 Lars Matthias Voll1* Robin Jonathan Horst1 Anna-Maria Voitsik1 Doreen Zajic1 Birgit Samans2,3 Jörn Pons-Kühnemann2,3 Gunther Doehlemann4 Steffen Münch5 Ramon Wahl6 Alexandra Molitor3,7 Jörg Hofmann1 Alfred Schmiedl1 Frank Waller3,7 Holger Bruno Deising5 Regine Kahmann4 Jörg Kämper6 Karl-Heinz Kogel3,7 Uwe Sonnewald1
Lars Matthias Voll1* Robin Jonathan Horst1 Anna-Maria Voitsik1 Doreen Zajic1 Birgit Samans2,3 Jörn Pons-Kühnemann2,3 Gunther Doehlemann4 Steffen Münch5 Ramon Wahl6 Alexandra Molitor3,7 Jörg Hofmann1 Alfred Schmiedl1 Frank Waller3,7 Holger Bruno Deising5 Regine Kahmann4 Jörg Kämper6 Karl-Heinz Kogel3,7 Uwe Sonnewald1
- 1 Division of Biochemistry, Friedrich-Alexander-University Erlangen-Nuremberg, Erlangen, Germany
- 2 Institute of Biometry and Population Genetics, Justus Liebig University, Giessen, Germany
- 3 Research Center for BioSystems, Land Use and Nutrition, Justus Liebig University, Giessen, Germany
- 4 Max Planck Institute for Terrestrial Microbiology, Marburg, Germany
- 5 Faculty of Agricultural and Nutritional Sciences, Phytopathology and Plant Protection, Martin-Luther-University Halle-Wittenberg, Halle, Germany
- 6 Department of Genetics, Institute of Applied Biosciences, University of Karlsruhe, Karlsruhe, Germany
- 7 Institute of Phytopathology and Applied Zoology, Justus Liebig University, Giessen, Germany
During compatible interactions with their host plants, biotrophic plant–pathogens subvert host metabolism to ensure the sustained provision of nutrient assimilates by the colonized host cells. To investigate, whether common motifs can be revealed in the response of primary carbon and nitrogen metabolism toward colonization with biotrophic fungi in cereal leaves, we have conducted a combined metabolome and transcriptome study of three quite divergent pathosystems, the barley powdery mildew fungus (Blumeria graminis f.sp. hordei), the corn smut fungus Ustilago maydis, and the maize anthracnose fungus Colletotrichum graminicola, the latter being a hemibiotroph that only exhibits an initial biotrophic phase during its establishment. Based on the analysis of 42 water-soluble metabolites, we were able to separate early biotrophic from late biotrophic interactions by hierarchical cluster analysis and principal component analysis, irrespective of the plant host. Interestingly, the corresponding transcriptome dataset could not discriminate between these stages of biotrophy, irrespective, of whether transcript data for genes of central metabolism or the entire transcriptome dataset was used. Strong differences in the transcriptional regulation of photosynthesis, glycolysis, the TCA cycle, lipid biosynthesis, and cell wall metabolism were observed between the pathosystems. However, increased contents of Gln, Asn, and glucose as well as diminished contents of PEP and 3-PGA were common to early post-penetration stages of all interactions. On the transcriptional level, genes of the TCA cycle, nucleotide energy metabolism and amino acid biosynthesis exhibited consistent trends among the compared biotrophic interactions, identifying the requirement for metabolic energy and the rearrangement of amino acid pools as common transcriptional motifs during early biotrophy. Both metabolome and transcript data were employed to generate models of leaf primary metabolism during early biotrophy for the three investigated interactions.
Introduction
Substantial effort is being devoted to gain insight into plant–pathogen interactions to improve crop plants for sustainable agriculture. Phytopathogenic bacteria and fungi drive their own cellular metabolism with substrates being diverted from the colonized and/or surrounding host cells. Nutrient acquisition from the host cells is crucial for the successful establishment of bacterial and fungal pathogens (reviewed by Divon and Fluhr, 2007). Plant–pathogens have evolved different strategies to divert nutrients from their plant hosts. While necrotrophic pathogens rapidly kill plant tissue usually by the secretion of highly efficient toxins and cell wall degrading enzymes (van Kan, 2006) – and thrive on the dead plant material, biotrophic pathogens strictly rely on living tissue to survive and complete their life cycle (Divon and Fluhr, 2007). In contrast, hemibiotrophs establish themselves during an initial biotrophic phase before necrotrophic growth is initiated (Mendgen and Hahn, 2002; Münch et al., 2008). In general, infection sites of biotrophic fungi represent strong local metabolic sinks that drain nutrients from the host environment. Evidence obtained for the rust fungus Uromyces fabae suggest that nutrients are mainly taken up as hexoses (generated by secreted fungal invertase) and amino acids (Hahn et al., 1997; Voegele et al., 2001; Struck et al., 2002, 2004). Recently, a novel high-affinity U. maydis sucrose transporter Srt1 has been characterized, which is required for full virulence (Wahl et al., 2010). Effective nutrient provision by host cells is necessary to establish a compatible interaction with biotrophs, as indicated by increased resistance of the variegated barley albostrians mutant toward powdery mildew fungus or by increased resistance of Arabidopsis overexpressing invertase inhibitors toward clubroot disease (Jain et al., 2004; Siemens et al., 2011). In addition, it was recently found that the induction of sugar efflux carriers in infected tissue by TAL-effectors of the bacterial rice pathogen Xanthomonas oryzae pv. oryzae (Xoo) is required for pathogenicity (Chen et al., 2010).
Vice versa, a vast array of fungal genes coding for metabolic enzymes was found to be induced upon host colonization, providing evidence that pathogen metabolism adapts to the host environment and nutrient availability (as reviewed by Divon and Fluhr, 2007). Despite its importance for hexose provision to the invaders, the induction of invertases, and the concomitant increase in free hexoses can serve as a signal for the repression of photosynthetic gene expression (as reviewed in Biemelt and Sonnewald, 2006). Furthermore, elevated hexose contents constitute an important cue in defense signaling (as reviewed by Bolton, 2009). Similarly, the support of the host defense response by the provision of reducing equivalents in the cytosol via glucose-6-phosphate dehydrogenase (G6PDH) seems to be an essential metabolic process that heightens defense effectiveness (Scharte et al., 2009). In Arabidopsis, strong evidence has been gathered that lipid metabolism in the chloroplast is involved in regulating the balance between SA- and JA-mediated defense responses and the induction of the hypersensitive response, HR (Kachroo et al., 2003; Chanda et al., 2008; Chaturvedi et al., 2008; Raffaele et al., 2008).
Although metabolic processes are important determinants of compatibility during plant–pathogen interactions, our knowledge on metabolic compatibility factors is scarce. Nevertheless, an increase in the sucrose/hexose ratio (Chou et al., 2000; Swarbrick et al., 2006) and elevated contents of nitrogen storage amino acids Gln and Asn (Olea et al., 2004; Tavernier et al., 2007; Horst et al., 2010a) have frequently been observed during biotrophic interactions, nourishing the hypothesis that a direct or indirect metabolic reprogramming of host metabolism occurs during the establishment of fungal biotrophs on their hosts. Employing comparative metabolome analysis, our study aims at identifying metabolic processes that are commonly altered during compatible interactions of biotrophic fungal leaf pathogens with agriculturally relevant cereal hosts. Pathosystems were selected to maximize biological diversity in the analyzed interactions and to minimize the chance of identifying effects specific to certain subclasses of pathogens. First, we have chosen to compare the response of barley, a C3-plant, with that of maize, a C4 plant, and second, the biotrophic lifestyle of the three fungal pathogens is quite diverse.
Ustilago maydis (Um), the causal agent of corn smut disease, is a biotrophic basidiomycete parasitizing maize and its natural ancestor teosinte. It can induce the formation of tumors on all aerial organs (Banuett, 1995) and exhibits a dimorphic lifestyle (Kahmann and Kämper, 2004): While haploid sporidia are not infectious and grow saprophytically in a yeast-like manner, filamentous growth is initiated upon mating of two compatible sporidia on the plant surface. Filamentous hyphae quickly form appressoria that penetrate host cells. Immediately upon host entry at around 24 h post inoculation, the invading biotrophic hyphae grow both inter- and intra-cellular without disrupting the host plasma membrane. About 4 days after penetration, the formation of hypertrophic host cells and concomitant tumor development are induced, while the fungal hyphae start proliferating in the apoplastic spaces that develop as a consequence of cell wall degradation and induced host cell enlargement (Doehlemann et al., 2008a,b).
Blumeria graminis f.sp. hordei (Bgh) is an obligate biotroph that causes powdery mildew disease on barley. Germination of wind-dispersed Bgh conidia on the barley leaf surface first produces a short primary germ tube prior to the formation of the infectious secondary germ tube, at the tip of which a hooked appressorium is formed. From the appressorium, a penetration peg is ejected within 15 h post inoculation (Hückelhoven et al., 1999; Both et al., 2005) that penetrates cuticle and wall of the host epidermis cell beneath and subsequently, a haustorium is established in the periplasmic space of the colonized host cell that serves as a strongly invaginated feeding organ. Unlike U. maydis hyphae that grow filamentously through the colonized maize tissue, only the haustoria of Bgh reside inside the infected leaf, while the predominant portion of fungal hyphae are growing epiphytically, occasionally forming secondary haustoria in adjacent epidermal cells. Eventually at 5 days post inoculation, conidiophores emerge from the epiphytic mycelium that shed series of conidiospores from their tips.
In contrast to U. maydis and Bgh, the maize pathogen Colletotrichum graminicola leads a hemibiotrophic lifestyle (as reviewed by Bergstrom and Nicholson, 1999; Mendgen and Hahn, 2002; Münch et al., 2008). Rain-dispersed conidia land on the leaf surface, produce germ tubes, which then differentiate sophisticated appressoria. During maturation, appressoria form rigid cell walls which melanize and synthesis of high concentrations of compatible solutes results in generation of enormous appressorial turgor pressure by diffusion of water into the appressorium. At the appressorial base, turgor pressure is translated into mechanical force that breaches the host cell wall. In the penetrated host epidermis cells, C. graminicola establishes itself as a biotroph within 36 h post inoculation by forming an infection vesicle that produces lobed biotrophic primary hyphae. During the subsequent colonization of neighboring cells at around 72 h post infection, the formation of narrow-bore secondary hyphae is initiated, which grow rapidly, are highly destructive and represent the necrotrophic lifestyle of the pathogen.
Thus, our set of fungal pathogens extends (i) an obligate biotroph that nourishes via epidermis-localized haustoria, Bgh, (ii) a biotroph that colonizes the entire leaf tissue by intra- and inter-cellularly growing hyphae, Um, and (iii) a hemibiotroph, Cg, that switches from biotrophic colonization of epidermis cells to vast proliferation by necrotrophic hyphae throughout the entire leaf.
Materials and Methods
Plant and Fungal Cultivation and Infection Conditions
For combined metabolite and transcript profiling experiments, Zea mays cv. Early Golden Bantam was cultivated as described in (Doehlemann et al., 2008a) and infected with U. maydis strain SG200 as described by Doehlemann et al. (2008a) or with C. graminicola strain CgM2 as described in Münch et al. (2011).
Combined metabolite and transcript profiling experiments with barley (cv. Golden Promise) after challenge with Bgh isolate B6 were conducted as described in Molitor et al. (2011).
Transcriptome Analysis by DNA Microarray
Transcriptome data from U. maydis-infected maize leaf tissue was obtained from the same set of material described in Doehlemann et al. (2008a), which is deposited in the Gene Expression Omnibus1 under the accession number GSE10023. The transcriptome dataset of Bgh infected barley leaves represents the same dataset as in Molitor et al. (2011). Transcriptome data for C. graminicola infected maize leaves (infection procedure as in Münch et al., 2011) were obtained as described in Doehlemann et al. (2008a) and are deposited in the Gene Expression Omnibus (see text footnote 1) under the accession number GSE31188. If not stated otherwise, a low stringent threshold of >1.5-fold change with no p-value filter was used for comparative analyses of transcriptome data.
Matching of Barley and Maize Microarray Data
To connect the transcripts from different microarray platforms, we used the microarray platform translator on the PlexDB homepage2. The transformation was performed with the default settings.
Calculation of Mapman Bin Enrichment and Subsequent HCA
The tool MapMan (Thimm et al., 2004) adapted for maize and barley Affymetrix microarrays was used to visualize the transcriptome data that was obtained as described above. For the analysis, the mean values from all three replicate experiments were employed. To calculate the percentage of regulated genes per MapMan BIN of primary carbon and nitrogen metabolism, the number of regulated features with fold change >2.0 was expressed as percentage of total number of features in the respective BINs, to enable a comparison of maize and barley data that do not share the same number of accessions per BIN. Percentage up-regulated and percentage down-regulated features were scored separately and used for hierarchical cluster analysis (HCA) analysis after log transformation, median centering, and normalization as described for metabolite data below.
Metabolite Quantification and Analysis
For all three interactions analyzed in this report, metabolite contents were determined in three independent experiments from subsets of the leaf material pools that were employed for transcriptome analysis, such that material of four independent samples for metabolite analysis were pooled to generate one sample pool for transcript analysis per time point. All metabolite assays were conducted as described by Horst et al. (2010a).
Multivariate Data Analysis
Mean values of the four biological replicates taken per time point and experiment were calculated for all individual metabolites prior to calculating the metabolite ratio between infected vs. non-infected tissue, which was employed for HCA. After log transformation of the data, median centered ratios were normalized and HCA was performed using the complete linkage algorithm of the program Cluster V2.11 (Eisen et al., 1998) and the results were visualized using Maple Tree3.
Principle component analysis of log-transformed metabolite ratios was performed with the MarkerView software (Version 1.1.0.7, Applied Biosystems, Foster City, CA, USA) using the autoscale algorithm for scaling.
Results
Experimental Design and Sampling Strategy
The sampling time points in all three pathosystems were carefully adjusted to the on planta development of the respective pathogen and to the diurnal light/dark cycles of the growth regimen (Figure 1). For every interaction, infected leaves were harvested at two crucial stages: (i) shortly after the establishment of biotrophy and (ii) at time points late in the biotrophic interaction, with a corresponding sampling time point during necrotrophic colonization by C. graminicola at 96hpi serving as a reference for non-biotrophic colonization. To minimize artifacts by diurnal oscillations of metabolite contents, leaf material harvested at the end of the subjective light phase was prioritized for comparative analysis described below.
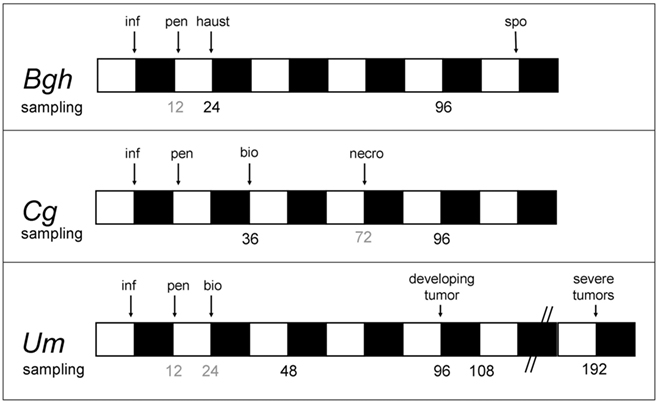
Figure 1. Mapping of sampling time points to respective infection stages in the three studied interactions. Bgh – barley (cv. Golden Promise) infected with Blumeria graminis f.sp. hordei isolate A6 (Wiberg, 1974); Cg – maize (cv. Early Golden Bantam) infected with Colletotrichum graminicola strain CgM2; Um – maize (cv. Early Golden Bantam) infected with the solopathogenic Ustilago maydis strain SG200 (Kämper et al., 2006). White bars indicate light period, and dark bars indicate dark period. Sampling time points analyzed in this study are indicated in black below the bars, while sampling time points that were disregarded are printed in gray. inf, Inoculation; pen, penetration, bio, initiation of biotrophic growth, haus, haustoria establishment; necro, commencement of necrotrophic growth; spo, formation of conidiospores begins.
To produce leaf infections of barley (cv. Golden Promise) and maize (cv. Early Golden Bantam) with Bgh and Cg, respectively, expanding leaves of young plants were inoculated with conidia of Bgh and Cg. In contrast, the infection of Early Golden Bantam with Um was performed by injecting sporidia suspension into the leaf canal with a syringe, giving rise to infections on meristematic tissue of developing leaves. For all three interactions studied, targeted analysis of 42 metabolites of central carbon and nitrogen metabolism as well as major low-molecular antioxidants was conducted in four biological replicates per time point and treatment in three independent experiments. For each of the independent experiments, material from all four biological replicates used for metabolite determination was pooled for subsequent transcriptome analysis (as described by Doehlemann et al., 2008).
Comparative Metabolome Analysis
Since the aim of our work was to assess, whether common metabolic signatures of biotrophy can be identified in cereal leaves during compatible interactions with fungal leaf pathogens, we first tried to identify similarities between the patterns of the 42 determined metabolites by HCA. As our goal was comparing the dynamics of host metabolism, we employed metabolite ratios between infected and mock control leaves for the HCA analysis, in order to avoid complications by species and experiment specific variation in steady state contents of metabolites. Table S1 in Supplementary Material contains a compilation of the individual metabolite contents ± SE and the calculated metabolite ratios infected/mock ± SE for all three replicate experiments for all time points and pathosystems analyzed. For the sake of clarity, only two of the three replicate datasets were used for subsequent multivariate data analysis, with the results remaining comparable.
In the HCA, three major clusters could be distinguished that correspond to three different types of interaction (Figure 2). The most prominent cluster contained samples derived from U. maydis-induced tumors, irrespective, whether the samples were taken at the beginning (Um 108hpi) or at the end of the subjective light phase (Um 96hpi and Um 192hpi), and independent of the developmental state of the tumors. This indicates that tumor development determines very profound changes in infected maize leaves (as already observed by Horst et al., 2010a) that even superimpose diurnal variations in metabolite contents. Consequently, these samples were not within the focus of our further analysis, as many metabolic changes specific to tumor formation occur at late stages of the U. maydis – maize interaction. However, the metabolite changes in maize leaves during the initial colonization phase at 48hpi, when no tumors had yet been formed, was most similar to that of barley leaves with strong powdery mildew colonization (Bgh 96hpi), suggesting that this cluster represents established biotrophic interactions. The third cluster is comprised of samples taken immediately after penetration (Cg 36hpi and Bgh 24hpi). For the two latter clusters, it is remarkable that the physiological situation of the samples, i.e., immediate post-penetration (Cg 36hpi and Bgh 24hpi) and established biotrophic interaction (Um 48hpi and Bgh 96hpi), respectively, appears to be more important for sample parsing than host or pathogen involved. All three clusters mentioned so far were separated from the samples obtained from the necrotrophic phase of C. graminicola infection (Cg 96hpi). Interestingly, replicate samples of pre-penetration stages (Bgh 12hpi, Um 12hpi) or from developing leaf tissue (Um 12hpi and Um 24hpi) did not cluster together when included in the HCA (not shown), indicating that despite strong transcriptional changes for genes involved in central metabolism during basal defense reaction (see corresponding publications by Horst et al., 2010a and Molitor et al., 2011), central metabolism itself was not strongly altered at post-penetration stages. This indicates that changes in central leaf metabolism only occur upon physical interaction with pathogens inside the host tissue, when the drainage of nutrients to the pathogen and the suppression of host defense is being established.
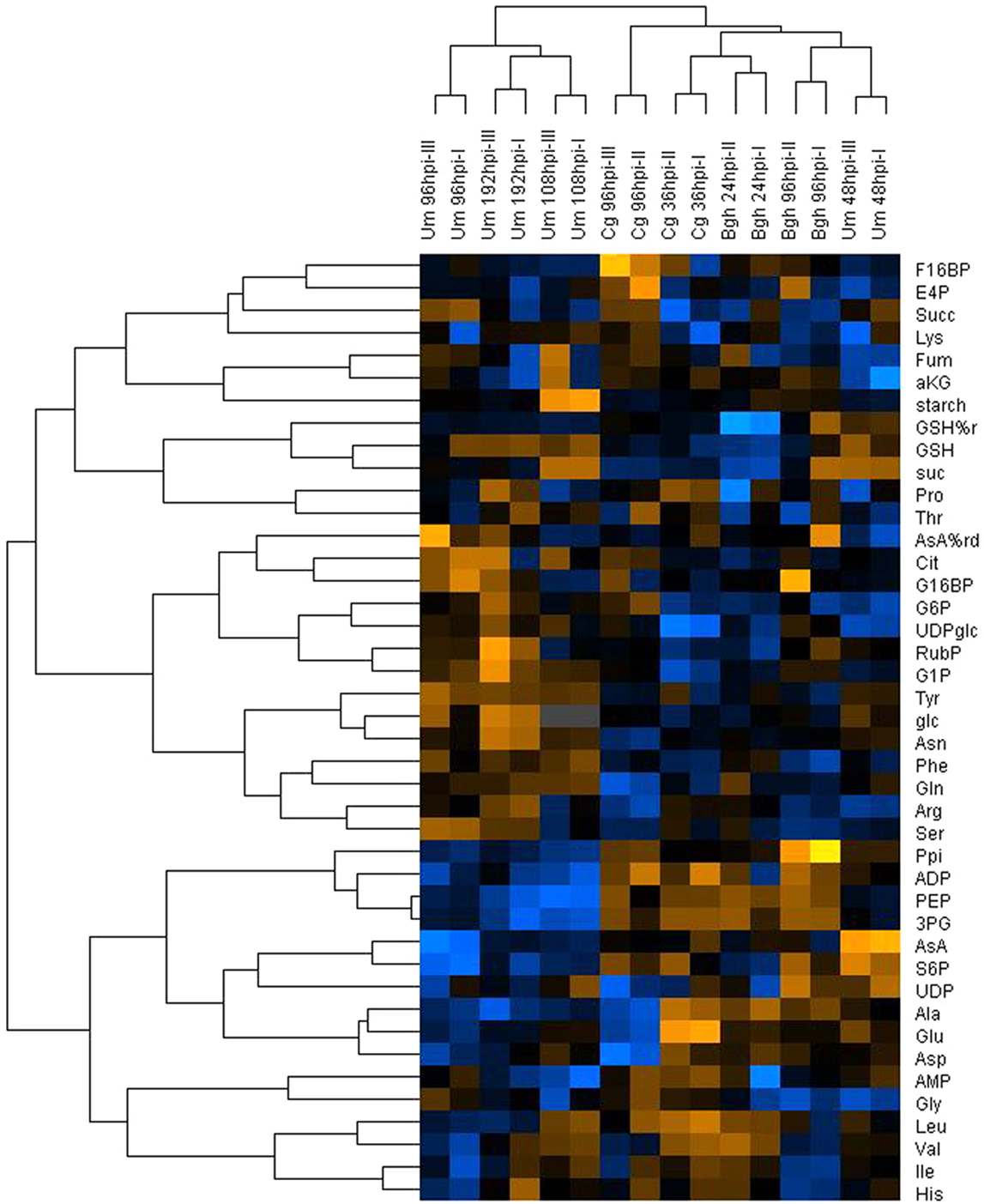
Figure 2. Hierarchical cluster analysis of metabolome data from infected leaves. Mean values of metabolite contents from four biological replicates of infected and mock control leaves harvested at the indicated time points after infection with the respective pathogens (Bgh, Blumeria graminis f.sp. hordei; Cg, Colletotrichum graminicola; Um, Ustilago maydis) were used to calculate the metabolite ratio infected/mock for the indicated experimental replicates. After log transformation of the data, median centered ratios were normalized and hierarchical clustering analysis (HCA) was performed using the complete linkage algorithm of the program Cluster v2.11 (Eisen et al., 1998 – www.eisenlab.org) and the results were visualized using Maple Tree (http://mapletree.sourceforge.net/). Metabolite ratios from two independent experiments (indicated by Roman numbers) of every pathosystem were used for HCA. Color intensity correlates with degree of increase (yellow) and decrease (blue) relative to the mean metabolite ratio. hpi, hours post infection. Amino acids and nucleotides are abbreviated according to three letter code, aKG, (α-ketoglutarate) Asc (ascorbate); %AsA red, (% reduced ascorbate); Cit, (citrate); E4P, (erythrose-4-phosphate); F16BP, (fructose-1,6-bisphosphate); F6P, (fructose-6-phosphate); frc, (fructose); Fum, (fumarate); G16BP, (glucose-1,6-bisphosphate); G1P, (glucose-1-phosphate); G6P, (glucose-6-phosphate); glc, (glucose); GSH, (glutathione); %GSH red, (% glutathione reduced); Icit, (isocitrate); PEP, (phosphoenol pyruvate); 3PG, (3-phosphoglycerate); 6PG, (6-phosphogluconate); Ppi, (pyrophosphate); Pyr, (pyruvate); RubP, (ribulose-1,5-bisphosphate); S6P, (sucrose-6-phosphate); suc, (sucrose); Succ, (succinate); T6P, (trehalose-6-phosphate); UDPglc, (UDP-glucose).
Identification of Metabolite Determinants Specific for Interaction Stages
The obtained results indicate that there must be certain metabolites, which can be used to discriminate the three major clusters produced in the HCA. Therefore, we conducted a principal component analysis (PCA) to identify those metabolite changes that contribute most to the distinction between early post-penetration (Cg 36hpi and Bgh 24hpi), established biotrophic interaction (Um 48hpi and Bgh 96hpi) and U. maydis-induced tumors. As already suggested by the HCA, principal component 1 (PC1), explaining 41% of the variation, distinguished U. maydis tumor samples from the rest (Figures 3A,B). Including the time point 108hpi sampled at dawn did not affect the clustering (not shown). As inferred from the metabolite loading scores, Glc, Asn, Ser, Tyr, Gln, and Arg showed the strongest positive distinction, while 3-PGA, PEP, and pyrophosphate exhibited the strongest negative loading in the U. maydis tumor samples. PC2, explaining 17% of the variance, separated the necrotrophic interaction (Cg 96hpi), indicating substantial differences in the metabolite pattern to all other samples (Figure 3B), which was reflected by a strong positive loading of the phosphorylated intermediate F16bP and negative loading of the major amino acids Asp, Ala, and Glu. Finally, PC3, corresponding to 13% of the overall variance, was able to subdivide the early biotrophic interaction time points (above the abscissa) from leaves with established biotrophy (below the abscissa, Figure 3B). Branched-chain amino acids, Gly, and His as well as phosphorylated intermediates of carbohydrate metabolism, G16bP, RubP, G1P, UDPglc, pyrophosphate, and the end product sucrose were the most important metabolites to separate these interaction stages from one another.
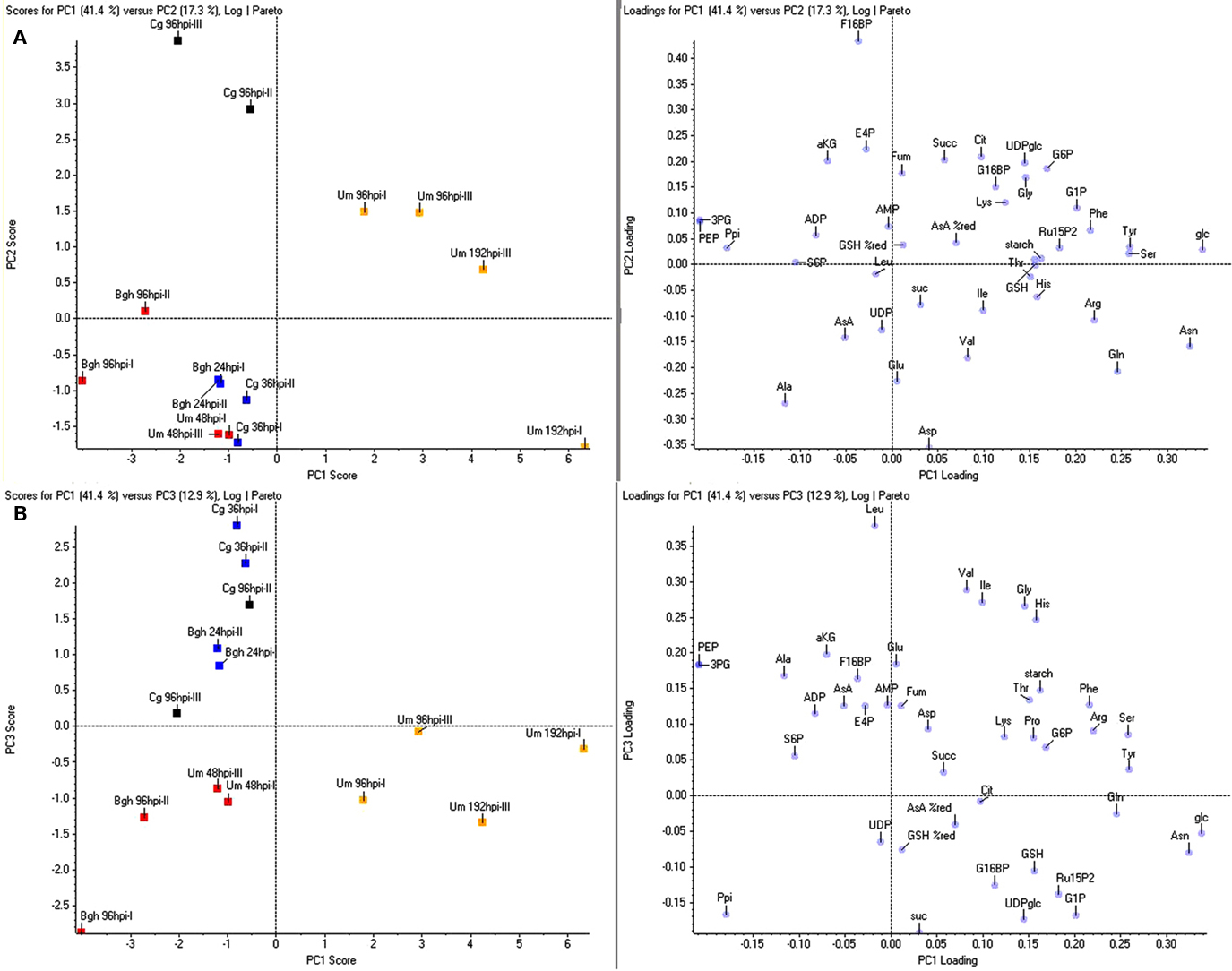
Figure 3. Principle component analysis of metabolite data. For principle component analysis (PCA), the same metabolite ratios as in Figure 1 were used, representing the ratio infected/mock calculated from the mean values of four sample replicates each. Per pathosystem, data from two independent experiments (designated by Roman numbers) were employed for PCA. (A) PC1 vs. PC2. (B) PC1 vs. PC3. Bgh, Blumeria graminis f.sp. hordei; Cg, Colletotrichum graminicola; Um, Ustilago maydis; yellow, tumor, red, established biotrophy, blue, early biotrophy, black, necrotrophy. Amino acids and nucleotides are abbreviated according to three letter code, aKG, (α-ketoglutarate) AsA (ascorbate); %AsA red, (% reduced ascorbate); Cit, (citrate); E4P, (erythrose-4-phosphate); F16BP, (fructose-1,6-bisphosphate); F6P, (fructose-6-phosphate); frc, (fructose); Fum, (fumarate); G16BP, (glucose-1,6-bisphosphate); G1P, (glucose-1-phosphate); G6P, (glucose-6-phosphate); glc, (glucose); GSH, (glutathione); %GSH red, (% glutathione reduced); Icit, (isocitrate); PEP, (phosphoenol pyruvate); 3PG, (3-phosphoglycerate); 6PG, (6-phosphogluconate); Ppi, (pyrophosphate); Pyr, (pyruvate); RubP, (ribulose-1,5-bisphosphate); S6P, (sucrose-6-phosphate); suc, (sucrose); Succ, (succinate); T6P, (trehalose-6-phosphate); UDPglc (UDP-glucose).
In general, we have observed numerous metabolite changes at most interaction stages (Tables 1 and 2), and therefore we analyzed not only the differences between the early interaction phase (Cg 36hpi and Bgh 24hpi) and established biotrophy (Um 48hpi and Bgh 96hpi), but also assessed common metabolite dynamics among these stages. Looking only at those metabolites that changed in average more than 1.4-fold in all four situations of interest (Cg 36hpi, Bgh 24hpi, Um 48hpi, and Bgh 96hpi), we could identify glucose and the nitrogen storage amino acids Glutamine and Asparagine being consistently increased, while the glycolytic intermediate PEP and the Calvin cycle intermediate 3-PGA were commonly decreased (Table 1). This might indicate that the balance between carbon and nitrogen metabolism and respiration is already readjusted early during compatible interactions. As indicated by the low number of metabolites that were consistently altered more than 1.4-fold in infected leaves in all three pathosystems, the stringency of the interspecies comparison needs to be low in order to identify common metabolic changes. For the vast majority of the regarded biotrophic interaction stages, changes in the abovementioned five metabolites were statistically significant in a Welch–Satterthwaite t-test, but not after Benjamini–Hochberg FDR correction.
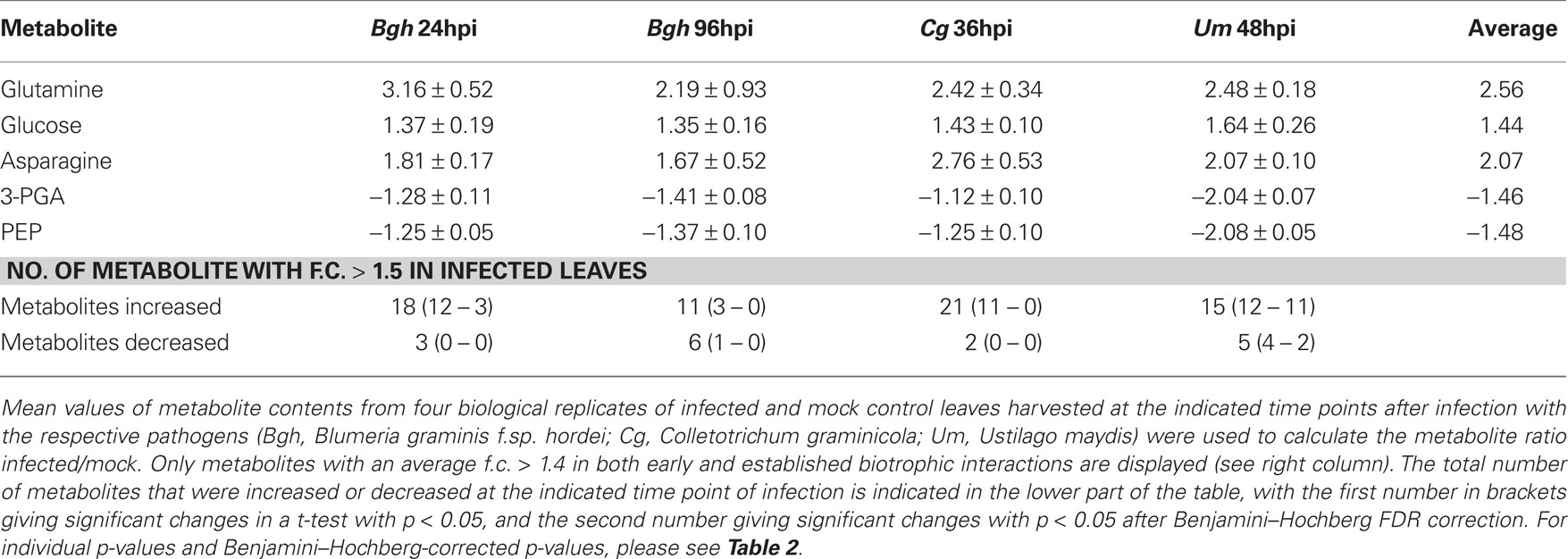
Table 1. Metabolites consistently altered in all biotrophic interactions.
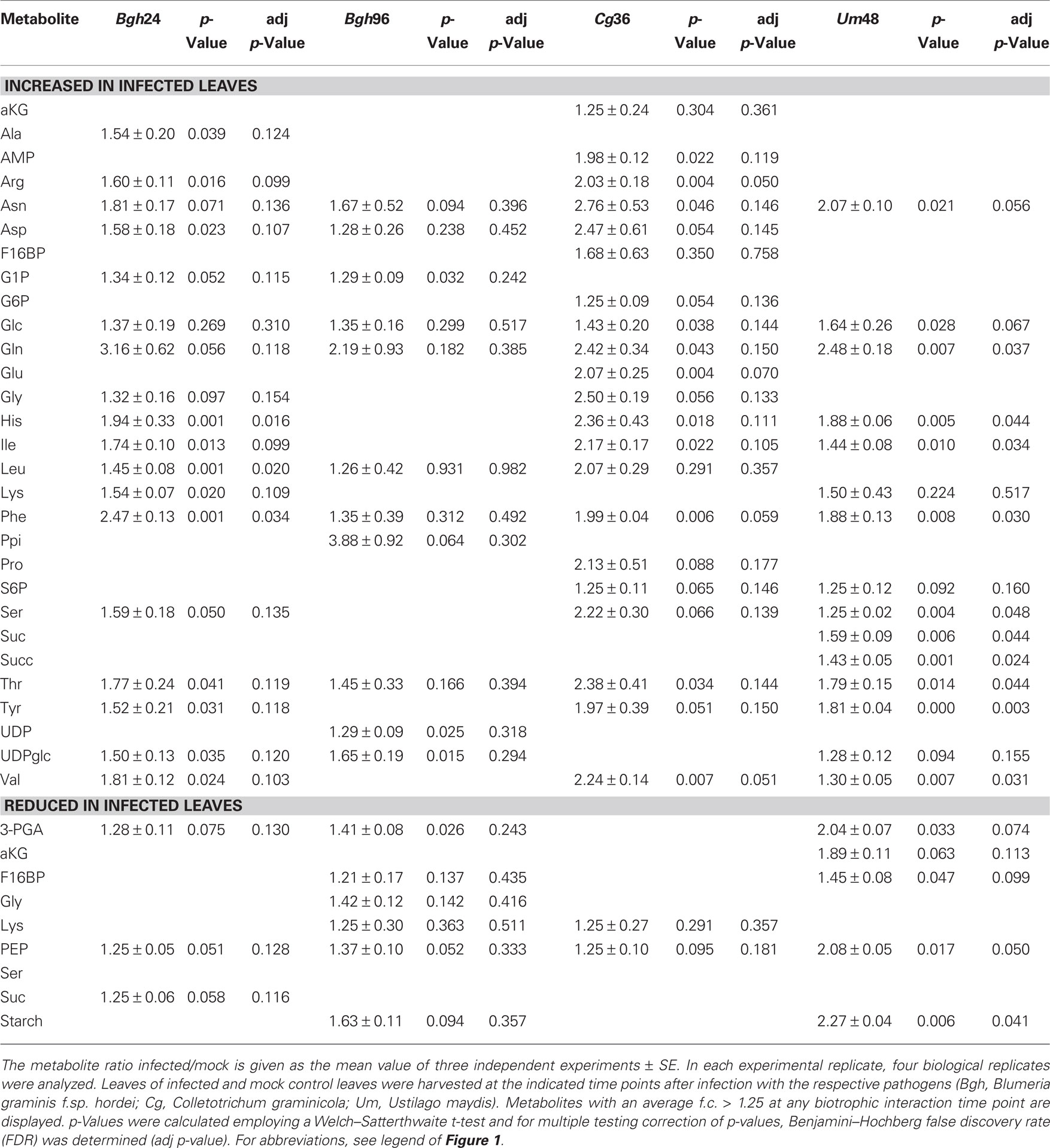
Table 2. Compilation of all substantial metabolite changes at biotrophic interaction time points.
Distinct Metabolite Responses are not Caused by Confined Transcriptional Programs
By multivariate data analysis, we were able to identify common and distinct metabolite changes associated with different phases of compatible biotrophic interactions, which could represent potential metabolic compatibility factors. To identify potential host targets of metabolic reprogramming by biotrophic fungi, we aimed at refining the underlying transcriptional changes governing the observed metabolic redirections.
We analyzed the corresponding transcriptome data obtained from the same pooled material that was used for metabolite analysis for transcriptional changes that could account for the observed dynamics in the metabolome. As not all genes in a pathway are subject to transcriptional regulation, it appeared instrumental to analyze the enrichment of transcriptional regulation within entire metabolic pathways. To avoid complications by annotation artifacts in the pairwise assignment of the maize and barley microarray features, we preferred to calculate the enrichment of regulated genes within MapMan BINs (Thimm et al., 2004) associated with central primary carbon and nitrogen metabolism, of which the functional annotations are quite robust (Table 3). If transcriptional reprogramming of metabolic pathways would account for the observed differences in the metabolome between early post-penetration and at established biotrophy, we would expect a similar clustering result of the transcript data as for the metabolite data. Surprisingly, an HCA comparing the fraction of regulated genes in MapMan BINs assigned to central carbon and nitrogen metabolism gave a completely different picture compared to the metabolome analysis (Figure 4). Only the samples reflecting established biotrophy (Um 48hpi and Bgh 96hpi) still clustered together. On the pathway level, deregulation of major carbon metabolism was consistent enough to parse the MapMan BINs Calvin cycle, sucrose, and starch biosynthesis into the same cluster.
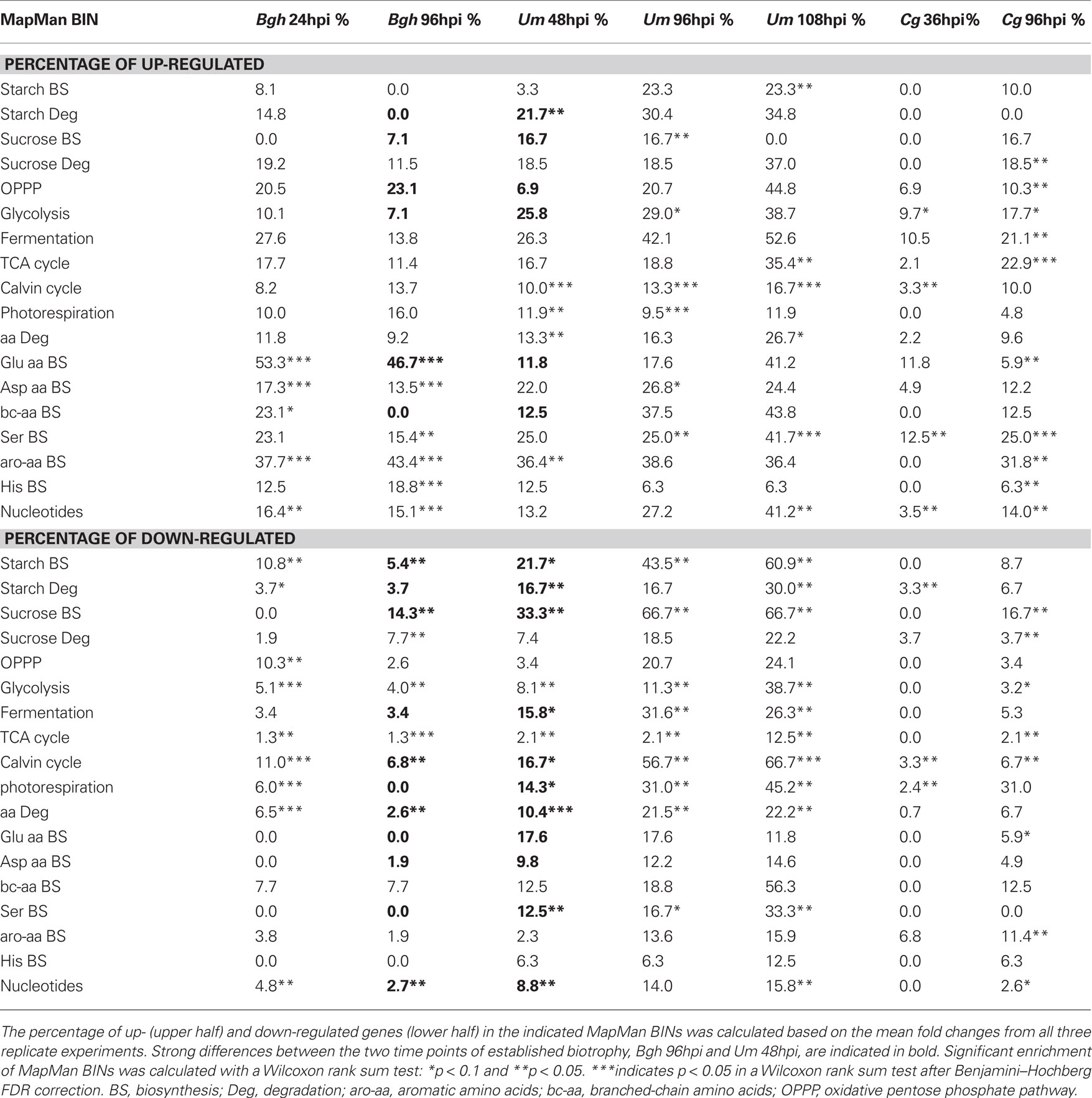
Table 3. Percentage of up- and down-regulated genes in MapMan BINs of central primary metabolism.
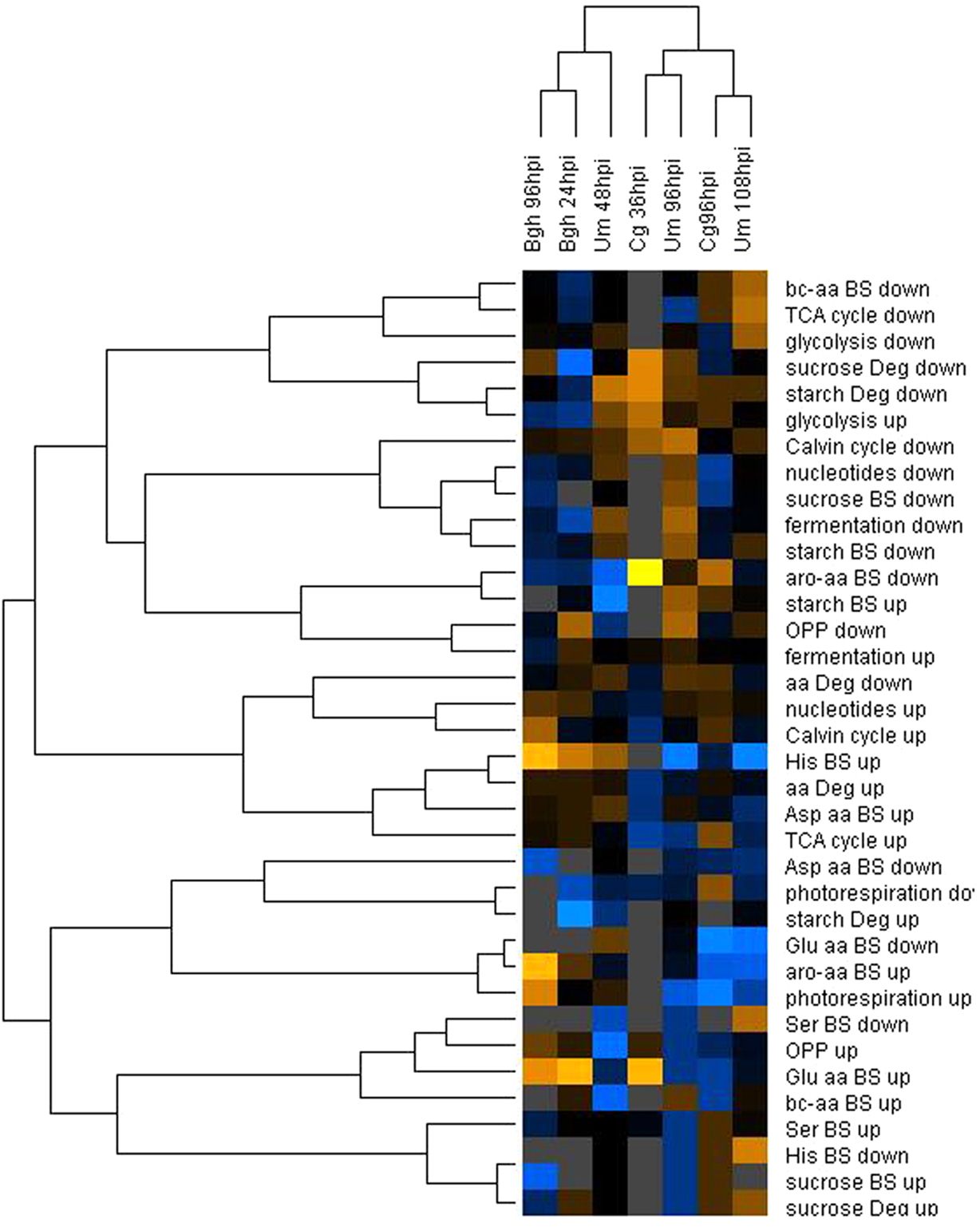
Figure 4. Hierarchical cluster analysis of MapMan BIN enrichment. The percentage of up- and down-regulated genes in each MapMan BIN was calculated based on the mean fold changes from all three replicate experiments. After log transformation of the data, median centered percentages were normalized and hierarchical clustering analysis (HCA) was performed using the complete linkage algorithm of the program Cluster v2.11 (Eisen et al., 1998 – www.eisenlab.org) and the results were visualized using Maple Tree (http://mapletree.sourceforge.net/). Color intensity correlates with degree of increase (yellow) and decrease (blue) relative to the BIN mean of all samples, while gray corresponds to 0% regulated genes in the MapMan BINs. Bgh, Blumeria graminis f.sp. hordei; Cg, Colletotrichum graminicola; Um, Ustilago maydis; hpi, hours post infection. Amino acids are abbreviated by three letter code; aro-aa, aromatic amino acids; bc-aa, branched-chain amino acids; BS, biosynthesis; Deg, degradation; OPPP, oxidative pentose phosphate pathway.
During early post-penetration biotrophy (Bgh 24hpi and Cg 36hpi), the most pronounced changes in metabolite contents had occurred in the accumulation of most free amino acids (see Figure 3B and Table 2) as well as by decreased contents of phosphorylated intermediates of starch and sucrose biosynthesis (see Figure 3B). While between 8 and 19% of genes annotated to central carbon metabolism and between 12 and 53% of genes annotated to amino acid biosynthesis are up-regulated at 24hpi after Bgh infection, most of these MapMan BINs are not regulated at all at 36hpi after Cg infection (Table 3), demonstrating that although both early post-penetration situations exhibit similar metabolite changes, transcriptional regulation of the corresponding metabolic pathways is utterly different. Likewise, the samples attributed to established biotrophy (Um 48hpi and Bgh 96hpi) were refined by PCA based on concomitant changes in phosphorylated intermediates of central carbon metabolism (see Figure 3B). In addition, the contents of the glycolytic intermediate PEP, and the Calvin cycle intermediates 3-PGA and F16bP (which are predominantly localized in the stroma in illuminated leaves, see Gerhardt et al., 1987; Heineke et al., 1994 and Leidreiter et al., 1995) were consistently diminished at 48hpi after Um infection and at 96hpi after Bgh infection (Table 2). MapMan BINs for sucrose and starch biosynthesis, the Calvin cycle, glycolysis, and major amino acid biosynthesis were much stronger deregulated in Um 48hpi than in Bgh 96hpi (highlighted in Table 3), again indicating a sincere difference on the transcriptional level despite similar metabolite changes as revealed by PCA.
To evaluate whether a more global transcriptome analysis would result in a similar outcome compared to the focused analysis of transcripts involved in central metabolism, we matched all features on the Barley1 and the maize Affymetrix arrays via the corresponding gene annotations deposited at PlexDB4. An HCA employing features with fold change >2 from the whole transcriptome dataset resulted in a different sample parsing than in the previous analysis employing data categorized into MapMan BINs. Mostly, clustering of samples occurred predominantly according to pathosystems, irrespective, whether only the previously analyzed samples were clustered or whether all available samples were used for the computation (Figure 5). Similar results were obtained when barley and maize genes were matched based on their closest homolog in rice (not shown). Although we still cannot rule out that part of the clustering is influenced by artifacts arising from matching the array annotations, the fact that Cg and Bgh samples form one cluster in the full transcriptome HCAs (Figure 5A) argues against a strong influence by such misinterpretations. At the bottom line, no infection stage specific clustering could be observed when transcriptome data were analyzed.
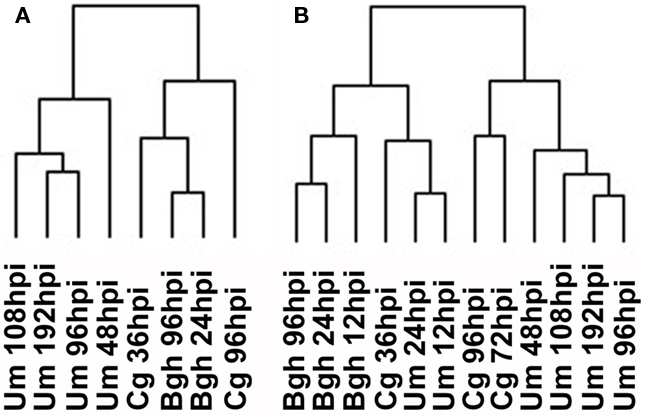
Figure 5. Hierarchical cluster analysis of whole transcriptome data. (A) HCA for the timepoints analyzed in this report. (B) HCA for all timepoints sampled. Barley and maize Affymetrix data were matched via PlexDB (www.plexDB.org) as described in the methods section and the mean transcript fold changes of infected vs. mock samples were calculated based on all three replicate experiments and were subsequently used for HCA. After log transformation of the data, median centered data were normalized and hierarchical clustering analysis (HCA) was performed using the complete linkage algorithm of the program Cluster v2.11 (Eisen et al., 1998 – www.eisenlab.org) and the results were visualized using Maple Tree (http://mapletree.sourceforge.net/). Bgh, Blumeria graminis f.sp. hordei; Cg, Colletotrichum graminicola; Um, Ustilago maydis; hpi, hours post infection.
Identification of Metabolic Genes Regulated in Response to Fungal Infection
Although we were unable to identify common motifs in the transcriptional response of metabolic pathways at the early post-penetration stage and during established biotrophy, we were surveying the transcriptome data for metabolic genes that were found to be regulated during biotrophic interactions in more than one pathosystem. To address this question, we had to decrease the fold change threshold down to 1.5-fold, as the standard twofold threshold appeared to be too stringent for such a cross-species comparison. Table 4 shows that genes involved in the TCA cycle and carboxylate metabolism as well as genes regulating the energy status of the nucleotide pool are consistently induced in more than one pathosystem. Similarly, remodeling of amino acid metabolism appears to be a common theme during compatible biotrophic interactions. Surprisingly, the number of targets in central carbohydrate metabolism is quite scarce. While there seems to be different ways of transcriptional regulation of fructose-2,6-bisphosphate homeostasis in all pathosystems, only few more genes in central carbohydrate metabolism were found, but not as consistent as genes involved in carboxylate, nucleotide, and amino acid metabolism, indicating that there is no strong regulation of carbohydrate flux on the transcriptional level.
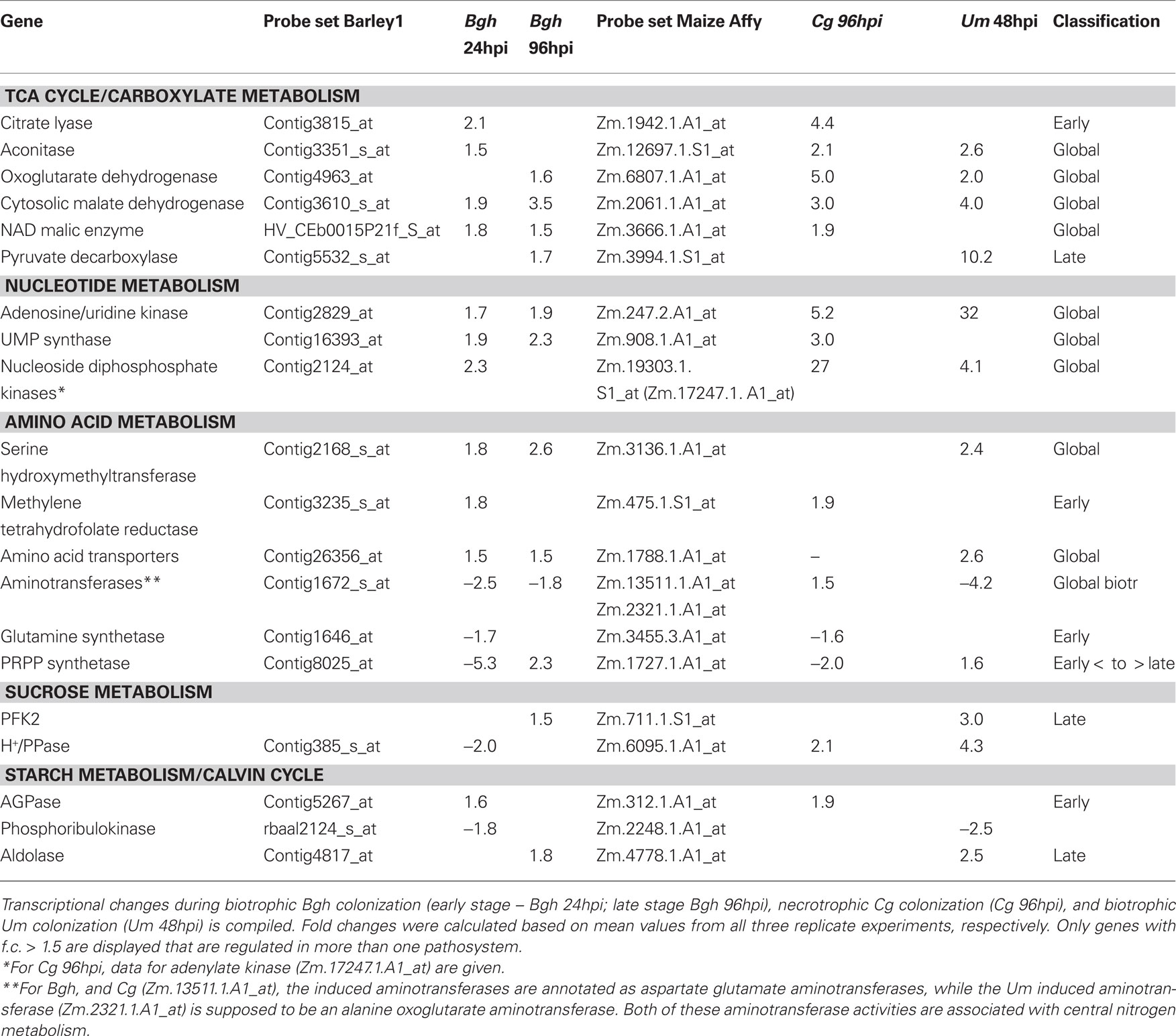
Table 4. Consistent transcriptional changes among the pathosystems.
Thus, a conserved transcriptional program that is activated to redirect primary metabolism during biotrophic interactions does not exist, indicating that the manipulation of host metabolism depends on the individual pathogen and the effector proteins it produces. Nevertheless, some metabolic pathways seem to be consistently addressed on the transcriptional level in all investigated pathosystems. Nitrogen metabolism and energy status appeared to be regulated more consistently on the transcriptional level than carbohydrate metabolism, which might be rather controlled on the post-translational level or by interaction-specific modulations.
Identification of Transcriptional Signatures in the Investigated Pathosystems
Our analysis has only revealed a few genes of central primary metabolism that were regulated in all investigated pathosystems. As stated above, a conserved transcriptional program that is activated to redirect primary metabolism during biotrophic interactions does not exist. Therefore, we set out to identify particular differences in the transcriptional responses of metabolic pathways between the pathosystems. In Figure 6, MapMan representations of those pathways were compiled that exhibit most pronounced differences between early biotrophy in the barley powdery mildew interaction (Bgh 24hpi), biotrophy in the U. maydis-maize interaction (Um 48hpi), necrotrophy (Cg 96hpi), and U. maydis-induced tumors (Um > 96hpi), each representing one cluster in the HCA of the metabolite data depicted in Figure 2. Please note that the transcriptional changes upon Bgh infection were very similar at 24 and 96hpi (also see Figure 5A).
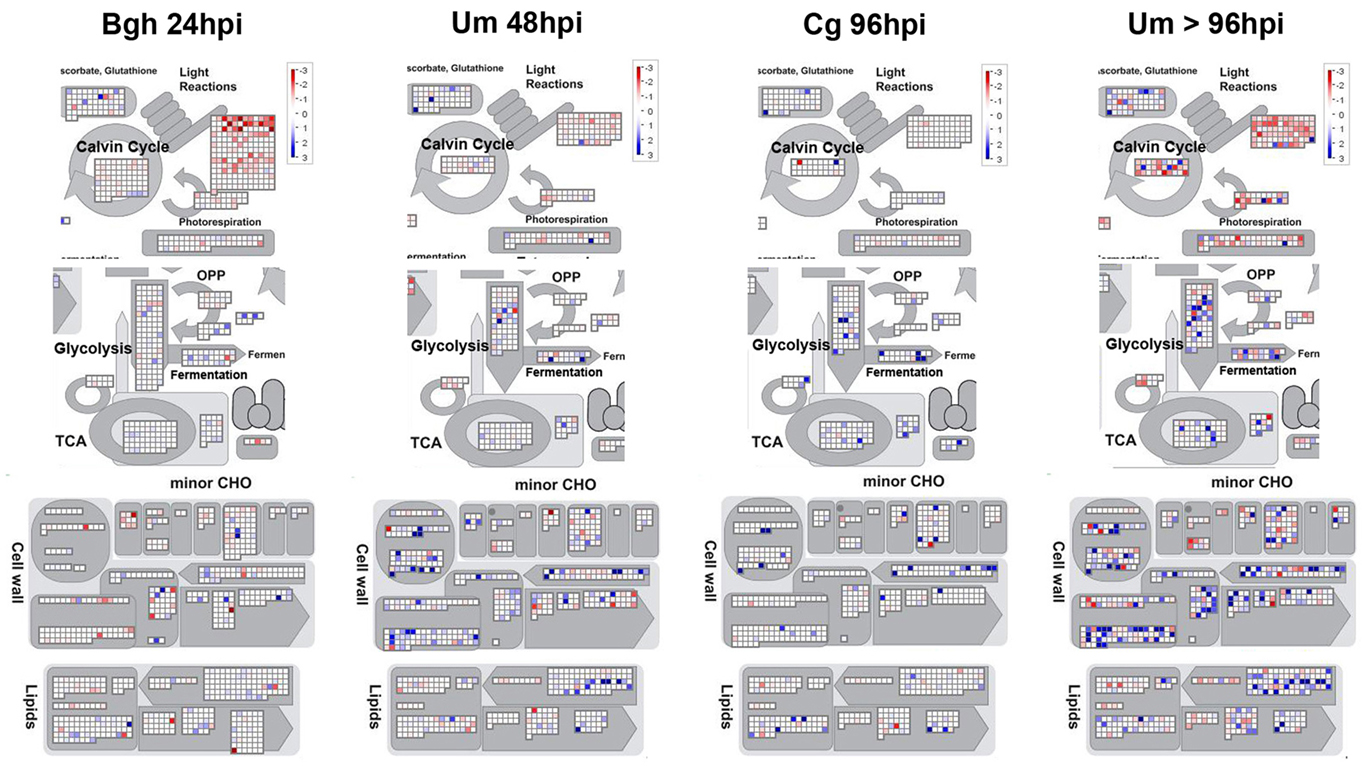
Figure 6. Visualization of specific transcriptional changes between biotrophy, necrotrophy, and tumors by MapMan analysis. Mean fold change of the transcripts in infected vs. mock treated samples was calculated based on all three replicate experiments per time point and log-scaled data were visualized using MapMan with all filters disabled. Data for established biotrophy (Bgh 96hpi and Um 48hpi), necrotrophy (Cg 96hpi), and Um tumors (Um > 96hpi) are displayed from left to right. The scale bar represents fold change and reaches from −3 (red) to +3 (blue) on the log scale, which corresponds to −8-fold (red) to +8-fold (blue) in the linear scale. Photosynthesis (Light reactions), Calvin cycle, and photorespiration are depicted in the upper panel, glycolysis, TCA cycle, and fermentation are shown in the second tier from the top, cell wall metabolism, and minor carbohydrate metabolism (minor CHO) are represented in the second panel from the bottom, while lipid metabolism is displayed in the bottom panel.
As already published (Doehlemann et al., 2008a; Horst et al., 2010a,b), U. maydis-induced tumors exhibit substantial transcriptional changes in almost all displayed metabolic pathways compared to mock control leaves (Figure 6; Table 3). While the majority of the genes involved in the light reaction, the Calvin cycle, and the photorespiratory C2 cycle were transcriptionally repressed in tumors, genes of lipid biosynthesis and remodeling, cell wall biosynthesis were significantly induced in comparison to mock control leaves >4 dpi. More subtle transcriptional differences at early stages of the three interactions could be identified. In Bgh infected leaves, transcriptional suppression of the light reaction is substantially more pronounced, while transcriptional regulation of other metabolic pathways is much weaker compared to Um biotrophy or in Cg necrotrophy (also see Table 3). This corroborates that changes in metabolic flux during Bgh infection are not predominantly caused by transcriptional regulation, but rather by post-translational fine-tuning. During the biotrophic (pre-tumor) colonization of maize by U. maydis, lipid biosynthesis, and cell wall biosynthesis are much stronger induced than in the other two interactions, already reflecting initial hypertrophic growth. Finally, necrotrophic colonization of maize leaves by C. graminicola results in a significant induction of glycolysis, TCA cycle, and fermentation, indicating that an increase in respiratory flux might occur during the challenge with the necrotroph.
Discussion
Consistent Motifs in Metabolic Reprogramming during Plant–Pathogen Interactions
Despite the extensive use of metabolomics for the analysis of plant metabolism (Bino et al., 2004), metabolomic studies of plant–pathogen interactions are rare, most of which rely on data acquisition by FIE-MS and NMR-based metabolite profiling and fingerprinting techniques and subsequent deconvolution by supervised or non-supervised multivariate data analysis (Widarto et al., 2006; Parker et al., 2009; Sana et al., 2010; Ward et al., 2010).
In these approaches, defense-associated metabolites like glucosinolates (Ward et al., 2010), indoles (Ward et al., 2010), and phenylpropanoids (Widarto et al., 2006; Parker et al., 2009; Sana et al., 2010; Ward et al., 2010) were commonly the most prominent metabolite changes that could be identified in infected leaf tissue. The pool sizes of branched-chain amino acids and aromatic amino acids fueling glucosinolate and phenylpropanoid biosynthesis with building blocks increased concomitantly (Parker et al., 2009; Sana et al., 2010; Ward et al., 2010; this study). Nevertheless, elevated Gln and Asn contents in infected leaves represented a major consistent change in primary metabolism at any stage during the biotrophic interactions in the cereal pathosystems investigated in our study (Table 1). Taken together with the reported results from the above cited and other studies (reviewed by Bolton, 2009), this indicates that a substantial reprogramming of central amino acid metabolism takes place already early during infection. It has to be stressed that early after the establishment of the three investigated interactions, only five metabolites exhibited consistent changes, most of which were only moderately significant. Likewise, only few genes coding for enzymes of primary metabolism were consistently altered on the transcriptional level during the early stages of infection. Most of these genes were only deregulated in two out of the three pathosystems. This indicates that the congruence of the metabolic response is rather low in the three examined cereal pathosystems.
Nevertheless, malate dehydrogenase (MDH) was found to be consistently induced early in all interactions we investigated and, with the exception of U. maydis-infected leaves, various isoforms of malic enzyme were also induced swiftly after inoculation (Table 4). Likewise, rice leaves challenged with Magnaporthe grisea (Parker et al., 2009) and Arabidopsis leaves in defense of the hemibiotroph Colletotrichum higginsianum (Voll et al., unpublished) exhibited an induction of malic enzyme activity that was shown to support the global defense response by providing reducing equivalents (Parker et al., 2009), identifying malic enzyme as a conserved player in early, i.e., basal plant defense. Like malic enzyme, MDH, would also produce reducing equivalents from the oxidation of malate in the cytosol, yet producting oxaloacetate instead of pyruvate, thereby competing with ME for the substrate malate.
Cell wall bound invertase is known to be involved in the defense response of several plant species (e.g., Bonfig et al., 2006; Swarbrick et al., 2006; Voegele et al., 2006; Essmann et al., 2008; Horst et al., 2008; Kocal et al., 2008; Siemens et al., 2011). Interestingly, we could only observe an induction of cell wall invertase (cw-INV) at late interaction stages of the two maize pathosystems, indicating that its induction might be slower in maize than in other species. As both malic enzyme (Parker et al., 2009) and invertase (see citations above) have been shown to be induced much stronger and faster in incompatible than in compatible interactions, we can rule out that their transcriptional induction represents a susceptibility factor.
An increase in TCA cycle intermediates Citrate, Malate, Succinate, and Fumarate had been observed during the necrotrophic phase of Magnaporthe grisea infection (Parker et al., 2009). Similarly, we have observed an accumulation of these carboxylates with isocitrate exhibiting the most pronounced increase in maize leaves during necrotrophic colonization with C. graminicola at 96hpi (Table S1 in Supplementary Material). In addition, TCA cycle, glycolysis, and respiration displayed the strongest induction on the transcriptional level in C. graminicola infected leaves at that time point (Figure 6), suggesting that necrotrophic growth in grass species might commonly provoke a strong induction of carboxylate metabolism via the TCA cycle. Increased metabolic flux into carboxylate production via the TCA cycle could either provide ample supply of reducing equivalents and ATP to the host cells or it could indicate increased respiratory flux as a result of impaired photoautotrophy during necrotrophic colonization. We also observed a strong induction of the TCA cycle in U. maydis-induced tumors (Figure 6). However, a thorough inspection disclosed that in leaf tumors, the TCA cycle will most probably provide nitrogen assimilation with carbon skeletons (Horst et al., 2010a,b).
Apart from only few very conserved responses of primary metabolism on the transcriptional and metabolic level between all three pathosystems, we could also identify changes that were quite specific to only one of the pathosystems investigated here. For instance, the induction of the TCA cycle and the accumulation of carboxylates was most pronounced during necrotrophic colonization of maize by Cg (as just discussed), whereas lipid and cell wall biosynthesis were most severely affected in the early interaction of maize leaves with Um, reflecting the initiation of hypertrophic growth. Genes involved in photosynthesis were most quickly suppressed in Bgh infected barley leaves. Given the connection of the Cg and the Um response to particular physiological situation described above, we can assume that these specific effects on the transcriptome and the metabolome of primary metabolism arise from the divergent strategies of the fungal pathogens to manipulate host metabolism. It appears likely, that besides specific targets in defense signaling (as reviewed by de Wit et al., 2009), also different enzymes and metabolic pathways are targeted by the fungi to match the metabolic requirements of the individual pathogens.
Metabolic Changes Are not Caused by a Conserved Transcriptional Reprogramming
Based on the comparison between pathosystems from our study and published data, we could resolve some recurring metabolic motifs in response to pathogen infection.
In the few references available to date, it remains controversial, however, whether the observed changes in metabolism fit to the corresponding transcriptome dynamics in infected leaf tissue. Sana et al. (2010) only reported a weak accordance of metabolome dynamics and the corresponding transcriptional changes in the assessed compatible and incompatible X. oryzae pv. oryzae (Xoo)-rice interactions. In contrast, Ward et al. (2010) stated a quite substantial congruence when aligning their metabolome data with the transcriptome analysis of publically available data for Pst infections on Arabidopsis (Truman et al., 2006). A specific re-assessment of the data by Ward et al. (2010) and Truman et al. (2006) did, however, not reveal a substantial number of regulated genes involved in central carbon and nitrogen metabolism, while the highest agreement of metabolome and transcriptome data was achieved for glucosinolate and phenylpropanoid metabolism (Ward et al., 2010). Similarly, we were also unable to identify a strong congruence between the observed changes in primary metabolism and the corresponding transcript data. Nevertheless, both Sana et al. (2010) and Ward et al. (2010) observed few, but consistent changes in some pathways of primary metabolism on the transcript and the metabolite level, which is in accordance with our results.
Although our metabolome dataset was restricted to quantitative data for only 42 metabolites of central primary metabolism obtained via targeted LC and LC-MS-based methods, we could separate discrete infection stages of the three interactions by HCA, i.e., early biotrophy, established biotrophy, necrotrophy, and tumors (Figure 3), indicating that the information in our dataset provided sufficient divergence. However, we cannot rule out that metabolic flux through certain pathways even differs between those interaction stages that clustered together in the HCA, because our metabolome data comprises of steady state contents that indicate individual metabolite accumulation, but do not reflect metabolic flux.
In addition, multivariate analysis of the metabolome data did not yield comparable results to any transcriptome based analysis in our study. Even if only transcripts coding for proteins involved in the corresponding pathways were regarded, no similarity to the metabolome data could be attained. This could be due to several reasons. First, secreted effectors of Bgh, Cg, and Um are very likely to target different molecular processes in their respective hosts, leading to interaction-specific variation in the observed transcriptional response that could mask common motifs in the defense response. From the complementary point of view, the defense reactions that are not suppressed by the pathogens during the investigated compatible interactions will diverge on the molecular level between the pathosystems. Second, transcript amounts and steady state contents of metabolites, which have been assessed in this study, are not directly correlated with metabolic flux. Primary carbon metabolism is strongly regulated on both, the post-transcriptional and the post-translational level throughout the diurnal cycle (e.g., Gibon et al., 2004), which could lead to a discrepancy between the assessed transcript amounts and actual in vivo activity of most enzymes in central carbon metabolism – which we did not determine. In addition, we have only measured steady state contents of the metabolites included in our metabolome dataset. As outlined above, despite similar steady state pools of most metabolites, flux could be utterly different between two specimen. Nevertheless, we have obtained evidence that allosteric regulation of key steps in central carbon and nitrogen metabolism is likely to account for some of the regulation of metabolic flux during fungal biotrophy, as indicated in the models shown below.
Models for the Redirection of Primary Metabolism during Early Biotrophic Interactions
By analyzing steady state contents of 42 metabolites in primary carbon and nitrogen metabolism, we were able to reveal similarities and differences in the response of host metabolism toward Bgh infection in barley leaves, Cg infection in maize leaves, and Um infection in maize leaves. Together with the transcriptome data obtained from the same samples, we integrated all the information into individual models of host metabolism at early time points in the investigated biotrophic interactions. We assumed that individual changes on the transcriptional and the metabolic level would not necessarily have to be comparable in strength. Therefore, we used a low stringent evaluation of our data for the generation of the presented models of primary metabolism, in order to better allow for comparisons between the pathosystems. When taken together, the integrated transcriptional and metabolite data were highly consistent for most of the depicted pathways in all three analyzed interactions.
Our survey for consistently regulated genes had already revealed that the TCA cycle, nucleotide energy status and amino acid metabolism represented strongly regulated pathways at early stages of all three interactions (Table 4). A concomitant induction of the TCA cycle and nucleotide diphosphate kinases apparently reflects an increase requirement for building blocks, reducing power, and energy in host leaves during the early interaction stage.
Consequently, a comparison of the models for Bgh, Cg, and Um infected leaves during early biotrophic colonization reveals quite similar gross tendencies between two or more pathosystems, despite all the singular differences discussed earlier (Figure 7): (i) the biosynthesis of the major amino acids Gln and Asn as well as of the defense-associated branched-chain and aromatic amino acids are commonly induced, (ii) the Calvin cycle and/or starch biosynthesis are reduced while (iii) glycolysis and the TCA cycle are more frequented. Mostly, (iv) photorespiration is elevated, while sucrose biosynthesis is hampered. Because these changes in primary metabolism are not specific to one particular pathosystem, it appears likely that they are part of a common response of cereal primary metabolism during the early infection phase rather than being associated with particular responses of the hosts toward targeted manipulation by individual pathogens.
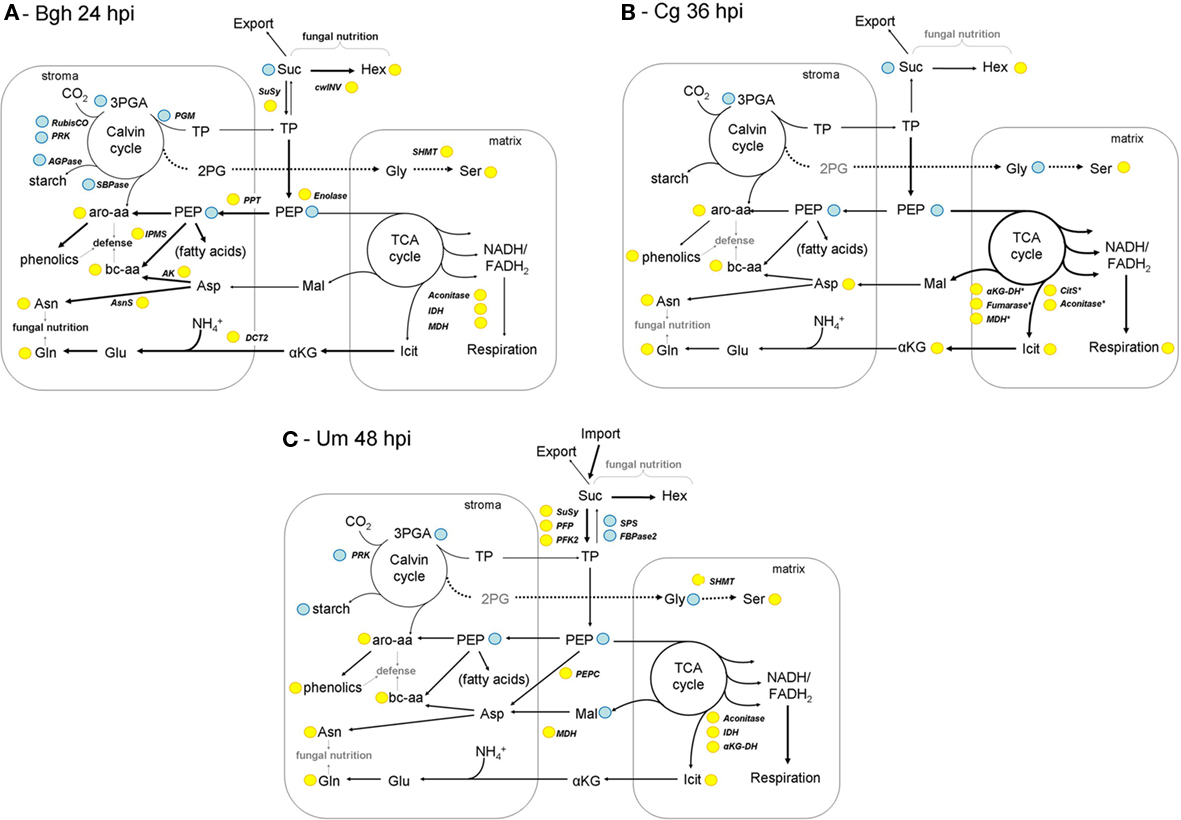
Figure 7. Models of leaf metabolism during early interaction stages. Based on the results of the combined metabolome and transcriptome analysis, models illustrating the reprogramming of host metabolism during early biotrophic interactions are depicted for Bgh infected barley leaves at 24hpi (A), Cg infected maize leaves at 36hpi (B) and Um infected maize leaves at 48hpi (C). Please note that for simplicity, C4 metabolism has been omitted from the maize models. Yellow – up compared to mock control; blue – down compared to mock control. Arrow thickness correlates with the proposed metabolic flux relative to the other depicted metabolic pathways. For explanations, please see the discussion text. Amino acids are abbreviated according to three letter code, 2PG, (2-phosphoglycolate); aKG, (α-ketoglutarate) Hex (hexoses); Icit, (isocitrate); PEP, (phosphoenol pyruvate); 3-PGA, (3-phosphoglycerate); Suc, (sucrose); TP (triose phosphates); αKG-DH, (α-ketoglutarate dehydrogenase); AK, (aspartate kinase); AsnS, (asparagine synthetase); CitS, (citrate synthase); cw-INV, (cell wall invertase); DCT2, (dicarboxylate translocator); FBPase2, (fructose-2,6-bisphosphatase); IDH, (isocitrate dehydrogenase); IPMS, (isopropylmalate synthase); MDH, (malate dehydrogenase); PEPC, (PEP carboxylase); PFK2, (phosphofructokinase 2); PFP, (pyrophosphate-dependent phosphofructokinase); PPT, (phosphoenolpyruvate/phosphate translocator); SHMT, (serine hydroxymethyl transferase); SPS, (sucrose phosphate synthase); SuSy, (sucrose synthase).
As suggested by our comparative analyses, there are important differences between the interactions. In the early powdery mildew (Bgh) infection (Figure 7A), the Calvin cycle genes RubisCO and phosphoribulokinase become transcriptionally repressed, which is also reflected by diminished 3-PGA contents that commonly correlate with activity of the Calvin cycle. Based on steady state metabolite contents and the transcriptome data, metabolic flux appears to be directed toward the biosynthesis of free amino acids, with the major amino acids Gln and Asn representing transient stores for organic N. In contrast, the TCA cycle is induced on the transcriptional level, but the involved carboxylates did not accumulate. Therefore, the depletion of the PEP pool (including the related transcriptional changes) can rather be interpreted due to its anaplerotic function of plastid-localized biosynthesis of branched-chain and aromatic amino acids (Schulze-Siebert et al., 1984; Herrmann and Weaver, 1999). As both plastidic and cytosolic protein biosynthesis are significantly induced processes in the barley–Bgh interaction, it appears likely that elevated production of amino acids will serve as building blocks for both PR proteins and low-molecular weight compounds like glucosinolates and phenylpropanoids. Sucrose breakdown by cw-INV and sucrose synthase (SuSy) could lead to an increase in the hexose/sucrose ratio. Due to the transcriptional repression of the Calvin cycle and the photosynthetic electron transport chain (Figure 6) diminished triose phosphate export and increased flux toward PEP could limit sucrose biosynthesis in turn. An accumulation of UDPglc further indicates diminished formation of sucrose in Bgh infected leaves.
During the biotrophic phase of the C. graminicola infection at 36hpi (Figure 7B), almost no transcriptional changes were observed, in contrast to numerous changes in steady state metabolite contents. The observed changes in metabolite contents are either the result of endogenous post-transcriptional, perhaps of allosteric regulation, or are due to altered flux through the respective pathways determined by substrate availability or substrate compartmentation, or it might be effectuated by the action of fungal effectors. As indicated by the high similarity to Bgh 24hpi in the HCA analysis of metabolite data (Figure 2), the changes in the metabolome of Cg 36hpi are almost congruent to those of Bgh infected barley leaves at 24hpi, rendering it unlikely that secreted effectors of Bgh and Cg exert identical effects on host metabolism. Furthermore, the sampled maize leaves perform C3–C4 intermediate photosynthesis, while barley is a C3-plant. The only substantial difference between the Bgh infected barley and the Cg infected maize leaves is an increased accumulation of the TCA cycle intermediates and amino acid building blocks α-ketoglutarate and isocitrate. At 96hpi, the majority of the genes involved in the TCA cycle are strongly induced on the transcriptional level in Cg infected leaves. Besides, an increased Gly/Ser ratio indicates increased photorespiration during biotrophic colonization of maize leaves with C. graminicola.
Maize leaves infected with U. maydis differ in two important aspects from the two previously regarded pathosystems (Figure 7C). First, the accumulation of free amino acids resembles the previously described situation for Bgh and Cg, except for the fact that anaplerotic provision of carbon skeletons by the TCA cycle does not appear to be substantially induced. Second, the balance of sucrose biosynthesis and sucrose degradation seems to be strongly regulated in favor of glycolytic utilization of sucrose on the transcriptional level. As discussed by Horst et al. (2008) for U. maydis-induced tumors, the altered regulation of sucrose metabolism indicates that the developing leaves at 48hpi might represent sink characteristics and cover part of their carbohydrate budget by import of sugar (as indicated in Figure 7C). In this light, increased amino acid contents without clearly elevated supply of carbon moieties by the TCA cycle might also indicate that part of the free amino acid pool is replenished by import from systemic leaves (as discussed for tumors in Horst et al., 2010b).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the Deutsche Forschungsgemeinschaft (DFG) in the framework of the priority program FOR 666.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/plant_physiology/10.3389/fpls.2011.00039/abstract/
Table S1|Complete metabolome data and calculated metabolite ratios.
Footnotes
References
Banuett, F. (1995). Genetics of Ustilago maydis, a fungal pathogen that induces tumors in maize. Annu. Rev. Genet. 29, 179–208.
Bergstrom, G. C., and Nicholson, R. L. (1999). The biology of corn anthracnose - knowledge for improved management. Plant Dis. 83, 596–608.
Biemelt, S., and Sonnewald, U. (2006). Plant–microbe interactions to probe the regulation of plant carbon metabolism. J. Plant Physiol. 163, 307–318.
Bino, R. J., Hall, R. D., Fiehn, O., Kopka, J., Saito, K., Draper, J., Nikolau, B. J., Mendes, P., Roessner-Tunali, U., Beale, M. H., Trethewey, R. N., Lange, B. M., Wurtele, E. S., and Sumner, L. W. (2004). Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 9, 418–425.
Bolton, M. D. (2009). Primary metabolism and plant defense – fuel for the fire. Mol. Plant Microbe Interact. 22, 487–497.
Bonfig, K. B., Schreiber, U., Gabler, A., Roitsch, T., and Berger, S. (2006). Infection with virulent and avirulent P. syringae strains differentially affects photosynthesis and sink metabolism in Arabidopsis leaves. Planta 225, 1–12.
Both, M., Csukai, M., Stumpf, M. P. H., and Spanu, P. D. (2005). Gene expression profiles of Blumeria graminis indicate dynamic changes to primary metabolism during development of an obligate biotrophic pathogen. Plant Cell 17, 2107–2122.
Chanda, B., Venugopal, S. C., Kulshrestha, S., Navarre, D. A., Downie, B., Vaillancourt, L., Kachroo, A., and Kachroo, P. (2008). Glycerol-3-phosphate levels are associates with basal resistance to the hemibiotrophic fungus Colletotrichum higginsianum in Arabidopsis. Plant Physiol. 147, 2017–2029.
Chaturvedi, R., Krothapalli, K., Makandar, R., Nandi, A., Sparks, A. A., Roth, M. R., Welti, R., and Shah, J. (2008). Plastid omega 3-fatty acid desaturase-dependent accumulation of a systemic acquired resistance inducing activity in petiole exudates of Arabidopsis thaliana is independent of jasmonic acid. Plant J. 54, 106–117.
Chen, L.-Q., Hou, B.-H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X.-Q., Guo, W.-J., Kim, J.-G., Underwood, W., Chaudhuri, B., Chermak, D., Antony, G., White, F. F., Sommerville, S. C., Mudgett, M. B., and Frommer, W. B. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532.
Chou, H. M., Bundock, N., Rolfe, S. A., and Scholes, J. D. (2000). Infection of Arabidopsis thaliana leaves with Albugo candida (white blister rust) causes a reprogramming of host metabolism. Mol. Plant Pathol. 1, 99–113.
de Wit, P. J. G. M., Mehrabi, R., van den Burg, H. A., and Stergiopoulos, I. (2009). Fungal effector proteins: past, present and future. Mol. Plant Pathol. 10, 735–747.
Divon, H. H., and Fluhr, R. (2007). Nutrition acquisition strategies during fungal infection of plants. FEMS Microbiol. Lett. 266, 65–74.
Doehlemann, G., Wahl, R., Horst, R. J., Voll, L., Usadel, B., Poree, F., Stitt, M., Pons-Kuehnemann, J., Sonnewald, U., Kahmann, R., and Kämper, J. (2008a). Reprogramming a maize plant: transcriptional and metabolic changes induced by the fungal biotroph Ustilago maydis. Plant J. 56, 181–195.
Doehlemann, G., Wahl, R., Vranes, M., de Vries, R. P., Kämper, J., and Kahmann, R. (2008b). Establishment of compatibility in the Ustilago maydis/maize pathosystem. J. Plant Physiol. 165, 29–40.
Eisen, M. B., Spellman, P. T., Brown, P. O., and Botstein, D. (1998). Cluster analysis and display of genome-wide expression patterns. PNAS 95, 14863–14868.
Essmann, J., Schmitz-Thom, I., Schon, H., Sonnewald, S., Weis, E., and Scharte, J. (2008). RNA interference-mediated repression of cell wall invertase impairs defense in source leaves of tobacco. Plant Physiol. 147,1288–1299.
Gerhardt, R., Stitt, M., and Heldt, H. W. (1987). Subcellular metabolite levels in spinach leaves: regulation of sucrose synthesis during diurnal alterations in photosynthetic partitioning. Plant Physiol. 83, 399–407.
Gibon, Y., Blaesing, O. E., Hannemann, J., Carillo, P., Höhne, M., Hendriks, J. H. M., Palacios, N., Cross, J., Selbig, J., and Stitt, M. (2004). A robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. Plant Cell 16, 3304–3325.
Hahn, M., Neef, U., Struck, C., Gottfert, M., and Mendgen, K. (1997). A putative amino acid transporter is specifically expressed in haustoria of the rust fungus Uromyces fabae. Mol. Plant Microbe Interact. 10, 438–445.
Heineke, D., Wildenberger, K., Sonnewald, U., Willmitzer, L., and Heldt, H.-W. (1994). Accumulation of hexoses in leaf vacuoles – studies with transgenic tobacco plants expressing yeast-derived invertase in the cytosol, vacuole or appoplasm. Planta 194, 29–33.
Herrmann, K. M., and Weaver, L. M. (1999). The shikimate pathway. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 473–503.
Horst, R. J., Doehlemann, G., Wahl, R., Kahmann, R., Kämper, J., Sonnewald, U., and Voll, L. M. (2010a). Ustilago maydis infection strongly alters organic nitrogen allocation in maize and stimulates productivity of systemic source leaves. Plant Physiol. 152, 293–308.
Horst, R. J., Doehlemann, G., Wahl, R., Hofmann, J., Schmiedl, A., Kahmann, R., Kämper, J., and Voll, L. M. (2010b). A model of Ustilago maydis leaf tumor metabolism. Plant Signal. Behav. 5, 1446–1449.
Horst, R. J., Engelsdorf, T., Sonnewald, U., and Voll, L. M. (2008). Infection of maize leaves with Ustilago maydis prevents establishment of C-4 photosynthesis. J. Plant Physiol. 165, 19–28.
Hückelhoven, R., Fodor, J., Preis, C., and Kogel, K. H. (1999). Hypersensitive cell death and papilla formation in barley attacked by the powdery mildew fungus are associated with h2o2 accumulation but are not accompanied by enhanced concentrations of salicylic acid. Plant Physiol. 119, 1251–1260.
Jain, S. K., Langen, G., Hess, W., Borner, T., Huckelhoven, R., and Kogel, K.-H. (2004). The white barley mutant albostrians shows enhanced resistance to the biotroph Blumeria graminis f. sp. hordei. Mol. Plant Microbe Interact. 17, 374–382.
Kachroo, A., Lapchyk, L., Fukushige, H., Hildebrand, D., Klessig, D., and Kachroo, P. (2003). Plastidial fatty acid signaling modulates salicylic acid- and jasmonic acid-mediated defense pathways in the Arabidopsis ssi2 mutant. Plant Cell 15, 2952–2965.
Kahmann, R., and Kämper, J. (2004). Ustilago maydis: how its biology relates to pathogenic development. New Phytol. 164, 31–42.
Kämper, J., Kahmann, R., Bolker, M., Ma, L.-J., Brefort, T., Saville, B. J., Banuett, F., Kronstad, J. W., Gold, S. E., Muller, O., Perlin, M. H., Wosten, H. A. B., de Vries, R., Ruiz-Herrera, J., Reynaga-Pena, C. G., Snetselaar, K., McCann, M., Perez-Martin, J., Feldbrugge, M., Basse, C. W., Steinberg, G., Ibeas, J. I., Holloman, W., Guzman, P., Farman, M., Stajich, J. E., Sentandreu, R., Gonzalez-Prieto, J. M., Kennell, J. C., Molina, L., Schirawski, J., Mendoza-Mendoza, A., Greilinger, D., Munch, K., Rossel, N., Scherer, M., Vranes, M., Ladendorf, O., Vincon, V., Fuchs, U., Sandrock, B., Meng, S., Ho, E. C. H., Cahill, M. J., Boyce, K. J., Klose, J., Klosterman, S. J., Deelstra, H. J., Ortiz-Castellanos, L., Li, W., Sanchez-Alonso, P., Schreier, P. H., Hauser-Hahn, I., Vaupel, M., Koopmann, E., Friedrich, G., Voss, H., Schluter, T., Margolis, J., Platt, D., Swimmer, C., Gnirke, A., Chen, F., Vysotskaia, V., Mannhaupt, G., Guldener, U., Munsterkotter, M., Haase, D., Oesterheld, M., Mewes, H.-W., Mauceli, E. W., DeCaprio, D., Wade, C. M., Butler, J., Young, S., Jaffe, D. B., Calvo, S., Nusbaum, C., Galagan, J., and Birren, B. W. (2006). Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444, 97–101.
Kocal, N., Sonnewald, U., and Sonnewald, S. (2008). Cell wall-bound invertase limits sucrose export and is involved in symptom development and inhibition of photosynthesis during compatible interaction between tomato and Xanthomonas campestris pv vesicatoria. Plant Physiol. 148, 1523–1536.
Leidreiter, K., Kruse, A., Heineke, D., Robinson, D. G., and Heldt, H.-W. (1995). Subcellular volumes and metabolite concentrations in potato (Solanum tuberosum cv. Désirée) leaves. Bot. Acta 108, 439–444.
Mendgen, K., and Hahn, M. (2002). Plant infection and the establishment of fungal biotrophy. Trends Plant Sci. 7, 352–356.
Molitor, A., Zajic, D., Voll, L. M., Pons-Kühnemann, J., Kogel, K. H., and Waller, F. (2011). Transcriptome and metabolite analysis of the barley-powdery mildew interaction reveals priming as a mechanism of P. indica-induced systemic resistance. Mol. Plant Microbe Interact. [Epub ahead of print].
Münch, S., Lingner, U., Floss, D. S., Ludwig, N., Sauer, N., and Deising, H. (2008). The hemibiotrophic lifestyle of Colletotrichum species. J. Plant Physiol. 165, 41–51.
Münch, S., Ludwig, N., Floß, D. S., Sugui, J. A., Koszucka, A. M., Voll, L. M., Sonnewald, U., and Deising, H. B. (2011). Identification of virulence genes in the corn pathogen Colletotrichum graminicola by Agrobacterium tumefaciens-mediated transformation. Mol. Plant Pathol. 12, 43–55.
Olea, F., Perez-Garcia, A., Canton, F. R., Rivera, M. E., Canas, R., Avila, C., Cazorla, F. M., Canovas, F. M., and de Vicente, A. (2004). Up-regulation and localization of asparagine synthetase in tomato leaves infected by the bacterial pathogen Pseudomonas syringae. Plant Cell Physiol. 45, 770–780.
Parker, D., Beckmann, M., Zubair, H., Enot, D. P., Caracuel-Rios, Z., Overy, D. P., Snowdon, S., Talbot, N. J., and Draper, J. (2009). Metabolomic analysis reveals a common pattern of metabolic re-programming during invasion of three host plant species by Magnaporthe grisea. Plant J. 59, 723–737.
Raffaele, S., Vailleau, F., Léger, A., Joubès, J., Miersch, O., Huard, C., Blée, E., Mongrand, S., Domergue, F., and Roby, D. (2008). A MYB transcription factor regulates very-long-chain fatty acid biosynthesis for activation of the hypersensitive cell death response in Arabidopsis. Plant Cell 20, 752–67.
Sana, T. R., Fischer, S., Wohlgemuth, G., Katrekar, A., Jung, K. H., Ronald, P. C., and Fiehn, O. (2010). Metabolomic and transcriptomic analysis of the rice response to the bacterial blight pathogen Xanthomonas oryzae pv. oryzae. Metabolomics 6,451–465.
Scharte, J., Schön, H., Tjaden, Z., Weis, E., and von Schaewen, A. (2009). Isoenzyme replacement of glucose-6-phosphate dehydrogenase in the cytosol improves stress tolerance in plants. Proc. Natl. Acad. Sci. U.S.A. 106, 8061–8066.
Schulze-Siebert, D., Heineke, D., Scharf, H., and Schulz, G. (1984). Pyruvate-derived amino acids in spinach chloroplasts: synthesis and regulation during photosynthetic carbon metabolism. Plant Physiol. 76, 465–71.
Siemens, J., González, M. C., Wolf, S., Hofmann, C., Greiner, S., Du, Y., Rausch, T., Roitsch, T., and Ludwig-Müller, J. (2011). Extracellular invertase is involved in the regulation of clubroot disease in Arabidopsis thaliana. Mol. Plant Pathol. 12, 247–262.
Struck, C., Ernst, M., and Hahn, M. (2002). Characterization of a developmentally regulated amino acid transporter (AAT1p) of the rust fungus Uromyces fabae. Mol. Plant Pathol. 3, 23–30.
Struck, C., Mueller, E., Martin, H., and Lohaus, G. (2004). The Uromyces fabae UfAAT3 gene encodes a general amino acid permease that prefers uptake of in planta scarce amino acids. Mol. Plant Pathol. 5, 183–189.
Swarbrick, P. J., Schulze-Lefert, P., and Scholes, J. D. (2006). Metabolic consequences of susceptibility and resistance (race-specific and broad-spectrum) in barley leaves challenged with powdery mildew. Plant Cell Environ. 29, 1061–1076.
Tavernier, V., Cadiou, S., Pageau, K., Lauge, R., Reisdorf-Cren, M., Langin, T., and Masclaux-Daubresse, C. (2007). The plant nitrogen mobilization promoted by Colletotrichum lindemuthianum in Phaseolus leaves depends on fungus pathogenicity. J. Exp. Bot. 58, 3351–3360.
Thimm, O., Bläsing, O., Gibon, Y., Nagel, A., Meyer, S., Krüger, P., Selbig, J., Müller, L. A., Rhee, S. Y., and Stitt, M. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939.
Truman, W., de Zabala, M. T., and Grant, M. (2006). Type III effectors orchestrate a complex interplay between transcriptional networks to modify basal defence responses during pathogenesis and resistance. Plant J. 46, 14–33.
van Kan, J. A. L. (2006). Licensed to kill: the lifestyle of a necrotrophic plant pathogen. Trends Plant Sci. 11, 247–253.
Voegele, R. T., Struck, C., Hahn, M., and Mendgen, K. (2001). The role of haustoria in sugar supply during infection of broad bean by the rust fungus Uromyces fabae. Proc. Natl. Acad. Sci. U.S.A. 98, 8133–8138.
Voegele, R. T., Wirsel, S., Möll, U., Lechner, M., and Mendgen, K. (2006). Cloning and characterization of a novel invertase from the obligate biotroph Uromyces fabae and analysis of expression patterns of host and pathogen invertases in the course of infection. Mol. Plant Microbe Interact. 19, 625–634.
Wahl, R., Wippel, K., Goos, S., and Kämper, J. (2010). Norbert Sauer a novel high-affinity sucrose transporter is required for virulence of the plant pathogen Ustilago maydis. PLoS Biol. 8, e1000303. doi: 10.1371/journal.pbio.1000303
Ward, J. L., Forcat, S., Beckmann, M., Bennett, M., Miller, S. J., Baker, J. M., Hawkins, N. D., Vermeer, C. P., Lu, C., Lin, W., Truman, W. M., Beale, M. H., Draper, J., Mansfield, J. W., and Grant, M. (2010). The metabolic transition during disease following infection of Arabidopsis thaliana by Pseudomonas syringae pv. tomato. Plant J. 63, 443–457.
Wiberg, A. (1974). Genetical studies of spontaneous sources of resistance to powdery mildew in barley. Hereditas 77, 89–148.
Widarto, H. T., Van Der Meijden, E., Lefeber, A. W., Erkelens, C., Kim, H. K., Choi, Y. H., and Verpoorte, R. (2006). Metabolomic differentiation of Brassica rapa following herbivory by different insect instars using two-dimensional nuclear magnetic resonance spectroscopy. J. Chem. Ecol. 32, 2417–2428.
Keywords: maize, barley, Ustilago maydis, Blumeria graminis f.sp. hordei, Colletotrichum graminicola, compatible interaction, metabolite analysis, transcriptome analysis
Citation: Voll LM, Horst RJ, Voitsik A-M, Zajic D, Samans B, Pons-Kühnemann J, Doehlemann G, Münch S, Wahl R, Molitor A, Hofmann J, Schmiedl A, Waller F, Deising HB, Kahmann R, Kämper J, Kogel K-H and Sonnewald U (2011) Common motifs in the response of cereal primary metabolism to fungal pathogens are not based on similar transcriptional reprogramming. Front. Plant Sci. 2:39. doi: 10.3389/fpls.2011.00039
Received: 19 May 2011; Accepted: 01 August 2011;
Published online: 22 August 2011.
Edited by:
Alisdair Fernie, Max Planck Institute for Plant Physiology, GermanyReviewed by:
Andreas P. M. Weber, University of Duesseldorf, GermanyVeronica Graciela Maurino, Heinrich-Heine-Universität Düsseldorf, Germany
Copyright: © 2011 Voll, Horst, Voitsik, Zajic, Samans, Pons-Kühnemann, Doehlemann, Münch, Wahl, Molitor, Hofmann, Schmiedl, Waller, Deising, Kahmann, Kämper, Kogel and Sonnewald. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Lars Matthias Voll, Division of Biochemistry, Friedrich-Alexander-University Erlangen-Nuremberg, Staudtstrasse 5, D-91058 Erlangen, Germany. e-mail: lvoll@biologie.uni-erlangen.de