 Nadim Tayeh1
Nadim Tayeh1 Grégoire Aubert1
Grégoire Aubert1 Marie-Laure Pilet-Nayel2
Marie-Laure Pilet-Nayel2 Isabelle Lejeune-Hénaut3
Isabelle Lejeune-Hénaut3 Thomas D. Warkentin4
Thomas D. Warkentin4 Judith Burstin1*
Judith Burstin1*- 1INRA, UMR1347 Agroécologie, Dijon, France
- 2INRA, UMR1349 Institut de Génétique Environment et Protection des Plantes, Le Rheu, France
- 3INRA, USC Institut Charles Viollette-Adaptation au Froid du Pois, Estrées-Mons, France
- 4Crop Development Centre, College of Agriculture and Bioresources, University of Saskatchewan, Saskatoon, SK, Canada
Pea (Pisum sativum L.) is an annual cool-season legume and one of the oldest domesticated crops. Dry pea seeds contain 22–25% protein, complex starch and fiber constituents, and a rich array of vitamins, minerals, and phytochemicals which make them a valuable source for human consumption and livestock feed. Dry pea ranks third to common bean and chickpea as the most widely grown pulse in the world with more than 11 million tons produced in 2013. Pea breeding has achieved great success since the time of Mendel's experiments in the mid-1800s. However, several traits still require significant improvement for better yield stability in a larger growing area. Key breeding objectives in pea include improving biotic and abiotic stress resistance and enhancing yield components and seed quality. Taking advantage of the diversity present in the pea genepool, many mapping populations have been constructed in the last decades and efforts have been deployed to identify loci involved in the control of target traits and further introgress them into elite breeding materials. Pea now benefits from next-generation sequencing and high-throughput genotyping technologies that are paving the way for genome-wide association studies and genomic selection approaches. This review covers the significant development and deployment of genomic tools for pea breeding in recent years. Future prospects are discussed especially in light of current progress toward deciphering the pea genome.
Introduction
Thanks to significant technological breakthroughs, pea genetics is rapidly evolving from conventional to large-scale molecular-assisted approaches to uncover the molecular bases of important traits and enhance breeding. Several reviews have been published recently that dealt with pea systematics, seed quality, and breeding (Burstin et al., 2011; Dahl et al., 2012; Bohra et al., 2014; Arnoldi et al., 2015; Smýkal et al., 2015; Varshney et al., 2015; Warkentin et al., 2015). The present review focuses on the genomic toolkit that was developed recently in pea thanks in part to next-generation sequencing technologies. This includes transcriptome, genotyping, and mapping resources that will pave the way to renewed pea breeding programs.
Economic Importance, Nutritive Value, Growing Regions
Pea (Pisum sativum L.) is a major cool-season pulse crop and an essential component of sustainable cropping systems (Nemecek et al., 2008; Duc et al., 2010; Jensen et al., 2012). Significant agro-ecological services linked with its ability to develop symbiotic nitrogen fixation as well as its role as a break crop for pest and pathogen pressure reduction have been described (Nemecek and Kägi, 2007; Hayer et al., 2010; Macwilliam et al., 2014). In 2013, the vegetable pea production amounted to 17.43 Mt worldwide (FAOSTAT)1 and dry pea represented the third most important pulse crop production after common bean and chickpea with 11.16 Mt produced worldwide (FAOSTAT).
Pea seeds are an important source of proteins and provide an exceptionally varied nutrient profile (for a review, Burstin et al., 2011): major constituents are starch (from 18.6 to 54.1%) and proteins (15.8–32.1%), followed by fibers (5.9–12.7%), sucrose (1.3–2.1%), and oil (0.6–5.5%). Seeds also contain minerals, vitamins, and micro-nutrients such as polyphenolics, saponins, α-galactosides, and phytic acids whose health-promoting effects are being tested (Bastianelli et al., 1998; Mitchell et al., 2009; Dahl et al., 2012; Marles et al., 2013; Arnoldi et al., 2015). Peas enter in human nutrition in a wide diversity of forms: fresh seedlings, immature pods, and seeds provide a green vegetable, and whole or ground dry seeds are cooked in various dishes. High quality starch, protein, or oligoside isolates are being extracted from dry pea seeds and whole seed structural and functional characteristics have been assessed for food improvement (Brummer et al., 2015). Because dry seeds contain little anti-nutritional factors, they are also introduced as a protein source mainly in monogastric diets without affecting growth and production traits (Stein et al., 2006; Laudadio et al., 2012; Dotas et al., 2014). Pea hay is used as fodder in ruminant diets (Bastida Garcia et al., 2011).
Pea is mainly cultivated in temperate regions of the world on well-drained and fertile soils. However, being distributed over all continents, the pea production area is characterized by a large range of pedo-climatic conditions. Indeed, China is the largest producer of vegetable peas (10.60 Mt, FAOSTAT) followed by India (4 Mt). Canada is the main producer of dry peas (3.85 Mt) followed by China (1.6 Mt), the Russian Federation (1.35 Mt), USA (0.71 Mt), India (0.60 Mt), France (0.50 Mt), and Ethiopia (0.40 Mt).
Variability of Germplasm used for Breeding
Pea was domesticated by Neolithic farmers in the Fertile Crescent some 10,000 years ago (Willcox et al., 2009; Weiss and Zohary, 2011; Smýkal, et al., 2014). Pea then spread rapidly toward south-west Asia, the Mediterranean basin, and Europe (Zohary, 1999). Probably linked with their large range of cultivation and the diversity of their use as food, feed, or fodder, pea landraces and varieties now exhibit an incredible diversity of forms and growing types, adapted to diverse environments, cropping systems, and end-uses (Burstin et al., 2015). This vast diversity of cultivated forms is the major reservoir for present crop improvement. Different types of pea varieties have been developed for vegetable pea production, varying at major genes controlling seed and plant traits. For example, wrinkled seeds are associated with significant changes of seed composition, linked with starch synthesis modification (Wang et al., 2003). Various types of dry peas are also available that differ by their cotyledon color, plant architecture, or flowering time. In addition to this cultivated reservoir of diversity, wild peas can be crossed with cultivated peas. Ben-Ze'Ev and Zohary (1973) showed that chromosomal rearrangements among accessions from the different Pisum species and subspecies could cause partial sterility in hybrids. Recently, Bogdanova et al. (2014) have identified a nucleo-cytoplasmic incompatibility between a P. sativum elatius accession and cultivated peas. However, within the Pisum genus, wild P. fulvum, wild subspecies P. sativum elatius, and P. sativum humile as well as P. abyssinicum, a taxon cultivated in Ethiopia, are in most cases inter-crossable with P. sativum sativum as long as the cultivated pea is used as female donor (Ben-Ze'Ev and Zohary, 1973; Ochatt et al., 2004). Different authors have thus used P. fulvum as well as wild P. sativum subspecies as a source of alleles for important breeding traits, such as resistance to various fungal diseases (Barilli et al., 2010; Fondevilla et al., 2011; Jha et al., 2012) or to Bruchus pisorum L. (Clement et al., 2009).
Target Traits and Achievements Through Conventional Breeding
Continued grain yield improvement is necessary for pea to remain an attractive option compared to cereals and oilseeds in crop rotations. Improving yield involves addressing many biotic and abiotic stresses, using a large set of strategies including diverse germplasm as parents, making many crosses, selecting for major gene traits under conditions conducive to selection, and yield testing of a large number of breeding lines. These stresses are specific to each growing region and/or growing type. However, fungal diseases are the major biotic stress in most cases, followed by various insects, viruses, and parasitic plants such as broomrape. Drought and heat stress at flowering are the main abiotic stresses, while frost, salinity, and early season flooding are diversely important according to growing types.
Important achievements were obtained in pea cultivars through conventional breeding over the past 20 years. Yield gains of approximately 2% per year have been achieved (Warkentin et al., 2015). Lodging resistance has been improved through deployment of the afila gene for semi-leafless type (Kujala, 1953; Goldenberg, 1965) and secondarily through selection for increased stem strength (Banniza et al., 2005). Powdery mildew resistance based on the single recessive gene er-1 (Harland, 1948) has been widely deployed. Partial resistance to the Ascochyta blight complex has been achieved through pyramiding of genes with minor effects (Kraft et al., 1998). Resistance to pea weevil (Bruchus pisorum L.) identified in the secondary gene pool (P. fulvum) (Clement et al., 2002) was transferred into cultivated pea through backcrossing (Clement et al., 2009; Aryamanesh et al., 2012). Cultivars adapted to winter sowing have been developed and deployed in Europe and north-west USA giving the potential for better yields because of a longer growing season, higher biomass production, and earlier maturity to avoid late season drought and heat stress (Hanocq et al., 2009). The introgression of the Hr allele which delays flower initiation until after the main winter freezing periods have passed (Lejeune-Hénaut et al., 2008) permitted to obtain some cultivars with notably improved winter hardiness. Field pea production for whole seed food markets requires appropriate seed visual quality. Quantitative inheritance, transgressive segregation, and moderately high heritability were observed for seed color, shape, and surface dimpling (Ubayasena et al., 2011) allowing for good progress in breeding. Seed protein concentration has been maintained in pea cultivars, even though overall seed yield has increased (Jha et al., 2013).
In some cases, achievements are at an earlier stage of deployment. Useful germplasms such as sources of resistance to various biotic and abiotic stresses have been identified and are currently being evaluated and introgressed. Research in France and USA has led to the identification and introgression of useful variation for resistance to Aphanomyces root rot; partial resistance controlled by several quantitative trait loci (QTLs) is being deployed (Pilet-Nayel et al., 2002, 2005; McGee et al., 2012). Improved stress tolerance has been identified in landrace accessions for toxicity to boron (Bagheri et al., 1994), salinity (Leonforte et al., 2013a), iron deficiency (Kabir et al., 2012), and for heat tolerance during flowering (Petkova et al., 2009). Selection for major gene resistance to pea seed-borne mosaic virus and potyviruses is now incorporated into breeding strategies (van Leur et al., 2007). Diversity in pea seed micronutrient concentration (Ray et al., 2014) and an approach to improving iron bioavailability for humans (Liu et al., 2015) have been described.
Future targets in pea breeding include (i) the optimization of pea interactions with Rhizobia, Mycorrhiza and other beneficial microorganisms in view of crop resilience against stresses, (ii) the adaptation of plant morphology and phenology to novel cropping systems, and (iii) the adaptation of seed composition to novel end-use application possibilities.
Available Genomic Resources in Pea
The pea genome is organized in 7 pairs of chromosomes (2n = 2x = 14). Its haploid size is estimated at 4.45 Gb (Dolezel et al., 1998; Dolezel and Greilhuber, 2010; Praca-Fontes et al., 2014). It is largely dominated by mobile elements, mainly of the Ty3/gypsy family (Macas et al., 2007). This large genome size and high transposable element content have undoubtedly contributed to delay the development and availability of genomic tools in pea. Recently, several national and international programs have developed diverse valuable genomic resources by taking advantage of cutting-edge sequencing and genotyping technologies. These programs indicate the determination of the pea community to make rapid progress toward targeted and efficient molecular breeding exploiting the rich diversity of pea germplasm and its wild relatives. An international consortium has been initiated in order to generate the full-sequence of the pea genome (Madoui et al., 2015).
Developing wide collections of mapped and easy-to-use molecular markers is among the first steps of gene tagging and gene introgression strategies. Pea genetic maps started to be developed early: Wellensiek (1925) constructed a six-linkage group (LG) map and Lamprecht (1948) published a full map with 7 LGs (see Rozov et al., 1999, for review). Later on, various marker types were developed and numerous linkage maps originating from intra- or inter-subspecific crosses have been generated (Table 1). Decisive progresses were achieved with the availability of mapped SSR and SNP markers. Thanks to their multi-allelic nature, genomic (Ford et al., 2002; Loridon et al., 2005; Sun et al., 2014) and EST-based SSR markers (Burstin et al., 2001; Gong et al., 2010; De Caire et al., 2012; Kaur et al., 2012; Mishra et al., 2012) have been widely used for studying germplasm diversity (Baranger et al., 2004; Smýkal et al., 2008; Zong et al., 2009; Sarikamis et al., 2010) and bridging between different genetic maps. Today, SNPs are the markers of choice because of their abundance, easy-scoring, and amenability to high-throughput genotyping. SNPs were identified based on sequencing data from 4 (Leonforte et al., 2013b), 6 (Sindhu et al., 2014), 8 (Duarte et al., 2014), and up to 16 (Tayeh et al., 2015a) pea genotypes. Illumina GoldenGate (Deulvot et al., 2010; Leonforte et al., 2013b; Duarte et al., 2014; Sindhu et al., 2014), Infinium (Tayeh et al., 2015a), and Sequenom MassARRAY (Cheng et al., 2015) platforms have been deployed for SNP genotyping (Table 2). In total, at least 52 genetic maps have been constructed for different F2 or recombinant inbred line (RIL) populations, comprising up to 8503 markers (Table 1). Consensus maps have been built in order to offer higher mapping resolution and better genome coverage (Table 1). These maps combine molecular data from 2 (Aubert et al., 2006; Hamon et al., 2011), 3 (Loridon et al., 2005; Sudheesh et al., 2014), 4 (Hamon et al., 2013; Duarte et al., 2014), 5 (Sindhu et al., 2014), 6 (Bordat et al., 2011), or 12 (Tayeh et al., 2015a) populations.
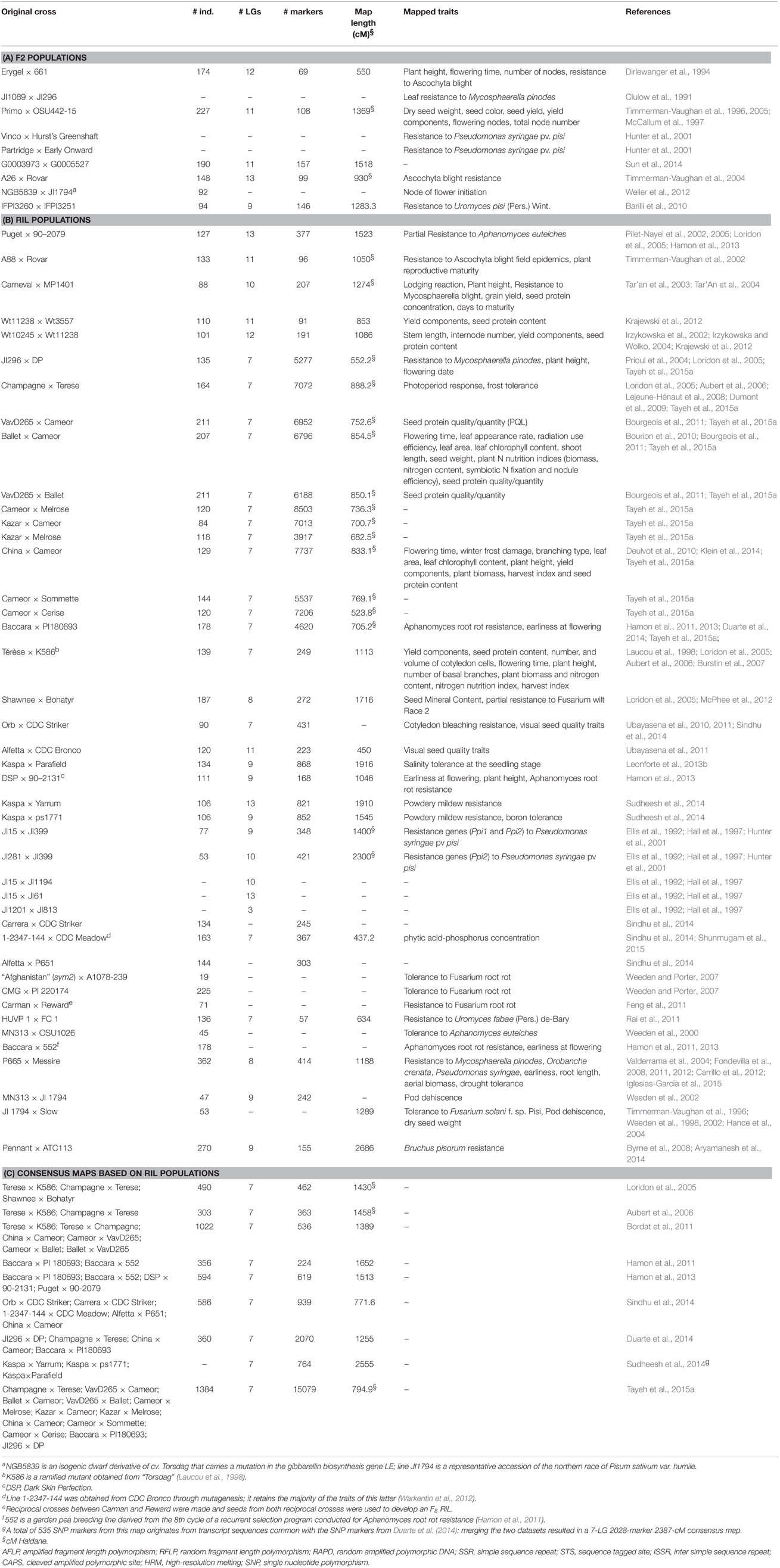
Table 1. Available individual and consensus genetic maps constructed for bi-parental populations and quantitative trait loci positioned on these maps.
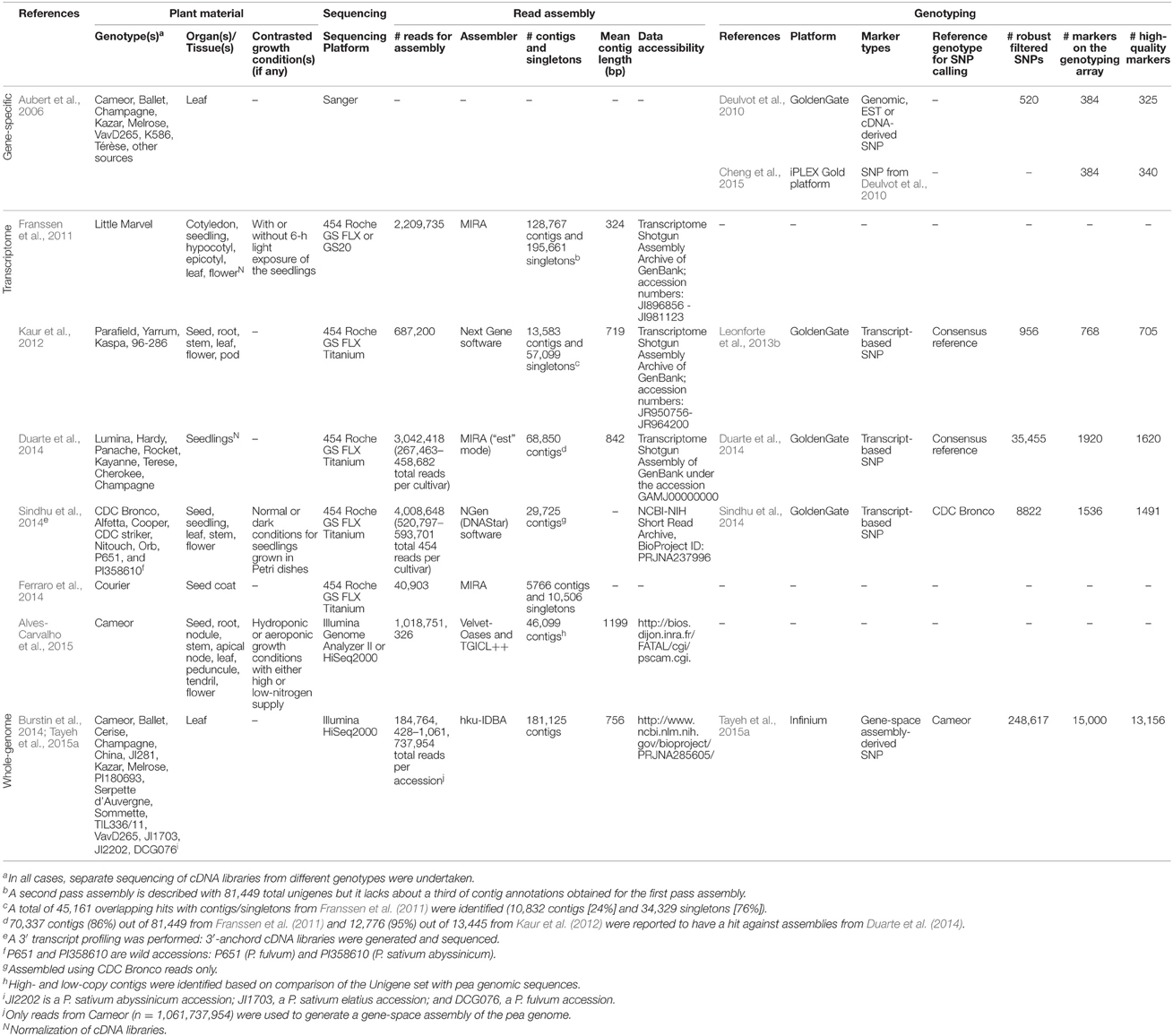
Table 2. Recently-developed resources in pea: transcriptome and whole-genome sequencing data, SNP collections, and genotyping platforms.
Uncovering the molecular bases underlying agriculturally important traits requires knowledge of the gene content of genomic regions controlling these traits of interest. Besides recently available genomic resources such as the pea gene atlas (Alves-Carvalho et al., 2015), whole-genome polymorphism data for multiple genotypes (see Table 2 for full description), BAC libraries developed for the genotypes Cameor (http://cnrgv.toulouse.inra.fr/fr) and PI 269818 (Coyne et al., 2007), researchers can count on the conserved synteny between pea and close species with available genome sequences. Gene-based rich individual and consensus maps have revealed connections between pea and Medicago truncatula (Choi et al., 2004; Aubert et al., 2006; Bordat et al., 2011; Leonforte et al., 2013b; Duarte et al., 2014; Sindhu et al., 2014; Tayeh et al., 2015a), Lotus japonicus, soybean (Bordat et al., 2011; Leonforte et al., 2013b; Tayeh et al., 2015a), pigeon pea (Leonforte et al., 2013b), chickpea (Leonforte et al., 2013b; Tayeh et al., 2015a), and lentil (Sindhu et al., 2014). Comprehensive understanding of shared syntenic blocks was, for instance, reported to be of great help to identify candidate genes for the control of seed size (D'Erfurth et al., 2012) and freezing tolerance (Tayeh et al., 2013a,b).
As other legume crops, pea is not easily amenable to genetic transformation (Warkentin et al., 2015): transformation occurs at low rates (Svabova and Griga, 2008) and plant regeneration is difficult. Fortunately, functional validation of candidate genes can benefit from a TILLING population developed from the genotype Cameor (Dalmais et al., 2008) and from the Virus Induced Gene Silencing (VIGS) methodology successfully adapted in pea (Grønlund et al., 2010; Pflieger et al., 2013).
The Use of Genomic Tools in Marker-trait Association Studies and Breeding Programs
Genetic maps have proven to be useful to uncover the molecular bases of monogenic characters such as Mendel's characters (see Ellis et al., 2011 for review) and also to decipher the determinism of complex agronomically-important traits (Table 1). QTLs responsible for the genetic control of yield-related traits, seed protein content, aerial and root architecture, and biotic and abiotic stress resistance have been detected under multiple environmental conditions and located on different maps (Table 1). In addition to QTL mapping analyses in bi-parental populations, association analyses have emerged as a complementary approach to dissect quantitative traits in pea by exploiting natural genetic diversity and ancestral recombination events characterizing germplasm collections. Diverse sets of cultivars with distinct geographic origins were used to determine associations between genetic markers and seed mineral nutrient concentration (Kwon et al., 2012; Cheng et al., 2015; Diapari et al., 2015), seed low-carbohydrate content (Cheng et al., 2015), seed lipid content (Ahmad et al., 2015), yield-related traits (Kwon et al., 2012), disease/pest resistance, and morphological traits such as flower color and seed coat color. Genome wide association mapping and subsequent allele mining could build on genomic resources reviewed herein and the phenotyping data available for diverse germplasm collections, as reviewed in Warkentin et al. (2015).
Specific markers linked to major genes were developed for use in breeding, especially for trypsin inhibitors in pea seeds (Page et al., 2002; Duc et al., 2004), flowering (Weller and Ortega, 2015), lodging resistance (Zhang et al., 2006) and resistance to diseases such as powdery mildew (Ghafoor and McPhee, 2012; Reddy et al., 2015), pea enation and seed borne mosaic virus (Frew et al., 2002; Jain et al., 2013), fusarium wilt (McClendon et al., 2002), Ascochyta blight (Jha et al., 2015), and rust (Barilli et al., 2010) (Supplementary Table 1). Some other resistance, flowering or seed composition genes were reviewed by Warkentin et al. (2015). Marker-assisted selection was conducted in early generation (F2) breeding populations using markers linked in coupling to two major QTLs controlling lodging resistance and was demonstrated more efficient than phenotypic selection (Zhang et al., 2006). Recently, marker-assisted backcrossing (MABC) was successfully used to introgress one to three of the seven main Aphanomyces root rot resistance QTLs (Hamon et al., 2013) into several recipient agronomic lines (Lavaud et al., 2015). Evaluation for resistance of the subsequent 157 BC5/6 Near Isogenic Lines (NILs) validated the effect of the major and some minor QTLs in controlled conditions and showed QTL x genetic background interactions. A MABC strategy was also used to introgress three frost tolerance QTLs among the main four QTLs identified by Lejeune-Hénaut et al. (2008). Field evaluations of 125 QTL-NILs validated the effect of these QTLs in the spring-type genetic background Eden (Hascoët et al., 2014). So far, marker-assisted construction of QTL-NILs has mainly allowed QTL effects to be validated. The rational use of these genetic regions in breeding strategies can now be considered in order to combine favorable alleles at complementary QTLs to improve multiple stress resistance in agronomic material. A list of the markers that should be useful for pea breeding is provided in Supplementary Table 1. In parallel to strategies considering the combination of individual QTLs, genomic selection seems a promising approach in pea, as first suggested by the prediction of the date of beginning of flowering and 1000 seed weight using a subset of 331 SNP markers genotyped in a reference collection of 372 pea accessions (Burstin et al., 2015). Increasing the marker coverage of the genome by using the newly-developed GenoPea 13.2K SNP Array (Tayeh et al., 2015a) further improved prediction accuracies (Tayeh et al., 2015b).
Perspectives
The gradually-developed genomic tools for pea now represent a rich resource for innovative strategies in both basic research and applied breeding. The large set of bi-parental interconnected populations segregating for diverse important agronomic traits, the individual and consensus genetic maps, the dense arrays of genetic markers, the high-throughput SNP genotyping tools, the BAC libraries, the TILLING population, and the whole-genome and transcriptome sequences from a large group of accessions should enhance significant advances in pea breeding in the next few years and foster the use of more diverse genetic resources for pea improvement. The genome sequence when released will further advance the pea genomic breeding revolution.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The present work of NT, GA, IL, MP, JB was done in the frame of the French National Research Agency (ANR) Project Investissements d'Avenir PeaMUST under the grant number ANR-11-BTBR-0002.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.01037
Footnotes
1. ^FAOSTAT. http://faostat3.fao.org/. 10/06/2015.
References
Ahmad, S., Kaur, S., Lamb-Palmer, N. D., Lefsrud, M., and Singh, J. (2015). Genetic diversity and population structure of Pisum sativum accessions for marker-trait association of lipid content. Crop J. 3, 238–245. doi: 10.1016/j.cj.2015.03.005
Alves-Carvalho, S., Aubert, G., Carrère, S., Cruaud, C., Brochot, A.-L., Jacquin, F., et al. (2015). Full-length de novo assembly of RNA-seq data in pea (Pisum sativum L.) provides a gene expression atlas and gives insights into root nodulation in this species. Plant J. 84, 1–19. doi: 10.1111/tpj.12967
Arnoldi, A., Zanoni, C., Lammi, C., and Boschin, G. (2015). The role of grain legumes in the prevention of hypercholesterolemia and hypertension. CRC. Crit. Rev. Plant Sci. 34, 144–168. doi: 10.1080/07352689.2014.897908
Aryamanesh, N., Byrne, O., Hardie, D. C., Khan, T., Siddique, K. H. M., and Yan, G. (2012). Large-scale density-based screening for pea weevil resistance in advanced backcross lines derived from cultivated field pea (Pisum sativum) and Pisum fulvum. Crop Pasture Sci. 63, 612–618. doi: 10.1071/CP12225
Aryamanesh, N., Zeng, Y., Byrne, O., Hardie, D. C., Al-Subhi, A. M., Khan, T., et al. (2014). Identification of genome regions controlling cotyledon, pod wall/seed coat and pod wall resistance to pea weevil through QTL mapping. Theor. Appl. Genet.127, 489–497. doi: 10.1007/s00122-013-2234-2
Aubert, G., Morin, J., Jacquin, F., Loridon, K., Quillet, M. C., Petit, A., et al. (2006). Functional mapping in pea, as an aid to the candidate gene selection and for investigating synteny with the model legume Medicago truncatula. Theor. Appl. Genet. 112, 1024–1041. doi: 10.1007/s00122-005-0205-y
Bagheri, A., Paull, J. G., and Rathjen, A. J. (1994). The response of Pisum sativum L. germplasm to high concentrations of soil boron. Euphytica 75, 9–17. doi: 10.1007/BF00024526
Banniza, S., Hashemi, P., Warkentin, T. D., Vandenberg, A., and Davis, A. R. (2005). The relationships among lodging, stem anatomy, degree of lignification, and resistance to mycosphaerella blight in field pea (Pisum sativum). Can. J. Bot. 83, 954–967. doi: 10.1139/b05-044
Baranger, A., Aubert, G., Arnau, G., Lainé, A. L., Deniot, G., Potier, J., et al. (2004). Genetic diversity within Pisum sativum using protein- and PCR-based markers. Theor. Appl. Genet. 108, 1309–1321. doi: 10.1007/s00122-003-1540-5
Barilli, E., Satovic, Z., Rubiales, D., and Torres, A. M. (2010). Mapping of quantitative trait loci controlling partial resistance against rust incited by Uromyces pisi (Pers.) Wint. in a Pisum fulvum L. intraspecific cross. Euphytica 175, 151–159. doi: 10.1007/s10681-010-0141-z
Bastianelli, D., Grosjean, F., Peyronnet, C., Duparque, M., and Regnier, J. M. (1998). Feeding value of pea (Pisum sativum, L.) - 1 Chemical composition of different categories of pea. Anim. Sci. 67, 609–619. doi: 10.1017/S1357729800033051
Bastida Garcia, J. L., González-Ronquillo, M., Domínguez Vara, I. A., Romero-Bernal, J., and Castelán Ortega, O. (2011). Effect of field pea (Pisum sativum L.) level on intake, digestion, ruminal fermentation and in vitro gas production in sheep fed maintenance diets. Anim. Sci. J. 82, 654–662. doi: 10.1111/j.1740-0929.2011.00884.x
Ben-Ze'Ev, N., and Zohary, D. (1973). Species relationships in the genus Pisum L. Isr. J. Bot. 22, 73–91.
Bogdanova, V. S., Kosterin, O. E., and Yadrikhinskiy, A. K. (2014). Wild peas vary in their cross-compatibility with cultivated pea (Pisum sativum subsp. sativum L.) depending on alleles of a nuclear-cytoplasmic incompatibility locus. Theor. Appl. Genet. 127, 1163–1172. doi: 10.1007/s00122-014-2288-9
Bohra, A., Pandey, M. K., Jha, U. C., Singh, B., Singh, I. P., Datta, D., et al. (2014). Genomics-assisted breeding in four major pulse crops of developing countries: present status and prospects. Theor. Appl. Genet. 127, 1263–1291. doi: 10.1007/s00122-014-2301-3
Bordat, A., Savois, V., Nicolas, M., Salse, J., Chauveau, A., Bourgeois, M., et al. (2011). Translational genomics in legumes allowed placing in silico 5460 unigenes on the pea functional map and identified candidate genes in Pisum sativum L. G3 1, 93–103. doi: 10.1534/g3.111.000349
Bourgeois, M., Jacquin, F., Cassecuelle, F., Savois, V., Belghazi, M., Aubert, G., et al. (2011). A PQL (protein quantity loci) analysis of mature pea seed proteins identifies loci determining seed protein composition. Proteomics 11, 1581–1594. doi: 10.1002/pmic.201000687
Bourion, V., Rizvi, S. M. H., Fournier, S., de Larambergue, H., Galmiche, F., Marget, P., et al. (2010). Genetic dissection of nitrogen nutrition in pea through a QTL approach of root, nodule, and shoot variability. Theor. Appl. Genet. 121, 71–86. doi: 10.1007/s00122-010-1292-y
Brummer, Y., Kaviani, M., and Tosh, S. M. (2015). Structural and functional characteristics of dietary fibre in beans, lentils, peas and chickpeas. Food Res. Int. 67, 117–125. doi: 10.1016/j.foodres.2014.11.009
Burstin, J., Alves-Carvalho, S., Aluome, C., Tayeh, N., Brochot, A. L., Carrère, S., et al. (2014). “Recent pea genomic resources will enhance complementary improvement strategies in this crop,” in IFLRC VI and ICLGG VII Conference (Saskatoon, SK).
Burstin, J., Deniot, G., Potier, J., Weinachter, C., Aubert, G., and Baranger, A. (2001). Microsatellite polymorphism in Pisum sativum. Plant Breed. 120, 311–317. doi: 10.1046/j.1439-0523.2001.00608.x
Burstin, J., Gallardo, K., Mir, R. R., Varshney, R. K., and Duc, G. (2011). “Improving protein content and nutrition quality,” in Biology and Breeding of Food Legumes, eds A. Pratap and J. Kumar (Wallingford, CT: CAB International), 314–328. doi: 10.1079/9781845937669.0314
Burstin, J., Marget, P., Huart, M., Moessner, A., Mangin, B., Duchene, C., et al. (2007). Developmental genes have pleiotropic effects on plant morphology and source capacity, eventually impacting on seed protein content and productivity in pea. Plant Physiol. 144, 768–781. doi: 10.1104/pp.107.096966
Burstin, J., Salloignon, P., Chabert-Martinello, M., Magnin-Robert, J. B., Siol, M., Jacquin, F., et al. (2015). Genetic diversity and trait genomic prediction in a pea diversity panel. BMC Genomics 16:105. doi: 10.1186/s12864-015-1266-1
Byrne, O. M., Hardie, D. C., Khan, T. N., Speijers, J., and Yan, G. (2008). Genetic analysis of pod and seed resistance to pea weevil in a Pisum sativum x P. fulvum interspecific cross. Aust. J. Agric. Res. 59, 854–862. doi: 10.1071/AR07353
Carrillo, E., Rubiales, D., Pérez-De-Luque, A., and Fondevilla, S. (2012). Characterization of mechanisms of resistance against Didymella pinodes in Pisum spp. Eur. J. Plant Pathol. 135, 761–769. doi: 10.1007/s10658-012-0116-0
Cheng, P., Holdsworth, W., Ma, Y., Coyne, C. J., Mazourek, M., Grusak, M. A., et al. (2015). Association mapping of agronomic and quality traits in USDA pea single-plant collection. Mol. Breed. 35, 75. doi: 10.1007/s11032-015-0277-6
Choi, H.-K., Mun, J.-H., Kim, D.-J., Zhu, H., Baek, J.-M., Mudge, J., et al. (2004). Estimating genome conservation between crop and model legume species. Proc. Natl. Acad. Sci. U.S.A. 101, 15289–15294. doi: 10.1073/pnas.0402251101
Clement, S. L., Hardie, D. C., and Elberson, L. R. (2002). Variation among accessions of Pisum fulvum for resistance to pea weevil. Crop Sci. 42, 2167–2173. doi: 10.2135/cropsci2002.2167
Clement, S. L., McPhee, K. E., Elberson, L. R., and Evans, M. A. (2009). Pea weevil, Bruchus pisorum L. (Coleoptera: Bruchidae), resistance in Pisum sativum x Pisum fulvum interspecific crosses. Plant Breed. 128, 478–485. doi: 10.1111/j.1439-0523.2008.01603.x
Clulow, S. A., Matthews, P., and Lewis, B. G. (1991). Genetic analysis of resistance to Mycosphaerella pinodes in pea seedlings. Euphytica 58, 183–189. doi: 10.1007/BF00022819
Coyne, C. J., McClendon, M. T., Walling, J. G., Timmerman-Vaughan, G. M., Murray, S., Meksem, K., et al. (2007). Construction and characterization of two bacterial artificial chromosome libraries of pea (Pisum sativum L.) for the isolation of economically important genes. Genome 50, 871–875. doi: 10.1139/G07-063
Dahl, W. J., Foster, L. M., and Tyler, R. T. (2012). Review of the health benefits of peas (Pisum sativum L.). Br. J. Nutr. 108, S3–S10. doi: 10.1017/s0007114512000852
Dalmais, M., Schmidt, J., Le Signor, C., Moussy, F., Burstin, J., Savois, V., et al. (2008). UTILLdb, a Pisum sativum in silico forward and reverse genetics tool. Genome Biol. 9:R43. doi: 10.1186/gb-2008-9-2-r43
De Caire, J., Coyne, C. J., Brumett, S., and Shultz, J. L. (2012). Additional pea EST-SSR markers for comparative mapping in pea (Pisum sativum L.). Plant Breed. 131, 222–226. doi: 10.1111/j.1439-0523.2011.01917.x
D'Erfurth, I., Le Signor, C., Aubert, G., Sanchez, M., Vernoud, V., Darchy, B., et al. (2012). A role for an endosperm-localized subtilase in the control of seed size in legumes. New Phytol. 196, 738–751. doi: 10.1111/j.1469-8137.2012.04296.x
Deulvot, C., Charrel, H., Marty, A., Jacquin, F., Donnadieu, C., Lejeune-Hénaut, I., et al. (2010). Highly-multiplexed SNP genotyping for genetic mapping and germplasm diversity studies in pea. BMC Genomics 11:468. doi: 10.1186/1471-2164-11-468
Diapari, M., Sindhu, A., Warkentin, T. D., Bett, K., and Tar'An, B. (2015). Population structure and marker-trait association studies of iron, zinc and selenium concentrations in seed of field pea (Pisum sativum L.). Mol. Breed 35, 30. doi: 10.1007/s11032-015-0252-2
Dirlewanger, E., Isaac, P. G., Ranade, S., Belajouza, M., Cousin, R., and de Vienne, D. (1994). Restriction fragment length polymorphism analysis of loci associated with disease resistance genes and developmental traits in Pisum sativum L. Theor. Appl. Genet. 88, 17–27. doi: 10.1007/BF00222388
Dolezel, J., and Greilhuber, J. (2010). Nuclear genome size: are we getting closer? Cytometry A 77A, 635–642. doi: 10.1002/cyto.a.20915
Dolezel, J., Greilhuber, J., Lucretti, S., Meister, A., Lysak, M. A., Nardi, L., et al. (1998). Plant genome size estimation by flow cytometry: inter-laboratory comparison. Ann. Bot. 82, 17–26. doi: 10.1006/anbo.1998.0730
Dotas, V., Bampidis, V. A., Sinapis, E., Hatzipanagiotou, A., and Papanikolaou, K. (2014). Effect of dietary field pea (Pisum sativum L.) supplementation on growth performance, and carcass and meat quality of broiler chickens. Livest. Sci. 164, 135–143. doi: 10.1016/j.livsci.2014.03.024
Duarte, J., Rivière, N., Baranger, A., Aubert, G., Burstin, J., Cornet, L., et al. (2014). Transcriptome sequencing for high throughput SNP development and genetic mapping in Pea. BMC Genomics 15:126. doi: 10.1186/1471-2164-15-126
Duc, G., Blancard, S., Hénault, C., Lecomte, C., Petit, M., Bernicot, M.-H., et al. (2010). Potentiels et leviers pour développer la production et l'utilisation des protéagineux dans le cadre d'une agriculture durable en Bourgogne. Innov. Agron. 11, 157–171.
Duc, G., Marget, P., Page, D., and Domoney, C. (2004). “Facile breeding markers to lower contents of vicine and convicine in faba bean seeds and trypsin inhibitors in pea seeds,” in Recent Advances of Research in Antinutritional Factors in Legume Seeds and Oilseeds, Proceedings of the Fourth International Workshop on Antinutritional Factors in Legume Seeds and Oilseeds, eds M. Muzquiz, G. D. Hill, C. Cuadrado, M. M. Pedrosa, and C. Burbano (Toledo), 281–285.
Dumont, E., Fontaine, V., Vuylsteker, C., Sellier, H., Bodèle, S., Voedts, N., et al. (2009). Association of sugar content QTL and PQL with physiological traits relevant to frost damage resistance in pea under field and controlled conditions. Theor. Appl. Genet. 118, 1561–1571. doi: 10.1007/s00122-009-1004-7
Ellis, T. H. N., Hofer, J. M. I., Timmerman-Vaughan, G. M., Coyne, C. J., and Hellens, R. P. (2011). Mendel, 150 years on. Trends Plant Sci. 16, 590–596. doi: 10.1016/j.tplants.2011.06.006
Ellis, T. H. N., Turner, L., Hellens, R. P., Lee, D., Harker, C. L., Enard, C., et al. (1992). Linkage maps in pea. Genetics 130, 649–663.
Feng, J., Hwang, R., Chang, K. F., Conner, R. L., Hwang, S. F., Strelkov, S. E., et al. (2011). Identification of microsatellite markers linked to quantitative trait loci controlling resistance to Fusarium root rot in field pea. Can. J. Plant Sci. 91, 199–204. doi: 10.4141/cjps09176
Ferraro, K., Jin, A. L., Trinh-Don, N., Reinecke, D. M., Ozga, J. A., and Ro, D.-K. (2014). Characterization of proanthocyanidin metabolism in pea (Pisum sativum) seeds. BMC Plant Biol. 14:238. doi: 10.1186/s12870-014-0238-y
Fondevilla, S., Almeida, N. F., Satovic, Z., Rubiales, D., Vaz Patto, M. C., Cubero, J. I., et al. (2011). Identification of common genomic regions controlling resistance to Mycosphaerella pinodes, earliness and architectural traits in different pea genetic backgrounds. Euphytica 182, 43–52. doi: 10.1007/s10681-011-0460-8
Fondevilla, S., Martin-Sanz, A., Satovic, Z., Dolores Fernandez-Romero, M., Rubiales, D., and Caminero, C. (2012). Identification of quantitative trait loci involved in resistance to Pseudomonas syringae pv. syringae in pea (Pisum sativum L.). Euphytica 186, 805–812. doi: 10.1007/s10681-011-0592-x
Fondevilla, S., Satovic, Z., Rubiales, D., Moreno, M. T., and Torres, A. M. (2008). Mapping of quantitative trait loci for resistance to Mycosphaerella pinodes in Pisum sativum subsp syriacum. Mol. Breed. 21, 439–454. doi: 10.1007/s11032-007-9144-4
Ford, R., Le Roux, K., Itman, C., Brouwer, J. B., and Taylor, P. W. J. (2002). Diversity analysis and genotyping in Pisum with sequence tagged microsatellite site (STMS) primers. Euphytica 124, 397–405. doi: 10.1023/A:1015752907108
Franssen, S. U., Shrestha, R. P., Bräutigam, A., Bornberg-Bauer, E., and Weber, A. P. M. (2011). Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing. BMC Genomics 12:227. doi: 10.1186/1471-2164-12-227
Frew, T. J., Russell, A. C., and Timmerman-Vaughan, G. M. (2002). Sequence tagged site markers linked to the sbm1 gene for resistance to pea seedborne mosaic virus in pea. Plant Breed. 121, 512–516. doi: 10.1046/j.1439-0523.2002.00761.x
Ghafoor, A., and McPhee, K. (2012). Marker assisted selection (MAS) for developing powdery mildew resistant pea cultivars. Euphytica 186, 593–607. doi: 10.1007/s10681-011-0596-6
Goldenberg, J. B. (1965). “Afila” a new mutation in pea (Pisum sativum L.). Bole I Genetico 1, 27–31.
Gong, Y. M., Xu, S. C., Mao, W. H., Hu, Q. Z., Zhang, G. W., Ding, J., et al. (2010). Developing new SSR markers from ESTs of pea (Pisum sativum L.). J. Zhejiang Univ. Sci. B 11, 702–707. doi: 10.1631/jzus.B1000004
Grønlund, M., Olsen, A., Johansen, E. I., and Jakobsen, I. (2010). Protocol: using virus-induced gene silencing to study the arbuscular mycorrhizal symbiosis in Pisum sativum. Plant Methods 6:28. doi: 10.1186/1746-4811-6-28
Hall, K. J., Parker, J. S., Ellis, T. H. N., Turner, L., Knox, M. R., Hofer, J. M. I., et al. (1997). The relationship between genetic and cytogenetic maps of pea.II. Physical maps of linkage mapping populations. Genome 40, 755–769. doi: 10.1139/g97-798
Hamon, C., Baranger, A., Coyne, C. J., McGee, R. J., Le Goff, I., L'Anthoëne, V., et al. (2011). New consistent QTL in pea associated with partial resistance to Aphanomyces euteiches in multiple French and American environments. Theor. Appl. Genet. 123, 261–281. doi: 10.1007/s00122-011-1582-z
Hamon, C., Coyne, C. J., McGee, R. J., Lesné, A., Esnault, R., Mangin, P., et al. (2013). QTL meta-analysis provides a comprehensive view of loci controlling partial resistance to Aphanomyces euteiches in four sources of resistance in pea. BMC Plant Biol. 13:45. doi: 10.1186/1471-2229-13-45
Hance, S. T., Grey, W., and Weeden, N. F. (2004). Identification of tolerance to Fusarium solani in Pisum sativum ssp. elatius. Pisum Genet. 36, 9–13.
Hanocq, E., Jeuffroy, M. H., Lejeune-Hénaut, I., and Munier-Jolain, N. (2009). Construire des idéotypes pour des systèmes de culture varies en pois d'hiver. Innov. Agron. 7, 14–28.
Harland, S. C. (1948). Inheritance of immunity to mildew in Peruvian forms of Pisum sativum. Heredity 2, 263–269. doi: 10.1038/hdy.1948.15
Hascoët, E., Jaminon, O., Devaux, C., Blassiau, C., Bahrman, N., Bochard, A.-M., et al. (2014). “Towards fine mapping of frost tolerance QTLs in pea,” in 2nd PeaMUST Annual Meeting (Dijon).
Hayer, F., Bonnin, E., Carrouee, B., Gaillard, G., Nemecek, T., Schneider, A., et al. (2010). “Designing sustainable crop rotations using Life Cycle assessment of crop combinations,” in 9th European IFSA Symposium (Vienna).
Hunter, P. J., Ellis, N., and Taylor, J. D. (2001). Association of dominant loci for resistance to Pseudomonas syringae pv. pisi with linkage groups II, VI and VII of Pisum sativum. Theor. Appl. Genet. 103, 129–135. doi: 10.1007/s001220100566
Iglesias-García, R., Prats, E., Fondevilla, S., Satovic, Z., and Rubiales, D. (2015). Quantitative trait loci associated to drought adaptation in pea (Pisum sativum L.). Plant Mol. Biol. Rep. 1–11. doi: 10.1007/s11105-015-0872-z
Irzykowska, L., and Wolko, B. (2004). Interval mapping of QTLs controlling yield-related traits and seed protein content in Pisum sativum. J. Appl. Genet. 45, 297–306.
Irzykowska, L., Wolko, B., and Swiecicki, W. (2002). Interval mapping of QTLs controlling some morphological traits in pea. Cell. Mol. Biol. Lett. 7, 417–422.
Jain, S., Weeden, N. F., Porter, L. D., Eigenbrode, S. D., and McPhee, K. (2013). Finding linked markers to en for efficient selection of pea enation mosaic virus resistance in pea. Crop Sci. 53, 2392–2399. doi: 10.2135/cropsci2013.04.0211
Jensen, E. S., Peoples, M. B., Boddey, R. M., Gresshoff, P. M., Hauggaard-Nielsen, H., Alves, B. J. R., et al. (2012). Legumes for mitigation of climate change and the provision of feedstock for biofuels and biorefineries. A review. Agron. Sust. Dev. 32, 329–364. doi: 10.1007/s13593-011-0056-7
Jha, A. B., Arganosa, G., Tar'An, B., Diederichsen, A., and Warkentin, T. D. (2013). Characterization of 169 diverse pea germplasm accessions for agronomic performance, Mycosphaerella blight resistance and nutritional profile. Genet. Resour. Crop Evol. 60, 747–761. doi: 10.1007/s10722-012-9871-1
Jha, A. B., Tar'An, B., Diapari, M., Sindhu, A., Shunmugam, A., Bett, K., et al. (2015). Allele diversity analysis to identify SNPs associated with ascochyta blight resistance in pea. Euphytica 202, 189–197. doi: 10.1007/s10681-014-1254-6
Jha, A. B., Warkentin, T. D., Gurusamy, V., Tar'An, B., and Banniza, S. (2012). Identification of mycosphaerella blight resistance in wild Pisum species for use in pea breeding. Crop Sci. 52, 2462–2468. doi: 10.2135/cropsci2012.04.0242
Kabir, A. H., Paltridge, N. G., Able, A. J., Paull, J. G., and Stangoulis, J. C. R. (2012). Natural variation for Fe-efficiency is associated with upregulation of Strategy I mechanisms and enhanced citrate and ethylene synthesis in Pisum sativum L. Planta 235, 1409–1419. doi: 10.1007/s00425-011-1583-9
Kaur, S., Pembleton, L. W., Cogan, N. O. I., Savin, K. W., Leonforte, T., Paull, J., et al. (2012). Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genomics 13:104. doi: 10.1186/1471-2164-13-104
Klein, A., Houtin, H., Rond, C., Marget, P., Jacquin, F., Boucherot, K., et al. (2014). QTL analysis of frost damage in pea suggests different mechanisms involved in frost tolerance. Theor. Appl. Genet. 127, 1319–1330. doi: 10.1007/s00122-014-2299-6
Kraft, J. M., Dunne, B., Goulden, D., and Armstrong, S. (1998). A search for resistance in peas to Mycosphaerella pinodes. Plant Dis. 82, 251–253. doi: 10.1094/PDIS.1998.82.2.251
Krajewski, P., Bocianowski, J., Gawłowska, M., Kaczmarek, Z., Pniewski, T., Święcicki, W., et al. (2012). QTL for yield components and protein content: a multienvironment study of two pea (Pisum sativum L.) populations. Euphytica 183, 323–336. doi: 10.1007/s10681-011-0472-4
Kujala, V. (1953). Felderbse, bei welcher die ganze Blattspreite in Ranken umgewandelt ist. Arch. Soc. Zool. Bot. Fenn. 8, 44–45.
Kwon, S.-J., Brown, A. F., Hu, J., McGee, R., Watt, C., Kisha, T., et al. (2012). Genetic diversity, population structure and genome-wide marker-trait association analysis emphasizing seed nutrients of the USDA pea (Pisum sativum L.) core collection. Genes Genomics 34, 305–320. doi: 10.1007/s13258-011-0213-z
Lamprecht, H. (1948). The variation of linkage and the course of crossingover. Agri Hort. Genet. 6, 10–48
Laucou, V., Haurogné, K., Ellis, N., and Rameau, C. (1998). Genetic mapping in pea. 1. RAPD-based genetic linkage map of Pisum sativum. Theor. Appl. Genet. 97, 905–915. doi: 10.1007/s001220050971
Laudadio, V., Nahashon, S. N., and Tufarelli, V. (2012). Growth performance and carcass characteristics of guinea fowl broilers fed micronized-dehulled pea (Pisum sativum L.) as a substitute for soybean meal. Poult. Sci. 91, 2988–2996. doi: 10.3382/ps.2012-02473
Lavaud, C., Lesné, A., Piriou, C., Le Roy, G., Boutet, G., Moussart, A., et al. (2015). Validation of QTL for resistance to Aphanomyces euteiches in different pea genetic backgrounds using near-isogenic lines. Theor. Appl. Genet. 128, 2273–2288. doi: 10.1007/s00122-015-2583-0
Lejeune-Hénaut, I., Hanocq, E., Bethencourt, L., Fontaine, V., Delbreil, B., Morin, J., et al. (2008). The flowering locus Hr colocalizes with a major QTL affecting winter frost tolerance in Pisum sativum L. Theor. Appl. Genet. 116, 1105–1116. doi: 10.1007/s00122-008-0739-x
Leonforte, A., Forster, J. W., Redden, R. J., Nicolas, M. E., and Salisbury, P. A. (2013a). Sources of high tolerance to salinity in pea (Pisum sativum L.). Euphytica 189, 203–216. doi: 10.1007/s10681-012-0771-4
Leonforte, A., Sudheesh, S., Cogan, N. O. I., Salisbury, P. A., Nicolas, M. E., Materne, M., et al. (2013b). SNP marker discovery, linkage map construction and identification of QTLs for enhanced salinity tolerance in field pea (Pisum sativum L.). BMC Plant Biol. 13:161. doi: 10.1186/1471-2229-13-161
Liu, X., Glahn, R. P., Arganosa, G. C., and Warkentin, T. D. (2015). Iron bioavailability in low phytate pea. Crop Sci. 55, 320–330. doi: 10.2135/cropsci2014.06.0412
Loridon, K., McPhee, K., Morin, J., Dubreuil, P., Pilet-Nayel, M. L., Aubert, G., et al. (2005). Microsatellite marker polymorphism and mapping in pea (Pisum sativum L.). Theor. Appl. Genet. 111, 1022–1031. doi: 10.1007/s00122-005-0014-3
Macas, J., Neumann, P., and Navrátilová, A. (2007). Repetitive DNA in the pea (Pisum sativum L.) genome: comprehensive characterization using 454 sequencing and comparison to soybean and Medicago truncatula. BMC Genomics 8:427. doi: 10.1186/1471-2164-8-427
Macwilliam, S., Wismer, M., and Kulshreshtha, S. (2014). Life cycle and economic assessment of Western Canadian pulse systems: the inclusion of pulses in crop rotations. Agric. Syst. 123, 43–53. doi: 10.1016/j.agsy.2013.08.009
Madoui, M. A., Labadie, K., Aury, J. M., Coyne, C. J., Warkentin, T., Kreplak, J., et al. (2015). “The international pea genome sequencing project: sequencing and assembly progresses,” in PLANT and ANIMAL Genome XXIII (San Diego, CA).
Marles, M. A. S., Warkentin, T. D., and Bett, K.E. (2013). Genotypic abundance of carotenoids and polyphenolics in the hull of field pea (Pisum sativum L.). J. Sci. Food Agric. 93, 463–470. doi: 10.1002/jsfa.5782
McCallum, J. A., Frew, T. J., Gilpin, B. J., and Timmerman-Vaughan, G. M. (1997). A linkage map of the pea (Pisum sativum L.) genome containing cloned sequences of known function and expressed sequence tags (ESTs). Theor. Appl. Genet. 95, 1289–1299. doi: 10.1007/s001220050695
McClendon, M. T., Inglis, D. A., McPhee, K. E., and Coyne, C. J. (2002). DNA markers linked to Fusarium wilt race 1 resistance in pea. J. Am. Soc. Hortic. Sci. 127, 602–607.
McGee, R. J., Coyne, C. J., Pilet-Nayel, M.-L., Moussart, A., Tivoli, B., Baranger, A., et al. (2012). Registration of pea germplasm lines partially resistant to aphanomyces root rot for breeding fresh or freezer pea and dry pea types. J. Plant Regist. 6, 203–207. doi: 10.3198/jpr2011.03.0139crg
McPhee, K. E., Inglis, D. A., Gundersen, B., and Coyne, C. J. (2012). Mapping QTL for Fusarium wilt Race 2 partial resistance in pea (Pisum sativum). Plant Breed. 131, 300–306. doi: 10.1111/j.1439-0523.2011.01938.x
Mishra, R. K., Gangadhar, B. H., Nookaraju, A., Kumar, S., and Park, S. W. (2012). Development of EST-derived SSR markers in pea (Pisum sativum) and their potential utility for genetic mapping and transferability. Plant Breed. 131, 118–124. doi: 10.1111/j.1439-0523.2011.01926.x
Mitchell, D. C., Lawrence, F. R., Hartman, T. J., and Curran, J. M. (2009). Consumption of dry beans, peas, and lentils could improve diet quality in the US population. J. Am. Diet. Assoc. 109, 909–913. doi: 10.1016/j.jada.2009.02.029
Nemecek, T., and Kägi, T. (2007). “Life Cycle Inventories of Swiss and European agricultural production systems,” in Final Report Ecoinvent V2. 0 No 15a. Agroscope Reckenholz-Tänikon Research Station ART. Zurich; Dübendorf, CH: Swiss Centre for Life Cycle Inventories. SimaPro Ph.D., 7.
Nemecek, T., Von Richthofen, J.-S., Dubois, G., Casta, P., Charles, R., and Pahl, H. (2008). Environmental impacts of introducing grain legumes into European crop rotations. Eur. J. Agron. 28, 380–393. doi: 10.1016/j.eja.2007.11.004
Ochatt, S. J., Benabdelmouna, A., Marget, P., Aubert, G., Moussy, F., Pontecaille, C., et al. (2004). Overcoming hybridization barriers between pea and some of its wild relatives. Euphytica 137, 353–359. doi: 10.1023/B:EUPH.0000040476.57938.81
Page, D., Aubert, G., Duc, G., Welham, T., and Domoney, C. (2002). Combinatorial variation in coding and promoter sequences of genes at the Tri locus in Pisum sativum accounts for variation in trypsin inhibitor activity in seeds. Mol. Genet. Genomics 267, 359–369. doi: 10.1007/s00438-002-0667-4
Petkova, V., Nikolova, V., Kalapchieva, S. H., Stoeva, V., Topalova, E., and Angelova, S. (2009). Physiological response and pollen viability of Pisum sativum genotypes under high temperature influence. Iv Balkan Symp.Veg. Potato. 830, 665–671. doi: 10.17660/actahortic.2009.830.96
Pflieger, S., Richard, M. M. S., Blanchet, S., Meziadi, C., and Geffroy, V. (2013). VIGS technology: an attractive tool for functional genomics studies in legumes. Funct. Plant Biol. 40, 1234–1248. doi: 10.1071/FP13089
Pilet-Nayel, L., Muehlbauer, F. J., McGee, R. J., Kraft, J. M., Baranger, A., and Coyne, C. J. (2002). Quantitative trait loci for partial resistance to Aphanomyces root rot in pea. Theor. Appl. Genet. 106, 28–39.
Pilet-Nayel, M. L., Muehlbauer, F. J., McGee, R. J., Kraft, J. M., Baranger, A., and Coyne, C. J. (2005). Consistent quantitative trait loci in pea for partial resistance to Aphanomyces euteiches isolates from the United States and France. Phytopathology 95, 1287–1293. doi: 10.1094/PHYTO-95-1287
Praca-Fontes, M. M., Carvalho, C. R., and Clarindo, W. R. (2014). Karyotype revised of Pisum sativum using chromosomal DNA amount. Plant Syst. Evol. 300, 1621–1626. doi: 10.1007/s00606-014-0987-y
Prioul, S., Frankewitz, A., Deniot, G., Morin, G., and Baranger, A. (2004). Mapping of quantitative trait loci for partial resistance to Mycosphaerella pinodes in pea (Pisum sativum L.), at the seedling and adult plant stages. Theor. Appl. Genet. 108, 1322–1334. doi: 10.1007/s00122-003-1543-2
Rai, R., Singh, A. K., Singh, B. D., Joshi, A. K., Chand, R., and Srivastava, C. P. (2011). Molecular mapping for resistance to pea rust caused by Uromyces fabae (Pers.) de-Bary. Theor. Appl. Genet. 123, 803–813. doi: 10.1007/s00122-011-1628-2
Ray, H., Bett, K., Tar'An, B., Vandenberg, A., Thavarajah, D., and Warkentin, T. (2014). Mineral micronutrient content of cultivars of field pea, chickpea, common bean, and lentil grown in Saskatchewan, Canada. Crop Sci. 54, 1698–1708. doi: 10.2135/cropsci2013.08.0568
Reddy, D. C. L., Preethi, B., Wani, M. A., Aghora, T. S., Aswath, C., and Mohan, N. (2015). Screening for powdery mildew (Erysiphe pisi DC) resistance gene-linked SCAR and SSR markers in five breeding lines of Pisum sativum L. J. Hortic. Sci. Biotechnol. 90, 78–82.
Rozov, S. M., Kosterin, O., Borisov, A. Y., and Tsyganov, V. (1999). The history of the pea gene map: last revolutions and the new symbiotic genes. Pisum Genet. 31, 55–56.
Sarikamis, G., Yanmaz, R., Ermis, S., Bakir, M., and Yüksel, C. (2010). Genetic characterization of pea (Pisum sativum) germplasm from Turkey using morphological and SSR markers. Genetics Mol. Res. 9, 591–600. doi: 10.4238/vol9-1gmr762
Shunmugam, A. S. K., Liu, X., Stonehouse, R., Tar'An, B., Bett, K. E., Sharpe, A. G., et al. (2015). Mapping seed phytic acid concentration and iron bioavailability in a pea recombinant inbred line population. Crop Sci. 55, 828–836. doi: 10.2135/cropsci2014.08.0544
Sindhu, A., Ramsay, L., Sanderson, L. A., Stonehouse, R., Li, R., Condie, J., et al. (2014). Gene-based SNP discovery and genetic mapping in pea. Theor. Appl. Genet. 127, 2225–2241. doi: 10.1007/s00122-014-2375-y
Smýkal, P., Coyne, C. J., Ambrose, M. J., Maxted, N., Schaefer, H., Blair, M. W., et al. (2015). Legume crops phylogeny and genetic diversity for science and breeding. CRC. Crit. Rev. Plant Sci. 34, 43–104. doi: 10.1080/07352689.2014.897904
Smýkal, P., Hybl, M., Corander, J., Jarkovsky, J., Flavell, A. J., and Griga, M. (2008). Genetic diversity and population structure of pea (Pisum sativum L.) varieties derived from combined retrotransposon, microsatellite and morphological marker analysis. Theor. Appl. Genet. 117, 413–424. doi: 10.1007/s00122-008-0785-4
Smýkal, P., Jovanović, Ž., Stanisavljević, N., Zlatković, B., Ćupina, B., Ðorđević, V., et al. (2014). A comparative study of ancient DNA isolated from charred pea (Pisum sativum L.) seeds from an Early Iron Age settlement in southeast Serbia: inference for pea domestication. Genet. Resour. Crop Evol. 61, 1533–1544. doi: 10.1007/s10722-014-0128-z
Stein, H. H., Everts, A. K. R., Sweeter, K. K., Peters, D. N., Maddock, R. J., Wulf, D. M., et al. (2006). The influence of dietary field peas (Pisum sativum L.) on pig performance, carcass quality, and the palatability of pork. J. Anim. Sci. 84, 3110–3117. doi: 10.2527/jas.2005-744
Sudheesh, S., Lombardi, M., Leonforte, A., Cogan, N. O. I., Materne, M., Forster, J. W., et al. (2014). Consensus genetic map construction for field pea (Pisum sativum L.), trait dissection of biotic and abiotic stress tolerance and development of a diagnostic marker for the er1 powdery mildew resistance gene. Plant Mol. Biol. Rep. 33, 1391–1403. doi: 10.1007/s11105-014-0837-7
Sun, X., Yang, T., Hao, J., Zhang, X., Ford, R., Jiang, J., et al. (2014). SSR genetic linkage map construction of pea (Pisum sativum L.) based on Chinese native varieties. Crop J. 2, 170–174. doi: 10.1016/j.cj.2014.03.004
Svabova, L., and Griga, M. (2008). The effect of cocultivation treatments on transformation efficiency in pea (Pisum sativum L.). Plant Celle Tissue Organ Culture 95, 293–304. doi: 10.1007/s11240-008-9443-4
Tar'an, B., Warkentin, T., Somers, D. J., Miranda, D., Vandenberg, A., Blade, S., et al. (2003). Quantitative trait loci for lodging resistance, plant height and partial resistance to mycosphaerella blight in field pea (Pisum sativum L.). Theor. Appl. Genet. 107, 1482–1491. doi: 10.1007/s00122-003-1379-9
Tar'an, B., Warkentin, T., Somers, D. J., Miranda, D., Vandenberg, A., Blade, S., et al. (2004). Identification of quantitative trait loci for grain yield, seed protein concentration and maturity in field pea (Pisum sativum L.). Euphytica 136, 297–306. doi: 10.1023/B:EUPH.0000032721.03075.a0
Tayeh, N., Aluome, C., Falque, M., Jacquin, F., Klein, A., Chauveau, A., et al. (2015a). Development of two major resources for pea genomics: the GenoPea 13.2K SNP Array and a high density, high resolution consensus genetic map. Plant J. doi: 10.1111/tpj.13070. [Epub ahead of print].
Tayeh, N., Bahrman, N., Devaux, R., Bluteau, A., Prosperi, J.-M., Delbreil, B., et al. (2013a). A high-density genetic map of the Medicago truncatula major freezing tolerance QTL on chromosome 6 reveals colinearity with a QTL related to freezing damage on Pisum sativum linkage group VI. Mol. Breed. 32, 279–289. doi: 10.1007/s11032-013-9869-1
Tayeh, N., Bahrman, N., Sellier, H., Bluteau, A., Blassiau, C., Fourment, J., et al. (2013b). A tandem array of CBF/DREB1 genes is located in a major freezing tolerance QTL region on Medicago truncatula chromosome 6. BMC Genomics 14:814. doi: 10.1186/1471-2164-14-814
Tayeh, N., Klein, A., Le Paslier, M. C., Jacquin, F., Houtin, H., Rond, C., et al. (2015b). Genomic prediction in pea: effect of marker density and training population size and composition on prediction accuracy. Front. Plant Sci. 6:941. doi: 10.3389/fpls.2015.00941
Timmerman-Vaughan, G. M., Frew, T. J., Butler, R., Murray, S., Gilpin, M., Falloon, K., et al. (2004). Validation of quantitative trait loci for Ascochyta blight resistance in pea (Pisum sativum L.), using populations from two crosses. Theor. Appl. Genet. 109, 1620–1631. doi: 10.1007/s00122-004-1779-5
Timmerman-Vaughan, G. M., Frew, T. J., Russell, A. C., Khan, T., Butler, R., Gilpin, M., et al. (2002). QTL mapping of partial resistance to field epidemics of ascochyta blight of pea. Crop Sci. 42, 2100–2111. doi: 10.2135/cropsci2002.2100
Timmerman-Vaughan, G. M., McCallum, J. A., Frew, T. J., Weeden, N. F., and Russell, A. C. (1996). Linkage mapping of quantitative trait loci controlling seed weight in pea (Pisum sativum L). Theor. Appl. Genet. 93, 431–439. doi: 10.1007/BF00223187
Timmerman-Vaughan, G. M., Mills, A., Whitfield, C., Frew, T., Butler, R., Murray, S., et al. (2005). Linkage mapping of QTL for seed yield, yield components, and developmental traits in pea. Crop Sci. 45, 1336–1344. doi: 10.2135/cropsci2004.0436
Ubayasena, L., Bett, K., Tar'an, B., Vijayan, P., and Warkentin, T. (2010). Genetic control and QTL analysis of cotyledon bleaching resistance in green field pea (Pisum sativum L.). Genome 53, 346–359. doi: 10.1139/G10-013
Ubayasena, L., Bett, K., Tar'an, B., and Warkentin, T. (2011). Genetic control and identification of QTLs associated with visual quality traits of field pea (Pisum sativum L.). Genome 54, 261–272. doi: 10.1139/g10-117
Valderrama, M. R., Roman, B., Satovic, Z., Rubiales, D., Cubero, J. I., and Torres, A. M. (2004). Locating quantitative trait loci associated with Orobanche crenata resistance in pea. Weed Res. 44, 323–328. doi: 10.1111/j.1365-3180.2004.00406.x
van Leur, J. A. G., Aftab, M., Leonforte, B., Moore, S., and Freeman, A. J. (2007). “Control of Pea seedborne mosaic virus in field pea through resistance breeding,” in Proceedings 16th Biennial Conference of the Australasian Plant Pathology Society (Adelaide, SA).
Varshney, R. K., Kudapa, H., Pazhamala, L., Chitikineni, A., Thudi, M., Bohra, A., et al. (2015). Translational genomics in agriculture: some examples in grain legumes. CRC. Crit. Rev. Plant Sci. 34, 169–194. doi: 10.1080/07352689.2014.897909
Wang, T. L., Domoney, C., Hedley, C. L., Casey, R., and Grusak, M. A. (2003). Can we improve the nutritional quality of legume seeds? Plant Physiol. 131, 886–891. doi: 10.1104/pp.102.017665
Warkentin, T. D., Delgerjav, O., Arganosa, G., Rehman, A. U., Bett, K. E., Anbessa, Y., et al. (2012). Development and characterization of low-phytate pea. Crop Sci. 52, 74–78. doi: 10.2135/cropsci2011.05.0285
Warkentin, T. D., Smykal, P., Coyne, C. J., Weeden, N., Domoney, C., Bing, D., et al. (2015). “Pea (Pisum sativum L.),” in Grain Legumes, Series Handbook of Plant Breeding, ed A. M. De Ron (New York, NY: Springer Science+Business Media), 37–83.
Weeden, N. F., Brauner, S., and Przyborowski, J. A. (2002). Genetic analysis of pod dehiscence in pea (Pisum sativum L.). Cell. Mol. Biol. Lett. 7, 657–663.
Weeden, N. F., Ellis, T. H. N., Timmerman-Vaughan, G. M., Swiecicki, W. K., Rozov, S. M., and Berdnikov, V. A. (1998). A consensus linkage map for Pisum sativum. Pisum Genet. 30, 1–4.
Weeden, N. F., McGee, R., Grau, C. R., and Muehlbauer, F. J. (2000). A gene influencing tolerance to common root rot is located on linkage group IV. Pisum Genet. 32, 53–55.
Weeden, N. F., and Porter, L. (2007). The genetic basis of Fusarium root rot tolerance in the ‘Afghanistan’ pea. Pisum Genet. 39, 35–36.
Weiss, E., and Zohary, D. (2011). The Neolithic Southwest Asian founder crops their biology and archaeobotany. Curr. Anthropol. 52, S237–S254. doi: 10.1086/658367
Weller, J. L., Liew, L. C., Hecht, V. F. G., Rajandran, V., Laurie, R. E., Ridge, S. et al. (2012). A conserved molecular basis for photoperiod adaptation in two temperate legumes. Proc. Natl. Acad. Sci. U.S.A. 109, 21158–21163. doi: 10.1073/pnas.1207943110
Weller, J. L., and Ortega, R. (2015). Genetic control of flowering time in legumes. Front. Plant Sci. 6:207. doi: 10.3389/fpls.2015.00207
Willcox, G., Buxo, R., and Herveux, L. (2009). Late Pleistocene and early Holocene climate and the beginnings of cultivation in northern Syria. Holocene 19, 151–158. doi: 10.1177/0959683608098961
Zhang, C., Tar'An, B., Warkentin, T., Tullu, A., Bett, K. E., Vandenberg, B., et al. (2006). Selection for lodging resistance in early generations of field pea by molecular markers. Crop Sci. 46, 321–329. doi: 10.2135/cropsci2005.0123
Zohary, D. (1999). Monophyletic vs. polyphyletic origin of the crops on which agriculture was founded in the Near East. Genet. Resour. Crop Evol. 46, 133–142. doi: 10.1023/A:1008692912820
Keywords: pea (Pisum sativum L.), breeding targets, genetic diversity, genomic resources, genotyping platforms, genetic maps, QTL and association mapping
Citation: Tayeh N, Aubert G, Pilet-Nayel M-L, Lejeune-Hénaut I, Warkentin TD and Burstin J (2015) Genomic Tools in Pea Breeding Programs: Status and Perspectives. Front. Plant Sci. 6:1037. doi: 10.3389/fpls.2015.01037
Received: 19 June 2015; Accepted: 09 November 2015;
Published: 27 November 2015.
Edited by:
Janila Pasupuleti, International Crops Research Institute for the Semi Arid Tropics, IndiaReviewed by:
Vikas Kumar Singh, International Crops Research Institute for the Semi-Arid Tropics, IndiaAntonio M. De Ron, Misión Biológica de Galicia, CSIC, Spain
Copyright © 2015 Tayeh, Aubert, Pilet-Nayel, Lejeune-Hénaut, Warkentin and Burstin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Judith Burstin, judith.burstin@dijon.inra.fr