 Alex D. Twyford
Alex D. Twyford
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Plant Sci., 06 April 2016
Sec. Plant Genetics and Genomics
Volume 7 - 2016 | https://doi.org/10.3389/fpls.2016.00433
An important experimental design consideration in plant genetics is the trade-off between number of individuals and number of loci that can be genotyped (Davey et al., 2011). For any given study, an investigator must choose how they partition research effort and resources, with the generation of many loci usually coming at the expense of many individuals, and vice versa. For example, for parentage and paternity analysis it is usually more important to sample many individuals (e.g., Andrew et al., 2013), while for comparative genome evolution the emphasis is firmly placed on recovering more loci (Figure 1). This trade-off still exits despite the plummeting costs of sequence data, with researchers having to decide the number of individuals feasible for a given sequencing strategy, and how the libraries will be multiplexed across lanes of a next generation sequencing (NGS) platform (Shen et al., 2011).
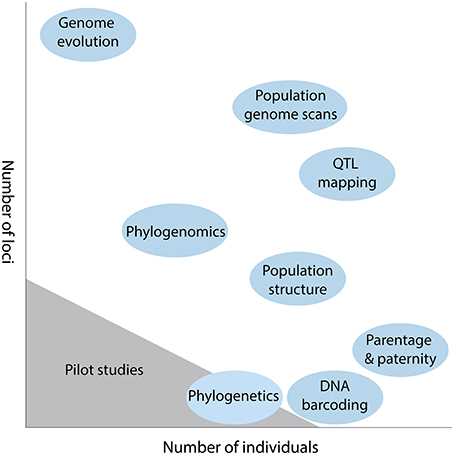
Figure 1. Diagrammatic representation of the trade-off between number of loci and number of individuals in some typical plant genetic studies. The scale of sequencing for pilot studies is also indicated.
NGS is well-suited to studies requiring large amounts of sequence data for few individuals, such as de novo genome assembly, or large-scale genome resequencing projects (e.g., Brandvain et al., 2014). At the other extreme, high-throughput sequencing is also ideal for single-locus studies of environmental variation, where universal primers are used to amplify a diverse mix of template DNA representing thousands of individuals (e.g., Shokralla et al., 2012). The sequencing trade-off space traditionally least well-served by NGS is where tens or hundreds of loci need to be generated for many individuals. While Restriction-site Associated DNA (RAD) sequencing and genotyping-by-sequencing (GBS, Elshire et al., 2011) partly fill this gap, there are many applications in population genetics, phylogenetics, DNA barcoding, and parentage analysis where a standard multiplexed RAD library run on a high-throughput sequencer would provide an excessive number of loci or unnecessarily high depth-of-coverage. Therefore, researchers wanting a modest number of loci would be more likely to consider either SNP chips, which can be costly to develop and may produce data with ascertainment bias (Albrechtsen et al., 2010), or continue using conventional markers such as microsatellites, or Sanger Sequencing of individual loci.
The uptake of NGS in small to medium-scale studies may be set to increase with the recent announcement of a new benchtop sequencing platform, the Illumina MiniSeq (http://www.illumina.com/miniseq). This sequencer has two key benefits over its larger cousins such as the Illumina HiSeq. Firstly, the MiniSeq fills a gap at the low-read production end of the market, generating 1.8–7.5 Gb of data [8–50 million (M) reads]. These data have a low error rate (>80% bases >Q30), and the platform offers some flexibility over read length configuration [36, 50, 75, 150 bp single end (SE) or paired end (PE) sequences]. Secondly, the MiniSeq is the first Illumina platform designed for smaller research institutions or individual laboratories. The instrument itself costs around $50,000, has a small footprint, relatively short run time, and the capacity to sequence a single sample (rather than the need to fill multiple lanes of a larger flow cell). As such, this sequencer may enable users to avoid queues and administration associated with large sequencing centers, and open up in-house genomics for the first time.
The MiniSeq joins a number of other NGS platforms capable of relatively small sequence runs (e.g., 400 Mb–15 Gb), such as the Illumina MiSeq and ThermoFisher's Ion Torrent, and third generation technologies such as Oxford NanoPore and Pacific BioSciences real-time sequencers (for full comparison see http://www.molecularecologist.com/next-gen-fieldguide-2016/). The MiniSeq's small footprint and low upfront cost make it a more attractive option for lab ownership than the MiSeq, and also boasts the lowest reagent costs for small Illumina sequencing runs (MiniSeq mid-output reagents $550 per run). However, the MiniSeq offers no cost benefits for higher-output runs, and has a shorter maximum read length than the MiSeq (150 bp as opposed to 300 bp). Ion Torrent systems such as the Ion S5 are another low-output benchtop alternative to the MiniSeq, and the fast run time make them the platform of preference for clinical diagnostics. Ion Torrent has not widely been used for non-model genomics (though see Recknagel et al., 2015), likely due to some sequence biases, moderate error rates, and difficulty reading homopolymer regions, particular with early release platforms (Loman et al., 2012; Quail et al., 2012; Salipante et al., 2014). Third generation sequencing options are Oxford NanoPore's MinION (Mikheyev and Tin, 2014) or Pacific BioSciences real-time sequencers (Jiao et al., 2013). While the long sequence reads (>5 Kb) make them extremely useful for de novo assembly of small genomes, and scaffolding non-model genomes (English et al., 2012), they have not been widely adopted for other research applications due to their high costs, error rates, and currently limited (but growing) number of bioinformatic pipelines.
The potential applications of low-output benchtop sequencers, such as the MiniSeq, are huge. The first important use would be in replacing panels of PCR-based markers in studies relying on modest numbers of loci. In phylogenetics, multiplexed tagged amplicons could be sequenced with sufficient sequencing depth, but at a cheaper cost and without the redundancy of higher-output platforms. For nuclear loci, this approach removes the time-consuming stage of cloning, and can provide directly phased sequences (O'Neill et al.,2013). Similarly, targeted enrichment studies such as those using hybridization-based probes are ideal for low-output sequencers, as sequencing effort is focused on a small subsection of the genome (e.g., Stull et al., 2013). In mating system studies, GBS libraries prepared with an infrequent cutting enzyme could be a time and cost effective way to generate a modest number of loci in many progeny derived from many seed families, leading to accurate estimates of outcrossing (Koelling et al., 2012). In all these cases, the output of the MiniSeq is optimized for part of the sequencing trade-off where many other platforms are not.
The second main use would be in genomic studies where few individuals need to be sequenced. MiniSeq runs would be suitable for sequencing small plant genomes (e.g., >50X coverage of 135 Mb Arabidopsis thaliana), or for characterizing features such as GC-content, transposon composition (Sveinsson et al., 2013), and genome size (Simpson, 2014) of non-model species. This output range could also be useful for multiplexed low coverage genome resequencing (“genome skimming,” Straub et al., 2012), which is proving a popular route for complete plastid assembly (e.g., Jackman et al., 2016). The low sequence run cost would also make this ideal for marker discovery and developing microsatellite primers (Zalapa et al., 2012).
The third use would be for pilot studies testing new sample assays and for validating libraries constructed from difficult samples. Low-output sequencing runs would be extremely valuable to verify the number of tags and the sequencing coverage in test RAD libraries. Similarly, targeted enrichment strategies could be tested at low coverage to check the efficacy of the enrichment and the proportion of off-bait targets. This information can then be used to pick the depth of coverage for large-scale sequencing efforts, with the same Illumina-compatible libraries being transferable across sequencing platforms. For validating samples, low-output sequencing runs could be used to assess the number of informative reads and the extent of sample contamination in dietary or environmental samples (e.g., Willerslev et al., 2014). In studies using degraded herbarium samples, the extent of C → T/G → A miscoding lesions caused by DNA degradation (Staats et al., 2011), could be assessed. This is particularly important as this may not be captured by other quality control metrics, such as those produced by the Agilent TapeStation or Bioanalyser. In all these cases, the small datasets would be able to address issues that would otherwise only come to light with greater sequencing effort.
NGS is providing a number of important solutions to the sequencing trade-off in plant genetic studies, with benchtop sequencers such as the Illumina MiniSeq potentially facilitating day-to-day low-output sequencing. However, the success of such platforms is far from guaranteed. The most cost-effective sequencing comes from high-output platforms such as the Illumina HiSeq 4000, and highly multiplexed libraries or pools of individuals (Pool-seq, Schlötterer et al., 2014) run on such systems have the lowest per-megabase costs. Therefore, current high-output systems may continue to meet most researcher's needs, leaving only a small gap in the market for these platforms. Another issue is the methodological challenges and costs associated with preparing NGS libraries (often $30–100/sample), and the bioinformatics involved in calling reliable variant sites, which may outweigh the benefits of conventional markers for some small-scale studies where these platforms could be useful. A final concern is whether research groups want to own and run their own sequencer, when technical assistance is available at larger sequencing hubs. As such, while the MiniSeq has great potential on paper, whether it really resolves the sequencing trade-off at the low-output end of the market remains to be seen.
The author confirms being the sole contributor of this work and approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Research by AT is supported by NERC Award NE/L011336/1.
Albrechtsen, A., Nielsen, F. C., and Nielsen, R. (2010). Ascertainment biases in SNP chips affect measures of population divergence. Mol. Biol. Evol. 27, 2534–2547. doi: 10.1093/molbev/msq148
Andrew, R. L., Bernatchez, L., Bonin, A., Buerkle, C. A., Carstens, B. C., Emerson, B. C., et al. (2013). A road map for molecular ecology. Mol. Ecol. 22, 2605–2626. doi: 10.1111/mec.12319
Brandvain, Y., Kenney, A. M., Flagel, L., Coop, G., and Sweigart, A. L. (2014). Speciation and introgression between Mimulus nasutus and Mimulus guttatus. PLoS Genet. 10:e1004410. doi: 10.1371/journal.pgen.1004410
Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Catchen, J. M., and Blaxter, M. L. (2011). Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 12, 499–510. doi: 10.1038/nrg3012
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. doi: 10.1371/journal.pone.0019379
English, A. C., Richards, S., Han, Y., Wang, M., Vee, V., Qu, J., et al. (2012). Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7:e47768. doi: 10.1371/journal.pone.0047768
Jackman, S. D., Warren, R. L., Gibb, E. A., Vandervalk, B. P., Mohamadi, H., Chu, J., et al. (2016). Organellar genomes of white spruce (Picea glauca): assembly and annotation. Genome Biol. Evol. 8, 29–41. doi: 10.1093/gbe/evv244
Jiao, X., Zheng, X., Ma, L., Kutty, G., Gogineni, E., Sun, Q., et al. (2013). A benchmark study on error assessment and quality control of CCS reads derived from the PacBio, RS.J. Data Mining Genomics Proteomics 4, 16008. doi: 10.4172/2153-0602.1000136
Koelling, V. A., Monnahan, P. J., and Kelly, J. K. (2012). A Bayesian method for the joint estimation of outcrossing rate and inbreeding depression. Heredity (Edinb). 109, 393–400. doi: 10.1038/hdy.2012.58
Loman, N. J., Misra, R. V., Dallman, T. J., Constantinidou, C., Gharbia, S. E., Wain, J., et al. (2012). Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotech. 30, 434–439. doi: 10.1038/nbt.2198
Mikheyev, A. S., and Tin, M. M. (2014). A first look at the Oxford Nanopore MinION sequencer. Mol. Ecol. Res. 14, 1097–1102. doi: 10.1111/1755-0998.12324
O'Neill, E. M., Schwartz, R., Bullock, C. T., Williams, J. S., Shaffer, H. B., Aguilar-Miguel, X., et al. (2013). Parallel tagged amplicon sequencing reveals major lineages and phylogenetic structure in the North American tiger salamander (Ambystoma tigrinum) species complex. Mol. Ecol. 22, 111–129. doi: 10.1111/mec.12049
Quail, M. A., Smith, M., Coupland, P., Otto, T. D., Harris, S. R., Connor, T. R., et al. (2012). A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13:341. doi: 10.1186/1471-2164-13-341
Recknagel, H., Jacobs, A., Herzyk, P., and Elmer, K. R. (2015). Double-digest RAD sequencing using Ion Proton semiconductor platform (ddRADseq-ion) with nonmodel organisms. Mol. Ecol. Res. 15, 1316–1329. doi: 10.1111/1755-0998.12406
Salipante, S. J., Kawashima, T., Rosenthal, C., Hoogestraat, D. R., Cummings, L. A., Sengupta, D. J., et al. (2014). Performance comparison of Illumina and Ion Torrent next-generation sequencing platforms for 16S rRNA-based bacterial community profiling. Appl. Environ. Microbiol. 80, 7583–7591. doi: 10.1128/AEM.02206-14
Schlötterer, C., Tobler, R., Kofler, R., and Nolte, V. (2014). Sequencing pools of individuals—mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 15, 49–763. doi: 10.1038/nrg3803
Shen, Y., Song, R., and Pe'er, I. (2011). Coverage tradeoffs and power estimation in the design of whole-genome sequencing experiments for detecting association. Bioinformatics 27, 1995–1997. doi: 10.1093/bioinformatics/btr305
Shokralla, S., Spall, J. L., Gibson, J. F., and Hajibabaei, M. (2012). Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 21, 1794–1805. doi: 10.1111/j.1365-294X.2012.05538.x
Simpson, J. T. (2014). Exploring genome characteristics and sequence quality without a reference. Bioinformatics 30, 1228–1235. doi: 10.1093/bioinformatics/btu023
Staats, M., Cuenca, A., Richardson, J. E., Vrielink-van Ginkel, R., Petersen, G., Seberg, O., et al. (2011). DNA damage in plant herbarium tissue. PLoS ONE 6:e28448. doi: 10.1371/journal.pone.0028448
Straub, S. C., Parks, M., Weitemier, K., Fishbein, M., Cronn, R. C., and Liston, A. (2012). Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am. J. Bot. 99, 349–364. doi: 10.3732/ajb.1100335
Stull, G. W., Moore, M. J., Mandala, V. S., Douglas, N. A., Kates, H.-R., and Qi, X. (2013). A targeted enrichment strategy for massively parallel sequencing of angiosperm plastid genomes. Appl. Plant Sci. 1:1200497. doi: 10.3732/apps. 1200497
Sveinsson, S., Gill, N., Kane, N. C., and Cronk, Q. (2013). Transposon fingerprinting using low coverage whole genome shotgun sequencing in Cacao (Theobroma cacao L.) and related species. BMC Genomics 14, 1–12. doi: 10.1186/1471-2164-14-502
Willerslev, E., Davison, J., Moora, M., Zobel, M., Coissac, E., Edwards, M. E., et al. (2014). Fifty thousand years of Arctic vegetation and megafaunal diet. Nature 506, 47–51. doi: 10.1038/nature12921
Keywords: genomics, next generation sequencing, benchtop sequencers, Illumina MiniSeq, population genetics, phylogenetics, parentage analysis
Citation: Twyford AD (2016) Will Benchtop Sequencers Resolve the Sequencing Trade-off in Plant Genetics? Front. Plant Sci. 7:433. doi: 10.3389/fpls.2016.00433
Received: 01 February 2016; Accepted: 21 March 2016;
Published: 06 April 2016.
Edited by:
Paula Casati, Centro de Estudios Fotosinteticos-CONICET, ArgentinaReviewed by:
Sean Warren Graham, University of British Columbia, CanadaCopyright © 2016 Twyford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alex D. Twyford, YWxleC50d3lmb3JkQGVkLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.