 Cun-dong Fan1†Jing-yi Sun2†Xiao-ting Fu1†Ya-jun Hou1Yuan Li1Ming-feng Yang1Xiao-yan Fu1*
Cun-dong Fan1†Jing-yi Sun2†Xiao-ting Fu1†Ya-jun Hou1Yuan Li1Ming-feng Yang1Xiao-yan Fu1* Bao-liang Sun1,3*
Bao-liang Sun1,3*- 1Key Lab of Cerebral Microcirculation in Universities of Shandong, Taishan Medical University, Taian, China
- 2Wonju Severance Christian Hospital, Yonsei University Wonju College of Medicine, Wonju, South Korea
- 3Department of Neurology, Affiliated Hospital of Taishan Medical University, Taian, China
Homocysteine (Hcy) as an independent risk factor contributes to the occurrence and development of human cardiovascular diseases (CVD). Induction of oxidative stress and apoptosis was commonly accepted as the major mechanism in Hcy-induced cardiotoxicity. Astaxanthin (ATX) as one of the most powerful antioxidants exhibits novel cardioprotective potential against Hcy-induced endothelial dysfunction. However, the protective effect and mechanism of ATX against Hcy-induced cardiotoxicity in cardiomyocytes have not been elucidated yet. Herein, H9c2 rat cardiomyocytes and Hcy-injured animal model were employed in the present study. The MTT, flow cytometry analysis (FCM), TUNEL-DAPI and western blotting results all demonstrated that ATX significantly alleviated Hcy-induced cytotoxicity in H9c2 cells through inhibition of mitochondria-mediated apoptosis. The JC-1 and Mito-tracker staining both revealed that ATX pre-treatment blocked Hcy-induced mitochondrial dysfunction by regulating Bcl-2 family expression. Moreover, DCFH-DA and Mito-SOX staining showed that ATX effectively attenuated Hcy-induced oxidative damage via scavenging intracellular reactive oxygen species (ROS). Importantly, the ELISA and immunohistochemical results indicated that Hcy-induced cardiotoxicity in vivo was also significantly inhibited by ATX through inhibition of oxidative damage and apoptosis, and improvement of the angiogenesis. Taken together, our results demonstrated that ATX suppressed Hcy-induced cardiotoxicity in vitro and in vivo by inhibiting mitochondrial dysfunction and oxidative damage. Our findings validated the strategy of using ATX may be a highly efficient way to combat Hcy-mediated human CVD.
Introduction
Cardiovascular diseases (CVD) as the leading causes of death globally represent one of the most challenges in clinic (Lee et al., 2016). Endothelial cells play key role in vascular homeostasis, and endothelial dysfunction contributed to the development of human CVD (Zhang et al., 2001; Ungvari et al., 2003; Austin et al., 2004). Homocysteine (Hcy) is an intermediate metabolite of cysteine and methionine. Large numbers of evidences have confirmed that elevated plasma levels of Hcy as an independent risk factor may induce endothelial dysfunction through oxidative stress and apoptosis, and eventually lead to the occurrence and development of human CVD (Almashhadany et al., 2015; Baggott and Tamura, 2015). However, Hcy-mediated toxicity toward cardiomyocytes was not well demonstrated, and the underlying mechanism remains elusive. Therefore, H9c2 rat cardiomyocytes and an Hcy-injured experimental animal model were employed to evaluate the potential cardiotoxicity and the underlying mechanism.
Astaxanthin (ATX) a red-orange carotenoid pigment represents one of the most powerful antioxidants and displays multiple biological activities, including anti-cancer, anti-inflammatory, anti-diabetic, immunomodulatory and neuroprotective activities (Hussein et al., 2006). Additionally, ATX also showed novel cardioprotective properities, and supplement of ATX in diet can decrease the risk of cardiovascular disease (Abdelzaher et al., 2016). Accumulated evidences have revealed that ATX had the potential to alleviate endothelial dysfunction through scavenging reactive oxygen species (ROS) and inhibiting oxidative damage (Sasaki et al., 2011; Zhao et al., 2011). Hence, ATX-mediated protection and mechanism in endothelial cells were well studied. But, little information about ATX-mediated protective potential in cardiomyocytes was available, and the protective mechanism was not well explored. Herein, the protective effects and mechanism of ATX against Hcy-induced cardiotoxicity in H9c2 rat cardiomyocytes and an experimental animal model were evaluated, and the results indicated that ATX attenuated Hcy-induced cardiotoxicity in vitro and in vivo by inhibiting mitochondrial dysfunction and oxidative damage, which validated its potential application in chemoprevention and chemotherapy of human CVD.
Materials and Methods
Chemicals
DMEM-F12 medium, phosphate buffered solution (PBS) fetal bovine serum (FBS), DCFH-DA probe and mitochondria-targeted MitoSOX probe (M36008) were purchased from Invitrogen. Hcy, ATX, MTT, propidium iodide (PI) were obtained from Sigma. TUNEL-DAPI kit, Mito-SOX probe and BCA assay kit were purchased from Beyotime Institute of Biotechnology (Shanghai, China). Primary antibodies, including cleaved PARP (#94885), active-caspase-3 (#9664), active-caspase-7 (#8438), active-caspase-9 (#9507), Bax (#2772), Bad (#9292), Bcl-xL (#2764), Ser428-ATR (#2853), Ser15-p53 (#9284), total-p53 (#2524), and Ser139-histone (#9718) were all obtained from Cell Signaling Technology (Beverly, USA). Bcl-2 (#14-6992-82), CD-34 (#MA1-10202) and Ser-1981-ATM (#14-9046-82) were bought from Invitrogen (Carlsbad, USA). All solvents used were of high performance liquid chromatography (HPLC) grade.
Cell Culture and Cell Viability Assay
H9c2 rat myocardial cells were obtained from ATCC company (USA). Cells were cultured with DMEM-F12 medium containing 10% FBS at 37° and 5% CO2 in a incubator. Cell viability with Hcy or/and ATX was detected by MTT assay. Briefly, H9c2 cells (8 × 103 cells/well) seeded in 96-well plate were treated with Hcy (1–16 mM) for 72 h, or cells were treated with 8 mM Hcy for 6, 12, 24, 48, and 72 h. For protective treatment, cells were pre-treated with 0.5–8 μM ATX for 6 h and co-incubated with Hcy for 72 h. After treatment, 20 μl of MTT solution was added and incubated for another 5 h. Then the medium was removed and 150 μl of dimethyl sulfoxide (DMSO) was added. The cell viability was measured by detecting the absorbance at 570 nm. H9c2 cells morphology was observed by phase microscope. All data and images were obtained from three independent trials and were conducted in accordance with the relevant guidelines and regulations of Taishan Medical University.
Flow Cytometry Analysis (FCM)
Cell apoptosis and cell cycle distribution in H9c2 cells were analyzed by FCM. Briefly, cells were pre-treated with or without 4 μM ATX for 6 and co-treated with 8 mM Hcy for 72 h. After treatment, cells were washed, collected and incubated with PI buffer at 37°C for 5 h in darkness. Then the stained cells were monitored by FCM. Cell cycle (G0/G1, S, and G2/M phases) was analyzed by Modfit software. The hypodiploid DNA content (Sub-G1 peak) was employed to quantify the apoptotic cell death. About 104 cells/sample were recorded. All data and images were obtained from three independent trials and were conducted in accordance with the relevant guidelines and regulations of Taishan Medical University.
TUNEL-DAPI Staining
Cells apoptosis in H9c2 cells were also detected by TUNEL-DAPI staining. Briefly, Treated H9c2 cells seeded on a cover glass were fixed with 4% formaldehyde, and permeabilized with 0.1% Triton X-100. Then cells were incubated with TUNEL reaction solution for 1 h in darkness at 37°C. After incubation, cells were stained with 10 μg/ml DAPI for 10 min, and washed with PBS for three times. The cells apoptosis was detected by an inverted fluorescence microscope. The TUNEL-positive cells represent the apoptotic cells. All data and images were obtained from three independent trials and were conducted in accordance with the relevant guidelines and regulations of Taishan Medical University.
Evaluation of Mitochondrial Dysfunction
Mitochondrial function was evaluated by the mitochondrial membrane potential (Δψm) and mitochondrial morphology, which were examined by JC-1 and Mito-Tracker probes, respectively. Briefly, cells seeded in 6-cm plate were treated with ATX or/and Hcy. Cells after treatment were washed and incubated with 10 μM JC-1 or Mito-Tracker for 15 min in darkness. Then cells were washed and imaged under an inverted fluorescence microscope. The green fluorescence intensity after JC-1 staining was quantified by ImagePlus Software. All data and images were obtained from three independent trials and were conducted in accordance with the relevant guidelines and regulations of Taishan Medical University.
Detection of ROS and Superoxide Anion
The intracellular ROS and superoxide anion were measured by DCFH-DA and Mito-SOX probes in live cells, respectively. Briefly, treated cells cultured on glass were washed and incubated with 10 μM DCFH-DA or 0.5 μM Mito-SOX. After reaction, cells were washed and observed by an inverted fluorescence microscope for detection of ROS (green fluorescence) and superoxide anion (red fluorescence). The images showed here were obtained from three independent experiments. All data and images were obtained from three independent trials and were conducted in accordance with the relevant guidelines and regulations of Taishan Medical University.
Western Blotting
Protein expression was examined by western blotting method. Briefly, cells after treatment were washed, collected and lysed by RIPA lysis buffer. The intracellular total protein was extracted by centrifugation at 11,000 g for 10 min at 4°C. Total protein after quantification was boiled and loaded (40 μg/lane) for separation by SDS-PAGE. Then protein was transferred onto a nitrocellulose membrane at 100 V for 1.5 h. Membrane subsequently was blocked, incubated with the primary antibody, and second antibody, respectively. Then the membrane was washed and the protein bands were visualized on an X-ray film using an enhanced ECL chemiluminescence system. The proteins expression was quantified by Quantity-One Software, and the proteins expression rate was labeled under the bands. All data and images were obtained from three independent trials and were conducted in accordance with the relevant guidelines and regulations of Taishan Medical University.
In Vivo Study
The therapeutic effect of ATX against Hcy-induced cardiotoxicity was also evaluated in vivo in mouse. Briefly, 40 mice were divided into four groups (10 mice/group), and administrated with 5 mg/kg/day ATX or/and 300 mg/kg/day Hcy for 4 weeks. The control group was given equal normal saline. After administration, mouse was given euthanasia and the body weight and heart weight were measured. The contents of reduced glutathione (GSH-Rs) and malondialdehyde (MDA) in heart tissue were examined by ELISA kits according to the manufacture's instruction. Part of heart tissue was cut into 4-μM section for immunohistochemical assay (IHC). All the animal experiments were conducted in accordance with the relevant guidelines and regulations of Taishan Medical University.
Statistical Analysis
All data and images were done from three independent experiments at least. The statistical analysis was carried out by SPSS013.0 software. The significance between two groups was analyzed by two-tailed Student's test. The difference among three or more groups was analyzed by multiple comparisons. Bars with “*” or “**” represent the P < 0.05 or P < 0.01, respectively. Bars with different characters indicates the statistical different at P < 0.05 level.
Results
ATX Alleviates Hcy-Induced Cytotoxicity in H9c2 Cells
The cytotoxicity of Hcy and possible protective effect of ATX on H9c2 cells were firstly examined by MTT assay. As shown in Figure 1 and Supplementary Figure 1, Hcy treatment alone significantly inhibited H9c2 cells viability in a dose- and time-dependent manner (Figures 1A,B). For instance, treatment of cells with 8 mM Hcy for 72 h significantly decreased the cell viability to 52.6%. However, pre-treatment of cells with ATX (1, 2 and 4 μM) for 6 h effectively blocked Hcy-induced cytotoxicity in H9c2 cells from 52.6% (Hcy) to 87.9, 95.4, and 99.8%, respectively (Figure 1D). ATX alone showed no obvious cytotoxicity toward H9c2 cells (Figure 1C). The improvement of cell morphology further confirmed this protective potential (Figure 1E). These results suggested that ATX has the potential to alleviate Hcy-induced cytotoxicity.
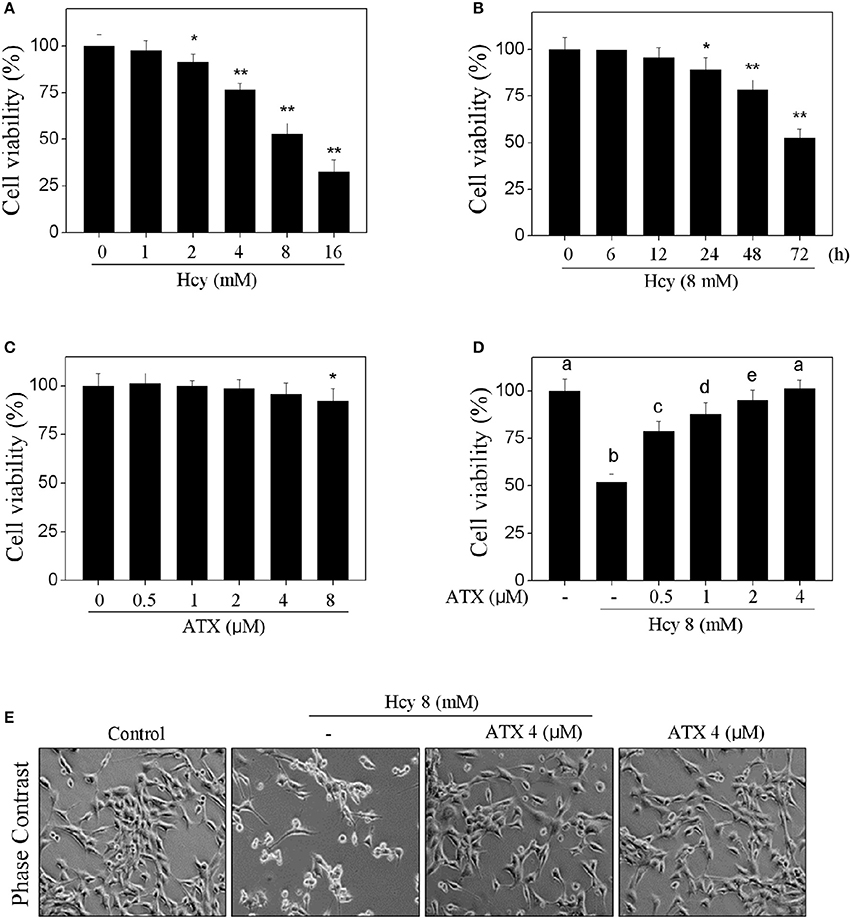
Figure 1. ATX alleviates Hcy-induced cytotoxicity in H9c2 cells. Dose-dependent (A) and time-dependent (B) cytotoxicity of Hcy toward H9c2 cells. H9c2 cells were treated with 0–16 mM Hcy for 72 h, or cells were exposed to 8 mM Hcy for 0–72 h. (C) Cytotoxicity of ATX toward H9c2 cells. Cells were treated with ATX (0–8 μM) for 72 h. (D) ATX alleviated Hcy-induced cytotoxicity in H9c2 cells. Cells were pre-treated with or without 0.5–4 μM ATX for 6 h and co-treated with 8 mM Hcy for 72 h. Cell viability after treatment was detected by MTT assay. (E) Morphological changes of H9c2 cells. Cells were treated with or without 4 μM ATX for 6 h, and co-incubated with 8 mM Hcy for 72 h. Cells after treatment were observed by phase contrast microscope (magnification, 200×). All data and images were obtained from three independent trials. Bars with “*” or “**” indicate the statistically different at the P < 0.05 and P < 0.01 level, respectively. Bars with different characters indicates the statistical different at P < 0.05 level.
ATX Suppresses Hcy-Induced Apoptosis in H9c2 Cells
We next evaluated Hcy-induced apoptosis and the possible protective mechanism of ATX on H9c2 cells by FCM. As shown in Figure 2A, Hcy alone caused apparent apoptosis, as reflected by the increase of Sub-G1 peak. However, the cell apoptosis in Hcy-treated cells was significantly attenuated by ATX pre-treatment from 52.6 to 4.1%. This protection of ATX on H9c2 cells was evaluated by TUNEL-DAPI co-staining. As shown in Figure 2B, Hcy treatment alone resulted in significant apoptosis, as convinced by the increase of TUNEL-positive cells. However, pre-treatment with ATX effectively prevented H9c2 cells from HCY-induced apoptosis. Moreover, the PARP cleavage and caspase activation were also conducted to investigate the underlying mechanism. As shown in Figure 2C, incubation of cells with Hcy alone induced dramatically poly-ADP-ribose polymerase (PARP) cleavage and the activation of caspase-3, -7, and -9. The activation of caspase-7 and caspase-9 indicated that Hcy activated the mitochondria-mediated apoptosis. The slight activation of capase-8 indicated that Hcy also triggered death receptor-mediated apoptosis, but not dominant (Supplementary Figure 2). As expected, ATX pre-treatment markedly inhibited Hcy-induced PARP cleavage and caspase activation. Taken together, these results above indicated that ATX suppressed the mitochondria-mediated apoptosis in Hcy-treated H9c2 cells.
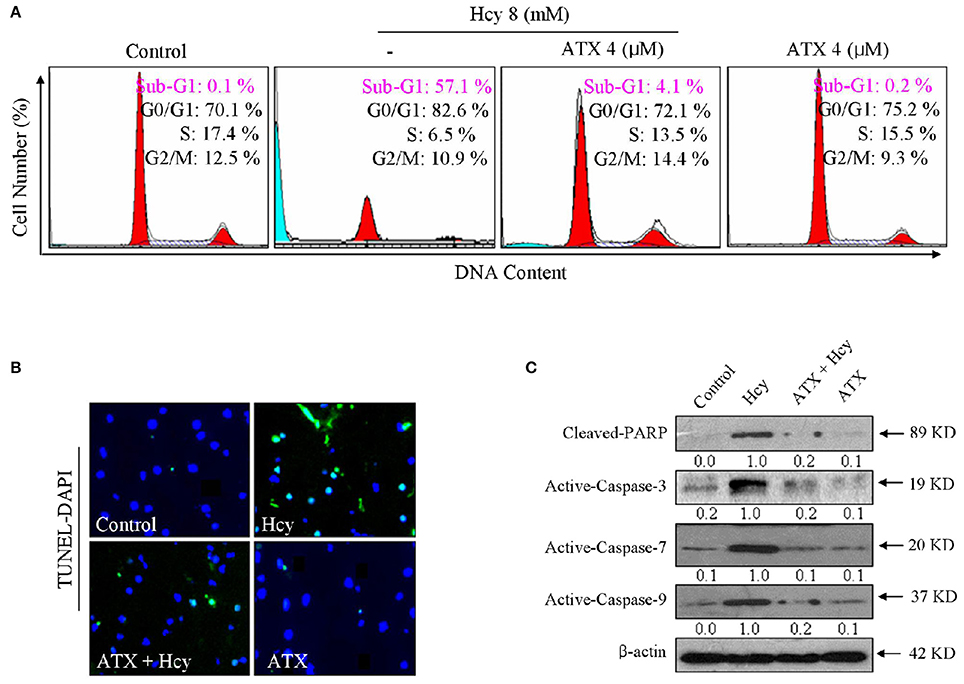
Figure 2. ATX suppresses Hcy-induced apoptosis in H9c2 cells. (A) FCM analysis of cell apoptosis and cell cycle distribution. H9c2 cells were treated with or without 4 μM ATX for 6 h, and co-incubated with 8 mM Hcy for 72 h. Cells after treatment were collected and fixed with 70% pre-cooled alcohol, and stained by PI solution and analyzed by FCM. The hypodiploid DNA content (Sub-G1 peak) was used to quantify the cell apoptosis. (B) TUNEL-DAPI staining. Cells after treatment were fixed with formaldehyde (4%) and administrated with the TUNEL staining kit as described in method section. The TUNEL-positive cells (green) indicated the apoptotic cells (magnification, 200×). (C) PARP and caspase expression. Total protein was prepared and the protein expression was examined by western blotting method. All data and images were obtained from three independent experiments.
ATX Blocks Hcy-Induced Mitochondrial Dysfunction by Balancing Bcl-2 Family
To elucidate the role of mitochondria in Hcy-induced apoptosis of H9c2 cells, the Δψm and mitochondrial morphology were detected by JC-1 and Mito-tracker probes, respectively. As shown in Figure 3A, Hcy treatment induced significant loss of Δψm, as reflected by the fluorescent shift from red to green. ATX pre-treatment completely improved the Δψm in Hcy-treated cells. The statistical result further confirmed this protective effect (Supplementary Figure 3). Moreover, Hcy treatment also caused obvious mitochondrial fragmentation, as demonstrated by the mitochondrial morphological changes from protonema to punctiform. Interestingly, ATX pre-treatment completely blocked Hcy-induced mitochondrial fragmentation.
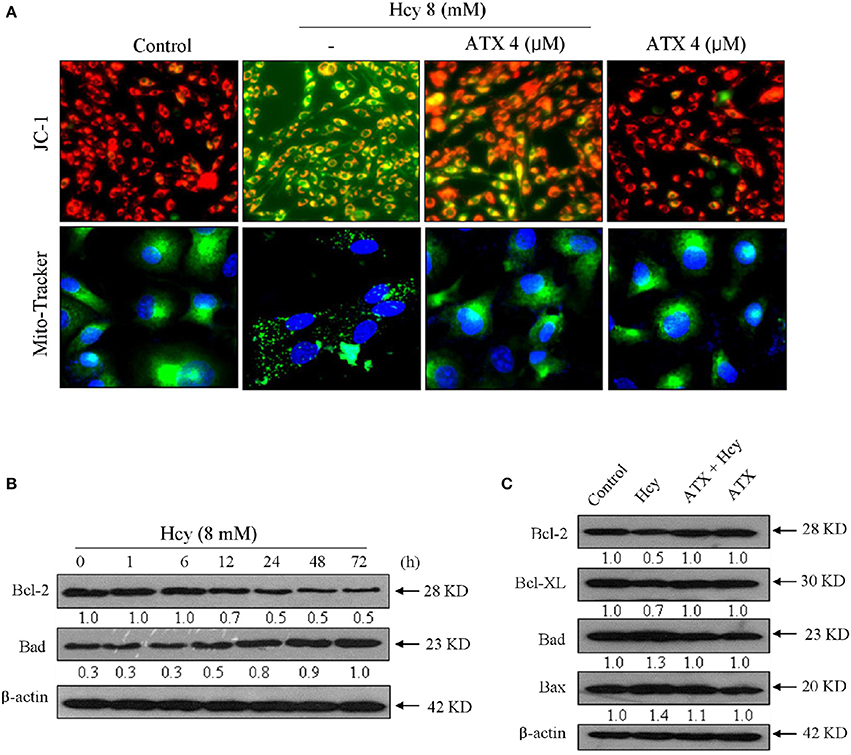
Figure 3. ATX blocks Hcy-induced mitochondrial dysfunction by regulating Bcl-2 family. (A) ATX blocked Hcy-induced the depletion of Δψm and mitochondrial fragmentation. The Δψm and mitochondrial morphology were detected by JC-1 and Mito-Tracker probes, respectively. The experiment details were conducted according to the method section. (B) Time-dependent inhibition of Hcy against Bcl-2 and Bad expression. Cells were treated with 8 mM Hcy for indicated time. (C) Protective effects of ATX on Bcl-2 family members in Hcy-treated cells. Protein expression was examined by western blotting method. All images were obtained from three independent trials.
Bcl-2 family plays important role in regulating mitochondrial permeability and inducing apoptosis. Therefore, a time-course effect of Hcy on Bcl-2 family members was investigated. As show in Figure 3B, Hcy treatment caused continuous down-regulation of Bcl-2 at 12 h. Bad expression in Hcy-treated H9c2 cells showed significant up-regulation at 12 h. However, the expression level of pro-apoptosis (Bax and Bad) and pro-survival (Blc-2 and Bcl-XL) in Hcy-treated cells were effectively normalized by ATX pre-treatment (Figure 3C). Taken together, these results revealed that ATX blocked Hcy-induced mitochondrial dysfunction by regulating Bcl-2 family proteins.
ATX Attenuates Hcy-Induced Oxidative Damage in H9c2 Cells
Accumulated evidences have confirmed that Hcy could trigger oxidative damage through inducing ROS accumulation. Therefore, the oxidative status in Hcy-treated H9c2 cells was investigated. Primarily, the intracellular superoxide anion and ROS were detected by Mito-SOX (a mitochondria-targeted red probe) and DCFH-DA (green), respectively. As shown in Figures 4A,B, Hcy treatment caused significant accumulation of superoxide anion and ROS, as demonstrated by the enhanced red fluorescence and green fluorescence in H9c2 cells, respectively. Secondly, Hcy-induced DNA damage was also detected. As shown in Figure 4C, cells treated with 8 mM Hcy showed significant phosphorylation activation of ataxia telangiectasia mutated (ATM) (Ser1981), and Rad3-related (ATR) (Ser428), p53 (Ser15), and histone (Ser139) in a time-dependent manner, indicating that Hcy induced oxidative damage in H9c2 cells. However, pre-treatment with ATX inhibited Hcy-induced generation of ROS and superoxide anion. The statistical results further confirmed ATX's anti-oxidative effect (Supplementary Figure 4). ROS inhibition by ATX eventually attenuated Hcy-induced oxidative damage (Figure 4D). Taken together, these results above demonstrated that ATX attenuated Hcy-induced oxidative damage through inhibition ROS overproduction.
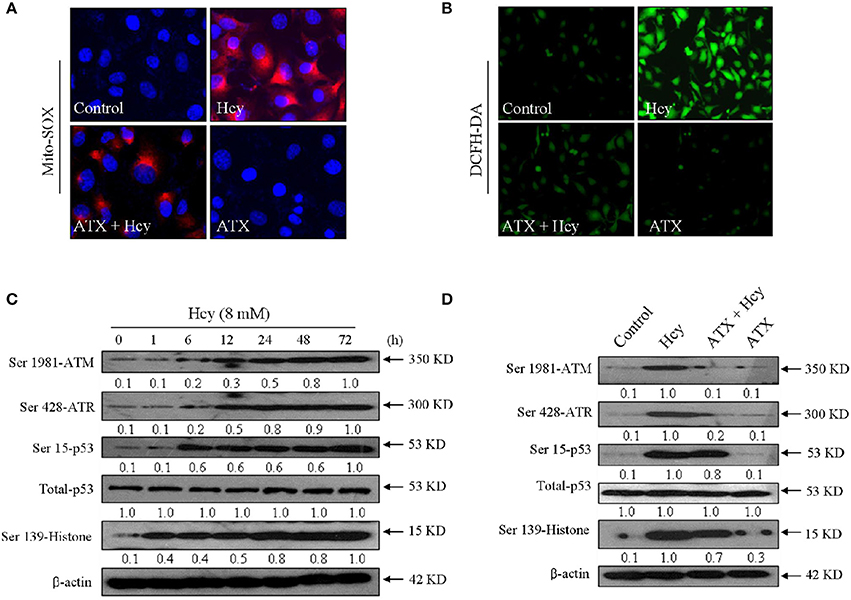
Figure 4. ATX attenuates Hcy-induced oxidative damage in H9c2 cells. ATX inhibited intracellular superoxide anion (A) and ROS (B) generation. The intracellular superoxide anion and ROS were measured by Mito-SOX and DCFH-DA probes, respectively. The experiment details were conducted according to the method section. (C) Time-dependent activation of Hcy on DNA damage. (D) ATX attenuated Hcy-induced DNA damage. Protein expression was examined by western blotting method. All images were obtained from three independent trials.
ATX Inhibits Hcys-Induced Cardiotoxicity in Vivo
To validate the in vivo protective effect and possible mechanism of ATX against Hcy-induced cardiotoxicity, mouse administrated with ATX or/and Hcy were employed. The results showed that Hcy treatment in vivo caused slight decrease in mice heart weight, but not affect the mice body weight (Figures 5A,B). Hcy treatment also decreased the content of reduced-glutathione (GSH-Rs) (Figure 5C) and increased the content of MDA (Figure 5D), indicating the induction of oxidative damage by Hcy. IHC analysis revealed that Hcy treatment in vivo induced significant myocardial apoptosis (active-caspase-3 staining) and inhibited angiogenesis (CD-34 staining). Importantly, ATX treatment in vivo effectively inhibited the oxidative damage and myocardial apoptosis, and improved the angiogenesis in Hcy-treated mouse (Figure 5E), which consisted with the in vitro mechanism. These results suggested that ATX inhibited Hcy-induced cardiotoxicity in vivo.
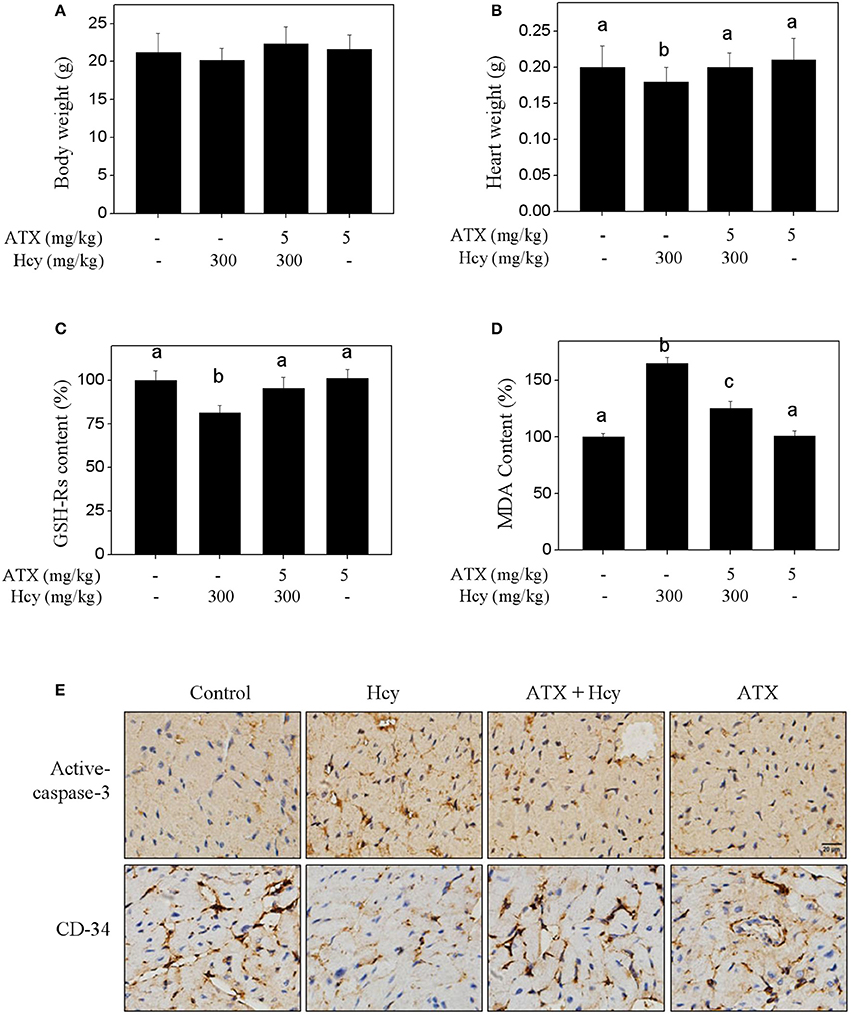
Figure 5. ATX inhibits Hcy-induced cardiotoxicity in vivo. Effects of ATX or/and Hcy on the mice body weight (A) and heart weight (B) in vivo. Mice were given ATX (5 mg/kg/day) or/and Hcy (300 mg/kg/day) for 2 weeks. After administration, the mice body weight and heart weight were both examined. Effects of ATX or/and Hcy on the content of GSH-Rs (C) and MDA (D). Total protein was extracted from mice heart tissue, and the content of GSH-Rs and MDA were examined by ELISA kits. (E) IHC assay of apoptosis and angiogenesis in vivo. Heart tissue were separated and cut into 4-μm section. The expressions of active-caspase-3 and CD-34 in heart tissue were detected by IHC method. All data and images were obtained from three independent trials. Bars with different characters indicates the statistical different at P < 0.05 level.
Discussion
Human CVD represents the leading causes of death globally, and therapy of human CVD has always remains one of the biggest challenges in clinic. High levels of plasma Hcy as an independent risk factor contributes to the occurrence and development of CVD (Feng and Xu, 2017; Markišić et al., 2017; Rudreshkumar et al., 2017). Accumulated studies have indicated that Hcy as an intermediate metabolite of cysteine and methionine can cause endothelial dysfunction (Zhang et al., 2017). For instance, patients with hyperhomocysteinemia usually show decreased numbers of endothelial cells with impaired endothelial activities in endothelial proliferation, migration and adhesion, which all do harm to human heart health (Jamaluddin et al., 2007; Almashhadany et al., 2015). Experimentally, Hcy has been proved to be associated with disturbed cardiac substrate metabolism, mitochondrial dysfunction and adverse cardiac remodeling with increased myocardial stiffness (Joseph et al., 2003; Devi et al., 2006; Suematsu et al., 2007). However, whether Hcy may show similar toxic effect on cardiomyocytes is unknown. The underlying mechanism has not been well elucidated yet. Hence, in the present study, Hcy-induced cardiotoxicity and underlying mechanism were evaluated in H9c2 rat cardiomyocytes and Hcy-injured animal model, which are both accepted as good models for exploring the cardiotoxicity in vitro and in vivo. The results indicated that Hcy displayed significant cardiotoxicity in vitro and in vivo, as reflected by the cytotoxicity and cells apoptosis of H9c2 cells in vitro, and inhibition of angiogenesis in vivo. Meanwhile, we revealed the toxic mechanism that Hcy-induced cardiotoxicity was mainly achieved by triggering mitochondrial dysfunction and oxidative damage.
ROS, including hydroxyl radical, hydrogen peroxide, and superoxide anion (Sun et al., 2013), all play important roles in mediating cell signaling and maintaining cell homeostasis (Fan et al., 2014). The balance of anti-antioxidant and pro-antioxidant system ultimately decides the intracellular ROS level (Li et al., 2015; Wang et al., 2016a,b; Fan et al., 2017a). Oxidative stress was accepted as a key index in the pathology of human CVD (Castardo-de-Paula et al., 2017; Ghosh et al., 2017; Paul et al., 2017; Ramirez-Lee et al., 2017). Inhibition of oxidative damage represents of one of the most efficient strategies in treatment of human CVD. Therefore, searching novel agents with potential antioxidant activity and low side effects to combat human CVD is urgently needed.
ATX exhibits excellent pharmacological properties, including anti-cancer, anti-inflammatory, anti-diabetic, immunomodulatory and neuroprotective activities, which are all based on its antioxidant activity (Hussein et al., 2006; Abdelzaher et al., 2016). Large number of evidences have supported that ATX had the potential in chemoprevention and chemotherapy of several human diseases through eliminating ROS and attenuating oxidative damage in all kinds of cell and animal models (Wu et al., 2015). However, ATX-mediated protective effects against Hcy-induced cardiotoxicity have not been reported, and the underlying mechanism remains unclear.
The underlying mechanism has not been well elucidated yet. Hence, in the present study, Hcy-induced cardiotoxicity and underlying mechanism were evaluated in H9c2 rat cardiomyocytes and Hcy-injured animal model. In the present study, Hcy treatment caused significantly ROS over-production and eventually triggered oxidative damage in vitro and in vivo. However, ATX as a powerful inhibitor of ROS effectively inhibited ROS accumulation and blocked Hcy-induced oxidative damage in vitro and in vivo, as convinced by the decreased level of phosphorylation activation of ATM (Ser1981), ATR (Ser428), p53 (Ser15), and histone (Ser139). These results indicated that ROS as early apoptotic event was involved in Hcy-induced H9c2 cells apoptosis, and ATX can act as ROS inhibitor to suppress Hcy-induced apoptosis and oxidative damage.
Mitochondria integrates the intrinsic and extrinsic signals and plays important role in lunching mitochondria-mediated apoptosis (Zhu et al., 2016). Mitochondrial membrane permeabilization acts as an important event in inducing cell apoptotic death in response to apoptotic stimuli (Kroemer et al., 2007; Zhu et al., 2016; Fan et al., 2017b). Bcl-2 family, including the pro-apoptotic and anti-apoptotic members, has been confirmed as essential factors in regulating mitochondria-mediated apoptosis pathway (Cory and Adams, 2002). Increasing studies supported that loss of Δψm was highly multiple associated with the capsases activation and Bcl-2 family expression (Kroemer et al., 2007). For instance, Bcl-2 and Bcl-XL can bind to the out membrane of mitochondria of healthy cells (Cory and Adams, 2002). Bax can form homo-oligomers with Bak to permeabilize the out membrane of mitochondria and cause the depletion of Δψm (Wei et al., 2001; Cory and Adams, 2002). In the present study, our result suggested that Hcy treatment caused significant mitochondria-mediated apoptosis with involvement of mitochondrial dysfunction. Immunofluorescent staining of mitochondria affirmed that the loss of Δψm and mitochondrial morphological changes both contributed to the mitochondrial dysfunction in Hcy-treated H9c2 cells. The western blotting results revealed that Hcy-induced mitochondrial dysfunction was associated with the imbalance of Bcl-2 family expression. However, ATX pre-treatment prevented H9c2 cells from Hcy-induced mitochondrial dysfunction and the imbalance of Bcl-2 family expression, and eventually reversed Hcy-induced apoptosis. Therefore, we concluded that ATX blocked Hcy-induced mitochondria-mediated apoptosis by stabilizing Bcl-2 family expression.
In summary, the present study demonstrated that ATX suppressed Hcy-induced cardiotoxicity in vitro and in vivo by inhibiting mitochondrial dysfunction and oxidative damage. Our findings validated the potential therapeutic role of ATX in chemoprevention and chemotherapy of Hcy-mediated human CVD.
Ethical Statement
The study entitled “Astaxanthin Attenuates Homocysteine-Induced Cardiotoxicity in Vitro and in Vivo by Inhibiting Mitochondrial Dysfunction and Oxidative Damage” was performed in Key Lab of Cerebral Microcirculation in Universities of Shandong, Taishan Medical University. All the in vitro and in vivo experiments were performed in accordance with the relevant guidelines and regulations of Taishan Medical University. Especially, the in vivo experiments were approved by the Taishan Medical University Ethics Committee. All surgery was performed under 10% chloral hydrate, and every effort was made to minimize suffering.
Author Contributions
C-dF and B-lS designed the experiments. J-yS, X-tF, Y-jH, and YL performed the in vitro and in vivo experiments. M-fY and X-yF analyzed the data and images. C-dF and X-yF wrote the manuscript. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the National Natural Science Foundation of China No. 81501106 to CF, No. 81701178 to XF and No. 81471212 to BS.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2017.01041/full#supplementary-material
References
Abdelzaher, L. A., Imaizumi, T., Suzuki, T., Tomita, K., Takashina, M., and Hattori, Y. (2016). Astaxanthin alleviates oxidative stress insults-related derangements in human vascular endothelial cells exposed to glucose fluctuations. Life Sci. 150, 24–31. doi: 10.1016/j.lfs.2016.02.087
Almashhadany, A., Shackebaei, D., Van der Touw, T., Jones, G. L., Suleiman, M. S., and King, N. (2015). Homocysteine exposure impairs myocardial resistance to ischaemia reperfusion and oxidative stress. Cell. Physiol. Biochem. 37, 2265–2274. doi: 10.1159/000438582
Austin, R. C., Lentz, S. R., and Werstuck, G. H. (2004). Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 11(Suppl. 1), S56–S64. doi: 10.1038/sj.cdd.4401451
Baggott, J. E., and Tamura, T. (2015). Homocysteine, iron and cardiovascular disease: a hypothesis. Nutrients 7, 1108–1118. doi: 10.3390/nu7021108
Castardo-de-Paula, J. C., de Campos, B. H., Amorim, E., da Silva, R. V., de Farias, C. C., Higachi, L., et al. (2017). Cardiovascular risk and the effect of nitric oxide synthase inhibition in female rats: the role of estrogen. Exp. Gerontol. 97, 38–48. doi: 10.1016/j.exger.2017.07.016
Cory, S., and Adams, J. M. (2002). The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer. 2, 647–656. doi: 10.1038/nrc883
Devi, S., Kennedy, R. H., Joseph, L., Shekhawat, N. S., Melchert, R. B., and Joseph, J. (2006). Effect of long-term hyperhomocysteinemia on myocardial structure and function in hypertensive rats. Cardiovasc. Pathol. 15, 75–82. doi: 10.1016/j.carpath.2005.11.001
Fan, C. D., Fu, X. Y., Zhang, Z. Y., Cao, M. Z., Sun, J. Y., Yang, M. F., et al. (2017a). Selenocysteine induces apoptosis in human glioma cells: evidence for TrxR1-targeted inhibition and signaling crosstalk. Sci. Rep. 7:6465. doi: 10.1038/s41598-017-06979-2
Fan, C. D., Li, Y., Fu, X. T., Wu, Q. J., Hou, Y. J., Yang, M. F., et al. (2017b). Reversal of beta-amyloid-induced neurotoxicity in PC12 cells by curcumin, the important role of ROS-mediated signaling and ERK pathway. Cell. Mol. Neurobiol. 37, 211–222. doi: 10.1007/s10571-016-0362-3
Fan, C., Zheng, W., Fu, X., Li, X., Wong, Y. S., and Chen, T. (2014). Strategy to enhance the therapeutic effect of doxorubicin in human hepatocellular carcinoma by selenocystine, a synergistic agent that regulates the ROS-mediated signaling. Oncotarget 5, 2853–2863. doi: 10.18632/oncotarget.1854
Feng, X., and Xu, Y. (2017). Hyperhomocysteinemia as a metabolic risk factor for glucose intolerance among high-risk groups of chinese adults. Med. Sci. Monit. 23, 2775–2781. doi: 10.12659/MSM.905002
Ghosh, A., Gao, L., Thakur, A., Siu, P. M., and Lai, C. (2017). Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 24:50. doi: 10.1186/s12929-017-0357-5
Hussein, G., Sankawa, U., Goto, H., Matsumoto, K., and Watanabe, H. (2006). Astaxanthin, a carotenoid with potential in human health and nutrition. J. Nat. Prod. 69, 443–449. doi: 10.1021/np050354+
Jamaluddin, M. S., Yang, X., and Wang, H. (2007). Hyperhomocysteinemia, DNA methylation and vascular disease. Clin. Chem. Lab. Med. 45, 1660–1666. doi: 10.1515/CCLM.2007.350
Joseph, J., Joseph, L., Shekhawat, N. S., Devi, S., Wang, J., and Melchert, R. B. (2003). Hyperhomocysteinemia leads to pathological ventricular hypertrophy in normotensive rats. Am. J. Physiol. Heart Circ. Physiol. 285, H679–H686. doi: 10.1152/ajpheart.00145.2003
Kroemer, G., Galluzzi, L., and Brenner, C. (2007). Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163. doi: 10.1152/physrev.00013.2006
Lee, W. W., Choi, K. C., Yum, R. W., Yu, D. S., and Chair, S. Y. (2016). Effectiveness of motivational interviewing on lifestyle modification and health outcomes of clients at risk or diagnosed with cardiovascular diseases: a systematic review. Int. J. Nurs. Stud. 53, 331–341. doi: 10.1016/j.ijnurstu.2015.09.010
Li, D. W., Sun, J. Y., Wang, K., Zhang, S., Hou, Y. J., Yang, M. F., et al. (2015). Attenuation of cisplatin-induced neurotoxicity by cyanidin, a natural inhibitor of ros-mediated apoptosis in PC12 cells. Cell. Mol. Neurobiol. 35, 995–1001. doi: 10.1007/s10571-015-0194-6
Markisic, M., Pavlovic, A. M., and Pavlovic, D. M. (2017). The impact of homocysteine, vitamin b12, and vitamin d levels on functional outcome after first-ever ischaemic stroke. Biomed Res. Int. 2017:5489057. doi: 10.1155/2017/5489057
Paul, S., Das, S., Tanvir, E. M., Hossen, M. S., Saha, M., Afroz, R., et al. (2017). Protective effects of ethanolic peel and pulp extracts of Citrus macroptera fruit against isoproterenol-induced myocardial infarction in rats. Biomed. Pharmacother. 94, 256–264. doi: 10.1016/j.biopha.2017.07.080
Ramirez-Lee, M. A., Espinosa-Tanguma, R., Mejía-Elizondo, R., Medina-Hernández, A., Martinez-Castañon, G. A., and Gonzalez, C. (2017). Effect of silver nanoparticles upon the myocardial and coronary vascular function in isolated and perfused diabetic rat hearts. Nanomedicine 13, 2587–2596. doi: 10.1016/j.nano.2017.07.007
Rudreshkumar, K. J., Majumdar, V., Nagaraja, D., and Christopher, R. (2017). Relevance of plasma levels of free homocysteine and methionine as risk predictors for ischemic stroke in the young. Clin. Nutr. doi: 10.1016/j.clnu.2017.07.005. [Epub ahead of print].
Sasaki, Y., Kobara, N., Higashino, S., Giddings, J. C., and Yamamoto, J. (2011). Astaxanthin inhibits thrombosis in cerebral vessels of stroke-prone spontaneously hypertensive rats. Nutr. Res. 31, 784–789. doi: 10.1016/j.nutres.2011.09.010
Suematsu, N., Ojaimi, C., Kinugawa, S., Wang, Z., Xu, X., and Koller, A. (2007). Hyperhomocysteinemia alters cardiac substrate metabolism by impairing nitric oxide bioavailability through oxidative stress. Circulation 115, 255–262. doi: 10.1161/CIRCULATIONAHA.106.652693
Sun, H., Leng, T., Zeng, Z., Gao, X., Inoue, K., and Xiong, Z. G. (2013). Role of TRPM7 channels in hyperglycemia-mediated injury of vascular endothelial cells. PLoS ONE 8:e79540. doi: 10.1371/journal.pone.0079540
Ungvari, Z., Csiszar, A., Edwards, J. G., Kaminski, P. M., Wolin, M. S., Kaley, G., et al. (2003). Increased superoxide production in coronary arteries in hyperhomocysteinemia: role of tumor necrosis factor-alpha, NAD(P)H oxidase, and inducible nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 23, 418–424. doi: 10.1161/01.ATV.0000061735.85377.40
Wang, K., Fu, X. T., Li, Y., Hou, Y. J., Yang, M. F., Sun, J. Y., et al. (2016a). Induction of s-phase arrest in human glioma cells by selenocysteine, a natural selenium-containing agent via triggering reactive oxygen species-mediated DNA damage and modulating MAPKs and AKT pathways. Neurochem Res. 41, 1439–1447. doi: 10.1007/s11064-016-1854-8
Wang, K., Fu, X. Y., Fu, X. T., Hou, Y. J., Fang, J., Zhang, S., et al. (2016b). DSePA Antagonizes high glucose-induced neurotoxicity: evidences for DNA damage-mediated p53 phosphorylation and MAPKs and AKT pathways. Mol. Neurobiol. 53, 4363–4374. doi: 10.1007/s12035-015-9373-1
Wei, M. C., Zong, W. X., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., et al. (2001). Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730. doi: 10.1126/science.1059108
Wu, H., Niu, H., Shao, A., Wu, C., Dixon, B. J., Zhang, J., et al. (2015). Astaxanthin as a potential neuroprotective agent for neurological diseases. Mar. Drugs. 13, 5750–5766. doi: 10.3390/md13095750
Zhang, C., Cai, Y., Adachi, M. T., Oshiro, S., Aso, T., Kaufman, R. J., et al. (2001). Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J. Biol. Chem. 276, 35867–35874. doi: 10.1074/jbc.M100747200
Zhang, Z., Wei, C., Zhou, Y., Yan, T., Wang, Z., Li, W., et al. (2017). Homocysteine induces apoptosis of human umbilical vein endothelial cells via mitochondrial dysfunction and endoplasmic reticulum stress. Oxid. Med. Cell. Longev. 2017:5736506. doi: 10.1155/2017/5736506
Zhao, Z. W., Cai, W., Lin, Y. L., Lin, Q. F., Jiang, Q., Lin, Z., et al. (2011). Ameliorative effect of astaxanthin on endothelial dysfunction in streptozotocin-induced diabetes in male rats. Arzneimittelforschung 61, 239–246. doi: 10.1055/s-0031-1296194
Zhu, L. Z., Hou, Y. J., Zhao, M., Yang, M. F., Fu, X. T., Sun, J. Y., et al. (2016). Caudatin induces caspase-dependent apoptosis in human glioma cells with involvement of mitochondrial dysfunction and reactive oxygen species generation. Cell Biol. Toxicol. 32, 333–345. doi: 10.1007/s10565-016-9338-9
Keywords: homocysteine, astaxanthin, cardiovascular diseases, mitochondrial dysfunction, oxidative damage
Citation: Fan C-d, Sun J-y, Fu X-t, Hou Y-j, Li Y, Yang M-f, Fu X-y and Sun B-l (2017) Astaxanthin Attenuates Homocysteine-Induced Cardiotoxicity in Vitro and in Vivo by Inhibiting Mitochondrial Dysfunction and Oxidative Damage. Front. Physiol. 8:1041. doi: 10.3389/fphys.2017.01041
Received: 07 August 2017; Accepted: 29 November 2017;
Published: 12 December 2017.
Edited by:
Jing-Yan Han, Peking University, ChinaReviewed by:
Qiaobing Huang, Southern Medical University, ChinaYunzeng Zou, Fudan University, China
Copyright © 2017 Fan, Sun, Fu, Hou, Li, Yang, Fu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-yan Fu, txyfu66@163.com
Bao-liang Sun, tblsun66@163.com
†These authors have contributed equally to this work.