 Mehanathan Muthamilarasan
Mehanathan Muthamilarasan Venkata S. Bonthala
Venkata S. Bonthala Rohit Khandelwal
Rohit Khandelwal Jananee Jaishankar
Jananee Jaishankar Shweta Shweta
Shweta Shweta Kashif Nawaz
Kashif Nawaz Manoj Prasad*
Manoj Prasad*- National Institute of Plant Genome Research, New Delhi, India
Transcription factors (TFs) are major players in stress signaling and constitute an integral part of signaling networks. Among the major TFs, WRKY proteins play pivotal roles in regulation of transcriptional reprogramming associated with stress responses. In view of this, genome- and transcriptome-wide identification of WRKY TF family was performed in the C4model plants, Setaria italica (SiWRKY) and S. viridis (SvWRKY), respectively. The study identified 105 SiWRKY and 44 SvWRKY proteins that were computationally analyzed for their physicochemical properties. Sequence alignment and phylogenetic analysis classified these proteins into three major groups, namely I, II, and III with majority of WRKY proteins belonging to group II (53 SiWRKY and 23 SvWRKY), followed by group III (39 SiWRKY and 11 SvWRKY) and group I (10 SiWRKY and 6 SvWRKY). Group II proteins were further classified into 5 subgroups (IIa to IIe) based on their phylogeny. Domain analysis showed the presence of WRKY motif and zinc finger-like structures in these proteins along with additional domains in a few proteins. All SiWRKY genes were physically mapped on the S. italica genome and their duplication analysis revealed that 10 and 8 gene pairs underwent tandem and segmental duplications, respectively. Comparative mapping of SiWRKY and SvWRKY genes in related C4 panicoid genomes demonstrated the orthologous relationships between these genomes. In silico expression analysis of SiWRKY and SvWRKY genes showed their differential expression patterns in different tissues and stress conditions. Expression profiling of candidate SiWRKY genes in response to stress (dehydration and salinity) and hormone treatments (abscisic acid, salicylic acid, and methyl jasmonate) suggested the putative involvement of SiWRKY066 and SiWRKY082 in stress and hormone signaling. These genes could be potential candidates for further characterization to delineate their functional roles in abiotic stress signaling.
Introduction
Plants are exposed to diverse environmental stresses, which significantly affect their growth and development leading to drastic decrease in productivity. Among the different environmental stimuli, abiotic stresses are predominant, which includes drought, heat, salinity, and submergence. Climate change due to global warming is another aggravating challenge that influences the sustainability and productivity of crop plants (Kole et al., 2015). Plants have developed broad-spectrum defense responses to circumvent these stresses and exhibit stress tolerance or stress avoidance through acclimation and adaptation mechanisms (Mickelbart et al., 2015). On perception of stress, a complex signal transduction pathway (either abscisic acid-dependent or -independent) is induced, which initiates molecular, physiological and metabolic responses that ultimately enhance stress tolerance (Lata et al., 2015). Transcription factors (TFs) are a class of genes that predominate as tolerance determinants in plants (Mickelbart et al., 2015) by regulating the expression of stress-inducible genes. The TFs may constitute gene networks or signaling cascades, by which they regulate other TFs and/or other regulatory and/or functional genes (Tran and Mochida, 2010). Approximately 7% of the plant genome encodes for TFs (Udvardi et al., 2007), which are classified into 58 TF families (Jin et al., 2014). Among these TFs, WRKY is the seventh largest TF family (http://planttfdb.cbi.pku.edu.cn/). WRKY TFs are characterized by their unique WRKYGQK motif followed by a metal chelating zinc finger motif (CX4−5CX22−23HXH or CX5−8CX25−28HX1−2C) (Eulgem et al., 2000). These WRKY proteins bind to a specific domain called W-box in the promoter region with consensus sequence (C/T)TGAC[T/C], resulting in the expression of downstream target genes (Eulgem et al., 2000). In addition to W-box, WRKY TFs can also interact with a sugar responsive cis-element called SURE and activate transcription of downstream genes (Sun et al., 2003).
Several reports have shown the regulatory role of WRKY TFs in signaling pathways and modulation of diverse molecular and physiological processes including pollen development and function (Guan et al., 2014), seed dormancy (Rushton et al., 2010; Ding et al., 2014), seed development (Johnson et al., 2002; Sun et al., 2003; Luo et al., 2005), flowering time and plant height (Cai et al., 2014b), somatic embryogenesis (Alexandrova and Conger, 2002), biomass (Wang et al., 2010; Yu et al., 2013), secondary metabolite biosynthesis (Sun et al., 2003; Xu et al., 2004; Ma et al., 2009; Suttipanta et al., 2011), hormone signaling (Zhang et al., 2004) and leaf senescence (Miao et al., 2004). More importantly, WRKY TFs have been shown to get activated in response to different biotic (Dong et al., 2003; Muthamilarasan and Prasad, 2013) and abiotic stresses (Tang et al., 2013), including heat and drought (Rizhsky et al., 2002; Wu et al., 2009; Ren et al., 2010), cold (Huang and Duman, 2002; Pnueli et al., 2002), salinity (Jiang and Deyholos, 2006), wounding (Hara et al., 2000; Yoo et al., 2014), bacterial infection (Dellagi et al., 2000; Du and Chen, 2000; Chen et al., 2002; Chen and Chen, 2002; Deslandes et al., 2002; Kim et al., 2008), fungal invasion (Chen et al., 2002; Zheng et al., 2006; Marchive et al., 2007), virus attack (Wang et al., 1998; Yang et al., 1999; Chen et al., 2002, 2013; Huh et al., 2012) and defense against oomycetes (Beyer et al., 2001; Kalde et al., 2003).
Thus, considering the vital role of WRKY TFs in various molecular, biological and physiological processes, the WRKY gene family has been extensively characterized in various crop plants (Zhang and Wang, 2005), such as rice (Ross et al., 2007), cucumber (Ling et al., 2011), maize (Wei et al., 2012), tomato (Huang et al., 2012), Castor bean (Li et al., 2012), physic nut (Xiong et al., 2013), barley (Liu et al., 2014), Brachypodium (Wen et al., 2014), Gossypium raimondii, G. hirsutum (Cai et al., 2014a; Dou et al., 2014), grapevine (Wang et al., 2014), G. arboretum (Ding et al., 2015), cabbage (Yao et al., 2015), and in trees including rubber (Li et al., 2014), poplar (He et al., 2012; Jiang et al., 2014) and willow (Rao et al., 2015), and in Arabidopsis (de Pater et al., 1996; Deslandes et al., 2002; Song and Gao, 2014). However, no such studies have been reported in C4 models, Setaria italica (foxtail millet) and S. viridis (green foxtail). Both S. italica and its wild progenitor S. viridis have collectively been accentuated as model crops for expediting functional genomics studies in Panicoideae, particularly C4 photosynthesis, biofuel traits and abiotic stress tolerance (Brutnell et al., 2010, 2015; Li and Brutnell, 2011; Wang et al., 2011; Lata et al., 2013; Diao et al., 2014; Muthamilarasan and Prasad, 2015).
In view of their importance, the U.S. Department of Energy Joint Genome Institute and Beijing Genomics Institute, China have independently sequenced the genomes of S. italica and S. viridis (Bennetzen et al., 2012; Zhang et al., 2012). The availability of genome sequence information of S. italica in public domain has facilitated the identification of 2297 putative TFs belonging to 55 families (Bonthala et al., 2014). Of these 55 families, NAC (Puranik et al., 2012), AP2/ERF (Lata et al., 2014), MYB (Muthamilarasan et al., 2014a) and C2H2 zinc fingers (Muthamilarasan et al., 2014b) have been extensively characterized and their expression patterns in response to different abiotic stresses and hormone treatments have been investigated. However, no such global analysis of TFs has been performed in S. viridis due to non-availability of genome sequence in public domain (Muthamilarasan and Prasad, 2015). Recently, Xu et al. (2013) pooled the RNA isolated from S. viridis at three developmental stages, namely seed germination, vegetative growth, and reproduction in different tissues including leaf, stem, node, crown, root, spikelet, floret, and seed tissues. Subsequently, cDNA library was constructed from the pooled RNA and sequenced using Illumina HiSeq 2000 platform (Xu et al., 2013). Transcriptome-wide analysis of TFs has been demonstrated in important crop plants, namely barley (Tombuloglu et al., 2013), bread wheat (Okay et al., 2014), Medicago sativa (Postnikova et al., 2014), and G. aridum (Fan et al., 2015). In the present study, similar computational approach has been used to identify WRKY encoding transcripts from S. viridis transcriptome and the identified transcripts were analyzed with WRKY encoding genes of S. italica. Being the first comprehensive study on WRKY TFs in S. italica and S. viridis, the present study provides insights into the functional aspects of these TFs in response to abiotic stress, and highlights potential candidates for further characterization toward delineating their functional role in abiotic stress signaling.
Materials and Methods
In Silico Mining of WRKY Proteins From Setaria Italica and S. viridis
The WRKY domain-containing protein sequences of Setaria italica and S. viridis were identified using the method of Plant Transcription Factor Database (Jin et al., 2014). S. italica protein sequences (v2.1) were retrieved from Phytozome v10.2 (Goodstein et al., 2012) and HMMER search was executed using the PFAM domain (PF03106) (Finn et al., 2011). The HMM profile generated with WRKY TFs of maize (Wei et al., 2012) and rice (Ross et al., 2007) were used to generate HMM profile and searched against the protein sequences of S. italica using HMMER (Finn et al., 2011). Both de novo and reference-based transcriptome sequences of S. viridis (kindly provided by Prof. Xin-Guang Zhu; Xu et al., 2013) were used to generate unique clusters using CD-Hit (Fu et al., 2012) with default parameters and the resultant sequences were subjected to ORF prediction using OrfPredictor (Min et al., 2005). The obtained peptide sequences were used for identification of WRKY domain-containing proteins using the methodology described for S. italica. The identified WRKY sequences were confirmed for the presence of PFAM domain PF03106 (WRKY DNA-binding domain) using HMMSCAN (http://www.ebi.ac.uk/Tools/hmmer/search/hmmscan) and ScanProsite (http://prosite.expasy.org/scanprosite/; de Castro et al., 2006). The identified SiWRKY protein sequences were searched using BLASTP against S. italica database (v2.1) of Phytozome v10.2 to retrieve corresponding genomic, transcripts and coding sequences along with their chromosomal positions.
Protein Features, Multiple Sequence Alignment, and Phylogenetic Analysis
Protein features including molecular weight, isoelectric point (pI) and instability index were predicted using ProtParam tool of ExPASy (Gasteiger et al., 2005). Amino acid sequences of WRKY TFs belonging to S. italica (SiWRKY) and S. viridis (SvWRKY) were imported into BioEdit v7.2.5 (Hall, 1999) and multiple sequence alignment was performed using ClustalW at default parameters. The SiWRKY and SvWRKY sequences along with maize sequences (ZmWRKY; Wei et al., 2012) were imported into MEGA v6.06 (Tamura et al., 2013) to construct a phylogenetic tree by Neighbor-Joining method and the bootstrap test was performed with 1000 iterations.
Prediction of Gene Structure and Chromosomal Locations
The coding sequences and genomic sequences of SiWRKY proteins were analyzed using GSDS web server v2.0 (Hu et al., 2015) to identify the positions of introns and exons. Gene structure analysis for SvWRKY genes was not performed due to non-availability of genomic sequence of S. viridis in public databases. The information about chromosomal position of each SiWRKY gene was imported into MapChart v2.2 (Voorrips, 2002) and a physical map was constructed by mapping the genes in ascending order from short-arm telomere to long-arm telomere. MCScanX was used to identify tandem and segmental duplications of SiWRKY genes (Wang et al., 2012).
Gene Ontology Annotation and Promoter Analysis
SiWRKY and SvWRKY amino acid sequences were analyzed using Blast2GO v3.0.10 (Conesa et al., 2005) to obtain gene ontology (GO) annotation. The sequences were screened using BLASTN against Oryza sativa protein sequences following which, mapping, InterProScan, and annotation were performed. GO enrichment was conducted using BiNGO plugin of Cytoscape v2.6 based on Benjamini and Hochberg false discovery correction value (Q-value) at 0.05 for the genes (Shannon et al., 2003; Maere et al., 2005). The SiWRKY gene sequences were searched using BLASTN against S. italica database in Phytozome to retrieve 2 kb upstream sequences. These sequences were screened for cis-regulatory elements using PLACE web server (Higo et al., 1999).
Identification of Orthologs in C4 Grass Genomes and Ks Dating
Orthologous genes of SiWRKY and SvWRKY in sequenced C4 grasses including switchgrass (Panicum virgatum), sorghum (Sorghum bicolor), and maize (Zea mays) were identified by BLAST analysis of the gene and protein sequences, respectively against these genomes. Sequences with >90% similarity were used for performing reciprocal BLAST and potential orthologs were identified. A comparative map was constructed using Circos (Krzywinski et al., 2009). Synonymous (Ks) and non-synonymous (Ka) substitution rates were calculated for paralogous and orthologous genes by PAL2NAL server (http://www.bork.embl.de/pal2nal/) and period of divergence was calculated using the equation T = Ks/2λ, where λ was taken as 6.5 × 10−9 (Mishra et al., 2013; Puranik et al., 2013).
In silico Expression Profiling of SiWRKY and SvWRKY Genes
The transcriptome data of root (SRX128223), stem (SRX128225), leaf (SRX128224), spica (SRX128226), dehydration stress library (SRR629694), and control library (SRR629695) of S. italica were retrieved from European Nucleotide Archive (http://www.ebi.ac.uk/ena) (Zhang et al., 2012; Qi et al., 2013). S. viridis transcriptome data of pooled RNA isolated from samples across three developmental stages, namely seed germination, vegetative growth, and reproduction in different tissues including leaf, stem, node, crown, root, spikelet, floret, and seed tissues available under the accession number SRP019744 (Xu et al., 2013) was retrieved from DNA Data Bank of Japan (Tateno et al., 2002). The reads were filtered using NGS Toolkit (Patel and Jain, 2012), mapped on S. italica genome using CLC Genomics Workbench v4.7.1 and normalized by RPKM method. A heatmap was generated using MultiExperiment Viewer (MeV) v4.9 (Saeed et al., 2003).
Expression Profiling of Candidate Genes under Abiotic Stress and Hormone Treatments
Candidate SiWRKY genes were chosen for qRT-PCR expression analysis based on their in silico expression patterns. Primers were designed for the 3′ UTR of each transcript using GenScript Real-time PCR Primer Design tool (https://www.genscript.com/ssl-bin/app/primer) (Supplementary Table S1). S. italica cv. “Prasad” was chosen for the study as the cultivar was reported to be tolerant to salinity and dehydration stress (Lata et al., 2010; Puranik et al., 2011). The seeds were grown in green house following conditions described by Lata et al. (2014). Twenty-one day old seedlings were treated with 250 mM NaCl (salinity) and 20% PEG 6000 (dehydration) for abiotic stress, and 100 μM methyl jasmonate (MJ), 100 μM salicylic acid (SA), and 100 μM abscisic acid (ABA) for hormone treatments (Lata et al., 2014). Samples were collected at 0 h (control), 1 h (early), and 24 h (late) intervals, immediately frozen in liquid nitrogen and stored at −80°C. Total RNA from each sample was isolated following the method described by Logemann et al. (1987) and treated with RNase-free DNase I (50 U/ml). The quality and purity of RNA was tested using NanoDrop Spectrophotometer (Thermo Fisher Scientific, USA) [OD260:OD280 nm absorption ratio (1.8–2.0)] and integrity was checked by resolving on 1.2% agarose gel containing 18% formaldehyde. First strand complementary DNA was synthesized with random primers from 1 μg total RNA using Thermo Scientific Verso cDNA Synthesis kit (Thermo Fisher Scientific, USA) following manufacturer's instructions. qRT-PCR was performed in StepOne Real-Time PCR Systems (Applied Biosystems, USA). A constitutive Act2 gene-based primer was used as the endogenous control (Kumar et al., 2013). The PCR mixtures and reactions followed by melting curve analysis and agarose gel electrophoresis were performed following Kumar et al. (2013). Three technical replicates for each biological replicate were maintained for qRT-PCR analysis.
Results
WRKY Transcription Factors of Setaria
HMM search for WRKY proteins in Setaria italica showed the presence of 113 WRKY proteins (SiWRKY), which was in agreement with the numbers reported in Plant Transcription Factor Database v3.0 (Jin et al., 2014) and Foxtail millet Transcription Factor Database (Bonthala et al., 2014). Among these, four SiWRKY proteins (Si031469 m, Si030012 m, Si029764 m, and Si036581 m) were found to be the products of alternate transcripts. In case of S. viridis, 50 WRKY TF sequences were identified (SvWRKY). Domain analysis of both SiWRKY and SvWRKY proteins using HMMSCAN and ScanProsite web tools revealed that four SiWRKY and six SvWRKY proteins did not possess the consensus WRKY DNA-binding domain (PF03106). The resultant 105 SiWRKY and 44 SvWRKY sequences (Supplementary Table S2) were used in further studies. Among the 105 SiWRKY proteins, SiWRKY099 was identified to be the smallest protein with 93 amino acids (aa), whereas the largest one was SiWRKY011 (1290 aa). The molecular weights of the proteins also varied according to protein size ranging from 10.3 kDa (SiWRKY099) to 145.8 kDa (SiWRKY011). In case of SvWRKY, the smallest proteins were SvWRKY008 (204 aa) and SvWRKY025 (207 aa), while the largest protein was SvWRKY031 (1290 aa). The molecular weight of SvWRKY proteins ranged from 21.6 kDa (SvWRKY008) to 145.8238 kDa (SvWRKY031). Isoelectric point (pI) of SiWRKY and SvWRKY proteins ranged from 4.8 (SiWRKY056) to 10.1 (SiWRKY037) and 5 (SvWRKY026) to 11.8 (SvWRKY006), respectively. The large variation in protein features might denote the presence of putative novel variants. Instability index of these proteins showed that most proteins (99 SiWRKY and 41 SvWRKY) were unstable (Supplementary Table S2).
Classification of SiWRKY and SvWRKY Proteins
WRKY proteins are classified into three major groups (I, II, and III) based on the conserved WRKY domain and zinc finger-like structure (Rushton et al., 1995). Group I has two WRKY domains as well as CX4−5CX22−23HXH structure, group II has one WRKY domain with conserved zinc-finger motif sequence, whereas group III has one WRKY domain and CX4−5CX22−23HXC structure (Eulgem et al., 2000). Group II proteins are further classified into five sub-groups (IIa–IIe) based on the conservation of amino acid motifs outside the WRKY domain (Park et al., 2005). Sequence alignment of SiWRKY and SvWRKY showed that all proteins, except SiWRKY044, SiWRKY063, SvWRKY005, SvWRKY007, SvWRKY008, and SvWRKY011, possess conserved WRKY domain and zinc finger-like structure. These exceptional WRKY proteins were classified as group IV. However, these proteins could represent pseudogenes or sequencing and assembly errors (Xie et al., 2005; Ross et al., 2007).
Among the remaining 103 SiWRKY proteins, 10 belong to group I, 54 to group II and 39 to group III, whereas in case of SvWRKY proteins, 6 belong to group I, 23 to group II and 11 to group III (Figure 1). The first WRKY domain of group I proteins possesses a conserved WRKYGQK amino acid motif, whereas the second domain lacked the GQK signature. Both the WRKY domains were followed by conserved CX4CX22−23HXH structure. Interestingly, SvWRKY004 was observed to possess three WRKY domains followed by zinc finger-like structures. In case of group IV proteins, the conserved WRKYGQK domain was present in the N-terminal region (Figure 1). Phylogenetic analysis of group I, II, and III proteins of SiWRKY, SvWRKY, and ZmWKRY (Wei et al., 2012) confirmed the group-wise classification and also enabled the sub-classification of group II proteins (Figure 2). Group IV proteins deduced through sequence alignment were not included in phylogenetic analysis as they represent the products of pseudogenes or sequencing and assembly errors (Xie et al., 2005; Ross et al., 2007; Wei et al., 2012). Among the 54 group II SiWRKY proteins, 5 belong to IIa, 8 to IIb, 20 to IIc, 9 to IId, and 12 to IIe. Similarly, two SvWRKY proteins belong to group IIa, 3 to IIb, 9 to IIc, 4 to IId, and 5 to IIe. Interestingly, group IIc was interrupted by the members of IIb and IIa (Figure 2). A similar observation was reported by Wei et al. (2012) in maize, wherein the phylogenetic tree of WRKY proteins from Arabidopsis, maize, rice, barley, and Physcomitrella patens showed the interruption in group IIc. Domain analysis using HMMSCAN and PROSITE tools revealed the presence of additional NB-ARC domain (PF00931) in SiWRKY011 and SvWRKY031, and domain of unknown function (PF12204) in SiWRKY011 and SiWRKY096 (Supplementary Table S3).

Figure 1. Multiple sequence alignment of three major groups of SiWRKY and SvWRKY proteins. Group I, II, III, and IV proteins have been aligned separately and the consensus motifs are highlighted in blue boxes. Highly conserved amino acids have been shown in black boxes, less conserved in gray boxes, while amino acids with no similarly were indicated in black texts. The red lines in group I proteins indicate that the intermittent sequences, which were less conserved were not shown.
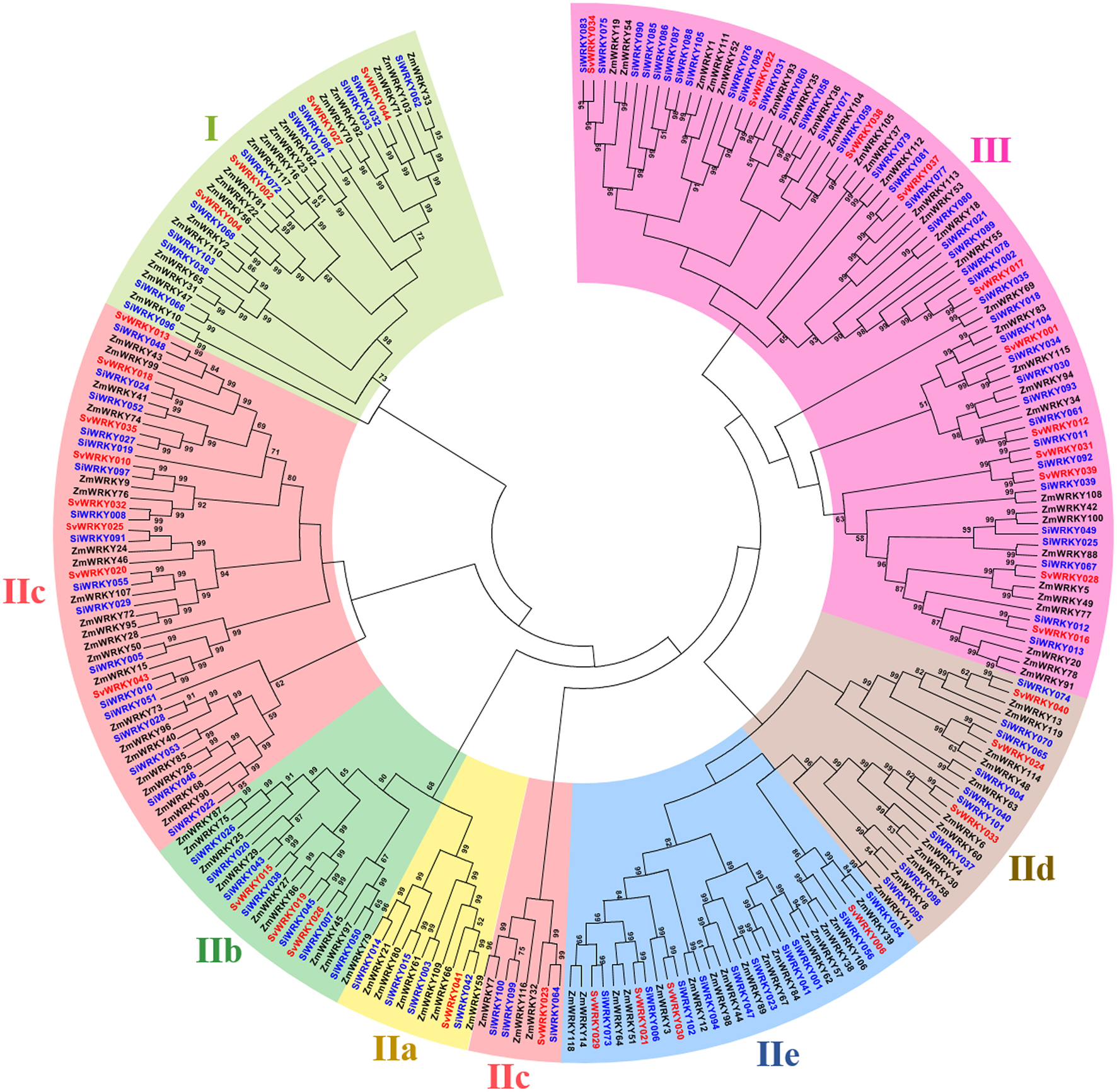
Figure 2. Unrooted Neighbor-Joining tree constructed with WRKY proteins of Setaria italica (SiWRKY), S. viridis (SvWRKY), and Zea mays (ZmWRKY). SiWRKY and SvWRKY protein IDs are highlighted in blue color and groups are differentiated with different colors.
Structure, Location, and Duplication of SiWRKY Genes
Positions of introns and exons within the SiWRKY genes and their chromosomal locations were determined. However, this could not be performed for SvWRKY genes since the genome sequence data of S. viridis is not released in public database, till date. Gene structure prediction showed the numbers and arrangement of introns and exons within the SiWRKY genes (Supplementary Figure S1). The majority of SiWRKY genes (59; ~56%) were found to contain two introns, whereas 22 genes (~21%) have a single intron. Thirteen SiWRKY genes (~12%) have three introns, while 5 (~5%) and 4 (~4%) genes have four and five introns, respectively. A maximum of 10 introns were found to be present in SiWRKY096 and the SiWRKY065 gene was intronless (Supplementary Figure S1). The length of SiWRKY genes was also observed to be variable ranging from 0.6 kb (SiWRKY019) to 7.5 kb (SiWRKY103). Physical mapping of all the 105 SiWRKY genes onto nine chromosomes of S. italica revealed an uneven distribution of these genes in the genome (Figure 3). Among the four groups, members of group II and III were present in all the nine chromosomes, whereas group I SiWRKY genes were not present in chromosomes 1 and 4. Two members of group IV, namely SiWRKY044 and SiWRKY063, were present in chromosome 5. Subsequently, the expansion of WRKY gene family in S. italica genome was examined using MCScanX tool, which showed that 10 and 8 SiWRKY gene pairs underwent tandem and segmental duplications, respectively (Figure 3). The tandemly duplicated genes include one pair of group I (in chromosome 3), two pairs of group II (in chromosomes 4 and 9), and seven pairs of group III genes (in chromosomes 1, 5, 7, and 8). Segmental duplication was found to occur between the SiWRKY genes of chromosome 3 and 5, and not in other chromosomes (Figure 3).

Figure 3. Physical map of SiWRKY genes showing their chromosomal locations. Vertical bars represent the chromosomes and numbers at the left indicate the position of genes (in Mb). Tandemly duplicated gene pairs are indicated with red vertical bars and segmentally duplicated gene pairs are connected with yellow bands. Colors of SiWRKY IDs correspond to the different groups.
Gene Ontology Annotation and Analysis of cis-acting Elements
Gene ontology (GO) annotation of SiWRKY and SvWRKY proteins was performed using Blast2GO and Cytoscape tools and showed the involvement of these proteins in different biological processes and molecular functions (Supplementary Table S4). A majority of these proteins were predicted to be involved in response to stress as well as cellular, metabolic and biosynthetic processes (biological process; P ≤ 2.2 × 10−6) (Figure 4). The molecular functions of these proteins corresponded to transcription regulator activity (P ≤ 4.2 × 10−13). Further, cellular component analysis revealed the localization of these gene products in nucleus (Figure 4). Promoter analysis of SiWRKY genes showed the presence of 284 cis-regulatory elements (CREs), of which some elements were present in all the 105 genes, whereas a few were unique to one or two genes of the entire family (Supplementary Table S5). ARR1AT (element involved in cytokinin responsiveness), CAATBOX1 (element in enhancer regions of the promoter), CACTFTPPCA1 (element involved in mesophyll-specific gene expression of C4 phosphoenolpyruvate carboxylase gene in C4 plants), DOFCOREZM (target binding site of Dof proteins), EBOXBNNAPA (target binding site of bHLH and MYB-transcription factor), GATABOX (light responsive element), MYCCONSENSUSAT (MYC recognition site), and WRKY71OS (binding site of WRKY TFs) were present in the upstream region of all SiWRKY genes. In contrast, few CREs were found to be present in only one SiWRKY gene (Supplementary Table S5). This includes ABADESI2 (Synthetic element related to response to abscisic acid and to desiccation; in SiWRKY009), ABRE2HVA1 (ABA responsive element; in SiWRKY053), ACGTSEED3 (bZIP transcription activator binding site; in SiWRKY015), AGL2ATCONSENSUS (MADS binding site; in SiWRKY034), AUXRETGA2GMGH3 (auxin responsive element; in SiWRKY017), EREGCC (ethylene responsive element; in SiWRKY051), HSE (heat shock responsive element; in SiWRKY047), MREATCHS (MYB Recognition Element; in SiWRKY094), POLLEN2LELAT52 (required for pollen specific expression; SiWRKY002), and S2FSORPL21 (leaf-specific, light-independent regulatory element; in SiWRKY094; Supplementary Table S5).
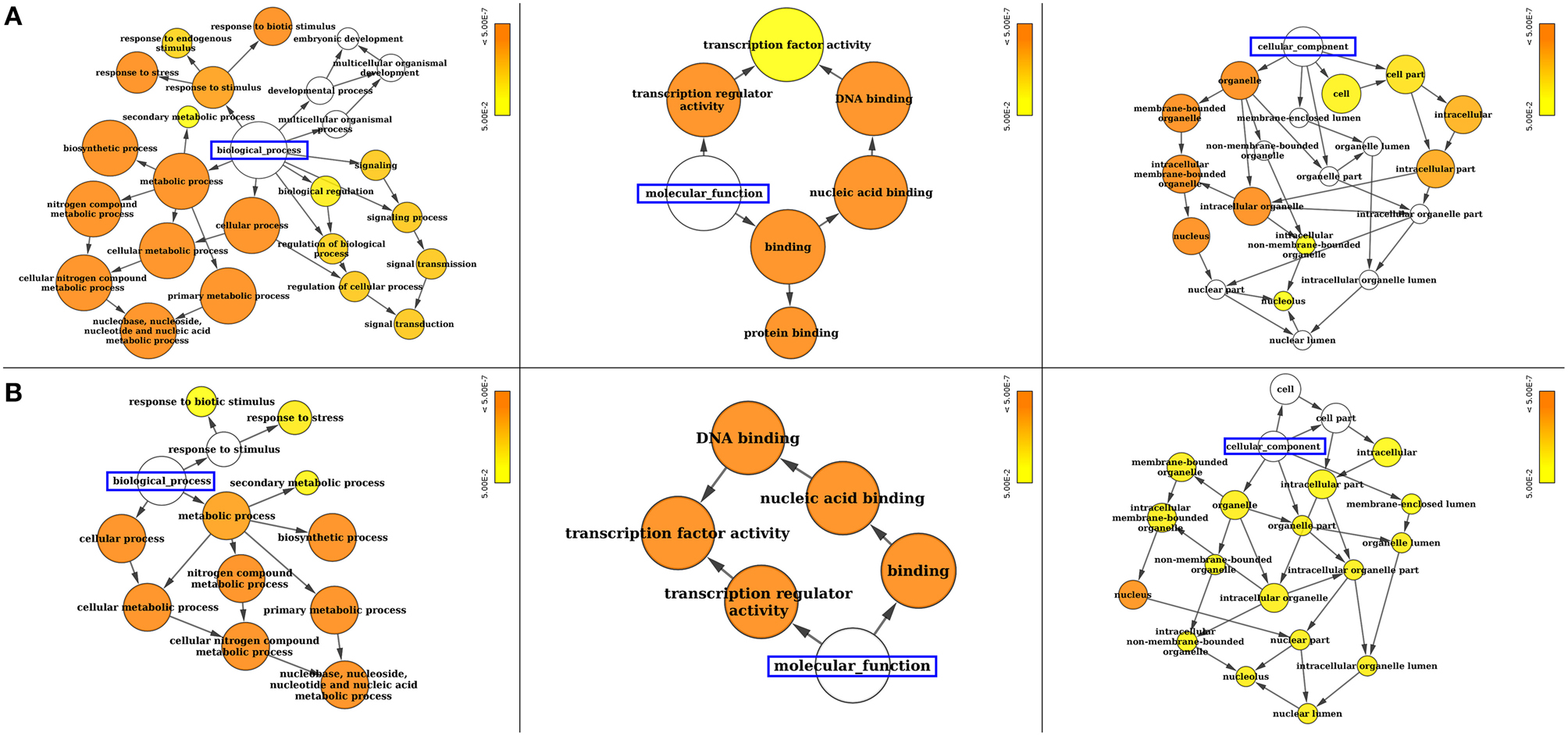
Figure 4. Gene ontology (GO) enrichment analysis of (A) SiWRKY and (B) SvWRKY genes. The number of genes falling in each GO category is directly proportional to the node size. The nodes are color shaded according to the significance level (corrected P-value).
Comparative Mapping in Related Grass Genomes and Ks Dating of Paralogs and Orthologs
All the 105 SiWRKY genes and 44 SvWRKY proteins were subjected to BLAST search against the database of switchgrass (Panicum virgatum), sorghum (Sorghum bicolor) and maize (Zea mays) to identify corresponding orthologs (>90% similarity). Potential orthologs were confirmed by reciprocal BLAST (Figure 5; Supplementary Tables S6–S11). A total of 60 SiWRKY genes (~57%) showed syntenic relationship with maize (Supplementary Table S8), followed by switchgrass (~54%; Supplementary Table S6) and sorghum (~40%) (Supplementary Table S7). In case of SvWRKY proteins, maximum synteny was observed with switchgrass (31, ~70%; Supplementary Table S9), followed by maize (24, ~55%; Supplementary Table S11) and sorghum (20, ~45%; Supplementary Table S10). Further, the effect of Darwinian positive selection in duplication and divergence of WRKY genes was examined by estimating the ratios of non-synonymous (Ka) vs. synonymous (Ks) substitution for paralogous as well as orthologous gene pairs. The Ka/Ks ratio for tandemly duplicated gene pairs ranged from 0.09 to 0.18 with an average of 0.13 (Supplementary Table S12), while for segmentally duplicated gene pairs, the ratio ranged from 0.06 to 0.14 with an average of 0.1 (Supplementary Table S13). Both the tandem and segmental duplications have been estimated to occur around 29 million years ago (mya) and 23 mya, respectively (Supplementary Tables S12, S13). Similarly, the average Ka/Ks ratios of orthologous gene pairs of S. italica - P. virgatum, S. italica - S. bicolor, and S. italica - Z. mays were estimated as 0.94, 0.19, and 0.19, respectively (Supplementary Tables S6–S8). In case of S. viridis - P. virgatum, S. viridis - S. bicolor, and S. viridis - Z. mays orthologs, the Ka/Ks ratios were 0.79, 0.21, and 0.18, respectively (Supplementary Tables S9–S11). This revealed that the orthologous gene pairs underwent natural selection (Ka/Ks < 1). The estimated time of divergence of S. italica and P. virgatum was 4.7 mya, whereas S. italica and S. bicolor as well as Z. mays diverged around 27 mya. Similar estimates were observed in case of S. viridis - P. virgatum (4.7 mya), S. viridis - S. bicolor (26.8 mya), and S. viridis - Z. mays (27.8 mya) orthologs.
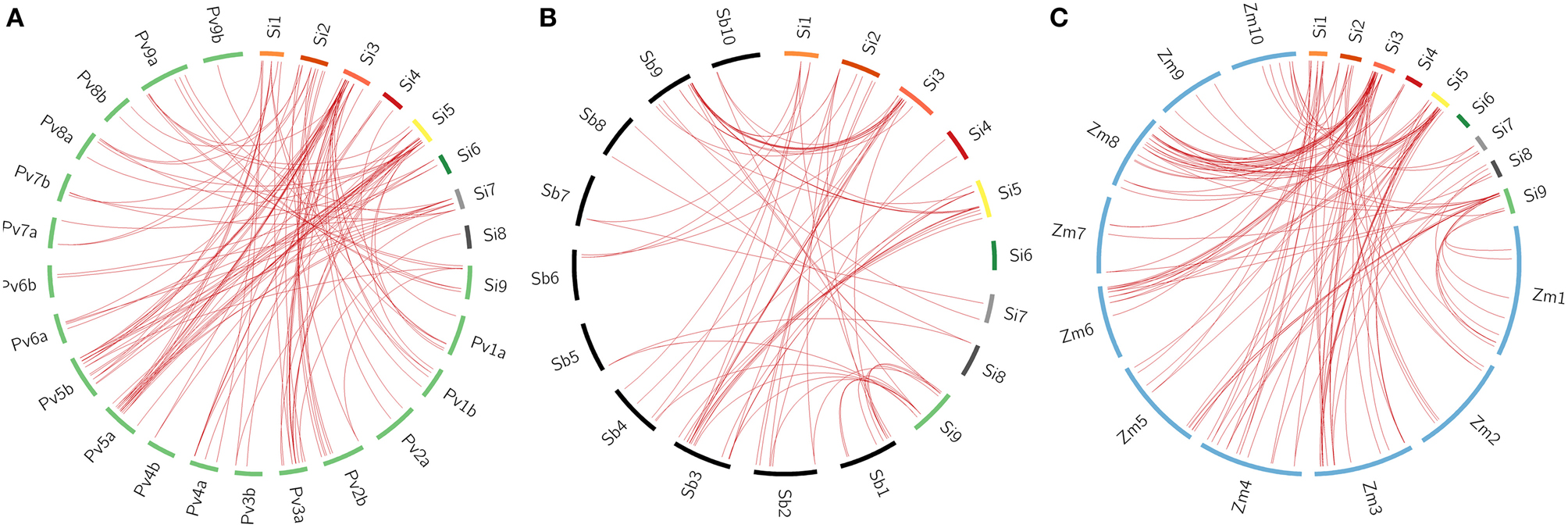
Figure 5. Synteny map showing the orthologous gene positions of WRKY genes in Setaria italica (Si) and (A) Panicum virgatum (Pv), (B) Sorghum bicolor (Sb), (C) Zea mays (Zm). Each block represents individual chromosome and the orthologous genomic regions are marked with red lines.
In silico Expression Profiling of SiWRKY and SvWRKY Genes
Expression pattern of SiWRKY genes in four tissues, namely root, leaf, spica, and stem revealed a differential expression pattern (Figure 6). A few genes including SiWRKY003, SiWRKY017, SiWRKY033, SiWRKY034, SiWRKY056, and SiWRKY101 were found to be highly expressed in all the tissues. Tissue-specific higher expression of SiWRKY028, SiWRKY032, SiWRKY042, SiWRKY045, SiWRKY060, SiWRKY062, SiWRKY078, and SiWRKY091 in root and SiWRKY044 in stem was also observed. Some genes such as SiWRKY006, SiWRKY019, SiWRKY026, SiWRKY039, SiWRKY057, etc. did not show any expression in all the four tissues (Figure 6). In case of expression profiles in dehydration stress library, relatively higher expression of SiWRKY004, SiWRKY024, SiWRKY046, and SiWRKY068 was observed in stressed sample as compared to control. Downregulation of a few genes viz., SiWRKY60, SiWRKY61, etc. was also seen (Figure 6). Expression patterns of SvWRKY genes in pooled RNA isolated from samples across three developmental stages, namely seed germination, vegetative growth, and reproduction in different tissues including leaf, stem, node, crown, root, spikelet, floret, and seed tissues showed higher transcript abundance of SvWRKY001, SvWRKY027, SvWRKY033, SvWRKY037, and SvWRKY039. However, the majority of SvWRKY genes showed no or negligible expression (Figure 6).
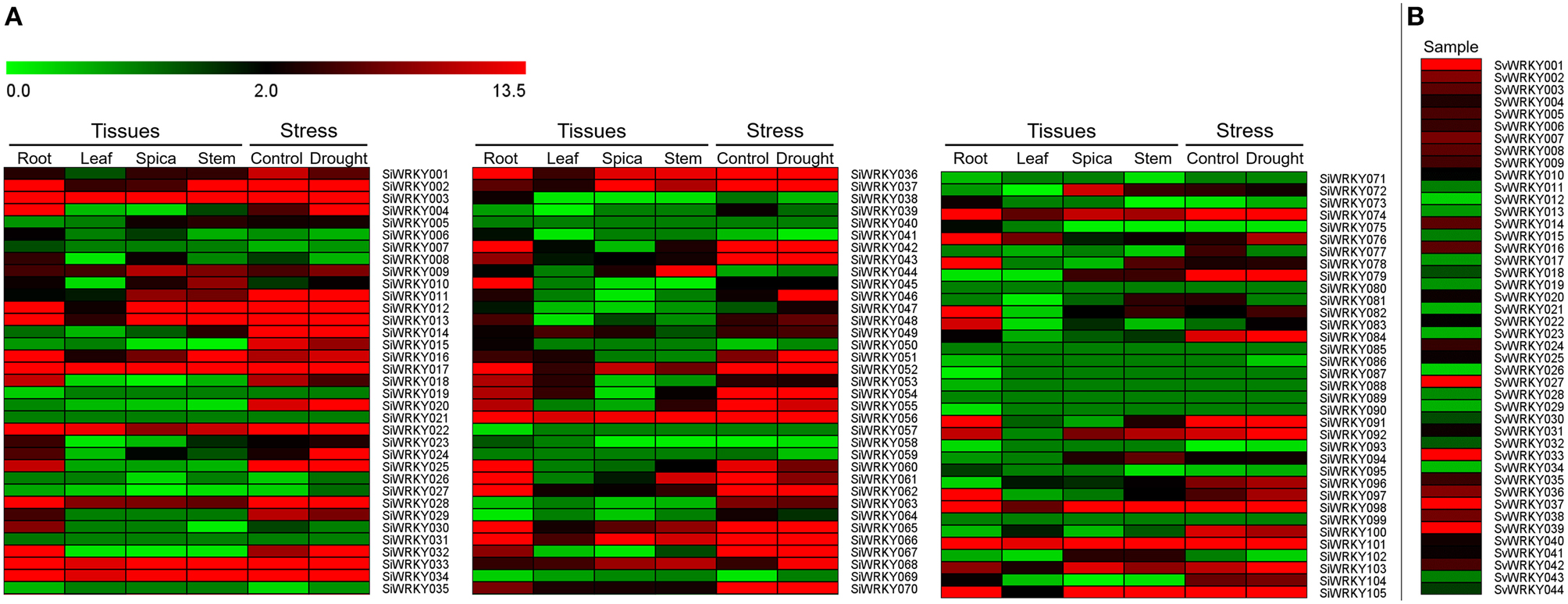
Figure 6. Heatmap showing the expression pattern of (A) SiWRKY genes in four tissues, namely root, leaf, spica, stem, and dehydration stress library of Setaria italica, and (B) SvWRKY genes in pooled transcriptome of S. viridis samples across three developmental stages, namely seed germination, vegetative growth, and reproduction in different tissues including leaf, stem, node, crown, root, spikelet, floret, and seed tissues. The colored bar at top left represents relative expression value, where 0.0, 2.0, and 13.5 denotes low, medium, and high expression, respectively.
Expression Pattern of SiWRKY Genes in Response to Stress and Hormone Treatments
To investigate the expression of SiWRKY genes in response to abiotic stress and hormone treatments, 12 genes were selected based on their differential expression pattern in RNA-seq libraries of four tissues and under drought stress (Figure 6). Additionally, the genes were resourced from the nine chromosomes of S. italica in order to provide a genome-wide coverage (Figure 2). The expression profiles of the 12 candidate genes were examined during early (1 h) and late (24 h) stages of dehydration, salinity, ABA, SA, and MeJA treatments. The relative transcript abundance assessed through qRT-PCR showed a differential expression pattern of all the SiWRKY genes (Figure 7). Few genes including SiWRKY003, SiWRKY017, SiWRKY033, SiWRKY042, and SiWRKY056 did not show any significant expression throughout the experiments, whereas SiWRKY034 was highly expressed only during the late phase of SA treatment. During dehydration and salinity stress, SiWRKY064, SiWRKY066, SiWRKY074, and SiWRKY082 were found to be upregulated at both the time points, in which, significant upregulation of SiWRKY064 and SiWRKY082 at late phase of salinity stress, and SiWRKY066 and SiWRKY074 at both the phases of dehydration were observed (Supplementary Figure S2). In case of hormone treatments, all these four genes were found to be highly expressed during late phase. In addition to these, SiWRKY101 was observed to be upregulated during late phase of dehydration and MeJA treatment. The fold expression of SiWRKY064 and SiWRKY082 were significantly higher during both the phases of stress and at late phase of hormone treatments, suggesting their potential as candidates for functional characterization.
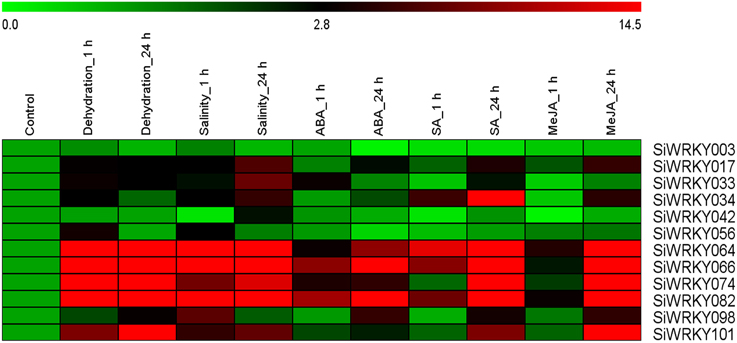
Figure 7. Relative transcript levels of candidate SiWRKY genes in response to treatment with stresses and hormones. Expression profiles of SiWRKY genes in Setaria italica seedlings exposed to dehydration, salinity, abscisic acid (ABA), salicylic acid (SA), and methyl jasmonate (MeJA) for two time points (1 h and 24 h) were analyzed by qRT-PCR and presented as heatmap. The scale bar at the top represents relative expression value. Relative fold expression values are presented as bar diagram in Supplementary Figure S2.
Discussion
WRKY transcription factors have been reported to play multiple roles in regulating normal growth and development, and in response to environmental stimuli in plants (Rushton et al., 2010). This class of TFs are one of the well-studied proteins whose mechanism of action, autoregulation and cross-regulation in signaling and evolution have been reported (Bakshi and Oelmüller, 2014). Though initially considered as vital players of biotic stress tolerance, WRKY TFs were later discovered to play significant roles in conferring tolerance to diverse abiotic stresses including salinity (Jiang and Yu, 2009; Chen et al., 2010), drought, heat (Li et al., 2009, 2011), cold (Zou et al., 2010), H2O2(Song et al., 2009), ozone oxidative stress (Jiang and Deyholos, 2009), UV radiation (Jiang and Deyholos, 2009), sugar starvation (Song et al., 2010), phosphate depreviation (Chen et al., 2009) and wounding (Shang et al., 2010). Further, numerous reports have indicated the response of a single WRKY gene to several stress factors, thus highlighting the diverse regulatory role of WRKY proteins in stress response (Wei et al., 2008; Jiang and Deyholos, 2009; Li et al., 2009, 2011; Chen et al., 2012). The expression of WRKY TFs in response to broad-spectrum abiotic stresses suggests their participation in regulation of signaling mechanisms associated with transcriptional reprogramming during environmental stress. Genome-wide identification of WRKY TFs has been performed in many crop plants and their expression profiling in response to various abiotic stresses have been studied.
Recently, C4 crops are gaining momentum in stress biology research owing to their improved water-use efficiency and nitrogen-use efficiency (Sadras et al., 2011). C4 photosynthesis also confers tolerance to crops against abiotic stress, particularly to drought and heat (Sadras et al., 2011). Setaria italica and its wild progenitor S. viridis, have recently been identified as model crops for studying C4 photosynthesis and abiotic stress tolerance due to their small genome, short life span, inbreeding nature and ability to withstand adverse environmental conditions (Brutnell et al., 2010; Wang et al., 2011; Diao et al., 2014; Muthamilarasan and Prasad, 2015). Furthermore, both the crops share maximum genetic synteny with various biofuel grasses such as switchgrass, napiergrass and pearl millet and therefore, S. italica and S. viridis have also been regarded as model systems for bioenergy research (Li and Brutnell, 2011; Lata et al., 2013; Brutnell et al., 2015; Muthamilarasan and Prasad, 2015) and nutrition studies (Muthamilarasan et al., in press). Therefore, in view of the importance of S. italica and S. viridis in abiotic stress biology, the present investigation was performed to identify and characterize WRKY TFs using computational tools and examine their expression patterns in response to abiotic stress and hormone treatments.
In this study, 105 WRKY genes from S. italica genome (SiWRKY) and 44 from S. viridis transcriptome (SvWRKY) were identified. Comparison of the number of WRKY genes in S. italica with other sequenced grass genomes namely maize (163 genes), sorghum (110 genes) and rice (O. sativa subsp. indica; 109 genes) has shown that S. italica has comparatively lesser number of genes. However, Brachypodium has a minimum of 87 genes, owing to its smaller genome size. Similar comparisons of the number of WRKY genes among all the sequenced plants showed that soybean has the maximum number of WRKY genes (233), followed by cotton (219), whereas the primitive plants of Chlorophyta have one to two genes. Interestingly, the genome of Physcomitrella patens has 41 WRKY genes. Only 44 WRKY proteins were identified from the transcriptome of S. viridis due to the non-availability of genome sequence information and this number is expected to increase when the whole genome sequence is released in public domain. Examining the protein properties of SiWRKY and SvWRKY TFs revealed large differences in amino acid length, molecular weight and isoelectric point of these proteins, and these variations could be attributed to the presence of putative novel variants, which needs to be validated.
Sequence alignment and phylogenetic analysis of SiWRKY and SvWRKY proteins classified them into three major groups (I, II, and III) based on the WRKY domain and conserved zinc finger-like motif. In addition, a distinct class of WRKY proteins classified as group IV has been identified with two members of SiWRKY and four members of SvWRKY. These proteins possess only the WRKY domain and not the zinc finger-like motif. Sequence alignment and phylogenetic analysis showed that a majority of SiWRKY proteins belong to group II (54) followed by group III (39) and group I (10). Similar case was observed in SvWRKY proteins, where a maximum of 23 proteins belong to group II, 11 to group III, and 6 to group I. This is in agreement with the distribution reported in maize (Wei et al., 2012). The position of WRKY domain and associated zinc finger-like structures in SiWRKY and SvWRKY was investigated through multiple sequence alignment and domain analyses tools, namely HMMSCAN and ScanProsite. The analyses revealed that the distribution of phylogenetic groups corresponds well with the domain structures and sequence conservation. It showed three interesting observations: (i) two proteins of SiWRKY (SiWRKY044 and SiWRKY063) and four SvWRKY proteins (SvWRKY005, SvWRKY007, SvWRKY008, and SvWRKY011) possess only WRKY domain and lack zinc finger-like structure. (ii) two additional domains, namely NB-ARC and DUF were present in SiWRKY011 and SvWRKY031, and SiWRKY011 and SiWRKY096, respectively. and (iii) SvWRKY004 has three WRKY domains followed by zinc finger-like structures.
Physical mapping of SiWRKY genes on the nine chromosomes of S. italica showed that maximum number of genes were present on chromosomes 5 (22 genes; ~21%) and 3 (19 genes; ~18%), and a minimum of 5 genes each (~5%) were present on chromosomes 4 and 6. The maximum number of genes on chromosomes 5 and 3 could be attributed to the occurrence of segmental duplication, as revealed by MCScanX analysis. Eight genes in these chromosomes were segmentally duplicated, and in addition, 10 gene pairs were identified to be tandem duplicates. The Ks dating and estimation of Ka/Ks ratios of duplicated gene pairs showed that these genes underwent intense purifying selection. The time of duplication of tandemly and segmentally duplicated gene pairs were estimated as ~26 and ~23 million years ago (mya), which were in congruence with the whole genome tandem and segmental duplication reported to have occurred around 25–27 and 18–22 mya (Zhang et al., 2012). This also demonstrates the effect of chromosomal duplication events in shaping the distribution and organization of WRKY genes in S. italica genome.
Comparative mapping of SiWRKY genes and SvWRKY proteins on the switchgrass, sorghum and maize databases was performed to understand the orthologous relationships between the grass genomes. SiWRKY genes showed maximum synteny with maize (~57%), followed by switchgrass (~54%) and sorghum (~40%), whereas SvWRKY proteins showed maximum orthology with switchgrass (~70%), followed by maize (~55%) and sorghum (~45%). Though higher percentage of orthology was expected between Setaria and switchgrass owing to their extensive gene-level synteny, SiWRKY genes were found to be more homologous to maize. However, SvWRKY revealed the syntenic pattern with respect to decrease in synteny with increase in phylogenetic distance, between these crops. Estimation of time of divergence of orthologous gene pairs revealed that S. italica and switchgrass WRKY genes diverged around 4.7 mya, whereas divergence between S. italica WRKY genes and those of maize and sorghum occurred around 27 and 27.5 mya, respectively. Similarly, S. viridis and switchgrass WRKY genes were predicted to have diverged around 4.7 mya, while S. viridis and maize, and sorghum WRKY genes diverged around 26.8 and 27.8 mya, respectively. These findings are in accordance with the period of divergence of Poaceae members as reported by Zhang et al. (2012). The comparative map constructed using orthologous WRKY genes demonstrated the frequent occurrence of nested chromosomal fusions in the grass genomes. Further, this comparative map would be useful in choosing candidate WRKY genes from these genomes for functional characterization.
The publicly available transcriptome data of four different tissues and dehydration stress library of S. italica, and pooled tissue library of S. viridis were processed using in-house perl scripts and computational tools to derive the RPKM expression values for SiWRKY and SvWRKY genes. The heatmap generated using these expression values showed tissue-specific and condition-specific expression patterns of WRKY genes. Relatively higher expression of few genes in all the tissues, or in any one tissue or only during dehydration stress suggested the multifaceted roles of WRKY genes in diverse molecular and physiological activities. This data could be exploited for selecting candidate genes showing distinct expression pattern for delineating their functional roles. Based on this heatmap and physical map data, twelve candidate SiWRKY genes were chosen for expression profiling under different abiotic stress (dehydration and salinity) and hormone (ABA, SA, and MeJA) treatments (at two time points). These genes showed differential expression pattern in the four tissues (root, stem, leaf, and spica) and drought stress library as deduced using RNA-seq expression data. Further, the genes were also chosen to represent all the nine chromosomes of foxtail millet, to provide a representative genome-wide coverage. The qRT-PCR analysis of these genes showed their differential expression patterns during exposure to stresses and hormones, and this suggested the putative involvement of WRKY genes in stress response mechanism and their regulation in response to phytohormones. Overall, the qRT-PCR analysis revealed that SiWRKY066 and SiWRKY082 could be potential candidates for further functional characterization and for delineating their roles in abiotic stress signaling.
Conclusions
With the advancement of high-throughput technologies and strategies, including physiology, chemical genetics, and computational approaches, the role of WRKY TFs in signal transduction and gene regulation has been well studied in all the major crops and tree species. However, no such study on WRKY TFs has been conducted in S. italica and S. viridis, which are now considered as model systems for investigating C4 photosynthesis, biofuel traits and abiotic stress tolerance mechanisms. Considering the importance of these crops and WRKY TFs, the present study used comprehensive computational approaches to identify and characterize WRKY gene family members. The identified members were used for construction of a physical map, duplication studies, phylogenetic analysis, gene ontology annotation, promoter analysis, comparative mapping, and evolutionary studies. In addition, in silico expression profiling of SiWRKY and SvWRKY genes were performed to understand the expression pattern of these genes in different tissues and dehydration stress conditions. Expression profiling of candidate SiWRKY genes under abiotic stress and hormone treatments showed differential expression pattern of these genes, thus providing an indication of their regulatory functions under stress conditions.
Author Contributions
MP conceived and designed the experiments. MM, VB, RK, JJ, SS, KN performed the experiments. MM analyzed the results and wrote the manuscript. MP approved the final version of the manuscript.
Funding
Research on foxtail millet genomics at MP's laboratory is funded by the Core Grant of National Institute of Plant Genome Research, New Delhi, India.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MM acknowledges University Grants Commission, New Delhi, India for providing Research Fellowship. The authors also thank Prof. Arun Jagannath, University of Delhi, India for critically reading the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00910
References
Alexandrova, K. S., and Conger, B. V. (2002). Isolation of two somatic embryogenesis-related genes from orchardgrass (Dactylis glomerata). Plant Sci. 162, 301–307. doi: 10.1016/S0168-9452(01)00571-4
Bakshi, M., and Oelmüller, R. (2014). WRKY transcription factors: Jack of many trades in plants. Plant Signal. Behav. 9:e27700. doi: 10.4161/psb.27700
Bennetzen, J. L., Schmutz, J., Wang, H., Percifield, R., Hawkins, J., Pontaroli, A. C., et al. (2012). Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 30, 555–561. doi: 10.1038/nbt.2196
Beyer, K., Binder, A., Boller, T., and Collinge, M. (2001). Identification of potato genes induced during colonization by Phytophthora infestans. Mol. Plant Pathol. 2, 125–134. doi: 10.1046/j.1364-3703.2001.00059.x
Bonthala, V. S., Muthamilarasan, M., Roy, R., and Prasad, M. (2014). FmTFDb: a foxtail millet transcription factors database for expediting functional genomics in millets. Mol. Biol. Rep. 41, 6343–6348. doi: 10.1007/s11033-014-3574-y
Brutnell, T. P., Bennetzen, J. L., and Vogel, J. P. (2015). Brachypodium distachyon and Setaria viridis: model genetic systems for the grasses. Annu. Rev. Plant Biol. 65, 715–741. doi: 10.1146/annurev-arplant-042811-105528
Brutnell, T. P., Wang, L., Swartwood, K., Goldschmidt, A., Jackson, D., Zhu, X. G., et al. (2010). Setaria viridis: a model for C4 Photosynthesis. Plant Cell 22, 2537–2544. doi: 10.1105/tpc.110.075309
Cai, C. P., Niu, E., Du, H., Zhao, L., Feng, Y., and Guo, W. Z. (2014a). Genome-wide analysis of the WRKY transcription factor gene family in Gossypium raimondii and the expression of orthologs in cultivated tetraploid. Crop J. 2, 87–101. doi: 10.1016/j.cj.2014.03.001
Cai, Y., Chen, X., Xie, K., Xing, Q., Wu, Y., Li, J., et al. (2014b). Dlf1, a WRKY transcription factor, is involved in the control of flowering time and plant height in rice. PLoS ONE 9:e102529. doi: 10.1371/journal.pone.0102529
Chen, C. H., and Chen, Z. X. (2002). Potentiation of developmentally regulated plant defense response by AtWRKY18, a pathogen-induced Arabidopsis transcription factor. Plant Physiol. 129, 706–716. doi: 10.1104/pp.001057
Chen, H., Lai, Z., Shi, J., Xiao, Y., Chen, Z., and Xu, X. (2010). Roles of Arabidopsis WRKY18, WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 10:281. doi: 10.1186/1471-2229-10-281
Chen, L., Song, Y., Li, S., Zhang, L., Zou, C., and Yu, D. (2012). The role of WRKY transcription factors in plant abiotic stresses. Biochim. Biophys. Acta. 1819, 120–128. doi: 10.1016/j.bbagrm.2011.09.002
Chen, T., Lv, Y., Zhao, T., Li, N., Yang, Y., Yu, W., et al. (2013). Comparative transcriptome profiling of a resistant vs. susceptible tomato (Solanum lycopersicum) cultivar in response to infection by tomato yellow leaf curl virus. PLoS ONE 8:e80816. doi: 10.1371/journal.pone.0080816
Chen, W., Provart, N. J., Glazebrook, J., Katagiri, F., Chang, H. S., Eulgem, T., et al. (2002). Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell 14, 559–574. doi: 10.1105/tpc.010410
Chen, Y. F., Li, L. Q., Xu, Q., Kong, Y. H., Wang, H., and Wu, W. H. (2009). The WRKY6 transcription factor modulates PHOSPHATE1 expression in response to low Pi stress in Arabidopsis. Plant Cell 21, 3554–3566. doi: 10.1105/tpc.108.064980
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
de Castro, E., Sigrist, C. J., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P. S., Gasteiger, E., et al. (2006). ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365. doi: 10.1093/nar/gkl124
Dellagi, A., Heilbronn, J., Avrova, A. O., Montesano, M., Palva, E. T., Stewart, H. E., et al. (2000). A potato gene encoding a WRKY-like transcription factor is induced in interactions with Erwinia carotovora subsp atroseptica and Phytophthora infestans and is coregulated with class I endochitinase expression. Mol. Plant Microbe Interact. 13, 1092–1101. doi: 10.1094/MPMI.2000.13.10.1092
de Pater, S., Greco, V., Pham, K., Memelink, J., and Kijne, J. (1996). Characterization of a zinc-dependent transcriptional activator from Arabidopsis. Nucleic Acids Res. 24, 4624–4631. doi: 10.1093/nar/24.23.4624
Deslandes, L., Olivier, J., Theulieres, F., Hirsch, J., Feng, D. X., Bittner-Eddy, P., et al. (2002). Resistance to Ralstonia solanacearum in Arabidopsis thaliana is conferred by the recessive RRS1-R gene, a member of a novel family of resistance genes. Proc. Natl. Acad. Sci. U.S.A. 99, 2404–2409. doi: 10.1073/pnas.032485099
Diao, X., Schnable, J., Bennetzen, J. L., and Li, J. (2014). Initiation of setaria as a model plant. Front. Agri. Sci. Eng. 1, 16–20. doi: 10.15302/J-FASE-2014011
Ding, M., Chen, J., Jiang, Y., Lin, L., Cao, Y., Wang, M., et al. (2015). Genome-wide investigation and transcriptome analysis of the WRKY gene family in Gossypium. Mol. Genet. Genomics 290, 151–171. doi: 10.1007/s00438-014-0904-7
Ding, Z. J., Yan, J. Y., Li, G. X., Wu, Z. C., Zhang, S. Q., and Zheng, S. J. (2014). WRKY41 controls Arabidopsis seed dormancy via direct regulation of ABI3 transcript levels not downstream of ABA. Plant J. 79, 810–823. doi: 10.1111/tpj.12597
Dong, J., Chen, C., and Chen, Z. (2003). Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol. Biol. 51, 21–37. doi: 10.1023/A:1020780022549
Dou, L., Zhang, X., Pang, C., Song, M., Wei, H., Fan, S., et al. (2014). Genome-wide analysis of the WRKY gene family in cotton. Mol. Genet. Genomics 289, 1103–1121. doi: 10.1007/s00438-014-0872-y
Du, L., and Chen, Z. (2000). Identification of genes encoding receptor-like protein kinases as possible targets of pathogen- and salicylic acid-induced WRKY DNA-binding proteins in Arabidopsis. Plant J. 24, 837–847. doi: 10.1046/j.1365-313x.2000.00923.x
Eulgem, T., Rushton, P. J., Robatzek, S., and Somssich, I. E. (2000). The WRKY superfamily of plant transcription factors. Trends Plant Sci. 5, 199–206. doi: 10.1016/S1360-1385(00)01600-9
Fan, X., Guo, Q., Xu, P., Gong, Y., Shu, H., Yang, Y., et al. (2015). Transcriptome-wide identification of salt-responsive members of the WRKY gene family in Gossypium aridum. PLoS ONE 10:e0126148. doi: 10.1371/journal.pone.0126148
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., et al. (2005). “Protein Identification and Analysis Tools on the ExPASy Server,” in The Proteomics Protocols Handbook, ed J. M. Walker (New York, NY: Springer), 571–607. doi: 10.1385/1-59259-890-0:571
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Guan, Y., Meng, X., Khanna, R., LaMontagne, E., Liu, Y., and Zhang, S. (2014). Phosphorylation of a WRKY transcription factor by MAPKs is required for pollen development and function in Arabidopsis. PLoS Genet. 10:e1004384. doi: 10.1371/journal.pgen.1004384
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids. Symp. Ser. 41, 95–98.
Hara, K., Yagi, M., Kusano, T., and Sano, H. (2000). Rapid systemic accumulation of transcripts encoding a tobacco WRKY transcription factor upon wounding. Mol. Gen. Genet. 263, 30–37. doi: 10.1007/PL00008673
He, H., Dong, Q., Shao, Y., Jiang, H., Zhu, S., Cheng, B., et al. (2012). Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 31, 1199–1217. doi: 10.1007/s00299-012-1241-0
Higo, K., Ugawa, Y., Iwamoto, M., and Korenaga, T. (1999). Plant cis-acting regulatory DNA elements (PLACE) database:1999. Nucleic Acids Res. 27, 297–300. doi: 10.1093/nar/27.1.297
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., and Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Huang, S., Gao, Y., Liu, J., Peng, X., Niu, X., Fei, Z., et al. (2012). Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mol. Genet. Genomics 287, 495–513. doi: 10.1007/s00438-012-0696-6
Huang, T., and Duman, J. G. (2002). Cloning and characterization of a thermal hysteresis (antifreeze) protein with DNA-binding activity from winter bittersweet nightshade, Solanum dulcamara. Plant Mol. Biol. 48, 339–350. doi: 10.1023/A:1014062714786
Huh, S. U., Choi, L. M., Lee, G. J., Kim, Y. J., and Paek, K. H. (2012). Capsicum annuum WRKY transcription factor d (CaWRKYd) regulates hypersensitive response and defense response upon Tobacco mosaic virus infection. Plant Sci. 197, 50–58. doi: 10.1016/j.plantsci.2012.08.013
Jiang, W. B., and Yu, D. Q. (2009). Arabidopsis WRKY2 transcription factor may be involved in osmotic stress response. Acta Bot. Yunnanica. 31, 427–432 doi: 10.3724/SP.J.1143.2009.09046
Jiang, Y., Duan, Y., Yin, J., Ye, S., Zhu, J., Zhang, F., et al. (2014). Genome-wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J. Exp. Bot. 65, 6629–6644. doi: 10.1093/jxb/eru381
Jiang, Y., and Deyholos, M. K. (2006). Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol. 6:25. doi: 10.1186/1471-2229-6-25
Jiang, Y., and Deyholos, M. K. (2009). Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 69, 91–105. doi: 10.1007/s11103-008-9408-3
Jin, J., Zhang, H., Kong, L., Gao, G., and Luo, J. (2014). PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187. doi: 10.1093/nar/gkt1016
Johnson, C. S., Kolevski, B., and Smyth, D. R. (2002). TRANSPARENT TESTA GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant Cell 14, 1359–1375. doi: 10.1105/tpc.001404
Kalde, M., Barth, M., Somssich, I. E., and Lippok, B. (2003). Members of the Arabidopsis WRKY group III transcription factors are part of different plant defense signaling pathways. Mol. Plant Microbe Interact. 16, 295–305. doi: 10.1094/MPMI.2003.16.4.295
Kim, K. C., Lai, Z., Fan, B., and Chen, Z. (2008). Arabidopsis WRKY38 and WRKY62 transcription factors interact with histone deacetylase 19 in basal defense. Plant Cell 20, 2357–2371. doi: 10.1105/tpc.107.055566
Kole, C., Muthamilarasan, M., Henry, R., Edwards, D., Sharma, R., Abberton, M., et al. (2015). Application of genomics-assisted breeding for generation of climate resilient crops: progress and prospects. Front. Plant Sci. 6:563. doi: 10.3389/fpls.2015.00563
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, K., Muthamilarasan, M., and Prasad, M. (2013). Reference genes for quantitative real-time PCR analysis in the model plant foxtail millet (Setaria italica L.) subjected to abiotic stress conditions. Plant Cell Tiss. Org. Cult. 115, 3–22. doi: 10.1007/s11240-013-0335-x
Lata, C., Gupta, S., and Prasad, M. (2013). Foxtail millet: a model crop for genetic and genomic studies in bioenergy grasses. Crit. Rev. Biotechnol. 33, 328–343. doi: 10.3109/07388551.2012.716809
Lata, C., Mishra, A. K., Muthamilarasan, M., Bonthala, V. S., Khan, Y., and Prasad, M. (2014). Genome-wide investigation and expression profiling of AP2/ERF transcription factor superfamily in foxtail millet (Setaria italica L.). PLoS ONE 9:e113092. doi: 10.1371/journal.pone.0113092
Lata, C., Muthamilarasan, M., and Prasad, M. (2015). “Drought stress responses and signal transduction in plants,” in Elucidation of Abiotic Stress Signaling in Plants, ed G. K. Pandey (New York, NY: Springer), 195–225 doi: 10.1007/978-1-4939-2540-7_7
Lata, C., Sahu, P. P., and Prasad, M. (2010). Comparative transcriptome analysis of differentially expressed genes in foxtail millet (Setaria italica L.) during dehydration stress. Biochem. Biophy. Res. Commun. 393, 720–727. doi: 10.1016/j.bbrc.2010.02.068
Li, H. L., Guo, D., Yang, Z. P., Tang, X., and Peng, S. Q. (2014). Genome-wide identification and characterization of WRKY gene family in Hevea brasiliensis. Genomics 104, 14–23. doi: 10.1016/j.ygeno.2014.04.004
Li, H. L., Zhang, L. B., Guo, D., Li, C. Z., and Peng, S. Q. (2012). Identification and expression profiles of the WRKY transcription factor family in Ricinus communis. Gene 503, 248–253. doi: 10.1016/j.gene.2012.04.069
Li, P., and Brutnell, T. P. (2011). Setaria viridis and Setaria italica, model genetic systems for the Panicoid grasses. J. Exp. Bot. 62, 3031–3037. doi: 10.1093/jxb/err096
Li, S., Fu, Q., Chen, L., Huang, W., and Yu, D. (2011). Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta 233, 1237–1252. doi: 10.1007/s00425-011-1375-2
Li, S., Fu, Q., Huang, W., and Yu, D. (2009). Functional analysis of an Arabidopsis transcription factor WRKY25 in heat stress. Plant Cell Rep. 28, 683–693. doi: 10.1007/s00299-008-0666-y
Ling, J., Jiang, W., Zhang, Y., Yu, H., Mao, Z., Gu, X., et al. (2011). Genome-wide analysis of WRKY gene family in Cucumis sativus. BMC Genomics 12:471. doi: 10.1186/1471-2164-12-471
Liu, D., Leib, K., Zhao, P., Kogel, K. H., and Langen, G. (2014). Phylogenetic analysis of barley WRKY proteins and characterization of HvWRKY1 and -2 as repressors of the pathogen-inducible gene HvGER4c. Mol. Genet. Genomics 289, 1331–1345. doi: 10.1007/s00438-014-0893-6
Logemann, J., Schell, J., and Willmitzer, L. (1987). Improved method for the isolation of RNA from plant tissues. Anal. Biochem. 163, 16–20. doi: 10.1016/0003-2697(87)90086-8
Luo, M., Dennis, E. S., Berger, F., Peacock, W. J., and Chaudhury, A. (2005). MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 102, 17531–17536. doi: 10.1073/pnas.0508418102
Ma, D., Pu, G., Lei, C., Ma, L., Wang, H., Guo, Y., et al. (2009). Isolation and characterization of AaWRKY1, an Artemisia annua transcription factor that regulates the amorpha-4,11-diene synthase gene, a key gene of artemisinin biosynthesis. Plant Cell Physiol. 50, 2146–2161. doi: 10.1093/pcp/pcp149
Maere, S., Heymans, K., and Kuiper, M. (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449. doi: 10.1093/bioinformatics/bti551
Marchive, C., Mzid, R., Deluc, L., Barrieu, F., Pirrello, J., Gauthier, A., et al. (2007). Isolation and characterization of a Vitis vinifera transcription factor, VvWRKY1, and its effect on responses to fungal pathogens in transgenic tobacco plants. J. Exp. Bot. 58, 1999–2010. doi: 10.1093/jxb/erm062
Miao, Y., Laun, T., Zimmermann, P., and Zentgraf, U. (2004). Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis. Plant Mol. Biol. 55, 853–867. doi: 10.1007/s11103-005-2142-1
Mickelbart, M. V., Hasegawa, P. M., and Bailey-Serres, J. (2015). Genetic mechanisms of abiotic stress tolerance that translate to crop yield stability. Nat. Rev. Genet. 16, 237–251. doi: 10.1038/nrg3901
Min, X. J., Butler, G., Storms, R., and Tsang, A. (2005). OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. 33, W677–W680. doi: 10.1093/nar/gki394
Mishra, A. K., Muthamilarasan, M., Khan, Y., Parida, S. K., and Prasad, M. (2013). Genome-wide investigation and expression analyses of WD40 protein family in the model plant foxtail millet (Setaria italica L.). PLoS ONE 9:e86852. doi: 10.1371/journal.pone.0086852
Muthamilarasan, M., Bonthala, V. S., Mishra, A. K., Khandelwal, R., Khan, Y., Roy, R., et al. (2014a). C2H2-type of zinc finger transcription factors in foxtail millet define response to abiotic stresses. Funct. Integr. Genomics 14, 531–543. doi: 10.1007/s10142-014-0383-2
Muthamilarasan, M., Dhaka, A., Yadav, R., and Prasad, M. (in press). Exploration of millet models for developing nutrient rich graminaceous crops. Plant Sci. doi: 10.1016/j.plantsci.2015.08.023
Muthamilarasan, M., Khandelwal, R., Yadav, C. B., Bonthala, V. S., Khan, Y., and Prasad, M. (2014b). Identification and molecular characterization of MYB Transcription Factor Superfamily in C4 model plant foxtail millet (Setaria italica L.). PLoS ONE 9:e109920. doi: 10.1371/journal.pone.0109920
Muthamilarasan, M., and Prasad, M. (2013). Plant innate immunity: an updated insight into defense mechanism. J. Biosci. 38, 433–449. doi: 10.1007/s12038-013-9302-2
Muthamilarasan, M., and Prasad, M. (2015). Advances in Setaria genomics for genetic improvement of cereals and bioenergy grasses. Theor. Appl. Genet. 128, 1–14. doi: 10.1007/s00122-014-2399-3
Okay, S., Derelli, E., and Unver, T. (2014). Transcriptome-wide identification of bread wheat WRKY transcription factors in response to drought stress. Mol. Genet. Genomics. 289, 765–781. doi: 10.1007/s00438-014-0849-x
Park, C. Y., Lee, J. H., Yoo, J. H., Moon, B. C., Choi, M. S., Kang, Y. H., et al. (2005). WRKY group IId transcription factors interact with calmodulin. FEBS Lett. 579, 1545–1550. doi: 10.1016/j.febslet.2005.01.057
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7:e30619. doi: 10.1371/journal.pone.0030619
Pnueli, L., Hallak-Herr, E., Rozenberg, M., Cohen, M., Goloubinoff, P., Kaplan, A., et al. (2002). Molecular and biochemical mechanisms associated with dormancy and drought tolerance in the desert legume Retama raetam. Plant J. 31, 319–330. doi: 10.1046/j.1365-313X.2002.01364.x
Postnikova, O. A., Shao, J., and Nemchinov, L. G. (2014). In silico identification of transcription factors in Medicago sativa using available transcriptomic resources. Mol. Genet. Genomics 289, 457–468. doi: 10.1007/s00438-014-0823-7
Puranik, S., Jha, S., Srivastava, P. S., Sreenivasulu, N., and Prasad, M. (2011). Comparative transcriptome analysis of contrasting foxtail millet cultivars in response to short-term salinity stress. J. Plant Physiol. 168, 280–287. doi: 10.1016/j.jplph.2010.07.005
Puranik, S., Sahu, P. P., Srivastava, P. S., and Prasad, M. (2012). NAC proteins: regulation and role in stress tolerance. Trends Plant Sci. 17, 369–381. doi: 10.1016/j.tplants.2012.02.004
Puranik, S., Sahu, P. P., Mandal, S. N., Venkata Suresh, B., Parida, S. K., and Prasad, M. (2013). Comprehensive genome-wide survey, genomic constitution and expression profiling of the NAC transcription factor family in foxtail millet (Setaria italica L.). PLoS ONE 8:e64594. doi: 10.1371/journal.pone.0064594
Qi, X., Xie, S., Liu, Y., Yi, F., and Yu, J. (2013). Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol. Biol. 83, 459–473. doi: 10.1007/s11103-013-0104-6
Rao, G., Sui, J., and Zhang, J. (2015). In silico genome-wide analysis of the WRKY gene family in Salix arbutifolia. Plant Omics J. 8, 353–360.
Ren, X., Chen, Z., Liu, Y., Zhang, H., Zhang, M., Liu, Q., et al. (2010). ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant J. 63, 417–429. doi: 10.1111/j.1365-313X.2010.04248.x
Rizhsky, L., Liang, H., and Mittler, R. (2002). The combined effect of drought stress and heat shock on gene expression in tobacco. Plant Physiol. 130, 1143–1151. doi: 10.1104/pp.006858
Ross, C. A., Liu, Y., and Shen, Q. J. (2007). The WRKY Gene Family in rice (Oryza sativa). J. Integ. Plant Biol. 49, 827–842. doi: 10.1111/j.1744-7909.2007.00504.x
Rushton, P. J., Macdonald, H., Huttly, A. K., Lazarus, C. M., and Hooley, R. (1995). Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of alpha-Amy2 genes. Plant Mol. Biol. 29, 691–702. doi: 10.1007/BF00041160
Rushton, P. J., Somssich, I. E., Ringler, P., and Shen, Q. J. (2010). WRKY transcription factors. Trends Plant Sci. 15, 247–258. doi: 10.1016/j.tplants.2010.02.006
Sadras, V., Grassini, P., and Steduto, P. (2011). The State of World's Land and Water Resources for Food and Agriculture (SOLAW). London: FAO.
Saeed, A. I., Sharov, V., White, J., Li, J., Liang, W., Bhagabati, N., et al. (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378.
Shang, Y., Yan, L., Liu, Z. Q., Cao, Z., Mei, C., Xin, Q., et al. (2010). The Mg-Chelatase H subunit of Arabidopsis antagonizes a group of transcription repressors to relieve ABA-responsive genes of inhibition. Plant Cell 22, 1909–1935. doi: 10.1105/tpc.110.073874
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Song, Y., Chen, L. G., Zhang, L. P., and Yu, D. Q. (2010). Overexpression of OsWRKY72 gene interferes in the ABA signal and auxin transport pathway of Arabidopsis. J. Biosci. 35, 459–471. doi: 10.1007/s12038-010-0051-1
Song, Y., and Gao, J. (2014). Genome-wide analysis of WRKY gene family in Arabidopsis lyrata and comparison with Arabidopsis thaliana and Populus trichocarpa. Chin. Sci. Bull. 59, 754–765. doi: 10.1007/s11434-013-0057-9
Song, Y., Jing, S. J., and Yu, D. Q. (2009). Overexpression of the stress induced OsWRKY08 improves the osmotic stress tolerance in Arabidopsis. Chin. Sci. Bull. 54, 4671–4678. doi: 10.1007/s11434-009-0710-5
Sun, C., Palmqvist, S., Olsson, H., Borén, M., Ahlandsberg, S., and Jansson, C. (2003). A novel WRKY transcription factor, SUSIBA2, participates in sugar signaling in barley by binding to the sugar-responsive elements of the iso1 promoter. Plant Cell 15, 2076–2092. doi: 10.1105/tpc.014597
Suttipanta, N., Pattanaik, S., Kulshrestha, M., Patra, B., Singh, S. K., and Yuan, L. (2011). The transcription factor CrWRKY1 positively regulates the terpenoid indole alkaloid biosynthesis in Catharanthus roseus. Plant Physiol. 157, 2081–2093. doi: 10.1104/pp.111.181834
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tang, J., Wang, F., Wang, Z., Huang, Z., Xiong, A., and Hou, X. (2013). Characterization and co-expression analysis of WRKY orthologs involved in responses to multiple abiotic stresses in Pak-choi (Brassica campestris ssp. chinensis). BMC Plant Biol. 13:188. doi: 10.1186/1471-2229-13-188
Tateno, Y., Imanishi, T., Miyazaki, S., Fukami-Kobayashi, K., Saitou, N., Sugawara, H., et al. (2002). DNA Data Bank of Japan (DDBJ) for genome scale research in life science. Nucleic Acids Res. 30, 27–30. doi: 10.1093/nar/30.1.27
Tombuloglu, H., Kekec, G., Sakcali, M. S., and Unver, T. (2013). Transcriptome-wide identification of R2R3-MYB transcription factors in barley with their boron responsive expression analysis. Mol. Genet. Genomics 288, 141–155. doi: 10.1007/s00438-013-0740-1
Tran, L. S., and Mochida, K. (2010). Identification and prediction of abiotic stress responsive transcription factors involved in abiotic stress signaling in soybean. Plant Signal Behav. 5, 255–257. doi: 10.4161/psb.5.3.10550
Udvardi, M. K., Kakar, K., Wandrey, M., Montanari, O., Murray, J., Andriankaja, A., et al. (2007). Legume transcription factors: global regulators of plant development and response to the environment. Plant Physiol. 144, 538–549. doi: 10.1104/pp.107.098061
Voorrips, R. E. (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78. doi: 10.1093/jhered/93.1.77
Wang, H., Avci, U., Nakashima, J., Hahn, M. G., Chen, F., and Dixon, R. A. (2010). Mutation of WRKY transcription factors initiates pith secondary wall formation and increases stem biomass in dicotyledonous plants. Proc. Natl. Acad. Sci. U.S.A. 107, 22338–22343. doi: 10.1073/pnas.1016436107
Wang, L., Peterson, R. B., and Brutnell, T. P. (2011). Regulatory mechanisms underlying C4 photosynthesis. New Phytol. 190, 9–20. doi: 10.1111/j.1469-8137.2011.03649.x
Wang, L., Zhu, W., Fang, L., Sun, X., Su, L., Liang, Z., et al. (2014). Genome-wide identification of WRKY family genes and their response to cold stress in Vitis vinifera. BMC Plant Biol. 14:103. doi: 10.1186/1471-2229-14-103
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi: 10.1093/nar/gkr1293
Wang, Z., Yang, P., Fan, B., and Chen, Z. (1998). An oligo selection procedure for identification of sequence- specific DNA-binding activities associated with the plant defence response. Plant J. 16, 515–522. doi: 10.1046/j.1365-313x.1998.00311.x
Wei, K. F., Chen, J., Chen, Y. F., Wu, L. J., and Xie, D. X. (2012). Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in maize. DNA Res. 19, 153–164. doi: 10.1093/dnares/dsr048
Wei, W., Zhang, Y., Han, L., Guan, Z., and Chai, T. (2008). A novel WRKY transcriptional factor from Thlaspi caerulescens negatively regulates the osmotic stress tolerance of transgenic tobacco. Plant Cell Rep. 27, 795–803. doi: 10.1007/s00299-007-0499-0
Wen, F., Zhu, H., Li, P., Jiang, M., Mao, W., Ong, C., et al. (2014). Genome-wide evolutionary characterization and expression analyses of WRKY family genes in Brachypodium distachyon. DNA Res. 21, 327–339. doi: 10.1093/dnares/dst060
Wu, X., Shiroto, Y., Kishitani, S., Ito, Y., and Toriyama, K. (2009). Enhanced heat and drought tolerance in transgenic rice seedlings overexpressing OsWRKY11 under the control of HSP101 promoter. Plant Cell Rep. 28, 21–30. doi: 10.1007/s00299-008-0614-x
Xie, Z., Zhang, Z. L., Zou, X., Huang, J., Ruas, P., Thompson, D., et al. (2005). Annotations and functional analyses of the rice WRKY gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiol. 137, 176–189. doi: 10.1104/pp.104.054312
Xiong, W., Xu, X., Zhang, L., Wu, P., Chen, Y., Li, M., et al. (2013). Genome-wide analysis of the WRKY gene family in physic nut (Jatropha curcas L.). Gene 524, 124–132. doi: 10.1016/j.gene.2013.04.047
Xu, J., Li, Y., Ma, X., Ding, J., Wang, K., Wang, S., et al. (2013). Whole transcriptome analysis using next-generation sequencing of model species Setaria viridis to support C4 photosynthesis research. Plant Mol. Biol. 83, 77–87. doi: 10.1007/s11103-013-0025-4
Xu, Y. H., Wang, J. W., Wang, S., Wang, J. Y., and Chen, X. Y. (2004). Characterization of GaWRKY1, a cotton transcription factor that regulates the sesquiterpene synthase gene (+)-delta-cadinene synthase-A. Plant Physiol. 135, 507–515. doi: 10.1104/pp.104.038612
Yang, P. Z., Chen, C. H., Wang, Z. P., Fan, B. F., and Chen, Z. X. (1999). A pathogen- and salicylic acid-induced WRKY DNA-binding activity recognizes the elicitor response element of the tobacco class I chitinase gene promoter. Plant J. 18, 141–149. doi: 10.1046/j.1365-313X.1999.00437.x
Yao, Q. Y., Xia, E. H., Liu, F. H., and Gao, L. Z. (2015). Genome-wide identification and comparative expression analysis reveal a rapid expansion and functional divergence of duplicated genes in the WRKY gene family of cabbage, Brassica oleracea var. capitata. Gene 557, 35–42. doi: 10.1016/j.gene.2014.12.005
Yoo, S. J., Kim, S. H., Kim, M. J., Ryu, C. M., Kim, Y. C., Cho, B. H., et al. (2014). Involvement of the OsMKK4-OsMPK1 cascade and its downstream transcription factor OsWRKY53 in the wounding response in rice. Plant Pathol. J. 30, 168–177. doi: 10.5423/PPJ.OA.10.2013.0106
Yu, Y., Hu, R., Wang, H., Cao, Y., He, G., Fu, C., et al. (2013). MlWRKY12, a novel Miscanthus transcription factor, participates in pith secondary cell wall formation and promotes flowering. Plant Sci. 212, 1–9. doi: 10.1016/j.plantsci.2013.07.010
Zhang, G., Liu, X., Quan, Z., Cheng, S., Xu, X., Pan, S., et al. (2012). Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 30, 549–554. doi: 10.1038/nbt.2195
Zhang, Y., and Wang, L. (2005). The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol. Biol. 5:1. doi: 10.1186/1471-2148-5-1
Zhang, Z. L., Xie, Z., Zou, X., Casaretto, J., Ho, T. H., and Shen, Q. J. (2004). A rice WRKY gene encodes a transcriptional repressor of the gibberellin signaling pathway in aleurone cells. Plant Physiol. 134, 1500–1513. doi: 10.1104/pp.103.034967
Zheng, Z., Qamar, S. A., Chen, Z., and Mengiste, T. (2006). Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 48, 592–605. doi: 10.1111/j.1365-313X.2006.02901.x
Keywords: WRKY transcription factors, Setaria italica, Setaria viridis, abiotic stress, stress signaling, expression profiling, comparative mapping
Citation: Muthamilarasan M, Bonthala VS, Khandelwal R, Jaishankar J, Shweta S, Nawaz K and Prasad M (2015) Global analysis of WRKY transcription factor superfamily in Setaria identifies potential candidates involved in abiotic stress signaling. Front. Plant Sci. 6:910. doi: 10.3389/fpls.2015.00910
Received: 14 July 2015; Accepted: 12 October 2015;
Published: 26 October 2015.
Edited by:
Laigeng Li, Institutes of Plant Physiology and Ecology, ChinaReviewed by:
Serena Varotto, University of Padova, ItalyEric Van Der Graaff, University of Copenhagen, Denmark
Copyright © 2015 Muthamilarasan, Bonthala, Khandelwal, Jaishankar, Shweta, Nawaz and Prasad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manoj Prasad, manoj_prasad@nipgr.ac.in