 Amit Rai1*
Amit Rai1* Mami Yamazaki1*
Mami Yamazaki1* Hiroki Takahashi2
Hiroki Takahashi2 Michimi Nakamura1
Michimi Nakamura1 Mareshige Kojoma3
Mareshige Kojoma3 Hideyuki Suzuki4
Hideyuki Suzuki4 Kazuki Saito1
Kazuki Saito1- 1Graduate School of Pharmaceutical Sciences, Chiba University, Chiba, Japan
- 2Medical Mycology Research Center, Chiba University, Chiba, Japan
- 3Faculty of Pharmaceutical Sciences, Health Sciences University of Hokkaido, Hokkaido, Japan
- 4Kazusa DNA Research Institute, Chiba, Japan
The Panax genus has been a source of natural medicine, benefitting human health over the ages, among which the Panax japonicus represents an important species. Our understanding of several key pathways and enzymes involved in the biosynthesis of ginsenosides, a pharmacologically active class of metabolites and a major chemical constituents of the rhizome extracts from the Panax species, are limited. Limited genomic information, and lack of studies on comparative transcriptomics across the Panax species have restricted our understanding of the biosynthetic mechanisms of these and many other important classes of phytochemicals. Herein, we describe Illumina based RNA sequencing analysis to characterize the transcriptome and expression profiles of genes expressed in the five tissues of P. japonicus, and its comparison with other Panax species. RNA sequencing and de novo transcriptome assembly for P. japonicus resulted in a total of 135,235 unigenes with 78,794 (58.24%) unigenes being annotated using NCBI-nr database. Transcriptome profiling, and gene ontology enrichment analysis for five tissues of P. japonicus showed that although overall processes were evenly conserved across all tissues. However, each tissue was characterized by several unique unigenes with the leaves showing the most unique unigenes among the tissues studied. A comparative analysis of the P. japonicus transcriptome assembly with publically available transcripts from other Panax species, namely, P. ginseng, P. notoginseng, and P. quinquefolius also displayed high sequence similarity across all Panax species, with P. japonicus showing highest similarity with P. ginseng. Annotation of P. japonicus transcriptome resulted in the identification of putative genes encoding all enzymes from the triterpene backbone biosynthetic pathways, and identified 24 and 48 unigenes annotated as cytochrome P450 (CYP) and glycosyltransferases (GT), respectively. These CYPs and GTs annotated unigenes were conserved across all Panax species and co-expressed with other the transcripts involved in the triterpenoid backbone biosynthesis pathways. Unigenes identified in this study represent strong candidates for being involved in the triterpenoid saponins biosynthesis, and can serve as a basis for future validation studies.
Introduction
Medicinal plants are a rich source of diverse bioactive compounds that play an important role in the management of human health. Availability and understanding of the phytogenome sequences, and transcriptomics information of medicinal plants provide a broad understanding of different ongoing metabolic processes, their localization across different plant tissues, and the cellular biosynthetic mechanisms involved in their biosynthesis (Rai and Saito, 2015). Complete genome sequencing of a medicinal plant has its own challenges which include the complexity of the genome and the sequencing cost, and the computational resources required, resulting in only few complete genomes of medicinal plants that are available. Recent advancements in the next generation sequencing (NGS) with reduced operational cost for such experiments, and development of computational resources to perform de novo transcriptome assembly, annotation, and characterization have revolutionized the field of natural medicine (Muranaka and Saito, 2013; Saito, 2013). The information from transcript sequences and their relative abundance across tissues in a medicinal plant provide key insight on important aspects of gene structure, function, and regulation including non-coding RNAs or differentially expressed genes.
Among medicinal plants, Panax japonicus CA Meyer, along with other Panax species such as P. notoginseng, P. quinquefolius, and particularly, P. ginseng are the most popular and among the list of the best-selling medicinal plants in the world (Briskin, 2000; Qi et al., 2011). ‘Panax’ is derived from a Greek word, ‘Panacea,’ which means cure for all. It includes 12 established species, P. japonicus being one of them, with useful medicinal properties (Han et al., 2013). P. japonicus, also known as ‘Chikusetsu-ninjin’ in Japan, belongs to the genus Panax of the family Araliaceae, and grows wild throughout Japan, China and Korea with a characteristic long bamboo-like rhizome (Morita et al., 1985). It has been used as a traditional medicinal herb and as a substitute to P. ginseng in minority ethnic group for 1000 years (Yang X. et al., 2014).
The rhizome extracts from P. japonicus have reportedly been used for the treatment of several life-style related diseases, such as arteriosclerosis, hyperlipidaemia, hypertension, and non-insulin dependent diabetes (Vogler et al., 1999; Han et al., 2005). Extracts from dried rhizome of P. japonicus have been used for 100s of years in Japan as a drug for gastroenteric disorder, anti-ulcer, expectorants, and anti-pyretic, while in China, it have been used as tonic, anti-inflammatory, and haemostatic agents (Zou et al., 2002). According to the description in the “Ben Cao Gong Mu Shi Yi,” rhizomes of P. japonicus possess combined medicinal properties of “conserving vitality” and “replenishing blood” activities attributed to P. ginseng and P. notoginseng, respectively, and therefore was named as the “king of herbs” in the traditional Tujia and Hmong medicine (Zhang et al., 2015). P. japonicus has been included in the Chinese and Japanese pharmacopeia for its various pharmacological properties, such as anti-ulcer, anti-obesity, analgesic, anti-fatigue, anti-oxidant, anti-cancer, and immuno-regulation (Yamahara et al., 1987; Yun, 2001; Han et al., 2005; Yang X. et al., 2014). Despite of well-documented medicinal properties of P. japonicus, little is known about its transcriptome, and its genome is not sequenced.
For Panax species, most of their therapeutic effects have been attributed to its metabolic extracts being rich in pharmacologically active constituents, known as ginsenosides (Shibata, 2001; Murthy et al., 2014; Yang W.Z. et al., 2014). Ginsenosides are the oligosaccharide glycosides of dammarane- or oleanane-type triterpenoids. Ginsenosides biosynthetic pathway derives its precursor from the triterpene backbone pathways, starting from acetyl-CoA toward the synthesis of 2,3-oxidosqualene, cyclization of which forms the branch point between dammarane- and oleanane-type terpenoids. These reactions are catalyzed by the cytochrome P450 enzymes, little of which involved in ginsenosides biosynthesis are known in Panax species (Han et al., 2011, 2013). Triterpenoids are synthesized through one of the major isoprenoid biosynthetic pathway, and are one of the most important classes of natural product, glycoside conjugation of which has important medicinal properties. Ginsenosides saponins are highly diversified, with 289 different types of saponins being isolated and characterized so far in pure form from either eleven different Panax species, or chemically derived from their extracts (Yang W.Z. et al., 2014). Ginsenosides composition varies significantly across Panax species, with P. ginseng, P. notoginseng, P. quinquefolius, and P. vietnamisis being grouped in the dammarane-type saponins rich species, while P. japonicus, P. zingiberensis, and P. stipuleanatus being grouped in the oleanane-type saponins rich species (Zhu et al., 2004). The total saponins content in P. japonicus rhizome can reach to 15% of the total content, which was 2- to 7-fold higher than that of P. ginseng, and threefold higher than that of P. quinquefolius (Yun, 2001). P. japonicus saponins content were found to be rich in oleanane-type saponins, with the ratio of dammarane- to oleanane-type saponins being less than 0.25 (Zhu et al., 2004). Several oleanane-type saponins, such as chikusetsusaponins-1b, -IV, -IVa, and -V, along with dammarane-type saponins, such as chikusetsusaponins-1a, and -111 among others have been isolated and characterized from P. japonicus (Morita et al., 1985; Zou et al., 2002; Yoshizaki et al., 2013).
Despite pharmacological importance of P. japonicus, the transcriptome and genome data are limited, with only 867 sequences being available in the NCBI database, out of which, 788 sequences are associated with chloroplast. The limited transcriptome data hinders identification of key factors that regulate saponins biosynthesis in P. japonicus. Further, genes encoding cytochrome P450 (CYP450s) and glycosyltransferases (GTs), and involved in the ginsenosides biosynthesis are not known. As ginsenosides are synthesized in all of the Panax species, transcriptome comparison across Panax genus will be useful to identify potential candidates for future validation.
In this study, we have established de novo transcriptome assembly for P. japonicus using five of its tissues, namely, flower, leaf, secRoot (secondary root), rhizome_Y (young rhizome tissue), and rhizome_O (old rhizome tissue). We identified putative genes from P. japonicus involved in the triterpene backbone biosynthetic pathways. Moreover, the comparative transcriptome analysis of P. japonicus with three major Panax species allowed us to identify potential candidate genes which may be involved in the synthesis of ginsenosides. This study, therefore, may serve as a basis for the future discoveries on functional genes involved in the ginsenosides biosynthesis and regulation.
Materials and Methods
Plant Materials
All five tissues, namely flower, leaf, secRoot, rhizome_Y, and rhizome_O were harvested in the month of June, 2014 from a 7-years-old P. japonicus plant, growing in the natural environment at the medicinal plant garden, Health Sciences University of Hokkaido, Japan. A voucher specimen (accession no. E4532-42-0003-3-C4) was deposited in the Herbarium of Faculty of Pharmaceutical Sciences, Health Sciences University of Hokkaido. For rhizome, we collected first bud scars, nearest to the stem, and called as rhizome_Y, and sixth bud from the stem, and called as rhizome_O. All tissues were cut into small pieces, frozen by liquid nitrogen, and stored at -80°C prior to RNA extraction.
RNA Isolation, and cDNA Synthesis
The frozen samples of P. japonicus were powdered using a Multi Beads Shocker (Yasui Kikai, Japan), and used for subsequent RNA extraction. Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, USA), according to manufacturer’s instruction. The extracted RNA was treated with RNase-free DNase (TaKaRa, Japan), purified on a RNA-purification column (Qiagen, USA), and finally collected by ethanol precipitation. The RNA quality was evaluated on the Agilent Bioanalyzer 2100 (Agilent Technologies, USA), and RNA samples with RIN (RNA Integrity Number) value above 8 was used for subsequent cDNA synthesis.
To create cDNA sequencing library, mRNAs with poly(A) tail were isolated from the total RNA using beads with oligo(dT). Fragmentation buffer was then added to shear mRNAs into short fragments, which then served as templates for the synthesis of first strand of cDNA using random hexamer primers. cDNA library was then prepared using SureSelect Strand Specific RNA Library kit (Agilent Technologies, USA) according to the manufacturer’s specifications.
Illumina Sequencing
Paired-end reads with an average length of 101 bps were generated by sequencing cDNA libraries on an Illumina HiSeqTM 2000 sequencer (Illumina, Inc., USA). Library construction and sequencing were performed at Kazusa DNA Research Institute, Chiba, Japan. After the removal of adapter sequences, empty reads, reads with ambiguous ‘N’ bases > 5%, raw reads of low quality (Q < 20), and raw reads with an average length less than 50 bases, Illumina sequencing resulted in a total of over 24 million paired end clean reads for P. japonicus. The raw read sequences for all five tissues of P. japonicus discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO; Edgar et al., 2002), and are accessible through GEO Series accession number GSE788931.
Data Pre-processing, and De novo Transcriptome Assembly
Filtering of raw reads were performed using Trimmomatic program (Bolger et al., 2014), resulting in paired end clean reads, and unpaired reads which lost its corresponding sequence partner due to quality control. Tissue-wise distribution of paired read counts, and resulting clean paired reads after pre-processing through Trimmomatic program is shown in the Supplementary Table S1. The Trinity program v2.0.6 was used for the de novo transcriptome assembly using clean reads from all the samples. We then processed these contigs for read alignment and abundance estimation using Bowtie 2.0 (Langmead et al., 2009), and RSEM (Li and Dewey, 2011), respectively. To calculate unigene expression, we used Fragments per Kilobase exon per Million mapped fragments (FPKM) method. GC content and basic statistics values for contigs and unigenes were calculated as described previously (Fukushima et al., 2015).
Functional Annotation and Classification of Unigenes
We performed BLASTx program based homology search for the P. japonicus de novo transcriptome assembly against the NCBI-nr protein database (2formatted on April 30, 2015) using a cutoff E-value < 10-5, and maximum number of allowed hit fixed at 20. The alignment results with the smallest E-value were used to annotate all the unigenes. For their further annotation and classification, we used the Blast2GO program v 3.0 to assign GO terms, an EC number, and KEGG information to the unigenes using associated BLASTx search results. Blast2GO v 3.0 (Biobam, Spain) was used for the visualization of the GO functional classification of all the unigenes and the distribution of the gene functions in different species. To perform k-mean clustering, we used Genespring 13.0 (Agilent Technologies, USA), with distance matrix being calculated by Euclidean similarity measurement with 10,000 iterations and number of clusters being set to six.
Simple Sequence Repeat (SSR) Detection
All P. japonicus transcripts were searched to determine the composition, frequency, and distribution of simple sequence repeats (SSRs) by using the microsatellite identification tool (MISA; Thiel et al., 20033). The search parameters for maximum motif length group were set to recognize hexamers with each SSRs length based category to have at least five repeats.
GO Enrichment Analysis
Unigenes with the FPKM value over 10 in each of the tissues were used as a test set, and compared against the whole transcriptome background of P. japonicus for GO enrichment analysis. GO enrichment analysis was performed on all of the tissues using Blast2GO v3.0 based on Fisher’s exact test, and a P-value cutoff of 0.05 (hypergeometric test with Benjamini and Hochberg false discovery rate correction) was applied.
Phylogenetic Analysis
Using Biology Workbench online tool4, we translated selected unigenes sequences from P. japonicus, and obtained corresponding protein sequences by selecting the translation frame that started with methionine and had longest amino acid sequences. Protein sequences from unigenes were combined together with protein sequences from the databases, aligned using MUSCLE program, and evolutionary distances were computed using Jones-Taylor-Thornton (JTT) method, and a Neighbor-Joining (NJ) tree was constructed with bootstrap values obtained after 1000 replications using MEGA6 software (Tamura et al., 2013).
Results
Sample Preparation and Illumina Sequencing
Panax japonicus plant is characterized by 3–5 leaflets, with a characteristic bamboo-like rhizome. As the stem (‘sympodium’) dies back each fall, it leaves a bud scar on the root collar or rhizome, which then are used as a means of assessing age of the plant. To study the transcriptomes of P. japonicus, five tissues, namely flower, leaf, secRoot (secondary root), rhizome_Y (young rhizome, first bud scar below the stem), and rhizome_O (old rhizome, sixth and seventh bud scar below the stem) were collected from 7-years-old plant (Figure 1). Total RNA for each sample were selected for the mRNA preparation, fragmentation, cDNA synthesis, and library preparation. Each library, thus prepared, was sequenced using the Illumina HiSeqTM 2000 platform. Adapter sequences, low-quality reads, and reads that were shorter than 50 base pairs (bps) were removed, yielding to a total of over 24 million 100 bps paired end clean reads, or approximately 2.4 Gbps in total. Mean phred score, a bench mark to access the quality of the short reads, across all tissues were above 36.5, indicating that our RNA sequencing was adequate for the de novo transcriptome assembly. The study overview is shown in the Supplementary Figure S1.

FIGURE 1. Five tissues of P. japonicus used for transcriptome study. Five tissues of P. japonicus, namely, leaf (a), flower (b), rhizome_Y (c), rhizome_O (d), and secRoot (e) were used to performed transcriptome profiling, and de novo transcriptome assembly. Scale bars for each panel measure to 0.5 cm.
De novo Transcriptome Assembly and Gene Expression Profiling
Clean reads from all samples were pooled together, and were further assembled de novo using Trinity program (Grabherr et al., 2011), resulting in 206,610 contigs (138,140,893 bps) with an average length of 668 bps and an N50-value of 950 bps (Table 1). The length and GC% distribution of all contigs have been shown in Figures 2A,B. We obtained 135,235 unigenes (77,297,613 bps) with an average length of 571 bps, and an N50-value of 889 bps, with length and GC% distribution as shown in Figures 2C,D. The length of assembled unigenes ranged from 200 to 11,387 bps, with 70,309 unigenes being shorter than 500 bps, and 1,258 unigenes sequence lengths being more than 3,000 bps. To estimate the expression abundance, clean reads were mapped to the P. japonicus de novo transcriptome assembly using Bowtie 2.0 (Langmead et al., 2009), and expression abundance was calculated using RSEM program (Li and Dewey, 2011).
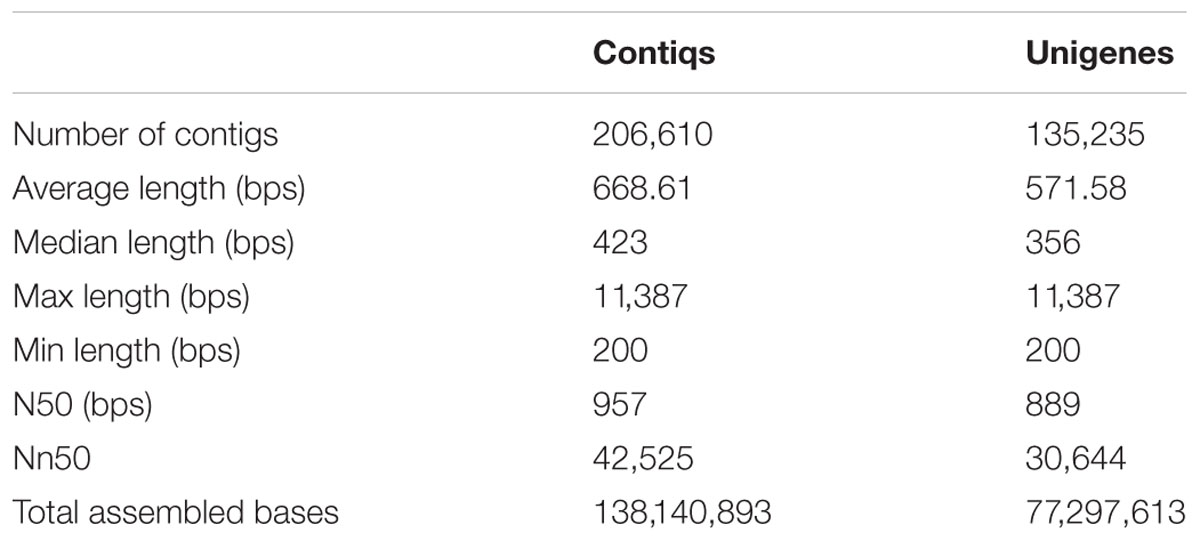
TABLE 1. Summary of the sequence assembly after Illumina sequencing.
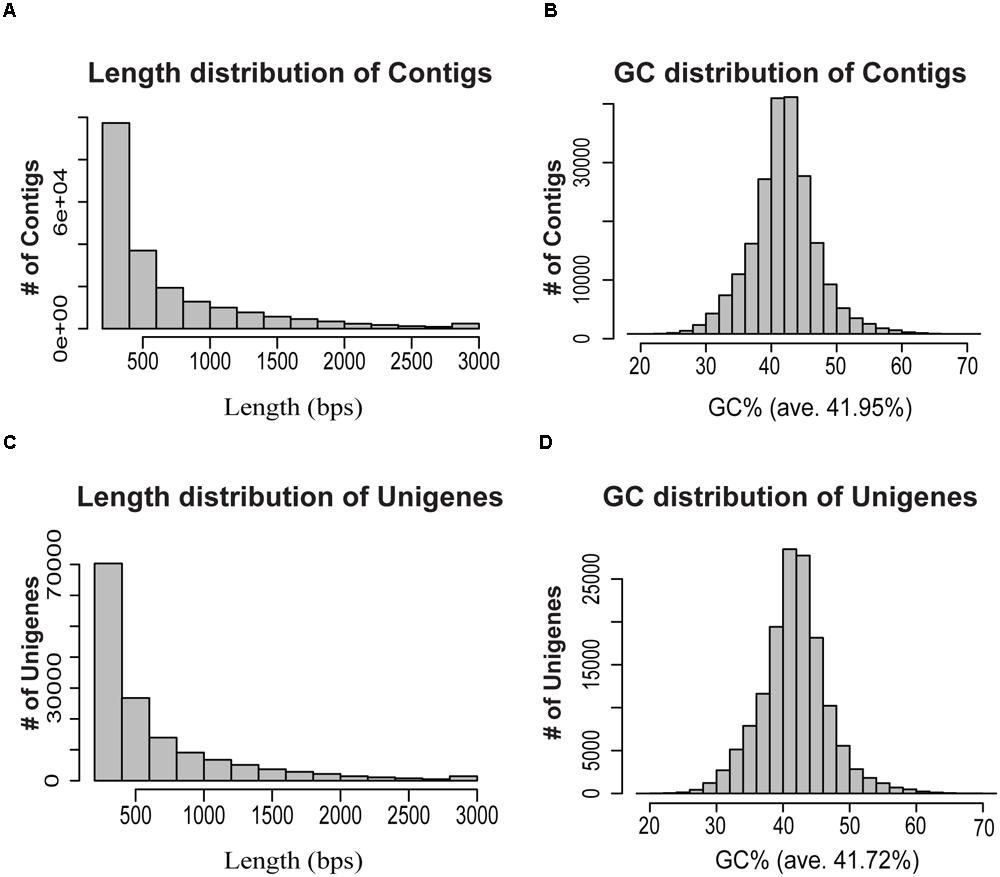
FIGURE 2. Overview of the de novo transcriptome assembly in P. japonicus. (A,B) represents length and GC distribution of contigs assembled from all cleaned reads from five tissues of P. japonicus using Trinity program (Grabherr et al., 2011), (C,D) represents length and GC distribution of unigenes generated from further contigs assembly.
Functional Annotation and Classification of P. japonicus Unigenes
The unigenes derived from the P. japonicus transcriptome assembly were subjected to additional validation and annotation. BLASTx program (Altschul et al., 1997) based homology search was conducted against an NCBI non-redundant (nr) protein database for all unigenes, and best aligning results were selected to annotate the unigenes. This resulted in 87.2% of aligned sequences displaying significant homology with the NCBI-nr database (Figure 3A). Based on the BLAST similarity distribution, over 80,000 sequences of P. japonicus exhibited alignment identity greater than 85% (Figure 3B). Blast2GO program v 3.0 (Conesa and Gotz, 2008) was then used to obtain gene ontology (GO), to assign an Enzyme Commission (EC) number, and Kyoto Encyclopedia of Gene and Genomes (KEGG; Kanehisa et al., 2014) based annotation to the unigenes based on the BLAST results. A total of 78,794 unigene (58.26%) were annotated with a minimum of one biological term from GO or KEGG pathway information (Figure 3C), while remaining 56,441 unigenes resulted in no BLAST hit. These unannotated unigenes may yet be uncharacterized genes or assembled sequences that were too small to produce hits in the search.
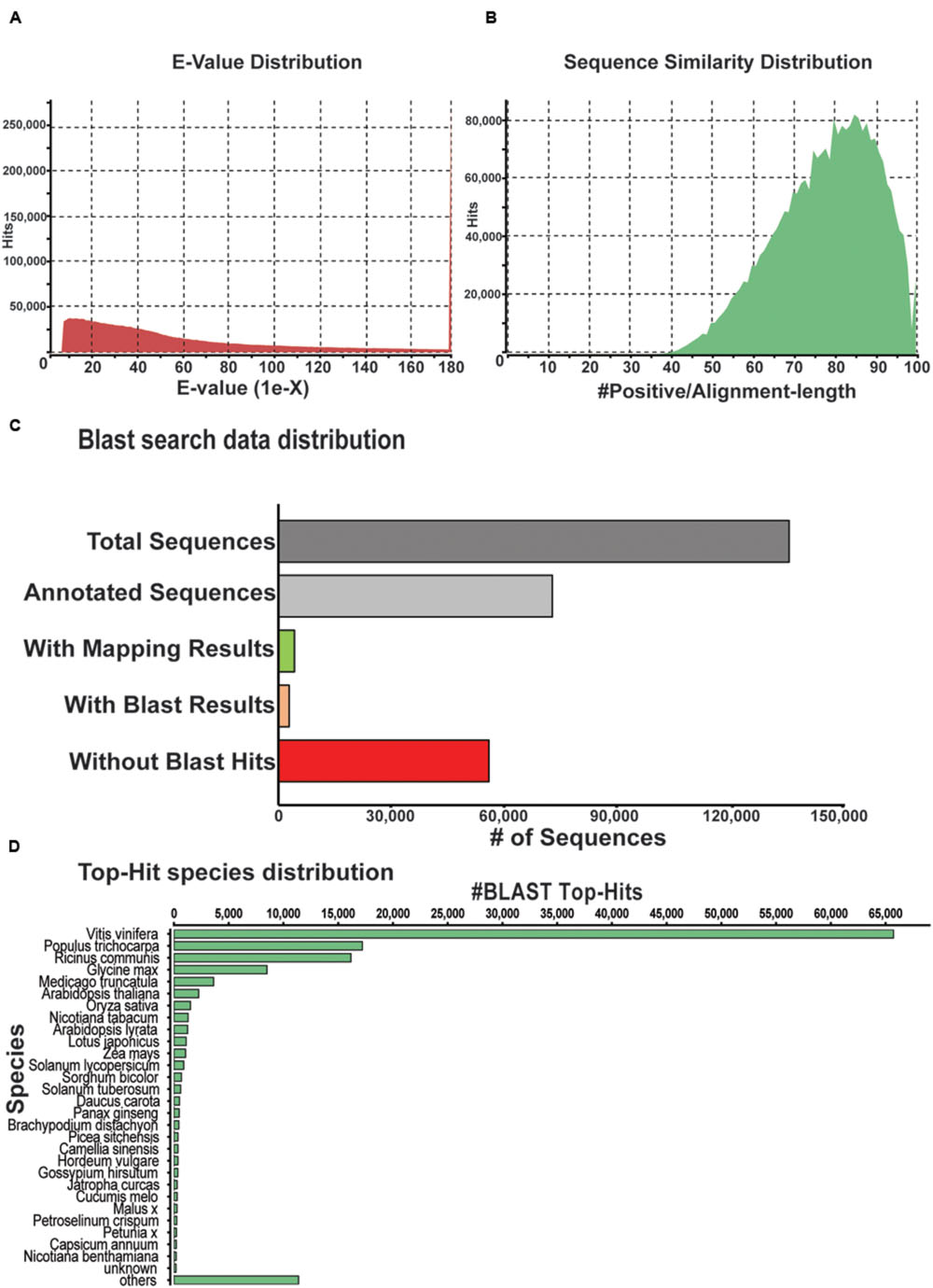
FIGURE 3. Characterization of P. japonicus unigenes based on NCBI non-redundant (nr) protein database search. (A) E-value distribution of Blast hits for the P. japonicus assembled transcriptome with a cutoff of E-value < 10-5. (B) Similarity score distribution plot of top Blast hits for the assembled unigenes. (C) Bar chart of the data distribution based on blast search and annotation using Blast2GO. (D) Species distribution of the top Blast hits for the assembled unigenes.
Top-hit species distribution analysis based on BLASTx results showed 75% of unigenes with blast hit sharing high sequence similarity with sequences of Vitis vinifera (47.6%), Populus trichocarpa (12.47%), Ricinus communis (11.71%), and Glycine max (6.14%; Figure 3D). Among other top-hit species, P. ginseng was placed at 16th position, with 470 unigenes from P. japonicus showing top sequences similarity. High sequence similarity of P. japonicus with that of P. ginseng was anticipated, and is consistent with earlier studies that suggests P. japonicus and P. ginseng to be closest among the rest of the Panax species (Zhu et al., 2003; Choi et al., 2011).
Gene ontology functional classification for the P. japonicus transcriptome was performed using Blast2GO at the annotation level 5, resulting in 66 GO categories; molecular function (25), biological process (25), and cellular component (16; Figure 4). Among molecular function category, most noticeably, GO terms corresponding to different hydrolase activities, hydrolase activity that involves hydrolysing o-glycosyl compounds, transferase activity that involves transferring hexosyl groups, and UDP-glycosyltransferase activity were abundant. Within the biological process category, metabolic processes, such as the cellular macromolecular metabolic process, protein metabolic process, nucleotide-containing compound metabolic process, macromolecule biosynthetic process, aromatic compound biosynthetic process, and organic acid metabolic process were among most abundant within biological processes category. Within cellular component category, GO terms corresponding to the intracellular part, plasma membrane, integral components of membrane, bounding membrane of organelle, external encapsulating structure and transferase complex were abundant among unigenes.
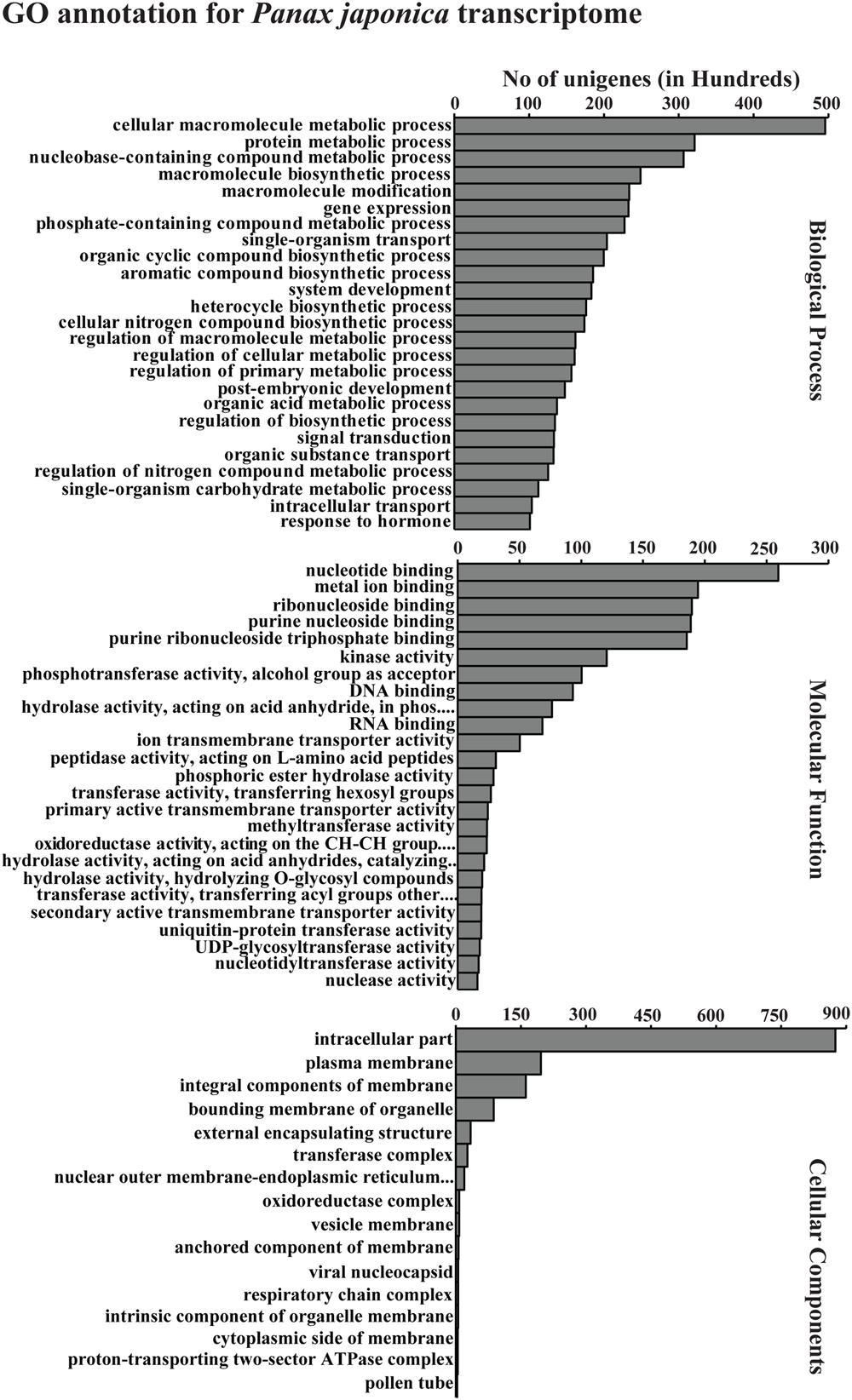
FIGURE 4. Gene ontology (GO) annotation for all assembled unigenes in P. japonicus. GO-terms for all unigenes were assigned based on Blast search results using Blast2GO program at GO annotation level 5. The results are summarized in terms of three functional categories, namely, biological process, molecular function, and cellular component.
Functional Classification of KEGG Pathways
The use of the KEGG pathway database facilitates the functional annotation of cellular components and their interactions within various biological pathways. This ”pathway” based annotation provides an overview of the different active metabolic processes within an organism, and therefore, enables further understanding of the biological functions of the unigenes. Using Blast2GO, all unigenes were mapped against the KEGG database, resulting in 17,394 unigenes grouped into 146 KEGG pathways (Supplementary Table S2). Purine metabolism, thiamine metabolism, starch and sucrose metabolism, aminobenzoate degradation, glycolysis, and phenylpropanoid biosynthesis pathways were the most represented pathways observed. Terpenoid backbone biosynthesis (map00900), a key pathway for the synthesis of saponins, was assigned to 322 of these unigenes.
Identification of Simple Sequence Repeats (SSRs)
Simple sequence repeats are ubiquitously distributed tandem iterations of short oligonucleotides within a genome, consisting of simple motifs of 1–6 nucleotides that are repeated from two to a few dozen times at a locus. SSRs being an indel mutational hotspots in genomes are important markers for determining genetic variations including paternity determination, genetic diversity assessment, population genetics studies, and for the development of genetic map (Hancock, 1996; Varshney et al., 2005). There have been several reports that implicate SSRs in affecting genes expression, and that polymorphism of SSRs tracts may be important in the evolution of gene regulation (Varshney et al., 2002; Jarvis et al., 2008; Senthilvel et al., 2008). To identify SSRs for P. japonicus, all the contigs for mono- to hexa-nucleotide motifs were searched, with a minimum of five repetitions using the software MISA (Thiel et al., 2003). We identified a total of 30,966 SSRs present across 26,557 contigs of P. japonicus, with 3,742 contigs showed presence of more than one SSRs (Table 2). Among the different SSRs repeat-type classes, the mono-nucleotide represented the largest fraction (48.64%) of identified SSRs, followed by di-nucleotide (23.94%) and tri-nucleotide (23.37%). Numbers of SSRs for tetra-, penta-, and hexa-nucleotide repeat classes were significant but relatively small compared to the mono-, di-, and tri-SSR repeats. All detected SSRs for P. japonicus are shown in Supplementary Table S3. Herein identified SSRs of P. japonicus may provide a potential genetic marker, which will be useful for performing population genetics and comparative genomics studies across the Panax species synthesizing saponins.

TABLE 2. Statistics of SSR detected in Panax japonicus.
Functional Classification and Comparison of Highly Expressed Transcripts across Five Tissues of P. japonicus
A comparative analysis of expression of the unigenes (presence or absence) across and within all the five tissues of P. japonicus showed 59,228, 63,757, 69,969, 60,819, and 69,222 unigenes with non-zero FPKM values for flower, leaf, secRoot, rhizome_Y, and rhizome_O, respectively. While numbers of unigenes with non-zero expression value were comparable across all five tissues, each showed several unique unigenes specific to that tissue. Among all unigenes, 9,534 unigenes were shared across all tissues (Figure 5A), while 2,381, 45,059, 3,256, 1,666, and 1,685 unigenes were specifically found in the flower, leaf, secRoot, rhizome_Y and rhizome_O, respectively. With the exception of leaf, other tissues of P. japonicus showed overlap of unigenes with non-zero expression values across all tissues. Overall, our results showed that the transcriptome profile for rhizome_Y and rhizome_O tissue were highly similar with sharing 48,808 unigenes, and were significantly similar to secRoot (Supplementary Figure S2).
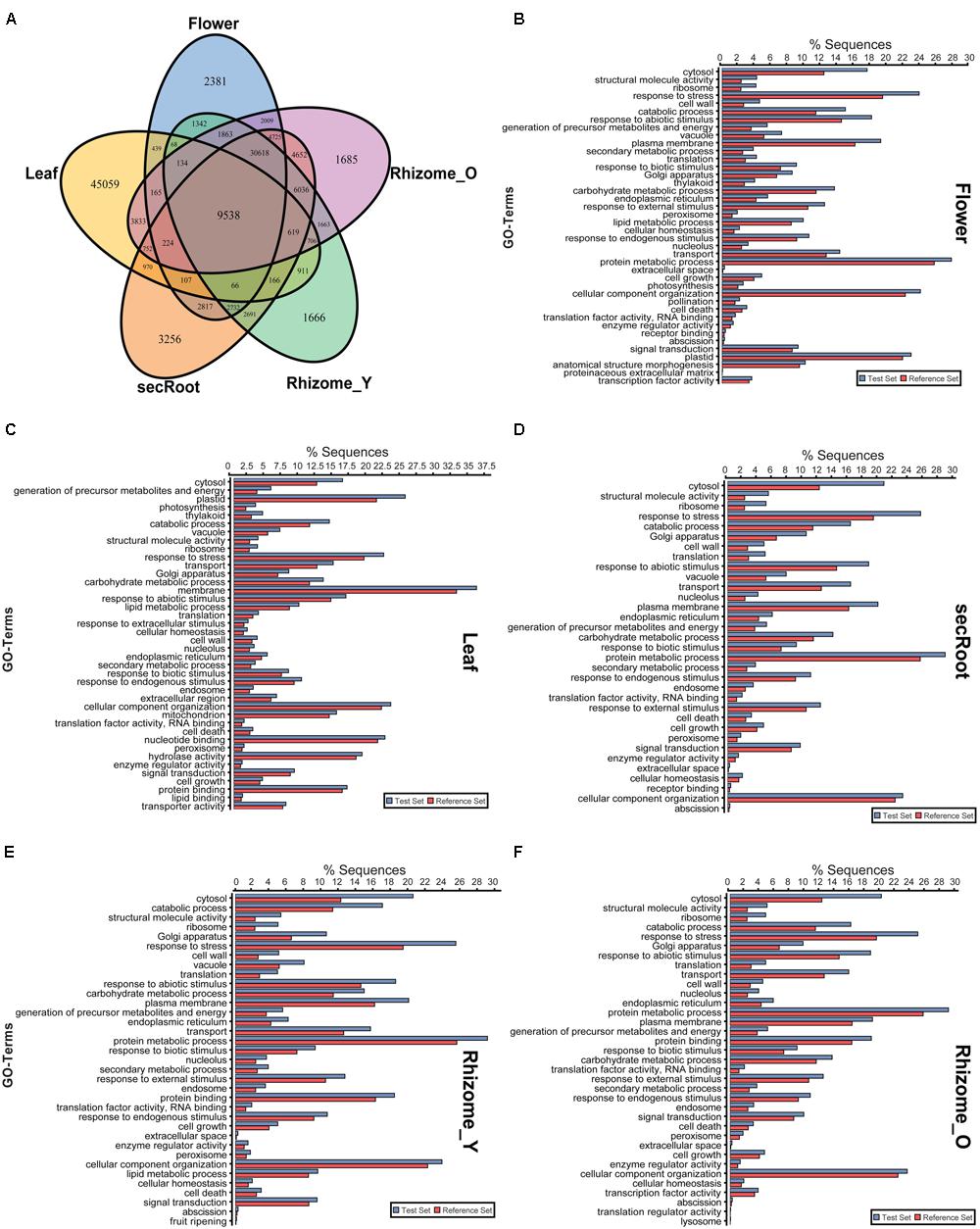
FIGURE 5. Distribution of unigenes and gene ontology enrichment analysis for five tissues of P. japonicus. (A) Venn diagram for unigenes with non-zero FPKM values in individual tissues. (B–F) Gene ontology enrichment analysis using Fisher’s exact test with P-value cutoff applied as 0.05. Unigenes for each tissue with FPKM value above 10 were selected as test set, and used against the P. japonicus transcriptome assembly as reference set to identify differential GO-terms enriched in the specific tissue, with P-value being calculated based on reference set.
To compare the distribution of differentially enriched GO-terms across five tissues of P. japonicus analyzed for the highly expressed transcripts, we performed GO-term enrichment analysis using Fisher’s exact test by selecting unigenes for each tissue with FPKM value greater than 10, and were further tested against P. japonicus transcriptome assembly (Figures 5B–F). Flower’s unigenes showed 40 GO-terms being enriched, with GO-terms corresponding to pollination, anatomical structure morphogenesis and proteinaceous extracellular matrix were specific to flower. GO-term enrichment analysis for the leaf resulted into 40 GO-terms, 8 of which were specific to leaf, where the top four enriched GO-terms for leaf were cytosol, generation of precursor metabolites and energy, plastid and photosynthesis. Enriched GO-terms for secRoot, rhizome_Y and rhizome_O were highly similar, with only one GO-term corresponding to fruit ripening was specific to rhizome_Y.
Comparison of Assembled Unigenes with P. ginseng, P. notoginseng, and P. quinquefolius Published Transcriptome
A comparative examination of the transcriptome assembly of P. japonicus with three Panax species, namely, P. ginseng, P. quinquefolius, and P. notoginseng was undertaken. Searching for Panax related nucleotides and ESTs from NCBI resulted in a total of 106,320 sequences being deposited for P. ginseng, while for the other Panax species, the number of sequences were limited. For P. ginseng, we obtained all nucleotide sequences deposited in NCBI and used for comparison with P. japonicus. For comparison with P. quinquefolius, we obtained its annotated transcriptome assembly (pqa_assembly_v_10072011) from medicinal plant genomics resource consortium5. As number of transcripts for P. notoginseng in the NCBI database were less than 300, we obtained raw sequence datasets from NCBI-SRA (sequence read archive) database as described in the material and methods, with specific experiments used in this study are listed in the Supplementary Table S4. Using this dataset, we performed de novo transcriptome assembly for P. notoginseng transcriptome with the Trinity software that resulted in 186,450 unigenes with N50-value 735 bps (Supplementary Table S4).
We performed two-way blastn search to compare P. japonicus with other Panax species (Supplementary Figure S1). In the first case, we used P. japonicus transcriptome as query against sequences from individual Panax species transcriptome selected for this study as blastn database, resulting in 51.56, 63.14, and 53.13% of its contigs having a hit against P. ginseng, P. notoginseng, and P. quinquefolius, respectively (Table 3). Overall, a total of 1,34,387 transcripts of P. japonicus got hit against at least one of the Panax species, of which, 90,951 contigs (67.67%) showed sequence similarity against corresponding sequences in all Panax species, while 72,223 contigs were unique for P. japonicus with no hit to any Panax species (Supplementary Figure S3).
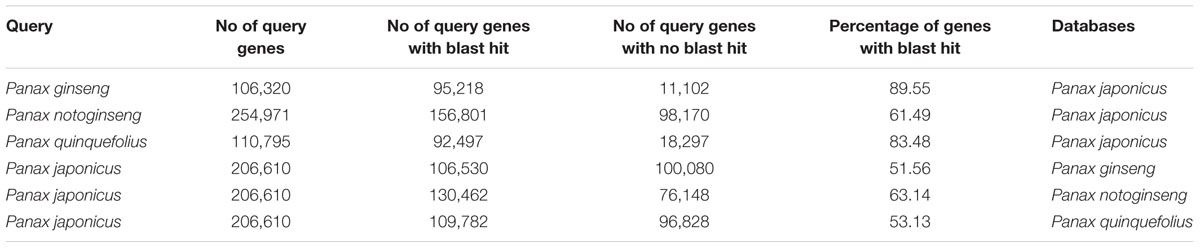
TABLE 3. Comparative transcriptomic analysis of P. japonicus with other three Panax species based on Blastn sequence similarity.
We next performed blastn search using each of the individual Panax species transcriptome as a query against the P. japonicus transcriptome as a blastn database. Among Panax species, 89.55, 61.49, and 83.48% of the sequences used as a query from P. ginseng, P. notoginseng, and P. quinquefolius, respectively, showed sequence similarity with at least one transcript of P. japonicus (Table 3). P. ginseng, and P. quinquefolius showed higher sequence similarity with P. japonicus, with majority of its sequences matched with the corresponding sequences in the P. japonicus, while P. notoginseng showed relatively less similarity with P. japonicus. Previous studies using SSR markers for the assessment of genetic diversity in the Panax ginseng cultivars and related species concluded that P. japonicus and P. ginseng are phylogenetically similar, while P. notoginseng and P. quinquefolius are relatively distant with P. japonicus (Choi et al., 2011). While high sequence similarity does not mean phylogenetic closeness, but if two species are phylogenetically closer, then we would expect higher sequence similarity compared to other species. Therefore, our results were consistent with previous reports, showing P. japonicus and P. ginseng sharing high sequence similarity.
Identification of Unigenes Putatively Involved in Triterpene Saponins Biosynthesis
Triterpene saponins are derived from terpenoid backbone biosynthesis, followed by sesquiterpenoid and triterpenoid biosynthesis, which are then used as precursors by specific CYP450s and GTs for the formation of various ginsenosides (Figure 6A). Using annotated P. japonicus transcriptome assembly and blastn results of P. japonicus transcripts against other Panax species, we identified 28 unigenes with sequence length more than 500 bps, annotated as known enzymes involved in the mevalonate (MVA) pathway for triterpene saponins biosynthesis, and showing above 90% sequences similarity against all Panax species (Figure 6B). A heat-map, representing gene expression of these unigenes, showed differential expression across the various P. japonicus tissues, with genes involved in the triterpene saponins biosynthesis displaying higher expression in the rhizome_Y tissue, followed by rhizome_O, and secRoot. A hierarchical clustering of unigenes for triterpene saponins biosynthesis pathways showed three distinct groups, with the rhizome_Y and rhizome_O being in one group, flower and secRoot in the second group, and the leaf being the third group, where the later showed the least expression of all selected unigenes.
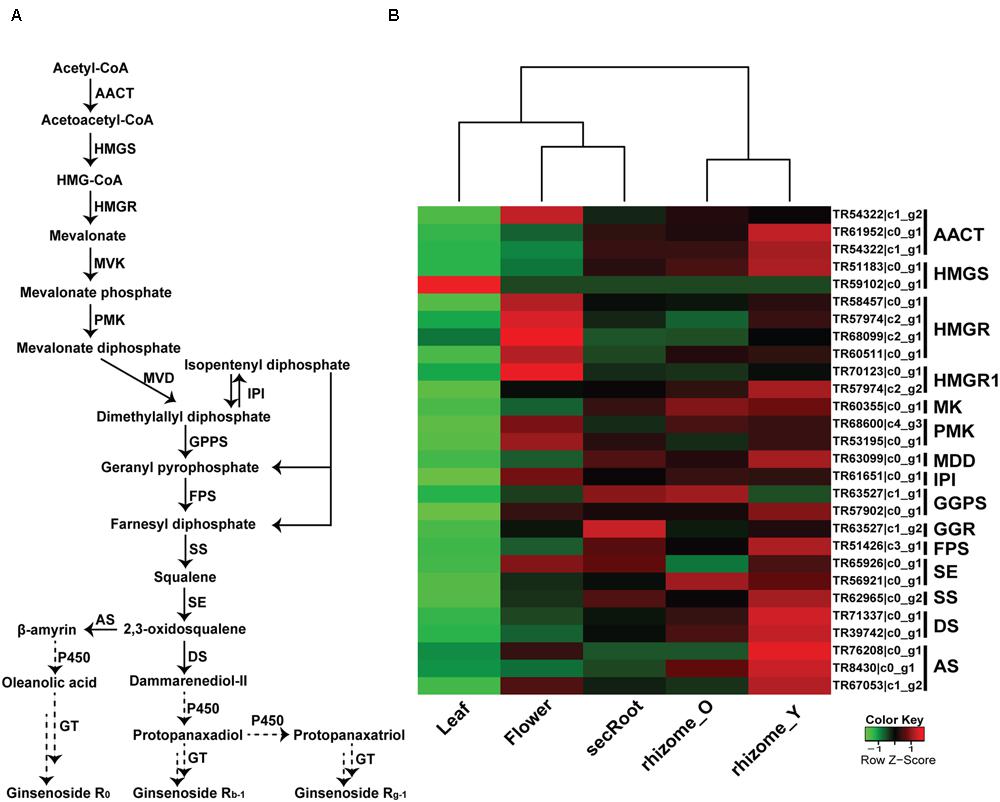
FIGURE 6. Putative triterpenoid saponins biosynthesis pathway and expression level of associated unigenes across five tissues in P. japonicus. (A) Proposed pathway for triterpenoid saponins biosynthesis. (B) Expression levels of the candidate unigenes that showed high sequence similarity with other Panax species, and were annotated as enzyme coding genes from triterpenoid backbone biosynthesis pathways. The expression value (FPKM) for unigenes across all tissues were log2 transformed and scaled across each row, and heatmap was generated using R-package heatmap2.0. AACT, Acetyl-CoA acetyltransferase; HMGS, hydroxymethylglutaryl-CoA synthase; HMGR, hydroxymethylglutaryl-CoA reductase; MVK, mevalonate kinase; PMK, phosphor mevalonate kinase; MVD, mevalonate diphosphate decarboxylase; IPI, isopentenyl diphosphate isomerase; GPPS, geranylgeranyl pyrophosphate synthase; FPS, farnesyl diphosphate synthase; SS, squalene synthase; SE, squalene epoxidase; DS, dammarendiol synthase; AS, beta-amyrin synthase; GTs, UDP glycosyltransferase; P450, cytochrome P450; FPKM, fragments per kilobase per million.
Identification of Candidate Unigenes Putatively Involved in Ginsenosides Biosynthesis
It has been suggested that putative genes involved in the triterpene saponins biosynthesis are mainly CYP450s and GTs enzyme coding genes, which may account for the synthesis and accumulation of triterpene saponins in specific tissues. In order to identify these putative genes associated with ginsenosides biosynthesis, we selected 870 P. japonicus unigenes annotated as CYP450s or associated with sugar conjugation, and have sequence similarity with all Panax species. These, together with the 28 unigenes associated with MVA pathway for triterpene saponins biosynthesis, were used to perform k-mean clustering using Euclidean similarity measurement with 10,000 iterations. This resulted in all unigenes being grouped in six different clusters (Figure 7 and Supplementary Table S5). Among gene clusters, cluster-2 and -3 (297 unigenes) included 22 out of 28 genes corresponding to triterpene saponins biosynthesis. Cluster-2 and -3 were unique with respect to other gene clusters, as expression of all unigenes were highest in the rhizome_Y and rhizome_O, while lowest in the leaf. Within cluster-2 and -3, the median expression of all unigenes in the rhizome_Y was 5.13 and 7.39, respectively. Although, the expression trend for cluster-4 was similar to cluster-2 and -3, and included three unigenes associated with MVA pathways, the median expression values for the unigenes were low. Inspection of the unigenes annotated as CYP450s within cluster-4 revealed a unigene TR49263_c0_g1 annotated as CYP716A53v2 (JX036031), a gene characterized as protopanaxadiol 6-hydroxylase producing protopanaxatriol triterpene aglycone from protopanaxadiol in P. ginseng (Han et al., 2012). The unigene TR49263_c0_g1 showed high sequence similarity with P. ginseng and P. quinquefolius, the highest expression level in root (in the order rhizome_Y > rhizome_O > and secRoot), but had lower expression levels when compared to unigenes within cluster-2 and -3. K-mean clustering showed that all unigenes associated with triterpene saponins biosynthesis were highly expressed in rhizome_Y, rhizome_O and secRoot, and all grouped within cluster-2, and -3. All these unigenes were conserved across three other Panax species along with P. japonicus, suggesting potential role in saponins biosynthesis. These unigenes, showing similar expression trend as that of those from triterpene saponins biosynthesis pathways would be strong candidates for potential role in ginsenosides biosynthesis.
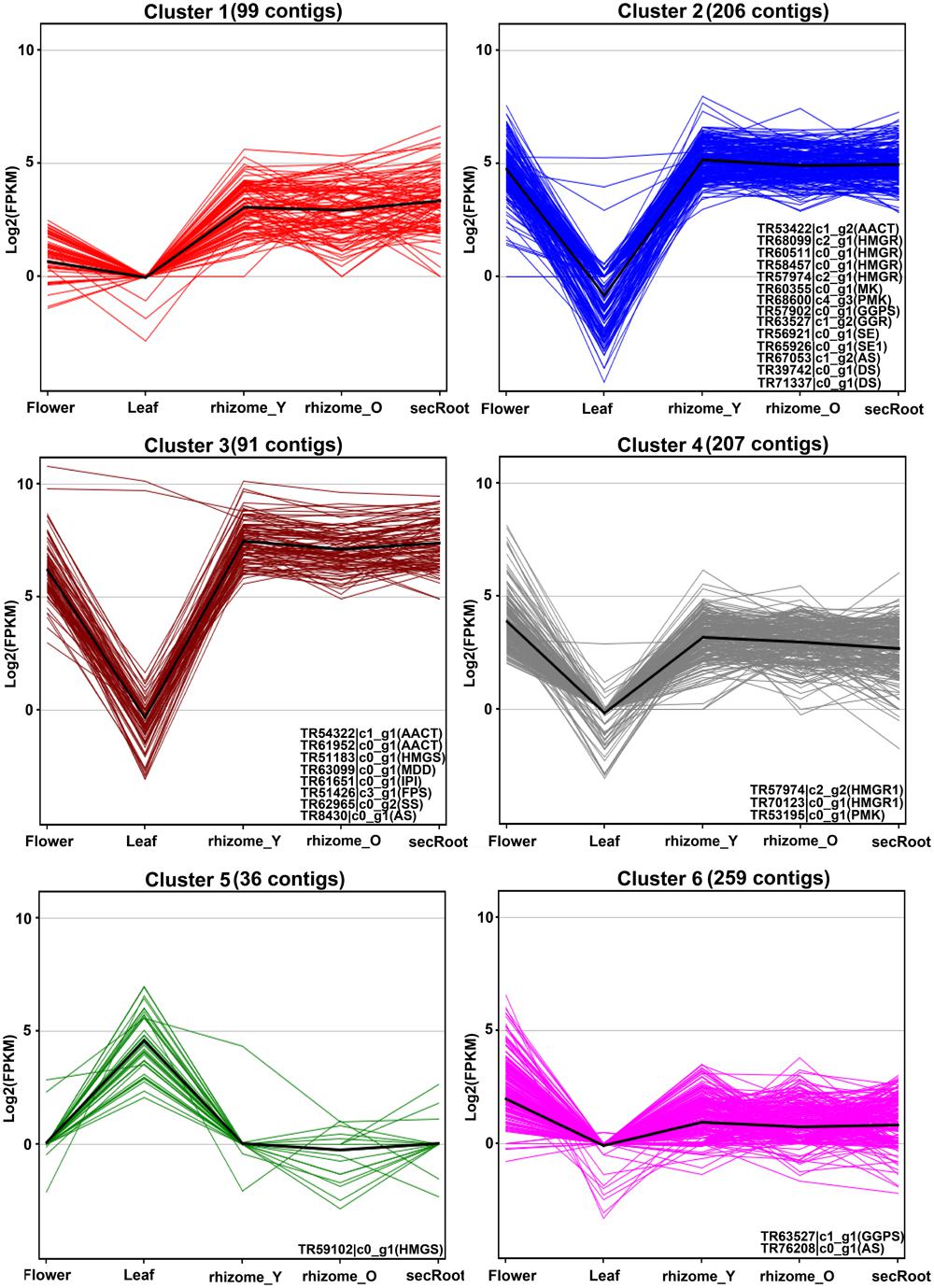
FIGURE 7. K-mean clustering for unigenes annotated as CYP450s or associated with sugar conjugation processes together with unigenes from triterpenoid backbone biosynthetic pathways. Unigenes annotated as cytochrome P450, or sugar conjugation associated processes were selected from the P. japonicus transcriptome assembly, resulting in total of 870 unigenes. These gene groups were used together with 28 unigenes annotated as enzyme coding genes from triterpenoid backbone biosynthetic pathways, and k-mean clustering analysis was performed based on gene expression values across all five tissues of P. japonicus. Distance matrix for k-mean clustering was calculated by Euclidean similarity measurement with 10,000 iterations, resulting in 6 gene clusters. Name of unigenes associated with triterpenoid backbone biosynthetic pathways and identified in a particular cluster is represented within each gene group.
For cluster-2 and -3, a total of 47 and 74 unigenes were identified being annotated as CYP450 and GTs, respectively. Unigenes with a length of 600 bps or above were selected from these gene clusters, with expression levels across all five tissues shown in the Figure 8. Among unigenes annotated as CYP450s, TR45427_c0_g1 showed over 95% of its length aligned with transcripts from all Panax species, and was annotated as CYP716A52v2. It has been reported that cytochrome P450 CYP716A52v2 participates in the formation of oleanane-type ginsenosides biosynthesis in P. ginseng (Han et al., 2012, 2013). Expression of TR45427_c0_g1 was highest in the rhizome_Y, followed by rhizome_O and secRoot, which was also the case for most of the unigenes from triterpene saponins biosynthesis. Identification of CYP716A52v2 coding unigene among the list of potential P450s involved in the ginsenosides biosynthesis thus validates our approach. Further, expression levels of unigene annotated as CYP716A52v2 were higher than that of unigenes annotated as CYP716A53v2. As CYP716A52v2 is required for oleanane-type ginsenosides, while CYP716A53v2 is required for dammarane-type ginsenosides, therefore, our transcriptome analysis for P. japonicus thus supported previous observations suggesting it as oleanane-type ginsenoside rich Panax species.
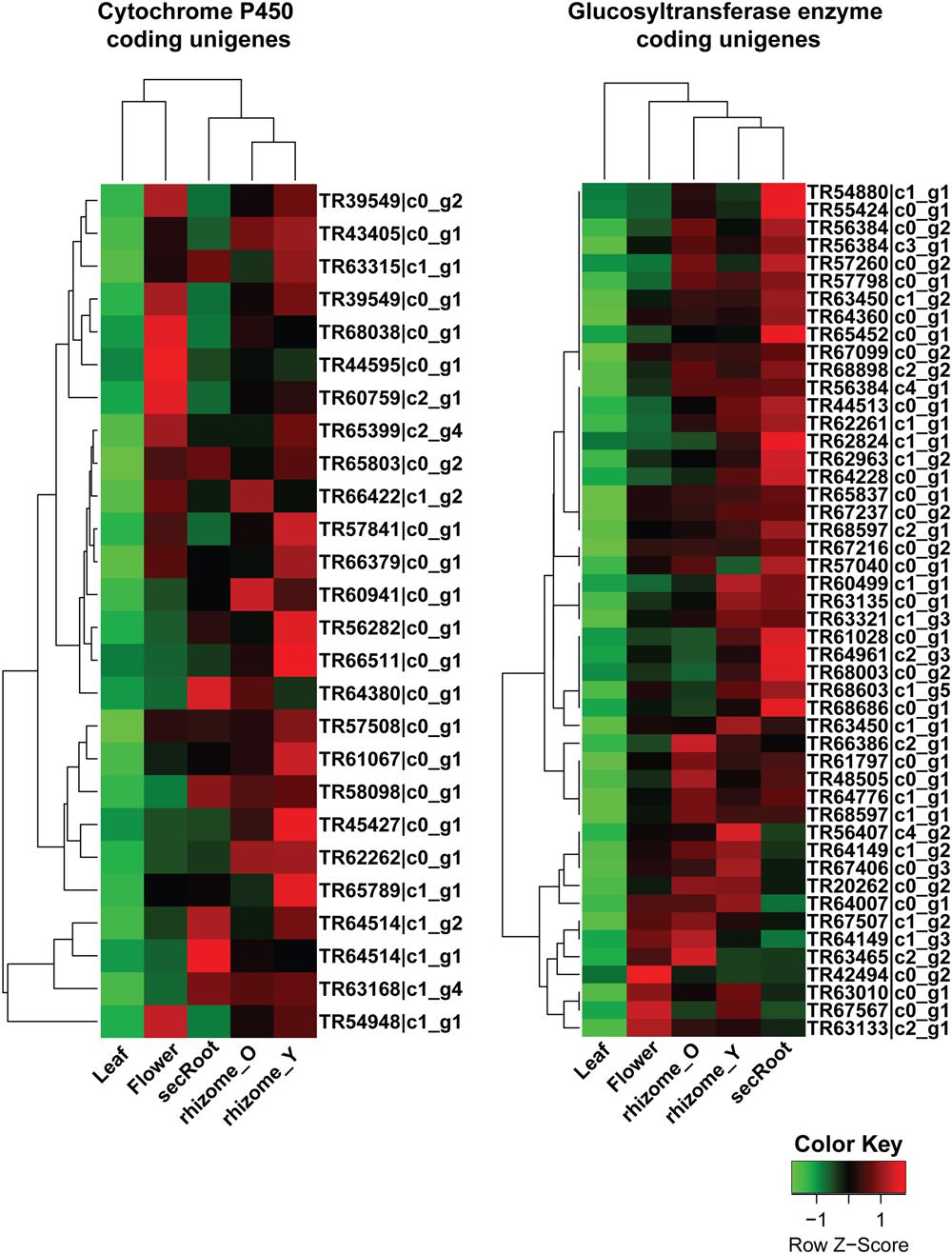
FIGURE 8. Expression profile for putative cytochrome P450 (CYP450s) and GTs unigenes from ginsenosides biosynthetic pathways across five tissues of P. japonicus. The expression value (FPKM) for unigenes, annotated as CYP450 or GTs and co-expressed with triterpenoid saponin biosynthetic pathways, were log2 transformed and scaled across each row, and heatmap was generated using R-package heatmap2.0.
Analyzing unigenes annotated as GTs identified 48 unigenes with a sequence length above 500 bps, and showed high sequence similarity across all Panax species studied. Expression trends for these unigenes were different from what we observed for CYP450s. For instance, while all CYP450s and MVA pathway annotated unigenes from cluster-2 and -3 showed high expression in the order of rhizome_Y > followed by rhizome_O > secRoot, unigenes annotated as GTs displayed high expression in the order of secRoot > rhizome_Y > rhizome_O.
Discussion
Ginsenosides, a pharmacologically active class of metabolites with useful medicinal properties, are the major constituents of root extracts from all 12 plant species from Panax genus, with P. ginseng and P. japonicus being the most popular. Among these Panax species, P. ginseng, P. notoginseng, and P. quinquefolius transcriptome have been studied extensively, while little is known about the transcriptome of P. japonicus, which is also considered as the “king of herbs” in the traditional Tujia and Hmong medicine. There are also limited comparative transcriptome studies being done on the Panax species, which may provide clues for the identification of conserved metabolic pathways of medicinal importance, particularly for the ginsenosides biosynthesis pathways. Here, we present deep transcriptome profiling and analysis of five tissues of P. japonicus, and compared its transcriptome with publically available transcriptome data for three popular Panax species, namely, P. ginseng, P. notoginseng and P. quinquefolius, leading to the identification of 24 CYP450s and 48 GTs as potential genes from the ginsenosides biosynthesis pathways.
Panax japonicus de novo transcriptome assembly resulted in 201,660 contigs with a mean length of 873 bps, N50-value as 947 bps, and total number of unigenes as 133,500. Annotation of the P. japonicus transcriptome assembly resulted in 67% of its contigs getting a hit against the NCBI-nr database, with the P. ginseng as one of the top-hit species. These results taken together suggest that our de novo assembly and annotation of transcripts were accurate and consistent with previous reports suggesting P. japonicus to be closest to the P. ginseng.
Analyzing gene product function using GO annotations for P. japonicus transcripts revealed processes related to metabolites oxidation and sugar conjugation being highly enriched. There were 312 unigenes being annotated as CYP450s, and 586 unigenes being assigned to several processes associated with glycosylation process including GTs in the P. japonicus transcriptome. For the molecular function categories, several GO categories corresponding to processes, such as UDP-glycosyl transferase activity, hydrolase activity-involving hydrolysing O-glycosyl compounds, oxido-reductase activity-while acting CH-CH groups, and transferase activity-transferring hexoyl groups were assigned to several of the P. japonicus transcriptome. Our GO annotation of P. japonicus transcriptome, therefore, suggests presence of several transcripts associated with active metabolism, especially in the enzymatic processes involving oxido-reduction reactions, and glycosylation processes, which are also very important for the synthesis of ginsenosides from terpenoids backbone.
Transcripts expression analysis revealed varying number of active transcripts across all five tissues of P. japonicus, with secRoot showing highest (69,969), and flower showing lowest (59,228) number of transcripts with non-zero expression value. Our results showed leaf to have the most different transcriptome profile (with approximately 70% of its active transcripts being unique) among rest of the tissues in P. japonicus, with rhizome_Y and rhizome_O showing highest number of common transcripts. To further get an insight on GO functional categories enriched with respect to the P. japonicus transcriptome assembly in different tissues, we selected unigenes with an expression value (FPKM) above 10 for each tissue to perform GO enrichment analysis. Results diplayed similar GO categories for rhizome_Y, rhizome_O, and secRoot, while leaf showed eight GO categories being specific to this tissue. Enriched GO categories were corroborating with the specific tissues type, and therefore, further validate our transcriptome assembly and annotation. For example, GO enrichment analysis using transcripts with FPKM values above 10 for flower as test set showed GO category corresponding to pollination being enriched and specific only to this tissue, while top four enriched GO categories for leaf included cytosol, generation of precursor metabolites and energy, plastid, and photosynthesis. As we expected, GO category corresponding to photosynthesis was not enriched in the secRoot, rhizome_Y and rhizome_O. Overall, out of the total 53 GO categories combined across five tissues, 25 GO categories were enriched and common across all tissues. This despite of the fact that majority of leaf transcriptome profile were unique, and were not transcriptionally active in the rest of the other tissues. These results thus suggest that while each tissue expressed unique genes, there are processes that are essential for maintaining normal activities of a cell, and therefore, are conserved across all tissues.
In order to identify conserved genes across Panax species, we compared P. japonicus transcriptome assembly with the transcripts from P. ginseng, P. notoginseng, and P. quinquefolius. Blastn based sequences alignment across Panax species showed high sequence similarity within and with P. japonicus transcriptome assembly. For the P. ginseng, over 89% of its deposited sequences in the NCBI database showed at least a hit against P. japonicus transcriptome assembly, while 83.48% of transcripts from P. quinquefolius showed high sequence similarity against P. japonicus. Over 65% of P. japonicus transcripts showed at least one hit, with 72,223 unique transcripts with no hit against any of the three Panax species. Previous studies reported that P. ginseng and P. japonicus were closest among other Panax species, while P. notoginseng and P. quinquefolius showed high similarity (Choi et al., 2011). Our results in principle does follow these observations. Our study also provided a comprehensive overview of conserved sequences across Panax species, which we also used to identify genes associated with ginsenosides biosynthetic pathways.
As triterpenoid saponins biosynthesis pathway is conserved across all Panax species, we selected unigenes with sequence length above 500 bps, and having high sequence similarity with all three Panax species selected for this study. This approach narrowed down candidate unigenes associated with MVA pathways to 28, and identified all known genes involved in the triterpenoid backbone biosynthesis pathways. All these genes were highly expressed in the rhizome_Y and rhizome_O, followed by secRoot, flower, and leaf, respectively. These results were consistent with previous studies on different Panax species, which also observed high expression of MVA associated genes in the roots.
In the ginsenosides biosynthesis, cyclization of 2,3-oxidosqulene, catalyzed by DS (dammarendiol synthase), is the rate limiting step. After the cyclization, the hydroxylation and glycosylation process, which are catalyzed by CYP450s and GTs in turn, are important for the production of triterpene saponins. So far, little is known about different CYP450s and GTs enzymes involved in the ginsenosides biosynthesis, particularly in the Panax species. As ginsenosides are derived from the triterpene backbone, we expected co-expression of genes associated with MVA pathways together with genes encoding CYP450s and GTs associated with the ginsenosides biosynthesis pathway. Therefore, we performed k-mean clustering using 28 unigenes associated with the MVA pathway together with 898 unigenes annotated as CYP450s (312 unigenes) or associated with sugar conjugation processes (586), which resulted in six gene clusters. Among these, cluster-2, and -3 included 22 out of 28 unigenes associated with the triterpene backbone synthesis, therefore, genes within these groups were considered as strong candidates to be associated with ginsenosides biosynthesis. Taken together all genes from cluster-2 and -3, resulting in 289 unigenes in total, we identified 24 and 51 unigenes annotated as CYP450 and GTs, respectively, showing high sequence similarity across all the Panax species with sequence length above 500 bps. All these unigenes showed high expression in the rhizome_Y and rhizome_O, followed by secRoot, flower, and leaf. As ginsenosides accumulate in the root, these unigenes, therefore, were considered as the potential candidates to be involved in ginsenosides biosynthesis. Our results also suggested that saponins aglycones are synthesized in roots, which is consistent with previous reports on other Panax species.
In order to further characterize CYP450s unigenes identified in this study, we performed phylogenetic analysis for all 24 unigenes together with 244 cytochrome P450 annotated genes in the Arabidopsis genome obtained from http://www.p450.kvl.dk/p450.shtml (Paquette et al., 2009), and classified genes in different clades as described (Bak et al., 2011). Majority of unigenes from P. japonicus were clustered in CYP71 (17 unigenes) and CYP85 (4 unigenes) clan (Figure 9). Previous reports have characterized CYP88D6 from Glycyrrhiza uralensis (Seki et al., 2008; CYP85 clan) and CYP93E1 from Glycine max (Shibuya et al., 2006; CYP71 clan) to be involved in the triterpene saponins biosynthesis, suggesting CYP450s within these two clan as the most likely candidates to be involved in ginsenosides biosynthesis. Within CYP85 clan, unigene TR45427_co_g1 showed high sequence similarity with all Panax species, and was annotated as CYP716A52v2. As V. vinifera emerged as the species with highest number of sequences matched to P. japonicus transcriptome assembly as top-hit (Figure 3D), we performed phylogenetic analysis using these 24 unigenes together with 325 cytochrome P450 genes from V. vinifera obtained from http://drnelson.uthsc.edu/vitis.htm, and classified genes in different clades as described (Nelson, 2009). Majority of unigenes were clustered in CYP71 clan (eight Unigenes) with unigene TR45427_co_g1 were grouped in CYP716A clan (Supplementary Figure S4). Previous reports have shown that CYP716A52v2 is involved in the ginsenosides biosynthesis. Therefore, identification of CYP716A52v2 annotated unigenes within our set of potential CYP450 candidates using an unbiased approach further indicates that other CYP450 identified in this study will most likely play a role in ginsenosides biosynthesis.
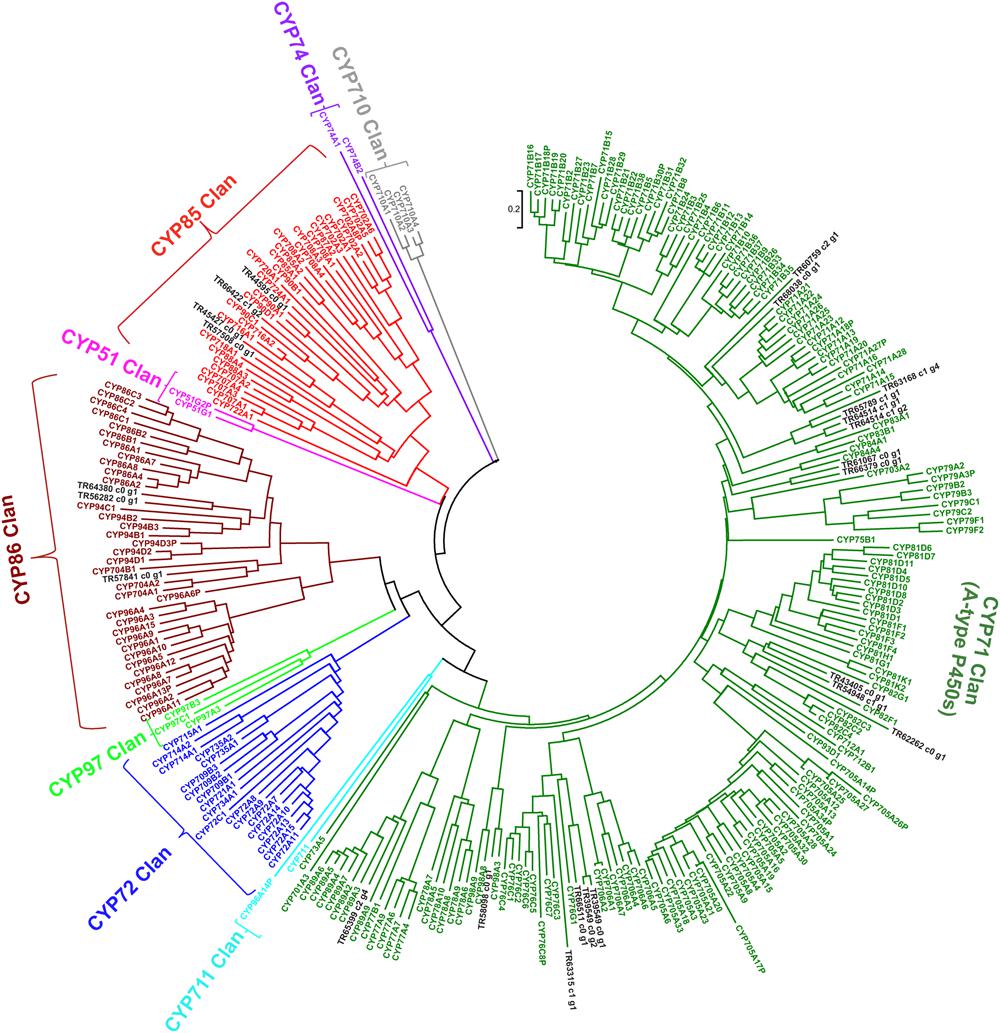
FIGURE 9. Phylogenetic analysis of putative CYP450s unigenes from P. japonicus transcriptome with all CYP450 genes from Arabidopsis genome. Protein sequences were aligned using MUSCLE program, and evolutionary distances were computed using Jones-Taylor-Thornton (JTT) method. A Neighbor-Joining (NJ) tree was constructed with bootstrap values obtained after 10,000 replications using MEGA6 program.
Expression analysis for unigenes annotated as GTs from cluster-2 and -3 were highly expressed in the roots, with highest expression level in the secRoot, followed by rhizome_Y and rhizome_O, respectively (Figure 8). This expression trend was different than what we observed for CYP450s and MVA pathways associated unigenes. Previous reports showed high saponins accumulation in the rhizome of Panax species, and roots as the main source of ginsenosides (Yang W.Z. et al., 2014). Our results are consistent with the previous findings, although, our transcriptome data suggested gene expression segregation across different parts of the root. Gene expression trend suggested that while saponins aglycones are synthesized in rhizome_Y and rhizome_O, most likely sugar conjugation occurs in the secRoot. It will be interesting to validate this hypothesis driven from our transcriptome analysis, and to further test if this is due to some transcriptional regulation or translocation of metabolites across these tissues. To further characterize unigenes annotated as GTs, we performed phylogenetic analysis together with 128 genes annotated as GTs from Arabidopsis genome, and as classified and obtained from http://www.p450.kvl.dk/p450.shtml (Paquette et al., 2009). Majority of these unigenes were grouped into UGT81 (19 unigenes), UGT71 (9 unigenes) and UGT78 (5 unigenes) families (Figure 10). Several genes from Arabidopsis within these GTs families have been characterized to conjugate sugar moieties to different classes of metabolites (Benning and Ohta, 2005; Priest et al., 2005; Yonekura-Sakakibara and Hanada, 2011). Our results indicate that these unigenes from P. japonicus annotated as GTs are most likely participating in sugar conjugation process of saponin aglycones. Further investigations will be required to ascertain roles of these potential unigenes for their role in ginsenosides biosynthesis.
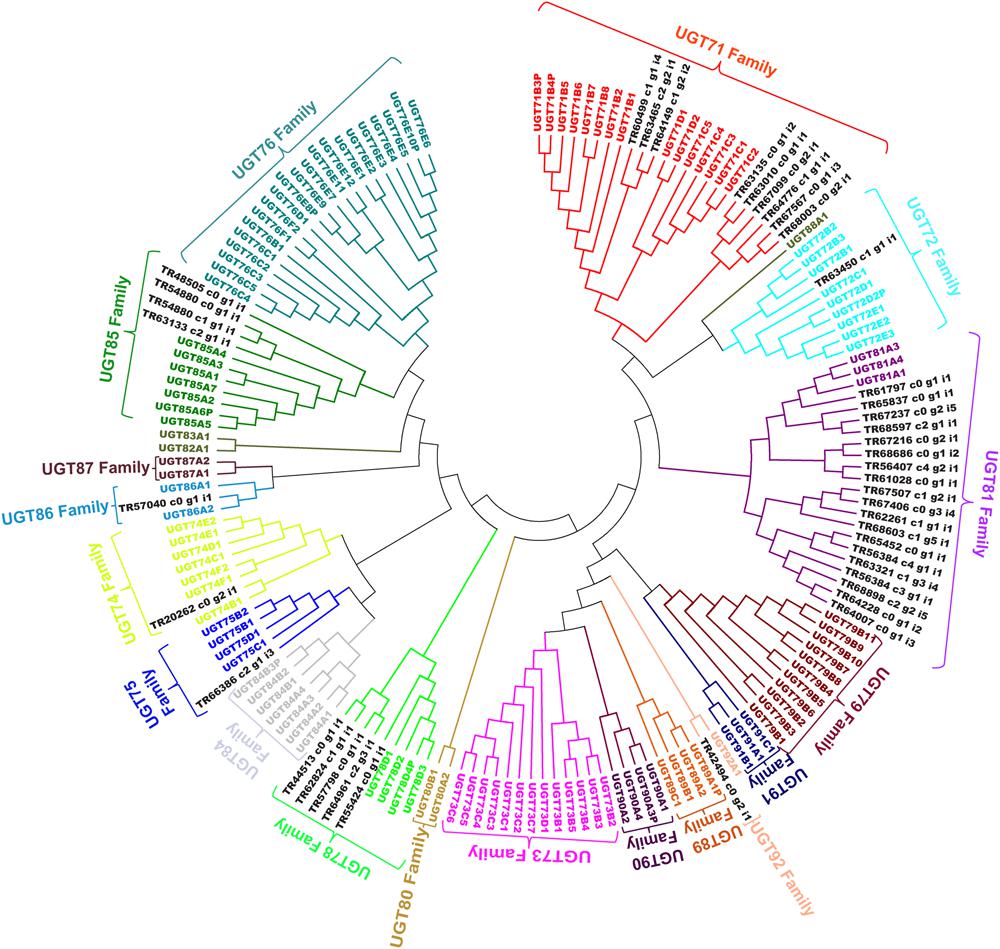
FIGURE 10. Phylogenetic analysis of putative GTs unigenes from P. japonicus transcriptome with 128 genes annotated as GTs from Arabidopsis genome. Protein sequences were aligned using MUSCLE program, and evolutionary distances were computed using JTT method. A NJ tree was constructed with bootstrap values obtained after 10,000 replications using MEGA6 program.
In summary, de novo transcriptome assembly of five tissues of P. japonicus, and its comparison with three key Panax species revealed great sequence similarity than known before within these major ginsenosides producing plants. Analysis derived from functional annotation and expression analysis can address many biological important questions. We believe that new candidate genes identified as a result of comparative transcriptomics approach will serve as strong candidates for future discoveries on ginsenosides biosynthetic pathways.
Author Contributions
KS, MY: Conceived and designed the experiments. MN, MK: Performed the experiments. HS: performed deep-transcriptome sequencing. MY, HS: Contributed reagents/materials/analysis tools. AR, MY, HT: Conducted the data interpretation and laboratory analysis. AR, MY, KS: Contributed to the manuscript preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported, in part, by Health and Labour Sciences Research Grant on the enhancement of ‘Comprehensive Medicinal Plant Database,’ by the Grants-in-Aid for Scientific Research of The Japan Society for the Promotion of Science (JSPS), and by Strategic Priority Research Promotion Program of Chiba University. HT was partially supported by MEXT KAKENHI (number 221S0002). The super-computing resource was provided by National Institute of Genetics, Research Organization of Information and Systems, Japan. The computing resources were provided by National Institute of Genetics, Research Organization of Information and Systems, and Medical Mycology Research Center, Chiba University, Japan. We are grateful to Dr. Ashfaq Mahmood (Chiba University), and Dr. Shivshankar Umashankar (National University of Singapore), and two anonymous reviewers for critical reading of the manuscript and many helpful suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00481
FIGURE S1 | Experimental workflow describing experimental design and analysis pipeline used for studying P. japonicus transcriptome, and its comparison with other Panax species.
FIGURE S2 | Correlation plot for all five tissues of P. japonicus based on expression value of unigenes with non-zero FPKM value. Unigenes from each tissue with non-zero FPKM expression value were use, and correlation values were calculated and plotted using corrplot from CRAN packages. The color and colored area of the pie chart for each tissue represent r2-value against rest of the tissues.
FIGURE S3 | Venn diagram for number of unigenes of P. japonicus with a blastn hit against P. ginseng, P. notoginseng, and P. quinquefolius sequences used as blastn search databases.
FIGURE S4 | Phylogenetic analysis of putative CYP450s unigenes from P. japonicus transcriptome with 325 genes annotated as CYP450 from Vitis vinifera. Protein sequences were aligned using MUSCLE program, and evolutionary distances were computed using JTT method.A Neighbor-Joining (NJ) tree was constructed with bootstrap values obtained after 10,000 replications using MEGA6 program.
TABLE S1 | Summary of Trimmomatic program based pre-processing of raw reads for all five tissues of P. japonicus.
TABLE S2 | List of KEGG pathways and assigned unigenes from the P. japonicus transcriptome assembly.
TABLE S3 | Identified SSRs motifs and its frequency in the P. japonicus transcriptome assembly.
TABLE S4 | Transcriptome data source for P. ginseng, P. notoginseng, and P. quinquefolius to perform comparative analysis with the P. japonicus transcriptome assembly.
TABLE S5 | Unigenes from the P. japonicus transcriptome assembly grouped in six different gene clusters by k-mean clustering analysis.
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE78893
- ^ http://www.ncbi.nlm.nih.gov
- ^ http://pgrc.ipk-gatersleben.de/misa/
- ^ http://workbench.sdsc.edu/
- ^ http://medicinalplantgenomics.msu.edu/
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bak, S., Beisson, F., Bishop, G., Hamberger, B., Hofer, R., Paquette, S., et al. (2011). Cytochromes p450. Arabidopsis Book 9:e0144. doi: 10.1199/tab.0144
Benning, C., and Ohta, H. (2005). Three enzyme systems for galactoglycerolipid biosynthesis are coordinately regulated in plants. J. Biol. Chem. 280, 2397–2400. doi: 10.1074/jbc.R400032200
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Briskin, D. P. (2000). Medicinal plants and phytomedicines. Linking plant biochemistry and physiology to human health. Plant Physiol. 124, 507–514. doi: 10.1104/pp.124.2.507
Choi, H. I., Kim, N. H., Kim, J. H., Choi, B. S., Ahn, I. O., Lee, J. S., et al. (2011). Development of reproducible EST-derived SSR markers and assessment of genetic diversity in panax ginseng cultivars and related species. J. Ginseng Res. 35, 399–412. doi: 10.5142/jgr.2011.35.4.399
Conesa, A., and Gotz, S. (2008). Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008:619832. doi: 10.1155/2008/619832
Edgar, R., Domrachev, M., and Lash, A. E. (2002). Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. doi: 10.1093/nar/30.1.207
Fukushima, A., Nakamura, M., Suzuki, H., Saito, K., and Yamazaki, M. (2015). High-throughput sequencing and de novo assembly of red and green forms of the Perilla frutescens var. crispa Transcriptome. PLoS ONE 10:e0129154. doi: 10.1371/journal.pone.0129154
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Han, J. Y., Hwang, H. S., Choi, S. W., Kim, H. J., and Choi, Y. E. (2012). Cytochrome P450 CYP716A53v2 catalyzes the formation of protopanaxatriol from protopanaxadiol during ginsenoside biosynthesis in Panax ginseng. Plant Cell Physiol. 53, 1535–1545. doi: 10.1093/pcp/pcs106
Han, J. Y., Kim, H. J., Kwon, Y. S., and Choi, Y. E. (2011). The Cyt P450 enzyme CYP716A47 catalyzes the formation of protopanaxadiol from dammarenediol-II during ginsenoside biosynthesis in Panax ginseng. Plant Cell Physiol. 52, 2062–2073. doi: 10.1093/pcp/pcr150
Han, J. Y., Kim, M. J., Ban, Y. W., Hwang, H. S., and Choi, Y. E. (2013). The involvement of beta-amyrin 28-oxidase (CYP716A52v2) in oleanane-type ginsenoside biosynthesis in Panax ginseng. Plant Cell Physiol. 54, 2034–2046. doi: 10.1093/pcp/pct141
Han, L. K., Zheng, Y. N., Yoshikawa, M., Okuda, H., and Kimura, Y. (2005). Anti-obesity effects of chikusetsusaponins isolated from Panax japonicus rhizomes. BMC Complement Altern. Med. 5:9. doi: 10.1186/1472-6882-5-9
Hancock, J. M. (1996). Simple sequences in a “minimal’ genome. Nat. Genet. 14, 14–15. doi: 10.1038/ng0996-14
Jarvis, D. E., Kopp, O. R., Jellen, E. N., Mallory, M. A., Pattee, J., Bonifacio, A., et al. (2008). Simple sequence repeat marker development and genetic mapping in quinoa (Chenopodium quinoa Willd.). J. Genet. 87, 39–51. doi: 10.1007/s12041-008-0006-6
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Morita, T., Tanaka, O., and Kohda, H. (1985). Saponin composition of rhizomes of Panax japonicus collected in South Kyushu, Japan, and its significance in oriental traditional medicine. Chem. Pharm. Bull. (Tokyo) 33, 3852–3858. doi: 10.1248/cpb.33.3852
Muranaka, T., and Saito, K. (2013). Phytochemical genomics on the way. Plant Cell Physiol. 54, 645–646. doi: 10.1093/pcp/pct058
Murthy, H. N., Georgiev, M. I., Kim, Y. S., Jeong, C. S., Kim, S. J., Park, S. Y., et al. (2014). Ginsenosides: prospective for sustainable biotechnological production. Appl. Microbiol. Biotechnol. 98, 6243–6254. doi: 10.1007/s00253-014-5801-9
Paquette, S. M., Jensen, K., and Bak, S. (2009). A web-based resource for the Arabidopsis P450, cytochromes b5, NADPH-cytochrome P450 reductases, and family 1 glycosyltransferases (http://www.P450.kvl.dk). Phytochemistry 70, 1940–1947. doi: 10.1016/j.phytochem.2009.08.024
Priest, D. M., Jackson, R. G., Ashford, D. A., Abrams, S. R., and Bowles, D. J. (2005). The use of abscisic acid analogues to analyse the substrate selectivity of UGT71B6, a UDP-glycosyltransferase of Arabidopsis thaliana. FEBS Lett. 579, 4454–4458. doi: 10.1016/j.febslet.2005.06.084
Qi, L. W., Wang, C. Z., and Yuan, C. S. (2011). Ginsenosides from American ginseng: chemical and pharmacological diversity. Phytochemistry 72, 689–699. doi: 10.1016/j.phytochem.2011.02.012
Rai, A., and Saito, K. (2015). Omics data input for metabolic modeling. Curr. Opin. Biotechnol. 37, 127–134. doi: 10.1016/j.copbio.2015.10.010
Saito, K. (2013). Phytochemical genomics–a new trend. Curr. Opin. Plant Biol. 16, 373–380. doi: 10.1016/j.pbi.2013.04.001
Seki, H., Ohyama, K., Sawai, S., Mizutani, M., Ohnishi, T., Sudo, H., et al. (2008). Licorice beta-amyrin 11-oxidase, a cytochrome P450 with a key role in the biosynthesis of the triterpene sweetener glycyrrhizin. Proc. Natl. Acad. Sci. U.S.A. 105, 14204–14209. doi: 10.1073/pnas.0803876105
Senthilvel, S., Jayashree, B., Mahalakshmi, V., Kumar, P. S., Nakka, S., Nepolean, T., et al. (2008). Development and mapping of simple sequence repeat markers for pearl millet from data mining of expressed sequence tags. BMC Plant Biol. 8:119. doi: 10.1186/1471-2229-8-119
Shibata, S. (2001). Chemistry and cancer preventing activities of ginseng saponins and some related triterpenoid compounds. J. Korean Med. Sci. 16(Suppl.), S28–S37. doi: 10.3346/jkms.2001.16.s.s28
Shibuya, M., Hoshino, M., Katsube, Y., Hayashi, H., Kushiro, T., and Ebizuka, Y. (2006). Identification of beta-amyrin and sophoradiol 24-hydroxylase by expressed sequence tag mining and functional expression assay. FEBS J. 273, 948–959. doi: 10.1111/j.1742-4658.2006.05120.x
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Thiel, T., Michalek, W., Varshney, R. K., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422.
Varshney, R. K., Graner, A., and Sorrells, M. E. (2005). Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 23, 48–55. doi: 10.1016/j.tibtech.2004.11.005
Varshney, R. K., Thiel, T., Stein, N., Langridge, P., and Graner, A. (2002). In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol. Biol. Lett. 7, 537–546.
Vogler, B. K., Pittler, M. H., and Ernst, E. (1999). The efficacy of ginseng. A systematic review of randomised clinical trials. Eur. J. Clin. Pharmacol. 55, 567–575. doi: 10.1007/s002280050674
Yamahara, J., Kubomura, Y., Miki, K., and Fujimura, H. (1987). Anti-ulcer action of Panax japonicus rhizome. J. Ethnopharmacol. 19, 95–101. doi: 10.1016/0378-8741(87)90141-3
Yang, W. Z., Hu, Y., Wu, W. Y., Ye, M., and Guo, D. A. (2014). Saponins in the genus Panax L. (Araliaceae): a systematic review of their chemical diversity. Phytochemistry 106, 7–24. doi: 10.1016/j.phytochem.2014.07.012
Yang, X., Wang, R., Zhang, S., Zhu, W., Tang, J., Liu, J., et al. (2014). Polysaccharides from Panax japonicus C.A. Meyer and their antioxidant activities. Carbohydr. Polym. 101, 386–391. doi: 10.1016/j.carbpol.2013.09.038
Yonekura-Sakakibara, K., and Hanada, K. (2011). An evolutionary view of functional diversity in family 1 glycosyltransferases. Plant J. 66, 182–193. doi: 10.1111/j.1365-313X.2011.04493.x
Yoshizaki, K., Devkota, H. P., Fujino, H., and Yahara, S. (2013). Saponins composition of rhizomes, taproots, and lateral roots of Satsuma-ninjin (Panax japonicus). Chem. Pharm. Bull. (Tokyo) 61, 344–350. doi: 10.1248/cpb.c12-00764
Yun, T. K. (2001). Panax ginseng–a non-organ-specific cancer preventive? Lancet Oncol. 2, 49–55. doi: 10.1016/S1470-2045(00)00196-0
Zhang, S., Wang, R., Zeng, W., Zhu, W., Zhang, X., Wu, C., et al. (2015). Resource investigation of traditional medicinal plant Panax japonicus (T.Nees) C.A. Mey and its varieties in China. J Ethnopharmacol. 166, 79–85. doi: 10.1016/j.jep.2015.02.051
Zhu, S., Fushimi, H., Cai, S., and Komatsu, K. (2003). Phylogenetic relationship in the genus Panax: inferred from chloroplast trnK gene and nuclear 18S rRNA gene sequences. Planta Med. 69, 647–653. doi: 10.1055/s-2003-41117
Zhu, S., Zou, K., Fushimi, H., Cai, S., and Komatsu, K. (2004). Comparative study on triterpene saponins of Ginseng drugs. Planta Med. 70, 666–677. doi: 10.1055/s-2004-827192
Keywords: Panax japonicas, RNA-seq, triterpenoid saponins, cytochrome P450, comparative transcriptomics analysis
Citation: Rai A, Yamazaki M, Takahashi H, Nakamura M, Kojoma M, Suzuki H and Saito K (2016) RNA-seq Transcriptome Analysis of Panax japonicus, and Its Comparison with Other Panax Species to Identify Potential Genes Involved in the Saponins Biosynthesis. Front. Plant Sci. 7:481. doi: 10.3389/fpls.2016.00481
Received: 26 January 2016; Accepted: 24 March 2016;
Published: 12 April 2016.
Edited by:
Danièle Werck, Centre National de la Recherche Scientifique, FranceCopyright © 2016 Rai, Yamazaki, Takahashi, Nakamura, Kojoma, Suzuki and Saito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amit Rai, amit.rai@chiba-u.jp; Mami Yamazaki, mamiy@faculty.chiba-u.jp