 10.1186/1471-2164-12-367
Wu, G. (2013). Plant microRNAs and development. J. Genet. Genomics 40, 217–230. doi: 10.1016/j.jgg.2013.04.002
Wu, H.-J., Ma, Y.-K., Chen, T., Wang, M., and Wang, X.-J. (2012). PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 40, W22–W28. doi: 10.1093/nar/gks554
Wu, M.-F., Tian, Q., and Reed, J. W. (2006). Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 133, 4211–4218. doi: 10.1242/dev.02602
Yakovlev, I. A., Carneros, E., Lee, Y., Olsen, J. E., and Fossdal, C. G. (2016). Transcriptional profiling of epigenetic regulators in somatic embryos during temperature induced formation of an epigenetic memory in Norway spruce. Planta 243, 1237–1249. doi: 10.1007/s00425-016-2484-8
Yakovlev, I. A., Fossdal, C. G., and Johnsen, Ø. (2010). MicroRNAs, the epigenetic memory and climatic adaptation in Norway spruce. N. Phytol. 187, 1154–1169. doi: 10.1111/j.1469-8137.2010.03341.x
Yakovlev, I. A., Fossdal, C. G., Skrøppa, T., Olsen, J. E., Jahren, A. H., and Johnsen, Ø. (2012). An adaptive epigenetic memory in conifers with important implications for seed production. Seed Sci. Res. 22, 63–76. doi: 10.1017/S0960258511000535
Yakovlev, I. A., Lee, Y., Rotter, B., Olsen, J. E., Skrøppa, T., Johnsen, Ø., et al. (2014). Temperature-dependent differential transcriptomes during formation of an epigenetic memory in Norway spruce embryogenesis. Tree Genet. Genomes 10, 355–366. doi: 10.1007/s11295-013-0691-z
Yoshikawa, M. (2013). Biogenesis of trans-acting siRNAs, endogenous secondary siRNAs in plants. Genes Genet. Syst. 88, 77–84. doi: 10.1266/ggs.88.77
Zeytuni, N., and Zarivach, R. (2012). Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure 20, 397–405. doi: 10.1016/j.str.2012.01.006
Zhang, J., Wu, T., Li, L., Han, S., Li, X., Zhang, S., et al. (2013). Dynamic expression of small RNA populations in larch (Larix leptolepis). Planta 237, 89–101. doi: 10.1007/s00425-012-1753-4
Zhang, J., Zhang, S., Han, S., Wu, T., Li, X., Li, W., et al. (2012). Genome-wide identification of microRNAs in larch and stage-specific modulation of 11 conserved microRNAs and their targets during somatic embryogenesis. Planta 236, 647–657. doi: 10.1007/s00425-012-1643-9
Keywords: conifers, Picea, epigenetic memory, epigenetic regulators; miRNAs, somatic embryos
Citation: Yakovlev IA and Fossdal CG (2017) In Silico Analysis of Small RNAs Suggest Roles for Novel and Conserved miRNAs in the Formation of Epigenetic Memory in Somatic Embryos of Norway Spruce. Front. Physiol. 8:674. doi: 10.3389/fphys.2017.00674
Received: 14 February 2017; Accepted: 23 August 2017;
Published: 08 September 2017.
Edited by:
Mahmood Yaish, Sultan Qaboos University, OmanReviewed by:
Lee Jeong Hwan, Chonbuk National University, South KoreaRui Shi, North Carolina State University, United States
Copyright © 2017 Yakovlev and Fossdal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Igor A. Yakovlev, yai@nibio.no