 Zhen-Ye Zhang
Zhen-Ye Zhang Ling-Ling Qian
Ling-Ling Qian Ru-Xing Wang
Ru-Xing Wang- Department of Cardiology, Wuxi People's Hospital Affiliated to Nanjing Medical University, Wuxi, China
Large-conductance calcium-activated potassium channels (BK channels) belong to a family of Ca2+-sensitive voltage-dependent potassium channels and play a vital role in various physiological activities in the human body. The renin-angiotensin-aldosterone system is acknowledged as being vital in the body's hormone system and plays a fundamental role in the maintenance of water and electrolyte balance and blood pressure regulation. There is growing evidence that the renin-angiotensin-aldosterone system has profound influences on the expression and bioactivity of BK channels. In this review, we focus on the molecular mechanisms underlying the regulation of BK channels mediated by the renin-angiotensin-aldosterone system and its potential as a target for clinical drugs.
Introduction
Large-conductance calcium-activated potassium channels (BK channels) are expressed on cell membranes and involved in diverse physiological processes including controlling membrane potential and Ca2+ homeostasis, setting vascular tone and blood pressure, controlling neuronal electrical activity and regulating endocrine hormone secretion. The renin-angiotensin-aldosterone system (RAAS) plays a key role in the maintenance of water and electrolyte balance and blood pressure regulation. In recent years, an increasing number of experiments have been conducted to explore the extremely complex relationship between BK channels and RAAS. This review provides an overview of the variety of mechanisms underlying the regulation of BK channels in different target tissues mediated by RAAS and its potential as a target for clinical drugs.
Structural Characteristics and Physiological Functions of BK Channels
Structural Characteristics of BK Channels
BK channels are widely expressed on various types of mammalian cell membranes. In some respects, BK channels are similar to the classic voltage-gated K+ channels, but differ in the mechanism of their activation (Salkoff et al., 2006). The minimal functional unit of the BK channel is a tetramer consisting of four α-subunits and auxiliary β-subunits (Lu et al., 2006). The α-subunit is encoded by KCNMA1 and almost ubiquitously expressed. The α-subunit is formed by seven putative transmembrane domains (S0-S6) starting with an extracellular N-terminus and four hydrophobic regions (S7-S10) connecting with an intracellular C-terminus (Lee and Cui, 2010) (Figure 1). The BK channel voltage sensor domains (VSDs) are formed by the first four transmembrane helices (S1-S4) and sense changes in membrane potentials. Each α-subunit contains one VSD; four identical VSDs arrange counter-clockwise in a bundle and confer depolarization-dependent gating to the BK channel (Ma et al., 2006). S5 and S6, together with the intervening amino acids, form a K+-selective pore and selectivity filter, referred to as a pore-gate domain (PGD). S0 is a unique additional N-terminal transmembrane segment serving as a site of coupling with β-subunits; this functional interaction is responsible for modulating the balance between resting and active states of the VSD (Koval et al., 2007). The C-terminal region contains four helices (S7-S10) and makes up nearly two-thirds of the total length of the primary amino acid sequence. S9 and S10 are the most highly conversed among species and can be expressed as a separate domain. The cytosolic domain is comprised of two tandem structurally homologous but non-identical regulators of K+ conductance domains: RCK1 and RCK2 (Yusifov et al., 2010). Each RCK domain has its own high-affinity Ca2+ sensing site. One is located in the RCK1 domain and includes the residue D367. The other is located in the RCK2 domain and functions as the high-affinity Ca2+ binding sites (Ca2+ bowl) (Yuan et al., 2010).
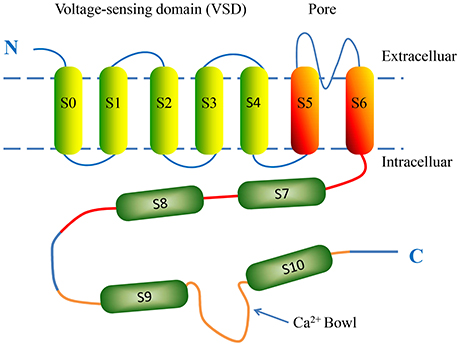
Figure 1. The putative structure of a single BK α-subunit. The α-subunit consists of seven putative transmembrane domains (S0-S6) and four hydrophobic regions (S7-S10). The voltage-sensing domain (VSD) is formed by four transmembrane helices S0-S4. Transmembrane helices S5 and S6 contribute to the K+-selective pore and selectivity filter. The intracellular C-terminal domain is comprised of two tandem structurally homologous regulator of conductance for K+ (RCK) domains, which possess BK intracellular ligand-binding sites. Regions S7-S8 and S9-S10 are located in the RCK1 (red) and RCK2 (orange) domains, respectively. The Ca2+ bowl (blue arrow) is a high-affinity Ca2+-binding site located in RCK2.
The β-subunits can be classified into four subtypes (β1-4) that are encoded by genes KCNMB1-4 respectively. The β-subunits are composed of two transmembrane helices, TM1 and TM2, with a large extracellular loop between them. TM1 lies close to S1 and S2, while TM2 is close to S0 (Liu et al., 2008). The β-subunits can alter the voltage and Ca2+ sensitivity of the α-subunits and regulate the kinetics and pharmacological properties of BK channels (Behrens et al., 2000). Recently, the four γ-subunits (γ1-4) of BK channels have been discovered and each contains a single-transmembrane domain. All four γ-subunits increase the voltage sensitivity of the BK α-subunit in the absence of Ca2+ (Li and Yan, 2016).
Physiological Functions of BK Channels
BK channels link chemical and electrical signaling through Ca2+-dependent voltage-operated activation (Pantazis and Olcese, 2016). Moreover, a wide range of intracellular ligands enable BK channels to be gated or modified (Piskorowski and Aldrich, 2006). BK channels are known to be widely distributed not only in the plasmalemma but also in the inner mitochondrial membrane and other organelles (Balderas et al., 2015) and exhibit different electrophysiological properties (Chen et al., 2005). BK channels seem to be critical regulators of vital physical functions such as blood flow, neurotransmission, urination and immunity.
In vascular smooth muscle cells, BK channels act as regulators of membrane potential and smooth muscle tone (Wang et al., 2012). With regards to their physiological activity, BK channels are viewed as negative-feedback regulators of electrical excitation (Sausbier et al., 2005). In neuronal cells, BK channels regulate brain physiology (Grunnet and Kaufmann, 2004), including shaping action potentials (Gorini et al., 2010), monitoring neurotransmitter homeostasis and signaling, and neurovascular coupling for the maintenance of vascular tone (Singh et al., 2016). In skeletal muscles, BK channels participate in modulating transmitter release by repolarization of the action potential (Maqoud et al., 2017). Tricarico et al. (2005) found that BK channels are involved in the Ca2+-dependent phenotype transition of the fibers, which contributes to the skeletal muscle fatigue. In renal tissue, BK channels are expressed in various kinds of renal tubular segments including distal convoluted tubule (Belfodil et al., 2003), connecting tubule (Frindt and Palmer, 2004), and cortical collecting duct. Woda et al. (2003) proposed that BK channels are involved in the regulation of K+ balance in the distal nephron and the cortical collecting duct. Moreover, BK channels are involved in other various body functions, including regulating circadian rhythm regulation (Whitt et al., 2016), urinary bladder function (Meredith et al., 2004), susceptibility to hearing damage (Pyott et al., 2007) and erectile dysfunction (Werner et al., 2005).
Main Constituents and Physiological Functions of RAAS
RAAS also referred to as the renin-angiotensin system (RAS), is a crucial hormone system that contributes to maintaining water-electrolyte balance and regulating arterial blood pressure. BK channels are known to associate with several RAAS factors, with each of these interactions having distinct physiological consequences. The interaction between BK channels and RAAS is emerging as a key regulator of BK channel function in both health and disease.
Renin, a protein and enzyme generated from cells in the kidney, plays a critical role in RAAS. It can cleave and activate another circulating protein, angiotensin, which represents the beginning of a chain reaction involving RAAS.
Ang II, an octapeptide hormone, plays a major role in regulating the body's blood pressure and maintaining water-electrolyte balance through sympathetic nervous activity, aldosterone-regulated sodium excretion, and thirst responses (King et al., 2013). Ang II contains two types of high-affinity plasma membrane receptors, AT1R and AT2R, and each binding respectively brings about a serious of physical effects (Carey et al., 2000). Ang II-activated receptors generate and convert downstream signals to multiple intricate reactions.
Aldosterone, the main mineralocorticoid hormone, is involved in regulating sodium and potassium homeostasis and body fluid equilibrium by means of influencing the re-absorption of Na+ and excretion of K+ in the kidney (Balakumar et al., 2017). Aldosterone is traditionally thought to play a key role in regulating salt re-absorption and potassium secretion in response to two apparently opposite conditions: hypovolemia and hyperkalemia (Ruilope and Tamargo, 2017). With regards to the decrease in blood volume, aldosterone is released and causes Na+ retention via salt transport mechanisms in the distal nephron, while K+ secretion remains unchanged. In other words, water and sodium are retained without losing K+. With regards to the increase of plasma K+, aldosterone is also activated and promotes K+ secretion in the distal nephron, while salt re-absorption remains unchanged. This contradictory phenomenon is referred to as the aldosterone paradox (Ming, 2011). In both hypovolemia and hyperkalemia, aldosterone causes two distinct physiological effects in order to maintain the salt and K+ balance, which indicates that aldosterone-mediated regulation is dependent on multiple mechanisms (Arroyo et al., 2011).
Regulation of BK Channels Mediated by RAAS
AT1R-Activated Signaling Pathways
An increasing number of studies apparently indicate that Ang II has an inhibitory influence on BK channels after binding to AT1R, whereas the molecular mechanisms are thought to be very complex and still unclear (Li et al., 2013). It has been reported that Ang II decreases the current amplitude of BK channels as well as the slope of the I-V curve and produces a left shift of the activation curves of BK channels in mouse podocyte cells. Ang II contributes to the pathogenesis of podocyte injury via the dysfunction of BK channels, which provides a theoretical basis for potential treatment of chronic glomerular disease in future (Gao et al., 2015). In recent years, the latest viewpoints have shown that Ang II-mediated inhibitive effects on BK channels are complicated, involving the redox-induced posttranslational modulation of BK α-subunits (Yoshimoto et al., 2004; Connelly et al., 2007), the regulation of BK β1-subunits expressions (Nieves-Cintrón et al., 2007), and a direct interaction between AT1R and BK α-subunit proteins (Zhang et al., 2014) (Figure 2).

Figure 2. The mechanisms of Ang II-induced AT1R-dependent regulation on BK channels. AT1R interacts with Gα after binding with Ang II, which leads to the activation of the PKC and enhances NOX-1 complex activity. In turn, the released Gβγ activates the c-Src/c-Abl-dependent signaling pathway, leading to the tyrosine phosphorylation of Cav-1. ROS enhances c-Src/protein tyrosine kinase (PTK) activity, which results in an increase in protein tyrosine phosphorylation. PKC also inhibits the anterograde trafficking of BK channels by influencing the function of Rab-GTPases. In addition, AT1R and BK channels closely co-localize at the cell membrane and have a close protein-protein interaction.
G-Protein-Dependent Signaling Pathways
AT1R belongs to a family of seven transmembrane G protein-coupled receptors (GPCR) and regulate most of the physiological activities of Ang II in various kinds of target cells (Moslemi et al., 2016). AT1R plays a predominant role in the control of AngII-induced vascular functions. There are two AT1R subtypes in rodents, AT1A and AT1B. G protein is a heterotrimer comprised of α, β, and γ subunits that enhances the combination and interaction between receptors and effector proteins. AT1R transmits signals through the activation of its downstream pathways that contains various kinds of signaling molecules (Burstein et al., 1997). There are two major downstream pathways stimulated by AT1R after binding with Ang II, consequently activating protein kinase C (PKC) and proto-oncogene tyrosine-protein kinase Src (c-Src) and leading to the activation of NADPH oxidase (NOX) (Thompson et al., 1998). NOX-1 and NOX-4 are the major NOX isoforms in human coronary arterial smooth muscle cells and have distinct subcellular distributions and regulation mechanisms (Hilenski et al., 2004). Caveolae-involved NOX-1 constitutes the major source of ROS generation in response to AT1R stimulation in vascular smooth muscle cells.
G-protein-involved signaling pathways
Gα-protein-mediated phospholipase C (PLC) phosphorylation is a primary mechanism through which Ang II exerts its function. The process leads to the activation of protein kinase C (PKC), a serine/threonine kinase (Touyz and Schiffrin, 2000). PKC consists of a multigene family of a few isoenzymes with diverse characteristics classified into three major groups. PKC is regarded as an important regulator of cellular functions (Wynne et al., 2009). A number of studies have demonstrated that PKC inhibits BK channels through various, complex mechanisms (Schubert et al., 1999; Xiao et al., 2014; Bo et al., 2015; Choi et al., 2015).
The expression of BK channels on the membrane is dependent on the balance of the dynamic change between transporting from the endoplasmic reticulum, recycling, endocytosis, and degradation (Conn and Ulloa-Aguirre, 2010; Simms and Zamponi, 2012). Rab-GTPases are known to control protein trafficking, including regulating vesicular transport and partaking in vesicle docking and fusion (Stenmark, 2009). The percentage of total β1-subunits localized on the plasma membrane is very small, and the intracullar β1-subunits are rich in mobile rab11A-positive recycling endosomes in smooth muscle cells (Leo et al., 2014). BK α-subunits are primarily transferred by rab4A-positive early endosomes to the plasma membrane. Leo et al. (2015) have found that PKC activated by Ang II inhibits BK channels not only by stimulating BK α-subunit degradation, but also by activating internalization and inhibiting anterograde trafficking of BK channels in cerebral artery myocytes. Experimental results of Leo et al. showed that the constriction to iberiotoxin (10 nM, IBTX) and dilation to NS-1619 (BK channel specific activator) (10 μM) of Ang II (100 nM, 8 h)-treated arteries were less than those in control untreated arteries, suggesting that Ang II stimulates BK channel internalization and degradation, leading to the construction of cerebral arteries. Recent studies demonstrate that the endocytosis and degradation of BK channel proteins are induced by glycosphingolipids or by PKC activation. The activation of PKC reduces channel recycling and diverts the channel to lysosomal degradation, which leads to a decrease in BK channel expression on the cell membrane. Ketsawatsomkron et al. (2012) demonstrated that the myogenic constriction is enhanced by PKC activation in endothelium denuded vessels. Zhou et al. (2010) have demonstrated that PKC-dependent phosphorylation of the α-subunit at S1151 and S695 in the channel pore is responsible for inhibiting BK channel function in HEK293 cells expressing recombinant channels. In addition, phosphorylation induced by PKC can also influence the stimulation of BK channel activity by protein kinase A (PKA) and protein kinase G (PKG), which results in the insensitivity of BK channels to the activation of PKG or PKA and subsequently inhibits cAMP-induced pulmonary vasodilatation (Barman et al., 2004). Hristov et al. (2014) have found that the reduction of BK channel activity in detrusor smooth muscle (DSM) is caused by PKC activation via a Ca2+-dependent mechanism, thus enhancing DSM contractility.
The Gβγ-subunits (Gβγ dimer), which form part of an inactivator of Gα subunits, play a significant role in AT1R-mediated regulation of BK channel activity. Gβγ activates c-Src, which in turn activates Abelson murine leukemia viral oncogene homolog (c-Abl) tyrosine kinase. C-Src, a non-receptor tyrosine kinase, is abundant in vascular tissues and can be activated by ROS as well, thereby forming a self-perpetuating activation loop and encouraging sustained generation of oxidative stress in response to Ang II stimulation. AT1R interacts with Gα after stimulation by Ang II, which in turn activates PKC and enhances NOX-1 complex activity through neutrophil cytosol factor 1 phosphorylation and Ras-related C3 botulinum toxin substrate 1 phosphorylation. In the meantime, the combination of Ang II and AT1R interacts with Gβγ, which activates c-Src/c-Abl-dependent signaling pathway. The process leads to the phosphorylation of Cav-1 tyrosine 14 mediated by c-Abl, which facilitates the trafficking of the receptor into relatively cav1-enriched lipid rafts and contributes to the Ang II-related BK channel dysfunction in diabetes. ROS not only increases c-Src/protein tyrosine kinase (PTK) activity but also inhibits protein tyrosine phosphatase (PTP) activity, which results in an increase of the phosphorylation in protein tyrosine. BK channel cysteine oxidation is directly caused by hydrogen peroxide (H2O2) and peroxynitrite (OONO−), the latter causing BK channel tyrosine nitration. Ang II-induced oxidative post-translational modification makes it possible to impair BK channel function in vascular smooth muscle (Ushio-Fukai and Alexander, 2006). In subsequent studies, Lu et al. (2016) proposed a molecular scheme of a receptor-enzyme-channel-caveolae microdomain complex and found that the colocalization between AT1R and BK α-subunits in vascular SMCs produces a physical disassociation between BK α and BK β1-subunits, enhancing Ang II-mediated inhibition of BK channels in STZ-induced diabetic rats. Caveolae-Ang II signaling is involved in vascular BK channel regulation and facilitates BK channel and coronary dysfunction in diabetes, indicating that BK channels may present a potential new clinical target in the prevention and treatment of cardiovascular complications in diabetes.
Roles of caveolae in Ang II signaling-dependent BK channel regulation
Caveolae are cell membrane flask-shaped invaginations and enriched in sphingomyelin and cholesterol, as a subset of lipid rafts. Caveolin, the signature protein of caveolae, is involved in the formation of caveolae and enabling caveolae to function as signaling organizing centers. Caveolin has emerged as an important scaffold protein that associates specific signaling complexes into caveolae and interacts with various kinds of signaling molecules (Krajewska and Maslowska, 2004). Among three mammalian isoforms of caveolin, caveolin-1 (Cav-1) is known to be a major structural protein and is widely expressed in vasculature (Cohen et al., 2004). Wang et al. (2005) have shown the co-localization of Cav-1 and BK channels in bovine aortic endothelial cells via immunofluorescence imaging. Moreover, a physical interaction between Cav-1 and BK channel proteins was confirmed by in vitro binding assay using GST-Cav-1 fusion proteins, showing that BK α-subunits directly interact with the Cav-1 scaffolding domains and producing an inhibitory effect on BK channel functions. In addition, the BK-Cav-1 interaction promotes the transition of the BK C-terminal end to the membrane and affects BK surface expression in native vascular tissue (Alioua et al., 2008).
Accumulating evidence suggests that caveolin and caveolae play a vital role in the regulation of AT1R-related and ROS-dependent Ang II signaling processes (Basset et al., 2009). Some studies demonstrate that Cav-1 is specifically involved in the ROS-dependent AT1R signaling events regulating SMC hypertrophy (Ushio-Fukai et al., 1998, 1999), which reveals the functional significance of Cav-1 in Ang II–mediated vascular pathophysiology and may provide new therapeutic strategies. Lu et al. (2010) have demonstrated that Ang II has an inhibitory effect on BK channels via Ang II-AT1R-caveolae oxidative stress signaling, which accelerates the development of vascular BK channel dysfunction in diabetes. Lu et al.'s experimental results have shown that Ang II-induced inhibition of BK channels was weakened in rat coronary vascular smooth muscle cells (SMC) with Cav-1 knock-down and in aortic SMC of Cav-1 knock-down mice, which indicates that Cav-1 plays an important role in this process. To determine the physiological relevance of these findings, the effects of Ang II and NS-1619 on the vasoreactivity of coronary arterial rings were measured and the results suggested that the components of IBTX-sensitivity BK channels and Ang II-induced vasoconstriction was reduced in STZ-induced diabetic rats, which may suggest an increased vascular tone in diabetic vessels. A recent study from same group has shown that genetic knockout of Cav-1 gene attenuated the myocardial infarction-induced by ischemia-reperfusion injury in diabetic mice (Lu et al., 2016). Such diabetic effects on myocardial infarction could be mimicked by 2 μM Ang II or 0.1 μM IBTX on the Langendorff-perfused hearts of non-diabetic wild type mice. Interestingly, 10 μM NS-1619 blocked the Ang II effects, but not IBTX effects, on myocardial infarction, suggesting that BK channels are the downstream target of Ang II signaling. Similar results were also found in the effects of forskolin on mouse myocardial infarct size reduction that was blocked by IBTX (Heinen et al., 2014). It is worthy to note that the protective effects of NS-1916 on myocardiac infarction are associated with mitochronfdral BK channnel activation (Sakamoto et al., 2008; Singh et al., 2013). Since IBTX is membrane impermeable, the authors argue that the vascular BK channel activation, at least in part, involves the protective roles of the experimental cardiac ischemia-reperfusion injury.
G-Protein-Independent Signaling Pathways
Ang II regulates various physiological effects by activating AT1R not only via a series of G-protein-dependent signaling pathways but also a number of G-protein-independent signaling cascades (Mehta and Griendling, 2007; Goette and Lendeckel, 2008). It has been reported that activation of calcineurin/the nuclear factor of activated T cells c3 (NFATc3) signaling pathway by Ang II downregulated BK β1-subunit expression in arterial smooth muscle cells, contributing to the BK channel dysfunction and the development of hypertension (Nieves-Cintrón et al., 2007). Recently, Zhang et al. (2014) have found that Ang II receptors and BK channels closely interact and co-localize at the cell membrane with high proximity indices in renal arterial smooth muscle. The Ang II-mediated regulation of BK channels is based on a close protein-protein interaction involving multiple BK regions. AT1R can diminish BK channel voltage sensitivity and inhibit BK activity independent of G-protein activation.
AT2R-Activated Signaling Pathways
AT2 receptors are highly expressed in fetal tissues and decreases rapidly after birth. They are much less abundant in adults and are found to be expressed in the pancreas, heart, kidney, adrenals, brain, and vasculature. The AT2R is a seven transmembrane G protein-coupled receptor and has 30% similarity with the AT1R. AT2R contributes to the regulation of renal, central nervous system and cardiovascular functions (De Gasparo et al., 2000). An increasing number of fundamental experiments indicate that AT2R may suppress AT1R-mediated biological effects due to triggering of activation of apoptosis via various signaling processes. Arima et al. (1997) reported that the activation of the AT2R may lead to endothelium-dependent vasodilation through a cytochrome P-450 pathway, which in turn opposes vasoconstriction induced by the AT1R in the rabbit glomerular afferent arteriole. Dimitropoulou et al. (2001) also demonstrated that the AT2R is able to enhance Ang II-induced endothelium-dependent vasodilation by stimulating BK channels in smooth muscle. The detailed mechanisms are still unknown and are expected to be further studied.
Regulation of the Hypovolemia State
Arroyo et al. (2011) proposed that Ang II plays a significant role in the distinct effects of aldosterone in hypovolemia and hyperkalemia, a process that involves WNK4 (Verissimo and Jordan, 2001). The WNK (with-no-lysine kinases) family is a serine/threonine kinase subfamily composed of four members known as WNK1 to WNK4, respectively encoded by genes WNK1-4. WNK1, WNK3, and WNK4 are primarily expressed in the kidney (Wilson et al., 2001). There is growing evidence that WNK4 functions as the “molecular switch” that alternates between different functional states to regulate Na+ and K+ transport in the distal nephron via mediating the activity of the Na+-Cl− cotransporter (NCC) (Arroyo et al., 2011). Under conditions of hypovolemia (high Ang II and aldosterone levels), WNK4 demonstrates antagonistic effects of K+ secretion in the renal distal nephron.
The mechanisms underlying WNK4-induced inhibition of BK channels by down-regulating their expression are still not fully understood, and several different hypotheses have been put forward. Zhuang et al. (2011) proposed that WNK4 kinase is involved in K+ secretion in the renal distal nephron and inhibits BK channel activity dependent on its kinase activity. The inhibition likely results from lysosomal degradation instead of clathrin-mediated endocytosis of BK channels. Yue et al. (2013) drew a similar conclusion, proposing that WNK4-mediated inhibition of BK channels is due to the activation of ERK and p38 mitogen-activated protein kinase (MAPK) via a mechanism other than the enhancement of endocytosis in the cortical collecting duct. Wang et al. (2013) suggested that WNK4 or a WNK4 mutant with a region containing a coiled coil domain and a autoinhibitory domain may decrease BK α-subunit plasma membrane expression as well as the whole cell expression, which suggests that WNK4 inhibits BK channel function by enhancing the degradation of BK channels via an ubiquitin-dependent pathway in the distal nephron.
Regulation of the High Dietary Potassium Intake State
Diets rich in potassium can result in an increase in plasma potassium and promote the release of aldosterone. An increasing number of experiments have shown that, in condition of hyperkalemia, apical membrane expression of BK channels in the cortical collecting duct (CCD) is apparently elevated, which enhances K+ secretion and maintains K+ homeostasis (Najjar et al., 2005). However, there is considerable controversy on the issue as to whether or not the enhancement of BK channel activity is directly dependent on the high level of plasma aldosterone.
Some investigators hold the view that the increase in aldosterone may enhance the activity of BK channels under conditions of high dietary potassium intake. Cornelius et al. (2015) tend towards this view and demonstrated that high aldosterone concentration in low Na+, high K+ diet mice contributes to a high rate of BK channel-mediated potassium secretion, which leads to an osmotic diuresis. The high urinary flow created by BK-mediated K+ secretion may be activated in a positive feedback manner, indicating that BK channels may present a therapeutic target for treating hyperkalemia.
In contrast, some studies deem that aldosterone has little or no influence on the expression or the activity of BK channels in response to dietary potassium intake. Estilo et al. (2008) hold the similar view that renal K+ excretion is associated with a kaliuretic pathway, rather than aldosterone directly affecting renal potassium ion transport due to changes in plasma potassium concentration. Recent studies have revealed a new understanding of the major role of BK channels activated by aldosterone-independent kaliuretic factors in which BK channel expression levels are closely related with flow-stimulated K+ secretion (Woda et al., 2001; Welling, 2013). In the distal nephron, renal K+ excretion is deemed to be dependent on tubular flow rate and BK channels are activated by an increase in the tubular flow rate (Woda et al., 2003). Liu et al. (2007) proposed that flow-stimulated BK channel-mediated potassium secretion is related to a signal pathway that involves an increase in Ca2+ concentration, which enables BK channels to translocate from the cell to the apical membrane. Through further studies, they found that PKA and PKC are involved in regulating the activity of BK channels in the mammalian distal nephron (Liu et al., 2009).
Effects of Ace Inhibitors on BK Channels
ACE inhibitors, which constitute a wide range of important clinical targeted drugs for RAAS, are a class of clinical drugs used mainly for the treatment of congestive heart failure and hypertension. Commonly prescribed ACE inhibitors include captopril, enalapril, zofenopril, perindopril, trandolapril, ramipril, and lisinopril. This group of drugs inhibits angiotensin-converting enzyme, an important component of the RAAS, which leads to hypovolemia as well as vasodilatation and causes decreased blood pressure and heart oxygen requirement. Albarwani et al. (2015) found that treatment with ACE inhibitor lisinopril increases the expression of BK channel proteins in mesenteric arteries. However, Li et al. (2014) obtained a contradictory result that the ACE inhibitor captopril fails to enhance the activity of BK channels. Captopril alleviates myocardial toxicity by reducing the generation of hemoglobin-based oxygen carriers (HBOCs), which is synergistically regulated by the reduction of NAD(P)H oxidase-induced ROS overproduction as well as increased NO bioavailability. The results of these studies show that the influences of captopril on BK channels are mediated by the endothelium instead of smooth muscle.
Conclusion
In summary, RAAS is deemed to have great influence on the expression and bioactivity of BK channels to regulate its physiological function via multiple mechanisms mediated by different components in this system. Ang II possesses a dual-aspect function that acts on BK channels after binding with the two types of receptors, AT1R and AT2R. The aldosterone paradox is a recently proposed concept in which aldosterone leads to two distinct physiological effects under conditions of hypovolemia and hyperkalemia to maintain the fluid and electrolyte balance. To some extent, this indicates that aldosterone-mediated regulation of BK channels is a very complex biological process, rather than a straightforward one mediated by a single mechanism. ACE inhibitors, such as the clinical commonly used RAAS-targeted drugs, contribute to the beneficial effects mediated by the endothelium on the cardiovascular system. There are conflicting study results as to whether the process is related to the decreased activity of BK channels induced by ACE inhibitors. Hence, more in-depth studies are required in the future, with any novel findings applied to developing new clinical treatments.
Author Contributions
ZZ wrote the initial drafts. LQ and RW revised the review and finalized the last version of the article. All authors checked and approved the submitted version.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81370303, 81500249) to RW and LQ and Natural Science Foundation of Jiangsu Province (BK20151110) to RW.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Albarwani, S., Al-Siyabi, S., Al-Husseini, I., Al-Ismail, A., Al-Lawati, I., Al-Bahrani, I., et al. (2015). Lisinopril alters contribution of nitric oxide and K(Ca) channels to vasodilatation in small mesenteric arteries of spontaneously hypertensive rats. Physiol. Res. 64, 39–49.
Alioua, A., Lu, R., Kumar, Y., Eghbali, M., Kundu, P., Toro, L., et al. (2008). Slo1 caveolin-binding motif, a mechanism of caveolin-1-Slo1 interaction regulating Slo1 surface expression. J. Biol. Chem. 283, 4808–4817. doi: 10.1074/jbc.M709802200
Arima, S., Endo, Y., Yaoita, H., Omata, K., Ogawa, S., Tsunoda, K., et al. (1997). Possible role of P-450 metabolite of arachidonic acid in vasodilator mechanism of angiotensin II type 2 receptor in the isolated microperfused rabbit afferent arteriole. J. Clin. Invest. 100, 2816–2823. doi: 10.1172/JCI119829
Arroyo, J. P., Ronzaud, C., Lagnaz, D., Staub, O., and Gamba, G. (2011). Aldosterone paradox: differential regulation of ion transport in distal nephron. Physiology (Bethesda) 26, 115–123. doi: 10.1152/physiol.00049.2010
Balakumar, P., Anand-Srivastava, M. B., and Jagadeesh, G. (2017). Renin-angiotensin-aldosterone: an inclusive, an invigorative, an interactive an interminable system. Pharmacol. Res. doi: 10.1016/j.phrs.2017.07.003. [Epub ahead of print].
Balderas, E., Zhang, J., Stefani, E., and Toro, L. (2015). Mitochondrial BKCa channel. Front. Physiol. 6:104. doi: 10.3389/fphys.2015.00104
Barman, S. A., Zhu, S., and White, R. E. (2004). Protein kinase C inhibits BKCa channel activity in pulmonary arterial smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 286, L149–L155. doi: 10.1152/ajplung.00259.2003
Basset, O., Deffert, C., Foti, M., Bedard, K., Jaquet, V., Ogier-Denis, E., et al. (2009). NADPH oxidase 1 deficiency alters caveolin phosphorylation and angiotensin II-receptor localization in vascular smooth muscle. Antioxid. Redox Signal. 11, 2371–2384. doi: 10.1089/ars.2009.2584
Behrens, R., Nolting, A., Reimann, F., Schwarz, M., Waldschutz, R., and Pongs, O. (2000). hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel beta subunit family. FEBS Lett. 474, 99–106. doi: 10.1016/S0014-5793(00)01584-2
Belfodil, R., Barriere, H., Rubera, I., Tauc, M., Poujeol, C., Bidet, M., et al. (2003). CFTR-dependent and -independent swelling-activated K+ currents in primary cultures of mouse nephron. Am. J. Physiol. Renal Physiol. 284, F812–F828. doi: 10.1152/ajprenal.00238.2002
Bo, L., Jiang, L., Zhou, A., Wu, C., Li, J., Gao, Q., et al. (2015). Maternal high-salt diets affected pressor responses and microvasoconstriction via PKC/BK channel signaling pathways in rat offspring. Mol. Nutr. Food Res. 59, 1190–1199. doi: 10.1002/mnfr.201400841
Burstein, E. S., Spalding, T. A., and Brann, M. R. (1997). Pharmacology of muscarinic receptor subtypes constitutively activated by G proteins. Mol. Pharmacol. 51, 312–319.
Carey, R. M., Wang, Z. Q., and Siragy, H. M. (2000). Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 35, 155–163. doi: 10.1161/01.HYP.35.1.155
Chen, L., Tian, L., Macdonald, S. H., Mcclafferty, H., Hammond, M. S., Huibant, J. M., et al. (2005). Functionally diverse complement of large conductance calcium- and voltage-activated potassium channel (BK) alpha-subunits generated from a single site of splicing. J. Biol. Chem. 280, 33599–33609. doi: 10.1074/jbc.M505383200
Choi, S., Kim, J. A., Kim, T. H., Li, H. Y., Shin, K. O., Lee, Y. M., et al. (2015). Altering sphingolipid composition with aging induces contractile dysfunction of gastric smooth muscle via K(Ca) 1.1 upregulation. Aging Cell 14, 982–994. doi: 10.1111/acel.12388
Cohen, A. W., Razani, B., Schubert, W., Williams, T. M., Wang, X. B., Iyengar, P., et al. (2004). Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes 53, 1261–1270. doi: 10.2337/diabetes.53.5.1261
Conn, P. M., and Ulloa-Aguirre, A. (2010). Trafficking of G-protein-coupled receptors to the plasma membrane: insights for pharmacoperone drugs. Trends Endocrinol. Metab. 21, 190–197. doi: 10.1016/j.tem.2009.11.003
Connelly, K. A., Boyle, A. J., and Kelly, D. J. (2007). Angiotensin II and the cardiac complications of diabetes mellitus. Curr. Pharm. Des. 13, 2721–2729. doi: 10.2174/138161207781662984
Cornelius, R. J., Wen, D., Li, H., Yuan, Y., Wang-France, J., Warner, P. C., et al. (2015). Low Na, high K diet and the role of aldosterone in BK-mediated K excretion. PLoS ONE 10:e0115515. doi: 10.1371/journal.pone.0115515
De Gasparo, M., Catt, K. J., Inagami, T., Wright, J. W., and Unger, T. (2000). International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 52, 415–472.
Dimitropoulou, C., White, R. E., Fuchs, L., Zhang, H., Catravas, J. D., and Carrier, G. O. (2001). Angiotensin II relaxes microvessels via the AT(2) receptor and Ca(2+)-activated K(+) (BK(Ca)) channels. Hypertension 37, 301–307. doi: 10.1161/01.HYP.37.2.301
Estilo, G., Liu, W., Pastor-Soler, N., Mitchell, P., Carattino, M. D., Kleyman, T. R., et al. (2008). Effect of aldosterone on BK channel expression in mammalian cortical collecting duct. Am. J. Physiol. Renal Physiol. 295, F780–F788. doi: 10.1152/ajprenal.00002.2008
Frindt, G., and Palmer, L. G. (2004). Apical potassium channels in the rat connecting tubule. Am. J. Physiol. Renal Physiol. 287, F1030–F1037. doi: 10.1152/ajprenal.00169.2004
Gao, N., Wang, H., Zhang, X., and Yang, Z. (2015). The inhibitory effect of angiotensin II on BKCa channels in podocytes via oxidative stress. Mol. Cell. Biochem. 398, 217–222. doi: 10.1007/s11010-014-2221-1
Goette, A., and Lendeckel, U. (2008). Electrophysiological effects of angiotensin II. Part I: signal transduction and basic electrophysiological mechanisms. Europace 10, 238–241. doi: 10.1093/europace/eum283
Gorini, G., Ponomareva, O., Shores, K. S., Person, M. D., Harris, R. A., and Mayfield, R. D. (2010). Dynamin-1 co-associates with native mouse brain BKCa channels: proteomics analysis of synaptic protein complexes. FEBS Lett. 584, 845–851. doi: 10.1016/j.febslet.2009.12.061
Grunnet, M., and Kaufmann, W. A. (2004). Coassembly of big conductance Ca2+-activated K+ channels and L-type voltage-gated Ca+ channels in rat brain. J. Biol. Chem. 279, 36445–36453. doi: 10.1074/jbc.M402254200
Heinen, A., Strothoff, M., Schmidt, A., Stracke, N., Behmenburg, F., Bauer, I., et al. (2014). Pharmacological options to protect the aged heart from ischemia and reperfusion injury by targeting the PKA-BK(Ca) signaling pathway. Exp. Gerontol. 56, 99–105. doi: 10.1016/j.exger.2014.03.029
Hilenski, L. L., Clempus, R. E., Quinn, M. T., Lambeth, J. D., and Griendling, K. K. (2004). Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 24, 677–683. doi: 10.1161/01.ATV.0000112024.13727.2c
Hristov, K. L., Smith, A. C., Parajuli, S. P., Malysz, J., and Petkov, G. V. (2014). Large-conductance voltage- and Ca2+-activated K+ channel regulation by protein kinase C in guinea pig urinary bladder smooth muscle. Am. J. Physiol. Cell Physiol. 306, C460–C470. doi: 10.1152/ajpcell.00325.2013
Ketsawatsomkron, P., Lorca, R. A., Keen, H. L., Weatherford, E. T., Liu, X., Pelham, C. J., et al. (2012). PPARgamma regulates resistance vessel tone through a mechanism involving RGS5-mediated control of protein kinase C and BKCa channel activity. Circ. Res. 111, 1446–1458. doi: 10.1161/CIRCRESAHA.112.271577
King, V. L., English, V. L., Bharadwaj, K., and Cassis, L. A. (2013). Angiotensin II stimulates sympathetic neurotransmission to adipose tissue. Physiol. Rep. 1:e00014. doi: 10.1002/phy2.14
Koval, O. M., Fan, Y., and Rothberg, B. S. (2007). A role for the S0 transmembrane segment in voltage-dependent gating of BK channels. J. Gen. Physiol. 129, 209–220. doi: 10.1085/jgp.200609662
Krajewska, W. M., and Maslowska, I. (2004). Caveolins: structure and function in signal transduction. Cell. Mol. Biol. Lett. 9, 195–220.
Lee, U. S., and Cui, J. (2010). BK channel activation: structural and functional insights. Trends Neurosci. 33, 415–423. doi: 10.1016/j.tins.2010.06.004
Leo, M. D., Bannister, J. P., Narayanan, D., Nair, A., Grubbs, J. E., Gabrick, K. S., et al. (2014). Dynamic regulation of beta1 subunit trafficking controls vascular contractility. Proc. Natl. Acad. Sci. U.S.A. 111, 2361–2366. doi: 10.1073/pnas.1317527111
Leo, M. D., Bulley, S., Bannister, J. P., Kuruvilla, K. P., Narayanan, D., and Jaggar, J. H. (2015). Angiotensin II stimulates internalization and degradation of arterial myocyte plasma membrane BK channels to induce vasoconstriction. Am. J. Physiol. Cell Physiol. 309, C392–C402. doi: 10.1152/ajpcell.00127.2015
Li, Q., and Yan, J. (2016). Modulation of BK Channel Function by Auxiliary beta and gamma subunits. Int. Rev. Neurobiol. 128, 51–90. doi: 10.1016/bs.irn.2016.03.015
Li, S., Fang, Q., Zhou, A., Wu, L., Shi, A., Cao, L., et al. (2013). Intake of high sucrose during pregnancy altered large-conductance Ca2+-activated K+ channels and vessel tone in offspring's mesenteric arteries. Hypertens. Res. 36, 158–165. doi: 10.1038/hr.2012.146
Li, T., Zhou, R., Yao, Y., Yang, Q., Zhou, C., Wu, W., et al. (2014). Angiotensin-converting enzyme inhibitor captopril reverses the adverse cardiovascular effects of polymerized hemoglobin. Antioxid. Redox Signal. 21, 2095–2108. doi: 10.1089/ars.2013.5606
Liu, G., Zakharov, S. I., Yang, L., Wu, R. S., Deng, S. X., Landry, D. W., et al. (2008). Locations of the beta1 transmembrane helices in the BK potassium channel. Proc. Natl. Acad. Sci. U.S.A. 105, 10727–10732. doi: 10.1073/pnas.0805212105
Liu, W., Morimoto, T., Woda, C., Kleyman, T. R., and Satlin, L. M. (2007). Ca2+ dependence of flow-stimulated K secretion in the mammalian cortical collecting duct. Am. J. Physiol. Renal Physiol. 293, F227–F235. doi: 10.1152/ajprenal.00057.2007
Liu, W., Wei, Y., Sun, P., Wang, W. H., Kleyman, T. R., and Satlin, L. M. (2009). Mechanoregulation of BK channel activity in the mammalian cortical collecting duct: role of protein kinases A and C. Am. J. Physiol. Renal Physiol. 297, F904–F915. doi: 10.1152/ajprenal.90685.2008
Lu, R., Alioua, A., Kumar, Y., Eghbali, M., Stefani, E., and Toro, L. (2006). MaxiK channel partners: physiological impact. J. Physiol. 570, 65–72. doi: 10.1113/jphysiol.2005.098913
Lu, T., Jiang, B., Wang, X. L., and Lee, H. C. (2016). Coronary arterial BK channel dysfunction exacerbates ischemia/reperfusion-induced myocardial injury in diabetic mice. Appl. Physiol. Nutr. Metab. 41, 992–1001. doi: 10.1139/apnm-2016-0048
Lu, T., Zhang, D. M., Wang, X. L., He, T., Wang, R. X., Chai, Q., et al. (2010). Regulation of coronary arterial BK channels by caveolae-mediated angiotensin II signaling in diabetes mellitus. Circ. Res. 106, 1164–1173. doi: 10.1161/CIRCRESAHA.109.209767
Ma, Z., Lou, X. J., and Horrigan, F. T. (2006). Role of charged residues in the S1-S4 voltage sensor of BK channels. J. Gen. Physiol. 127, 309–328. doi: 10.1085/jgp.200509421
Maqoud, F., Cetrone, M., Mele, A., and Tricarico, D. (2017). Molecular structure and function of big calcium-activated potassium channels in skeletal muscle: pharmacological perspectives. Physiol. Genomics 49, 306–317. doi: 10.1152/physiolgenomics.00121.2016
Mehta, P. K., and Griendling, K. K. (2007). Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 292, C82–C97. doi: 10.1152/ajpcell.00287.2006
Meredith, A. L., Thorneloe, K. S., Werner, M. E., Nelson, M. T., and Aldrich, R. W. (2004). Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J. Biol. Chem. 279, 36746–36752. doi: 10.1074/jbc.M405621200
Ming, C. H. (2011). Aldosterone paradox. Physiology (Bethesda) 26, 307; author reply 308. doi: 10.1152/physiol.00026.2011
Moslemi, F., Taheri, P., Azimipoor, M., Ramtin, S., Hashemianfar, M., Momeni-Ashjerdi, A., et al. (2016). Effect of angiotensin II type 1 receptor blockade on kidney ischemia/reperfusion; a gender-related difference. J. Renal Inj. Prev. 5, 140–143. doi: 10.15171/jrip.2016.29
Najjar, F., Zhou, H., Morimoto, T., Bruns, J. B., Li, H. S., Liu, W., et al. (2005). Dietary K+ regulates apical membrane expression of maxi-K channels in rabbit cortical collecting duct. Am. J. Physiol. Renal Physiol. 289, F922–F932. doi: 10.1152/ajprenal.00057.2005
Nieves-Cintrón, M., Amberg, G. C., Nichols, C. B., Molkentin, J. D., and Santana, L. F. (2007). Activation of NFATc3 down-regulates the beta1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension. J. Biol. Chem. 282, 3231–3240. doi: 10.1074/jbc.M608822200
Pantazis, A., and Olcese, R. (2016). Biophysics of BK Channel Gating. Int. Rev. Neurobiol. 128, 1–49. doi: 10.1016/bs.irn.2016.03.013
Piskorowski, R. A., and Aldrich, R. W. (2006). Relationship between pore occupancy and gating in BK potassium channels. J. Gen. Physiol. 127, 557–576. doi: 10.1085/jgp.200509482
Pyott, S. J., Meredith, A. L., Fodor, A. A., Vazquez, A. E., Yamoah, E. N., and Aldrich, R. W. (2007). Cochlear function in mice lacking the BK channel alpha, beta1, or beta4 subunits. J. Biol. Chem. 282, 3312–3324. doi: 10.1074/jbc.M608726200
Ruilope, L. M., and Tamargo, J. (2017). Aldosterone a relevant factor in the beginning and evolution of arterial hypertension. Am. J. Hypertens. 30, 468–469. doi: 10.1093/ajh/hpx010
Sakamoto, K., Ohya, S., Muraki, K., and Imaizumi, Y. (2008). A novel opener of large-conductance Ca2+-activated K+ (BK) channel reduces ischemic injury in rat cardiac myocytes by activating mitochondrial KCa channel. J. Pharmacol. Sci. 108, 135–139. doi: 10.1254/jphs.08150SC
Salkoff, L., Butler, A., Ferreira, G., Santi, C., and Wei, A. (2006). High-conductance potassium channels of the SLO family. Nat. Rev. Neurosci. 7, 921–931. doi: 10.1038/nrn1992
Sausbier, M., Arntz, C., Bucurenciu, I., Zhao, H., Zhou, X. B., Sausbier, U., et al. (2005). Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation 112, 60–68. doi: 10.1161/01.CIR.0000156448.74296.FE
Schubert, R., Noack, T., and Serebryakov, V. N. (1999). Protein kinase C reduces the KCa current of rat tail artery smooth muscle cells. Am. J. Physiol. 276, C648–C658.
Simms, B. A., and Zamponi, G. W. (2012). Trafficking and stability of voltage-gated calcium channels. Cell. Mol. Life Sci. 69, 843–856. doi: 10.1007/s00018-011-0843-y
Singh, H., Li, M., Hall, L., Chen, S., Sukur, S., Lu, R., et al. (2016). MaxiK channel interactome reveals its interaction with GABA transporter 3 and heat shock protein 60 in the mammalian brain. Neuroscience 317, 76–107. doi: 10.1016/j.neuroscience.2015.12.058
Singh, H., Lu, R., Bopassa, J. C., Meredith, A. L., Stefani, E., and Toro, L. (2013). MitoBK(Ca) is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc. Natl. Acad. Sci. U.S.A. 110, 10836–10841. doi: 10.1073/pnas.1302028110
Stenmark, H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525. doi: 10.1038/nrm2728
Thompson, J. B., Wade, S. M., Harrison, J. K., Salafranca, M. N., and Neubig, R. R. (1998). Cotransfection of second and third intracellular loop fragments inhibit angiotensin AT1a receptor activation of phospholipase C in HEK-293 cells. J. Pharmacol. Exp. Ther. 285, 216–222.
Touyz, R. M., and Schiffrin, E. L. (2000). Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 52, 639–672.
Tricarico, D., Mele, A., and Conte Camerino, D. (2005). Phenotype-dependent functional and pharmacological properties of BK channels in skeletal muscle: effects of microgravity. Neurobiol. Dis. 20, 296–302. doi: 10.1016/j.nbd.2005.03.011
Ushio-Fukai, M., and Alexander, R. W. (2006). Caveolin-dependent angiotensin II type 1 receptor signaling in vascular smooth muscle. Hypertension 48, 797–803. doi: 10.1161/01.HYP.0000242907.70697.5d
Ushio-Fukai, M., Alexander, R. W., Akers, M., and Griendling, K. K. (1998). p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by Angiotensin II. J. Biol. Chem. 273, 15022–15029. doi: 10.1074/jbc.273.24.15022
Ushio-Fukai, M., Alexander, R. W., Akers, M., Yin, Q., Fujio, Y., Walsh, K., et al. (1999). Reactive oxygen species mediate the activation of Akt/protein kinase B by Angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 274, 22699–22704. doi: 10.1074/jbc.274.32.22699
Verissimo, F., and Jordan, P. (2001). WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene 20, 5562–5569. doi: 10.1038/sj.onc.1204726
Wang, R. X., Shi, H. F., Chai, Q., Wu, Y., Sun, W., Ji, Y., et al. (2012). Molecular mechanisms of diabetic coronary dysfunction due to large conductance Ca2+-activated K+ channel impairment. Chin. Med. J. 125, 2548–2555. doi: 10.3760/cma.j.issn.0366-6999.2012.14.028
Wang, X. L., Ye, D., Peterson, T. E., Cao, S., Shah, V. H., Katusic, Z. S., et al. (2005). Caveolae targeting and regulation of large conductance Ca(2+)-activated K+ channels in vascular endothelial cells. J. Biol. Chem. 280, 11656–11664. doi: 10.1074/jbc.M410987200
Wang, Z., Subramanya, A. R., Satlin, L. M., Pastor-Soler, N. M., Carattino, M. D., and Kleyman, T. R. (2013). Regulation of large-conductance Ca2+-activated K+ channels by WNK4 kinase. Am. J. Physiol. Cell Physiol. 305, C846–C853. doi: 10.1152/ajpcell.00133.2013
Welling, P. A. (2013). Regulation of renal potassium secretion: molecular mechanisms. Semin. Nephrol. 33, 215–228. doi: 10.1016/j.semnephrol.2013.04.002
Werner, M. E., Zvara, P., Meredith, A. L., Aldrich, R. W., and Nelson, M. T. (2005). Erectile dysfunction in mice lacking the large-conductance calcium-activated potassium (BK) channel. J. Physiol. 567, 545–556. doi: 10.1113/jphysiol.2005.093823
Whitt, J. P., Montgomery, J. R., and Meredith, A. L. (2016). BK channel inactivation gates daytime excitability in the circadian clock. Nat. Commun. 7:10837. doi: 10.1038/ncomms10837
Wilson, F. H., Disse-Nicodeme, S., Choate, K. A., Ishikawa, K., Nelson-Willams, C., Desitter, I., et al. (2001). Human hypertension caused by mutations in WNK kinases. Science 293, 1107–1112. doi: 10.1126/science.1062844
Woda, C. B., Bragin, A., Kleyman, T. R., and Satlin, L. M. (2001). Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am. J. Physiol. Renal Physiol. 280, F786–F793.
Woda, C. B., Miyawaki, N., Ramalakshmi, S., Ramkumar, M., Rojas, R., Zavilowitz, B., et al. (2003). Ontogeny of flow-stimulated potassium secretion in rabbit cortical collecting duct: functional and molecular aspects. Am. J. Physiol. Renal Physiol. 285, F629–F639. doi: 10.1152/ajprenal.00191.2003
Wynne, B. M., Chiao, C. W., and Webb, R. C. (2009). Vascular smooth muscle cell signaling mechanisms for contraction to Angiotensin II and Endothelin-1. J. Am. Soc. Hypertens. 3, 84–95. doi: 10.1016/j.jash.2008.09.002
Xiao, D., Zhu, R., and Zhang, L. (2014). Gestational hypoxia up-regulates protein kinase C and inhibits calcium-activated potassium channels in ovine uterine arteries. Int. J. Med. Sci. 11, 886–892. doi: 10.7150/ijms.9338
Yoshimoto, T., Fukai, N., Sato, R., Sugiyama, T., Ozawa, N., Shichiri, M., et al. (2004). Antioxidant effect of adrenomedullin on angiotensin II-induced reactive oxygen species generation in vascular smooth muscle cells. Endocrinology 145, 3331–3337. doi: 10.1210/en.2003-1583
Yuan, P., Leonetti, M. D., Pico, A. R., Hsiung, Y., and Mackinnon, R. (2010). Structure of the human BK channel Ca2+-activation apparatus at 3.0 A resolution. Science 329, 182–186. doi: 10.1126/science.1190414
Yue, P., Zhang, C., Lin, D. H., Sun, P., and Wang, W. H. (2013). WNK4 inhibits Ca(2+)-activated big-conductance potassium channels (BK) via mitogen-activated protein kinase-dependent pathway. Biochim. Biophys. Acta 1833, 2101–2110. doi: 10.1016/j.bbamcr.2013.05.004
Yusifov, T., Javaherian, A. D., Pantazis, A., Gandhi, C. S., and Olcese, R. (2010). The RCK1 domain of the human BKCa channel transduces Ca2+ binding into structural rearrangements. J. Gen. Physiol. 136, 189–202. doi: 10.1085/jgp.200910374
Zhang, Z., Li, M., Lu, R., Alioua, A., Stefani, E., and Toro, L. (2014). The angiotensin II type 1 receptor (AT1R) closely interacts with large conductance voltage- and Ca2+-activated K+ (BK) channels and inhibits their activity independent of G-protein activation. J. Biol. Chem. 289, 25678–25689. doi: 10.1074/jbc.M114.595603
Zhou, X. B., Wulfsen, I., Utku, E., Sausbier, U., Sausbier, M., Wieland, T., et al. (2010). Dual role of protein kinase C on BK channel regulation. Proc. Natl. Acad. Sci. U.S.A. 107, 8005–8010. doi: 10.1073/pnas.0912029107
Keywords: large-conductance calcium-activated potassium channels, renin, angiotensin ii, aldosterone, angiotensin-converting enzyme inhibitors
Citation: Zhang Z-Y, Qian L-L and Wang R-X (2017) Molecular Mechanisms Underlying Renin-Angiotensin-Aldosterone System Mediated Regulation of BK Channels. Front. Physiol. 8:698. doi: 10.3389/fphys.2017.00698
Received: 02 May 2017; Accepted: 30 August 2017;
Published: 13 September 2017.
Edited by:
Gaetano Santulli, Columbia University, United StatesReviewed by:
Rudolf Schubert, Heidelberg University, GermanyGayathri Krishnamoorthy, Nutriflour International Inc. LLC, United States
Copyright © 2017 Zhang, Qian and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ru-Xing Wang, ruxingw@aliyun.com