 Dong An
Dong An Feng Hao2
Feng Hao2 Xuemin Xu
Xuemin Xu Mei-Zhen Cui
Mei-Zhen Cui- 1School of Life Sciences, Jilin University, Changchun, China
- 2Department of Biomedical and Diagnostic Sciences, College of Veterinary Medicine, University of Tennessee, Knoxville, TN, United States
Foam cell formation is the key process in the development of atherosclerosis. The uptake of oxidized low-density lipoprotein (oxLDL) converts macrophages into foam cells. We recently reported that lipopolysaccharide (LPS)-induced foam cell formation is regulated by CD14 and scavenger receptor AI (SR-AI). In this study, we employed pharmaceutical and gene knockdown approaches to determine the upstream molecular mediators, which control LPS-induced foam cell formation. Our results demonstrated that the specific c-Jun N-terminal kinase (JNK) pathway inhibitor, SP600125, but neither the specific inhibitor of extracellular signaling-regulated kinase (ERK) kinase MEK1/2, U0126, nor the specific inhibitor of p38 MAPK, SB203580, significantly blocks LPS-induced oxLDL uptake, suggesting that the JNK pathway is the upstream mediator of LPS-induced oxLDL uptake/foam cell formation. To address whether JNK pathway mediates LPS-induced oxLDL uptake is due to JNK pathway-regulated CD14 and SR-AI expression, we assessed whether the pharmaceutical inhibitor of JNK influences LPS-induced expression of CD14 and SR-AI. Our results indicate that JNK pathway mediates LPS-induced CD14 and SR-AI expression. To conclusively address the isoform role of JNK family, we depleted JNK isoforms using the JNK isoform-specific siRNA. Our data showed that the depletion of JNK1, but not JNK2 blocked LPS-induced CD14/SR-AI expression and foam cell formation. Taken together, our results reveal for the first time that JNK1 is the key mediator of LPS-induced CD14 and SR-AI expression in macrophages, leading to LPS-induced oxLDL uptake/foam cell formation. We conclude that the novel JNK1/CD14/SR-AI pathway controls macrophage oxLDL uptake/foam cell formation.
Introduction
Atherosclerosis is a leading disease, which causes death and disability in developed countries. The accumulation of cholesterol esters within artery cells, especially in macrophages, converting them into foam cells, is fundamental to the pathogenesis of atherosclerosis (Brown and Goldstein, 1983; Steinberg et al., 1989). In early fatty streak lesions, macrophages that are differentiated from monocytes become matured and gather low-density lipoprotein (LDL) and modified LDL, such as oxidized low-density lipoprotein (oxLDL). Uptake of oxLDL through scavenging receptors (SRs) leads to the formation of mature lipid-laden macrophages, which are called foam cells. SR-AI has been reported to be dominantly expressed in mouse macrophages and plays an important role in modified LDL uptake (Kzhyshkowska et al., 2012).
Lipopolysaccharide (LPS), is also referred as an endotoxin, which has numerous biological activities and is responsible for many pathological conditions caused by Gram-negative bacteria. As the highly inflammatory constituent of the outer membrane of Gram-negative bacteria, LPS induces macrophage foam cell formation in vitro (Howell et al., 2011; Morishita et al., 2013) and leads to the formation of aortic lesions in vivo (Li et al., 2002; Westerterp et al., 2007; Gitlin and Loftin, 2009). The binding of LPS to its co-receptor, CD14, activates inflammatory toll-like receptor pathways, initiating the release of pro-inflammatory cytokines into the vasculature (Wright et al., 1990; Dunzendorfer et al., 2004; Levy et al., 2009). It has been reported that the expression of CD14 is associated with thrombosis in carotid plaques (Hermansson et al., 2014). Our recent study revealed that CD14 is a key mediator in LPS-induced foam cell formation in macrophages (An et al., 2017). We further demonstrated that SR-AI is the downstream mediator of CD14 in regulating LPS-induced foam cell formation (An et al., 2017). In the current study, we aimed to understand the upstream pathway, leading to the LPS-induced oxLDL uptake by macrophages/foam cell formation.
Mitogen-activated protein (MAP) kinases have been implicated in the development of atherosclerosis (Muslin, 2008). Conventional MAPKs include the extracellular signal-regulated kinase 1 and 2 (Erk1/2 or p44/42), the c-Jun N-terminal kinases 1–3 (JNK1-3) and the p38 isoforms. In this study, we evaluated whether MAPK activity mediates LPS-induced oxLDL uptake in macrophages/foam cell formation and how MAPKs mediate LPS-induced foam cell formation. In particular, we determined whether activation of these kinases influences the expression of CD14 and SR-AI. Understanding the signaling pathways that control foam cell formation will significantly contribute to designing therapeutic strategies for curing atherosclerosis.
Materials and Methods
Reagents
Lipopolysaccharide (LPS) was obtained from Sigma-Aldrich Co (St. Louis, MO). Antibodies against mouse CD14 and SR-AI were from R&D system (Minneapolis, MN). Antibodies against mouse Phospho-Erk1/2, Phospho-p38, Phospho-JNK, JNK1 and JNK2 were from Cell Signaling Technology (Danvers, MA). Antibody against mouse GAPDH was from EMD Millipore (St. Charles, MO). The MEK1/2-specific inhibitor U0126, the p38-specific inhibitor SB203580 and the JNK-specific inhibitor SP600125 were from Cayman Chemical (Ann Arbor, MI). The RNeasy kit, Non-silencing siRNA, and JNK1 and JNK2 siRNAs were from Qiagen (Gaithersburg, MD). Human LDL and Dil-labeled oxLDL was purchased from Biomedical Technologies (Stoughton, MA).
Cell Culture
Bone-marrow progenitor cells were harvested from the femur section of 8–10 weeks old C57B/6 mice. Total bone marrow cells were washed once with PBS, resuspended in LADMAC conditioned medium, and plated in tissue culture dishes. To produce the LADMAC-conditioned medium, the LADMAC cell line (ATCC, Manassas, VA) was grown to confluence in 75-cm2 flasks for 5 days, followed by harvesting and filtering these culture supernatants. DMEM supplemented with 10% FBS and 20% LADMAC supernatant was used as the conditioned medium to foster the growth and differentiation of bone marrow macrophages. The conditioned medium, prepared in a similar manner, has been used to support the growth of bone marrow-derived macrophages because the LADMAC cell line is a source of M-CSF (Sklar et al., 1985; Olivas et al., 1995). After 6 days in culture, 95% of bone-marrow progenitor cells were differentiated into macrophages (bone marrow-derived macrophages, BMDMs) (An et al., 2017).
Western Blot Analysis
Cultured mouse BMDMs were rinsed with cold PBS and lysed in Western blot lysis buffer (50 mM Tris-HCl, pH 6.8, 8 M urea, 5% mercaptoethanol, 2% SDS, and protease/phosphatase inhibitors) with sonication for 30 s on ice. Cellular proteins were separated by 10% SDS-PAGE and transferred to an Immobilon-P PVDF transfer membrane (Billerica, MA). Membranes were then probed with the specific antibodies, and the specific protein bands were viewed using Luminata Forte Western HRP substrate (Billerica, MA).
siRNA Treatment
BMDMs were transfected with non-silencing or specific siRNA (Qiagen) for 48 h, using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific) following the instructions provided by the manufacturer. On day 3, cells were cultured in serum free medium for 24 h, followed by treatment either with or without LPS.
LDL Oxidation
Oxidized LDL was prepared by dialyzing native LDL against 10 μM CuSO4 in PBS at room temperature for 24 h. The oxidation was stopped, and the lipoprotein bound metal ion was removed by dialyzing against PBS containing 0.5 mM EDTA at 4°C overnight (Chai et al., 1996).
Oil Red O Staining
BMDMs were cultured on microscope cover glasses in 12-well plates and starved for 24 h. After the LPS treatment, the cells were incubated with native LDL or oxidized LDL at 37°C for 3.5 h. After the completion of treatments, cells were rinsed once with PBS, fixed in 4% paraformaldehyde at 4°C for 30 min. Subsequently, the cells were rinsed once with PBS and then stained with Oil Red O solution (Sigma Aldrich) at room temperature for 10 min. After staining, the cells were rinsed two times with PBS then stained with hematoxylin (Sigma Aldrich) for 1 min. Afterward, they were rinsed two times with PBS and observed by light microscopy (Nikon Eclipse E600 microscope) (Xu et al., 2010).
Analysis of Dil-oxLDL Uptake
Cellular uptake of Dil-oxLDL was measured using both fluorescence microscopy and plate reader. BMDMs were cultured in 24-well plates and then starved for 24 h. After LPS treatment for 18 h, cells were incubated with Dil-labeled oxLDL (Biomedical Technologies, Stoughton, MA) for 3.5 h and then rinsed with cold PBS containing 5% BSA two times. For fluorescence microscopy, cells grown on cover glass slides were fixed in 4% ice-cold paraformaldehyde solution for 30 min. Then the cells were rinsed with cold PBS containing 5% BSA two times. Afterwards, the coverslips were mounted on slides with permanent aqueous mounting medium (Biogenex, Fremont, CA), and the labeled oxLDL were analyzed by fluorescence microscopy with a Nikon Eclipse E600 microscope. For plate reader quantification, cells were further rinsed with PBS for two more times. Cells were then detached from the culture plate with 0.5% Triton X-100 in PBS. The fluorescence value was measured by the Synergy HT plate reader (BioTek, Winooski, VT) at 530 nm of excitation wavelength and 590 nm of emission wavelength (Barlic et al., 2009; Park et al., 2012).
Statistical Analysis
Results are means ± S.E. Comparisons between multiple groups were performed using one-way analysis of variance with post-hoc t-tests. Single comparisons were made using two-tailed, unpaired Student t-tests. A p-value of 0.05 was considered statistically significant.
Results
LPS Activates ERK, p38 and JNK Pathways in BMDMs
To pursue the upstream molecular determinants that control LPS-induced foam cell formation, we examined the possible signaling pathways involved. It has been demonstrated that the MAPKs signaling cascades play important roles in the development of atherosclerosis (Muslin, 2008). Whether these pathways are involved in LPS-induced oxLDL uptake/foam cell formation has been currently unrevealed. We isolated mouse bone marrow cells and then differentiated them into macrophages. We observed that the addition of LPS to the BMDMs highly and time dependently induced the phosphorylation of ERK, p38 and JNK MAPKs (Figures 1A,B). The activation peak for ERK is around 30–60 min; the activation peak for p38 is around 15–60 min and JNK at 30 min. These data suggest that the MAPK pathways might be involved in mediating LPS-induced oxLDL uptake by BMDMs.
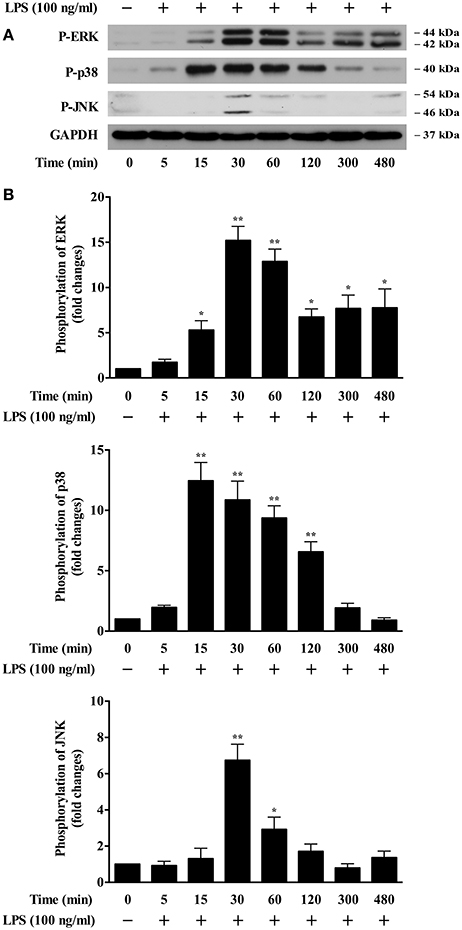
Figure 1. LPS induction of MAPK activation in BMDMs. (A) Western blotting data showed time courses of LPS induction of phosphorylation of ERK, p38, and JNK in BMDMs. Cultured BMDMs were starved for 24 h prior to LPS (100 ng/ml) stimulation for various times. Cell lysates were analyzed by Western blotting, using specific antibodies against p-ERK, p-p38, and p-JNK. GAPDH served as the loading control. The representative Western blot results shown were from three independent experiments. (B) results of the Western blot analysis were quantified as the densitometry value analyzed by UN-SCAN-IT gel 6.1 software. *p < 0.05; **p < 0.01 vs. control.
JNK Pathway Is Involved in LPS-Induced oxLDL Uptake in BMDMs
The experimental results of the Oil Red O staining demonstrated that LPS dose dependently increased the uptake of oxLDL but not native LDL (Figure 2A). To determine whether these MAPK pathways contribute to LPS-induced oxLDL uptake in BMDMs, we examined the influence of MAPK inhibitors on LPS-induced oxLDL uptake. As shown in Figure 2B, the visualized fluorescence microscopy data reveal that the specific JNK inhibitor, SP600125 (1 μM), nearly completely blocked LPS-induced oxLDL uptake in BMDMs. However, pretreatment of either the specific MEK1/2 inhibitor, U0126 or the specific p38 inhibitor, SB203580, had no noticeable effect on LPS-induced oxLDL uptake. To confirm the role of JNK in LPS-induced oxLDL uptake, we quantified the Dil-oxLDL uptake by using a fluorescence microplate reader. Fluorescence at 530 nm of excitation wavelength and 590 nm of emission wavelength was assessed. As shown in Figure 2C, the JNK inhibitor SP600125 dose-dependently inhibited LPS-induced oxLDL uptake, but neither the specific MEK1/2, U0126 nor the p38 inhibitor, SB203580, in all of these doses tested, had an effect on LPS-induced oxLDL uptake. These doses have been previously demonstrated dose-dependently blocking the activation of ERK and p38 (Cuenda et al., 1995; Favata et al., 1998). These data suggest a regulatory role of JNK in LPS-induced oxLDL uptake.
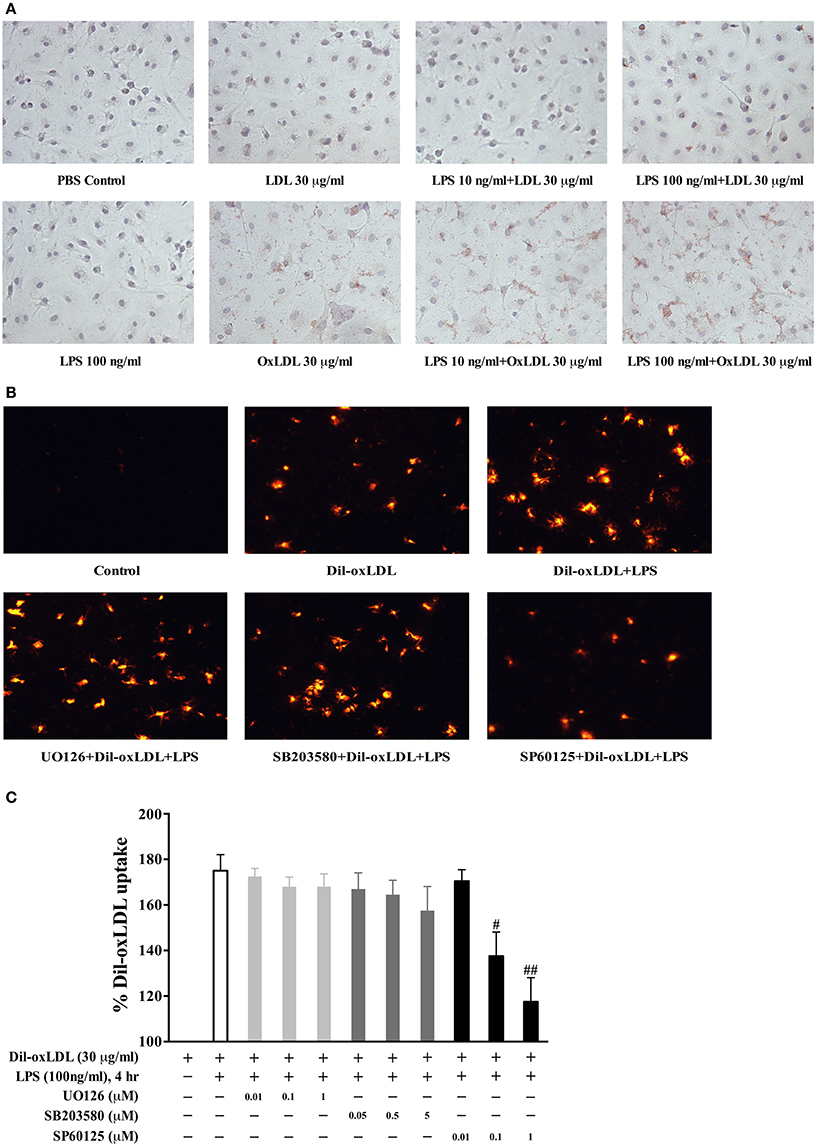
Figure 2. The involvement of JNK pathway in LPS induction of foam cell formation. (A) LPS effects on the uptake of oxLDL and LDL were examined by the Oil Red O staining with Nikon Eclipse E600 microscopy. BMDMs were stimulated with LPS for 18 h prior to the treatment of oxLDL or LDL for 3.5 h. (B) fluorescence microscopy results showed that pretreatment with the specific JNK inhibitor SP600125, but not the specific MEK inhibitor U0126 or p38 inhibitor SB203580, significantly blocked LPS-induced Di-oxLDL uptake. Twenty-four-hour serum free starved BMDMs were pretreated with various MAPK inhibitors (SP600125, 1 μM; SB203580, 5 μM; U0126, 1 μM) for 45 min prior to LPS treatment, followed by the addition of 30 μg/ml DiI-oxLDL for 3.5 h. The results were examined by fluorescence microscopy (Nikon Eclipse E600 microscopy); the Dil-oxLDL signals were shown in red. (C) pretreatment with specific JNK inhibitor SP600125, but not the specific MEK inhibitor U0126 or p38 inhibitor SB203580, dose-dependently blocked LPS-induced Dil-oxLDL uptake. The uptake of Dil-oxLDL fluorescence value was measured in Synergy HT plate reader at 530 nm (excitation) and 590 nm (emission). The Y axis represents the increased oxLDL uptake levels by LPS compared to the basal oxLDL uptake (uptake of oxLDL alone was considered as 100%). Quantified results were from three independent experiments. #p < 0.05; ##p < 0.01 vs. LPS alone group.
JNK Pathway Mediates the LPS-Induced Expression of CD14 and SR-AI in BMDMs
We recently identified that the CD14 and SR-AI axis mediates LPS-induced oxLDL uptake/foam cell formation (An et al., 2017). Our results demonstrated that LPS dose-dependently induced CD14 and SR-AI protein expression (Figure 3A). To identify the specific MAPK that mediates the expression of CD14 and SR-AI, we evaluated the influences of various MAPK inhibitors on the expression of CD14 and SR-AI. As shown in Figures 3B,C, the specific JNK inhibitor, SP60125, dose-dependently blocked LPS-induced expression of CD14 and SR-AI. However, neither the MEK specific inhibitor U0126 nor the p38 specific inhibitor SB203580 has a significant effect on the expression of CD14 and SR-AI. These data suggest that the novel JNK-CD14-SR-AI pathway mediates LPS-induced oxLDL uptake/foam cell formation.
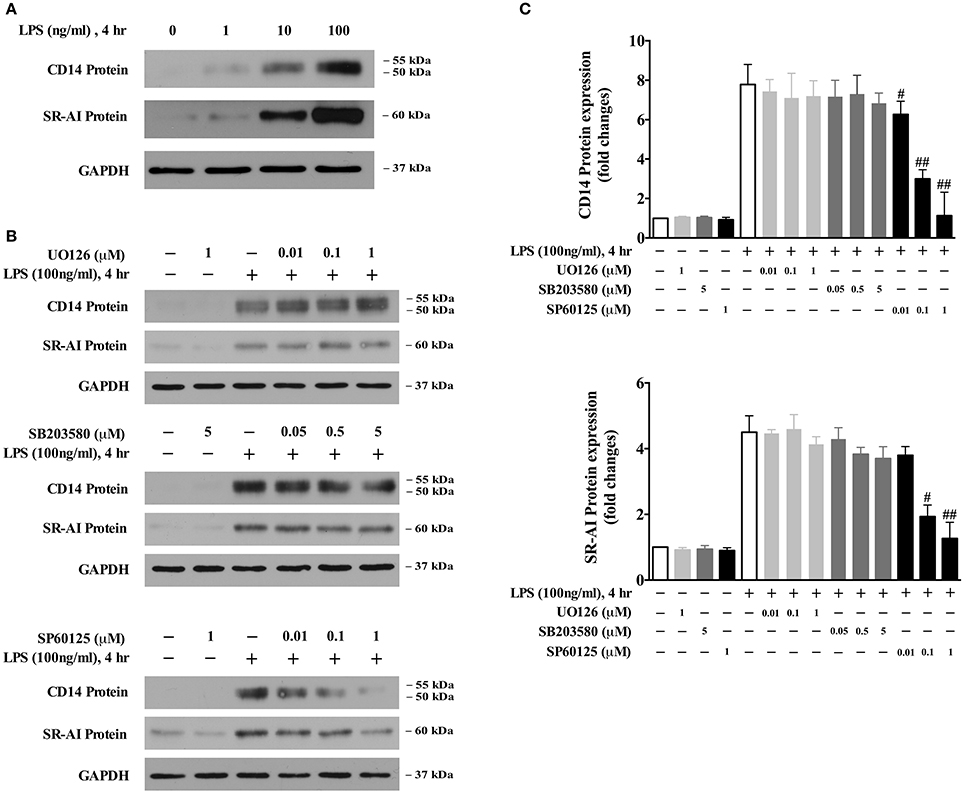
Figure 3. The JNK pathway contributes to CD14 and SR-AI expression. (A) Western blotting data showed that LPS dose-dependently induces CD14 and SR-AI expression in BMDMs. (B) Pretreatment with the specific JNK inhibitor SP600125, but not the specific MEK inhibitor U0126 or p38 inhibitor SB203580, dose-dependently blocked LPS-induced CD14 and SR-AI expression. Twenty-four-hour serum starved BMDMs were pretreated with various inhibitors for 45 min, and then 100 ng/ml LPS was added for 4 h. CD14 and SR-AI protein levels were determined by Western blotting. Data shown were from three independent experiments. (C) results of the Western blot analysis were quantified as the densitometry value analyzed by UN-SCAN-IT gel 6.1 software. #p < 0.05; ##p < 0.01 vs. LPS alone group.
JNK1, but Not JNK2, Mediates LPS-Induced Expression of CD14 and SR-AI, Which Leads to oxLDL Uptake in BMDMs
The JNK family consists of ten isoforms derived from three genes: JNK1, JNK2, and JNK3 (Gupta et al., 1996). It has been shown that JNK1 and JNK2 are widely expressed in various tissues; however, JNK3 is mainly expressed in the nervous system (Martin et al., 1996). To identify which JNK isoform is responsible for LPS-induced CD14 and SR-AI expression, specific siRNA of JNK1 or JNK2 were applied in BMDMs. As shown in Figures 4A,B, depletion of JNK1 largely abolished LPS-induced CD14 and SR-AI expression; however, knockdown of JNK2 with the specific siRNA of JNK2, has no significant effect on the expression of CD14 or SR-AI induced by LPS. Depletion of both JNK1 and JNK2 almost completely blocked LPS-induced expression of CD14 and SR-AI. These results demonstrate for the first time that JNK1 is required for LPS-induced CD14 and SR-AI expression.
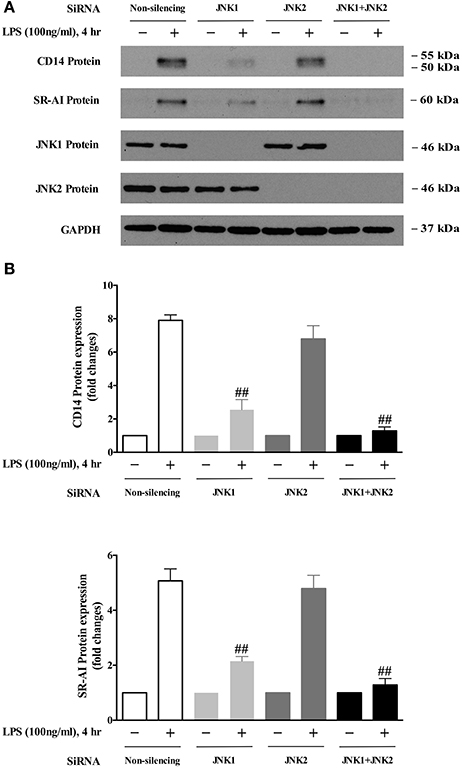
Figure 4. JNK 1 is the key mediator of LPS-induced CD14 and SR-AI expression. (A) Western blotting data showed that knockdown of JNK1 expression, but not JNK2, blocked LPS-induced CD14 and SR-AI expression in BMDMs. GAPDH served as the loading control. The representative Western blot results shown were from three independent experiments. (B) results of the Western blot analysis were quantified as the densitometry value analyzed by UN-SCAN-IT gel 6.1 software. ##p < 0.01 vs. Non-silencing group.
JNK1 Mediates LPS-Induced oxLDL Uptake
We next examined whether JNK1 is required for LPS induction of oxLDL uptake. As shown in Figure 5A, the expression of JNK1 or JNK2 was successfully depleted using the specific JNK1 or JNK2 siRNA. We observed that the depletion of JNK1 blocked LPS-induced oxLDL uptake, whereas the depletion of JNK2 had no significant effect on oxLDL uptake (microscopy analysis, Figure 5B), indicating a key role of JNK1 in oxLDL uptake in macrophages. We substantiated our observation by quantifying the Dil-oxLDL uptake in a fluorescence microplate reader as described in Figure 2B. Our data show that depletion of JNK1 but not JNK2 nearly completely blocked oxLDL uptake (Figure 5C). Together, the results indicate that the specific isoform JNK1, but not JNK2 is required for LPS-induced oxLDL uptake in BMDMs.
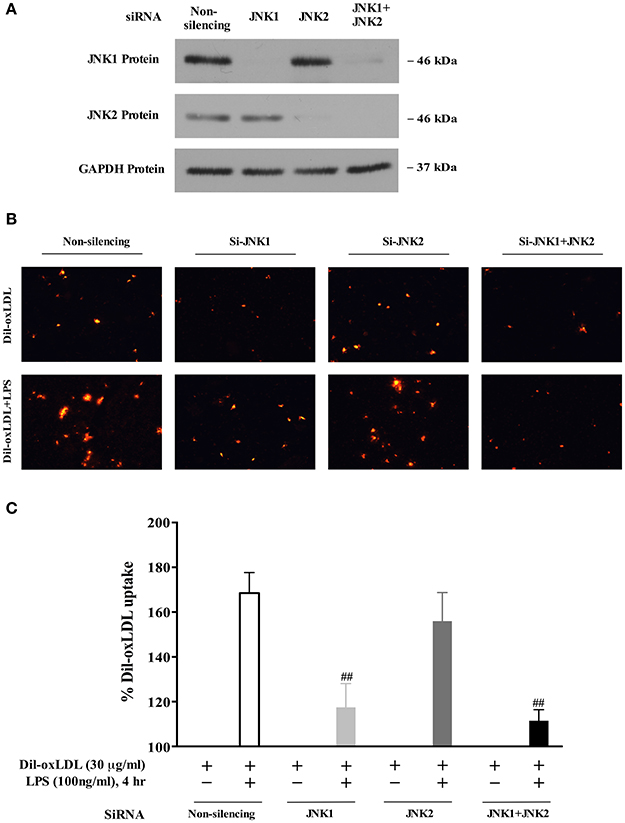
Figure 5. The JNK1 activity is required for LPS-induced oxLDL uptake in macrophages/foam cell formation. (A) knockdown efficiency of JNK1 and JNK2 with specific siRNAs in BMDMs was assessed in a Western blot analysis. (B) Visualized fluorescence microscopy data showed that knockdown of expression of JNK1 with specific siRNA blocked LPS-induced Dil-oxLDL uptake in BMDMs. The results were examined by fluorescence microscopy (Nikon Eclipse E600 microscopy); the Dil-oxLDL signals were shown in red. (C) quantitative fluorescence data indicated that knockdown of JNK1 expression using the specific JNK1 siRNA blocked LPS-induced Dil-oxLDL uptake in BMDMs. The quantitative analyses of oxLDL uptake using a fluorescence microplate reader were described in Figure 2. The Y axis represents the increased oxLDL uptake levels by LPS compared to the basal oxLDL uptake (uptake of oxLDL alone was considered as 100%). Quantified results were from three independent experiments. ##p < 0.01 vs. Non-silencing group.
Discussion
Atherosclerosis is a chronic inflammatory disease, in which the formation of foam cells from macrophages is a crucial event, leading to the thrombotic complications. The unrestricted uptake of lipoproteins through scavenger receptor pathways, which is not limited by intracellular cholesterol levels, eventually leads to the formation of foam cells, the initial step in atherosclerosis (de Winther et al., 2000; Lusis, 2000). LPS, usually found in the outer membrane of Gram-negative bacteria, is a potential mediator of inflammatory responses. Chlamydiae-derived chlamydial LPS has been detected in atherosclerotic lesions (Hu et al., 1999). Repeated intravenous and intraperitoneal administration of LPS accelerates atherosclerosis in rabbits and apoe-/- mice (Lehr et al., 2001; Ostos et al., 2002; Engelmann et al., 2004; Westerterp et al., 2007). It has been shown that common chronic infection plays an important role in human atherogenesis (Kiechl et al., 2001). In a recent study, we reported that LPS via CD14 and SR-AI mediates oxLDL uptake in macrophages/foam cell formation (An et al., 2017). Our data also demonstrated that lysophosphatidic acid (LPA) synergistically enhances LPS-induced macrophage foam cell formation (An et al., 2017). LPA highly accumulates in human atherosclerotic lesion (Siess et al., 1999). A long-term, high-fat diet elevates LPA levels in rabbit plasma/serum (Tokumura et al., 2002) and in small intestines in atherosclerotic mice (Navab et al., 2015). Therefore, LPA may worsen atherosclerosis via enhancing macrophage foam cell formation. On the other hand, a recent in vitro evidence has shown that LPS enhances LPA secretion and autotaxin levels, which converts lysophosphatidylcholine to LPA (Awada et al., 2014), suggesting that LPS may trigger to increase LPA levels in vivo. Therefore, it is possible that LPS may deteriorate atherosclerosis via an increase of LPA secretion in atherosclerotic lesions.
In the current study, we identified that JNK pathway mediates LPS-induced oxLDL uptake via JNK cascade-triggered upregulation of CD14 and SR-AI. We further report that the JNK1 MAPK, but not JNK2 MAPK, is the key mediator for LPS-induced CD14 and SR-AI expression, which leads to the oxLDL uptake in BMDMs. This conclusion was drawn based on the following lines of evidence: (1) pretreatment of the specific JNK MAPK inhibitor SP600125 blocked LPS-induced oxLDL uptake/foam cell formation, (2) pretreatment of the specific JNK MAPK inhibitor SP600125 blocked LPS-induced CD14 and SR-AI expression and (3) depletion of the specific isoform JNK1, but not JNK2 abolished LPS-induced CD14 and SR-AI expression and LPS-induced oxLDL uptake/foam cell formation. Therefore, our data reveal that the novel JNK1/CD14/SR-AI pathway contributes to macrophage oxLDL uptake/foam cell formation.
Besides the recognition of bacterial LPS function in the innate immune system, which leads to uncontrollable cytokine production and results in fatal sepsis syndrome in humans (Triantafilou and Triantafilou, 2002), LPS-triggers oxLDL uptake in macrophages causing foam cell formation (Howell et al., 2011; Morishita et al., 2013). LPS has also been reported to be involved in cardiovascular disease via activated proinflammatory pathways (Mehta et al., 1998; Becker et al., 2001). LPS induces expression of inflammatory cytokines IL-6, MCP-1 and tumor necrosis factor-α (TNF-α) (Jatta et al., 2005; Yin et al., 2013). LPS induction of inflammatory cytokine expression is mediated by the NF-κB pathway, which contributes to intima hyperplasia (Vink et al., 2002; He et al., 2017). In addition, LPS induces tissue factor expression, contributing to thrombosis (Fei et al., 1993; Brand et al., 1994; Nakagomi et al., 2000). Therefore, LPS is an important inflammatory trigger for various inflammatory diseases.
In this study, we identified that the specific isoform JNK1 is required for mediating LPS-induced CD14 and SR-AI expression and foam cell formation. These data also indicate that JNK pathway is essential for the development of atherosclerosis. This conclusion is supported by previous studies, which demonstrated that inhibition of JNK pathway, but not the ERK pathway abolished oxLDL-induced foam cell formation (Rahaman et al., 2006) and that inhibition of JNK activation attenuated low shear stress-induced atherosclerosis in ApoE-deficient mice (Wang et al., 2011).
LPS-induced JNK1 activation may contribute to CD14 and SR-AI expression via transcriptional regulation. It has been shown that once JNKs have been activated, these kinases translocate to the nucleus (Cavigelli et al., 1995), where they phosphorylate c-Jun and thereby trigger the target gene transcriptional activity (Karin, 1995). Jun and Fos are family members of transcription factor AP1 (activator protein-1) (Angel and Karin, 1991). It has been reported that AP-1 is critical for both CD14 and SR-AI expression (Mietus-Snyder et al., 1998; Iwahashi et al., 2000). Our data suggest that, JNK1 may, via the activation of AP1 family, activate the transcriptional machinery of CD14 and SR-AI genes to increase their expression, which in turn contributes to oxLDL uptake in macrophages/foam cell formation, resulting in atherosclerosis.
Ethics Statement
This study was carried out in accordance with the guidelines of the National Institutes of Health. The protocol was approved by the University of Tennessee Institutional Animal Care and Use Committee.
Author Contributions
M-ZC conceived the idea and coordinated the research. DA, FH, and CH designed and performed experiments and analyzed data. DA prepared the figures. DA, XX, WK, and M-ZC analyzed and interpreted data. DA and M-ZC wrote the manuscript. All authors edited, revised, and approved the final version of the manuscript.
Funding
This work was support by National Institutes of Health Grants HL107466 (to M-ZC) and NS095256 (to XX).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
An, D., Hao, F., Zhang, F., Kong, W., Chun, J., Xu, X., et al. (2017). CD14 is a key mediator of both lysophosphatidic acid and lipopolysaccharide induction of foam cell formation. J. Biol. Chem. 292, 14391–14400. doi: 10.1074/jbc.M117.781807
Angel, P., and Karin, M. (1991). The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1072, 129–157. doi: 10.1016/0304-419X(91)90011-9
Awada, R., Saulnier-Blache, J. S., Grès, S., Bourdon, E., Rondeau, P., Parimisetty, A., et al. (2014). Autotaxin downregulates LPS-induced microglia activation and pro-inflammatory cytokines production. J. Cell. Biochem. 115, 2123–2132. doi: 10.1002/jcb.24889
Barlic, J., Zhu, W., and Murphy, P. M. (2009). Atherogenic lipids induce high-density lipoprotein uptake and cholesterol efflux in human macrophages by up-regulating transmembrane chemokine CXCL16 without engaging CXCL16-dependent cell adhesion. J. Immunol. 182, 7928–7936. doi: 10.4049/jimmunol.0804112
Becker, A. E., de Boer, O. J., and van Der Wal, A. C. (2001). The role of inflammation and infection in coronary artery disease. Annu. Rev. Med. 52, 289–297. doi: 10.1146/annurev.med.52.1.289
Brand, K., Banka, C. L., Mackman, N., Terkeltaub, R. A., Fan, S. T., and Curtiss, L. K. (1994). Oxidized LDL enhances lipopolysaccharide-induced tissue factor expression in human adherent monocytes. Arterioscler. Thromb. 14, 790–797. doi: 10.1161/01.ATV.14.5.790
Brown, M. S., and Goldstein, J. L. (1983). Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu. Rev. Biochem. 52, 223–261. doi: 10.1146/annurev.bi.52.070183.001255
Cavigelli, M., Dolfi, F., Claret, F. X., and Karin, M. (1995). Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J. 14, 5957–5964.
Chai, Y. C., Howe, P. H., DiCorleto, P. E., and Chisolm, G. M. (1996). Oxidized low density lipoprotein and lysophosphatidylcholine stimulate cell cycle entry in vascular smooth muscle cells. Evidence for release of fibroblast growth factor-2. J. Biol. Chem. 271, 17791–17797. doi: 10.1074/jbc.271.30.17791
Cuenda, A., Rouse, J., Doza, Y. N., Meier, R., Cohen, P., Gallagher, T. F., et al. (1995). SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 364, 229–233. doi: 10.1016/0014-5793(95)00357-F
de Winther, M. P., van Dijk, K. W., Havekes, L. M., and Hofker, M. H. (2000). Macrophage scavenger receptor class A: a multifunctional receptor in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 20, 290–297. doi: 10.1161/01.ATV.20.2.290
Dunzendorfer, S., Lee, H. K., Soldau, K., and Tobias, P. S. (2004). TLR4 is the signaling but not the lipopolysaccharide uptake receptor. J. Immunol. 173, 1166–1170. doi: 10.4049/jimmunol.173.2.1166
Engelmann, M. G., Redl, C. V., and Nikol, S. (2004). Recurrent perivascular inflammation induced by lipopolysaccharide (endotoxin) results in the formation of atheromatous lesions in vivo. Lab. Invest. 84, 425–432. doi: 10.1038/labinvest.3700065
Favata, M. F., Horiuchi, K. Y., Manos, E. J., Daulerio, A. J., Stradley, D. A., Feeser, W. S., et al. (1998). Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273, 18623–18632. doi: 10.1074/jbc.273.29.18623
Fei, H., Berliner, J. A., Parhami, F., and Drake, T. A. (1993). Regulation of endothelial cell tissue factor expression by minimally oxidized LDL and lipopolysaccharide. Arterioscler. Thromb. 13, 1711–1717. doi: 10.1161/01.ATV.13.11.1711
Gitlin, J. M., and Loftin, C. D. (2009). Cyclooxygenase-2 inhibition increases lipopolysaccharide-induced atherosclerosis in mice. Cardiovasc. Res. 81, 400–407. doi: 10.1093/cvr/cvn286
Gupta, S., Barrett, T., Whitmarsh, A. J., Cavanagh, J., Sluss, H. K., Dérijard, B., et al. (1996). Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15, 2760–2770.
He, M., Zhang, W., Dong, Y., Wang, L., Fang, T., Tang, W., et al. (2017). Pro-inflammation NF-kappaB signaling triggers a positive feedback via enhancing cholesterol accumulation in liver cancer cells. J. Exp. Clin. Cancer Res. 36:15. doi: 10.1186/s13046-017-0490-8
Hermansson, C., Lundqvist, A., Magnusson, L. U., Ullström, C., Bergstrom, G., and Hultén, L. M. (2014). Macrophage CD14 expression in human carotid plaques is associated with complicated lesions, correlates with thrombosis, and is reduced by angiotensin receptor blocker treatment. Int. Immunopharmacol. 22, 318–323. doi: 10.1016/j.intimp.2014.07.009
Howell, K. W., Meng, X., Fullerton, D. A., Jin, C., Reece, T. B., and Cleveland, J. C. Jr. (2011). Toll-like receptor 4 mediates oxidized LDL-induced macrophage differentiation to foam cells. J. Surg. Res. 171, e27–e31. doi: 10.1016/j.jss.2011.06.033
Hu, H., Pierce, G. N., and Zhong, G. (1999). The atherogenic effects of chlamydia are dependent on serum cholesterol and specific to Chlamydia pneumoniae. J. Clin. Invest. 103, 747–753. doi: 10.1172/JCI4582
Iwahashi, H., Takeshita, A., and Hanazawa, S. (2000). Prostaglandin E2 stimulates AP-1-mediated CD14 expression in mouse macrophages via cyclic AMP-dependent protein kinase A. J. Immunol. 164, 5403–5408. doi: 10.4049/jimmunol.164.10.5403
Jatta, K., Wågsäter, D., Norgren, L., Stenberg, B., and Sirsjö, A. (2005). Lipopolysaccharide-induced cytokine and chemokine expression in human carotid lesions. J. Vasc. Res. 42, 266–271. doi: 10.1159/000085721
Karin, M. (1995). The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270, 16483–16486. doi: 10.1074/jbc.270.28.16483
Kiechl, S., Egger, G., Mayr, M., Wiedermann, C. J., Bonora, E., Oberhollenzer, F., et al. (2001). Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation 103, 1064–1070. doi: 10.1161/01.CIR.103.8.1064
Kzhyshkowska, J., Neyen, C., and Gordon, S. (2012). Role of macrophage scavenger receptors in atherosclerosis. Immunobiology 217, 492–502. doi: 10.1016/j.imbio.2012.02.015
Lehr, H. A., Sagban, T. A., Ihling, C., Zähringer, U., Hungerer, K. D., Blumrich, M., et al. (2001). Immunopathogenesis of atherosclerosis: endotoxin accelerates atherosclerosis in rabbits on hypercholesterolemic diet. Circulation 104, 914–920. doi: 10.1161/hc3401.093153
Levy, E., Xanthou, G., Petrakou, E., Zacharioudaki, V., Tsatsanis, C., Fotopoulos, S., et al. (2009). Distinct roles of TLR4 and CD14 in LPS-induced inflammatory responses of neonates. Pediatr. Res. 66, 179–184. doi: 10.1203/PDR.0b013e3181a9f41b
Li, L., Messas, E., Batista, E. L. Jr., Levine, R. A., and Amar, S. (2002). Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation 105, 861–867. doi: 10.1161/hc0702.104178
Martin, J. H., Mohit, A. A., and Miller, C. A. (1996). Developmental expression in the mouse nervous system of the p493F12 SAP kinase. Brain Res. Mol. Brain Res. 35, 47–57. doi: 10.1016/0169-328X(95)00181-Q
Mehta, J. L., Saldeen, T. G., and Rand, K. (1998). Interactive role of infection, inflammation and traditional risk factors in atherosclerosis and coronary artery disease. J. Am. Coll. Cardiol. 31, 1217–1225. doi: 10.1016/S0735-1097(98)00093-X
Mietus-Snyder, M., Glass, C. K., and Pitas, R. E. (1998). Transcriptional activation of scavenger receptor expression in human smooth muscle cells requires AP-1/c-Jun and C/EBPbeta: both AP-1 binding and JNK activation are induced by phorbol esters and oxidative stress. Arterioscler. Thromb. Vasc. Biol. 18, 1440–1449. doi: 10.1161/01.ATV.18.9.1440
Morishita, M., Ariyoshi, W., Okinaga, T., Usui, M., Nakashima, K., and Nishihara, T. (2013). A. Actinomycetemcomitans LPS enhances foam cell formation induced by LDL. J. Dent. Res. 92, 241–246. doi: 10.1177/0022034512473309
Muslin, A. J. (2008). MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin. Sci. 115, 203–218. doi: 10.1042/CS20070430
Nakagomi, A., Freedman, S. B., and Geczy, C. L. (2000). Interferon-gamma and lipopolysaccharide potentiate monocyte tissue factor induction by C-reactive protein: relationship with age, sex, and hormone replacement treatment. Circulation 101, 1785–1791. doi: 10.1161/01.CIR.101.15.1785
Navab, M., Chattopadhyay, A., Hough, G., Meriwether, D., Fogelman, S. I., Wagner, A. C., et al. (2015). Source and role of intestinally derived lysophosphatidic acid in dyslipidemia and atherosclerosis. J. Lipid Res. 56, 871–887. doi: 10.1194/jlr.M056614
Olivas, E., Chen, B. B., and Walker, W. S. (1995). Use of the pannell-milstein roller bottle apparatus to produce high concentrations of the CSF-1, the mouse macrophage growth factor. J. Immunol. Methods 182, 73–79. doi: 10.1016/0022-1759(95)00024-5
Ostos, M. A., Recalde, D., Zakin, M. M., and Scott-Algara, D. (2002). Implication of natural killer T cells in atherosclerosis development during a LPS-induced chronic inflammation. FEBS Lett. 519, 23–29. doi: 10.1016/S0014-5793(02)02692-3
Park, Y. M., Kashyap, S. R., Major, J. A., and Silverstein, R. L. (2012). Insulin promotes macrophage foam cell formation: potential implications in diabetes-related atherosclerosis. Lab. Invest. 92, 1171–1180. doi: 10.1038/labinvest.2012.74
Rahaman, S. O., Lennon, D. J., Febbraio, M., Podrez, E. A., Hazen, S. L., and Silverstein, R. L. (2006). A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 4, 211–221. doi: 10.1016/j.cmet.2006.06.007
Siess, W., Zangl, K. J., Essler, M., Bauer, M., Brandl, R., Corrinth, C., et al. (1999). Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. U.S.A. 96, 6931–6936. doi: 10.1073/pnas.96.12.6931
Sklar, M. D., Tereba, A., Chen, B. D., and Walker, W. S. (1985). Transformation of mouse bone marrow cells by transfection with a human oncogene related to c-myc is associated with the endogenous production of macrophage colony stimulating factor 1. J. Cell. Physiol. 125, 403–412. doi: 10.1002/jcp.1041250307
Steinberg, D., Parthasarathy, S., Carew, T. E., Khoo, J. C., and Witztum, J. L. (1989). Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 320, 915–924.
Tokumura, A., Majima, E., Kariya, Y., Tominaga, K., Kogure, K., Yasuda, K., et al. (2002). Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 277, 39436–39442. doi: 10.1074/jbc.M205623200
Triantafilou, M., and Triantafilou, K. (2002). Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol. 23, 301–304. doi: 10.1016/S1471-4906(02)02233-0
Vink, A., Schoneveld, A. H., van der Meer, J. J., van Middelaar, B. J., Sluijter, J. P., Smeets, M. B., et al. (2002). In vivo evidence for a role of toll-like receptor 4 in the development of intimal lesions. Circulation 106, 1985–1990. doi: 10.1161/01.CIR.0000032146.75113.EE
Wang, J., An, F. S., Zhang, W., Gong, L., Wei, S. J., Qin, W. D., et al. (2011). Inhibition of c-Jun N-terminal kinase attenuates low shear stress-induced atherogenesis in apolipoprotein E-deficient mice. Mol. Med. 17, 990–999. doi: 10.2119/molmed.2011.00073
Westerterp, M., Berbée, J. F., Pires, N. M., van Mierlo, G. J., Kleemann, R., Romijn, J. A., et al. (2007). Apolipoprotein C-I is cruciallyinvolved in lipopolysaccharide-induced atherosclerosis development in apolipoprotein E-knockout mice. Circulation 116, 2173–2181. doi: 10.1161/CIRCULATIONAHA.107.693382
Wright, S. D., Ramos, R. A., Tobias, P. S., Ulevitch, R. J., and Mathison, J. C. (1990). CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249, 1431–1433. doi: 10.1126/science.1698311
Xu, S., Huang, Y., Xie, Y., Lan, T., Le, K., Chen, J., et al. (2010). Evaluation of foam cell formation in cultured macrophages: an improved method with Oil Red O staining and DiI-oxLDL uptake. Cytotechnology 62, 473–481. doi: 10.1007/s10616-010-9290-0
Keywords: vascular biology, macrophage, lipopolysaccharide, signal transduction, CD14
Citation: An D, Hao F, Hu C, Kong W, Xu X and Cui M-Z (2018) JNK1 Mediates Lipopolysaccharide-Induced CD14 and SR-AI Expression and Macrophage Foam Cell Formation. Front. Physiol. 8:1075. doi: 10.3389/fphys.2017.01075
Received: 01 November 2017; Accepted: 06 December 2017;
Published: 05 January 2018.
Edited by:
Zhongkui Hong, University of South Dakota, United StatesReviewed by:
Shiyou Chen, University of Georgia, United StatesXiao-feng Yang, Temple University, United States
Copyright © 2018 An, Hao, Hu, Kong, Xu and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mei-Zhen Cui, cuim@utk.edu