 Srivignesh Sundaresan1,2
Srivignesh Sundaresan1,2 Sonia Philosoph-Hadas1Joseph Riov2
Sonia Philosoph-Hadas1Joseph Riov2 Raja Mugasimangalam3
Raja Mugasimangalam3 Nagesh A. Kuravadi3Bettina Kochanek1
Nagesh A. Kuravadi3Bettina Kochanek1 Shoshana Salim1
Shoshana Salim1 Mark L. Tucker4
Mark L. Tucker4 Shimon Meir1*
Shimon Meir1*- 1Department of Postharvest Science of Fresh Produce, Agricultural Research Organization, The Volcani Center, Bet-Dagan, Israel
- 2The Robert H. Smith Faculty of Agriculture, Food and Environment, The Robert H. Smith Institute of Plant Sciences and Genetics in Agriculture, The Hebrew University of Jerusalem, Rehovot, Israel
- 3Department of Bioinformatics, QTLomics Technologies Pvt. Ltd, Bangalore, India
- 4Soybean Genomics and Improvement Laboratory, United States Department of Agriculture, Agricultural Research Service, Beltsville, MD, USA
Abscission of flower pedicels and leaf petioles of tomato (Solanum lycopersicum) can be induced by flower removal or leaf deblading, respectively, which leads to auxin depletion, resulting in increased sensitivity of the abscission zone (AZ) to ethylene. However, the molecular mechanisms that drive the acquisition of abscission competence and its modulation by auxin gradients are not yet known. We used RNA-Sequencing (RNA-Seq) to obtain a comprehensive transcriptome of tomato flower AZ (FAZ) and leaf AZ (LAZ) during abscission. RNA-Seq was performed on a pool of total RNA extracted from tomato FAZ and LAZ, at different abscission stages, followed by de novo assembly. The assembled clusters contained transcripts that are already known in the Solanaceae (SOL) genomics and NCBI databases, and over 8823 identified novel tomato transcripts of varying sizes. An AZ-specific microarray, encompassing the novel transcripts identified in this study and all known transcripts from the SOL genomics and NCBI databases, was constructed to study the abscission process. Multiple probes for longer genes and key AZ-specific genes, including antisense probes for all transcripts, make this array a unique tool for studying abscission with a comprehensive set of transcripts, and for mining for naturally occurring antisense transcripts. We focused on comparing the global transcriptomes generated from the FAZ and the LAZ to establish the divergences and similarities in their transcriptional networks, and particularly to characterize the processes and transcriptional regulators enriched in gene clusters that are differentially regulated in these two AZs. This study is the first attempt to analyze the global gene expression in different AZs in tomato by combining the RNA-Seq technique with oligonucleotide microarrays. Our AZ-specific microarray chip provides a cost-effective approach for expression profiling and robust analysis of multiple samples in a rapid succession.
Introduction
Abscission is a systematically regulated process in plant development, by which subtended organs, leaves, flowers, fruit, and seed, separate from the parent plant in response to various physiological cues. This process is required to recycle nutrients for continuous growth, develop appropriate organs, survive diseases, and facilitate reproduction (Addicott, 1982; Sexton and Roberts, 1982; Roberts et al., 2002; Lewis et al., 2006). Since the domestication of crops was started, a great emphasis has been put forth on selection for abrupted abscission to improve crop qulaity and yield. For example, reduced seed shaterring in rice (Li et al., 2006), wheat (Tanno and Willcox, 2006), maize (Doebley, 2004), and fruit tree species (Bangerth, 2000) was obtained. In an agricultural perspective, both enhanced and delayed abscission are highly relevant for growers.
In plants, the abscission process occurs at a predetermined region called abscission zone (AZ), composed of few layers of small and dense cytoplasmic cells, which lack large vacuoles and any maturation characteristics (Osborne and Morgan, 1989), resembling undifferentiated cells (Van Nocker, 2009). AZ cells belong to type II cells, in which extended growth can be enhanced by ethylene, but not by auxin (McManus, 2008), conferring their meristematic potential (Roberts et al., 2000). Physiological studies revealed that ethylene and auxin control the cell separation process. The abscission process is initiated or timed by changes in the auxin gradient across the AZ, and is acceltrated by ethylene (Roberts et al., 2002; McManus, 2008; Meir et al., 2010). The four key steps in the abscission process are: differentiation of the AZ, acquisition of the competence to respond to abscission signals, execution of organ abscission, and formation of a protective layer (Meir et al., 2011; Wang et al., 2013). During the late abscission stages, the cell wall and middle lamella are the major targets for degradation, which is operated by many cell wall modifiying enzymes, including polygalacturonases (PGs), xyloglucan endotransglucosylase/ hydrolase (XTH), β-1,4-glucanase (cellulase, Cel), and expansins (EXP) (Lashbrook et al., 1994; del Campillo and Bennett, 1996; Cho and Cosgrove, 2000; Taylor and Whitelaw, 2001; Roberts et al., 2002; Ogawa et al., 2009; Meir et al., 2010).
Tomato (Solanum lycopersicum) is one of the most important vegetable crops, whose genome sequence was recently published (The Tomato Genome Consortium, 2012). It serves as a model crop for studying fruit development (Klee and Giovannoni, 2011) and abscission, as it posses a distinct joint-like structure in the AZ of the flower pedicels. The molecular mechanisms underlying the abscission progress in tomato are still evolving, even though the abscission physiology was studied long ago (Sexton and Roberts, 1982; Bleecker and Patterson, 1997; Roberts et al., 2000). The genes affecting AZ development have been identified by studying abscission-impaired mutants, such as jointless, jointless2, macrocalyx, blind (bl), and lateral supressor (ls) (Butler, 1936; Rick, 1956; Schumacher et al., 1999; Mao et al., 2000; Szymkowiak and Irish, 2006; Shalit et al., 2009; Nakano et al., 2012; Liu et al., 2014). However, information regarding the expression of AZ-associated genes in tomato is still lacking.
Micro/oligo nucleotide arrays have been used until recently to study semi-global gene expression in AZs of citrus leaves (Agustí et al., 2008, 2009, 2012) and shoot tips (Zhang et al., 2014), tomato flowers (Meir et al., 2010, 2011; Nakano et al., 2012, 2013; Wang et al., 2013; Ma et al., 2015), Arabidopsis stamen (Cai and Lashbrook, 2008), and apple fruit and fruitlets (Botton et al., 2011; Zhu et al., 2011). The information on gene specific resources of tomato AZs during the abscission process obtained by microarrays is limited. The new era of high throughput next generation sequencing (NGS) technologies and bioinformatics tools to analyze and integrate the vast data, led to a significant rapid progress in the genomic research. RNA-Sequencing (RNA-Seq) involves direct sequencing of complementary DNAs (cDNAs), followed by mapping of reads to the reference genome or gene sets, to obtain a direct information from transcribed regions (Wang et al., 2009), gene expression profiles, and polymorphism detection in the genome. RNA-Seq provides a more comprehensive understanding of the transcriptome at a specific developmental stage of a tissue (Marioni et al., 2008; Wang et al., 2009; Parchman et al., 2010; Mäder et al., 2011), and an ability to detect novel transcripts, sense and antisense transcripts, single nucleotide polymorphism, small RNAs, alternate splice transcripts, and transcription initiation sites (Ozsolak and Milos, 2011). In comparison to other technologies, such as microarrays and Sanger-based sequencing technologies, RNA-Seq has additional advantages in terms of speed, depth, and accuracy. Recently, few RNA-Seq studies for studying the abscission process were carried out in various plant systems, such as olive (Gil-Amado and Gomez-Jimenez, 2013; Parra et al., 2013) and melon (Corbacho et al., 2013) fruit, Arabidopsis stamen AZ (Niederhuth et al., 2013), tomato flower AZ (FAZ) (Liu et al., 2014), and rose petals (Singh et al., 2013). However, a complete transcriptome study of the tomato FAZ and leaf AZ (LAZ) at various stages of the abscission process was not carried out, because of the higher cost of RNA-Seq analysis as compared to microarray. Considering the clear advantages of the RNA-Seq technology, the aim of the present research was to study the tomato FAZ and LAZ transcriptome, using RNA-Seq, followed by de novo transcriptome assembly and annotation.
In the last few years, customized-made expression arrays became more affordable to the scientific community due to the number of total features, complete coverage of genomic regions, and automated data analysis of microarrays. The information regarding the new transcripts, such as unannotated transcripts, splice variants, naturally occurring antisense transcripts (NATs), and novel genes in particular tissues, is less emphasized on the traditional arrays (Bertone et al., 2005; Mockler et al., 2005). More complexity is added due to methylation sites on the 3′ end that might interfere with transcription initiation and termination (Carninci et al., 2005, 2006). To overcome these problems, new customized microarray approaches for various needs, such as tilling arrays were developed, (Johnson et al., 2005). The custom-made arrays have a complete control of the number, expression, and distribution of probes specific to the studied system. In order to achieve higher hybridization and quality data, many considerations have to be taken while designing the customized microarrays, including probe length, density, melting temperatures, placement, level of cross hybridization, complexity, and mismatch levels to achieve the consensus property (Mei et al., 2003). The RNA-Seq information will be a useful tool to update the design of microarray probes for transcriptome analysis of large samples (Bellin et al., 2009).
In the present study, we used the Illumina sequencing technology to obtain a comprehensive transcriptome profile of the tomato FAZ and LAZ pooled samples taken at of various time points during organ abscission induced by auxin depletion, thereby expanding the tomato transcript catalog. We focused on comparing the transcriptomes generated from the FAZ and the LAZ tissues to establish the divergences and similarities in their transcriptional networks, and particularly to characterize the biological processes and transcriptional regulators enriched in gene clusters that are differentially regulated in these two AZs. The RNA-Seq data were used as a major source to design an AZ-specific microarray for tomato abscission studies. Additionally, in this chip the probes were designed in both sense and antisense orientations for transcripts, which enable future analyses of expression profiles of NATs in the AZs. The unique design of this chip allowed us to accurately quantify global changes in the transcriptome of the tomato AZs during the abscission process. Results from this study will help to identify target genes for further understanding and manipulating of abscission, as well as markers for breeding. We are currently using this chip to quantify molecular shifts in gene expression in transgenic plants that display altered abscission phenotypes.
Materials and Methods
Plant Material and Treatments
Tomato (S. lycopersicum) cv. “VF-36” flower bunches were harvested from greenhouse grown 4-month-old plants between 08:00 and 10:00 a.m. Bunches bearing at least 2–4 freshly open flowers were brought to the laboratory under high humidity conditions. All procedures were performed in a controlled observation room maintained at 20°C, 12 h photoperiod, and 60% RH. Closed young flower buds and senesced flowers were removed, and the stem ends of the bunches were trimmed. Groups of 2–3 flower explants were placed in vials containing double distilled water. The pedicel abscission assays were performed after flower removal, as previously described (Meir et al., 2010). For RNA extraction from the FAZ, 30 sections, <2-mm-thick, were excised, about 1 mm from each side of the visible AZ fracture. The samples were collected at predefined time points: 0 h—before flower removal, and 2, 4, 8, and 14 h after flower removal (Figure 1A).
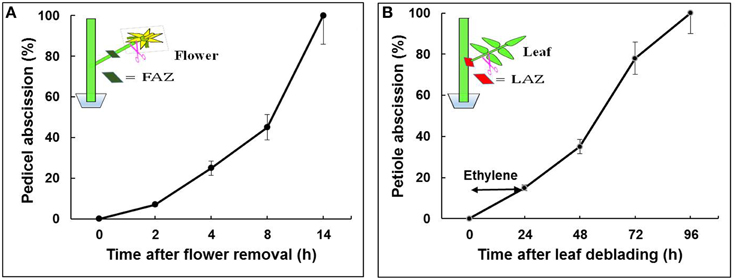
Figure 1. Effect of flower removal (A) or leaf deblading and ethylene treatment (B) on the kinetics of pedicel and petiole abscission, respectively, in tomato explants. Flowers and leaves were excised as indicated in the schematic illustrations. The debladed-leaf explants held in vials with water were prepared as previously described for the flower explants (Meir et al., 2010), and exposed to ethylene (10 μL L−1 for 24 h). The percentage of accumulated pedicel or petiole abscission were monitored at the indicated time intervals following organ removal. The results are means of four replicates (n = 30 explants) ± SE.
Shoots containing at least 5–6 expanded leaves were brought to the laboratory under high humidity conditions. The shoot cut ends were trimmed, and the shoots were placed in jars containing double distilled water and incubated for 3 h to avoid dehydration before starting leaf deblading. The fully expanded leaves were debladed using a sharp razor by leaving a subtended 2-cm long petiole. To accelerate petiole abscission, the debladed leaf explants were exposed to 10 μL L−1 ethylene for 24 h in an air-tight chamber at 23°C. The number of abscising petioles was then recorded. The ethylene concentration was determined by a gas chromatograph (Varian 3300), equipped with an alumina column and a flame ionization detector, in a 5-mL air samples withdrawn from the chamber with a gas tight syringe. Tissue samples for RNA extraction were taken from 20 LAZ sections, as described above for the FAZ. The samples were collected at the following predefined time points: 0 h—before leaf deblading, and 24, 48, 72, and 96 h after leaf deblading and ethylene treatment (Figure 1B). All the collected FAZ and LAZ samples were placed in RNA-later solution for further analyses. Three biological replicates of equal tissue weight were taken from each sample for RNA extraction. Equal RNA quantities from all the time point samples of the FAZ or the LAZ were pooled to create one RNA sample of FAZ and one of the LAZ.
RNA Isolation and Quality Controls
Total RNA was extracted from the FAZ and the LAZ samples, using an Agilent plant RNA isolation mini kit (Agilent, USA). The concentration and purity of the RNA samples were examined by a Nanodrop instrument, and validated for quality by running an aliquot on a Bioanalyzer with RNA 6000 Nano Kit (Agilent Technologies, California, USA). Only samples having an RNA integrity number >8.0 were selected for cDNA preparation. RNA samples extracted from the FAZ and the LAZ at five specific time points were pooled to make individual transcriptome libraries for RNA-Seq study.
Illumina Sequencing and Quality Controls
Transcriptome libraries for sequencing were constructed according to the Illumina TruSeq RNA library protocol outlined in “TruSeq RNA Sample Preparation Guide” (Part # 15008136; Rev. A, Illumina, USA). Briefly, mRNA was purified from 1 μg of intact total RNA using oligodT beads (TruSeq RNA Sample Preparation Kit, Illumina, USA). The purified mRNA was fragmented at an elevated temperature in the presence of divalent cations, and reverse transcribed with Superscript II Reverse transcriptase (Invitrogen, USA) by priming with random hexamers. Second strand cDNA was synthesized in the presence of DNA polymerase I and RNase H enzymes. The cDNA was cleaned up using Agencourt Ampure XP Solid Phase Reversible Immobilization beads (Beckman Coulter, Switzerland). Illumina Adapters were ligated to the cDNA molecules after end repair and addition of A base. Solid Phase Reversible Immobilization cleanup was performed after ligation. The library was amplified using PCR for enrichment of adapter ligated fragments. The prepared libraries were quantified by a Nanodrop instrument and validated for quality by running an aliquot on High Sensitivity Bioanalyzer Chip (Agilent Technologies, California, USA).
The DNA obtained from the prepared libraries was denatured and sequenced by the Illumina Genome Analyzer IIX, using the sequencing by synthesis method to read 72 bases PE. The raw sequencing data were then extracted from the server using the proprietary Illumina software to obtain Fastq format. Quality check (QC) of raw data was performed using SeqQC-V2.0 program (NGS data QC).
De novo Transcriptome Assembly and Differentially Expressed Genes
The following methodology for transcriptome assembly was used for each of the pooled samples of the FAZ and the LAZ. Raw reads were assembled using the Velvet-1.1.05 software and transcripts were generated using Oases assembler (Schulz et al., 2012). The transcripts were then subjected to Basic Local Alignment Search Tool (BLAST) analysis against S. lycopersicum mRNA and protein sequences from the International Tomato Annotation Group 2 (ftp://ftp.sgn.cornell.edu/genomes/Solanum_lycopersicum/annotation/ITAG2.3_release/). The transcripts with more than 50% identity and 70% query coverage were used for further analysis. Raw reads of the FAZ and the LAZ were aligned to reference cDNA using the Bowtie-0.12.7 program (Langmead et al., 2009). Expression studies were performed based on enrichment calculation of each gene. Enrichment = average read depth × coverage. Genes were considered to be upregulated, down-regulated, or neutral based on the LAZ/FAZ enrichment ratio. The parameters definitions were as follows: Up, log fold change >1; Down, log fold change <-1; Neutral, log fold change >-1 to <1.
Design of the AZ-Specific Microarray (Pooled Data of the FAZ and the LAZ)
The following steps were performed: Raw reads of both the FAZ and the LAZ samples were pooled and assembled as described above for de novo assembly (steps 1 and 2); Unannotated transcripts were subjected to BLAST analysis against Arabidopsis thaliana and Nicotiana tabacum mRNA and protein sequences, with references derived from The National Center for Biotechnology Information (NCBI) and The Arabidopsis Information Resource (TAIR) databases (step 3); Transcripts with more than 50% identity and 70% query coverage were considered as annotated, and all the rest were categorized as novel transcripts (step 4); Pooled reads were further aligned to mRNA sequences of S. lycopersicum and unaligned reads were assembled to derive contigs. These contigs were further filtered by E. coli, Cestrum elegans, A. thaliana, and N. tabacum mRNA sequences, with references derived from the NCBI database (step 5); Unannotated contigs from the above analysis (step 5) were added to the novel transcripts category (step 6); The unassembled reads from step 5 were further filtered by aligning them to E. coli and C. elegans mRNA sequences. Unaligned reads were assembled and added to the category of novel transcripts (step 7); Transcripts from steps 4, 6, and 7 were pooled together as novel transcripts, and transcripts which were annotated by S. lycopersicum, A. thaliana, and N. tabacum as performed in steps 2 and 3 were considered as known transcripts (step 8); To further filter the novel transcripts, BLAST was performed between novel and known transcripts, and only transcripts showing <10% query coverage with known transcripts were considered as novel transcripts (step 9). The novel (step 9) and known (step 8) transcripts were used for the design of the AZ-specific microarray. Probes were categorized as specific and cross hybridizing on the basis of their BLAST results. The criteria for a specific probe were as follows: a probe with a single hit against the target, a probe alignment length of 60–31 bp, a probe with allowed mismatches <3 and gaps <2, and a probe with a minimum length of 28 bases. Thus, out of a total number of 176,026 designed probes, 88,445 were specific probes, and 5363 were cross hybridized probes. For 429 transcripts, no probes were designed as filtered due to repeats and vector masked.
Gene Ontology (GO) Term Enrichment and Biological Pathways
Distribution of transcripts into various biological pathways in Kyoto Encyclopedia of Genes and Genomes (KEGG) was done through the KEGG Automatic Annotation Server (http://www.genome.jp/tools/kaas/) to obtain the KEGG IDs for the transcriptome sequences, and to identify the genes involved in plant hormone signal transduction.
Validation of Gene Expression by Real Time PCR (qPCR)
Primers for qPCR analysis were designed manually using the Gene Runner V 3.05 software (Hastings Software Inc. Hastings, USA; http://www.generunner.net). The primers were validated using one of the samples, and the amplicon sizes were confirmed on a 2% agarose gel. The sequences, amplicon length, and melting temperature (Tm) of the primers used are detailed in Table S1. The RNA samples used for the qPCR assay were the same samples used for the microarray analysis. Samples of 400 ng of DNase-treated RNA were reverse transcribed to synthesize 20 ng/μl of cDNA, using an oligo (dT) primer with Affinity Script QPCR cDNA synthesis kit (Agilent Technologies, USA). Relative quantification by qPCR was then performed using Brilliant II SYBR Green qPCR Master mix (Agilent Technologies, USA) according to the manufacturer's instructions. The experiment was conducted using a Stratagene Mx3005P QPCR machine platform (Mx3005P system software, Stratagene, CA). The relative expression levels of the genes were determined after normalizing with ACTIN as the reference gene, using the Delta Ct method. The PCR program consisted of an initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 60°C for 1 min, and 72°C for 1 min. A melt curve was also performed after the assay to check for specificity of the reaction. Duplicates of each gene with 20 ng cDNA input per reaction from two independent experiments were used. The relative quantification of gene expression level was determined by the comparative CT method 2−ΔΔCT.
Results
Kinetics of Flower Pedicel and Leaf Petiole Abscission
The abscission of flower pedicels and leaf petioles was induced by removing the auxin sources, flowers or leaves, respectively. The selected two sets of time points analyzed, 0, 2, 4, 8, and 14 h for flower pedicels and 0, 24, 48, 72, and 96 h for leaf petioles, were based on the abscission kinetics of these organs after flower removal or leaf deblading followed by ethylene treatment, respectively. Flower pedicels completely abscised 14 h after flower removal (Figure 1A), while leaf petioles completely abscised only 96 h after leaf deblading and exposure to ethylene (Figure 1B). The exposure of the leaf debladed explants to ethylene was required to enhance the abscission process, since the petioles abscised very slowly (8–12 days) without ethylene treatment, with a large variation between replicates (data not shown). It should be noted that a similar percentage of organ abscission was obtained in both systems, but on a different time scale (Figure 1). Based on these similar abscission percentages, we have chosen the indicated time points for RNA sampling from both AZs for the RNA-Seq experiments, which correspond to steps 2–4 of the abscission process, namely acquisition of the competence of AZ cells to respond to abscission signals, execution of organ abscission, and beginning of formation of a protective layer. The kinetics of tomato flower pedicel abscission in response to flower removal, without or with exogenous application of ethylene, is well documented (Meir et al., 2010; Wang et al., 2013).
Illumina Paired-End Sequencing and De novo Assembly
The RNA extracts of the FAZ and the LAZ sampled at specified time points after flower or leaf blade removal, respectively, were prepared as described in Figure 1, and sequenced using the Illumina sequencing platform. We used a pooled sequencing strategy by mixing RNA samples of the FAZ and the LAZ from various abscission stages. Individual cDNA libraries for de novo transcriptome sequencing were constructed for these pooled samples, in order to obtain a wide range of expressed transcript sequences and to reduce the sequencing costs (Sangwan et al., 2013; Xu et al., 2013). By PE sequencing of the tomato AZs in separate lanes, we obtained a total of 84,626,974 and 80,837,288 reads with 73 bp in length encompassing about 9.05 GB and 9.47 GB of sequence data for the LAZ and the FAZ, respectively (Table 1), in Fastq format after the initial quality filtering was performed with the default parameters. High quality reads were obtained by trimming primers/adapters and filtering by stringent parameters to increase the analysis reliability by trimming the reads with more than 70% of the bases having Phred quality score >20. This resulted in an average of 79,570,559 and 75,843,448 PE reads, which represent 94.02 and 93.82% of high quality read percentage for the LAZ and the FAZ, respectively (Table 1). In addition, this procedure enhanced the average quality score at each base position of the sequence reads by 94.61 and 94.71% (Table S2) of high quality base percentage for the FAZ and the LAZ, respectively (Figures S1, S2).
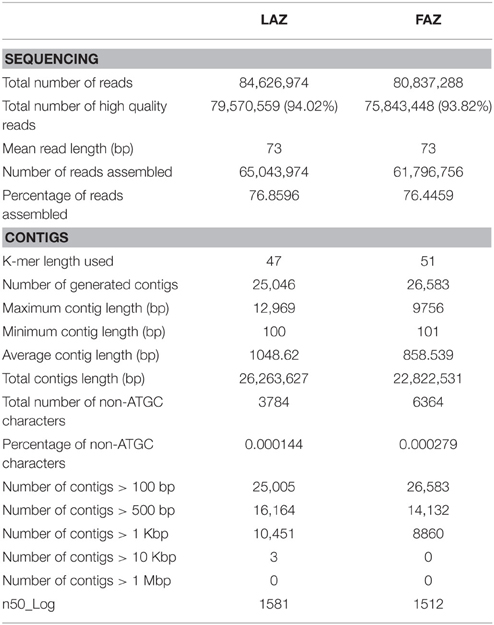
Table 1. Assembly statistics of pooled transcriptome of the tomato LAZ and FAZ.
Initially, we performed a de novo assembly for the transcripts, and later on the reference-based assembly was performed using the tomato genome sequence (The Tomato Genome Consortium, 2012) as reference genome. Both the de novo and reference assemblies were merged to generate the final assembly, which was used for all further analyses. We also carried out an analysis of differentially expressed genes (DEG) for the FAZ and the LAZ. De novo assembly was optimized using different criteria, such as the numbers of used reads, total number of contigs, contigs length in bp (>100, 500, 1000 bp and so on), n50_Log, maximum, average, and minimum contigs length (100 bp), based on the function of K-mer length (Hash Length). The inversed relation between the k-mer and the number of contigs (Sangwan et al., 2013) led us to optimize the assembly with various k-mer values ranging from 33 to 65 (Table S2). At K-mer = 33 the highest number of contigs, 49,849 and 57,398, and a higher number of longer coting length (>10 Kb) was obtained in both the FAZ and the LAZ samples, respectively (Table S2). We assembled most of the high quality reads, 78.74 and 78.41%, into longer contigs at k-mer = 39 and 37 in the FAZ and the LAZ samples, respectively. These recorded the highest percentage of assembled reads (Table 1 and Table S2), thereby implying a high coverage for these sequencing data. The generated assembly had total sequences of 33,025 and 41,592, with an average sequence length of ~1233 and 1255 bp, and a minimum sequence length of 100 bp in the FAZ and the LAZ samples, respectively. Ultimately, FASTA files containing the assembled contigs were obtained for the FAZ and the LAZ, respectively. Table S2 presents a general view of the sequencing and assembly processes, which provides the length distribution for these high-quality reads. The statistics for the two assemblies is detailed in Table 1.
Differentially Expressed Gene Analysis for the FAZ and the LAZ
To investigate abscission distinctions, we compared the transcriptomes of tomato FAZ and LAZ using Illumina DEG analysis. Specifically, we analyzed variations in gene expression between the FAZ and the LAZ in pooled samples, which resulted from two independent DEG libraries. Raw reads of the FAZ and the LAZ were aligned to reference cDNA using the Bowtie-0.12.7 program. The expression study was performed based on enrichment calculation of each gene, when enrichment = average read depth × coverage. The total contigs were classified into five groups, based on their differentially expression levels in the pooled LAZ and FAZ samples, expressed by the LAZ/FAZ enrichment ratio, as follows: Group A, contigs over-expressed in the LAZ; Group B, contigs over-expressed in the FAZ; Group C, contigs equally expressed in the FAZ and the LAZ; Group A1, contigs exclusively expressed in the LAZ; Group B1, contigs exclusively expressed in the FAZ. The parameters used for this classification included: Group A, genes which were up-regulated in the LAZ samples compared to FAZ samples with log fold change >1; Group B, genes which were down-regulated in the LAZ samples compared to the FAZ samples, with log fold change <-1; Group C, genes which were equally expressed in both AZs with log fold change ranging between −1 to +1. The results presented in Figure 2 show that out of 22,650 total genes, 4997 genes were classified in Group A, 2899 genes in Group B, 14,754 genes in Group C, 1349 genes in Group A1, and 1259 genes in Group B1. The number of genes expressed in the LAZ pooled sample was approximately twice the number of genes expressed in the FAZ pooled sample. Table S3 presents the detailed lists of genes expressed in each category.
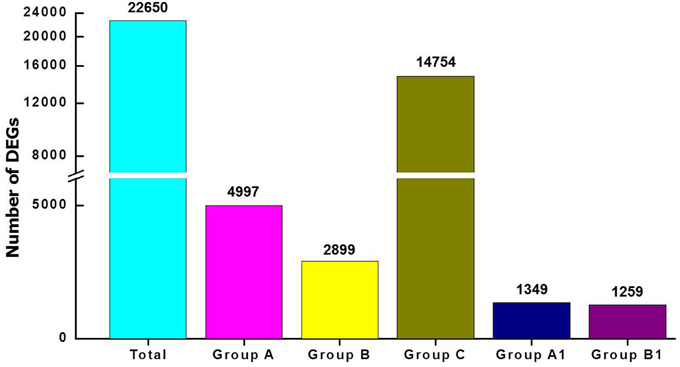
Figure 2. Analysis of differentially expressed genes (DEG) in pooled samples of FAZ and LAZ. The number of transcripts indicated above the bars was obtained after performing the Illumina DEG analysis and dividing into groups. Total, transcripts expressed in both tissues regardless of the expression pattern; Group A, transcripts over-expressed in LAZ samples; Group B, transcripts over-expressed in FAZ samples; Group C, transcripts equally expressed in FAZ and LAZ samples; Group A1, transcripts exclusively expressed only in LAZ samples; Group B1, transcripts exclusively expressed only in FAZ samples. The detailed lists of genes in each category are summarized in Table S2.
Annotation and Gene Ontology Functional Enrichment Analysis
The tomato genome (The Tomato Genome Consortium, 2012) was recently published, and the functional annotation and assignment of GO by the ITAG enabled us to clearly designate the differences in gene expression between the two tomato AZs. However, the complete transcriptome information on the tomato FAZ and LAZ has not yet been determined. Annotation of assembled genes was performed using the Velvet-1.1.05 and Oases assembler programs and subjected to BLAST analysis against the S. lycopersicum mRNA and protein sequences from ITAG2.3 (ftp://ftp.sgn.cornell.edu/genomes/Solanum_lycopersicum/annotation/ITAG2.3_release/). Transcripts showing more than 50% identity and 70% query coverage were taken for further analysis. The average sequence length was 11,840 and 15,794 bp for the FAZ and the LAZ, respectively, with a minimum sequence length of 100 bp. The annotation description of mRNA and proteins is detailed in Table 2. The results indicate that out of 41,592 and 33,025 total contigs generated for the LAZ and the FAZ, respectively, 41 and 49% compared well with the annotated genes and proteins of tomato in the LAZ and the FAZ, respectively. On the other hand, we found that ~58.9 and 50.5% contigs of the LAZ and the FAZ, respectively, failed to map to ITAG2.3 identities of both mRNA and proteins (Table 2). This pool may serve as a good source for discovering new genes. The lists of mRNA-annotated, protein-annotated, mRNA-overlapping, and protein-overlapping contigs are detailed in Tables S4, S5. Once again the assembly was annotated with the recent available genome assembly (The Tomato Genome Consortium, 2012), which resulted in 32,949 and 41,502 transcripts (FASTA file) for the FAZ and the LAZ, respectively (data not shown).
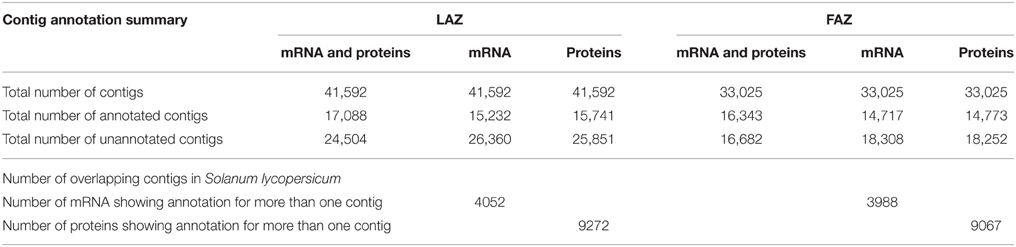
Table 2. Summary of the modules for contig annotation in the LAZ and FAZ.
We have further carried out an enrichment GO analysis for the total annotated proteins (complete list), including 15,741 and 14,773 LAZ and FAZ proteins, respectively (Table 2). Among them, 12,409 FAZ and 13,106 LAZ proteins were designated with at least one GO term (Tables S6, S7). The GO terms of “protein binding,” “oxidation-reduction process,” and “membrane” were the most represented ones among the categories of molecular function, biological process, and cellular component, respectively, in both AZs (Figure 3). The analysis represents the top 10 GO in molecular function, biological process, and cellular component for the FAZ (Figure 3A) and the LAZ (Figure 3B). These data show that both the FAZ and the LAZ share a similar type of gene enrichments when samples of all-time points following abscission induction were pooled together.
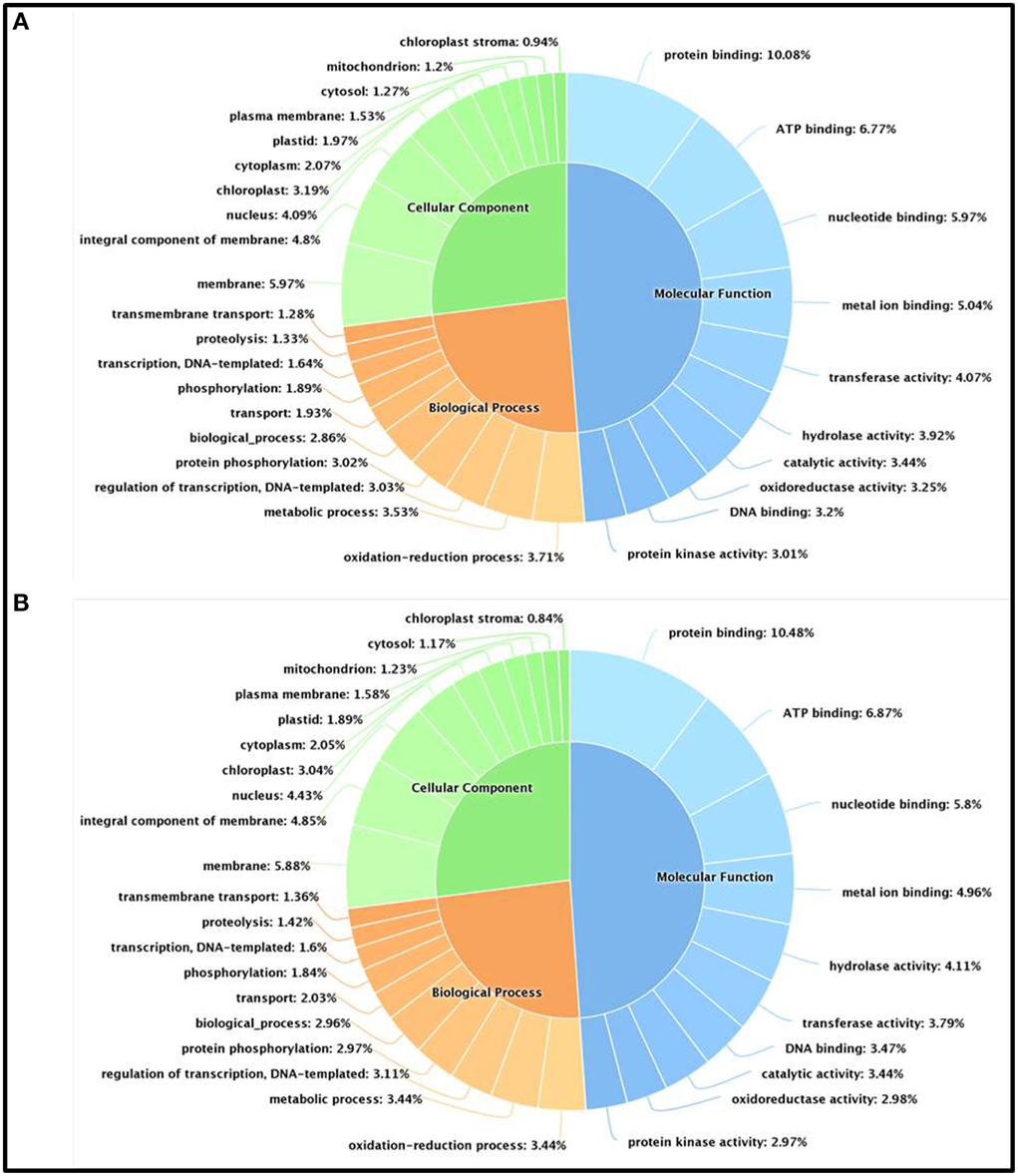
Figure 3. Enrichment Gene Ontology (GO) terms in the FAZ (A) and LAZ (B). The enrichment analysis included 14,773 genes for the FAZ and 15,741 genes for the LAZ. The chart represents the top 10 GO listed in Tables S5, S6 in the categories of Molecular Function (blue), Biological Process (orange), and Cellular Components (green).
In the search of cues for explanation of the different abscission rates of petioles and pedicels, we examined the overexpression categories (Groups A and B) in both AZs, as the complete list resulted in similar types of gene enrichments (Figure 3). Out of 4997 and 2899 annotated contigs found in Groups A and B, respectively, 2066 and 1135 were designated with at least one GO term (Tables S8, S9) in the LAZ and the FAZ, respectively. The GO terms identified in Groups A and B (Figures 4–6) showed the enrichment of GO categories of highly represented contigs, as demonstrated in the complete annotation charts for both AZs (Figure 3).
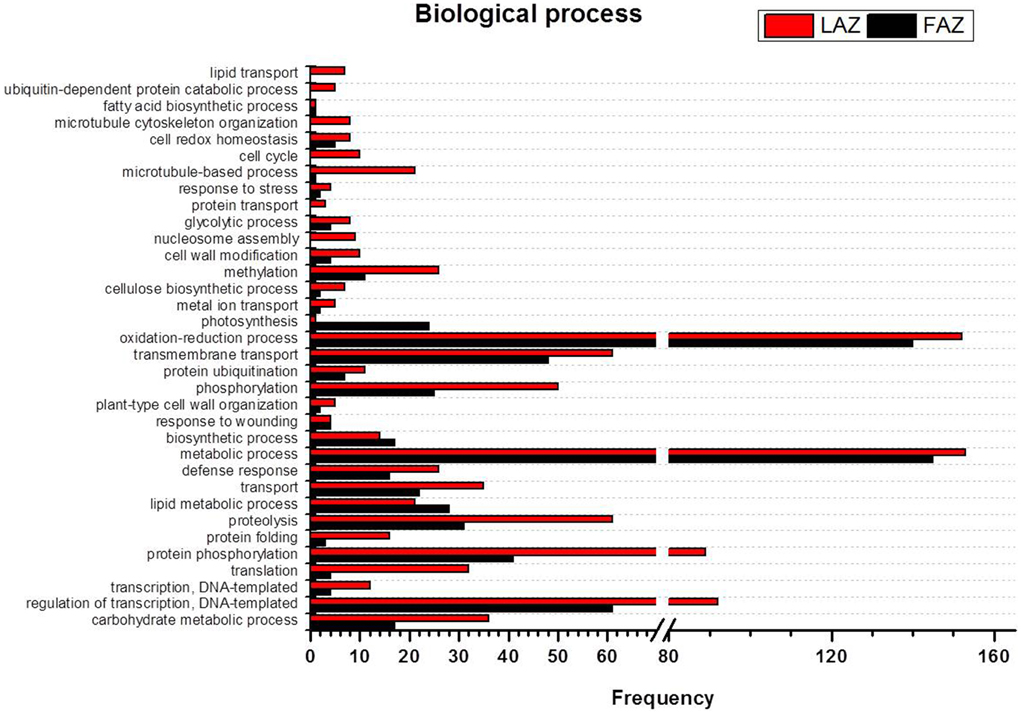
Figure 4. Comparison of frequencies of the GO “biological process” term of over-expressed transcripts in the LAZ (Group A) and FAZ (Group B). The bars represent the comparison of the occurrence frequencies of the GO “biological process” term in the GO annotations of 1135 and 2066 over-expressed transcripts in the FAZ and LAZ, respectively. The frequencies are given for the most abundant biological processes.
The GO terms include indicators of the various biological processes operating in the two AZs during abscission. In the category of biological process, most of the DEG in Group B over-expressed in the FAZ were classified as associated with metabolic processes, oxidation-reduction, regulation of transcription, transmembrane transport, protein amino acid phosphorylation, and proteolyis. Interestingly, Group A (over-expressed in the LAZ) also showed enrichment in a similar list of genes (Figure 4), indicating that the same biological processes might require the operation of the same gene sets in the two AZs during abscission execution. Nevertheless, some differences were found between the two lists of enriched GO terms. In Group A, the GO terms were associated with phosphorylation, transport, carbohydrate metabolic processes, translation, methylation, defense response, microtubule-based processes, protein folding, and cell cycle (Figure 4). This suggests that such biological processes may be associated with leaf abscission. On the other hand, in Group B the enriched GO terms included the processes of lipid metabolism, photosynthesis, and biosynthesis (Tables S8, S9).
In the category of molecular function, the abundant transcripts in Groups A and B showed the predominant expression of genes associated with metal-ion binding, catalytic, transferase, hydrolase activities, and transferring phosphorus-containing groups. Apart of these similar gene categories, the most over-represented GO terms in Group A also included genes associated with protein and ATP binding, while Group B also included genes associated with oxidoreductase activity (Figure 5, Tables S8, S9). Finally, within the category of cellular component, the GO terms of membrane, nucleus, cytoplasm, and integral to membrane constituted the most over-represented categories of the genes with increased transcript accumulation in both Groups A and B (Figure 6).
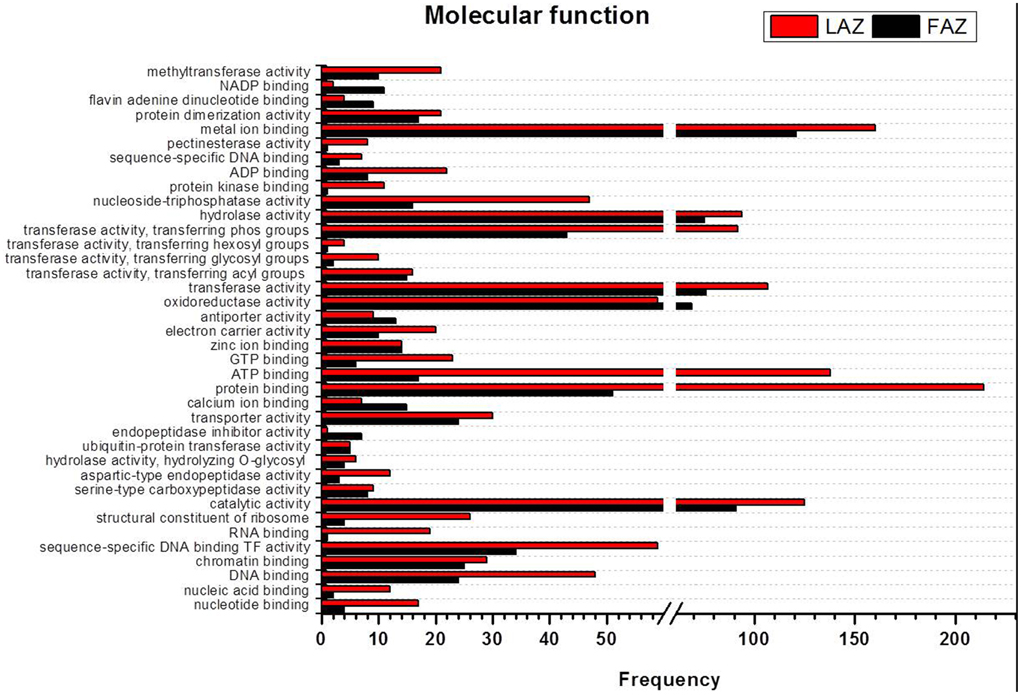
Figure 5. Comparison of frequencies of the GO “molecular function” term of over-expressed transcripts in the LAZ (Group A) and FAZ (Group B). The bars represent the comparison of the occurrence frequencies of the GO “molecular function” term in the GO annotations of 1135 and 2066 over-expressed transcripts in the FAZ and LAZ, respectively. The frequencies are given for the most abundant molecular functions.
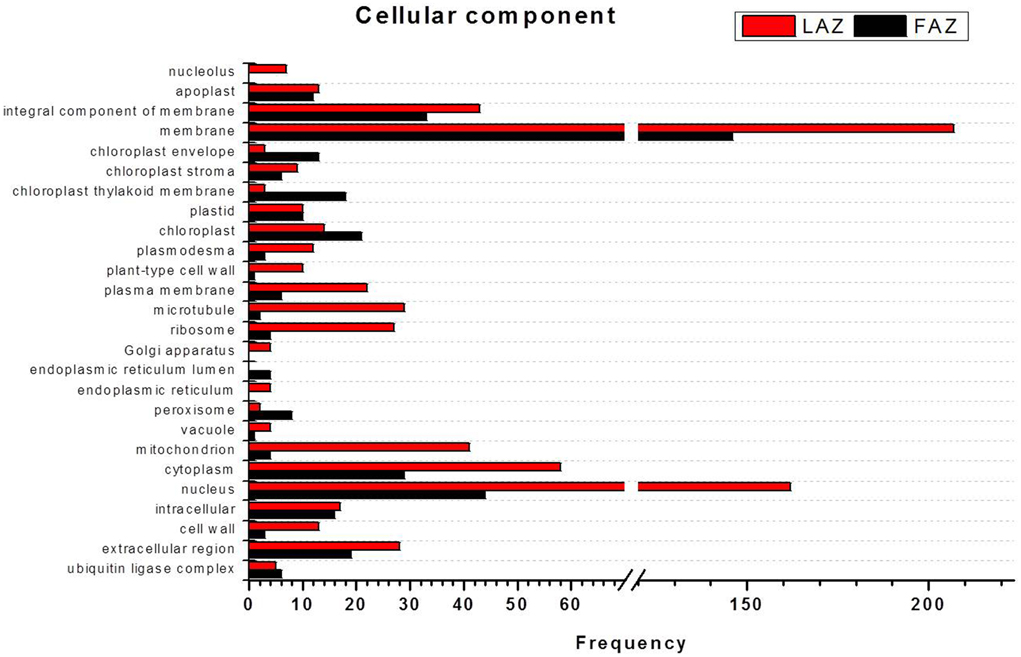
Figure 6. Comparison of frequencies of the GO “cellular component” term of over-expressed transcripts in the LAZ (Group A) and FAZ (Group B). The bars represent the comparison of the occurrence frequencies of the GO “cellular component” term in the GO annotations of 1135 and 2066 over-expressed transcripts in the FAZ and LAZ, respectively. The frequencies are given for the most abundant cellular components.
Differential Regulation of Genes Encoding Transcription Factors in the FAZ and the LAZ
Among the total 7896 differentially regulated genes of Groups A and B, diverse families of genes putatively encoding transcription factors (TFs) were differentially expressed (Table S10) in the pooled samples of the LAZ and the FAZ. Thus, 336 TF genes were expressed in Group A and 215 TF genes were expressed in Group B. Changes in the abundance of these 551 differentially over-expressed TF genes, belonging to the top 20 highly represented TF families, were determined in both the LAZ and the FAZ (Figure 7A). Additionally, diverse families of TF genes were also equally expressed in the FAZ and the LAZ (Group C). Apart from the differentially overexpressed TF genes, we also found exclusively expressed TF genes in each of the AZs. Thus, 71 and 70 TF genes, which belong to various TF families, were specifically expressed in the LAZ and the FAZ, respectively (Group A1, B1) (Table S10, Figure 7B).
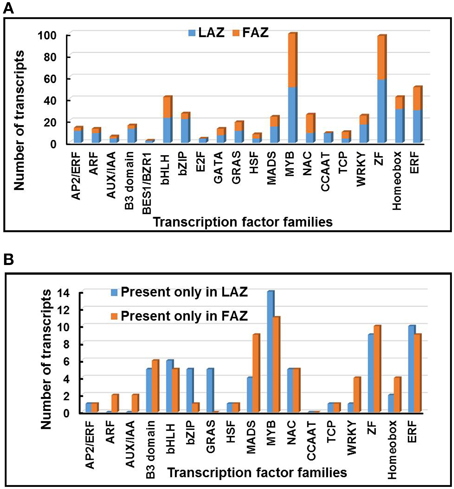
Figure 7. Distribution of abscission-regulated transcription factor (TF) families over-expressed in the LAZ or in the FAZ (A), and exclusively expressed only in the LAZ or FAZ (B) during abscission. The changes in the abundance of 551 TF transcripts belonging to 20 families were determined in graph (A) for Group A (LAZ) and Group B (FAZ). The changes in the abundance of 141 TF transcripts belonging to 20 families were determined in graph (B) for Group A1 (LAZ) and Group B1 (FAZ). The Groups were classified according to the categories presented in Figure 2.
The most abundant TF genes overexpressed in the LAZ (Group A) were: ZINC FINGER (ZF) proteins, MYB, Homeobox domain proteins, Ethylene Responsive Factor (ERF), basic leucine zipper (bZIP), basic helix–loop–helix (bHLH), and WRKY. In the FAZ (Group B) the most abundant TF genes were: MYB, ZF proteins, ERF, bHLH, NAC, Homeobox domain proteins, and MADS box (Figure 7A). The most abundant TF genes found in the exclusively expressed genes in the LAZ (Group A1) included: MYB, ERF, ZF proteins, bHLH, GRAS, bZIP, and B3 domain families, whereas the exclusively expressed genes in the FAZ (Group B1) was enriched with the TF genes of MYB, ZF proteins, ERF, MADS box, and B3 domain (Figure 7B). Although the two sub-groups (A1, B1) contained members of several TF families, in each primary Group (A, B), significant differences were found in the proportion of the TF families. Moreover, distinct TF families were differentially expressed in each Group, including AP2/ERF, Auxin Response Factor (ARF), Aux/IAA, TF E2F, and CCAAT-binding protein families in Group A, and NAC, TCP, GATA TF—Zinc finger GATA-type, and GRAS families in Group B (Figure 7A). The enrichment of sequence elements in different gene groups from each cluster, in combination with the data on transcript abundance offer a tenable set of TFs which bind these elements, and that could be examined in future research.
Our results show that the most abundant TF family in Groups A and A1 or Groups B and B1 was MYB (Figure 7). The MYB gene (Solyc11g069030—Bl) was specifically expressed in the tomato FAZ compared to proximal and distal NAZ tissues (Nakano et al., 2013), and was overexpressed in the LAZ (Group A) as shown by our analysis (Table S10). Several MYB blind-like (bli) genes (bli1,3,4,5,7) (Solyc09g008250, Solyc04g077260, Solyc12g008670, Solyc08g065910, Solyc02g091980, respectively) were also expressed in all groups (Table S10). Our analysis show that a total number of 98 ZF TF genes were overexpressed in both AZs, being the second most highly expressed family in Groups A and B (Figure 7A, Table S10).
The MADS-box and GRAS TFs gene families were expressed in both AZs represented by Groups A and B (Figure 7A, Table S10). While the GRAS family was highly expressed in Group A1, the MADS-box family was much more highly expressed in Group B1 (Figure 7B, Table S10). The MADS-box and GRAS TF genes, MACROCALYX (Solyc05g012020), JOINTLESS (Solyc11g010570), SEPALLATA MADS-box Protein21 (SLMBP21), and Ls (Solyc07g066250), which were shown to regulate the differentaion and development of the tomato FAZ (Schumacher et al., 1999; Mao et al., 2000; Nakano et al., 2012; Liu et al., 2014), were expressed in both the FAZ and the LAZ (Table S10). The MACROCALYX gene was highly expressed in the tomato FAZ (Group B), whereas the JOINTLESS gene was expressed at a similar level in both the FAZ and the LAZ (Group C) (Table S10). Therefore, we speculate that a similar type of organ identity specification, reported for the FAZ, might also operate in the LAZ.
The MADS-box gene Tomato AGAMOUS-LIKE12 (TAGL12), which is known to be expressed during tomato seed and fruit development (Busi et al., 2003), was upregulated in the FAZ after flower removal (Meir et al., 2010). Our analysis data show that TAGL12 was equally present in both the FAZ and the LAZ (Group C), while TAGL2 (Solyc02g089200—LeSEP1) was highly over-expressed in the FAZ (Group B) compared to the LAZ (Group A) (Table S10), in accordance with our previous report (Meir et al., 2010). Several GRAS TFs, including GRAS2 (Solyc07g063940), GRAS5 (Solyc09g018460), GRAS7 (Solyc07g065270), and GRAS9 (Solyc06g036170), were exclusively over-expressed in the FAZ (Group B), whereas GRAS4 (Solyc01g100200) was overexpressed in the LAZ (Group A) (Table S10). This suggests that different GRAS TF family members probably mediate abscission-responsive transcription in both flowers and leaves.
The WRKY TF family identified in multiple crop species, was implicated to operate in various biological processes in plants, especially in regulating defense mechanisms against biotic and abiotic stresses (Rushton et al., 2010). So far, 137, 89, and 81 WRKY genes were identified in rice, Arabidopsis, and tomato, respectively (Zhang et al., 2011; Huang et al., 2012). Our analyses revealed that 17 and 8 WRKY genes were overexpressed in the tomato LAZ and FAZ, respectively (Table S10). These results are consistent with previous studies showing upregulation of several WRKY TF genes in fruit AZ during abscission of mature melon and olive fruit (Corbacho et al., 2013; Gil-Amado and Gomez-Jimenez, 2013). WRKY1 and WRKYIId-1 (AY157063) genes were upregulated in the tomato FAZ at the early and late stages of pedicel abscission (Meir et al., 2010). In the present study, we show that the WRKYII (Solyc01g079360) gene was expressed in both tomato AZs, but was highly expressed in the FAZ (Group B) compared to the LAZ (Table S10). This suggests that the WRKY TFs have a role in both AZs in mediating the late events of the abscission process.
Recently, 159 and 152 bHLH TFs genes were identified in the tomato genome (Sun et al., 2015; Wang et al., 2015). Out of the 159 identified bHLH genes in tomato, we could detect 137 bHLH TFs in both AZs, which were distributed among all the groups presented in Figure 2, indicating that this TF family is associated with the abscission process. Out of these 137 AZ-associated bHLH genes, 30 genes were expressed in Group A, 28 genes in Group B, 67 genes in Group C, and 7 and 5 genes were exclusively present in the LAZ (Group A1) and the FAZ (Group B1), respectively (Figure 7, Table S10). Most of the bHLH TFs were overexpressed in both the FAZ and the LAZ (Group A and B) (Table S10).
Most of the bZIP TFs genes (22) were present and ove-expressed in the LAZ (Group A), compared to only five genes in the FAZ (Group B) (Figure 7A, Table S10). In the exclusively expressed categories, five bZIP TF genes were present in the LAZ compared to one gene in the FAZ (Figure 7B, Table S10). One of the bZIP TF gene (BG631669) was reported to be downregulated in the FAZ at an early stage of tomato pedicel abscission (Meir et al., 2010). Most members of the AP2/ERF, B3 domain, bHLH, bZIP, MADS, MYB, WRKY, ZF, Homeobox, and Ethylene Responsive Factor (ERF) gene families were overexpressed in the tomato LAZ samples compared to the FAZ, while most members of the NAC families were less expressed in the LAZ samples (Group A) (Figure 7).
In the present study, we identified common and distinct TFs that were not previously related to abscission. Our results also show that distinct patterns of transcriptional regulation occur in the tomato FAZ and LAZ.
Key Meristem Genes in the AZs
A long list of shoot apical meristem (SAM) genes were similarly expressed in both the LAZ and the FAZ (Group C) and in the differentially regulated groups (Group A and B) (Table S3). The key Arabidopsis SAM activity genes and their orthologs were preferentially expressed in tomato FAZ (Nakano et al., 2012; Wang et al., 2013). Our data show that the well-documented key meristem genes, KNOTTED2 protein/Tkn3/KNAT6 (Solyc05g005090), BEL1-like homeodomain protein4 (TBL4)/BELL-like homeodomain protein3 (BLH) (Solyc08g065420), along with the LATERAL ORGAN BOUNDARIES DOMAIN PROTEIN1 (LBD1) (Solyc11g072470), and the tomato auxiliary meristem gene, Bl (Solyc11g069030), which encodes a MYB TF, were highly expressed in the tomato LAZ (Group A) compared to the FAZ (Table 3). On the other hand, the WUSCHEL-related homeobox-containing protein4 (WUS) gene (Solyc02g083950), OVATE FAMILIY PROTEIN (OFP) gene (Solyc02g085500), another MYB-Cpm10/MYB78 gene (Solyc05g053330), Goblet (Solyc07g062840), a NAC domain TF gene, and the Ls (Solyc07g066250), a GRAS family TF gene, were highly expressed in the tomato FAZ compared to the LAZ (Group B) (Table 3).
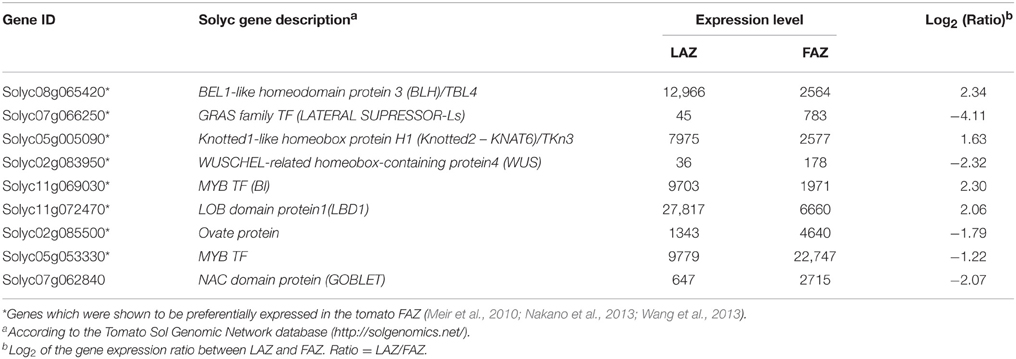
Table 3. Differential expression patterns of shoot meristem genes in the tomato LAZ and FAZ.
Cell Wall Related Genes in the FAZ and the LAZ
Our RNA-Seq analyses show that several genes encoding cell wall and middle lamella degradation and remodeling factors, which are the main targets at the late stages of the abscission process, were expressed in both the tomato FAZ and LAZ. These include PG, Cel, XTH, and EXP genes (Table 4). These genes were previously demonstrated to be highly expressed in the AZs of a large number of abscising organs (Lashbrook et al., 1994; del Campillo and Bennett, 1996; Cho and Cosgrove, 2000; Taylor and Whitelaw, 2001; Roberts et al., 2002; Ogawa et al., 2009; Meir et al., 2010). The previously characterized tomato AZ-specific PG genes (TAPGs), TAPG1,2,3,4,5, were expressed in both the FAZ and the LAZ, with TAPG4 expression being the highest and TAPG3 expression being the lowest in both AZs (Table 4). These results are in agreement with previous works showing that TAPG1,2,4 were specifically expressed in tomato FAZ (Lashbrook et al., 1994; Kalaitzis et al., 1997; Wang et al., 2013), and their expression was inhibited by 1-methylcyclopropene (1-MCP) in a positive correlation with its inhibitory effect on pedicel abscission (Meir et al., 2010).
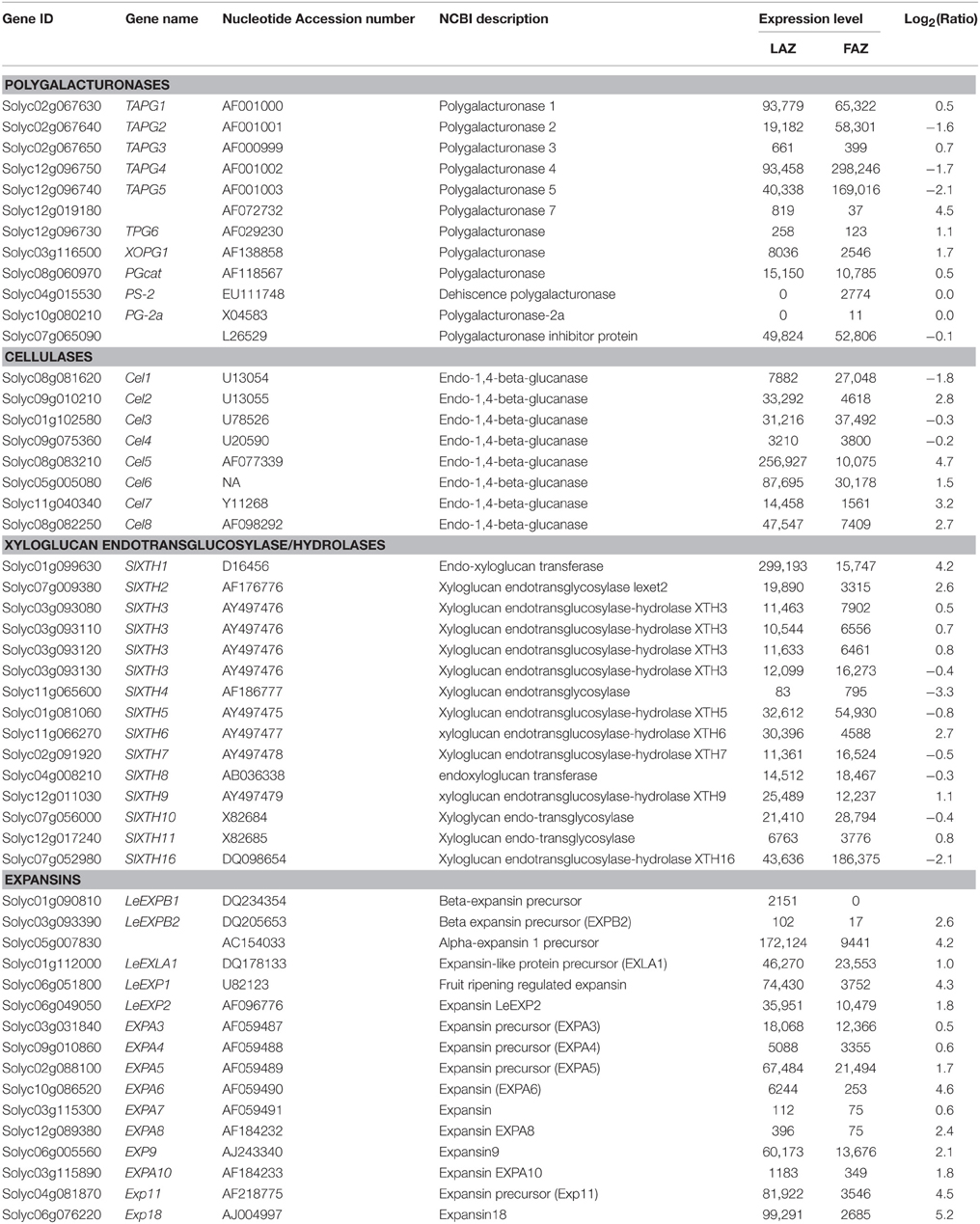
Table 4. Differential expression patterns of cell wall-related genes in the tomato LAZ and FAZ.
The expression levels of genes encoding cellulase enzymes, such as Cel1,2,3,4,5,6,7,8, are detailed in Table 4. Cel2,5,6,7,8 were highly expressed in the LAZ (Group A), with Cel5 showing the highest expression among all the Cel family genes in both AZs (Table 4). Only the expression of Cel1 was higher in the FAZ (Group B) than in the LAZ, while Cel3 and Cel4 had similar expression levels in both AZs (Table 4). The expression levels of several XTH genes, SlXTH1,2,6,9, was higher in the LAZ than in the FAZ samples, while the other genes of this family were similarly expressed in both AZs (Table 4). It was previously reported that XTH8,9 were highly and specifically expressed in tomato FAZ following ethylene treatment (Wang et al., 2013), and XTH6 was specifically up-regulated in the tomato FAZ following flower removal, and this increased expression was inhibited by 1-MCP pretreatment (Meir et al., 2010). XTH1,2 genes were also over-expressed in the LAZ during ethylene-induced citrus leaf abscission (Agustí et al., 2008). These results further confirm the role of ethylene in inducing abscission via increased expression of XTH genes in both the LAZ and the FAZ.
In general, all the members of the EXP gene family, which were expressed in both tomato AZs, were highly upregulated in the LAZ compared to the FAZ (Table 4). The expression level of several EXP genes, LeEXP1,5,9,11,18, was particularly high in the LAZ.
Differential Regulation of Genes Associated with Hormonal Signal Transduction
The RNA-Seq data for both the FAZ and the LAZ samples were analyzed for the KEGG pathway database to examine the potential involvement of consensus sequences in hormonal signal transduction pathways. The pathway-based analysis helped us to understand the biological functions and their interactions (Figures S3, S4). The KEGG categories and list of transcripts in each sample with their expression values in the LAZ and the FAZ are listed in Table 5. The genes related to the different plant hormones, auxin, ethylene, jasmonic acid (JA), abscisic acid (ABA), brassinosteroids, cytokinins, and gibberellins (GA), and other signaling-related factors (protein kinases), were present in both AZs (Table 5). Most of the genes were equally expressed in both AZs (Group C), but some genes of each hormone category were differentially expressed in the LAZ or the FAZ, belonging to Groups A or B, respectively (Figure 2). The data suggest specific roles for this family members in each AZ.
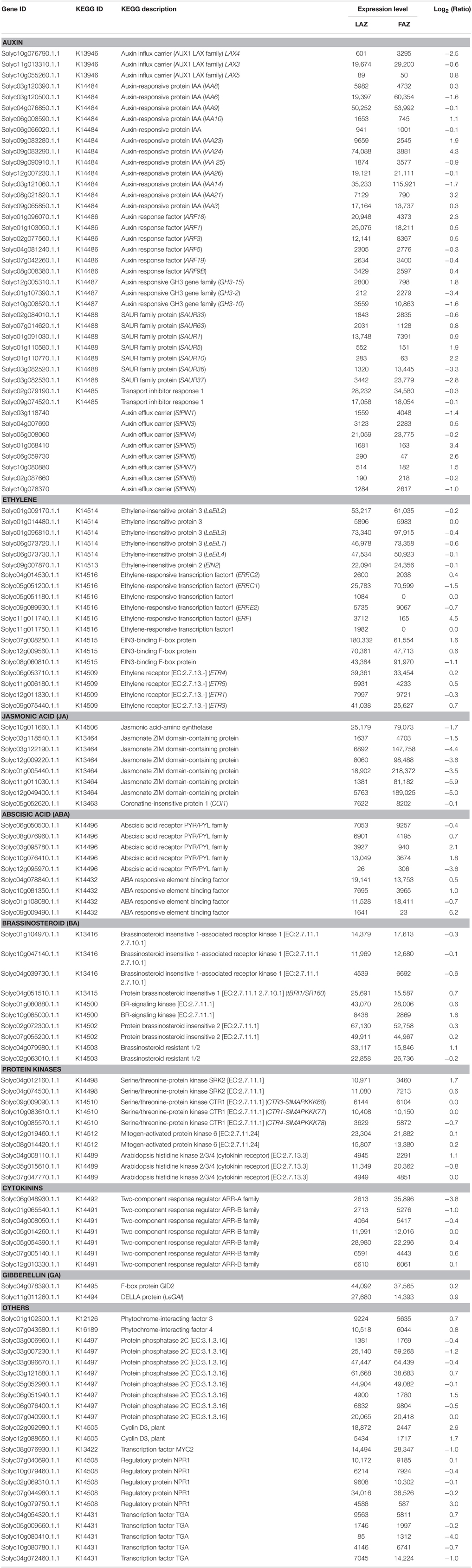
Table 5. The KEGG categories and their expression values for plant hormone signaling-related genes in the tomato LAZ and FAZ.
Auxin
The results presented in Table 5 show that 19 auxin-related genes had a higher expression level in the FAZ (Group B), and 20 auxin-related genes had a higher expression level in the LAZ (Group A). This included auxin responsive genes, such as Aux/IAA, Gretchen Hagen3 (GH3), and Small Auxin Upregulated RNA (SAUR), auxin response factor (ARFs) genes, and auxin transport-related genes, such as Like Auxin (LAX) influx carriers and Pin-formed (PIN) efflux carrier genes. Aux/IAA proteins are negative repressors which bind to the Auxin-Responsive Elements (AREs) of the target gene promoters, leading to activation or repression of the target genes, and their degradation is promoted by auxin (Worley et al., 2000; Overvoorde et al., 2005). The members of the tomato Aux/IAA gene family were differentially regulated in the tomato FAZ and LAZ (Table 5). The SlIAA6,9,25,26,14 genes were overexpressed in the FAZ, with SlIAA14 showing the highest expression among this family members. On the other hand, SlIAA8,10,23,24,21,3 were overexpressed in the LAZ, with SlIAA24 showing the highest expression level among this family members. The Aux/IAA1,3,4,7,8,9,10 genes were downregulated in tomato FAZ and served as good markers for auxin depletion after flower removal (Meir et al., 2010). Similarly, CitAux/IAA3,4,18,19 genes were downregulated in citrus fruit AZ during fruitlet abscission (Xie et al., 2015). Genetic mutation and expression analysis demonstrated that ARF genes could regulate plant organ abscission (Ellis et al., 2005; Guan et al., 2014). The results presented in Table 5 show that SlARF18,1,3,9B genes were over-expressed in the LAZ, whereas SlARF5,19 genes were over-expressed in the FAZ. The expression of SlARF1 was the highest in both AZs compared to the other family members of this gene.
The tomato homolog of METHYLESTERASE1 (MES1) (AK328818, Solyc03g070380) was highly expressed in the LAZ (Group B) compared to the FAZ (Group A), whereas the DWARF IN LIGHT1 (DFL1)/auxin-inducible GH3.9 gene (AK319847, Solyc07g063850) was expressed equally at very high levels in both AZs (Group C) (Table S3). MES1 converts the storage form of IAA to the active free form, whereas DWARF IN LIGHT1 does the opposite, i.e., it converts the active free IAA to the inactive conjugated form (Staswick et al., 2005; Woodward and Bartel, 2005; Yang et al., 2008; Ludwig-Müller, 2011). In addition, conjugation of IAA to amino acids provides a negative feedback loop for controlling auxin homoeostasis, and the SlGH3-2,10,15 genes, which control IAA conjugation, were found to respond quickly to exogenous auxin application (Kumar et al., 2012; Meir et al., 2015). Our KEGG analysis revealed that SlGH3-2,10 genes were overexpressed in the FAZ, whereas SlGH3-15 was over-expressed in the LAZ (Table 5).
The auxin influx and efflux transporters PIN and AUX/LAX proteins mediate the auxin polar transport (Vanneste and Friml, 2009), resulting in directional auxin flow and creation of auxin gradients (Bainbridge et al., 2008; Petrásek and Friml, 2009). The auxin influx carrier genes, SlLAX3,4, were overexpressed in the tomato FAZ compared to the LAZ, whereas SlLAX5 was expressed at low levels in both AZs, but was comparatively expressed higher in the LAZ (Table 5). The auxin efflux carrier genes, SlPIN1,4,9 were over-expressed in the FAZ compared to the LAZ, whereas SlPIN3,5,6,7 were over-expressed (by ~2- to 10-fold) in the LAZ compared to the FAZ (Table 5). Reduced auxin levels were attributed to increased activity of auxin efflux transporters in Arabidopsis and other systems (Sorefan et al., 2009; Meir et al., 2015).
Ethylene
Many genes related to different steps of the ethylene signaling transduction pathway were expressed in both the tomato AZs following flower removal and leaf deblading (Table 5). Out of six genes encoding for ethylene receptors in tomato (Klee, 2002, 2004), four genes were presented in both AZs. Among them, ETR3,4,5 were expressed at very high levels in the LAZ, whereas ETR1 was expressed higher in the FAZ (Table 5). These data confirm previous results demonstrating the expression of ETR1,4 genes in tomato FAZ during pedicel abscission (Payton et al., 1996; Meir et al., 2010). The Constitutive Triple Response1 (CTR1) gene product acts downstream of the ethylene receptors, and belongs to the Arabidopsis RAF Mitogen-Activated Protein Kinase Kinase (MAPKKK) subfamily, which negatively regulates ethylene signal transduction (Kieber et al., 1993). Recent studies of genome-wide analysis of the Mitogen-Activated Protein Kinase (MAPKK) and MAPKKK gene families in tomato revealed five and 89 genes, respectively (Wu et al., 2014). The predicted transmitter domain of ETR1 and the regulatory domain of CTR1 were found to interact directly (Clark et al., 1998; Huang et al., 2003; Binder, 2008). Our analysis data show that three CTR genes, CTR1-SlMAPKKK77, CTR3-SlMAPKKK68, and CTR4- SlMAPKKK78, were expressed in both AZs, with CTR1 showing the highest expression (Table 5). All the CTR genes identified in this study belong to the RAF MAPKKK sub-family according to the classification detailed previously (Wu et al., 2014).
Downstream ethylene signaling events are mediated by ERFs, which are plant-specific TFs which belong to the large AP2/ERF super-family (Riechmann et al., 2000), containing a cis-acting ethylene-responsive element named the GCC-box in their promoter regions (Fujimoto et al., 2000). The GCC-box interacts with trans-acting factors termed ethylene-responsive element-binding proteins, which are required for ethylene regulation in many plant species (Ohme-Takagi and Shinshi, 1995). Our data show that SlERF.C1 and SlERF.E2 genes were highly expressed in the FAZ, while SlERF.C2 and SlERF (Solyc11g011740) genes were highly expressed in the LAZ (Table 5). Interestingly, two other ERF genes (Solyc05g051180, Solyc11g011750) were present only in the LAZ (Group A1) (Table 5). The ERF1c gene (AY044236, SlERF.C1) was proposed to be involved in the late stages of flower pedicel abscission (Meir et al., 2010). Several other tomato ERF genes, SlERF52 (Solyc03g117130), SlERF56 (Solyc09g066360), SlERF68 (Solyc08g078180), ERF1 (AF502085.1, SlERF.E2), and ERF2 (AI776626, Solyc09g089910), were overexpressed in both the FAZ and the LAZ (Table S3).
Ethylene Insensitive2 (EIN2) was reported to act downstream of CTR1 as a positive regulator of the ethylene signaling pathway (Alonso et al., 1999). LeEIN2 was expressed in both the FAZ and the LAZ at similar levels (Table 5). EIN3, which acts downstream of EIN2, belongs to a multi-gene family designated as Ethylene Insensitive-Like (EIL) in tomato. EIN3 encodes a downstream component of the ethylene signal transduction pathway, which ultimately activates ethylene-responsive genes (Ecker, 1995; Roman et al., 1995). LeEILs are functionally redundant and positive regulators of multiple ethylene responses throughout plant development. (Tieman et al., 2001). Our data show that all the EIL genes, LeEIL1,2,3,4, were expressed at high levels in both AZs, with the highest levels in the FAZ (Table 5).
Other Hormones
Several genes involved in pathways related to other phytohormones were expressed in both the FAZ and the LAZ (Table 5). Thus, a gene involved in JA perception and signaling COI1 was highly expressed in the FAZ (Table 5). This gene encodes an F-box protein, which is required for JA signaling in Arabidopsis (Xie et al., 1998). A gene involved in brassinosteroid biosynthesis and signaling encoding for the BRI1 protein—tBRI1/SR160 (Koka et al., 2000; Montoya et al., 2002), showed a high expression in both the FAZ and the LAZ, with the highest level in the LAZ (Table 5). A gene involved in GA signaling, LeGAI (Martí et al., 2007; Bassel et al., 2008), also showed a higher expression in the LAZ compared to the FAZ (Table 5).
Designing and Preparation of an AZ-Specific Microarray for Transcriptomic Abscission Research in Tomato
The above comparisons between gene expression in the FAZ and the LAZ were based on samples pooled from individual samples taken at the different time points during pedicel and petiole abscission following abscission induction (Figure 1). The pooled samples limit the identification of specific gene expression profiles at the timing of the spatial events of the abscission process. To perform an analysis that enables to reveal the sequences of the molecular events involved in petiole and pedicel abscission following induction by artificial auxin depletion (IAA source removal), it is necessary to develop a low-cost method than the RNA-Seq, but with comparable robustness, to perform transcriptomic analyses. For this purpose, we developed an AZ-specific microarray based on the RNA-Seq results.
The RNA-Seq analysis of the tomato FAZ and LAZ samples revealed a total number of 40,959 transcripts, including 31,298 transcripts analyzed for sequence similarity to the known database of sequences, 8823 novel tomato transcripts (novel ORF predictions), and 838 transcripts annotated with A. thaliana and N. tabacum (Table 6). The NGS annotated and the novel transcripts from the tomato FAZ and LAZ, together with sequences of known abscission-related genes originated from various sources detailed in Table 6, were used to design the AZ-specific microarray chip. We included all these available transcripts to enrich this customized microarray chip with previously reported AZ-related genes. As a result, the eArray was finalized with 100,276 probes for the 41,315 transcripts (Table 6).
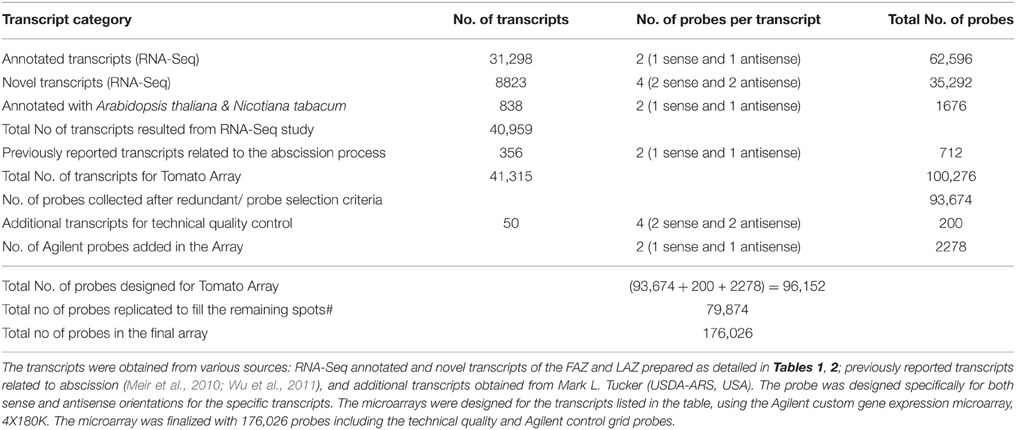
Table 6. Probe design summary for the AZ-specific microarray chip.
A 4x180K gene expression array was designed with the probes having 60-mer oligonucleotides from annotated and novel transcriptome sequenced data and gene sequence related to Nicotiana tabcum and A. thaliana. The 4x180K array comprised of 180,880 features including 176,026 probes and 4854 Agilent controls. All the oligonucleotides were designed and synthesized in situ according to the standard algorithms and methodologies used by Agilent Technologies for 60 mer in situ oligonucleotide DNA microarray (Table 6). The probes were designed in both sense and antisense orientations and with multiple probes for each transcripts (Table 6, Figure S5). Blast analyses were performed against the complete set of sequence databases to check the specificity of the probes. Finally, 96,152 probes were designed, and 79,874 specific probes were duplicated to fill the remaining spots (Table 6). The detailed list of transcripts, probes, and cross hybrid probe details is presented in Table S11. The designed AZ-specific microarray chip (AMADID: 043310; Genotypic Technology Private Limited, India) is now available from the company for abscission research upon our approval.
Validation of Differentially Regulated Genes in the LAZ and the FAZ
To verify the results of the RNA-Seq analyses, we characterized the expression patterns of nine arbitrarily selected genes, which were differentially regulated in the LAZ and the FAZ, using quantitative reverse transcription-PCR (qRT-PCR). We randomly selected the genes from the four differentially expressed groups, Group A, Group A1, Group B, and Group B1 (Figure 2). In the tomato AZs, the expression levels of the auxin-related genes, SlIAA24, SlARF18, GH3-15, and NPR1-like protein (Solyc10g079750) mRNAs, highly increased in the LAZ (Group A) (Figures 8A–D). In Group A1, the LOB domain proteins (Solyc02g086480), Peroxidase1 (Solyc10g076210), 1-aminocyclopropane-1-carboxylate oxidase (Solyc06g060070), were exclusively expressed in the LAZ (Figures 8G–I). The auxin influx carrier gene SlLAX4 (Solyc10g076790) was highly expressed in the FAZ (Group B) (Figure 8F). In the group of genes exclusively expressed in the FAZ (Group B1), a MYB TF, SlMYB21 (Solyc02g067760) was highly expressed, but it also exhibited a low expression in the LAZ (Figure 8E), which was not spotted by the RNA-Seq analysis. The qPCR performed in this study for the selected genes confirms of the RNA-Seq data. The slight variations obtained between the qRT-PCR and RNA-Seq results are due to the newly pooled samples used for the qRT-PCR, similar to the method used for the RNA-Seq analysis. It seems, therefore, that in general the qRT-PCR technique validated the RNA-Seq data of expression patterns of all the selected transcripts in pooled samples of the LAZ and the FAZ during the abscission process.
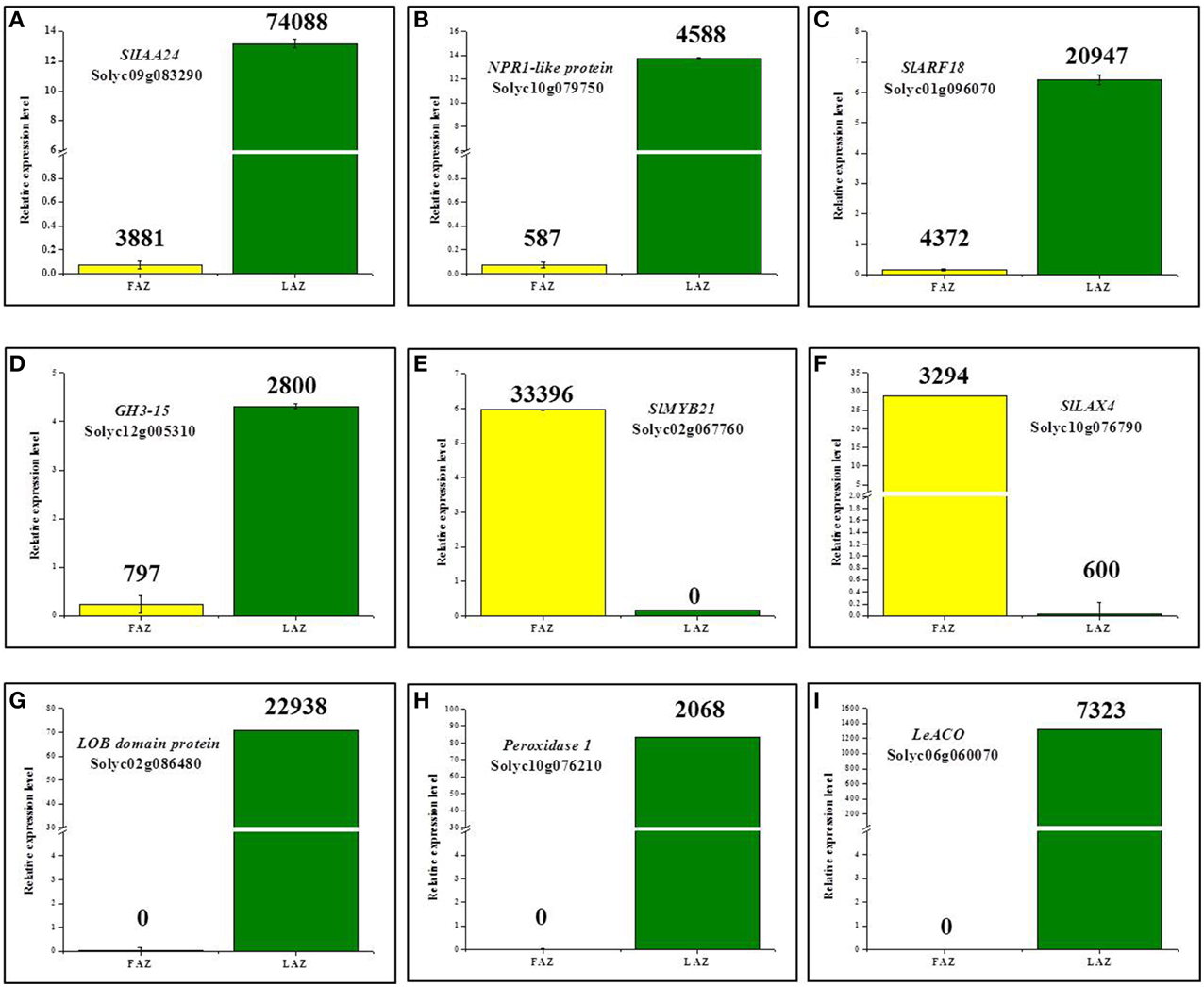
Figure 8. Validation by qPCR of differential expression patterns of selected genes in the FAZ and LAZ pooled samples following abscission induction. Expression levels were measured for tomato Aux/IAA24 (SlIAA24) (A), NPR1-like protein (B), Auxin response factor18 (SlARF18) (C), Gretchen Hagen3-15 (GH3-15) (D), SlMYB21 (E), Like Aux4 (SlLAX4) (F), LOB domain protein (G), Peroxidase1 (H), and 1-Aminocyclopropane-1-Carboxylate-Oxidase (LeACO) (I). The relative quantification of the gene expression level in the qPCR assay was determined by the comparative CT method 2−ΔΔCT, using ACTIN as a reference gene. The results are means of three biological replicates ± SD. The values presented on top of each bar indicate the expression levels derived from the RNA-Seq data. Transcript identities are indicated in the graphs by their gene ID. The qPCR and RNA-Seq analyses were performed with different samples taken from independent biological replicates of two separate experiments.
Discussion
The present study was based on examination of the effects of auxin depletion, obtained by organ removal, on the molecular changes occurring during the abscission process in tomato FAZ and LAZ (Figure 1). Quantitative measurements of the changes in endogenous auxin content following flower removal were already performed in tomato FAZ, and indeed showed a significant reduction in auxin content with time (Guan et al., 2014). Basically, our study included two parts: (1) Analysis of the RNA-Seq transcriptome with the pooled samples strategy and the preparation of the customized AZ-specific microarray based on it; (2) Use of the transcriptome data for comparative analysis of global gene expression in the tomato FAZ and LAZ. The customized AZ-specific chip was further used for specific analyses at each time point with the appropriate replicates. Part of the microarray results was recently published elsewhere (Kim et al., 2015; Meir et al., 2015), and part are still being analyzed. Hence, the present study does not include the microarray data, as its main aim was to expose the occurrence of global changes.
Transcriptome Assembly and Design of an AZ-Specific Array
Transcriptome studies were previously performed in the tomato FAZ and flower NAZ using microarrays (Meir et al., 2010, 2011; Nakano et al., 2012, 2013; Wang et al., 2013; Ma et al., 2015). However, a comparative transcriptome profiling of the FAZ and the LAZ was not performed, and no comprehensive cell separation stages in the FAZ (0–14 h after flower removal) and the LAZ (0–96 h after leaf deblading) were addressed by RNA–Seq. The aim of the present research was to study the transcriptome of tomato FAZ and LAZ pooled samples of the various time points, using Illumina PE sequencing and de novo assembly to permit assessment of expression across the entire genome. In addition, the data generated were used to create an AZ-specific microarray chip that enables to study the specific abscission stages. We have utilized both the de novo as well as the reference-based assemblies of tomato transcriptome to generate a more comprehensive and unbiased representation of the FAZ and the LAZ pooled samples during tomato flower and leaf abscission process. Stringent categories were applied for both the generated de novo assembly (BLAST with 50% identity and 70% query coverage) (Tables S5, S6) and the reference-based assembly (BLASTN with E-value of e−5). It should be noted, that after the genome alignment with the current tomato reference genome, the analysis revealed only 76 and 90 unannotated transcripts (FASTA file) for the FAZ and the LAZ, respectively. These results suggest that the sequences generated are of tomato genes rather than sequences of non-coding RNAs, UTRs, unannotated introns, or contaminated sequences from other organisms, such as fungi, bacteria, and viruses. The assembly statistics revealed that the merged assembly had better characteristics, such as higher N50 value, average, and longest contig length, total number of contigs, and etc., which usually serve as hallmarks to assess the assembly quality (Table S2). The coverage of the transcriptome was comprehensive enough to discover major genes of several metabolic and regulatory pathways. Particularly, genes associated with plant hormonal signaling components could be mapped to their relevant pathways.
We used the strategy of transcriptome sequencing (RNA-Seq) of pooled samples (RNA pooled equally from all time points), since there are specific transcripts of various transcriptional networks and genes of various functions which are induced at specific time points in the AZ (Meir et al., 2010). The pooling strategy ensures that a given transcript will be present in abundance during the RNA-Seq analysis, rather than analyzing it at a specific time point in which it might not be highly expressed. In the present study, we compared the global transcriptomes generated from the FAZ and the LAZ rather than performing a direct quantitation of the expressed genes in each abscission stage. The differential expression levels described in this study are related to the fold changes of the transcripts in the FAZ and the LAZ pooled samples. However, by pooling samples from different time points during the abscission process we lost the resolution to detect dynamic and specific changes in gene expression at specific time points. Hence, for the differential expression analysis, we utilized the transcriptome data to generate an AZ-specific microarray chip, which allowed a cost effective way to detect all the dynamic changes in gene expression at all stages of the abscission process. We had to design this AZ-specific chip, since the commercial Affymetrix Tomato Gene Chip used in our previous studies (Meir et al., 2010) contained only about 10,000 genes, and many AZ-specific genes were not included in this array. Transcriptome sequencing data were previously utilized to create customized microarrays suitable for various research needs in Syrian golden hamsters (Ying et al., 2015) and Camellia sinensis (Wang et al., 2014a). Customized microarrays are used for global transcriptome analysis in various organisms/systems (Lipovich et al., 2014; Yasuike et al., 2015), and strand-specific customized arrays were designed for studying NATs in the Human genome (Yelin et al., 2003) and mouse (Kiyosawa et al., 2005).
The additional novelty of our AZ-specific microarray is that the probes were designed in both sense and antisense orientations, and multiple probes were used for each transcripts (Table 6, Figure S5). Thus, this developed tomato AZ-specific chip contains much more transcripts than any other commercial microarray chip. Therefore, it can serve as an excellent means to further study the tomato AZ transcriptome. We have utilized the AZ-specific microarray chip (GSE45355; GSE45356; GSE64221) for the transcriptome analysis of several genes in the tomato FAZ and LAZ samples. Part of these analyses at specific time points following abscission induction, which represent various abscission stages, was already published. These publications showed changes in expression of specific groups of tomato genes, such as auxin-related genes in the FAZ and LAZ (Meir et al., 2015), and genes associated with cell wall, boundary layer, pathogen-related, and lipid transport in the FAZ and NAZ (Kim et al., 2015; Meir et al., 2015). Regarding the clear advantages of this AZ-specific microarray, these recent studies enabled the exploration of genes responsible for abscission induction, execution and synthesis of the defense layer processes in the tomato LAZ and FAZ.
The rapid technological progress in NGS, which led to the generation of the Strand-specific RNA-Seq, can provide a bp resolution. Thus, several cheap library construction protocols are already available (Zhong et al., 2011), and can be utilized for similar research needs. Our choice of a customized microarray platform was driven by cost advantages relative to RNA-Seq, for robustness of its own kind with well-standardized methods, and by the known inadequacies of commercial microarray platforms, which under-represents genomic complexity.
Annotation Analysis Characterizes Abscission as a Dynamic Process
The GO terms of “protein binding,” “oxidation-reduction process,” and “membrane” were the most represented ones among the categories of molecular function, biological process, and cellular component, respectively, in both tomato AZs (Figure 3). The data show that both the FAZ and the LAZ share a similar type of gene enrichments in all the three categories of biological process, molecular function, and cellular component, with only few differences, when samples of all-time points following abscission induction were pooled together. These annotations of the over-expressed categories provide a useful basis for studying gene functions, cellular structures, and processes in the two examined AZs (Tables S8, S9). Our data are consistent with the data of the Arabidopsis stamen AZ (Cai and Lashbrook, 2008) and olive fruit AZ transcriptomes (Gil-Amado and Gomez-Jimenez, 2013) in the category of cellular component, where the GO term “membrane” is the most represented. In the olive fruit AZ transcriptome, the most represented term in the category of biological process was metabolic process; whereas in the tomato FAZ and LAZ our data show that the terms of “oxidation-reduction process” and “metabolic process” were represented at similar levels (Figure 3). In the laminar AZ of citrus leaves, the most important GO terms represented cell organization, biogenesis/metabolic process, metabolism of fatty acids, carbohydrate metabolism, response to biotic and abiotic stimuli, and transport (Agustí et al., 2012). Our data in tomato (Tables S8, S9, Figure 4) confirm that these terms were highlighted in the over-expressed groups in the LAZ (Group A). This is in agreement with earlier studies of the abscission process in Arabidopsis and olive (Cai and Lashbrook, 2008; Gil-Amado and Gomez-Jimenez, 2013), suggesting that most genes involved in AZ functions are being shared at all stages of the abscission process of various organs, although there might be significant changes in the transcriptional activities at each stage of the abscission process.
Comparative Gene Expression in the Tomato FAZ and LAZ
Transcription Factors Gene Families
TFs play key roles in plant development and act as major switches of various transcriptional regulatory networks by temporarily and spatially regulating the transcription of their target genes. Recent reports highlighted the involvement of various TFs in organ abscission and dehiscence processes, FAZ development, and formation of protective layers (Meir et al., 2010, 2011; Nakano et al., 2012; Corbacho et al., 2013; Gil-Amado and Gomez-Jimenez, 2013; Parra et al., 2013; Liu et al., 2014). Since many genes are involved in the abscission process, it is not feasible to manipulate such a complex and dynamic process by modifying a single gene expression, and therefore efforts have been focused on specific TFs that control the entire pathways (Nath et al., 2007; Van Nocker, 2009; Ma et al., 2015). Many TFs families associated with FAZ development during tomato flower abscission process was already characterized, and it was interesting to study if these TFs families are also involved in tomato leaf abscission.
MYB is one of the largest plant TF families (Riechmann et al., 2000), which is involved in various regulatory pathways (Jin and Martin, 1999). Our results show that several MYB genes were upregulated and exclusively present in the FAZ and LAZ (Table S10). In tomato, it was shown that the Blind (Bl) gene, which encodes the R2R3 class MYB TF, was upregulated in the AZ-forming lines (Nakano et al., 2012), and it interacted with jointless in controlling the meristem cell fate (Schmitz et al., 2002; Quinet et al., 2011). In rice, a single non-synonymous substitution (G to T) in the shattering gene sh4, encoding a MYB3 DNA-binding domain, resulted in AZ disfunction and incomplete development, leading to reduced seed shattering (Li et al., 2006). Most members of the MYB families were shown to be abundantly present in melon fruit AZ (Corbacho et al., 2013) and were upregulated in olive fruit AZ (Gil-Amado and Gomez-Jimenez, 2013) during fruit abscission. Additionally, our data show that SlMYB43—THM16 (Solyc11g011050), which is a homolog to AtMYB43 and PtrMYB152, was present in both tomato AZs and overexpressed in group A (Table S10). This indicates that this gene regulates secondary cell wall biosynthesis similarly to Arabidopsis MYBs (Wang et al., 2014b). Other MYB genes, such as SlMYB3 (Solyc06g065100)—At1g22640 (salicylic acid- and ABA-inducible TF), SlMYB108 (Solyc12g099130)—AT3G06490 (ethylene- and JA-inducible TF), were expressed in both tomato AZs (Table S10). This supports the idea that these MYB proteins act as critical components of multiple hormone-mediated transcriptional cascades and cell wall biogenesis, which regulate tomato flower and leaf abscission.
ZF proteins regulate many developmental and stress responses (Takatsuji, 1998, 1999). Our results show that 98 ZF TF genes were overexpressed in both AZs, being the second most highly expressed family in Groups A and B (Figure 7A, Table S10). In Arabidopsis, ZINC FINGER PROTEIN2 was upregulated in the stamen AZ, and its overexpression delayed the abscission process and contributed to the AZ development (Cai and Lashbrook, 2008). The C2C2 type ZF genes were upregulated in the tomato FAZ upon ethylene-induced abscission (Wang et al., 2013).
Most of the MADS-box and GRAS TFs gene families were expressed in both tomato AZ tissues represented by Groups A and B (Figure 7A, Table S10). Since our pooled LAZ and FAZ samples were taken when the AZs were already well defined, the higher expression of MADS-box and GRAS TFs gene families in these samples suggest that these TF genes also regulate the late stages of the abscission process, when the AZ is already differentiated. It was previously proposed that MADS box TFs might substantially contribute to the specificity of the identity of the pedicel regions (Nakano et al., 2012), and might form region-specific protein complexes similar to the floral quartets model of flower organ identification (Theissen and Saedler, 2001). Our data show that the MADS-box gene MACROCALYX (Solyc05g012020) is highly expressed in the tomato FAZ (Group B), whereas the JOINTLESS (Solyc11g010570) gene was expressed at a similar level in both the FAZ and the LAZ (Group C) (Table S10). Therefore, we speculate that a similar type of organ identity specification, reported for the FAZ, might also operate in the LAZ.
Our results show that 137 bHLH TFs genes, out of 159 identified bHLH genes in tomato, were overexpressed in both the FAZ and the LAZ (Table S10). These genes were distributed among all the groups presented in Figure 2, indicating that the bHLH TF family is associated with the abscission process. It should be emphasized that our expression studies were performed with pooled samples, and therefore they do not represent the pattern of abscission progress. Most members of the bHLH TF gene family were downregulated during abscission of mature olive fruit (Gil-Amado and Gomez-Jimenez, 2013). Similarly, the bHLH TF gene (AW648468) was sharply downregulated in the tomato FAZ after flower removal (Meir et al., 2010). Mutation in the myc/bHLH gene ALCATRAZ in Arabidopsis delayed fruit dehiscence by blocking the separation of the valve cells from the replum (Rajani and Sundaresan, 2001). MYB and bHLH proteins (MYC type bHLH) interact to form multi protein complexes to regulate gene transcription (Wolberger, 1999; Zimmermann et al., 2004). It seems therefore, that bHLH and MYB TFs manifest a potential interaction necessary for the regulation of genes operating in downstream events in the FAZ and the LAZ during flower and leaf abscission.
The majority of the bZIP TFs were present and overexpressed in the tomato LAZ (Group A, A1) (Figure 7, Table S10). Among the bZIP TFs, the HY5 (Solyc08g061130) gene was present and equally expressed in both AZs (Group C). Most members of the bZIP family genes and HY5 were specifically induced and abundantly present in melon fruit AZ (Corbacho et al., 2013), and were upregulated in olive fruit AZ during abscission (Gil-Amado and Gomez-Jimenez, 2013; Parra et al., 2013). The bZIP gene (BG631669) was downregulated at an early stage of tomato pedicel abscission (Meir et al., 2010). The TGA type bZIP genes were found to be involved in plant development (Izawa et al., 1993), auxin-induced stress responses (Pascuzzi et al., 1998), and regulation of abscission-specific Cel gene expression (Tucker et al., 2002). This suggests that these TF genes might act as positive regulators of abscission signaling. Our results suggest that different bZIP TFs probably mediate the abscission-responsive transcription processes in flowers and mainly in leaves. Taken together, our data corroborate that in the tomato FAZ and LAZ, TFs belonging to these families may potentially act to trigger the transcriptional cascade during abscission and formation of the defense layer. Additional research is needed to reveal the molecular basis of the regulation of expression of these genes.
Key Meristem Genes
Reports demonstrating the expression of key meristem genes and their functional association with the AZ are emerging, implying that the undifferentiated AZ cells have the capability to differentiate in response to various stimulators. Most of the key meristem genes were expressed in both tomato AZs (Table 3). The Tkn3/KNAT6, TBL4, LBD1, Bl were highly expressed in the tomato LAZ (Group A) compared to the FAZ (Table 3). The KNOX family genes regulate the size and proliferation of the AZ cells during floral organ abscission (Shi et al., 2011). Moreover, TKN3 and BL4, encoding KNOX and BELL family TFs, form a heterodimer required for SAM functioning (Rutjens et al., 2009). On the other hand, the WUS, OFP, MYB-Cpm10/MYB78, Goblet, Ls were highly expressed in the tomato FAZ compared to the LAZ (Group B) (Table 3). WUSCHEL-related homeobox-containing protein (WUS) and KNOX gene families are key genes in the regulation of the maintenance of undifferentiated cells (Long et al., 1996; Mayer et al., 1998; Lenhard et al., 2002). It was previously reported that the homologs of WUS and its potential functional partner OFP were downregulated during flower abscission (Meir et al., 2010; Wang et al., 2013). More interestingly, the family members of Arabidopsis KNAT6, BELL-like homeodomain protein 1, and OFP, can potentially form ternary complexes, which are critical for meristem activities (Hake et al., 2004; Hackbusch et al., 2005; Hamant and Pautot, 2010; Li et al., 2011). Similarly, homologs of LBD1 (Wang et al., 2013) and the SAM gene, NAC-domain TF GLOBLET (Berger et al., 2009), were also highly expressed in the tomato FAZ (Hu et al., 2014). WUS, Bl, and Ls genes displayed differential expressions between tomato wild type and the mc mutant, and hence showed a jointless phenotype (Nakano et al., 2012). On the other hand, Bl and AGL12 were continuously and specifically induced in the tomato FAZ during the pedicel abscission process (Meir et al., 2010; Wang et al., 2013). The REVOLUTA gene is involved in apical meristem development for limiting cell divisions in Arabidopsis (Talbert et al., 1995). Overexpression of a microRNA166-resistant version of the tomato REVOLUTA (SlREV) (35S::REVRis) caused dramatic reproductive alterations, including continuous production of flowers at the FAZ (Hu et al., 2014). Our data show that SlREV (Solyc11g069470) was expressed equally (Group C) in both the FAZ and the LAZ samples (Table S3), indicating its importance for the development of the apical meristem in both AZs.
Our findings showing that most of the SAM and auxiliary meristem genes are preferentially expressed at differential levels in both the tomato FAZ and LAZ and interact with each other, support the idea that meristem activity genes play important roles in maintaining the undifferentiated status of cells in both AZs. The transcriptome data show that there are substantial differences between the tomato FAZ and LAZ. In particular, genes involved in key meristem functions show distinct expression patterns. These differences between the FAZ and the LAZ might explain the significant differences observed in the rate of pedicel and petiole abscission, respectively, when petiole abscission took a much longer time and had to be enhanced by ethylene (Figure 1). Apart of the key meristem genes, many other floral meristem genes were preferentially expressed in both the tomato FAZ and LAZ (Table S3).
Cell Wall Related Genes
Our transcriptome analyses showed that the majority of cell wall degrading and remodeling factors, including PG, Cel, XTH, and EXP genes (Table 4) were highly expressed in both tomato AZs. These genes were previously demonstrated to be highly expressed also in the AZs of a large number of abscission systems including tomato AZ (Lashbrook et al., 1994; del Campillo and Bennett, 1996; Cho and Cosgrove, 2000; Taylor and Whitelaw, 2001; Roberts et al., 2002; Agustí et al., 2008; Ogawa et al., 2009; Meir et al., 2010). TAPG1 was highly expressed in the LAZ following exposure of the debladed leaf explants to ethylene (Table 4), thereby confirming previous reports showing that TAPG1 transcript in the LAZ was induced by ethylene (Jiang et al., 2008) and inhibited by auxin (Hong et al., 2000). In addition, TAPG4 showed the highest expression level in the FAZ, and therefore its promoter was used for specific silencing of genes in the tomato FAZ (Ma et al., 2015). Previous microarray experiments performed with the tomato pedicel abscission system showed that Cel1 and Cel5 were strongly and specifically upregulated in the FAZ, but the expression levels of Cel2,3,7,8 were very low in the FAZ and were not affected by flower removal (Meir et al., 2010). However, our results demonstrate that the Cel2,5,6,7,8 genes were present and expressed at high levels in the LAZ pooled samples (Table 4). The functions of Cel1 and Cel2 were already demonstrated by antisense suppression in tomato flower and leaf abscission (Lashbrook et al., 1998; Brummell et al., 1999). Cel1,6,9 genes were upregulated in the LAZ of soybean (Glycine max) explants after ethylene treatment (Tucker et al., 2007). Expansins are involved in cell wall enlargement and pectin remodifications (Lee et al., 2001; Cosgrove et al., 2002; Zenoni et al., 2011), and were reported to regulate pedicel abscission in Arabidopsis and soybean and leaflet abscission in elderberry (Sambucus nigra) (Cho and Cosgrove, 2000; Belfield et al., 2005; Tucker et al., 2007). We observed that the expression levels of several EXP genes, LeEXP1,5,9,11,18, were particularly high in the LAZ (Table 4). It was previously reported that the expression levels of EXP3,4,5,9 genes in tomato, which were specifically and highly expressed in the FAZ, decreased during the process of pedicel abscission, and were not affected by the 1-MCP pretreatment (Meir et al., 2010). LeEXP1,11 genes were also specifically upregulated in tomato FAZ (Wang et al., 2013).
Taken together, our data regarding the changes in the expression of genes encoding cell wall modifying enzymes, confirmed previous reports on various abscission systems. The results also indicate that both the FAZ and the LAZ have similar types of cell wall-related genes, but with different expression levels, which were generally higher in the LAZ than in the FAZ. This may be ascribed to the ethylene treatment applied to the debladed leaf explants in order to enhance the rate of leaf petiole abscission (Figure 1B). Ethylene treatment might also induce an increased expression of cell wall modifying genes in the LAZ.
Hormonal Signal Transduction Genes
The interplay of plant hormones during the abscission process has been widely reported. In tomato, the pedicel abscission process is inhibited by a continuous auxin flow from the flowers, and is triggered by ethylene following auxin depletion in the FAZ (Roberts et al., 1984; Meir et al., 2010). Ethylene also inhibits auxin transport, thereby enhancing auxin depletion in the AZ (Meir et al., 2015). Therefore, it was expected that genes of auxin and ethylene signal transduction cascades would be expressed predominantly in both the FAZ and the LAZ as compared to the other plant hormones. A continuous auxin flow to the AZ is required for preventing the acquisition of ethylene sensitivity by the AZ cells, which leads in turn to organ abscission (Taylor and Whitelaw, 2001). The molecular basis of auxin transport in tomato was elucidated in sympodial growth, compound leaves, fleshy fruit, and whole plants (Giovannoni, 2004; Kimura and Sinha, 2008; Nishio et al., 2010; Pattison and Catalá, 2012). This auxin flow is presumably regulated by auxin-responsive genes, belonging to three major groups, Aux/IAA, SAUR, and GH3. These auxin-responsive genes, which are regulated by ARFs (Guilfoyle and Hagen, 2007), are well characterized in tomato (Kumar et al., 2011, 2012; Wu et al., 2012). Our data show that various auxin-related genes, such as Aux/IAA, SAUR, GH3, ARFs, LAX, and PINs, are expressed in both the tomato FAZ and LAZ at various levels (Table 5).
The expression level of Aux/IAA genes was used as a marker for auxin activity associated with inhibition of floret abscission by auxin application in C. elegans (Abebie et al., 2008), as well as a marker of auxin depletion in tomato FAZ following flower removal (Meir et al., 2010) and in citrus AZ during fruitlet abscission (Xie et al., 2015). We previously showed that the auxin responsive genes IAA1,3,4,7,8,9,10 were downregulated following abscission induction in tomato flowers (Meir et al., 2010), and our present data showed that IAA3,8,9,10 were presented and expressed in both the tomato FAZ and LAZ pooled samples at various levels (Table 5). The remaining genes which were already shown to be present in the FAZ, IAA1,4,7 (Solyc06g053840, Solyc04g076850, Solyc06g053830, respectively), were shown to have equal expression in the FAZ and the LAZ (Table S3). These genes are not KEGG-annotated in the auxin hormonal pathway, which might indicate that that they are not significant for the auxin signaling cascade.
ARFs are TFs that bind to AREs in promoters of early auxin responsive genes and play central roles in many auxin-mediated processes, leading to activation or suppression of the selected genes. The expression pattern and the possible role of the ARF gene family in the tomato FAZ, as well as auxin- and ethylene-induced changes during flower abscission were comprehensively studied (Guan et al., 2014). We showed that multiple ARF genes were expressed in both tomato AZs (Table 5, Table S3). Upregulation of the SlARF1,3,5,19 delayed the abscission process in the FAZ (Guan et al., 2014), indicating that these ARFs have a similar function in both AZs.
Several reports suggested that GH3 genes are involved in flower or fruitlet abscission (Kuang et al., 2012; Wang et al., 2013; Meir et al., 2015). MES1 converts the storage form of IAA into its active free form, whereas Dwarf in Light1 does the opposite, i.e., converts active free IAA to an inactive conjugated form (Staswick et al., 2005; Woodward and Bartel, 2005; Yang et al., 2008; Ludwig-Müller, 2011). Our results demonstrate that MES1 and DFL1 (Table S3) as well as other GH3 genes (Table 5) were expressed in both tomato AZs. Recently, we reported (Meir et al., 2015) that GH3 genes in the FAZ were upregulated in response to auxin depletion, confirming that increased IAA conjugation is involved in the process of auxin depletion, while their expression decreased in the LAZ after leaf deblading, further confirming that GH3 is an auxin-induced gene. In addition, conjugation of IAA to amino acids provides a negative feedback loop to control auxin homoeostasis, and the SlGH3-2,10,15 genes, which control IAA conjugation, were found to respond quickly to exogenous auxin application (Kumar et al., 2012; Meir et al., 2015).
Reduced auxin levels in Arabidopsis were attributed to increased activity of auxin efflux transporters (Sorefan et al., 2009). Recently, it was demonstrated that the KD1 gene played a role in modulating auxin levels in the tomato FAZ, by altering the expression profiles of the auxin efflux transporters, PIN9 (HQ127075; SlPIN9) and PIN-like3 (SL_TC197872) (Ma et al., 2015). Our data show that the auxin influx carrier genes, SlLAX3,4,5 and the auxin efflux carrier genes SlPIN1,3,4,5,6,7,9, were present in both the tomato FAZ and LAZ (Table 5). In addition, SlPIN3,5,6,7 genes were overexpressed (by ~2- to 10-fold) in the LAZ compared to the FAZ (Table 5). Most of the auxin-related gene families are expressed in both AZs, but some members of different gene families are expressed specifically in the FAZ or the LAZ (Meir et al., 2015). The only study so far on the effect of leaf deblading on expression of auxin-related genes in the LAZ was performed in Mirabilis jalapa (Meir et al., 2006). The differential expression of the auxin influx and efflux carrier genes between the two AZs suggests that different genes of these families play important roles in different AZs. This might further provide us with means for selective manipulation of leaf and flower abscission by specifically manipulating auxin transport in these two AZs. Such a manipulation might be important for agricultural purposes, for example to reduce olive fruit strength before harvest without affecting leaf abscission.
Ethylene response factors (ERFs) are plant transcriptional regulators that specifically bind the GCC motif of the promoter region of ethylene-regulated genes, thereby mediating ethylene-dependent gene expressions (Ohme-Takagi and Shinshi, 1995; Solano et al., 1998). These SlERF genes were reported to be specifically overexpressed in the tomato FAZ compared to the proximal (basal) and distal (apical) NAZ regions (Nakano et al., 2013; Wang et al., 2013). It was recently reported that in SlERF52-suppressed tomato plants, flower pedicel abscission was significantly delayed compared to the wild type, and accordingly, a reduced expression of the genes encoding for the abscission-associated enzymes, cellulase and PGs, was detected (Nakano et al., 2014). Our data show that several SlERF genes, and particularly SlERF52, which were reported to be essential for flower pedicel abscission, operate also in the tomato LAZ. The downstream components (positive) of ethylene signaling, such as EIN2 and EILS, were expressed in both the tomato AZs (Table 5). Antisense lines of LeEIL plants exhibited delayed flower abscission (Tieman et al., 2001), which suggests that the LeEILS has the same function in the tomato LAZ.
Many genes related to different steps of the ethylene signaling transduction pathway were expressed in both the tomato AZs following flower removal and leaf deblading (Table 5). The tomato ethylene receptor genes ETR1,3,4,5 were expressed in both AZs, confirming previous reports that they were expressed in the FAZ (Payton et al., 1996; Meir et al., 2010). The Constitutive Triple Response1 (CTR1) gene product acts downstream of the ethylene receptors, and negatively regulates ethylene signal transduction (Kieber et al., 1993). It was previously reported that CTR1 was upregulated in early stages of the tomato pedicel abscission process, and was specifically expressed in the FAZ in the late stages of the abscission process (Meir et al., 2010). Our analysis data show that CTR1,3,4 genes were expressed in both tomato AZs, with CTR1 showing the highest expression (Table 5). This indicates that a similar cascade of events operates in both AZs.
Conclusions
This study presents a de novo assembly of AZ-associated transcripts and provides valuable information about their functional annotation by using an integrated approach to enrich the transcriptome of S. lycopersicum FAZ and LAZ. To the best of our knowledge, this is the first comprehensive report on RNA-Seq of tomato FAZ and LAZ, which we believe would contribute to the understanding of the expression differences in these AZs. The present research identified genes that may be involved in the abscission process of tomato flowers and leaves, including genes encoding for TFs, hormone transduction components, cell wall-related enzymes, and key meristem factors. Similar gene family members were expressed in both the FAZ and the LAZ following flower or leaf removal, respectively, suggesting a similar regulation of the abscission process of these organs with few exceptions. This provides a significant improvement in our understanding of the abscission process.
We have utilized the RNA-Seq data for the development of an AZ-specific microarray chip. The AZ microarray is more comprehensive than other commercially available arrays, having more transcripts with multiple probes and probes designed in antisense direction, which might be further used to explore the roles of NATs. The AZ-specific microarray can be used for further examination of detailed gene expression in various abscission stages, and is applicable to other transcriptomic studies in tomato. The AZ-specific microarray chip provides a cost-effective approach for analysis of multiple samples in a rapid succession. Hence, this study can serve as a foundation for characterization of candidate genes, which would not only provide novel insights into understanding of the AZ development, early and late abscission events, but also will provide resources for improved tomato breeding for preventing abscission and for specific manipulation of flower or leaf abscission for horticultural uses.
Sequence Deposition
NGS data is submitted to the NCBI sequence read archive (NCBI-SRA) under the study ID: PRJNA192557. Individual SRS ID = SRS399193 entitled with FAZ transcriptome analysis of VF-36, and SRS401162 entitled with LAZ transcriptome analysis of VF-36. The AZ-specific microarray (AMADID: 043310) was validated and used to analyze the FAZ, flower NAZ and LAZ at various time points and deposited in the NCBI Gene Expression Omnibus (GEO) databases (http://www.ncbi.nlm.nih.gov/geo/) under the following locators: GSE45355; GSE45356; GSE64221. These data will be released for public access upon acceptance of this publication.
Author Contributions
SS, SM and SP were responsible for the conception, design of the experiments and interpretation of data for work. SS, RM and NK were responsible for acquisition of the data and the bioinformatics analysis. SS, BK and ShS performed the laboratory experiments and analyses. SS, RJ, and MT were responsible for the design of the microarray chip. SS, SP, JR and SM were responsible for final approval of the version to be published.
Funding
This work was supported by the United States-Israel Binational Agricultural Research and Development Fund (BARD) [grant number US-4571-12C to SM, MT and SP], and the Chief Scientist of the Israeli Ministry of Agriculture Fund [grant number 203-0898-10 to SM and SP].
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer Autar Krishen Mattoo declares that, despite being affiliated with the same institute as the author Mark L. Tucker, the review process was conducted objectively.
Acknowledgments
Contribution No. 730/15 from the ARO, The Volcani Center, Bet Dagan, Israel. SS would like to thank the Indian Council of Agricultural Research for providing him with an International Fellowship (ICAR-IF), New Delhi, India, as partial support of his PhD studies. The authors would like to acknowledge the Genomics core unit of Genotypic Technology, Bangalore, India, for preparation of the library and generating the sequence data reported in this publication.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.01258
Abbreviations
AREs, auxin-responsive elements; ARF, auxin response factor; Aux/IAA, auxin resistant/indole-3-acetic acid-inducible; AZ, abscission zone; bHLH, basic helix–loop–helix; Bl, blind; BLAST, basic local alignment search tool; bZIP, basic leucine zipper; Cel, cellulase; CTR, constitutive triple response; DEG, differentially expressed genes; EIL, ethylene insensitive-like; EIN, ethylene insensitive; ERF, ethylene responsive factor; EXP, expansin; FAZ, flower abscission zone; GH, Gretchen Hagen3; GO, gene ontology; IAA, indole-3-acetic acid; JA, jasmonic acid; KEGG, Kyoto encyclopedia of genes and genomes; LAX, auxin influx carrier (Like Aux); LAZ, leaf abscission zone; LBD1, lateral organ boundaries domain protein1; Ls, lateral suppressor; MAPKK, mitogen- activated protein kinase kinase; MAPKKK, MAPKK Kinase; 1-MCP, 1-methylcyclopropene; NAT, naturally occurring antisense transcripts; NAZ, non-AZ; NCBI, national center for biotechnology information; NGS, next generation sequencing; OFP, ovate family protein; PG, polygalacturonase; PIN, pin-formed; PE, paired end; QC, quality check; qRT-PCR, quantitative real time PCR; RNA-Seq, RNA sequencing; SAM, shoot apical meristem; SAUR, small auxin up-regulated RNA; TAGL, tomato AGAMOUS-like; TAPG, tomato abscission polygalacturonase; TF, transcription factor; WUS, WUSCHEL-related homeobox-containing protein; XTH, xyloglucan endotransglucosylase-hydrolase; ZF, zinc finger.
References
Abebie, B., Lers, A., Philosoph-Hadas, S., Goren, R., Riov, J., and Meir, S. (2008). Differential effects of NAA and 2,4-D in reducing floret abscission in cestrum (Cestrum elegans) cut flowers are associated with their differential activation of Aux/IAA homologous genes. Ann. Bot. 101, 249–259. doi: 10.1093/aob/mcm115
Agustí, J., Gimeno, J., Merelo, P., Serrano, R., Cercós, M., Conesa, A., et al. (2012). Early gene expression events in the laminar abscission zone of abscission-promoted citrus leaves after a cycle of water stress/rehydration: involvement of CitbHLH1. J. Exp. Bot. 63, 6079–6091. doi: 10.1093/jxb/ers270
Agustí, J., Merelo, P., Cercós, M., Tadeo, F. R., and Talón, M. (2008). Ethylene-induced differential gene expression during abscission of citrus leaves. J. Exp. Bot. 59, 2717–2733. doi: 10.1093/jxb/ern138
Agustí, J., Merelo, P., Cercós, M., Tadeo, F. R., and Talón, M. (2009). Comparative transcriptional survey between laser-microdissected cells from laminar abscission zone and petiolar cortical tissue during ethylene-promoted abscission in citrus leaves. BMC Plant Biol. 9:127. doi: 10.1186/1471-2229-9-127
Alonso, J. M., Hirayama, T., Roman, G., Nourizadeh, S., and Ecker, J. R. (1999). EIN2, a bifunctional transducer of ethylene and stress responses in Arabidopsis. Science 284, 2148–2152. doi: 10.1126/science.284.5423.2148
Bainbridge, K., Guyomarc'h, S., Bayer, E., Swarup, R., Bennett, M., Mandel, T., et al. (2008). Auxin influx carriers stabilize phyllotactic patterning. Genes Dev. 22, 810–823. doi: 10.1101/gad.462608
Bangerth, F. (2000). Abscission and thinning of young fruit and thier regulation by plant hormones and bioregulators. Plant Growth Regul. 31, 43–59. doi: 10.1023/A:1006398513703
Bassel, G. W., Mullen, R. T., and Bewley, J. D. (2008). Procera is a putative DELLA mutant in tomato (Solanum lycopersicum): effects on the seed and vegetative plant. J. Exp. Bot. 59, 585–593. doi: 10.1093/jxb/erm354
Belfield, E. J., Ruperti, B., Roberts, J. A., and McQueen-Mason, S. (2005). Changes in expansin activity and gene expression during ethylene-promoted leaflet abscission in Sambucus nigra. J. Exp. Bot. 56, 817–823. doi: 10.1093/jxb/eri076
Bellin, D., Ferrarini, A., Chimento, A., Kaiser, O., Levenkova, N., Bouffard, P., et al. (2009). Combining next-generation pyrosequencing with microarray for large scale expression analysis in non-model species. BMC Genomics 10:555. doi: 10.1186/1471-2164-10-555
Berger, Y., Harpaz-Saad, S., Brand, A., Melnik, H., Sirding, N., Alvarez, J. P., et al. (2009). The NAC-domain transcription factor GOBLET specifies leaflet boundaries in compound tomato leaves. Development 136, 823–832. doi: 10.1242/dev.031625
Bertone, P., Gerstein, M., and Snyder, M. (2005). Applications of DNA tiling arrays to experimental genome annotation and regulatory pathway discovery. Chromosome Res. 13, 259–274. doi: 10.1007/s10577-005-2165-0
Binder, B. M. (2008). The ethylene receptors: complex perception for a simple gas. Plant Sci. 175, 8–17. doi: 10.1016/j.plantsci.2007.12.001
Bleecker, A. B., and Patterson, S. E. (1997). Last exit: senescence, abscission, and meristem arrest in Arabidopsis. Plant Cell 9, 1169–1179. doi: 10.1105/tpc.9.7.1169
Botton, A., Eccher, G., Forcato, C., Ferrarini, A., Begheldo, M., Zermiani, M., et al. (2011). Signaling pathways mediating the induction of apple fruitlet abscission. Plant Physiol. 155, 185–208. doi: 10.1104/pp.110.165779
Brummell, D. A., Hall, B. D., and Bennett, A. B. (1999). Antisense suppression of tomato endo-1,4-beta-glucanase Cel2 mRNA accumulation increases the force required to break fruit abscission zones but does not affect fruit softening. Plant Mol. Biol. 40, 615–622. doi: 10.1023/A:1006269031452
Busi, M. V., Bustamante, C., D'Angelo, C., Hidalgo-Cuevas, M., Boggio, S. B., Valle, E. M., et al. (2003). MADS-box genes expressed during tomato seed and fruit development. Plant Mol. Biol. 52, 801–815.
Cai, S., and Lashbrook, C. C. (2008). Stamen abscission zone transcriptome profiling reveals new candidates for abscission control: enhanced retention of floral organs in transgenic plants overexpressing Arabidopsis ZINC FINGER PROTEIN2. Plant Physiol. 146, 1305–1321. doi: 10.1104/pp.107.110908
Carninci, P., Kasukawa, T., Katayama, S., Gough, J., Frith, M. C., Maeda, N., et al. (2005). The transcriptional landscape of the mammalian genome. Science 309, 1559–1563. doi: 10.1126/science.1112014
Carninci, P., Sandelin, A., Lenhard, B., Katayama, S., Shimokawa, K., Ponjavic, J., et al. (2006). Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 38, 626–635. doi: 10.1038/ng1789
Cho, H. T., and Cosgrove, D. J. (2000). Altered expression of expansin modulates leaf growth and pedicel abscission in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 97, 9783–9788. doi: 10.1073/pnas.160276997
Clark, K. L., Larsen, P. B., Wang, X., and Chang, C. (1998). Association of the Arabidopsis CTR1 Raf-like kinase with the ETR1 and ERS ethylene receptors. Proc. Natl. Acad. Sci. U.S.A. 95, 5401–5406. doi: 10.1073/pnas.95.9.5401
Corbacho, J., Romojaro, F., Pech, J. C., Latché, A., and Gomez-Jimenez, M. C. (2013). Transcriptomic events involved in melon mature-fruit abscission comprise the sequential induction of cell-wall degrading genes coupled to a stimulation of endo and exocytosis. PLoS ONE 8:e58363. doi: 10.1371/journal.pone.0058363
Cosgrove, D. J., Li, L. C., Cho, H. T., Hoffmann-Benning, S., Moore, R. C., and Blecker, D. (2002). The growing world of expansins. Plant Cell Physiol. 43, 1436–1444. doi: 10.1093/pcp/pcf180
del Campillo, E., and Bennett, A. B. (1996). Pedicel breakstrength and cellulase gene expression during tomato flower abscission. Plant Physiol. 111, 813–820. doi: 10.1104/pp.111.3.813
Doebley, J. (2004). The genetics of maize evolution. Annu. Rev. Genet. 38, 37–59. doi: 10.1146/annurev.genet.38.072902.092425
Ecker, J. R. (1995). The ethylene signal transduction pathway in plants. Science 268, 667–675. doi: 10.1126/science.7732375
Ellis, C. M., Nagpal, P., Young, J. C., Hagen, G., Guilfoyle, T. J., and Reed, J. W. (2005). AUXIN RESPONSE FACTOR1 and AUXIN RESPONSE FACTOR2 regulate senescence and floral organ abscission in Arabidopsis thaliana. Development 132, 4563–4574. doi: 10.1242/dev.02012
Fujimoto, S. Y., Ohta, M., Usui, A., Shinshi, H., and Ohme-Takagi, M. (2000). Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box-mediated gene expression. Plant Cell 12, 393–404. doi: 10.1105/tpc.12.3.393
Gil-Amado, J. A., and Gomez-Jimenez, M. C. (2013). Transcriptome analysis of mature fruit abscission control in olive. Plant Cell Physiol. 54, 244–269. doi: 10.1093/pcp/pcs179
Giovannoni, J. J. (2004). Genetic regulation of fruit development and ripening. Plant Cell 16(Suppl.), S170–S180. doi: 10.1105/tpc.019158
Guan, X., Xu, T., Gao, S., Qi, M., Wang, Y., Liu, X., et al. (2014). Temporal and spatial distribution of auxin response factor genes during tomato flower abscission. J. Plant Growth Regul. 33, 317–327. doi: 10.1007/s00344-013-9377-x
Guilfoyle, T. J., and Hagen, G. (2007). Auxin response factors. Curr. Opin. Plant Biol. 10, 453–460. doi: 10.1016/j.pbi.2007.08.014
Hackbusch, J., Richter, K., Müller, J., Salamini, F., and Uhrig, J. F. (2005). A central role of Arabidopsis thaliana ovate family proteins in networking and subcellular localization of 3-aa loop extension homeodomain proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 4908–4912. doi: 10.1073/pnas.0501181102
Hake, S., Smith, H. M., Holtan, H., Magnani, E., Mele, G., and Ramirez, J. (2004). The role of knox genes in plant development. Annu. Rev. Cell Dev. Biol. 20, 125–151. doi: 10.1146/annurev.cellbio.20.031803.093824
Hamant, O., and Pautot, V. (2010). Plant development: a TALE story. C. R. Biol. 333, 371–381. doi: 10.1016/j.crvi.2010.01.015
Hong, S. B., Sexton, R., and Tucker, M. L. (2000). Analysis of gene promoters for two tomato polygalacturonases expressed in abscission zones and the stigma. Plant Physiol. 123, 869–881. doi: 10.1104/pp.123.3.869
Hu, G., Fan, J., Xian, Z., Huang, W., Lin, D., and Li, Z. (2014). Overexpression of SlREV alters the development of the flower pedicel abscission zone and fruit formation in tomato. Plant Sci. 229, 86–95. doi: 10.1016/j.plantsci.2014.08.010
Huang, S., Gao, Y., Liu, J., Peng, X., Niu, X., Fei, Z., et al. (2012). Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mol. Genet. Genomics 287, 495–513. doi: 10.1007/s00438-012-0696-6
Huang, Y., Li, H., Hutchison, C. E., Laskey, J., and Kieber, J. J. (2003). Biochemical and functional analysis of CTR1, a protein kinase that negatively regulates ethylene signaling in Arabidopsis. Plant J. 33, 221–233. doi: 10.1046/j.1365-313X.2003.01620.x
Izawa, T., Foster, R., and Chua, N. H. (1993). Plant bZIP protein DNA binding specificity. J. Mol. Biol. 230, 1131–1144. doi: 10.1006/jmbi.1993.1230
Jiang, C. Z., Lu, F., Imsabai, W., Meir, S., and Reid, M. S. (2008). Silencing polygalacturonase expression inhibits tomato petiole abscission. J. Exp. Bot. 59, 973–979. doi: 10.1093/jxb/ern023
Jin, H., and Martin, C. (1999). Multifunctionality and diversity within the plant MYB-gene family. Plant Mol. Biol. 41, 577–585. doi: 10.1023/A:1006319732410
Johnson, J. M., Edwards, S., Shoemaker, D., and Schadt, E. E. (2005). Dark matter in the genome: evidence of widespread transcription detected by microarray tiling experiments. Trends Genet. 21, 93–102. doi: 10.1016/j.tig.2004.12.009
Kalaitzis, P., Solomos, T., and Tucker, M. L. (1997). Three different polygalacturonases are expressed in tomato leaf and flower abscission, each with a different temporal expression pattern. Plant Physiol. 113, 1303–1308. doi: 10.1104/pp.113.4.1303
Kieber, J. J., Rothenberg, M., Roman, G., Feldmann, K. A., and Ecker, J. R. (1993). CTR1, a negative regulator of the ethylene response pathway in Arabidopsis, encodes a member of the raf family of protein kinases. Cell 72, 427–441. doi: 10.1016/0092-8674(93)90119-B
Kim, J., Sundaresan, S., Philosoph-Hadas, S., Yang, R., Meir, S., and Tucker, M. L. (2015). Examination of the abscission-associated transcriptomes for soybean, tomato and Arabidopsis highlights the conserved biosynthesis of an extensible extracellular matrix and boundary layer. Front. Plant Sci. 6:1109. doi: 10.3389/fpls.2015.01109
Kimura, S., and Sinha, N. (2008). Tomato (Solanum lycopersicum): a model fruit-bearing crop. CSH Protoc. 2008:pdb.emo105. doi: 10.1101/pdb.emo105
Kiyosawa, H., Mise, N., Iwase, S., Hayashizaki, Y., and Abe, K. (2005). Disclosing hidden transcripts: mouse natural sense-antisense transcripts tend to be poly(A) negative and nuclear localized. Genome Res. 15, 463–474. doi: 10.1101/gr.3155905
Klee, H. J. (2002). Control of ethylene-mediated processes in tomato at the level of receptors. J. Exp. Bot. 53, 2057–2063. doi: 10.1093/jxb/erf062
Klee, H. J. (2004). Ethylene signal transduction. Moving beyond Arabidopsis. Plant Physiol. 135, 660–667. doi: 10.1104/pp.104.040998
Klee, H. J., and Giovannoni, J. J. (2011). Genetics and control of tomato fruit ripening and quality attributes. Annu. Rev. Genet. 45, 41–59. doi: 10.1146/annurev-genet-110410-132507
Koka, C. V., Cerny, R. E., Gardner, R. G., Noguchi, T., Fujioka, S., Takatsuto, S., et al. (2000). A putative role for the tomato genes DUMPY and CURL-3 in brassinosteroid biosynthesis and response. Plant Physiol. 122, 85–98. doi: 10.1104/pp.122.1.85
Kuang, J. F., Wu, J. Y., Zhong, H. Y., Li, C. Q., Chen, J. Y., Lu, W. J., et al. (2012). Carbohydrate stress affecting fruitlet abscission and expression of genes related to auxin signal transduction pathway in litchi. Int. J. Mol. Sci. 13, 16084–16103. doi: 10.3390/ijms131216084
Kumar, R., Agarwal, P., Tyagi, A. K., and Sharma, A. K. (2012). Genome-wide investigation and expression analysis suggest diverse roles of auxin-responsive GH3 genes during development and response to different stimuli in tomato (Solanum lycopersicum). Mol. Genet. Genomics 287, 221–235. doi: 10.1007/s00438-011-0672-6
Kumar, R., Tyagi, A. K., and Sharma, A. K. (2011). Genome-wide analysis of auxin response factor (ARF) gene family from tomato and analysis of their role in flower and fruit development. Mol. Genet. Genomics 285, 245–260. doi: 10.1007/s00438-011-0602-7
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Lashbrook, C. C., Giovannoni, J. J., Hall, B. D., Fischer, R. L., and Bennett, A. B. (1998). Transgenic analysis of tomato endo-β-1,4-glucanase gene function. Role of cel1 in floral abscission. Plant J. 13, 303–310. doi: 10.1046/j.1365-313X.1998.00025.x
Lashbrook, C. C., Gonzalez-Bosch, C., and Bennett, A. B. (1994). Two divergent endo-beta-1,4-glucanase genes exhibit overlapping expression in ripening fruit and abscising flowers. Plant Cell 6, 1485–1493. doi: 10.1105/tpc.6.10.1485
Lee, Y., Choi, D., and Kende, H. (2001). Expansins: ever-expanding numbers and functions. Curr. Opin. Plant Biol. 4, 527–532. doi: 10.1016/S1369-5266(00)00211-9
Lenhard, M., Jürgens, G., and Laux, T. (2002). The WUSCHEL and SHOOTMERISTEMLESS genes fulfil complementary roles in Arabidopsis shoot meristem regulation. Development 129, 3195–3206.
Lewis, M. W., Leslie, M. E., and Liljegren, S. J. (2006). Plant separation: 50 ways to leave your mother. Curr. Opin. Plant Biol. 9, 59–65. doi: 10.1016/j.pbi.2005.11.009
Li, C., Zhou, A., and Sang, T. (2006). Rice domestication by reducing shattering. Science 311, 1936–1939. doi: 10.1126/science.1123604
Li, E., Wang, S., Liu, Y., Chen, J. G., and Douglas, C. J. (2011). OVATE FAMILY PROTEIN4 (OFP4) interaction with KNAT7 regulates secondary cell wall formation in Arabidopsis thaliana. Plant J. 67, 328–341. doi: 10.1111/j.1365-313X.2011.04595.x
Lipovich, L., Tarca, A. L., Cai, J., Jia, H., Chugani, H. T., Sterner, K. N., et al. (2014). Developmental changes in the transcriptome of human cerebral cortex tissue: long noncoding RNA transcripts. Cereb. Cortex 24, 1451–1459. doi: 10.1093/cercor/bhs414
Liu, D., Wang, D., Qin, Z., Zhang, D., Yin, L., Wu, L., et al. (2014). The SEPALLATA MADS-box protein SLMBP21 forms protein complexes with JOINTLESS and MACROCALYX as a transcription activator for development of the tomato flower abscission zone. Plant J. 77, 284–296. doi: 10.1111/tpj.12387
Long, J. A., Moan, E. I., Medford, J. I., and Barton, M. K. (1996). A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of Arabidopsis. Nature 379, 66–69. doi: 10.1038/379066a0
Ludwig-Müller, J. (2011). Auxin conjugates: their role for plant development and in the evolution of land plants. J. Exp. Bot. 62, 1757–1773. doi: 10.1093/jxb/erq412
Ma, C., Meir, S., Xiao, L., Tong, J., Liu, Q., Reid, M. S., et al. (2015). A KNOTTED1-LIKE HOMEOBOX protein regulates abscission in tomato by modulating the auxin pathway. Plant Physiol. 167, 844–853. doi: 10.1104/pp.114.253815
Mäder, U., Nicolas, P., Richard, H., Bessières, P., and Aymerich, S. (2011). Comprehensive identification and quantification of microbial transcriptomes by genome-wide unbiased methods. Curr. Opin. Biotechnol. 22, 32–41. doi: 10.1016/j.copbio.2010.10.003
Mao, L., Begum, D., Chuang, H. W., Budiman, M. A., Szymkowiak, E. J., Irish, E. E., et al. (2000). JOINTLESS is a MADS-box gene controlling tomato flower abscission zone development. Nature 406, 910–913. doi: 10.1038/35022611
Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., and Gilad, Y. (2008). RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18, 1509–1517. doi: 10.1101/gr.079558.108
Martí, C., Orzáez, D., Ellul, P., Moreno, V., Carbonell, J., and Granell, A. (2007). Silencing of DELLA induces facultative parthenocarpy in tomato fruits. Plant J. 52, 865–876. doi: 10.1111/j.1365-313X.2007.03282.x
Mayer, K. F., Schoof, H., Haecker, A., Lenhard, M., Jürgens, G., and Laux, T. (1998). Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 95, 805–815. doi: 10.1016/S0092-8674(00)81703-1
McManus, M. T. (2008). Further examination of abscission zone cells as ethylene target cells in higher plants. Ann. Bot. 101, 285–292. doi: 10.1093/aob/mcm269
Mei, R., Hubbell, E., Bekiranov, S., Mittmann, M., Christians, F. C., Shen, M. M., et al. (2003). Probe selection for high-density oligonucleotide arrays. Proc. Natl. Acad. Sci. U.S.A. 100, 11237–11242. doi: 10.1073/pnas.1534744100
Meir, S., Hunter, D. A., Chen, J. C., Halaly, V., and Reid, M. S. (2006). Molecular changes occurring during acquisition of abscission competence following auxin depletion in Mirabilis jalapa. Plant Physiol. 141, 1604–1616. doi: 10.1104/pp.106.079277
Meir, S., Philosoph-Hadas, S., Sundaresan, S., Selvaraj, K. S., Burd, S., Ophir, R., et al. (2010). Microarray analysis of the abscission-related transcriptome in the tomato flower abscission zone in response to auxin depletion. Plant Physiol. 154, 1929–1956. doi: 10.1104/pp.110.160697
Meir, S., Philosoph-Hadas, S., Sundaresan, S., Selvaraj, K. S., Burd, S., Ophir, R., et al. (2011). Identification of defense-related genes newly-associated with tomato flower abscission. Plant Signal. Behav. 6, 590–593. doi: 10.4161/psb.6.4.15043
Meir, S., Sundaresan, S., Riov, J., Agarwal, I., and Philosoph-Hadas, S. (2015). Role of auxin depletion in abscission control. Stewart Posthar. Rev. 11, 1–15. doi: 10.2212/spr.2015.2.2
Mockler, T. C., Chan, S., Sundaresan, A., Chen, H., Jacobsen, S. E., and Ecker, J. R. (2005). Applications of DNA tiling arrays for whole-genome analysis. Genomics 85, 1–15. doi: 10.1016/j.ygeno.2004.10.005
Montoya, T., Nomura, T., Farrar, K., Kaneta, T., Yokota, T., and Bishop, G. J. (2002). Cloning the tomato curl3 gene highlights the putative dual role of the leucine-rich repeat receptor kinase tBRI1/SR160 in plant steroid hormone and peptide hormone signaling. Plant Cell 14, 3163–3176. doi: 10.1105/tpc.006379
Nakano, T., Fujisawa, M., Shima, Y., and Ito, Y. (2013). Expression profiling of tomato pre-abscission pedicels provides insights into abscission zone properties including competence to respond to abscission signals. BMC Plant Biol. 13:40. doi: 10.1186/1471-2229-13-40
Nakano, T., Fujisawa, M., Shima, Y., and Ito, Y. (2014). The AP2/ERF transcription factor SlERF52 functions in flower pedicel abscission in tomato. J. Exp. Bot. 65, 3111–3119. doi: 10.1093/jxb/eru154
Nakano, T., Kimbara, J., Fujisawa, M., Kitagawa, M., Ihashi, N., Maeda, H., et al. (2012). MACROCALYX and JOINTLESS interact in the transcriptional regulation of tomato fruit abscission zone development. Plant Physiol. 158, 439–450. doi: 10.1104/pp.111.183731
Nath, P., Sane, A. P., Trivedi, P. K., Sane, V. A., and Asif, M. H. (2007). Role of transcription factors in regulating ripening, senescence and organ abscission in plants. Stewart Posthar. Rev. 3, 1–14. doi: 10.2212/spr.2007.2.6
Niederhuth, C. E., Patharkar, O. R., and Walker, J. C. (2013). Transcriptional profiling of the Arabidopsis abscission mutant hae hsl2 by RNA-Seq. BMC Genomics 14:37. doi: 10.1186/1471-2164-14-37
Nishio, S., Moriguchi, R., Ikeda, H., Takahashi, H., Takahashi, H., Fujii, N., et al. (2010). Expression analysis of the auxin efflux carrier family in tomato fruit development. Planta 232, 755–764. doi: 10.1007/s00425-010-1211-0
Ogawa, M., Kay, P., Wilson, S., and Swain, S. M. (2009). ARABIDOPSIS DEHISCENCE ZONE POLYGALACTURONASE1 (ADPG1), ADPG2, and QUARTET2 are polygalacturonases required for cell separation during reproductive development in Arabidopsis. Plant Cell 21, 216–233. doi: 10.1105/tpc.108.063768
Ohme-Takagi, M., and Shinshi, H. (1995). Ethylene-inducible DNA binding proteins that interact with an ethylene-responsive element. Plant Cell 7, 173–182. doi: 10.1105/tpc.7.2.173
Osborne, D. J., and Morgan, P. W. (1989). Abscission. CRC Crit. Rev. Plant Sci. 8, 103–129. doi: 10.1080/07352688909382272
Overvoorde, P. J., Okushima, Y., Alonso, J. M., Chan, A., Chang, C., Ecker, J. R., et al. (2005). Functional genomic analysis of the AUXIN/INDOLE-3-ACETIC ACID gene family members in Arabidopsis thaliana. Plant Cell 17, 3282–3300. doi: 10.1105/tpc.105.036723
Ozsolak, F., and Milos, P. M. (2011). RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 12, 87–98. doi: 10.1038/nrg2934
Parchman, T. L., Geist, K. S., Grahnen, J. A., Benkman, C. W., and Buerkle, C. A. (2010). Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics 11:180. doi: 10.1186/1471-2164-11-180
Parra, R., Paredes, M. A., Sanchez-Calle, I. M., and Gomez-Jimenez, M. C. (2013). Comparative transcriptional profiling analysis of olive ripe-fruit pericarp and abscission zone tissues shows expression differences and distinct patterns of transcriptional regulation. BMC Genomics 14:866. doi: 10.1186/1471-2164-14-866
Pascuzzi, P., Hamilton, D., Bodily, K., and Arias, J. (1998). Auxin-induced stress potentiates trans-activation by a conserved plant basic/leucine-zipper factor. J. Biol. Chem. 273, 26631–26637. doi: 10.1074/jbc.273.41.26631
Pattison, R. J., and Catalá, C. (2012). Evaluating auxin distribution in tomato (Solanum lycopersicum) through an analysis of the PIN and AUX/LAX gene families. Plant J. 70, 585–598. doi: 10.1111/j.1365-313X.2011.04895.x
Payton, S., Fray, R. G., Brown, S., and Grierson, D. (1996). Ethylene receptor expression is regulated during fruit ripening, flower senescence and abscission. Plant Mol. Biol. 31, 1227–1231. doi: 10.1007/BF00040839
Petrásek, J., and Friml, J. (2009). Auxin transport routes in plant development. Development 136, 2675–2688. doi: 10.1242/dev.030353
Quinet, M., Kinet, J. M., and Lutts, S. (2011). Flowering response of the uniflora:blind:self-pruning and jointless:uniflora:self-pruning tomato (Solanum lycopersicum) triple mutants. Physiol. Plant. 141, 166–176. doi: 10.1111/j.1399-3054.2010.01426.x
Rajani, S., and Sundaresan, V. (2001). The Arabidopsis myc/bHLH gene ALCATRAZ enables cell separation in fruit dehiscence. Curr. Biol. 11, 1914–1922. doi: 10.1016/S0960-9822(01)00593-0
Rick, C. M. (1956). Genetic and systematic studies on accessions of Lycospersicon from the Galapagos Islands. Am. J. Bot. 43, 687–696. doi: 10.2307/2438834
Riechmann, J. L., Heard, J., Martin, G., Reuber, L., Jiang, C., Keddie, J., et al. (2000). Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290, 2105–2110. doi: 10.1126/science.290.5499.2105
Roberts, J. A., Elliott, K. A., and Gonzalez-Carranza, Z. H. (2002). Abscission, dehiscence, and other cell separation processes. Annu. Rev. Plant Biol. 53, 131–158. doi: 10.1146/annurev.arplant.53.092701.180236
Roberts, J. A., Schindler, C. B., and Tucker, G. A. (1984). Ethylene-promoted tomato flower abscission and the possible involvement of an inhibitor. Planta 160, 159–163. doi: 10.1007/BF00392864
Roberts, J. A., Whitelaw, C. A., Gonzalez-Carranza, Z. H., and McManus, M. T. (2000). Cell separation processes in plants—models, mechanisms and manipulation. Ann. Bot. 86, 223–235. doi: 10.1006/anbo.2000.1203
Roman, G., Lubarsky, B., Kieber, J. J., Rothenberg, M., and Ecker, J. R. (1995). Genetic analysis of ethylene signal transduction in Arabidopsis thaliana: five novel mutant loci integrated into a stress response pathway. Genetics 139, 1393–1409.
Rushton, P. J., Somssich, I. E., Ringler, P., and Shen, Q. J. (2010). WRKY transcription factors. Trends Plant Sci. 15, 247–258. doi: 10.1016/j.tplants.2010.02.006
Rutjens, B., Bao, D., van Eck-Stouten, E., Brand, M., Smeekens, S., and Proveniers, M. (2009). Shoot apical meristem function in Arabidopsis requires the combined activities of three BEL1-like homeodomain proteins. Plant J. 58, 641–654. doi: 10.1111/j.1365-313X.2009.03809.x
Sangwan, R. S., Tripathi, S., Singh, J., Narnoliya, L. K., and Sangwan, N. S. (2013). De novo sequencing and assembly of Centella asiatica leaf transcriptome for mapping of structural, functional and regulatory genes with special reference to secondary metabolism. Gene 525, 58–76. doi: 10.1016/j.gene.2013.04.057
Schmitz, G., Tillmann, E., Carriero, F., Fiore, C., Cellini, F., and Theres, K. (2002). The tomato Blind gene encodes a MYB transcription factor that controls the formation of lateral meristems. Proc. Natl. Acad. Sci. U.S.A. 99, 1064–1069. doi: 10.1073/pnas.022516199
Schulz, M. H., Zerbino, D. R., Vingron, M., and Birney, E. (2012). Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092. doi: 10.1093/bioinformatics/bts094
Schumacher, K., Schmitt, T., Rossberg, M., Schmitz, G., and Theres, K. (1999). The Lateral suppressor (Ls) gene of tomato encodes a new member of the VHIID protein family. Proc. Natl. Acad. Sci. U.S.A. 96, 290–295. doi: 10.1073/pnas.96.1.290
Sexton, R., and Roberts, J. A. (1982). Cell biology of abscission. Annu. Rev. Plant Physiol. 33, 133–162. doi: 10.1146/annurev.pp.33.060182.001025
Shalit, A., Rozman, A., Goldshmidt, A., Alvarez, J. P., Bowman, J. L., Eshed, Y., et al. (2009). The flowering hormone florigen functions as a general systemic regulator of growth and termination. Proc. Natl. Acad. Sci. U.S.A. 106, 8392–8397. doi: 10.1073/pnas.0810810106
Shi, C. L., Stenvik, G. E., Vie, A. K., Bones, A. M., Pautot, V., Proveniers, M., et al. (2011). Arabidopsis class I KNOTTED-like homeobox proteins act downstream in the IDA-HAE/HSL2 floral abscission signaling pathway. Plant Cell 23, 2553–2567. doi: 10.1105/tpc.111.084608
Singh, A. P., Dubey, S., Lakhwani, D., Pandey, S. P., Khan, K., Dwivedi, U. N., et al. (2013). Differential expression of several xyloglucan endotransglucosylase/hydrolase genes regulates flower opening and petal abscission in roses. AoB Plants 5:plt030. doi: 10.1093/aobpla/plt030
Solano, R., Stepanova, A., Chao, Q., and Ecker, J. R. (1998). Nuclear events in ethylene signaling: a transcriptional cascade mediated by ETHYLENE-INSENSITIVE3 and ETHYLENE-RESPONSE-FACTOR1. Genes Dev. 12, 3703–3714. doi: 10.1101/gad.12.23.3703
Sorefan, K., Girin, T., Liljegren, S. J., Ljung, K., Robles, P., Galván-Ampudia, C. S., et al. (2009). A regulated auxin minimum is required for seed dispersal in Arabidopsis. Nature 459, 583–586. doi: 10.1038/nature07875
Staswick, P. E., Serban, B., Rowe, M., Tiryaki, I., Maldonado, M. T., Maldonado, M. C., et al. (2005). Characterization of an Arabidopsis enzyme family that conjugates amino acids to indole-3-acetic acid. Plant Cell 17, 616–627. doi: 10.1105/tpc.104.026690
Sun, H., Fan, H. J., and Ling, H. Q. (2015). Genome-wide identification and characterization of the bHLH gene family in tomato. BMC Genomics 16:9. doi: 10.1186/s12864-014-1209-2
Szymkowiak, E. J., and Irish, E. E. (2006). JOINTLESS suppresses sympodial identity in inflorescence meristems of tomato. Planta 223, 646–658. doi: 10.1007/s00425-005-0115-x
Takatsuji, H. (1998). Zinc-finger transcription factors in plants. Cell. Mol. Life Sci. 54, 582–596. doi: 10.1007/s000180050186
Takatsuji, H. (1999). Zinc-finger proteins: the classical zinc finger emerges in contemporary plant science. Plant Mol. Biol. 39, 1073–1078. doi: 10.1023/A:1006184519697
Talbert, P. B., Adler, H. T., Parks, D. W., and Comai, L. (1995). The REVOLUTA gene is necessary for apical meristem development and for limiting cell divisions in the leaves and stems of Arabidopsis thaliana. Development 121, 2723–2735.
Tanno, K., and Willcox, G. (2006). How fast was wild wheat domesticated? Science 311, 1886. doi: 10.1126/science.1124635
Taylor, J. E., and Whitelaw, C. A. (2001). Signals in abscission. New Phytol. 151, 323–340. doi: 10.1046/j.0028-646x.2001.00194.x
The Tomato Genome Consortium (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641. doi: 10.1038/nature11119
Theissen, G., and Saedler, H. (2001). Plant biology. Floral quartets. Nature 409, 469–471. doi: 10.1038/35054172
Tieman, D. M., Ciardi, J. A., Taylor, M. G., and Klee, H. J. (2001). Members of the tomato LeEIL (EIN3-like) gene family are functionally redundant and regulate ethylene responses throughout plant development. Plant J. 26, 47–58. doi: 10.1046/j.1365-313x.2001.01006.x
Tucker, M. L., Burke, A., Murphy, C. A., Thai, V. K., and Ehrenfried, M. L. (2007). Gene expression profiles for cell wall-modifying proteins associated with soybean cyst nematode infection, petiole abscission, root tips, flowers, apical buds, and leaves. J. Exp. Bot. 58, 3395–3406. doi: 10.1093/jxb/erm188
Tucker, M. L., Whitelaw, C. A., Lyssenko, N. N., and Nath, P. (2002). Functional analysis of regulatory elements in the gene promoter for an abscission-specific cellulase from bean and isolation, expression, and binding affinity of three TGA-type basic leucine zipper transcription factors. Plant Physiol. 130, 1487–1496. doi: 10.1104/pp.007971
Van Nocker, S. (2009). Development of the abscission zone. Stewart Posthar. Rev. 5, 1–6. doi: 10.2212/spr.2009.1.5
Vanneste, S., and Friml, J. (2009). Auxin: a trigger for change in plant development. Cell 136, 1005–1016. doi: 10.1016/j.cell.2009.03.001
Wang, J., Hu, Z., Zhao, T., Yang, Y., Chen, T., Yang, M., et al. (2015). Genome-wide analysis of bHLH transcription factor and involvement in the infection by yellow leaf curl virus in tomato (Solanum lycopersicum). BMC Genomics 16:39. doi: 10.1186/s12864-015-1249-2
Wang, L., Wang, X., Yue, C., Cao, H., Zhou, Y., and Yang, Y. (2014a). Development of a 44 K custom oligo microarray using 454 pyrosequencing data for large-scale gene expression analysis of Camellia sinensis. Sci. Hortic. 174, 133–141. doi: 10.1016/j.scienta.2014.05.024
Wang, S., Li, E., Porth, I., Chen, J. G., Mansfield, S. D., and Douglas, C. J. (2014b). Regulation of secondary cell wall biosynthesis by poplar R2R3 MYB transcription factor PtrMYB152 in Arabidopsis. Sci. Rep. 4:5054. doi: 10.1038/srep05054
Wang, X., Liu, D., Li, A., Sun, X., Zhang, R., Wu, L., et al. (2013). Transcriptome analysis of tomato flower pedicel tissues reveals abscission zone-specific modulation of key meristem activity genes. PLoS ONE 8:e55238. doi: 10.1371/journal.pone.0055238
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wolberger, C. (1999). Multiprotein-DNA complexes in transcriptional regulation. Annu. Rev. Biophys. Biomol. Struct. 28, 29–56. doi: 10.1146/annurev.biophys.28.1.29
Woodward, A. W., and Bartel, B. (2005). Auxin: regulation, action, and interaction. Ann. Bot. 95, 707–735. doi: 10.1093/aob/mci083
Worley, C. K., Zenser, N., Ramos, J., Rouse, D., Leyser, O., Theologis, A., et al. (2000). Degradation of Aux/IAA proteins is essential for normal auxin signalling. Plant J. 21, 553–562. doi: 10.1046/j.1365-313x.2000.00703.x
Wu, J., Peng, Z., Liu, S., He, Y., Cheng, L., Kong, F., et al. (2012). Genome-wide analysis of Aux/IAA gene family in Solanaceae species using tomato as a model. Mol. Genet. Genomics 287, 295–211. doi: 10.1007/s00438-012-0675-y
Wu, J., Wang, F., Cheng, L., Kong, F., Peng, Z., Liu, S., et al. (2011). Identification, isolation and expression analysis of auxin response factor (ARF) genes in Solanum lycopersicum. Plant Cell Rep. 30, 2059–2073. doi: 10.1007/s00299-011-1113-z
Wu, J., Wang, J., Pan, C., Guan, X., Wang, Y., Liu, S., et al. (2014). Genome-wide identification of MAPKK and MAPKKK gene families in tomato and transcriptional profiling analysis during development and stress response. PLoS ONE 9:e103032. doi: 10.1371/journal.pone.0103032
Xie, D. X., Feys, B. F., James, S., Nieto-Rostro, M., and Turner, J. G. (1998). COI1: an Arabidopsis gene required for jasmonate-regulated defense and fertility. Science 280, 1091–1094. doi: 10.1126/science.280.5366.1091
Xie, R., Pang, S., Ma, Y., Deng, L., He, S., Yi, S., et al. (2015). The ARF, AUX/IAA and GH3 gene families in citrus: genome-wide identification and expression analysis during fruitlet drop from abscission zone A. Mol. Genet. Genomics 290, 2089–2105. doi: 10.1007/s00438-015-1063-1
Xu, P., Liu, Z., Fan, X., Gao, J., Zhang, X., Zhang, X., et al. (2013). De novo transcriptome sequencing and comparative analysis of differentially expressed genes in Gossypium aridum under salt stress. Gene 525, 26–34. doi: 10.1016/j.gene.2013.04.066
Yang, Y., Xu, R., Ma, C. J., Vlot, A. C., Klessig, D. F., and Pichersky, E. (2008). Inactive methyl indole-3-acetic acid ester can be hydrolyzed and activated by several esterases belonging to the AtMES esterase family of Arabidopsis. Plant Physiol. 147, 1034–1045. doi: 10.1104/pp.108.118224
Yasuike, M., Fujiwara, A., Nakamura, Y., Iwasaki, Y., Nishiki, I., Sugaya, T., et al. (2015). A functional genomics tool for the Pacific bluefin tuna: development of a 44K oligonucleotide microarray from whole-genome sequencing data for global transcriptome analysis. Gene 576, 603–609. doi: 10.1016/j.gene.2015.10.023
Yelin, R., Dahary, D., Sorek, R., Levanon, E. Y., Goldstein, O., Shoshan, A., et al. (2003). Widespread occurrence of antisense transcription in the human genome. Nat. Biotechnol. 21, 379–386. doi: 10.1038/nbt808
Ying, B., Toth, K., Spencer, J. F., Aurora, R., and Wold, W. S. (2015). Transcriptome sequencing and development of an expression microarray platform for liver infection in adenovirus type 5-infected Syrian golden hamsters. Virology 485, 305–312. doi: 10.1016/j.virol.2015.07.024
Zenoni, S., Fasoli, M., Tornielli, G. B., Dal Santo, S., Sanson, A., de Groot, P., et al. (2011). Overexpression of PhEXPA1 increases cell size, modifies cell wall polymer composition and affects the timing of axillary meristem development in Petunia hybrida. New Phytol. 191, 662–677. doi: 10.1111/j.1469-8137.2011.03726.x
Zhang, H., Jin, J., Tang, L., Zhao, Y., Gu, X., Gao, G., et al. (2011). PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 39, D1114–D1117. doi: 10.1093/nar/gkq1141
Zhang, J. Z., Zhao, K., Ai, X. Y., and Hu, C. G. (2014). Involvements of PCD and changes in gene expression profile during self-pruning of spring shoots in sweet orange (Citrus sinensis). BMC Genomics 15:892. doi: 10.1186/1471-2164-15-892
Zhong, S., Joung, J. G., Zheng, Y., Chen, Y. R., Liu, B., Shao, Y., et al. (2011). High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 940–949. doi: 10.1101/pdb.prot5652
Zhu, H., Dardick, C. D., Beers, E. P., Callanhan, A. M., Xia, R., and Yuan, R. (2011). Transcriptomics of shading-induced and NAA-induced abscission in apple (Malus domestica) reveals a shared pathway involving reduced photosynthesis, alterations in carbohydrate transport and signaling and hormone crosstalk. BMC Plant Biol. 11:138. doi: 10.1186/1471-2229-11-138
Keywords: auxin, ethylene, flower pedicel abscission, leaf petiole abscission, oligonucleotide microarray, RNA-Sequencing, tomato (Solanum lycopersicum), transcriptome
Citation: Sundaresan S, Philosoph-Hadas S, Riov J, Mugasimangalam R, Kuravadi NA, Kochanek B, Salim S, Tucker ML and Meir S (2016) De novo Transcriptome Sequencing and Development of Abscission Zone-Specific Microarray as a New Molecular Tool for Analysis of Tomato Organ Abscission. Front. Plant Sci. 6:1258. doi: 10.3389/fpls.2015.01258
Received: 22 September 2015; Accepted: 24 December 2015;
Published: 14 January 2016.
Edited by:
Richard S. Winder, Natural Resources Canada, CanadaReviewed by:
Antonio Ferrante, Università degli Studi di Milano, ItalyAutar Krishen Mattoo, United States Department of Agriculture, USA
Copyright © 2016 Sundaresan, Philosoph-Hadas, Riov, Mugasimangalam, Kuravadi, Kochanek, Salim, Tucker and Meir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shimon Meir, shimonm@volcani.agri.gov.il