 Dmitriy Shevela
Dmitriy Shevela Johannes Messinger
Johannes Messinger- Department of Chemistry, Chemistry Biology Centre, Umeå University, Umeå, Sweden
Monitoring isotopic compositions of gaseous products (e.g., H2, O2, and CO2) by time-resolved isotope-ratio membrane-inlet mass spectrometry (TR-IR-MIMS) is widely used for kinetic and functional analyses in photosynthesis research. In particular, in combination with isotopic labeling, TR-MIMS became an essential and powerful research tool for the study of the mechanism of photosynthetic water-oxidation to molecular oxygen catalyzed by the water-oxidizing complex of photosystem II. Moreover, recently, the TR-MIMS and 18O-labeling approach was successfully applied for testing newly developed catalysts for artificial water-splitting and provided important insight about the mechanism and pathways of O2 formation. In this mini-review we summarize these results and provide a brief introduction into key aspects of the TR-MIMS technique and its perspectives for future studies of the enigmatic water-splitting chemistry.
Introduction
In nature, the splitting of water to molecular oxygen (O2) is catalyzed by the membrane-bound pigment-protein photosystem II (PSII) of plants, algae, and cyanobacteria (Vinyard et al., 2013). The catalytic site of the water-splitting reaction is an inorganic tetra-manganese mono-calcium penta-oxygen (Mn4CaO5) cluster (Figure 1) that forms, together with its protein ligands, the water-oxidizing complex (WOC) of PSII (Yano et al., 2006; Umena et al., 2011). Water-splitting by the Mn4CaO5 cluster is energetically driven by the strongest biological oxidant, P680+ (with a midpoint potential of ~1.25 V), generated by the light-induced charge separation within the Chl-containing reaction center (RC) of PSII (Diner and Rappaport, 2002; Ishikita et al., 2005). A redox-active tyrosine residue (YZ) is the essential electron transfer intermediate between the photoactive RC and the Mn4CaO5 cluster of PSII. Following light absorption, the Mn4CaO5 cluster is oxidized step-wise (one electron at a time) and thereby cycles through five redox states, known as Si states (where i reflects the number of oxidizing equivalents stored by the cluster) (Figure 1). The four-electron four-proton oxidation chemistry of two water molecules is completed when the four oxidizing equivalents are accumulated within the WOC, and the highly reactive S4 state relaxes into the most reduced S0 state with the concomitant O–O bond formation and release of O2 (Messinger et al., 2012; Cox and Messinger, 2013). This reaction cycle of water oxidation is also known as the Kok cycle (Kok et al., 1970).
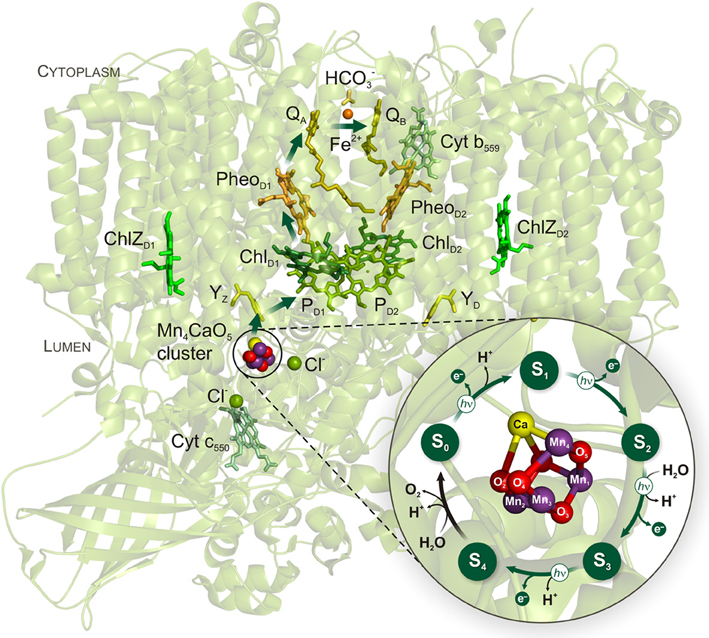
Figure 1. Cyanobacterial PSII structure and Kok cycle of photosynthetic water oxidation by the Mn4CaO5 cluster. The arrows within PSII indicate the direction of electron transfer which comprises the following redox-active cofactors: inorganic Mn4CaO5 cluster, redox-active tyrosine Z (YZ), the primary electron donor P680 that includes a pair of Chls a (PD1 and PD2) and two accessory Chls (ChlD1 and ChlD2), the primary pheophytin (PheoD1) acceptor, the primary (QA) and the secondary (QB) quinone acceptors. The phytyl tails of the Chl's and Pheo's, and the isoprenyl chains of the quinones have been cut for clarity. The light-induced S state transitions of the Mn4CaO5 cluster are indicated by arrows with “hν” labels. The PSII structure and the zoomed structural model of the Mn4CaO5 cluster in the center of the Kok cycle are based on the recent PSII crystal structure at a resolution of 1.9 Å (PDB entry 3ARC; Umena et al., 2011).
During the last few decades an enormous progress in elucidation of the WOC structure and in understanding the mechanism of the water-splitting became possible due to employment of numerous biophysical techniques (summarized in Aartsma and Matysik, 2008; Messinger et al., 2009a,b; also see refs therein). Among them, time-resolved isotope-ratio membrane-inlet mass spectrometry (TR-IR-MIMS) in combination with isotope labeling (Konermann et al., 2008; Beckmann et al., 2009) provided the most direct information on the Si state dependent substrate water binding to the WOC (Messinger et al., 1995; Wydrzynski et al., 1996). These findings were recently reviewed in detail by Hillier and Wydrzynski (2008), Messinger et al. (2012), and Cox and Messinger (2013) and are, therefore, only briefly discussed here. However, TR-MIMS has also been successfully employed and yielded important data on other structural and mechanistic aspects of the water-splitting chemistry in both natural PSII and in variously designed artificial O2-evolving catalysts. In this mini-review, we summarize these recent investigations and also provide some comments on perspectives of the TR-MIMS technique for future studies of water-splitting and O2 evolution.
Key Concepts of TR-MIMS
The concept of TR-MIMS was first applied in 1963, when Georg Hoch and Bessel Kok began to use mass spectrometer with a semipermeable membrane as inlet system (Hoch and Kok, 1963). This allowed to separate the liquid sample from the high vacuum space of the mass spectrometer, while at the same time it was permeable to the gaseous analytes. This excellent solution allowed continuous on-line measurements of dissolved gaseous analytes (either dissolved in solution or directly from the gas phase) with a temporal resolution of a few seconds. Therefore, the TR-MIMS technique is ideally suited for investigations of photosynthetic and artificial water-oxidation/O2 evolution (for instance, see Konermann et al., 2008; Beckmann et al., 2009). For an outline of other TR-MIMS applications in biological and in industrial systems, see reviews by Lauritsen and Kotiaho (1996) and Johnson et al. (2000). Recent technological advances in MIMS instrumentation are summarized in Davey et al. (2011).
A schematic view of a TR-MIMS set-up employing an isotope ratio mass spectrometer is shown in Figure 2. This type of mass spectrometers is normally equipped with an electron-impact ion source, magnetic sector field analyzer, and individual detectors (Faraday cups) that provide simultaneous detection of several masses (ions) with high sensitivity and signal stability. For its ability to monitor and to selectively analyze all isotopologues (molecules that differ only in their isotopic composition) of gaseous products with one instrument, the TR-MIMS approach in combination with isotope enrichments became indispensable tool for kinetic and functional analyses of photosynthetic enzymes (Konermann et al., 2008; Beckmann et al., 2009). The key part of the TR-MIMS instrument is a gas inlet system that is integrated within a MIMS cell. The design of MIMS cells may vary depending on the measuring purposes (Konermann et al., 2008; Beckmann et al., 2009), but all of them contain a gas-permeable membrane functioning as analyte inlet system into the vacuum of the mass spectrometer. The coupling of such a cell to various light sources (e.g., Xenon lamps or lasers) allows carrying out the measurements of light-induced O2 evolution in photosynthetic samples or light-driven O2-evolving artificial catalysts. Before entering the ion source of the mass spectrometer the analytes pass through a cryogenic trap (Figure 2), which freezes out water vapor that inadvertently pervaporate through the membrane in trace amounts.
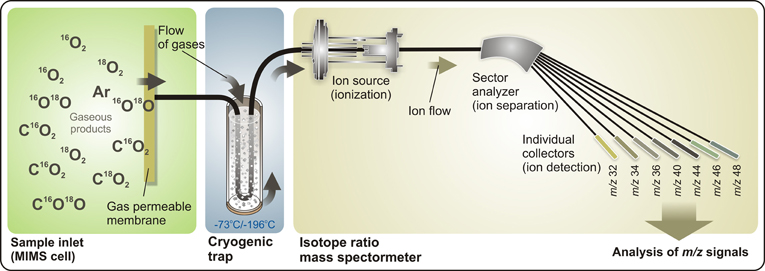
Figure 2. Representation of a TR-IR-MIMS set-up. Gaseous products, produced by sample suspension (for instance, by PSII samples) in the cell, penetrate through a gas-permeable membrane into a high-vacuum space, pass through a cryogenic trap (which removes water vapor from a flow of gaseous analytes), and enter the isotope ratio mass spectrometer. Here, gaseous analytes are first ionized in the ion source by electron impact, and are then separated according to their m/z ratios by a magnetic field in the sector analyzer that allows simultaneous online detection by individual collector cups (e.g., a 7-cup Faraday detector array). MS signals are monitored and analyzed using a personal computer. See text for further details.
Enrichment of aqueous sample suspension with oxygen's heavy isotope (18O) for isotope ratio measurements of O2 (and/or CO2) isotopologues is a powerful and commonly used tool in studies of water-splitting chemistry and/or related reactions. Therefore, most of the experiments are carried out in H182O-labeled sample suspensions/solutions.
Is Water the Immediate Substrate of Photosynthetic O2 Evolution?
It is widely accepted that water is the immediate substrate for photosynthetic O2 production. However, Metzner (1978) suggested that instead hydrogen carbonate (bicarbonate; HCO−3) is the immediate substrate for O2 formation that is subsequently replenished by the reaction of CO2 with H2O. This hypothesis was discounted for long because the isotopic equilibration between 18O-water and HCO−3 is too slow to account for early isotope labeling studies (Ruben et al., 1941; Stemler and Radmer, 1975; Stevens et al., 1975; Radmer and Ollinger, 1980). Due to the discovery that a carbonic anhydrase (CA) activity is associated with PSII (Lu and Stemler, 2002; Villarejo et al., 2002; Moskvin et al., 2004) the “bicarbonate-as-substrate hypothesis” needed to be re-investigated with refined expriments. Indeed, due to rapid exchange of HCO−3 and CO2 species by CA, the 18O-label could “escape” from HCO−3 to water (which has a several orders higher concentration than the added 18O-labeled HCO−3), and, thus, lead to the lack of O2 yield from HCO−3 (Stemler and Radmer, 1975; Radmer and Ollinger, 1980).
Two different TR-MIMS approaches were taken recently and both exclude that HCO−3 is a physiologically significant substrate (Clausen et al., 2005a; Hillier et al., 2006). Clausen et al. (2005a) reported that under H182O-labeled and CO2/HCO−3-depleted conditions the typical oscillation pattern of 18O-enriched O2 evolution is obtained in response to single light flashes, but didn't find any evidence for CO2 release. The latter would be expected in case the Metzner's hypothesis would be correct. Hillier et al. (2006), in their TR-MIMS study, employed 18O/13C-labeled HCO−3 to probe the capability of PSII (from higher plants and cyanobacteria) to oxidize HCO−3. The authors were able to detect an extremely small (and, thus, non-physiological) flux of 18O from HCO−3 into O2 similar to that observed in an early TR-MIMS study of Radmer and Ollinger (1980). Moreover, no relationship between O2 evolution and PSII-associated CA activity was found by McConnell et al. (2007) in their TR-MIMS examination of PSII preparations from higher plants.
Is Hydrogen Carbonate A Ligand to the WOC?
Hydrogen carbonate had been proposed to bind as integral cofactor to the Mn4CaO5 cluster after accumulation of many experimental results indicating: (i) the requirement of HCO−3 ions for optimal stability and functionality of the WOC (Van Rensen and Klimov, 2005), and (ii) it's important role for the photoassembly reaction of the Mn4CaO5 cluster (Dasgupta et al., 2008). Moreover, in the PSII crystal structure from Thermosynechococcus elongatus at 3.5 Å resolution, HCO−3 was modeled as a non-protein ligand bridging Mn and Ca ions within the WOC (Ferreira et al., 2004).
Earlier, interesting TR-MIMS experiments were performed by Stemler (1989) and Govindjee et al. (1991), in which formate was tested to induce the release of HCO−3 (that can be detected by TR-MIMS as CO2) from PSII. Although Govindjee et al. (1991) provided clear evidence for the formate-induced release of CO2/HCO−3, the HCO−3 binding site was not specified in this study. Based on numerous previous data indicating that HCO−3 is a ligand to the non-heme iron (NHI) at the electron acceptor side of PSII, the released CO2 was suggested to derive from this binding site. However, later formate was shown to bind both at the acceptor and the donor (water-splitting) side of PSII (Feyziev et al., 2000), and therefore, the released CO2/HCO−3 could also originate from the water-splitting side.
In order to specifically probe the possible binding of HCO−3 to the Mn4CaO5 cluster at the donor side of PSII, Shevela et al. (2008a,b) re-examined and extended the earlier TR-MIMS investigations. Thus, a comparison of the formate-induced C16O18O yields (Figure 3A), under H182O-enriched conditions, in intact PSII and Mn-depleted PSII was performed. This was complemented by experiments in which the gaseous products produced by a quick reductive destruction of the of the Mn4CaO5 cluster by 15N-labeleld NH2OH (Figure 3B) were studied. Both approaches clearly demonstrated that the detected CO2/HCO−3 does not originate from the inorganic core of the water-splitting site of PSII (Shevela et al., 2008a,b). Independent evidence for absence of HCO−3 bound to the WOC was provided by FTIR and GS-MS experiments (Aoyama et al., 2008; Ulas et al., 2008). Moreover, in recent x-ray crystallography studies at resolutions of 1.9-3.0 Å no HCO−3 was found in the vicinity of the Mn4CaO5 cluster, while they all clearly show HCO−3 bound to the NHI on the acceptor side of PSII (Guskov et al., 2010; Umena et al., 2011) (also, see Figure 1). Thus, it can be excluded that HCO−3 is a tightly bound ligand of the Mn4CaO5 cluster.
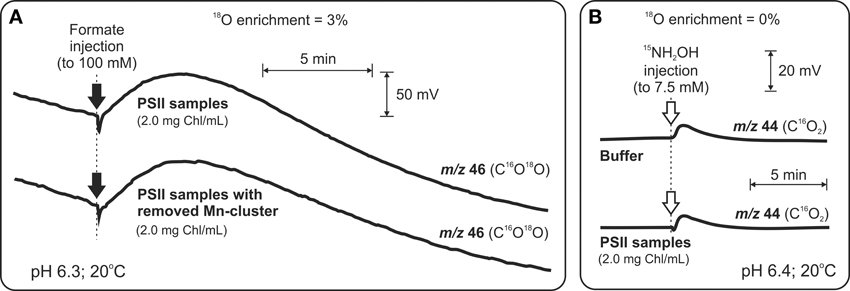
Figure 3. TR-MIMS experiments demonstrating that HCO−3 is not a tightly bound ligand to the Mn4CaO5 cluster in spinach PSII membrane fragments. (A) Amount of released CO2 upon formate addition (black arrows) to intact PSII membranes is the same as in the case of PSII membranes without the Mn4CaO5 cluster (due to 75-min pre-incubation with 80 mM N2H4). Due to enrichment of sample suspension with H182O (3%) CO2 was detected as C16O18O at m/z 46. (B) Addition of the strong reductant NH2OH (white arrows) at concentrations known to cause rapid reduction of the Mn4CaO5 cluster and release of Mn ions as MnII into the solution didn't lead to a release of CO2/HCO−3 above background. In order to avoid the overlay of CO2 and N2O signals (the latter is known to be produced during interaction of NH2OH with the Mn4CaO4 cluster), the N2O signal was shifted from m/z 44 to m/z 46 by employing the 15N-labeled NH2OH for these experiments. To facilitate equilibration between CO2 and HCO−3 all measurements were performed in the presence of externally added CA (to a final concentration of 3 μg ml−1). Modified from Shevela et al. (2008b).
However, none of the mentioned studies negates the option that a mobile, weakly bound, and rapidly exchanging HCO−3 affects the activity of the WOC. Thus, in case of the TR-MIMS measurements, weakly bound HCO−3 may have been removed during the required degassing of the MIMS cell prior to formate or NH2OH additions to PSII samples. Therefore, the possible loss of weakly bound HCO−3 and the amount of HCO−3 associated with PSII under air-saturated condition remain to be established in future TR-MIMS experiments.
When and How does Substrate Water Bind to the WOC?
Indisputably, the most significant and unique contribution of the TR-MIMS instrumentation in understanding of water-oxidation mechanism in photosynthesis was its application for studying substrate binding in the different Si states of the WOC. In these experiments the binding of water to the WOC is probed by the rapid injection of H182O into the PSII samples which were preset into the desired Si state by pre-illumination with 0, 1, 2, or 3 flashes. Then, after desired incubation times, O2 evolution is induced by a sequence of additional flashes. The exchange rates of the two substrate molecules are then calculated from the change of the 16O18O and 18O18O yields as a function of incubation time (see Figure 4A for protocol of the TR-MIMS measurements of substrate water exchange in the S3 state). The mixing time of H182O with the PSII samples after injection and a very low level of dissolved O2 in H182O are highly important for these experiments since they determine the time resolution of the TR-MIMS measurements. In the first H216O/H182O-exchange TR-MIMS experiments the water exchange kinetics could not be resolved (Radmer and Ollinger, 1986; Bader et al., 1993). The development of the MIMS cell by Messinger et al. (1995), which allowed for fast mixing of H182O with the sample and also implemented O2 removal from the labeled water by the glucose—glucose oxidase—catalase method, greatly improved the time resolution down to the milliseconds scale and allowed measurements of substrate water exchange in all semistable Si states (Messinger et al., 1995; Hillier et al., 1998; Hillier and Wydrzynski, 2000).
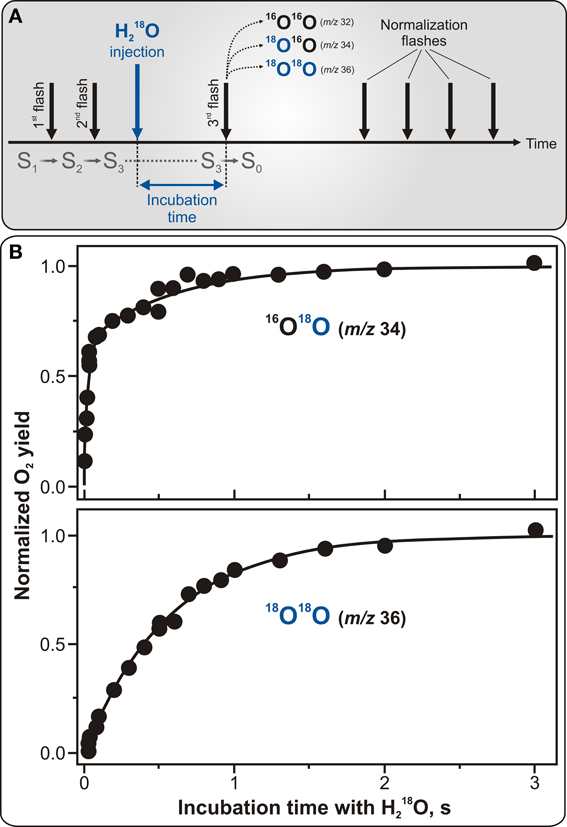
Figure 4. Protocol for TR-MIMS measurements of H216O/H182O exchange in the S3 state of PSII (A) and experimentally obtained substrate water exchange rates in spinach thylakoids (B). (A) The S3 state is populated by two pre-flashes given at 2 Hz (shown by the two first black vertical arrows). This is followed by the rapid injection of H182O into the PSII sample (shown by blue vertical arrow) and subsequent fast mixing of the injected H182O with the sample. Evolution of O2 isotopologues is then induced by a 3rd flash, given at varying delay times (from 0 to 10 s) after the H182O injection (signified as incubation time). Finally, a series of four flashes is given at 2 Hz to induce O2 yield used for normalization. (B) TR-MIMS measurements of substrate H216O/H182O exchange kinetics were performed at m/z 34 (top plot) for singly-labeled isotopologue 16O18O, and at m/z 36 (bottom plot) for doubly-labeled 18O18O in the S3 state in spinach thylakoids at 10°C and pH 6.8. Symbols in both plots are experimental data, and the lines in the top and bottom plots are biexponential and monoexponential fits, respectively. The biexponential fit yields rate constants of ~40 s−1 for the fast phase and ~2 s−1 for the slow phase. The slow phase in the 16O18O data is matching the rate found in the monoexponential fit of the 18O18O data (Messinger et al., 1995; Hillier et al., 1998; Hillier and Wydrzynski, 2000, 2004). Adapted from Cox and Messinger (2013).
Figure 4B illustrates characteristic water exchange kinetics in the S3 state as measured in spinach thylakoids with the time resolution of 8 ms. In this figure, the yields of the singly-labeled (16O18O) and doubly-labeled (18O18O) isotopologues of molecular oxygen are plotted as a function of H182O incubation time in the S3 state. While the former plot reflects the result when only one of the two possible 18O-water substrates is exchanged, the latter one is for the case when both 18O-waters are exchanged. The biphasic behavior of the 16O18O rise (detected at m/z 34) (see Figure 4B) is known to represent the exchange rates of two independent slowly and fast exchanging substrate water molecules bound at separate sites within the WOC. In contrast, the 18O18O product (monitored at m/z 36) exhibits a mono-exponential rise with a rate equal to that of the slow phase kinetic of the 16O18O data, thus-reflecting the exchange of the same “slowly” exchanging substrate water as observed at m/z 34. This finding clearly confirms that the two phases of the 16O18O data are an intrinsic feature of the WOC and do not originate from PSII heterogeneity (Messinger et al., 1995; Hillier et al., 1998).
Further TR-MIMS experiments also revealed that the “slowly” exchanging water is bound to the WOC in all semi-stable Si states, while the “fast” exchanging water was detected only in the S2 and S3 states (Hillier et al., 1998; Hillier and Wydrzynski, 2000, 2004; Hendry and Wydrzynski, 2002). Thus, the TR-MIMS technique provides not only the most direct evidence for independent substrate water binding within the WOC, but also allows to monitor the change in their binding affinities throughout the reaction cycle. For a complete overview of the TR-MIMS findings in this field, we refer the readers to reviews by Hillier and Messinger (2005), Hillier and Wydrzynski (2008), Beckmann et al. (2009), Messinger et al. (2012), and Cox and Messinger (2013).
The 16O/18O Isotope Effect and Photosynthetic Water-Splitting
Up to now there is no final agreement on whether isotopic discrimination during O2 production by photosynthetic water-splitting in PSII contributes to the so-called Dole effect, which describes the finding that the percentage of the 18O isotope in atmosphere is higher (by 23‰) than in oceanic waters (Dole, 1936; Tcherkez and Farquhar, 2007). While many gas isotope ratio studies clearly showed that oxygen produced by O2-evolving organisms is isotopically identical to the water they are suspended in (Dole and Jenks, 1944; Stevens et al., 1975; Guy et al., 1993; Helman et al., 2005), recent 18O-enriched TR-MIMS experiments indicated that the 18O isotope is favored by the WOC for O2 production, thus-suggesting a significant 16O/18O isotope effect in the photosynthetic water-splitting (Burda et al., 2001, 2003). This finding was challenged by recent theoretical estimations that suggest a very small isotope effect (Tcherkez and Farquhar, 2007). Undoubtedly, a resolution of these conflicting results can be provided by revisiting TR-MIMS studies. These future studies should be designed to account for: (i) technical limitations/drawbacks of the previous TR-MIMS experiments (for instance, the absence of fast H182O mixing upon its addition to sample suspension inside the MIMS cuvette Bader et al., 1987; Burda et al., 2003), (ii) possible contribution of isotopic fractionation due to transfer of O2 isotopologues through the membrane inlet toward the high vacuum of mass spectrometer recently reported by Hillier et al. (2006), and (iii) current knowledge of the S-state dependent substrate water binding and exchange rates as derived from TR-MIMS measurements (for reviews, see Hillier and Wydrzynski, 2008; Cox and Messinger, 2013). However, we note here, that without specific investigations of the 18O isotope effect in photosynthetic water-oxidation, our TR-MIMS studies do not reveal any oxygen isotope discrimination in photosynthetically produced O2 (for instance, see Figure 6C and text below for explanations), indicating that any such effect must be small at best.
In Search for Intermediates of Water Splitting by TR-MIMS Approach
While most states of the Kok cycle (S0, S1, S2, S3) are semistable, the S3Y•Z and S4 state are known to be a highly reactive intermediates that until very recently were not characterized. Clausen and Junge (2004) attempted to stabilize and identify putative intermediate(s) of the S4 state by applying a high partial O2 pressure in order to shift the equilibrium of the terminal S4 → S0 + O2 + nH+ reaction backwards. Based on their UV-absorption transients the authors observed half suppression of Mn oxidation under only 10-fold increase of ambient O2 pressure (2.3 bar). These results were considered to be the first indication for an intermediate in the S3 → S4 → S0 transition and as a possible route for stabilizing it (Clausen and Junge, 2004). Although a further delayed Chl fluorescence study corroborated these results (Clausen et al., 2005b), experiments by time-resolved X-ray absorption spectroscopy (TR-XAS) (Haumann et al., 2008) and by visible fluorescence study (Kolling et al., 2009) shed doubt on the existence of accessible S4 intermediate(s) that can be populated by inhibition of the terminal step of O2 release from the WOC by elevated O2 concentrations. These controversial studies prompted application of the TR-MIMS technique, which allowed investigation of the effect of elevated O2 pressure on photosynthetic O2 release by direct O2 detection (Shevela et al., 2011a). In these experiments direct monitoring of 18O2 evolution from 18O-labeled water against a high level of 16O2 in a suspension of PSII complexes became possible due to a specially designed high pressure MIMS cell (for details, see Figure 5A). This study demonstrated that neither an inhibition nor altered flash-induced pattern of O2 evolution take place under up to 50-fold increased concentration of dissolved O2 around PSII (Figure 5B). These findings show that the terminal water-splitting reaction/O2 release in PSII is highly exothermic, and are in line with the results obtained by TR-XAS (Haumann et al., 2008) and variable fluorescence (Kolling et al., 2009) studies.
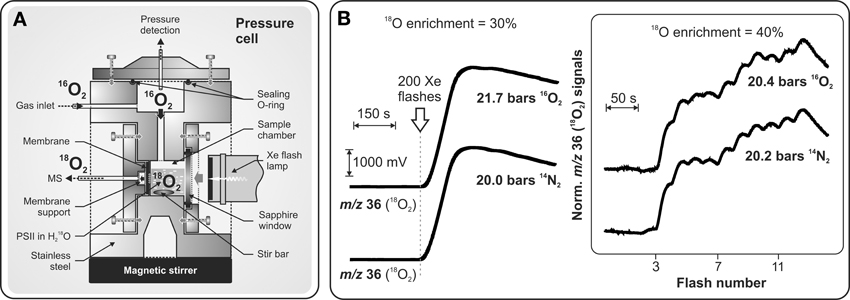
Figure 5. Schematic representation of the pressure cell (A) specially designed for TR-MIMS measurements of light-induced 18O2 evolution of PSII under high 16O2/N2 pressures (up to 20 bars). (B) MIMS signals in panel (B); Left: 18O2 production of PSII core complexes from Synechocystis sp. PCC 6803 induced by a series of 200 saturating Xenon flashes (given at 2 Hz; indicated by arrow) at 21.7 bars O2, or 20 bars N2. Other conditions: 30% H182O enrichment; [Chl] = 50 μM; 250 μM DCBQ, pH 6.7, 20°C. Right: Flash-induced 18O2 evolution patterns of PSII membrane fragments from spinach induced by a series of saturating laser flashes (separated by dark times of 25 s) at 20.4 bars O2, or 20.2 bars N2. Other conditions: as above, but with 40% H182O. Adapted from Shevela et al. (2011a).
Applications in Artificial Photosynthesis
One of the central goals of artificial photosynthesis is the development of bio-inspired, efficient and robust catalysts that are able to split water employing the energy of sunlight in a fashion similar to the water-oxidizing Mn4CaO5 cluster in PSII (Concepcion et al., 2012; Nocera, 2012; Wiechen et al., 2012). Therefore, data concerning catalytic rates and turnover numbers (stability) of newly synthetized O2-evolving catalysts are highly important for their further development. In this regard, in addition to traditionally used amperometric methods for O2 detection (Renger and Hanssum, 2009), TR-MIMS can be applied as a highly sensitive method for studying the O2-evolving capability of these complexes. However, a major advantage and uniqueness of the TR-MIMS technique in this field is that, in combination with 18O-labeling experiments, it can be employed for studying the pathways of O2 formation in reactions catalyzed by the ‘potential’ solar water-oxidation catalysts (Poulsen et al., 2005; Beckmann et al., 2008; Sala et al., 2010; Shevela et al., 2011b; Najafpour et al., 2012; Vigara et al., 2012). Thus, TR-MIMS detection of the isotopologues of O2 (16O2, 16O18O, 18O2) during catalytic O2-formation in the 18O-enriched aqueous solutions allows to analyze the 18O-fraction (18α) of the evolved O2 with good time resolution and very high accuracy. A correlation of the 18O-fraction in the substrate water (18αtheor; reflects the H182O-enrichment of the solvent water) and in the product O2 (18αexp) gives important information about the origin of the O atoms in the produced molecular oxygen. For instance, the incorporation of exactly half of the possible 18O-fraction into the evolved O2 may indicate that only one of the two O atoms of the O2 product originates from the bulk water as it has been monitored by TR-MIMS in the reactions of O2-evolving catalysts with oxygen-transferring oxidizing agent, oxone (HSO−5) (Poulsen et al., 2005; Beckmann et al., 2008; Shevela et al., 2011b) (see Figure 6A). In the case of “true” water-splitting, 18O-fractions in bulk water and in evolved O2 are expected to be same (i.e., 18αtheor = 18αexp) as depicted in Figure 6B for the reaction of a synthetic catalyst (CaMn2O4 · H2O oxide) with photogenerated oxidizing agent [RuIII(bipy)3]3+ (RuIIIphoto), and in Figure 6C for natural light-induced water-splitting reaction performed by PSII. It's worth mentioning here, that the initial phase of the presented traces until stable 18α values (Figure 6) is a technical artefact, merely caused by the response time of the membrane-inlet system of the mass spectrometer which seems to be related to the overall O2 concentration. However, the difference in time needed to reach final 18α value in two water-splitting reactions shown in Figure 6 also reflects a much slower reaction rate for the reaction of the oxide with RuIIIphoto. Thus, O2 evolution for this reaction was detected only after 1 min of illumination since this time is required to build up a sufficient concentration of photosensitizer RuIIIphoto (data not shown here; for details, see Shevela et al., 2011b). We note that one of the attractive extensions to the described TR-MIMS approach for the characterization of water-splitting catalysts is the coupling of the TR-MIMS instrument to an electrochemical cell (for further details, see Konermann et al., 2008 and refs therein).
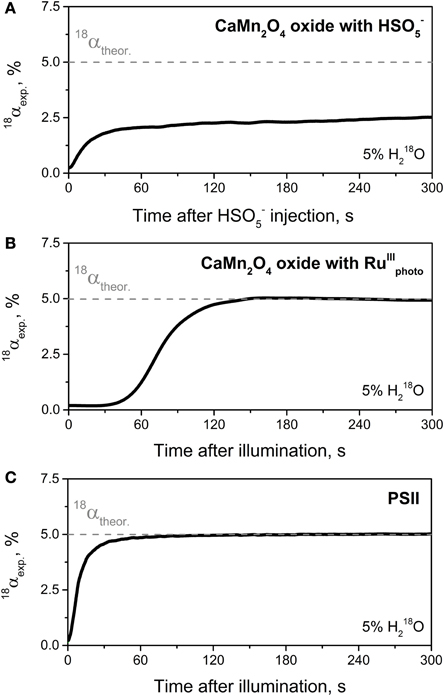
Figure 6. Development of the 18O-isotope fraction (18α) over time for the course of the catalytic O2-formation in reactions catalyzed by synthesized CaMn2O4 · H2O oxide (A,B) and by the WOC of PSII (C). (A) Change in 18α-value for the reaction of CaMn2O4 · H2O with HSO−5 (oxone) indicating that only one of the two oxygen atoms of O2 evolved originates from the bulk water. A solution of HSO−5 in H182O-enriched water was injected at t = 0 into the MIMS cell filled in with a non-enriched oxide suspension (1 mg ml−1; pH ~4.5) to give a final HSO−5 concentration of 3.7 mM and an H182O enrichment of 5%. Note that the rise of 18α to the value of 2.5% corresponds to half the percentage of the 18O-labeled water. (B) Change in 18α-value for the reaction of CaMn2O4 · H2O with photogenerated [RuIII(bipy)3]3+. Shortly before illumination (started at t = 0) the reaction mixture (H182O (5%), CaMn2O4 · H2O (1 mg ml−1), [Ru(bipy)3]2+ (1.5 mM), and [Co(NH3)5Cl]2+ (12.5 mM); pH ~4) inside the MIMS cell was purged with N2 until “zero” O2 level was reached. (C) Change in 18α-value for O2 production by PSII membrane fragments isolated from spinach. O2 evolution was induced by actinic continuous light at t = 0. Other conditions: 5% H182O, [Chl] = 0.03 mg ml−1, 0.6 mM PPBQ, 2 mM K3[Fe(CN)6], pH 6.0, and 20°C. Gray dashed lines in all panels indicates the theoretical 18α value expected for reaction of the “true” water-splitting, i.e., when both oxygen atoms of formed O2 originate from water. In all cases O2 production was detected by TR-MIMS as 16O2 (at m/z 32), 16O18O (m/z 34), and 18O2 (m/z 36), and the 18α was calculated according to the following equation: 18α = ([18O2] + 1/2[16O18O])/[O2]total. Adapted from Shevela et al. (2011b).
Conclusions and Future Perspectives
Application of the TR-MIMS technique and isotope labeling for studies of various biophysical aspects of photosynthetic water-splitting and O2 production is continuously growing. It provides not only insightful and unique information (which is sometimes not accessible by other methods) about this fundamental biological process, but also becomes an essential and highly precise tool for testing artificial water-oxidizing catalysts. Future applications of TR-MIMS for studies of water-splitting chemistry and O2 production could follow from advances in membrane materials, different designs of the membrane-inlet systems, coupling with electrochemistry and spectroscopy, and technological developments of the mass spectrometers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge financial support by the Kempe Foundation, the Swedish Research Council, the Energimyndigheten, the Strong Research Environment Solar Fuels (Umeå University), and the Artificial Leaf Project (K&A Wallenberg Foundation).
Abbreviations
Ci, inorganic carbon (CO2, H2CO3, HCO−3, CO32−); Chl, chlorophyll; PSII, photosystem II; RC, reaction center; WOC, water-oxidizing complex.
References
Aartsma, T. J., and Matysik, J. (eds.). (2008). Biophysical Techniques in Photosynthesis. Dordrecht: Springer. doi: 10.1007/978-1-4020-8250-4
Aoyama, C., Suzuki, H., Sugiura, M., and Noguchi, T. (2008). Flash-induced FTIR difference spectroscopy shows no evidence for the structural coupling of bicarbonate to the oxygen-evolving Mn cluster in photosystem II. Biochemistry 47, 2760–2765. doi: 10.1021/bi702241t
Bader, K. P., Renger, G., and Schmid, G. H. (1993). A mass-spectrometric analysis of the water splitting reaction. Photosynth. Res. 38, 355–361. doi: 10.1007/BF00046761
Bader, K. P., Thibault, P., and Schmid, G. H. (1987). Study on the properties of the S3 state by mass-spectrometry in the filamentous cyanobacterium Oscillatoria chalybea. Biochim. Biophys. Acta 893, 564–571. doi: 10.1016/0005-2728(87)90108-3
Beckmann, K., Messinger, J., Badger, M. R., Wydrzynski, T., and Hillier, W. (2009). On-line mass spectrometry: membrane inlet sampling. Photosynth. Res. 102, 511–522. doi: 10.1007/s11120-009-9474-7
Beckmann, K., Uchtenhagen, H., Berggren, G., Anderlund, M. F., Thapper, A., Messinger, J., et al. (2008). Formation of stoichiometrically 18O-labelled oxygen from the oxidation of 18O-enriched water mediated by a dinuclear manganese complex-a mass spectrometry and EPR study. Energy Environ. Sci. 1, 668–676. doi: 10.1039/b811806j
Burda, K., Bader, K. P., and Schmid, G. H. (2001). An estimation of the size of the water cluster present at the cleavage site of the water splitting enzyme. FEBS Lett. 491, 81–84. doi: 10.1016/S0014-5793(01)02175-5
Burda, K., Bader, K. P., and Schmid, G. H. (2003). 18O isotope effect in the photosynthetic water splitting process. Biochim. Biophys. Acta 1557, 77–82. doi: 10.1016/S0005-2728(02)00395-X
Clausen, J., Beckmann, K., Junge, W., and Messinger, J. (2005a). Evidence that bicarbonate is not the substrate in photosynthetic oxygen evolution. Plant Physiol. 139, 1444–1450. doi: 10.1104/pp.105.068437
Clausen, J., Junge, W., Dau, H., and Haumann, M. (2005b). Photosynthetic water oxidation at high O2 backpressure monitored by delayed chlorophyll fluorescence. Biochemistry 44, 12775–12779. doi: 10.1021/bi051183a
Clausen, J., and Junge, W. (2004). Detection of an intermediate of photosynthetic water oxidation. Nature 430, 480–483. doi: 10.1038/nature02676
Concepcion, J. J., House, R. L., Papanikolas, J. M., and Meyer, T. J. (2012). Chemical approaches to artificial photosynthesis. Proc. Natl. Acad. Sci. U.S.A. 109, 15560–15564. doi: 10.1073/pnas.1212254109
Cox, N., and Messinger, J. (2013). Reflections on substrate water and dioxygen formation. Biochim. Biophys. Acta 1827, 1020–1030. doi: 10.1016/j.bbabio.2013.01.013
Dasgupta, J., Ananyev, G. M., and Dismukes, G. C. (2008). Photoassembly of the water-oxidizing complex in photosystem II. Coord. Chem. Rev. 252, 347–360. doi: 10.1016/j.ccr.2007.08.022
Davey, N. G., Krogh, E. T., and Gill, C. G. (2011). Membrane-introduction mass spectrometry (MIMS). Trends Anal. Chem. 30, 1477–1485. doi: 10.1016/j.trac.2011.05.003
Diner, B. A., and Rappaport, F. (2002). Structure, dynamics, and energetics of the primary photochemistry of photosystem II of oxygenic photosynthesis. Annu. Rev. Plant Biol. 53, 551–580. doi: 10.1146/annurev.arplant.53.100301.135238
Dole, M. (1936). The relative atomic weight of oxygen in water and in air. J. Chem. Phys. 4, 268–275. doi: 10.1063/1.1749834
Dole, M., and Jenks, G. (1944). Isotopic composition of photosynthetic oxygen. Science 3, 409. doi: 10.1126/science.100.2601.409
Ferreira, K. N., Iverson, T. M., Maghlaoui, K., Barber, J., and Iwata, S. (2004). Architecture of the photosynthetic oxygen-evolving center. Science 303, 1831–1838. doi: 10.1126/science.1093087
Feyziev, Y. M., Yoneda, D., Yoshii, T., Katsuta, N., Kawamori, A., and Watanabe, Y. (2000). Formate-induced inhibition of the water-oxidizing complex of photosystem II studied by EPR. Biochemistry 39, 3848–3855. doi: 10.1021/bi992479h
Govindjee, Weger, H. G., Turpin, D. H., Van Rensen, J. J. S., Devos, O. J., and Snel, J. F. H. (1991). Formate releases carbon dioxide/bicarbonate from thylakoid membranes - measurements by mass spectroscopy and infrared gas analyzer. Naturwissenschaften 78, 168–170. doi: 10.1007/BF01136204
Guskov, A., Gabdulkhakov, A., Broser, M., Glöckner, C., Hellmich, J., Kern, J., et al. (2010). Recent progress in the crystallographic studies of photosystem II. ChemPhysChem 11, 1160–1171. doi: 10.1002/cphc.200900901
Guy, R. D., Fogel, M. L., and Berry, J. A. (1993). Photosynthetic fractionation of the stable isotopes of oxygen and carbon. Plant Physiol. 101, 37–47.
Haumann, M., Grundmeier, A., Zaharieva, I., and Dau, H. (2008). Photosynthetic water oxidation at elevated dioxygen partial pressure monitored by time-resolved X-ray absorption measurements. Proc. Natl. Acad. Sci. U.S.A. 105, 17384–17389. doi: 10.1073/pnas.0802596105
Helman, Y., Barkan, E., Eisenstadt, D., Luz, B., and Kaplan, A. (2005). Fractionation of the three stable oxygen isotopes by oxygen-producing and oxygen-consuming reactions in photosynthetic organisms. Plant Physiol. 138, 2292–2298. doi: 10.1104/pp.105.063768
Hendry, G., and Wydrzynski, T. (2002). The two substrate water molecules are already bound to the oxygen evolving complex in the S2 state of photosystem II. Biochemistry 41, 13328–13334. doi: 10.1021/bi026246t
Hillier, W., McConnell, I., Badger, M. R., Boussac, A., Klimov, V. V., Dismukes, G. C., et al. (2006). Quantitative assessment of intrinsic carbonic anhydrase activity and the capacity for bicarbonate oxidation in photosystem II. Biochemistry 45, 2094–2102. doi: 10.1021/bi051892o
Hillier, W., Messinger, J., and Wydrzynski, T. (1998). Kinetic determination of the fast exchanging substrate water molecule in the S3 state of photosystem II. Biochemistry 37, 16908–16914. doi: 10.1021/bi980756z
Hillier, W., and Messinger, J. (2005). “Mechanism of photosynthetic oxygen production,” in Photosystem II. The Light-Driven Water:Plastoquinone Oxidoredutase, eds T. Wydrzynski and K. Satoh (Dordrecht: Springer), 567–608.
Hillier, W., and Wydrzynski, T. (2000). The affinities for the two substrate water binding sites in the O2 evolving complex of photosystem II vary independently during S-state turnover. Biochemistry 39, 4399–4405. doi: 10.1021/bi992318d
Hillier, W., and Wydrzynski, T. (2004). Substrate water interactions within the photosystem II oxygen evolving complex. Phys. Chem. Chem. Phys. 6, 4882–4889. doi: 10.1039/b407269c
Hillier, W., and Wydrzynski, T. (2008). 18O-Water exchange in photosystem II: Substrate binding and intermediates of the water splitting cycle. Coord. Chem. Rev. 252, 306–317. doi: 10.1016/j.ccr.2007.09.004
Hoch, G., and Kok, B. (1963). A mass spectrometer inlet system for sampling gases dissolved in liquid phases. Arch. Biochem. Biophys. 101, 160–170. doi: 10.1016/0003-9861(63)90546-0
Ishikita, H., Loll, B., Biesiadka, J., Saenger, W., and Knapp, E.-W. (2005). Redox potentials of chlorophylls in the photosystem II reaction center. Biochemistry 44, 4118–4124. doi: 10.1021/bi047922p
Johnson, R. C., Cooks, R. G., Allen, T. M., Cisper, M. E., and Hemberger, P. H. (2000). Membrane introduction mass spectrometry: Trends and applications. Mass Spectrom. Rev. 19, 1–37. doi: 10.1002/(SICI)1098-2787(2000)19:1<1::AID-MAS1>3.0.CO;2-Y
Kok, B., Forbush, B., and McGloin, M. (1970). Cooperation of charges in photosynthetic O2 evolution. Photochem. Photobiol. 11, 457–476. doi: 10.1111/j.1751-1097.1970.tb06017.x
Kolling, D. R. J., Brown, T. S., Ananyev, G., and Dismukes, G. C. (2009). Photosynthetic oxygen evolution is not reversed at high oxygen pressures: mechanistic consequences for the water-oxidizing complex. Biochemistry 48, 1381–1389. doi: 10.1021/bi801774f
Konermann, L., Messinger, J., and Hillier, W. (2008). “Mass spectrometry based methods for studying kinetics and dynamics in biological systems,” in Biophysical Techniques in Photosynthesis, eds J. Amesz and A. J. Hoff (Dordrecht: Springer), 167–190. doi: 10.1007/978-1-4020-8250-4_9
Lauritsen, F. R., and Kotiaho, T. (1996). Advances in membrane inlet mass spectrometry (MIMS). Rev. Anal. Chem. 15, 237–264. doi: 10.1515/REVAC.1996.15.4.237
Lu, Y. K., and Stemler, A. J. (2002). Extrinsic photosystem II carbonic anhydrase in maize mesophyll chloroplasts. Plant Physiol. 128, 643–649. doi: 10.1104/pp.010643
McConnell, I. L., Badger, M. R., Wydrzynski, T., and Hillier, W. (2007). A quantitative assessment of the carbonic anhydrase activity in photosystem II. Biochim. Biophys. Acta 1767, 639–647. doi: 10.1016/j.bbabio.2007.01.019
Messinger, J., Alia, A., and Govindjee, (2009a). Special educational issue on “Basics and application of biophysical techniques in photosynthesis and related processes.” Photosynth. Res. 101, 89–92. doi: 10.1007/s11120-009-9471-x
Messinger, J., Alia, A., and Govindjee, (2009b). Special educational issue on “Basics and application of biophysical techniques in photosynthesis and related processes”—Part B. Photosynth. Res. 102, 103–106. doi: 10.1007/s11120-009-9494-3
Messinger, J., Badger, M. R., and Wydrzynski, T. (1995). Detection of one slowly exchanging substrate water molecule in the S3 state of photosystem II. Proc. Natl. Acad. Sci. U.S.A. 92, 3209–3213. doi: 10.1073/pnas.92.8.3209
Messinger, J., Noguchi, T., and Yano, J. (2012). “Photosynthetic O2 evolution,” in Molecular Solar Fuels, eds T. Wydrzynski and W. Hillier (Cambridge: RSC Publishing), 163–207.
Moskvin, O. V., Shutova, T. V., Khristin, M. S., Ignatova, L. K., Villarejo, A., Samuelsson, G., et al. (2004). Carbonic anhydrase activities in pea thylakoids—A photosystem II core complex-associated carbonic anhydrase. Photosynth. Res. 79, 93–100. doi: 10.1023/B:PRES.0000011925.93313.db
Najafpour, M. M., Hillier, W., Shamkhali, A. N., Amini, M., Beckmann, K., Jaglicic, Z., et al. (2012). Synthesis, characterization, DFT studies and catalytic activities of manganese(ii) complex with 1,4-bis(2,2′:6,2″-terpyridin-4′-yl) benzene. Dalton Trans. 41, 12282–12288. doi: 10.1039/c2dt31544k
Poulsen, A. K., Rompel, A., and McKenzie, C. J. (2005). Water oxidation catalyzed by a dinuclear Mn complex: a functional model for the oxygen-evolving center of photosystem II. Angew. Chem. Int. Ed. 44, 6916–6920. doi: 10.1002/anie.200502114
Radmer, R., and Ollinger, O. (1980). Isotopic composition of photosynthetic O2 flash yields in the presence of H182O and HC18O−3. FEBS Lett. 110, 57–61. doi: 10.1016/0014-5793(80)80022-6
Radmer, R., and Ollinger, O. (1986). Do the higher oxidation states of the photosynthetic O2 evolving system contain bound water? FEBS Lett. 195, 285–289. doi: 10.1016/0014-5793(86)80178-8
Renger, G., and Hanssum, B. (2009). Oxygen detection in biological systems. Photosynth. Res. 102, 487–498. doi: 10.1007/s11120-009-9434-2
Ruben, S., Randall, M., Kamen, M., and Hyde, J. L. (1941). Heavy oxygen (O18) as a tracer in the study of photosynthesis. J. Am. Chem. Soc. 63, 877–879. doi: 10.1021/ja01848a512
Sala, X., Ertem, M. Z., Vigara, L., Todorova, T. K., Chen, W., Rocha, R. C., et al. (2010). The cis-[RuII(bpy)2(H2O)2]2+ water-oxidation catalyst revisited. Angew. Chem. Int. Ed. 49, 7745–7747. doi: 10.1002/anie.201002398
Shevela, D., Beckmann, K., Clausen, J., Junge, W., and Messinger, J. (2011a). Membrane-inlet mass spectrometry reveals a high driving force for oxygen production by photosystem II. Proc. Natl. Acad. Sci. U.S.A. 108, 3602–3607. doi: 10.1073/pnas.1014249108
Shevela, D., Koroidov, S., Najafpour, M. M., Messinger, J., and Kurz, P. (2011b). Calcium manganese oxides as oxygen evolution catalysts: O2 formation pathways indicated by 18O-labelling studies. Chem. Eur. J. 17, 5415–5423. doi: 10.1002/chem.201002548
Shevela, D., Klimov, V., and Messinger, J. (2008a). “Formate-induced release of carbon dioxide/hydrogencarbonate from photosystem II,” in Photosynthesis. Energy From the Sun, eds J. F. Allen, E. Gantt, J. H. Golbeck, and B. Osmond (Glasgow: Springer), 497–501.
Shevela, D., Su, J. H., Klimov, V., and Messinger, J. (2008b). Hydrogencarbonate is not a tightly bound constituent of the water-oxidizing complex in photosystem II. Biochim. Biophys. Acta 1777, 532–539. doi: 10.1016/j.bbabio.2008.03.031
Stemler, A. (1989). Absence of a formate-induced release of bicarbonate from photosystem 2. Plant Physiol. 91, 287–290. doi: 10.1104/pp.91.1.287
Stemler, A., and Radmer, R. (1975). Source of photosynthetic oxygen in bicarbonate-stimulated Hill reaction. Science 190, 457–458. doi: 10.1126/science.190.4213.457
Stevens, C. L. R., Schultz, D., Van Baalen, C., and Parker, P. L. (1975). Oxygen isotope fractionation during photosynthesis in a blue-green and a green alga. Plant Physiol. 56, 126–129. doi: 10.1104/pp.56.1.126
Tcherkez, G., and Farquhar, G. D. (2007). On the 16O/18O isotope effect associated with photosynthetic O2 production. Funct. Plant Biol. 34, 1049–1052. doi: 10.1071/FP07168
Ulas, G., Olack, G., and Brudvig, G. W. (2008). Evidence against bicarbonate bound in the O2-evolving complex of photosystem II. Biochemistry 47, 3073–3075. doi: 10.1021/bi8000424
Umena, Y., Kawakami, K., Shen, J.-R., and Kamiya, N. (2011). Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60. doi: 10.1038/nature09913
Van Rensen, J. J. S., and Klimov, V. V. (2005). “Bicarbonate interactions,” in Photosystem II. The Light-Driven Water:Plastoquinone Oxidoreductase, eds T. Wydrzynski and K. Satoh (Dordrecht: Springer), 329–346.
Vigara, L., Ertem, M. Z., Planas, N., Bozoglian, F., Leidel, N., Dau, H., et al. (2012). Experimental and quantum chemical characteriazation of the water oxidation cycle catalysed by [RuII(damp)(bpy)(H2O)]2+. Chem. Sci. 3, 2576–2586. doi: 10.1039/c2sc20399e
Villarejo, A., Shutova, T., Moskvin, O., Forssen, M., Klimov, V. V., and Samuelsson, G. (2002). A photosystem II-associated carbonic anhydrase regulates the efficiency of photosynthetic oxygen evolution. EMBO J. 21, 1930–1938. doi: 10.1093/emboj/21.8.1930
Vinyard, D. J., Ananyev, G. M., and Dismukes, G. C. (2013). Photosystem II: the reaction center of oxygenic photosynthesis*. Annu. Rev. Biochem. 82, 577–606. doi: 10.1146/annurev-biochem-070511-100425
Wiechen, M., Berends, H.-M., and Kurz, P. (2012). Water oxidation catalysed by manganese compounds: from complexes to ‘biomimetic rocks’. Dalton Trans. 41, 21–31. doi: 10.1039/c1dt11537e
Wydrzynski, T., Hillier, W., and Messinger, J. (1996). On the functional significance of substrate accessibility in the photosynthetic water oxidation mechanism. Physiol. Plant. 96, 342–350. doi: 10.1111/j.1399-3054.1996.tb00224.x
Keywords: isotope-ratio membrane-inlet mass spectrometry, isotope labeling, O2 evolution, photosynthetic and artificial water-splitting, photosystem II, water-oxidizing complex
Citation: Shevela D and Messinger J (2013) Studying the oxidation of water to molecular oxygen in photosynthetic and artificial systems by time-resolved membrane-inlet mass spectrometry. Front. Plant Sci. 4:473. doi: 10.3389/fpls.2013.00473
Received: 04 October 2013; Accepted: 01 November 2013;
Published online: 26 November 2013.
Edited by:
Suleyman I. Allakhverdiev, Russian Academy of Sciences, RussiaReviewed by:
Mohammad M. Najafpour, Institute for Advanced Studies in Basic Sciences, IranHarvey J. M. Hou, Alabama State University, USA
Copyright © 2013 Shevela and Messinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dmitriy Shevela and Johannes Messinger, Department of Chemistry, Chemistry Biology Centre, Umeå University, Linnaeus Väg 6, S-90187 Umeå, Sweden e-mail: dmitriy.shevela@chem.umu.se; johannes.messinger@chem.umu.se