 Esther López1†L. Gómez-Gordo2†
Esther López1†L. Gómez-Gordo2† Carlos Cantonero1Nuria Bermejo3
Carlos Cantonero1Nuria Bermejo3 Jorge Pérez-Gómez4María P. Granados5
Jorge Pérez-Gómez4María P. Granados5 Gines M. Salido6
Gines M. Salido6 Juan A. Rosado Dionisio1
Juan A. Rosado Dionisio1 Pedro C. Redondo Liberal1*
Pedro C. Redondo Liberal1*- 1Department of Physiology (PHYCELL) of the Veterinary Faculty, University of Extremadura, Cáceres, Spain
- 2Department of Animal Medicine, Veterinary Faculty University of Extremadura, Cáceres, Spain
- 3Hematology Unit, San Pedro de Alcantara Hospital, Cáceres, Spain
- 4Faculty of Sport Sciences, University of Extremadura, Cáceres, Spain
- 5Aldea Moret Health Center, Extremadura Health Service, Cáceres, Spain
- 6Institute of Molecular Pathology Biomarkers, Cáceres, Spain
Stanniocalcin 2 (STC2) is a fish protein that controls body Ca2+ and phosphate metabolism. STC2 has also been described in mammals, and as platelet function highly depends on both extracellular and intracellular Ca2+, we have explored its expression and function in these cells. STC2−/− mice exhibit shorter tail bleeding time than WT mice. Platelets from STC2-deficient mice showed enhanced aggregation, as well as enhanced Ca2+ mobilization in response to the physiological agonist thrombin (Thr) and the diacylglycerol analog, OAG, a selective activator of the non-capacitative Ca2+ entry channels. Interestingly, platelets from STC2−/− mice exhibit attenuated interaction between STIM1 and Orai1 in response to Thr, thus suggesting that STC2 is required for Thr-evoked STIM1-Orai1 interaction and the subsequent store-operated Ca2+ entry (SOCE). We have further assessed possible changes in the expression of the most relevant channels involved in non-capacitative Ca2+ entry in platelets. Then, protein expression of Orai3, TRPC3 and TRPC6 were evaluated by Western blotting, and the results revealed that while the expression of Orai3 was enhanced in the STC2-deficient mice, others like TRPC3 and TRPC6 remains almost unaltered. Summarizing, our results provide for the first time evidence for a role of STC2 in platelet physiology through the regulation of agonist-induced Ca2+ entry, which might be mediated by the regulation of Orai3 channel expression.
Introduction
Platelets are anucleated cells that play a relevant role in haemostasis. Attenuation of platelet function causes spontaneous browsing and bleeding. In contrast, hyperaggregability might lead to both macrothrombosis (observed in ictus or stroke) and microthrombosis (as detected in certain forms of retinopathy and nephropathy), as well as other cardiovascular disorders (Gue and Gorog, 2017; Malerba et al., 2017; Mancuso and Santagostino, 2017; Scherlinger et al., 2017). Hence, understanding the mechanisms underlying platelet function would be of relevance to prevent platelet-related complications linked to certain diseases. It is well known that platelet function strongly depends on changes in the intracellular calcium concentration ([Ca2+]c). Agonist might increase [Ca2+]c either by releasing Ca2+ from the intracellular stores or inducing Ca2+ entry from the extracellular medium. Regarding Ca2+ entry, store-operated calcium entry (SOCE) is a major mechanism activated by receptor occupation (Redondo and Rosado, 2015; Berna-Erro et al., 2016). With respect to SOCE activation, it is well known that a reduction in the endoplasmic reticulum Ca2+ content is detected by STIM1, which undergoes a conformational change and translocation to endoplasmic reticulum/plasma membrane (ER/PM) junctions in order to communicate with and activates Orai1 (Schindl et al., 2009; Scrimgeour et al., 2014; Shim et al., 2015). Since new regulators of the core components conforming the structure of SOCE channels has been described, more research on this topic is required for fully understanding the mechanism that controls Ca2+ entry from the extracellular medium. Orai1 is the pore-forming subunit of the highly Ca2+ selective calcium release-activated calcium (CRAC) channels (Prakriya et al., 2006). Additionally, STIM1 also activates a non-selective current mediated by store-operated calcium (SOC) channels, which consist on Orai1 and TRPC1 subunits (Desai et al., 2015). SOCE has been found to be modulated by a number of regulatory proteins that fine-tune Ca2+ influx in order to generate appropriate Ca2+ signals, and can be regulated by direct protein interaction (like the cytosolic Ca2+ sensor, calmodulin; Galan et al., 2011; Li et al., 2017).
In addition to SOCE, non-capacitative Ca2+ entry has also been reported in platelets, where TRPC3, TRPC6 and Orai3 have been found to play an important role (Berna-Erro et al., 2012; Harper et al., 2013). Concerning Orai3, this channel is able to form store independent channels with Orai1 regulated by arachidonate, the arachidonic acid-regulated calcium-selective (Mignen et al., 2008; Demuro et al., 2011; Zhang et al., 2014; Albarran et al., 2016) as well as the supposedly homomeric channels regulated by 2-aminoethoxydiphenyl borate (Demuro et al., 2011; Amcheslavsky et al., 2014). TRPC6 has been found to be involved in both SOCE and non-capacitive Ca2+ entry in human platelets and TRPC3 was reported to participate in non-capacitative Ca2+ entry in human and murine platelets (Zbidi et al., 2009; Harper et al., 2013).
Stanniocalcin 2 (STC2) was first identified as a fish protein involved in calcium and phosphate homeostasis (Wagner et al., 1998a,b). In mammals, indirect evidence suggest that STC2 has a relevant regulatory role on phosphate physiology. Contrary, its regulatory function in Ca2+ is limited to the intracellular environments, as evidenced in bone tissue (Ishibashi and Imai, 2002). In addition, STC2 is overexpressed by cancer cells (Na et al., 2015; Wang et al., 2015). Mammalian STC2 has been reported to play a protective role against oxidative stress or hypoxia (Law and Wong, 2010; Kim et al., 2015). Both stressing conditions evoke the accumulation of unfolded proteins in the ER leading to the activation of the unfolded protein response (UPR) to which STC2 belongs (Ito et al., 2004). Ca2+ mobilizing agents, such as thapsigargin (TG) and Ca2+ ionophores, have been found to activate UPR in culture cells (Ito et al., 2004), thus promoting the transcription of a variety of genes, including NUCB2, ATP2A2, CGRP2, and STC2 (Ito et al., 2004). In mouse embryonic fibroblasts STC2 overexpression results in reduced SOCE probably mediated by interaction with STIM1 (Zeiger et al., 2011), which strongly suggests a possible role for STC2 in the regulation of SOCE. We have recently reported that STC2 is expressed in human and mouse platelets (Lopez et al., 2017). Here we provide for the first time evidence for a role of STC2 in the regulation of Ca2+ entry and aggregation in mouse platelets. Our results indicate that in STC2-deficient mice thrombin-stimulated platelet aggregation and Ca2+ mobilization is enhanced, which might be associated to the overexpression of Orai3 channels.
Materials and Methods
Materials
Fura-2/AM was from Invitrogen Molecular probes (California, USA). Mouse anti-STIM1 [Clone 44 Clone 44–GOK (RUO)] antibody was from BD Biosciences® (Madrid, Spain). Rabbit polyclonal anti-TRPC1 (against peptide QLYDKGYTSKEQKDC) antibody was supplied by OriGene® (Rockville (MD) USA). Anti-TRPC6 extracellular antibody was from Alomone (Jerusalem, Israel). Anti-TRPC3 was from Abcam (Cambridge, UK). Mouse and rabbit HRP-conjugated secondary antibodies were from Jackson Immunoresearch® (West Grove, PA, USA). Rabbit polyclonal anti-Orai1 (against sequence amino acids 288–301) antibody, anti-Orai3 antibody, anti-actin antibody, thrombin (Thr), ADP, thapsigargin (TG), 1-oleoyl-2-acetyl-sn-glycerol (OAG), apyrase, aspirin, as well as other reagents of analytic grade were purchased from Sigma-Aldrich® (Madrid, Spain).
Animals
C57BL/6J WT mice were provided by Harlam®, while C57BL/6J STC2−/− mice were kindly provided by Dr. Roger Reddel from the Children's Medical Research Institute (CMRI) (Australia) (Chang et al., 2008). Animals were breed under controlled environmental conditions (25°C), which consisted on a regular 12 h light-dark photoperiod and free access to drink and food. The two mice conolies were bred separately, and after two or three generations, animals were backcrossed in order to minimize genetic drift. STC2 deficiency was regularly assessed by PCR and Western blotting. Mice of age range between 8 and 10 weeks were bled three times under isoflurane anesthesia, leaving a resting period of a week between each extraction.
All procedures were performed in agreement with the Helsinki Declaration, the guidelines from the Council of Europe N° 123 (Strasbourg, 1985) and have been approved by the Local Ethical Committee.
Tail Bleeding
Anesthetized animals were subjected to surgical sectioning of the tip of the tail (sections of 1–4 mm were cut off, approximately), and a whatman filter paper was imbibed with the blood at regularly periods of 30 s until the bleeding stopped (Vermeersch et al., 2017). In case of excessive bleeding, lasting more than 25 min, the experiments were artificially interrupted by cauterizing the injury. Average of the bleeding time was considered in these experiments, and the possible differences between WT and STC2-deficient mice were analyzed.
Blood Extraction Procedures and Platelet Isolation
Isoflurane anesthetized mice were bled from the retroorbital plexus (Berna-Erro et al., 2014), and the blood was mixed with 300 μL of acid citrate dextrose buffer containing: 85 mM sodium citrate, 78 mM citric acid, 111 mM glucose, pH 7.3. Blood was centrifuged at 300 × g for 5 min, and upon collected the supernatants, they were centrifuged again at 100 × g for 5 min in order to obtain the platelet-rich plasma (PRP). PRP was supplemented with aspirin (100 μM) and apyrase (40 U/mL) (except those used for aggregation, to which only apyrase was added), and PRP was centrifuged again at 600 × g for 5 min. The pellet was suspended in Ca2+-free Tyrode's buffer containing: NaCl 137 mM, KCl 2.7 mM, NaHCO3 12 mM, NaH2PO4 0.43 mM, Glucose 0.1%, HEPES 5 mM, BSA 0.35%, MgCl2 1 mM, pH 7.13 and supplemented with apyrase (40 μg/mL).
Platelet Aggregation
Platelet aggregation was monitored using a Cronologh® aggregometer according to the protocols described elsewhere (Lopez et al., 2013a). Briefly, isolated platelets were suspended in fresh-Tyrode's buffer containing 40 U/mL of apyrase. At the moment of the experiment 1 mM CaCl2 was added and following aggregation was stimulated by using physiological agonists such as thrombin (Thr, 0.1 U/mL) and ADP (100 μM). Percentage of aggregation at two different time points (3 and 8 min after stimulation with Thr, and 4 and 10 min after treatment with ADP) as well as the percentage of the slope (in case of Thr) and percentage of delay time to initiate the irreversible phase of aggregation (in case of ADP) were used to compare platelet aggregability in both mice strains.
[Ca2+]c Determination
Platelets suspended in Tyrode's buffer were loaded with fura-2 by incubating for 30 min at room temperature with fura-2/AM (2 μM) (Lopez et al., 2013b; Berna-Erro et al., 2014). Fluorescence was recorded from aliquots of magnetically stirred platelet suspensions at 37°C using a fluorescence spectrophotometer (RF-5301PC, Shimadzu Corporation, Kyoto, Japan) with excitation wavelengths of 340 and 380 nm and emission at 505 nm. Changes in [Ca2+]c were monitored using the fura-2 340/380 fluorescence ratio and calibrated according to the method of Grynkiewicz and coworkers (Grynkiewicz et al., 1985). Thr-, OAG- and TG-evoked Ca2+ mobilization was estimated as the integral of the rise in [Ca2+]c above basal for 2½ min after the addition of the stimuli, taking a sample every second and expressed as nM•s (Lopez et al., 2005). To calculate the initial rate of Ca2+ elevation after the addition of TG to the medium, the traces were fitted to the equation y = A + KX, where K is the slope (Redondo et al., 2003).
Immunoprecipitation and Western Blotting
Murine platelets were either mixed with RIPA (pH 7.2, containing 474 mM NaCl, 30 mM Tris, 3 mM EGTA, 0.3% SDS, 3% sodium deoxycholate, 3% triton X-100, 3 mM Na3VO4 and protease inhibitors) or Laemmli's buffer as required (Alcaraz et al., 1990; Karlsson et al., 1994). Platelet samples in RIPA were immunoprecipitated by incubation with 2 μg/mL of anti-Orai1 and anti-STIM1, and protein A-agarose beads overnight at 4°C. The immunoprecipitates were resolved by 10% SDS/PAGE, and separated proteins were electrophoretically transferred onto nitrocellulose membranes using a semi-dry blotter (for 2 h at 0.8 mA/cm2; Hoefer Scientific, Newcastle under-Lyme, U.K.). Blots were incubated overnight with 10% (w/v) BSA in Tris-buffered saline with 0.1% Tween 20 (TBST) to block residual protein-binding sites. Detection of STIM1, Orai1, TRPC1, TRPC3, TRPC6, and Orai3 was achieved by incubation for 1 h with anti-STIM1 antibody diluted 1:1,000 in TBST, for 2 h with anti-Orai1, anti-TRPC1, or anti-TRPC6 antibody diluted 1:500 in TBST, and overnight with anti-TRPC3 or anti-Orai3 antibody diluted 1:1,000 and 1:250 in TBST, respectively. To detect the primary antibody, membranes were incubated with the appropriate secondary antibody (diluted either 1:5,000 or 1:10,000 in TBST for 1 h) washed six times in TBST, and exposed to enhanced chemiluminescence reagents for 1 min. Optical density of the bands was detected using the C-Digit System (Licor®, Spain), and differences between samples were analyzed using the Image J software (N.I.H. USA). Data are normalized using the amount of actin (for Western blotting) or the result obtained after reprobing the membranes with the immunoprecipitating antibody (for the immunoprecipitations).
Statistical Analysis
Analysis of statistical significance was performed using the Student's unpaired t-test or one-way analysis of variance (ANOVA) combined with the Dunnet's post-test. The significance level was P < 0.05.
Results
STC2-Deficient Mice Exhibit Shorter Bleeding Times
Wild type (WT) and STC2-deficient mice (KO) were subjected to tail bleeding as described in the Material and Methods section. As depicted in Figure 1, STC2−/− mice presented shorter mean bleeding time than WT mice (5:10 ± 1:31 vs. 12:28 ± 2:40 min:s, respectively; n = 10–14, P < 0,001). These findings suggest that STC2 might play a role in the regulation of haemostasia.
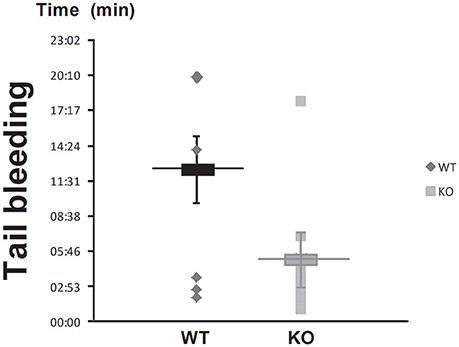
Figure 1. STC2 is involved in blood clotting. The tip of the tail of anesthetized STC2-deficient (KO) and WT mice was sectioned as described in Materials and Methods, and the bleeding time was determined. Box graph represents mean ± SEM of 10 to 14 mice.
STC2 Is Involved in Murine Platelet Aggregation
We next explored the possible role of STC2 in platelets aggregation. As depicted in Figure 2A, platelets from STC2-deficient mice exhibited enhanced rate and amplitude of aggregation in response to thrombin (Thr, 0.1 U/mL) compared to WT. The percentage of aggregation was 50% and 70% at 3 min, and 77% and 84% at 8 min in WT and STC2-deficient cells stimulated with Thr, respectively (P < 0,05); while the slope was 75 and 95% at 3 min and 63 and 71% at 8 min in platelets from WT and STC2-deficient mice, respectively. Similarly, platelets from STC2−/− mice exhibited a significant difference in the percentage of aggregation 4 min after the addition of the weak agonist, ADP (100 μM; Figure 2B); while 10 min after the addition of ADP a slightly greater percentage of aggregation was still evident in STC2-defficient mice compared to WT mice. As depicted in the aggregation traces of Figure 2B and the below histogram (delay time), the initial phase of irreversible aggregation occurred significantly earlier in platelets from STC2-deficient mice, while the reversible aggregation almost occurred simultaneously in both strains. These findings indicate a certain degree of hyperactivity and hyperaggregability in platelets from STC2-deficient mice, which is consistent with the reduced bleeding time reported in Figure 1.
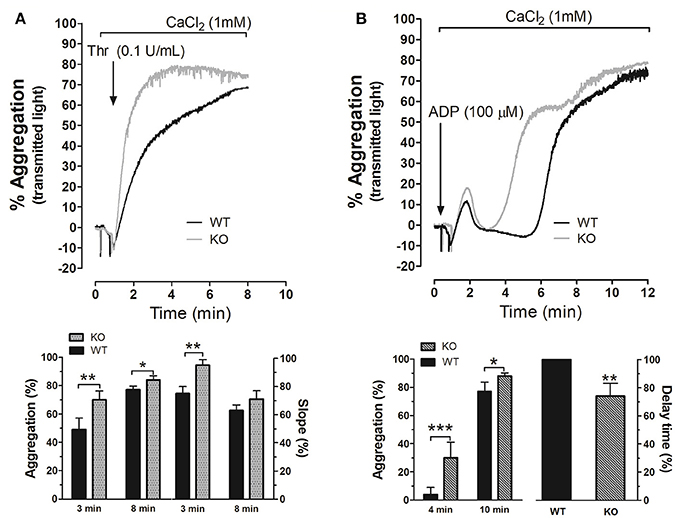
Figure 2. STC2 regulates platelet aggregation. Platelets drawn from WT (black traces) and STC2-deficient mice (gray traces) were isolated and suspended in Tyrode's buffer containing 1 mM Ca2+ and then stimulated with Thr (A) or ADP (B). Aggregation of mice platelets was induced at a shear rate of 1,200 r.p.m. at 37°C in an aggregometer as described in the Materials and Methods section. Traces shown are representative of 14 separate experiments. Histograms represent the percentage of aggregation and slope for Thr-induced platelet aggregation or the percentage of aggregation and delay time for ADP-evoked response expressed as mean ± SEM. Delay time was presented as percentage of the value obtained in platelets from WT mice. *,**,***: represent P < 0.05, <0.01, and <0.001, respectively as compared to the corresponding WT value.
Platelets From STC2−/− Mice Presented Altered Ca2+ Homeostasis
As previously described by others, platelet function is regulated by several mechanisms, including intracellular Ca2+ mobilization. Hence, we have analyzed agonist-induced Ca2+ mobilization in platelets from WT and STC2−/− mice. As shown in Figure 3A, in the presence of 1 mM extracellular CaCl2, the integral of Thr-evoked Ca2+ mobilization, estimated as described in Methods, in platelets from STC2-deficient mice was 308% greater than in platelets from WT mice (P < 0.001). Next, we have explored the mechanism underlying the exacerbated response to thrombin. First of all, we assessed Ca2+ release from the TG-sensitive stores using the SERCA inhibitor thapsigargin (TG), which induces passive Ca2+ efflux from the stores. The treatment of mice platelets with 1 μM TG in a Ca2+-free medium resulted in a sustained increase in [Ca2+]c due to Ca2+ release from the internal stores. As shown in Figure 3B, TG-induced response was 27% greater in STC2-deficient cells. Inhibition of SERCA by TG reveals passive Ca2+ efflux from the ER, an event that is influenced by both the Ca2+ gradient across the ER membrane, which provides the driving force for Ca2+ efflux, and also the ER membrane permeability to Ca2+. As the response to TG, estimated as the integral of the rise in [Ca2+]c over the resting level for 2½ min after the addition of TG, was found to be greater in STC2-deficient mice, thus suggesting that these cells have a greater ability to store Ca2+, we have further investigated the Ca2+ leakage rate in platelets from both strains. Our results indicate that slope of the rise in [Ca2+]c upon stimulation with TG was similar in platelets from WT and STC2-deficient mice (1.05 ± 0.27 and 1.34 ± 0.18 nM/s in platelets from WT and STC2-deficient mice, respectively, P > 0.05; n = 8), thus suggesting that the leakage rate is similar in both strains.
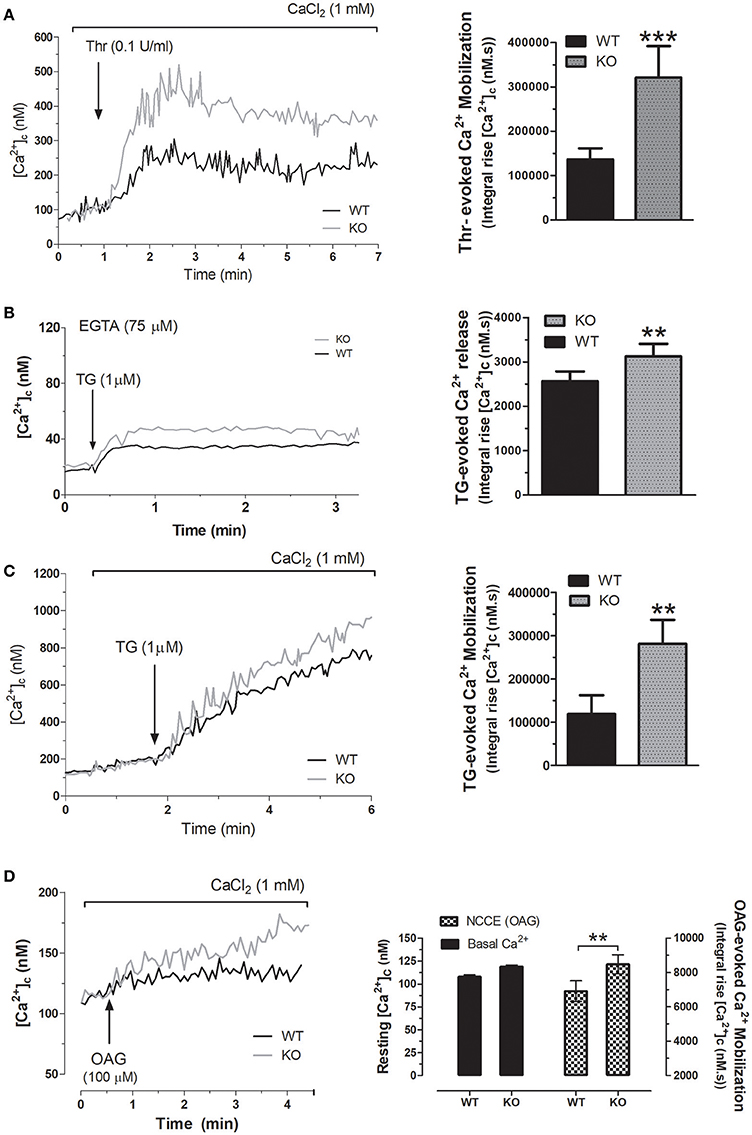
Figure 3. STC2 negatively regulates calcium entry. (A) Fura-2-loaded platelets from WT and STC2-deficient mice were suspended in a medium containing 1 mM Ca2+ (A, C, and D) or in a Ca2+-free medium (75 μM EGTA added; B). Cells were stimulated with Thr (A), TG (B,C) or OAG (D), as indicated. Calcium mobilization was determined as described in Material and Methods. Traces are representative of 8-10 independent experiments. Bar graphs represent agonist-evoked Ca2+ mobilization expressed as the mean ± SEM and presented as the integral of the rise in [Ca2+]c above basal for 2½ min after the addition of the stimuli as described in Material and Methods. **, ***, P < 0.01, and 0.001, respectively, as compared to the response in platelets from WT mice.
Thr-evoked Ca2+-entry in human platelets mainly occurs via two different pathways, SOCE and non-capacitative Ca2+ influx (Jardin et al., 2009). Hence, we have further explored the mechanism involved in the exacerbated Thr mobilization in platelets from STC2-deficient mice. SOCE was assessed by depletion of the intracellular calcium stores using TG in the presence of extracellular calcium. Our results indicate that TG-evoked Ca2+ entry was significantly greater in STC2-deficient cells (Figure 3C); however, the differences in SOCE in platelets from both strains does not completely explain the magnitude of the different Ca2+ entry evoked by Thr. Therefore, our findings indicate that, in addition to the negative regulatory role of STC2 in SOCE, previously documented by others and confirmed by us in mice platelets, the exacerbated Thr-evoked Ca2+ mobilization might involve the enhancement of non-capacitative Ca2+ entry. To assess this possibility, platelets were stimulated with the diacylglycerol analog, OAG, a selective activator of non-capacitative Ca2+ entry channels (Hassock et al., 2002; Berna-Erro et al., 2012). As depicted in Figure 3D, treatment of platelets from WT mice with OAG (100 μM) in a medium containing 1 mM CaCl2 resulted in a sustained increase in [Ca2+]c. As expected, platelets from the STC2-deficient mice exhibited a greater increase in [Ca2+]c in response to OAG; while we did not detect changes in the resting [Ca2+]c.
STC2 Modulates the Interaction Between STIM1 and Orai1
As mentioned above, SOCE requires physical interaction between the ER sensor STIM1 and the Ca2+ channel Orai1. Previous studies have revealed that STC2 regulates STIM1 expression and function, and subsequently, negatively modulates SOCE. Therefore, we have explored whether STC2 might be involved in the interaction between STIM1 and Orai1. As depicted in Figure 4, in platelets from STC2-deficient mice the interaction between STIM1 and Orai1 in response to Thr was significantly reduced. In addition, some authors have reported the existence of the interaction between STIM1 and TRPC1 in the absence of Orai1 in vascular smooth muscle cells. This mechanism was elicited by activation of PLCβ1, which requires changes in the [Ca2+]c (Shi et al., 2017). Previously, we have also demonstrated interaction between STIM1 and TRPC1 in human platelets (Lopez et al., 2006). In platelets from STC2-deficient mice we have detected a 10% increase in the interaction between STIM1 and TRPC1 over the resting level after stimulation with Thr; therefore, a detectable interaction between STIM1 and TRPC1 takes place in platelets from STC2-deficient mice (see right hand side image of Figure 4). Furthermore, no differences were detected in the expression of the components of the SOCE mechanism in the platelet of STC2-deficient mice compared to those of WT. Therefore, Thr-evoked Ca2+ entry might involve STIM1-TRPC1 interaction in platelets from STC2-deficient mice.
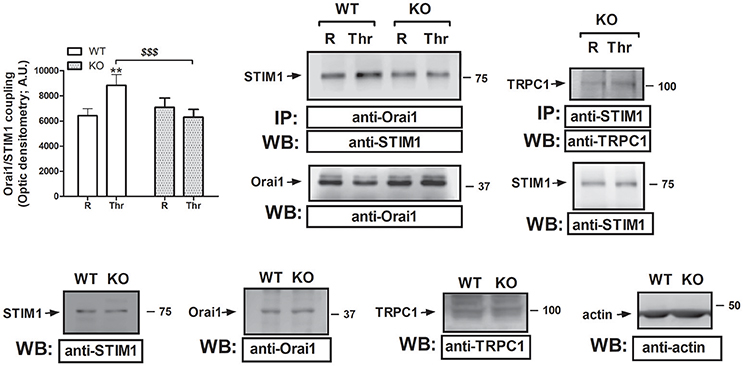
Figure 4. STC2 silencing impairs STIM1/Orai1 interaction. Murine platelets drawn from STC2-deficient (KO) and WT mice were either left untreated (R) or stimulated for 1 min with Thr (0.1 U/mL), and then lysed. Samples were then subjected to immunoprecipation using an anti-Orai1 or anti-STIM1 antibody, and subsequent Western blotting was performed using anti-STIM1 and anti-TRPC1 antibodies, as indicated. Membranes were reprobed with the immunoprecipitating antibody for protein loading control. Bottom panels, Platelet lysates from WT and STC2-deficient (KO) mice were subjected to Western blotting with anti-STIM1, anti-Orai1 or anti-TRPC1 antibody, as indicated. Membranes were reprobed with an anti-actin antibody as loading control. Blots are representative of 8-12 separate experiments. Bar graph represents Orai1-STIM1 coupling expressed as the mean ± SEM of the optical density in a.u. **P < 0.01 as compared to the response in resting platelets. $$$P < 0.001 as compared to Thr-induced Orai1-STIM1 association in WT mice.
STC2 Modulates the Expression of Orai3 in Mouse Platelets
In an attempt to assess the mechanism underlying the increase in non-capacitative Ca2+ entry in platelets from STC2-deficient mice, we have analyzed the expression of the non-capacitative channels at the protein level in the platelets of both strains. Previous studies have reported that TRPC3, TRPC6 and Orai3 are involved in non-capacitative entry (Berna-Erro et al., 2012; Harper et al., 2013). Hence, we have examined the expression of these channels in platelet lysates from WT and STC2-deficient mice by Western blotting using specific antibodies. As depicted in Figure 5, Western blotting of platelet lysates with anti-TRPC3 or anti-TRPC6 antibodies revealed that the expression of these proteins is not altered in the platelets of STC2-deficient mice with respect to WT. Interestingly, our results indicate that Orai3 expression is enhanced by 75% in platelets from STC2−/− mice as detected by Western blotting with the anti-Orai3 antibody. This finding strongly suggests that STC2 modulates Orai3 expression in mice platelets. Furthermore, this observation might provide a mechanism for the enhanced non-capacitative Ca2+ influx in platelets from STC2-deficient mice.
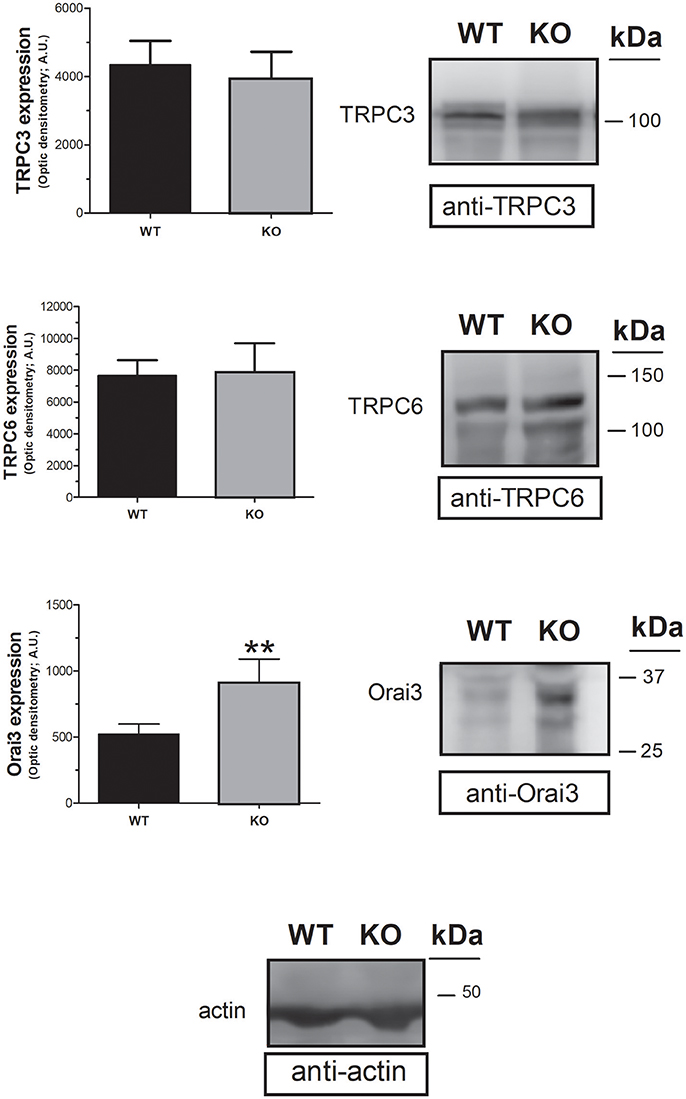
Figure 5. STC2 silencing upregulates Orai3 expression. Platelet lysates from WT and STC2-deficient (KO) mice were subjected to Western blotting with anti-TRPC3, anti-TRPC6 or anti-Orai3 antibody, and reprobed with anti-actin antibody, as indicated. Blots are representative of 6 separate experiments. Bar graph represents TRPC3, TRPC6 or Orai3 expression as the mean ± SEM of the optical density in a.u. **P < 0.01 as compared to the expression in platelets from WT mice.
Discussion
Here we provide for the first time evidence supporting a role for STC2 in platelet aggregation and haemostasis. Using the murine model lacking STC2, we have found that these mice exhibited enhanced platelet response to strong and weak agonists, such as thrombin and ADP, respectively, and subsequently reduced bleeding time. Our results indicate that STC2 might modulate platelet aggregation through the regulation of Ca2+ homeostasis in these cells, as it was previously reported in other mammal cells (Zeiger et al., 2011). We have found that platelets from STC2-deficient mice exhibit a greater ability to store Ca2+ in the ER, although the leakage rate across the ER membrane is not significantly enhanced. Furthermore, our results indicate that STC2 modulate agonist-induced Ca2+ entry, so that the deficiency of STC2 results in exacerbated Ca2+ signals and, most likely, platelet hyperaggregability.
Thrombin evokes Ca2+ influx in platelets via SOCE and non-capacitative Ca2+ entry (Jardin et al., 2009). Our results indicate that STC2 plays a role supporting stimulated STIM1/Orai1 interaction, as Thr was unable to enhance STIM1/Orai1 association in platelets from STC2 KO mice. Although we have found a detectable increase in the STIM1-TRPC1 interaction upon stimulation with Thr, our findings indicate that it is unlikely that SOCE is enhanced, and, therefore, the enhanced Ca2+ influx induced by Thr in platelets from STC2-deficient mice is more likely mediated by enhanced non-capacitative Ca2+ entry. Our findings indicate that STC2 deficiency results in an impaired STIM1/Orai1 interaction, but also we have found that TG-induced Ca2+ entry in platelets from STC2 KO mice is significantly enhanced. This apparent discrepancy might be explained by the fact that TG has been found to activate non-capacitative Ca2+ entry in platelets through the release of autocrine factors (Harper et al., 2009); therefore, as for thrombin, the enhanced Ca2+ entry induced by TG in platelets from STC2-deficient mice might involve reduced SOCE and enhanced non-capacitative Ca2+ entry. In support of this hypothesis, we have found that non-capacitative Ca2+ entry evoked by OAG, a diacylglycerol analog, is enhanced in STC2-deficient mice, which provides evidence for a role of STC2 in the regulation of non-capacitative Ca2+ influx in these cells. In murine platelets, two main TRPC channels have been found to be activated in response to OAG, TRPC3 and TRPC6 (Harper et al., 2013). However, we have not detected significant changes in the protein expression of TRPC3 or TRPC6 channels in platelet from STC2-deficient mice. Orai3 has also been found to be involved in non-capactitative Ca2+ entry (Berna-Erro et al., 2012), and we have found that Orai3 expression is significantly enhanced in platelets from STC2-deficient mice. Although we have not directly addressed the involvement of Orai3 in Thr-induced non-capacitative Ca2+ entry, these findings suggest a possible mechanism for the enhanced activation of non-capacitative Ca2+ entry in these cells. The enhanced Orai3 expression in platelets from STC2-deficient mice is unlikely to be responsible for the enhanced Ca2+ release in response to TG, as a previous study has revealed that Orai3 is not involved in the TG-revealed ER Ca2+ leak (Leon-Aparicio et al., 2017).
We have recently suggested that STC2 might be involved in the pathophysiology of type 2 diabetes mellitus, where STC2 is downregulated (Lopez et al., 2017). We have previously reported that platelets from patients with type 2 diabetes mellitus exhibit both hyperactivity and hyperaggregability (Saavedra et al., 2004; Redondo et al., 2005; Lopez et al., 2013b). In the type 2 diabetic patients we found that non-capacitative Ca2+ entry is enhanced, while SOCE might be attenuated (Jardin et al., 2009, 2011). These findings resemble our current observations. Thus, our present results might provide an explanation to the platelet complications in diabetic patients as a reduction in STC2 expression might lead to platelet hyperaggregability due to enhanced Orai3 expression, although at present we cannot discard other possible mechanisms for the increase in non-capacitative Ca2+ entry in these cells. Summarizing, we provide for the first time evidence for a role of STC2 in platelet aggregation through the modulation of Ca2+ homeostasis.
Author Contributions
EL and LG-G performed most of the experiments with the same grade of responsibility. JP-G, CC, NB, and MG provided us with the mice blood samples, as well as they were in charge of the statistical analysis of data. GS performed a critical review of the manuscript, while JR and PR were in charge of conceptual and protocol design of the manuscript and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been supported by MINECO (BFU2013-45564C2-1-P and BFU2016-74932-C2-1-P) and Junta de Extremadura-FEDER (IB16046 and GR15029). We thank to Dr. Reddel for providing the STC2-deficient mice. CC is supported by Junta de Extremadura-FEDER.
References
Albarran, L., Lopez, J. J., Woodard, G. E., Salido, G. M., and Rosado, J. A. (2016). Store-operated Ca2+ Entry-Associated Regulatory factor (SARAF) plays an important role in the regulation of Arachidonate-Regulated Ca2+ (ARC) Channels. J. Biol. Chem. 291, 6982–6988. doi: 10.1074/jbc.M115.704940
Alcaraz, C., De Diego, M., Pastor, M. J., and Escribano, J. M. (1990). Comparison of a radioimmunoprecipitation assay to immunoblotting and ELISA for detection of antibody to African swine fever virus. J. Vet. Diagn. Invest. 2, 191–196. doi: 10.1177/104063879000200307
Amcheslavsky, A., Safrina, O., and Cahalan, M. D. (2014). State-dependent block of Orai3 TM1 and TM3 cysteine mutants: insights into 2-APB activation. J. Gen. Physiol. 143, 621–631. doi: 10.1085/jgp.201411171
Berna-Erro, A., Albarran, L., Dionisio, N., Redondo, P. C., Alonso, N., Gomez, L. J., et al. (2014). The canonical transient receptor potential 6 (TRPC6) channel is sensitive to extracellular pH in mouse platelets. Blood Cells Mol. Dis. 52, 108–115. doi: 10.1016/j.bcmd.2013.08.007
Berna-Erro, A., Galan, C., Dionisio, N., Gomez, L. J., Salido, G. M., and Rosado, J. A. (2012). Capacitative and non-capacitative signaling complexes in human platelets. Biochim. Biophys. Acta 1823, 1242–1251. doi: 10.1016/j.bbamcr.2012.05.023
Berna-Erro, A., Jardín, I., Smani, T., and Rosado, J. A. (2016). Regulation of platelet function by orai, STIM and TRP. Adv. Exp. Med. Biol. 898, 157–181. doi: 10.1007/978-3-319-26974-0_8
Chang, A. C., Hook, J., Lemckert, F. A., Mcdonald, M. M., Nguyen, M. A., Hardeman, E. C., et al. (2008). The murine stanniocalcin 2 gene is a negative regulator of postnatal growth. Endocrinology 149, 2403–2410. doi: 10.1210/en.2007-1219
Demuro, A., Penna, A., Safrina, O., Yeromin, A. V., Amcheslavsky, A., Cahalan, M. D., et al. (2011). Subunit stoichiometry of human Orai1 and Orai3 channels in closed and open states. Proc. Natl. Acad. Sci. U.S.A. 108, 17832–17837. doi: 10.1073/pnas.1114814108
Desai, P. N., Zhang, X., Wu, S., Janoshazi, A., Bolimuntha, S., Putney, J. W., et al. (2015). Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci. Signal 8, ra74. doi: 10.1126/scisignal.aaa8323
Galan, C., Dionisio, N., Smani, T., Salido, G. M., and Rosado, J. A. (2011). The cytoskeleton plays a modulatory role in the association between STIM1 and the Ca2+ channel subunits Orai1 and TRPC1. Biochem. Pharmacol. 82, 400–410. doi: 10.1016/j.bcp.2011.05.017
Grynkiewicz, G., Poenie, M., and Tsien, R. Y. (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450.
Gue, Y. X., and Gorog, D. A. (2017). Importance of endogenous fibrinolysis in platelet thrombus formation. Int. J. Mol. Sci. 18:1850. doi: 10.3390/ijms18091850
Harper, A. G., Mason, M. J., and Sage, S. O. (2009). A key role for dense granule secretion in potentiation of the Ca2+ signal arising from store-operated calcium entry in human platelets. Cell Calcium 45, 413–420. doi: 10.1016/j.ceca.2009.02.003
Harper, M. T., Londoño, J. E., Quick, K., Londoño, J. C., Flockerzi, V., Philipp, S. E., et al. (2013). Transient receptor potential channels function as a coincidence signal detector mediating phosphatidylserine exposure. Sci Signal 6, ra50. doi: 10.1126/scisignal.2003701
Hassock, S. R., Zhu, M. X., Trost, C., Flockerzi, V., and Authi, K. S. (2002). Expression and role of TRPC proteins in human platelets: evidence that TRPC6 forms the store-independent calcium entry channel. Blood 100, 2801–2811. doi: 10.1182/blood-2002-03-0723
Ishibashi, K., and Imai, M. (2002). Prospect of a stanniocalcin endocrine/paracrine system in mammals. Am. J. Physiol. Renal Physiol. 282, F367–F375. doi: 10.1152/ajprenal.00364.2000
Ito, D., Walker, J. R., Thompson, C. S., Moroz, I., Lin, W., Veselits, M. L., et al. (2004). Characterization of stanniocalcin 2, a novel target of the mammalian unfolded protein response with cytoprotective properties. Mol. Cell. Biol. 24, 9456–9469. doi: 10.1128/MCB.24.21.9456-9469.2004
Jardin, I., Gómez, L. J., Salido, G. M., and Rosado, J. A. (2009). Dynamic interaction of hTRPC6 with the Orai1-STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca(2+) entry pathways. Biochem. J. 420, 267–276. doi: 10.1042/BJ20082179
Jardín, I., López, J. J., Zbidi, H., Bartegi, A., Salido, G. M., and Rosado, J. A. (2011). Attenuated store-operated divalent cation entry and association between STIM1, Orai1, hTRPC1 and hTRPC6 in platelets from type 2 diabetic patients. Blood Cells Mol. Dis. 46, 252–260. doi: 10.1016/j.bcmd.2010.12.008
Karlsson, J. O., Ostwald, K., Kåbjörn, C., and Andersson, M. (1994). A method for protein assay in Laemmli buffer. Anal. Biochem. 219, 144–146. doi: 10.1006/abio.1994.1243
Kim, P. H., Na, S. S., Lee, B., Kim, J. H., and Cho, J. Y. (2015). Stanniocalcin 2 enhances mesenchymal stem cell survival by suppressing oxidative stress. BMB Rep. 48, 702–707. doi: 10.5483/BMBRep.2015.48.12.158
Law, A. Y., and Wong, C. K. (2010). Stanniocalcin-2 is a HIF-1 target gene that promotes cell proliferation in hypoxia. Exp. Cell Res. 316, 466–476. doi: 10.1016/j.yexcr.2009.09.018
Leon-Aparicio, D., Pacheco, J., Chavez-Reyes, J., Galindo, J. M., Valdes, J., Vaca, L., et al. (2017). Orai3 channel is the 2-APB-induced endoplasmic reticulum calcium leak. Cell Calcium 65, 91–101. doi: 10.1016/j.ceca.2017.01.012
Li, X., Wu, G., Yang, Y., Fu, S., Liu, X., Kang, H., et al. (2017). Calmodulin dissociates the STIM1-Orai1 complex and STIM1 oligomers. Nat. Commun. 8, 1042. doi: 10.1038/s41467-017-01135-w
López, E., Berna-Erro, A., Bermejo, N., Brull, J. M., Martinez, R., Garcia Pino, G., et al. (2013a). Long-term mTOR inhibitors administration evokes altered calcium homeostasis and platelet dysfunction in kidney transplant patients. J. Cell. Mol. Med. 17, 636–647. doi: 10.1111/jcmm.12044
López, E., Berna-Erro, A., Hernández-Cruz, J. M., Salido, G. M., Redondo, P. C., and Rosado, J. A. (2013b). Immunophilins are involved in the altered platelet aggregation observed in patients with type 2 diabetes mellitus. Curr. Med. Chem. 20, 1912–1921. doi: 10.2174/0929867311320140008
Lopez, J. J., Camello-Almaraz, C., Pariente, J. A., Salido, G. M., and Rosado, J. A. (2005). Ca2+ accumulation into acidic organelles mediated by Ca2+- and vacuolar H+ ATPases in human platelets. Biochem. J. 390, 243–252. doi: 10.1042/BJ20050168
López, J. J., Jardín, I., Chamorro, C. C., Duran, M. L., Tarancón Rubio, M. J., Reyes Panadero, M., et al. (2017). Involvement of stanniocalcins in the deregulation of glycaemia in obese mice and type 2 diabetic patients. J. Cell. Mol. Med. 22, 684–694. doi: 10.1111/jcmm.13355
Lopez, J. J., Salido, G. M., Pariente, J. A., and Rosado, J. A. (2006). Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J. Biol. Chem. 281, 28254–28264. doi: 10.1074/jbc.M604272200
Malerba, M., Nardin, M., Radaeli, A., Montuschi, P., Carpagnano, G. E., and Clini, E. (2017). The potential role of endothelial dysfunction and platelet activation in the development of thrombotic risk in COPD patients. Expert Rev. Hematol. 10, 821–832. doi: 10.1080/17474086.2017.1353416
Mancuso, M. E., and Santagostino, E. (2017). Platelets: much more than bricks in a breached wall. Br. J. Haematol. 178, 209–219. doi: 10.1111/bjh.14653
Mignen, O., Thompson, J. L., and Shuttleworth, T. J. (2008). Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. J. Physiol. 586, 185–195. doi: 10.1113/jphysiol.2007.146258
Na, S. S., Aldonza, M. B., Sung, H. J., Kim, Y. I., Son, Y. S., Cho, S., et al. (2015). Stanniocalcin-2 (STC2): a potential lung cancer biomarker promotes lung cancer metastasis and progression. Biochim. Biophys. Acta 1854, 668–676. doi: 10.1016/j.bbapap.2014.11.002
Prakriya, M., Feske, S., Gwack, Y., Srikanth, S., Rao, A., and Hogan, P. G. (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. doi: 10.1038/nature05122
Redondo, P. C., Jardin, I., Hernandez-Cruz, J. M., Pariente, J. A., Salido, G. M., and Rosado, J. A. (2005). Hydrogen peroxide and peroxynitrite enhance Ca2+ mobilization and aggregation in platelets from type 2 diabetic patients. Biochem. Biophys. Res. Commun. 333, 794–802. doi: 10.1016/j.bbrc.2005.05.178
Redondo, P. C., Lajas, A. I., Salido, G. M., González, A., Rosado, J. A., and Pariente, J. A. (2003). Evidence for secretion-like coupling involving pp60src in the activation and maintenance of store-mediated Ca2+ entry in mouse pancreatic acinar cells. Biochem. J. 370, 255–263. doi: 10.1042/bj20021505
Redondo, P. C., and Rosado, J. A. (2015). Store-operated calcium entry: unveiling the calcium handling signalplex. Int. Rev. Cell Mol. Biol. 316, 183–226. doi: 10.1016/bs.ircmb.2015.01.007
Saavedra, F. R., Redondo, P. C., Hernandez-Cruz, J. M., Salido, G. M., Pariente, J. A., and Rosado, J. A. (2004). Store-operated Ca2+ entry and tyrosine kinase pp60src hyperactivity are modulated by hyperglycemia in platelets from patients with non insulin-dependent diabetes mellitus. Arch. Biochem. Biophys. 432, 261–268. doi: 10.1016/j.abb.2004.09.034
Scherlinger, M., Sisirak, V., Richez, C., Lazaro, E., Duffau, P., and Blanco, P. (2017). New insights on platelets and platelet-derived microparticles in systemic lupus erythematosus. Curr. Rheumatol. Rep. 19, 48. doi: 10.1007/s11926-017-0678-0
Schindl, R., Muik, M., Fahrner, M., Derler, I., Fritsch, R., Bergsmann, J., et al. (2009). Recent progress on STIM1 domains controlling Orai activation. Cell Calcium 46, 227–232. doi: 10.1016/j.ceca.2009.08.003
Scrimgeour, N. R., Wilson, D. P., Barritt, G. J., and Rychkov, G. Y. (2014). Structural and stoichiometric determinants of Ca2+ release-activated Ca2+ (CRAC) channel Ca2+-dependent inactivation. Biochim. Biophys. Acta 1838, 1281–1287. doi: 10.1016/j.bbamem.2014.01.019
Shi, J., Miralles, F., Kinet, J. P., Birnbaumer, L., Large, W. A., and Albert, A. P. (2017). Evidence that Orai1 does not contribute to store-operated TRPC1 channels in vascular smooth muscle cells. Channels 11, 329–339. doi: 10.1080/19336950.2017.1303025
Shim, A. H., Tirado-Lee, L., and Prakriya, M. (2015). Structural and functional mechanisms of CRAC channel regulation. J. Mol. Biol. 427, 77–93. doi: 10.1016/j.jmb.2014.09.021
Vermeersch, E., Denorme, F., Maes, W., De Meyer, S. F., Vanhoorelbeke, K., Edwards, J., et al. (2017). The role of platelet and endothelial GARP in thrombosis and hemostasis. PLoS ONE 12:e0173329. doi: 10.1371/journal.pone.0173329
Wagner, G. F., Haddad, M., Fargher, R. C., Milliken, C., and Copp, D. H. (1998a). Calcium is an equipotent stimulator of stanniocalcin secretion in freshwater and seawater salmon. Gen. Comp. Endocrinol. 109, 186–191. doi: 10.1006/gcen.1997.7010
Wagner, G. F., Jaworski, E. M., and Haddad, M. (1998b). Stanniocalcin in the seawater salmon: structure, function, and regulation. Am. J. Physiol. 274, R1177–R1185. doi: 10.1152/ajpregu.1998.274.4.R1177
Wang, Y., Gao, Y., Cheng, H., Yang, G., and Tan, W. (2015). Stanniocalcin 2 promotes cell proliferation and cisplatin resistance in cervical cancer. Biochem. Biophys. Res. Commun. 466, 362–368. doi: 10.1016/j.bbrc.2015.09.029
Zbidi, H., López, J. J., Amor, N. B., Bartegi, A., Salido, G. M., and Rosado, J. A. (2009). Enhanced expression of STIM1/Orai1 and TRPC3 in platelets from patients with type 2 diabetes mellitus. Blood Cells Mol. Dis. 43, 211–213. doi: 10.1016/j.bcmd.2009.04.005
Zeiger, W., Ito, D., Swetlik, C., Oh-Hora, M., Villereal, M. L., and Thinakaran, G. (2011). Stanniocalcin 2 is a negative modulator of store-operated calcium entry. Mol. Cell. Biol. 31, 3710–3722. doi: 10.1128/MCB.05140-11
Keywords: STC2, platelets, non-SOCE, Orai3, TRPC6, TRPC3
Citation: López E, Gómez-Gordo L, Cantonero C, Bermejo N, Pérez-Gómez J, Granados MP, Salido GM, Rosado Dionisio JA and Redondo Liberal PC (2018) Stanniocalcin 2 Regulates Non-capacitative Ca2+ Entry and Aggregation in Mouse Platelets. Front. Physiol. 9:266. doi: 10.3389/fphys.2018.00266
Received: 08 November 2017; Accepted: 08 March 2018;
Published: 23 March 2018.
Edited by:
Agustín Guerrero-Hernández, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional (CINVESTAV-IPN), MexicoReviewed by:
Nicolas Demaurex, Université de Genève, SwitzerlandOlivier Mignen, Université de Bretagne Occidentale, France
Copyright © 2018 López, Gómez-Gordo, Cantonero, Bermejo, Pérez-Gómez, Granados, Salido, Rosado Dionisio and Redondo Liberal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro C. Redondo Liberal, pcr@unex.es
†These authors have contributed equally to this work.