 Erich M. Schwarz
Erich M. Schwarz Adrienne H. K. Roeder
Adrienne H. K. Roeder- 1Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY, USA
- 2Weill Institute for Cell and Molecular Biology and Section of Plant Biology, School of Integrative Plant Sciences, Cornell University, Ithaca, NY, USA
Endoreduplication is a specialized cell cycle in which DNA replication occurs, but mitosis is skipped creating enlarged polyploid cells. Endoreduplication is associated with the differentiation of many specialized cell types. In the Arabidopsis thaliana sepal epidermis endoreduplicated giant cells form interspersed between smaller cells. Both the transcription factor Arabidopsis thaliana MERISTEM LAYER1 (ATML1) and the plant-specific cyclin dependent kinase inhibitor LOSS OF GIANT CELLS FROM ORGANS (LGO)/SIAMESE RELATED1 (SMR1) are required for the formation of giant cells. Overexpression of LGO is sufficient to produce sepals covered in highly endoreduplicated giant cells. Here we ask whether overexpression of LGO changes the transcriptome of these mature sepals. We show that overexpression of LGO in the epidermis (LGOoe) drives giant cell formation even in atml1 mutant sepals. Using RNA-seq we show that LGOoe has significant effects on the mature sepal transcriptome that are primarily ATML1-independent changes of gene activity. Genes activated by LGOoe, directly or indirectly, predominantly encode proteins involved in defense responses, including responses to wounding, insects (a predator of Arabidopsis), and fungus. They also encode components of the glucosinolate biosynthesis pathway, a key biochemical pathway in defense against herbivores. LGOoe-activated genes include previously known marker genes of systemic acquired resistance such as PR1 through PR5. The defensive functions promoted by LGOoe in sepals overlap with functions recently shown to be transcriptionally activated by hyperimmune cpr5 mutants in a LGO-dependent manner. Our findings show that the cell cycle regulator LGO can directly or indirectly drive specific states of gene expression; in particular, they are consistent with recent findings showing LGO to be necessary for transcriptional activation of many defense genes in Arabidopsis.
Introduction
During development, particular cell types often have specialized cell cycles. In plants, many differentiating cell types undergo endoreduplication, a cell cycle in which cells grow and replicate their DNA but do not undergo mitosis or cytokinesis (Lee et al., 2009; Breuer et al., 2010). Likewise, in animals, endoreduplication is linked with differentiation of several key cell types, such as megakaryocytes that generate platelets in mammalian blood and salivary gland cells in Drosophila that contain polytene chromosomes (Ullah et al., 2009). Endoreduplication allows cells to become enlarged, and the endopolyploidy level (i.e., DNA content) is directly proportional to cell size (Melaragno et al., 1993; Roeder et al., 2010).
The Arabidopsis sepal epidermis is a new model system in which to investigate the role of endoreduplication in the formation of specialized giant cells. The sepal is the outermost green floral organ, which encloses and protects the developing reproductive organs. The cells in the outer/abaxial epidermis of Arabidopsis sepals are diverse in size, ranging from giant cells stretching to an average of 360 μm, to the smallest cells reaching only about 10 μm (Figures 1A–C) (Roeder et al., 2010). Giant cells are also found on the abaxial epidermis of leaves (Melaragno et al., 1993; Roeder et al., 2010, 2012). A key function of giant cells is precise control of the curvature of sepals, which is necessary for sepals to form a closed shell protecting immature flowers (Roeder et al., 2010, 2012). In the sepal epidermis, cell types are correlated with variations in cell cycles. Giant cells generally undergo three rounds of endoreduplication to become endopolyploid 16C cells, whereas small cells undergo mitotic divisions and remain generally 2C or 4C (Roeder et al., 2010). Two enhancer trap markers drive cell type-specific expression within the sepal, one in giant cells and the other in small cells; these enhancers demonstrate that giant cells and small cells can have distinct patterns of gene expression, as well as distinct cell sizes and DNA contents (Roeder et al., 2012). Moreover, study of these enhancers in mutant backgrounds has shown that the balance between giant and small cells in sepals depends both on the transcription factor gene ATML1 and on the cell cycle regulator gene LGO.
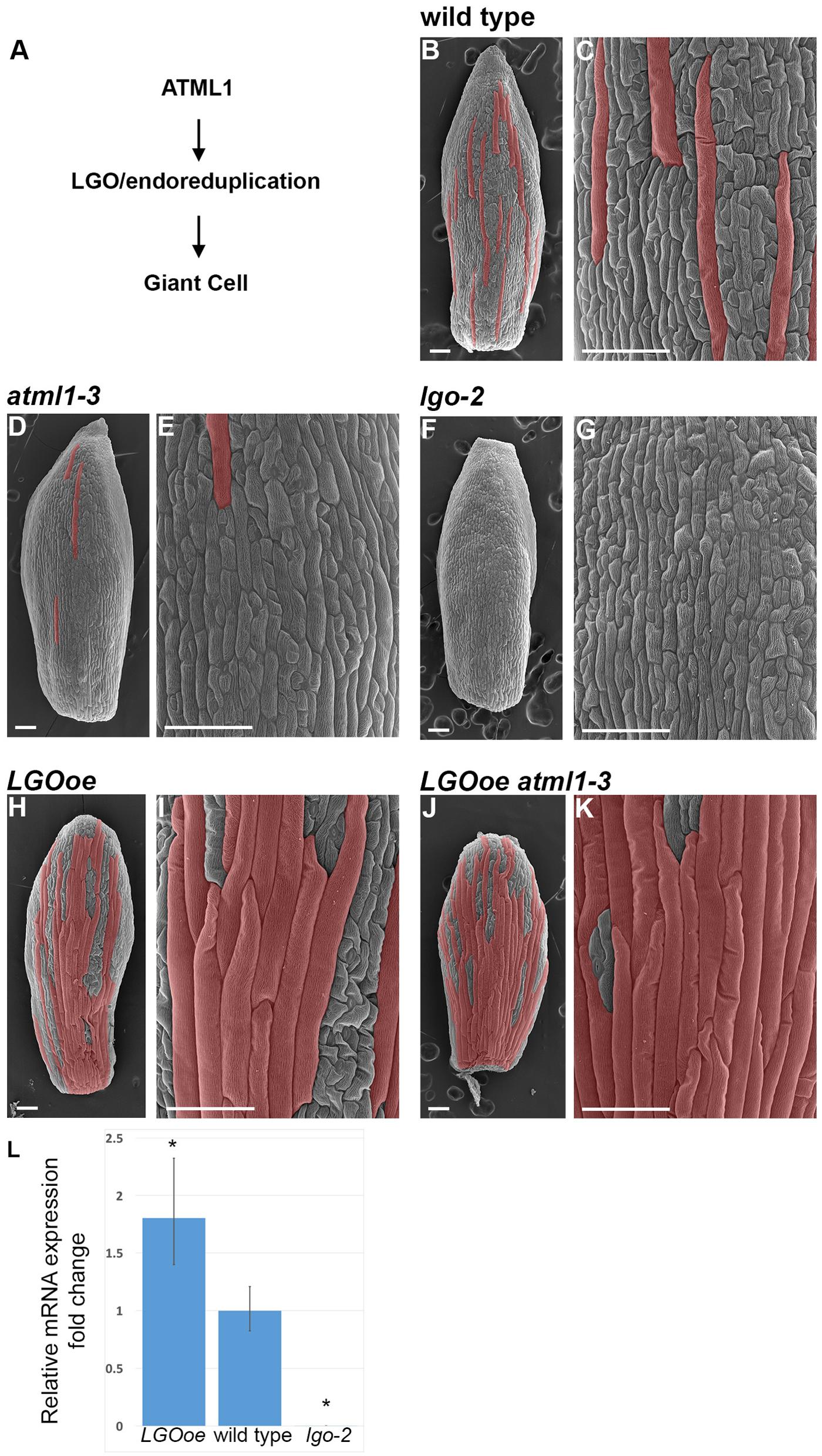
FIGURE 1. Cell size patterning phenotypes. (A) A genetic diagram depicting our current understanding of the pathway leading to giant cell formation. The HD-ZIP class IV transcription factor ATML1 directly or indirectly activates endoreduplication through the CKI LGO. Endoreduplication causes the enlargement and differentiation of the cell. It is not yet known to what extent LGO, endoreduplication, or both regulate gene expression in the sepal. (B,C) Scanning electron micrograph (SEM) of a wild-type sepal at stage 12. Giant cells, as assessed by their size and outward bulging from the surface of the sepal, have been false-colored red in Photoshop. (C) Is a magnified view of the cells in (B). (D,E) SEM of atml1-3 stage 12 mutant sepal (D) and magnified view of the cells (E). Giant cells are strongly reduced in this allele, although the phenotype is not as strong as lgo-2. (F,G) SEM of lgo-2 mutant sepal (F) at stage 12 with magnified view of the cells (G). Note the absence of giant cells. (H,I) SEM of a stage 12 sepal in which LGO is overexpressed throughout the epidermis under control of the ATML1 promoter (ATML1p::LGO); this genotype is referred to throughout the text as LGOoe. Many highly enlarged cells are formed that appear similar to giant cells in phenotype. (J,K) SEM of a sepal in which LGO is overexpressed in the epidermis of an atml1-3 mutant (LGOoe atml1-3). Many highly enlarged cells form, suggesting that LGO acts downstream of ATML1 in the formation of giant cells. This genotype allowed us to determine what genes LGO could regulate independently of ATML1 activity. Sepals in all of the images were taken from replicate 1 plants. (L) qPCR measurement of the 1.8-fold increase of LGO expression in inflorescences from LGOoe plants relative to Col_WT inflorescences. With these primers which flank the t-DNA insertion site, no lgo-2 transcript is detected. ∗ indicates p-value < 0.05 when compared to wild type. Error bars represent the standard deviation.
Mutations in atml1 result in the reduction or absence of giant cells in sepals, and the corresponding loss of 16C cells in the epidermis (Figures 1D,E) (Roeder et al., 2012). ATML1 encodes a HD-ZIP IV transcription factor and is important for establishing epidermal identity together with its paralog, PROTODERMAL FACTOR2 (PDF2) (Abe et al., 2003; Nakamura et al., 2006). The epidermis is absent in atml1 pdf2 double mutants, exposing the mesophyll cells, whereas atml1 single mutants have an intact epidermis, but lack giant cells. Overexpression of ATML1 or the related HD-ZIP protein HDG2 in internal cell layers of the cotyledon is sufficient to induce the ectopic formation of epidermal cell types including giant cells and stomata (Peterson et al., 2013; Takada et al., 2013). ATML1 promotes expression of the giant cell molecular marker: in atml1 sepals, its expression significantly diminishes (Roeder et al., 2012). Conversely, ATML1 has little effect on expression of the small cell molecular marker, which remains largely unchanged in atml1 sepals.
Similarly to atml1, lgo mutants fail to form giant cells because all the epidermal cells in sepals and leaves divide instead of endoreduplicating, creating numerous small cells in the place of giant cells (Figures 1F,G) (Roeder et al., 2010). Ploidy measurements confirm that 16C giant cells are absent in lgo mutants. Conversely, overexpression of LGO throughout the epidermis (herein referred to as LGOoe) causes the abaxial sepal epidermis to be nearly covered with highly endoreduplicated giant cells (Figures 1H,I) (Roeder et al., 2010), because epidermal cells that would normally divide to make small cells instead endoreduplicate to make giant cells. LGO, also known as SIAMESE RELATED1 (SMR1), encodes a cyclin dependent kinase inhibitor (CKI) in the plant-specific family of SIAMESE RELATED (SMR) homologs, whose founding member is SIAMESE (SIM). SMRs bind to and inhibit cyclins and CDKs, promoting endoreduplication (Churchman et al., 2006; Peres et al., 2007; Roeder et al., 2010; Kumar et al., 2015). LGO has little effect on expression of the giant cell molecular marker, which is still expressed in lgo mutant sepals (that lack overt giant cells) and whose expression does not increase in LGOoe sepals (that are dominated by excess giant cells). Conversely, LGO has strong effects on expression of the small cell molecular marker: in lgo sepals, its expression spreads to most cells in the sepal; in LGOoe sepals, its expression shrinks to fewer cells.
There are at least two ways that CKIs such as LGO could affect gene expression. First, CKIs can affect gene expression through their regulation of the cell cycle. CKIs inhibit the activity of cyclin dependent kinases (CDKs) (Kumar et al., 2015). The LGO paralog SIM has been shown to interact with both CYCLIN Ds and CYCLIN DEPENDENT KINASE A;1 (CDKA;1) which regulate the G1 to S transition in the cell cycle (Churchman et al., 2006; Kumar et al., 2015). CYCLIND/CDKA;1 complexes phosphorylate RETINOBLASTOMA RELATED1 (RBR1), releasing E2F transcription factors to activate cell cycle gene transcription (Desvoyes et al., 2006, 2014). RBR1 can bind to and regulate the activity of other transcription factors including the key developmental regulators FAMA and SCARECROW (Cruz-Ramírez et al., 2012; Matos et al., 2014). In addition, CDKs can phosphorylate transcription factors to regulate their activity (Reindl et al., 1997; Yang et al., 2015). Second, in animals it has been found that CKIs can directly control gene expression (Lim and Kaldis, 2013) by binding transcription factors and altering their activity by either activating or inhibiting target genes (Li et al., 2012; Pippa et al., 2012; Lawrence et al., 2014; Hardwick et al., 2015). Similar mechanisms are likely in plants. In a large-scale survey of Arabidopsis cell cycle protein complexes, four p27Kip1-related CKIs (KRP2, KRP3, KRP4, and KRP5) were found to bind four different transcription factors; LGO was found to bind the transcription factor bZIP69 (Van Leene et al., 2010). ChIP-seq has demonstrated that one of these Arabidopsis CKIs, KRP5, binds 264 genes that are enriched 23-fold for functions in cell wall organization, and that may be transcriptionally activated by KRP5 (Jegu et al., 2013). Likewise, a CKI in tomato forms part of a protein complex with the bZIP transcription factor SPGB (Pnueli et al., 2001; Churchman et al., 2006). Meanwhile, genetic analysis of the CKI SIAMESE (SIM) in Arabidopsis has demonstrated that it is required for cell fate: some siamese mutant trichomes (leaf hair cells) that fail to undergo endoreduplication lose their trichome identity, and revert to pavement cells (Bramsiepe et al., 2010).
Here, we use RNA-seq to determine to what extent LGO controls sepal gene expression directly or indirectly in mature sepals, independently of ATML1.
Results
Overexpression of LGO Promotes Giant Cell Formation Even in the Absence of ATML1
To determine to what degree LGO affects gene expression in mature sepals independently of ATML1, we first generated plants in which LGO was ectopically expressed under the epidermal-specific ATML1 promoter (referred to as LGOoe) (Sessions et al., 1999; Roeder et al., 2010, 2012) in the wild-type Columbia (Col_WT) background. LGO expression increased by 1.8-fold in inflorescences of the LGOoe plants (Figure 1L). We crossed the LGOoe plants to atml1-3 mutants and isolated plants homozygous for both LGOoe and atml1-3 (Figures 1J,K). We have previously shown that overexpressing KRP1 in a atml1-3 mutant background is sufficient to induce large, highly endoreduplicated cells (Roeder et al., 2012). Similarly, overexpressing LGO throughout the epidermis in either wild-type (Figures 1H,I) or an atml1-3 mutant background (Figures 1J,K) produced plants with highly enlarged cells, morphologically indistinguishable from giant cells, that largely covered the outer sepal epidermis. The existence of giant cells in LGOoe atml1-3 sepals (Figure 1A) confirmed previous work indicating that LGO exerts its effects on sepal endoreduplication and differentiation downstream of ATML1 and other epidermal specification genes (Roeder et al., 2012).
Transcriptomic Analysis Identifies Genes Regulated by LGO Independently of ATML1
We then used RNA-seq to observe and compare the transcriptomes of wild-type Columbia sepals (Col_WT; Figures 1B,C), atml1-3 sepals (Figures 1D,E), lgo-2 sepals (Figures 1F,G), LGOoe sepals (Figures 1H,I), and LGOoe atml1-3 sepals (Figures 1J,K). Three batches of these five genotypes were grown and harvested for RNA-seq, providing three biological replicates per genotype. For each replicate, we performed RNA-seq on 250 dissected whole stage 12 sepals. At stage 12, cells in the sepal have fully differentiated and cell division and endoreduplication has been completed (Roeder et al., 2012; Hervieux et al., 2016; Hong et al., 2016). This strategy allowed us to assess sepal transcriptomes in their final, differentiated state: expressing terminal genes that might be driven by LGO in mature sepals, rather than others (e.g., cell-cycle genes, or developmental regulatory genes) that might be driven by LGO in earlier stages of sepal development.
We used DESeq2 (Love et al., 2014) to identify 1,341 genes with significant differences in expression between genotypes, taking into account the three biological replicate batches of the samples (Figure 2; Supplementary File S01). Between genotypes, the strongest differences in gene expression were caused by LGO overexpression (Figure 2; Table 1; Supplementary File S02). More genes were significantly upregulated by LGO overexpression than downregulated (Figure 2; Table 1; Supplementary File S02). Expression trends were similar in LGO overexpression and LGO overexpression in the atml1 mutant background (LGOoe versus LGOoe atml1-3), suggesting that LGO regulates gene expression largely independently of ATML1 (Figure 2; see further analysis below). In addition, there was a substantial variability in expression of these significantly differentially expressed genes between replicates; however, all of these genes had significant p-values for differential expression based on genotypes controlling for variation between replicate batches (Figure 2; see further analysis below).
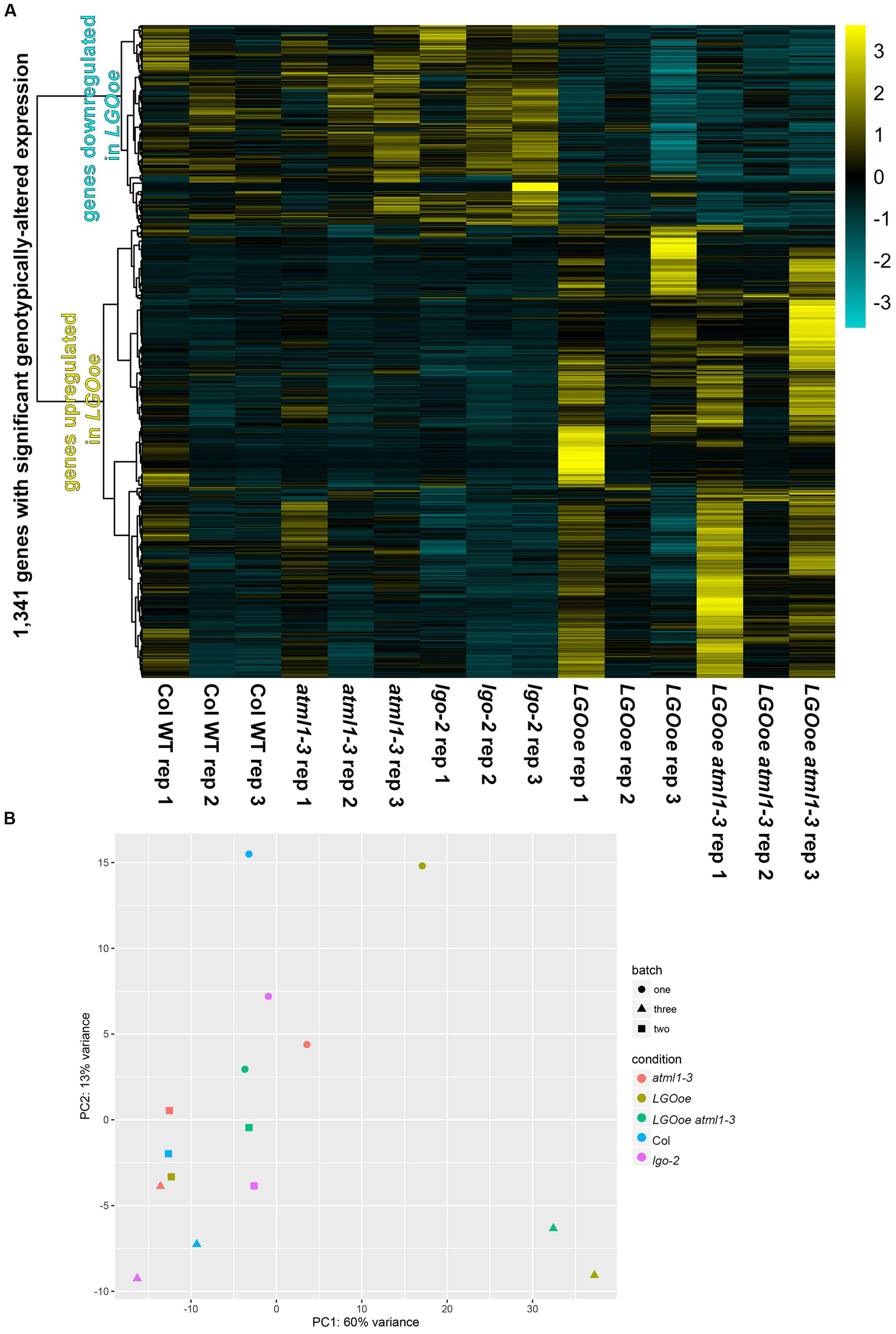
FIGURE 2. Gene expression in sepals. (A) An overview of relative gene activity for the 1,341 genes with significant differences in expression between genotypes. For each gene, expression levels were centered and scaled, so that the relative expression levels between conditions for the gene (rather than absolute changes of its expression levels) are shown. On the left, a dendrogram showing the clustering of these relative expression patterns is given. Relative expression is graded from maximum (yellow) to minimum (dark turquoise). The most obvious distinction in gene expression is between LGOoe and non-LGOoe genotypes; other changes in genotype have much weaker effects. Meanwhile, there are also visible differences between batches, with replicate 1 being obviously different from replicates 2 and 3. This probably reflects the effects of subtle environmental variations upon gene activity in sepals. (B) A principal component analysis (PCA) of each genotype (represented by color) in each batch (represented by shape) showing the differences in gene expression between genotypes and replicates. Note that samples in which LGO is overexpressed tend to fall toward the right of their respective batch on PC1.
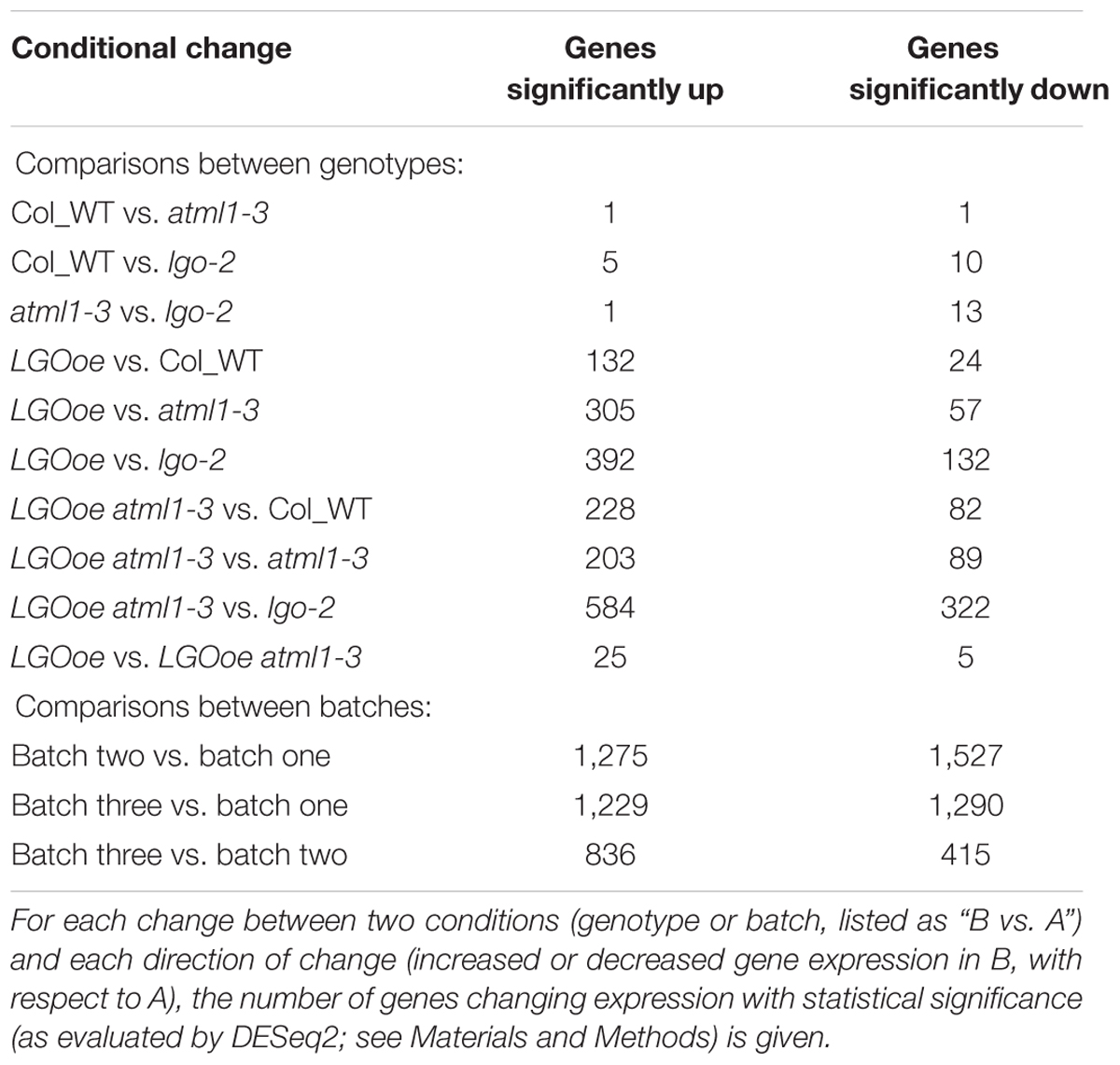
TABLE 1. Genes with conditionally changed expression.
In contrast, only two genes were significantly differentially expressed between Col_WT and atml1-3 mutants (Table 1; Supplementary File S02). Likewise only 15 genes were significantly differentially expressed between Col_WT and lgo-2 mutants (Table 1; Supplementary File S02). Given that there are only about 20 giant cells per sepal out of about 1,600 outer epidermal cells and many more cells in the other cell layers, it is not surprising that transcriptomic differences between Col_WT, atml1-3 and lgo-2 sepals would be small (Figure 1).
To double-check our analysis and ensure its accuracy, given that in some comparisons we found very few significantly differentially expressed genes, we used a second statistical analysis software package (edgeR) to reanalyze the RNA-seq data. edgeR identified exactly the same two genes and only those two genes as differentially expressed between Col_WT and atml1-3 mutant sepals. Furthermore, for each comparison, edgeR identified fewer genes as significantly differentially expressed between the genotypes. There was nearly complete overlap between the genes identified as differentially expressed by edgeR and the larger set of genes identified by DESeq2 (Supplementary File S03). This reanalysis validated the low number of differentially expressed genes identified by DESeq2; therefore, we used the DESeq2-derived genes for further analysis.
In the analyses below, we have focused on differential gene expression caused by LGO overexpression. In many cases, this led us to compare a gain-of-function mutant genotype (e.g., LGOoe) to a loss-of-function mutant genotype (e.g., lgo-2), rather than to Col_WT alone.
We next asked to what extent LGO affects transcription in the mature sepal independently of ATML1. We found that 292 genes were differentially expressed when LGO was overexpressed in the atml1-3 mutant when compared to the atml1-3 mutant alone (LGOoe atml1-3 versus atml1-3; Table 1; Supplementary File S02), indicating that the change of expression of these genes was independent of ATML1. 149 of these genes were also significantly differentially regulated when LGOoe was compared with lgo-2, confirming that these genes are regulated by LGO (Figure 3). Thus, many genes are directly or indirectly regulated by LGO independent of ATML1. Conversely, only 30 genes were significantly differentially regulated between LGOoe and LGOoe in the atml1-3 mutant (LGOoe versus LGOoe atml1-3), suggesting that very few genes that are regulated by LGO depend on ATML1 activity (Table 1; Figure 3). Therefore, a large majority of genes whose transcription was either up- or down-regulated by LGOoe did not depend on ATML1 activity, which is consistent with the role of LGO downstream of ATML1 in the giant cell formation pathway (Figure 1A).
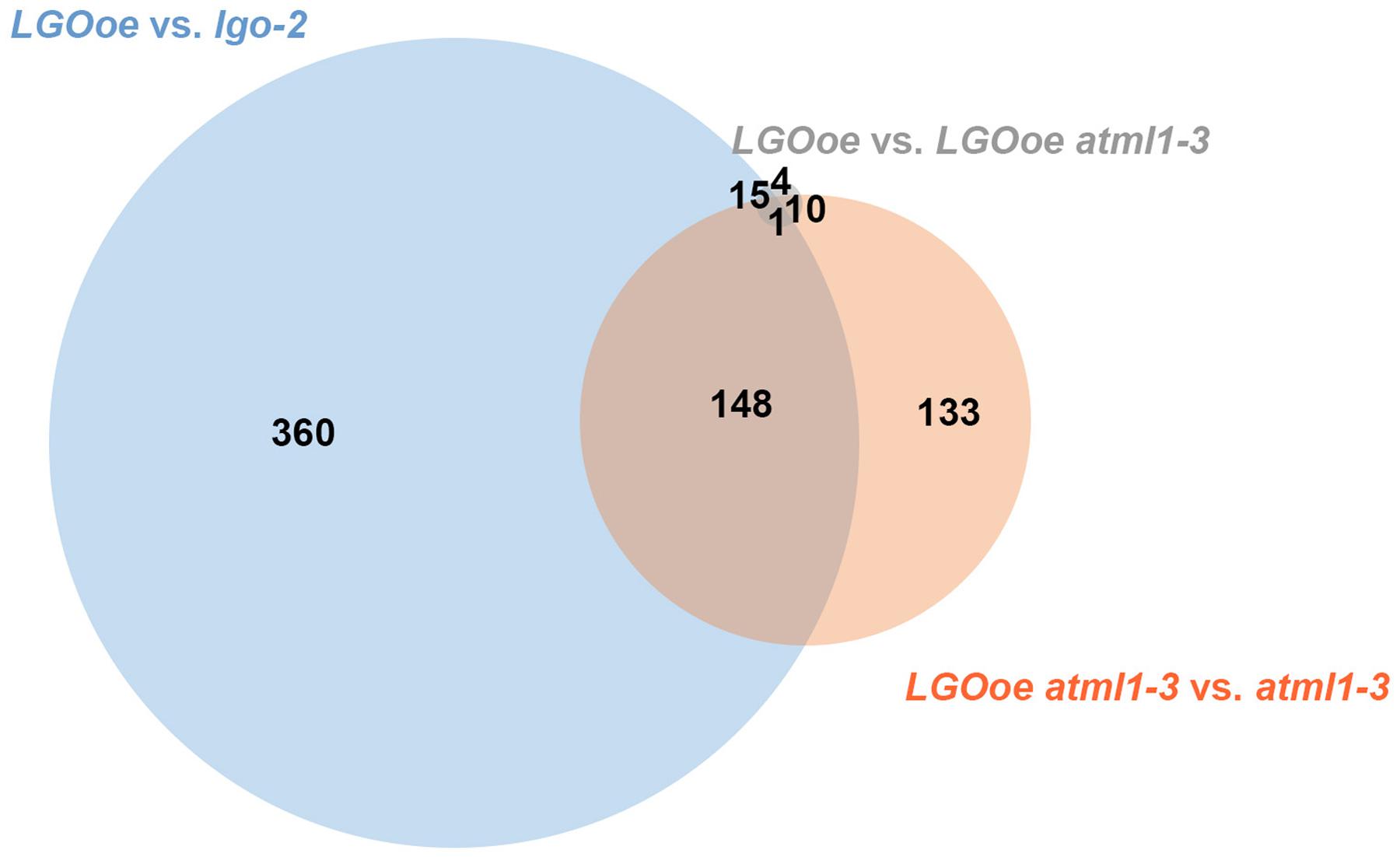
FIGURE 3. LGOoe differentially expressed genes. Venn diagram showing the number of differentially expressed genes shared between three different genotypic comparisons: LGOoe versus lgo-2 (blue), LGOoe atml1-3 versus atml1-3 (orange), and LGOoe versus LGOoe atml1-3 (gray). Note that only 30 genes are differentially expressed between LGOoe versus LGOoe atml1-3, suggesting that very few of the genes regulated by LGOoe require ATML1 activity.
As a positive control, we examined expression of AT5G17700, a MATE transporter gene that we have previously shown contains an enhancer driving giant cell-specific expression in sepals (Table 2) (Roeder et al., 2012). As expected, AT5G17700 expression was significantly lower in lgo-2 mutant sepals than wild type (2.4-fold higher expression in Col_WT versus lgo-2, padj = 2.3 ⋅ 10-2; Supplementary File S01), consistent with the diminished levels of giant cell enhancer expression observed in the small cells of lgo-2 mutant sepals (Roeder et al., 2012). A slight downregulation of AT5G17700 in atml1 mutants (1.9-fold higher expression in Col_WT versus atml1) did not reach statistical significance (p = 1.2 ⋅ 10-3, but padj = 1 after multiple testing correction). However, it did match previous observations of decreased giant enhancer expression in atml1 mutant sepals. AT5G17700 expression in either LGOoe or LGOoe atml1 genotypes was somewhat lower than its expression in Col_WT (LGOoe, 1.5-fold lower, p = 4.4 ⋅ 10-2, padj = 0.37; LGOoe atml1, 1.3-fold lower, p = 0.21, padj = 0.87); this fit our previous finding that endoreduplication alone does not activate expression of the giant cell enhancer (Roeder et al., 2012).

TABLE 2. Expression of giant cell enhancer gene AT5G17700.
We noticed considerable variability in expression of genes regulated by LGOoe between replicates (Figure 2) and many of the genes differentially regulated in LGOoe sepals were associated with response to the biotic environment (see below), so we examined the differences between biological replicates (batches). When we examined the expression of all genes, our biological replicates were highly reproducible with high correlation (R2) in gene expression between replicates (Supplementary File S04). However, we found 3,954 genes with significant changes of expression between batches but not genotypes, 598 genes with significant changes between genotypes but not batches, and 743 genes with significant changes between both batches and genotypes (Supplementary File S01); importantly, DESeq2 enabled statistical analyses in which changes in one condition (e.g., genotype) were controlled for simultaneous changes in a different condition (e.g., batch). Despite large changes in gene activity between the three batches, we observed no obvious differences in their health or growth conditions; nor did we observe any obvious sources of stress, such as wounding. Sepals are protective organs, and plants are thought to use transcriptional responses to cope with the challenges of their sessile lifestyle; thus, the changes in gene activity between batches may have been authentic in vivo responses to subtle environmental variations. In fact, these environmental variations were advantageous, allowing us to identify genes whose expression responded both to LGOoe and the environment (e.g., PR1 through PR5; see below).
Defense Responses are Upregulated by LGO Overexpression
To identify biological and molecular functions driven by LGO overexpression in sepal transcriptomes, we used FUNC (Prüfer et al., 2007) to identify GO terms that were statistically overrepresented among genes whose expression significantly changed between either genotypes or batches. To filter out environmental effects, we sorted the resulting GO terms into three groups (Supplementary File S05): those for which the most significant effect (as measured by p-value) came from a changed genotype rather than a changed batch (used for further analysis); those for which the most significant effect came from a changed batch comparison, but that did have significant p-values for changed genotypes as well; and those GO terms that only had significant p-values for changed batches. Because the first group of GO terms described functions more strongly affected by genotypic than environmental changes, we considered it most likely to describe the effects of LGO on sepal transcriptomes. Within this group, we identified several distinct subsets of GO terms that were associated with different genotypic changes and varying degrees of dependence on ATML1 (Tables 3–5; Supplementary Files S06–S10). In our sample harvesting technique, sepals were snap-frozen immediately upon dissection; we therefore consider it unlikely that the biological effects described below were induced by our experimental procedures.
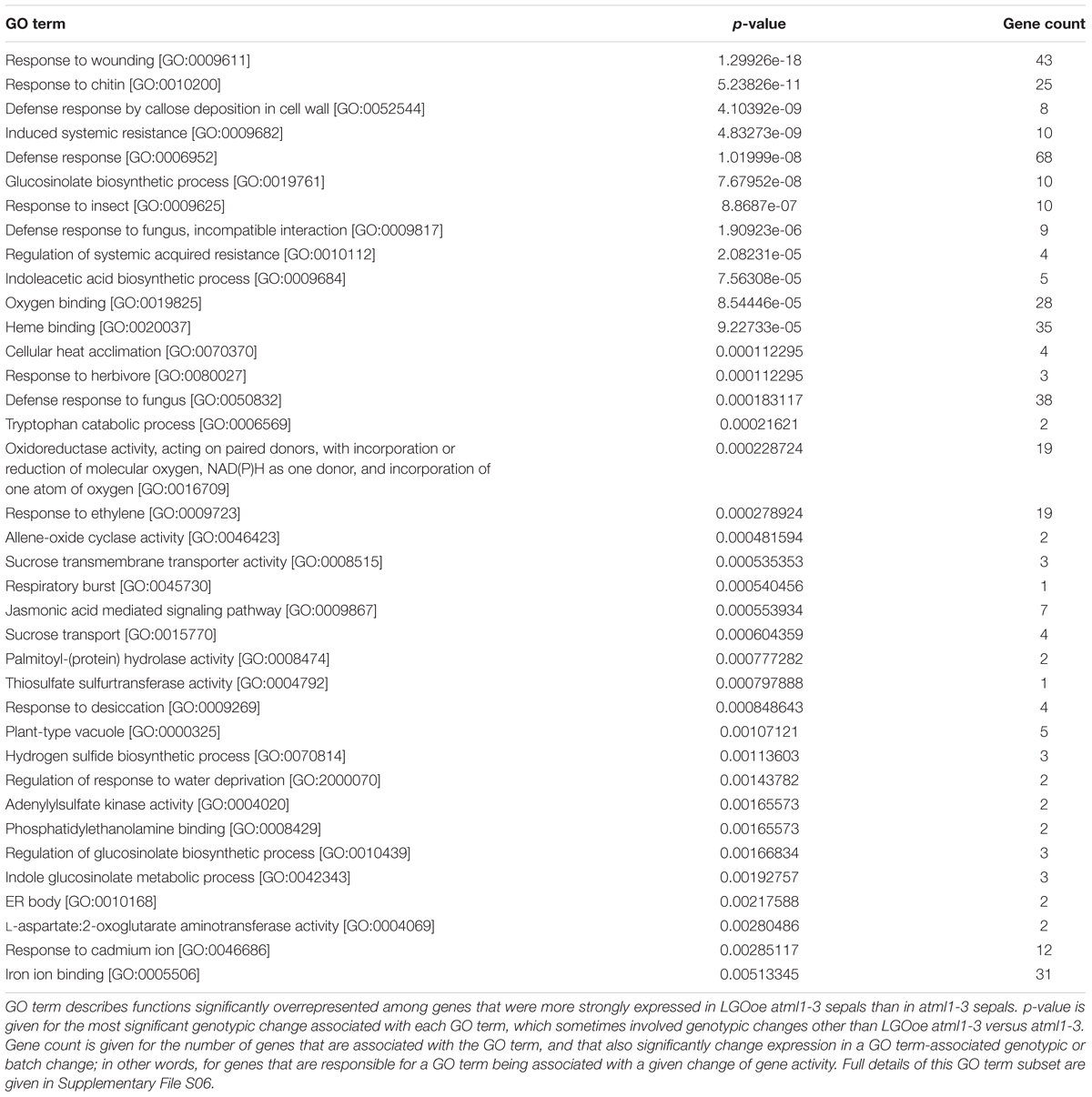
TABLE 3. Functions upregulated in sepals by LGOoe despite the absence of ATML1.
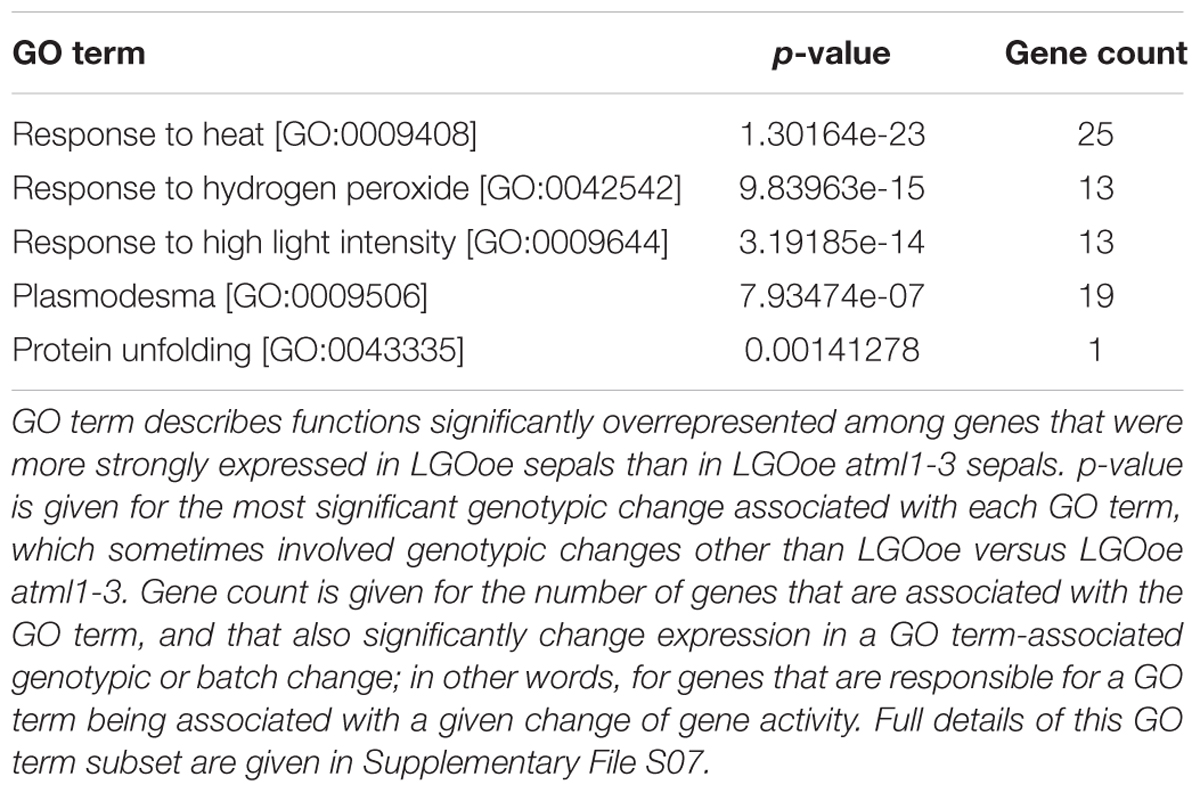
TABLE 4. Functions expressed in LGOoe sepals in an ATML1-dependent manner.

TABLE 5. Functions more expressed in LGOoe sepals than in either lgo-2 or atml1-3 sepals.
Genes with functions in defense response [GO:0006952] to many different pathogens including response to insect [GO:0009625] and defense response to fungus [GO:0050832] were all significantly upregulated in sepals by LGOoe versus non-LGOoe genotypes (i.e. Col_WT, atml1-3, or lgo-2; Tables 3–4; Figure 4A; Supplementary Files S06, S07, S11, S12). Many GO terms associated with the defense response hormone signaling pathways salicylic acid (response to salicylic acid [GO:0009751]), jasmonic acid (jasmonic acid mediated signaling pathway [GO:0009867]), and ethylene (response to ethylene [GO:0009723]) (Pieterse et al., 2009) have significantly higher representation among the genes upregulated by LGOoe versus non-LGOoe genotypes than among all protein-coding genes in the Arabidopsis genome (Figure 4; Table 3). Modification of the cell wall at the site of pathogen attack is a common response to infection (Clay et al., 2009; Luna et al., 2011) and defense response by callose deposition in the cell wall [GO:0052544] is significantly associated with both LGOoe versus lgo-2 and LGOoe atml1-3 versus atml1-3 (Figure 4A; Tables 3–4). PAMP-triggered immunity initiates upon recognition of a common feature of pathogens such as chitin, which is found in the cell wall of fungi and in the exoskeleton of insects (Shibuya and Minami, 2001); response to chitin [GO:0010200] is significantly upregulated in both LGOoe versus lgo-2 and LGOoe atml1-3 versus atml1-3 (Figure 4B; Table 3; Supplementary File S06) (Shibuya and Minami, 2001). On the other hand, response to herbivore [GO:0080027] and response to wounding [GO:0009611] that occurs during insect predation induce the jasmonic acid mediated signaling pathway [GO:0009867], which are all significantly associated with genes upregulated in LGOoe atml1-3 versus atml1-3 (Figure 4A; Table 3; Supplementary File S06) (Leon et al., 2001; Acosta and Farmer, 2010). Plants also recognize pathogen effectors and initiate effector-triggered immunity, which induces a respiratory burst [GO:0045730] of reactive oxygen species and response to hydrogen peroxide [GO:0042542]; both of these functions are significantly associated with genes upregulated by LGOoe versus non-LGOoe genotypes (Figure 4A; Tables 3–4; Supplementary Files S06 and S07) (Torres et al., 2006; Suzuki et al., 2011).
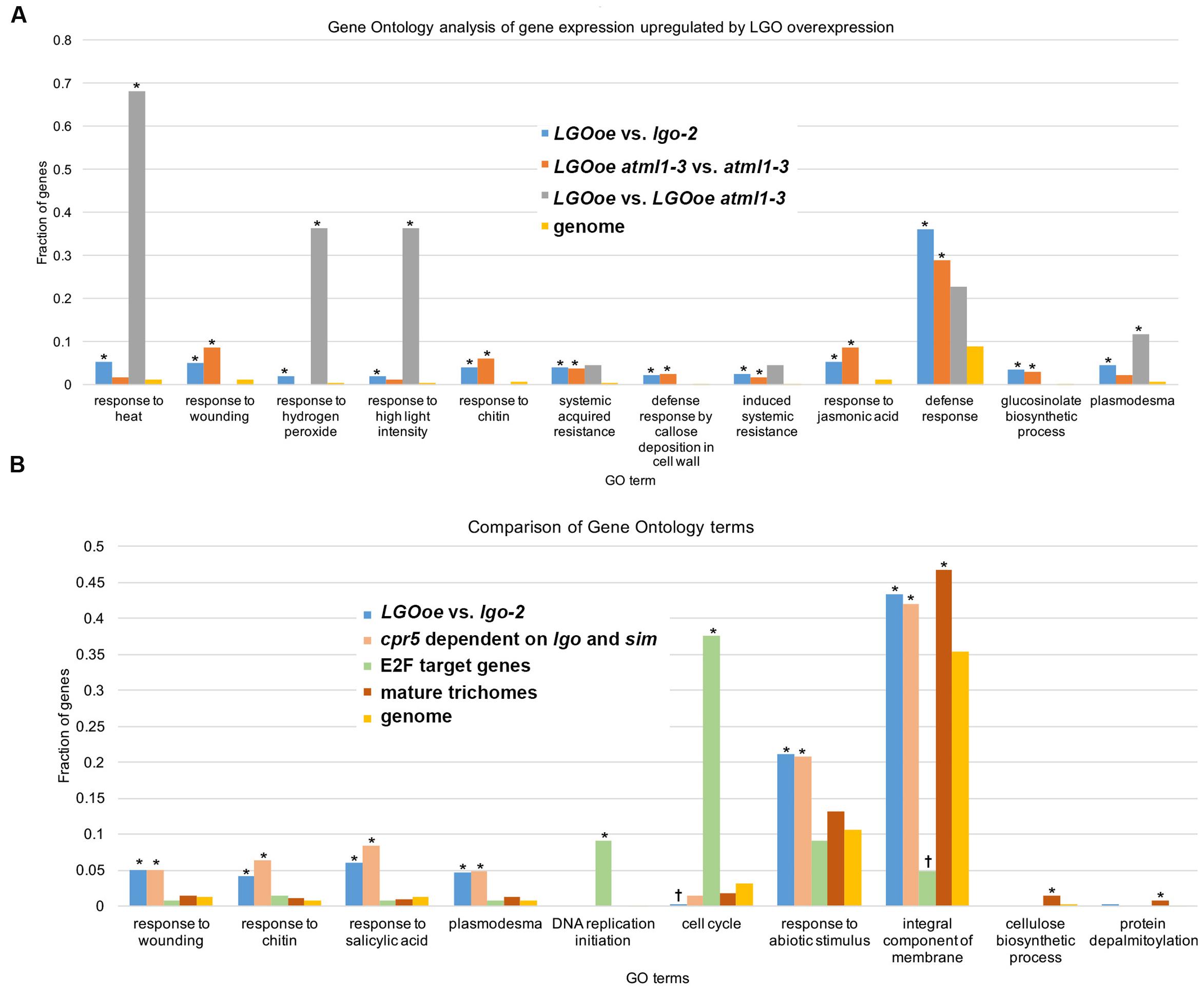
FIGURE 4. Defense response is upregulated by LGO overexpression in the mature sepal epidermis. (A) Bar graph showing the fraction of genes associated with the GO term in each genotype comparison: LGOoe versus lgo-2 (blue), LGOoe atml1-3 versus atml1-3 (orange), and LGOoe versus LGOoe atml1-3 (gray). “Genome” (yellow) reports the frequency of genes associated with that term in the Arabidopsis genome, which would be the frequency expected by chance for a randomly selected subset of genes. The fraction of genes was calculated as the number of genes associated with that term divided by the total number of genes in the set that can be associated with any GO term as determined by FUNC. Asterisks indicate significant enrichment (p-values ≤ 0.01). Note that many defense response-related GO terms were associated with LGOoe, and that only a few terms (such as response to heat [GO:0009408] and response to hydrogen peroxide [GO:0042542]) required ATML1 activity. (B) Bar graph showing the fraction of genes associated with the GO term in each set: LGOoe versus lgo-2 (blue), cpr5 versus cpr5 sim lgo (salmon; genes whose expression in cpr5 depends on sim and lgo; Wang et al., 2014), known E2F target genes (green; Vandepoele et al., 2005), mature trichomes (brick red; wild type trichome versus wild type shoot and gl3-sst sim mutant; Marks et al., 2009), and genome (yellow). The fraction of genes was calculated as the number of genes associated with that term divided by the total number of genes in the set that can be associated with any GO term as determined by FUNC. Asterisks indicate significant enrichment (p-values ≤ 0.01) and daggers (†) indicate significant depletion (p-values ≤ 0.01). Note that the frequencies of GO terms associated with LGOoe versus lgo-2 match well with those associated with cpr5 versus cpr5 sim lgo, but not with known E2F targets, or with mature trichomes.
This pathway leads to the expression of WRKY transcription factors, which generally modulate transcription of defense response genes, with both positive and negative effects on defense responses (Pandey and Somssich, 2009). WRKY25 and WRKY33 were both significantly upregulated in both LGOoe versus lgo-2 and LGOoe atml1 versus atml1, WRKY51 and WRKY53 were significantly upregulated in LGOoe versus lgo-2, and WRKY26 and WRKY48 were significantly upregulated in LOGoe atml1-3 versus atml1-3 (Table 6; Supplementary File S01). WRKY33 is required for normal resistance both to necrotrophic fungal pathogens and to abiotic stresses such as salinity and heat (Zheng et al., 2006; Jiang and Deyholos, 2008; Liu et al., 2015; Zhou et al., 2015). WRKY25 and WRKY26 also promote resistance to heat stress (Li et al., 2011). However, WRKY genes upregulated by LGOoe also have negative or complex effects on defense responses. wrky53 mutants show increased resistance to Ralstonia solanacearum, but decreased resistance to Pseudomonas syringae (Murray et al., 2007; Hu et al., 2008), and both WRKY25 and WRKY48 negatively regulate resistance to the bacterial pathogen Pseudomonas syringae (Zheng et al., 2007; Xing et al., 2008). WRKY51 functions in the salicylic acid pathway to inhibit the jasmonic acid pathway in response to low oleic acid, and thus to inhibit basal resistance to the necrotrophic fungus Botrytis cinerea (Gao et al., 2011).
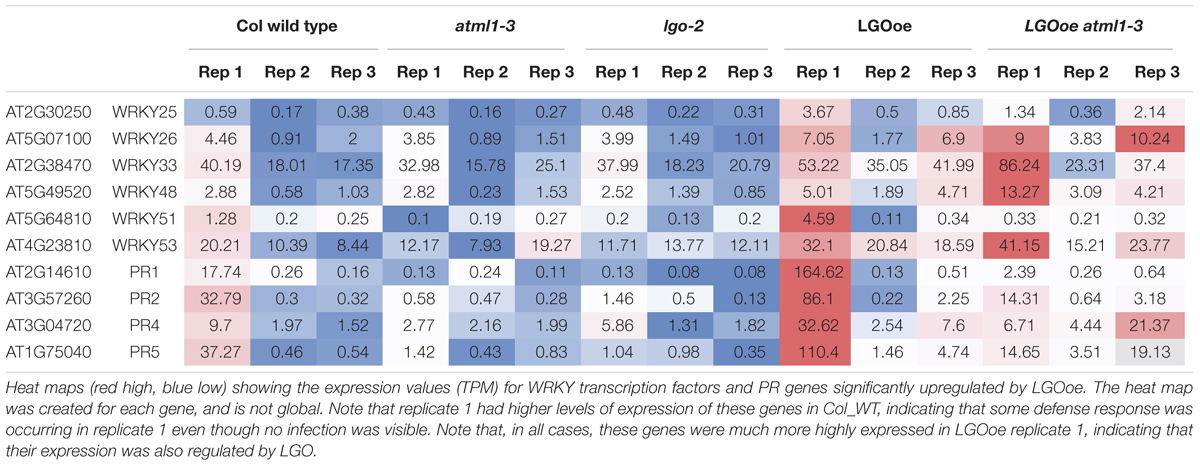
TABLE 6. Heat map of WRKY transcription factors and PR genes.
After the plant activates defense responses at the local site of infection, it also sends a signal to activate regulation of systemic acquired resistance [GO:0010112] (SAR), increasing defense responses throughout the plant to protect undamaged tissues from pathogen invasion (Pieterse et al., 2009) (Table 5; Supplementary Files S08 and S13). PR genes, which generally encode antimicrobial proteins, are associated with SAR, and PR1, PR2, PR4, and PR5 are all significantly upregulated in LGOoe versus lgo-2 (Table 6; Supplementary Files S01 and S08) (Van Loon et al., 2006). Interestingly, these PR genes were all significantly upregulated in Col_WT replicate 1 versus replicates 2 and 3, but their upregulation was significantly stronger in LGOoe replicate 1 plants than in wild type Col_WT replicate 1, indicating that they were regulated by both environment and genotype (Table 6; Supplementary File S01). No infection was observed in replicate 1 plants; however, given the transcriptomic results, replicate 1 may have had a minor infection that was not visible. In addition, biotic interactions with beneficial microorganisms cause induced systemic resistance [GO:0009682] to pathogens through jasmonic acid and ethylene signaling (Pieterse et al., 2009); this function is associated with 10 genes upregulated by LGOoe in the absence of ATML1 (LGOoe atml1-3 versus atml1-3; Figure 4A; Table 3; Supplementary File S06).
Glucosinolate Biosynthesis Is Upregulated by LGO Independently of ATML1
As part of the defense response, genes involved in glucosinolate biosynthetic process [GO:0019761] are significantly overrepresented among the genes upregulated by LGOoe in the absence of ATML1 (Figure 4A; Table 3). Glucosinolates are defensive secondary metabolites produced in Brassicaceae species, including Arabidopsis, that are responsible for their mustard flavor (Ratzka et al., 2002; Celenza et al., 2005). When the plant tissue is disrupted (e.g., because an insect eats the plant), the damage exposes glucosinolates to myrosinase enzymes that cleave them to make toxic and reactive compounds including nitriles, isothiocyanates, and thiocyanates (Wittstock and Halkier, 2002). In the intact plant, glucosinolates and myrosinases are compartmentalized so that they do not come into contact (Burow et al., 2007), with myrosinases accumulating in distinct cell types from glucosinolates (Koroleva et al., 2000; Thangstad et al., 2004; Li and Sack, 2014). There are three types of glucosinolates: aliphatic, aromatic and indolic (Wittstock and Halkier, 2002). Indole glucosinolate metabolic process [GO:0042343] was specifically overrepresented among genes that are more strongly expressed in LGOoe atml1 versus atml1-3 sepals (Table 3). Glucosinolates contain sulfur, and the sulfur metabolism GO terms thiosulfate sulfurtransferase activity [GO:0004792] and hydrogen sulfide biosynthetic process [GO:0070814] were both significantly overrepresented among genes upregulated in LGOoe atml1 versus atml1-3 (Table 3) (Kopriva et al., 2015). In addition to its role in defense against herbivores, the glucosinolate biosynthetic pathway is required for defense response by callose deposition in the cell wall [GO:0052544] at the site of pathogen infection (Clay et al., 2009), which was associated with genes upregulated in LGOoe atml1 versus atml1-3.
Regulation of glucosinolate biosynthetic process [GO:0010439] was significantly associated with genes upregulated in LGOoe atml1-3 versus atml1-3 (Table 3). The transcription factor gene MYB51 (also known as HIGH INDOLIC GLUCOSINOLATE1, or HIG1) regulates indolic glucosinolate biosynthesis (Gigolashvili et al., 2007) and was significantly upregulated in LGOoe versus Col_WT, lgo-2 or atml1-3 sepals (Supplementary File S01). Although both MYB34 and MYB122 regulate indole glucosonolate biosynthesis together with MYB51 (Celenza et al., 2005; Frerigmann and Gigolashvili, 2014; Frerigmann et al., 2016), these genes were not differentially expressed in our RNA-seq data. We observed 10 genes that were upregulated in LGOoe atml1-3 versus atml1-3 and that were associated with glucosinolate biosynthetic process [GO:0019761]. To determine whether MYB51 or its paralogs might regulate these 10 genes, we searched for short DNA motifs (6–12 nt long) that were significantly overrepresented in the 500-nt 5′-flanking promoter regions of these 10 genes (regions ranging from 1 to 500 nt 5′-ward of their start codons). We selected a 500-nt sequence range for motif prediction because a majority of conserved non-coding DNA elements (which are likely to include cis-regulatory sequences) have been shown to reside within 0.5 kb of the 5′ ends of Arabidopsis genes (Haudry et al., 2013). This search identified a motif (5′-TAGGTAGGTGA-3′) that most closely resembled one of two alternative binding sites for MYB111 (Franco-Zorrilla et al., 2014) (Figures 5A,B; Supplementary File S14). To determine the specificity of this motif, we then used it to search the 500-nt 5′-flanks for all 27,416 protein-coding genes in the Arabidopsis genome: this rediscovered 9 out of the 10 original genes in which we had found the motif, along with 317 other genes in Arabidopsis. Thus, the motif was found 75 times more frequently in the LGOoe-upregulated glucosinolate biosynthetic genes than in the Arabidopsis genome as a whole (p = 2.2 ⋅ 10-16). These data are consistent with the hypothesis that LGOoe upregulates MYB51, whose product in turn upregulates the glucosinolate biosynthesis transcriptional network in sepals.
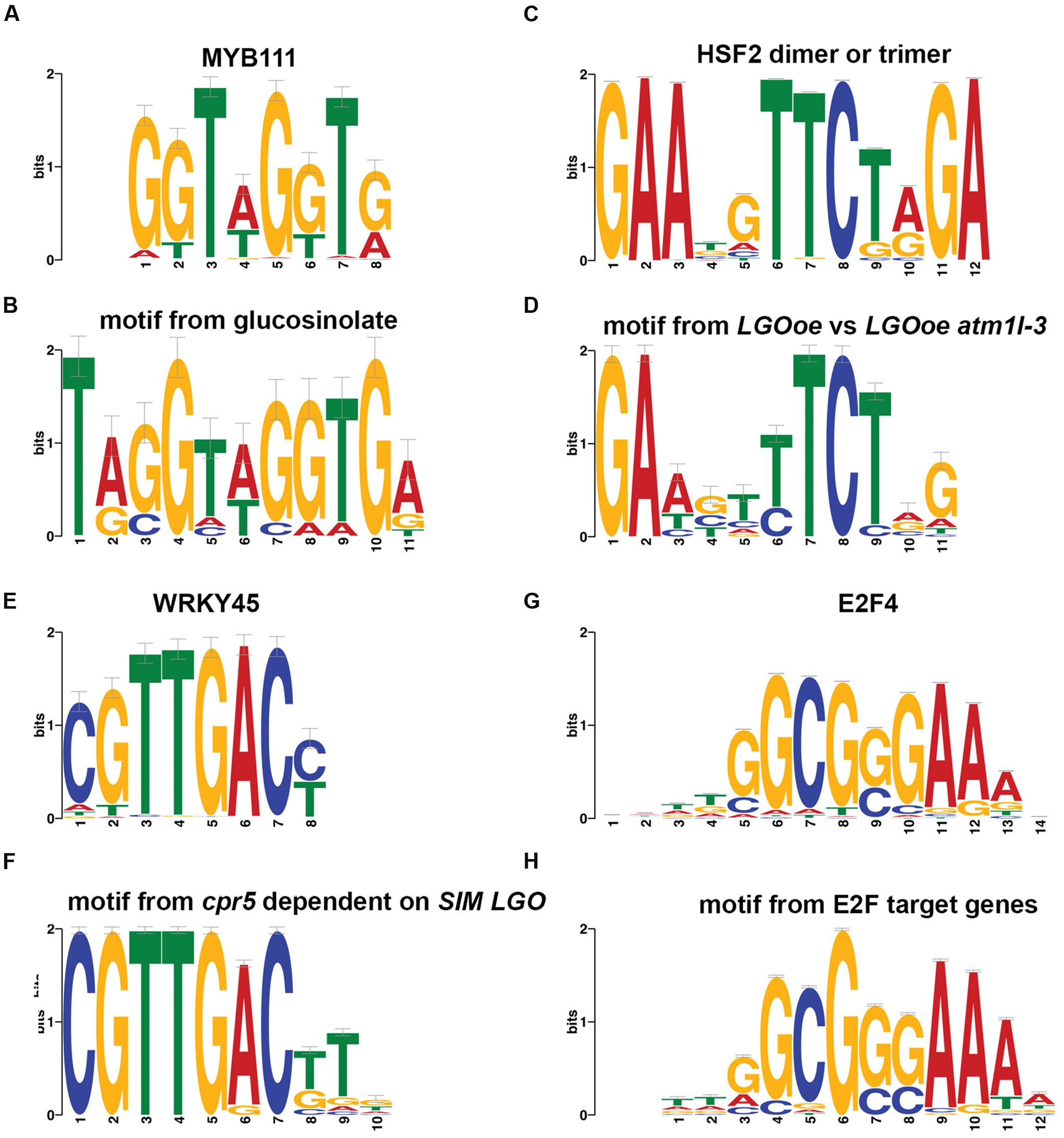
FIGURE 5. Promoter motifs. (A,B) A motif found in the 500-nt regions 5′-ward from the start codons of genes differentially expressed in our sepal dataset that were associated with the GO term glucosinolate biosynthetic process [GO:0019761] (B) matched a binding site previously observed in vitro for MYB111 (A) (Franco-Zorrilla et al., 2014). MYB51 was upregulated by LGOoe, and is likely to have a similar binding site. (C,D) A motif found in the 500-nt regions 5′-ward from the start codons of genes differentially expressed in LGOoe versus LGOoe atml1-3 (D) matched with a binding site previously observed in vitro for human HSF2 dimers or trimers (C) (Jolma et al., 2010). (E,F) A motif in the 500-nt regions 5′-ward from the start codons of genes differentially expressed in cpr5 versus cpr5 sim lgo (F) matched a binding site previously observed in vitro for WRKY45 (E) (Franco-Zorrilla et al., 2014). This motif was also significantly overrepresented among all 1,341 genes with significant differences in sepal expression between genotypes. (G,H) A motif in the 500-nt regions 5′-ward from the start codons of known E2F target genes (H) matched a binding site previously observed via ChIP-seq for human E2F4 (G) (Kulakovskiy et al., 2016). This motif was significantly underrepresented among all 1,341 differentially expressed sepal genes.
Response to Heat Is Upregulated in LGOoe Sepals in an ATML1-Dependent Manner
Although there were few genes upregulated by LGOoe in an ATML1-dependent manner, we still investigated their functions. To identify the functions of genes upregulated by LGOoe that required ATML1 for upregulation, we examined GO terms associated with genes more strongly expressed in LGOoe than in LGOoe atml1 sepals (Figure 4A; Table 4; Supplementary Files S07 and S12). Two of the defense responses upregulated by LGOoe also depended on ATML1. Response to hydrogen peroxide [GO:0042542] depended entirely on ATML1; no genes in that GO term were differentially expressed in LGOoe atml1-3 versus atml1-3 (Figure 4A; Table 4; Supplementary File S07). Response to heat [GO:0009408] and response to high light intensity [GO:0009644] were highly overrepresented among genes upregulated in LGOoe versus LGOoe atml1, relative to all genes in the genome (Figure 4A; Table 4; Supplementary File S07). Searching for short DNA motifs in the 500-nt 5′-flanks of genes more strongly expressed in LGOoe than LGOoe atml1, we identified one motif (5′-GAAGTTTCTGG-3’) that matched the binding site in vitro for human heat shock factor (HSF) dimers or trimers (Figures 5C,D; Supplementary File S14) (Jolma et al., 2010). The Arabidopsis HSF gene ATHSF4 (AT4G36990) is upregulated in LGOoe versus atml1-3 or lgo-2 (Supplementary File S01). Thus, ATML1 contributes to some defense responses and environmental responses driven by LGOoe, and part of this contribution may be indirectly affected via ATHSF4.
Overlap of Genes Upregulated by LGOoe With Genes Upregulated by the Constitutive Defense Response Mutant cpr5 in a SIM- and LGO-Dependent Manner
Consistent with our results that LGOoe promotes expression of defense response genes, it has recently been shown that LGO/SMR1 and SIM play a non-canonical role in effector-triggered immunity (Wang et al., 2014). Wang et al. showed that a constitutive defense response found in plants mutant for the gene constitutive expresser of pathogenesis-related gene5 (cpr5) was lost in cpr5 sim lgo triple mutants (Wang et al., 2014). They further found that most of the genes differentially expressed between cpr5 and wild type were dependent on SIM and LGO; in other words, these genes were no longer differentially expressed in cpr5 sim lgo compared to wild type. From their published data, we identified 698 genes differentially expressed in cpr5 versus cpr5 sim lgo. Comparing them with 392 genes more strongly expressed in LGOoe than in lgo-2 sepals, we found that 151 genes overlapped, a 15-fold higher overlap than would have been expected by chance (p < 2.2 ⋅ 10-16). To determine what biological functions might be mediated in common by the two gene sets, we identified GO terms disproportionately associated with the 698 genes differentially expressed in cpr5 versus cpr5 sim lgo (Supplementary File S15) and compared them with the GO terms associated with LGOoe-upregulated genes (Supplementary Files S06–S08); this revealed substantial overlap, including response to chitin [GO:0010200], response to wounding [GO:0009611], systemic acquired resistance [GO:0009627], defense response [GO:0006952], defense response to fungus [GO:0050832], plasmodesma [GO:0009506], and induced systemic resistance [GO:0009682] (Figure 4B; Supplementary File S15). To detect possible cis-regulatory elements that might mediate these common functions, we searched the 500-nt 5′-flanks of the 698 cpr5-upregulated genes for non-coding DNA short motifs, and found a motif (5′-CGTTGACTTG-3′) that matched the binding site for WRKY45 (Franco-Zorrilla et al., 2014) (Figures 5E,F). Using this motif to scan the 500-nt 5′-flanks of all 27,416 protein-coding genes in Arabidopsis, we found it to be present 1.9-fold more often among all 1,341 genes expressed in sepals with significantly genotype-altered expression than it was present genomewide (47 genes, sepalwide; 506 genes, genomewide; p = 6.2 ⋅ 10-5; Supplementary File S14). For the 392 genes more strongly expressed in LGOoe than in lgo-2 sepals, the overlap was 24 genes, 3.3-fold higher than the genomewide rate (p = 4.8 ⋅ 10-7). As noted above, WRKY transcription factors play important roles in defense response (Pandey and Somssich, 2009) and several WRKY transcription factors were significantly upregulated by LGO overexpression (Table 6; Supplementary File S01). Hamdoun et al. (2015) have recently shown that transcriptional activation of defense response primarily requires LGO, with SIM having only a minor role, due to differences in their expression patterns. The overlap of genes upregulated by LGOoe with SIM- and LGO-dependent cpr5-driven genes expressed provides independent evidence that genes driven by transgenic LGOoe in the sepal are indeed regulated by LGO in vivo.
LGO Does Not Regulate Transcription in the Mature Sepal Directly Through E2Fs
One mechanism through which LGO could regulate transcription is by modulating the activity of E2F cell-cycle transcription factors. Furthermore, Wang et al. demonstrated that e2f mutants suppress constitutive defense response in cpr5 mutants similarly to lgo sim, suggesting that LGO and SIM promote defense response through their regulation of the E2F pathway (Wang et al., 2014). Because Wang et al. (2014) found that several genes known to be regulated by E2F were upregulated in cpr5 mutants (Vandepoele et al., 2005), we tested whether there was overlap between the 1,341 genes expressed in sepals with significantly genotype-altered expression and the known set of 180 E2F target genes in Arabidopsis (Vandepoele et al., 2005). Unexpectedly, we found that the overlap between known E2F target genes and differentially expressed sepal genes was almost non-existent (only 4 genes, 2.2-fold lower than expected by chance; p = 0.125; AT1G05490, chromatin remodeling 31; AT3G57260, BETA-1,3-GLUCANASE 2; AT4G14365, XBAT34; and AT5G49520, ATWRKY48). We determined the GO terms associated with the known 180 E2F target genes, and found them to be generally associated with DNA replication initiation [GO:0006270] and cell cycle [GO:0007049] (Supplementary File S15); however, when we compared them with GO terms associated with LGOoe, we observed no overlap at all (Figure 4B; Tables 3–5; Supplementary Files S06–S08). Finally, we searched for short DNA motifs associated with the 500-nt 5′-flanks of the 180 known E2F target genes and recovered a motif (5′-TTGGCGGGAAAA-3′), that matched the binding site for human E2F4 (Figures 5G,H) (Kulakovskiy et al., 2016). Scanning all 27,416 protein-coding genes for occurrences of this motif in their 500-nt 5′-flanks yielded a set of 786 predicted E2F targets genomewide. The overlap of these predicted E2F target genes to the 1,341 differentially expressed sepal genes was 2.0-fold lower than expected by chance, more significantly than with known E2F target genes (19 genes; p = 7.4 ⋅ 10-4; AT1G03080, NET1D; AT1G07970; AT1G14420; AT1G15825; AT1G15830; AT1G54215; AT2G17840, EARLY-RESPONSIVE TO DEHYDRATION 7 [ERD7]; AT2G36570, PXC1; AT2G42890, MEI2-like 2; AT2G43865; AT3G19620; AT4G05553; AT4G13000; AT4G23230, CRK15; AT4G34440, AtPERK5; AT4G35610; AT5G25930; AT5G54960, pyruvate decarboxylase-2; AT5G64510, and tunicamycin induced 1 [TIN1]). These results indicate that genes driven by LGOoe in differentiated sepals are not regulated directly through E2F transcription factors and the standard cell cycle pathway.
KRP5 Target Genes Do Not Overlap With Genes Differentially Expressed in LGOoe Sepals
Jégu et al. (2013) identified 264 genes bound by KRP5 using ChIP-seq. To examine the specificity of our results we examined the overlap between these 264 genes and the 1,340 genes that we found to be significantly differentially expressed (up- or down- regulated) in response to overexpression of LGO in some genotype (LGOoe). We found that only six genes belonged to both sets; this represents a modestly significant decrease of KRP5 targets in our LGOoe-regulated gene set. Specifically, this is a 2.1-fold underrepresentation, compared to the overlap of 13 genes that would be expected by chance; it has a p-value of 0.05 by a two-sided exact binomial test with a 99% confidence interval. We conclude that there is no evidence that KRP5 preferentially binds our LGOoe-regulated genes; indeed, the evidence suggests that it may be less likely to do so than one would expect randomly.
Limited Similarity between LGOoe Sepals and Mature Trichomes
We looked for similarities between the genes upregulated in sepals by LGOoe, where the sepal is covered with giant cells (Figures 1H,I), and genes specifically expressed in mature trichomes, because sepal giant cells and trichomes are both highly endoreduplicated cell types. However, each of two published mature trichome gene sets were only modestly enriched for overlaps with the 1,341 genes differentially expressed between different sepal genotypes: out of 1,143 trichome-specific genes from Jakoby et al. (2008), only 82 overlapped with the sepal genes (1.5-fold more than expected by chance; p = 7.8 ⋅ 10-4); and out of 788 trichome-specific genes from Marks et al. (2009), only 84 overlapped with the sepal-expressed genes (2.2-fold more than expected by chance; p = 8.4 ⋅ 10-11). Moreover, when we analyzed GO terms associated with two different sets of genes found to be specifically expressed in mature trichomes (Supplementary File S15) (Jakoby et al., 2008; Marks et al., 2009), few overlapped with the GO terms associated with LGOoe-driven genes (Figure 4B; Supplementary Files S06–S08). However, metabolic pathway analysis has previously shown that glucosinolate biosynthesis is upregulated in mature trichomes (Jakoby et al., 2008) and we find glucosinolate biosynthesis upregulated in LGOoe sepals, indicating one important similarity.
Discussion
Motivated by evidence that cell cycle regulators can directly and indirectly control transcription, we used transcriptomics to determine to what extent the CKI LGO affects gene expression in mature Arabidopsis sepals. While the primary role of LGO is to promote endoreduplication at early stages of sepal development (Figure 1), we found that in mature sepals overexpression of LGO throughout the sepal epidermis mainly increased expression of genes associated with defense responses to fungi, insects, and bacteria. Notably, the indolic glucosinolate biosynthesis pathway, whose products defend against insect herbivores, was upregulated by overexpression of LGO. Other genes that were upregulated in the sepal by LGO overexpression, but that are currently uncharacterized, may encode new components of the defense response. The RNA-seq data generated here provide a high-quality, public transcriptomic resource for future study of the interaction between cell cycle genes and the defense response in Arabidopsis.
Our results support the emerging connection between the cell cycle and plant defense response (Bao and Hua, 2015). Many of the SIAMESE related CKIs are induced in response to pathogens or abiotic stress (Peres et al., 2007). For example, in rice, the EL2 gene is rapidly induced upon treatment with N-acetylchitoheptaose (a biotic elicitor for phytoalexin biosynthesis) or with bacterial flagellin (Minami et al., 1996; Che et al., 2000). Furthermore, Bao et al. (2013) recently found that an activation-tagging allele upregulating expression of OMISSION OF THE SECOND DIVISION (OSD1), which encodes a negative regulator of the anaphase-promoting complex/cyclosome (APC/C), showed both increased immunity to a bacterial pathogen and increased expression of resistance (R) genes. Our results similarly show that overexpression of the cell cycle regulator LGO induces expression of many classes of defense response genes. Wang et al. (2014) have previously shown that LGO and SIM participate in the defense response, that lgo sim double mutants are abnormally susceptible to infection with Pseudomonas syringae, and that many genes upregulated in the cpr5 constitutive defense response mutant were no longer upregulated in the cpr5 lgo sim triple mutant. In other words, the cpr5-activated genes characterized by Wang et al. (2014) depended on LGO and SIM for expression. Hamdoun et al. (2015) have subsequently demonstrated that LGO is more strongly required for the defense response than its paralog SIM. We have found that genes upregulated by the cpr5 mutation in a SIM- and LGO-dependent manner disproportionately overlap with genes that are upregulated when LGO is overexpressed in sepals, indicating that LGO is not only necessary but sufficient to drive the expression of defense response genes. Although our data is based on LGOoe, and thus may not reflect the endogenous functions of the LGO gene, the overlap with genes no longer upregulated in the cpr5 lgo sim triple mutant increases credence that LGO directly or indirectly regulates these genes. Conversely, we find that E2F transcription factors, which have been demonstrated to act downstream of LGO and SIM in cpr5 plants, are unlikely to be direct transcriptional activators of defense genes in LGOoe sepals. In our RNAseq data the expression of the defense response genes is highly variable, although significant differences in expression due to LGOoe were detected within the noise. Some of the variability may be due to unseen pathogen attack on the plants from which the RNAseq samples were isolated. It is also possible that expression of some pathogen response genes is further enhanced by LGOoe.
The mechanism through which overexpression of LGO regulates gene activity requires further investigation, but the transcriptomic results themselves provide some insight. In LGOoe plants, a LGO transgene is driven by the ATML1 promoter, which is expressed throughout the epidermis of sepals from early stages through maturity (Lu et al., 1996; Sessions et al., 1999; Roeder et al., 2010). Therefore, LGO overexpression likely drives downstream genes through more than one mechanism at more than one stage of sepal development. This is further supported by our finding that the transcription factors MYB51, ATHSF4, and WRKY33 are upregulated in LGOoe sepals, along with putative downstream targets encoding components of glucosinolate biosynthesis, heat shock response, and defense response, and that these downstream targets share distinct cis-regulatory motifs for binding transcription factors (for MYB, HSF, and WRKY) in their proximal promoter regions.
Based on similarity of the LGOoe sepal phenotype to ATML1::KRP1 sepals, in which live imaging has shown induction of endoreduplication in sepal primordia to form giant cells (Roeder et al., 2010), induction of endoreduplication and formation of giant cells occurs at early stages of sepal development (stages 6–10) before our assay of the transcriptome at stage 12, which explains why we do not observe significant differences in expression of any cyclins or CDKs. In this context, there are several ways one might explain LGO’s effects on gene activity. First, it might act through endoreduplication. In the budding yeast Saccharomyces cerevisiae, it has been shown that a change in ploidy alone is sufficient to alter gene expression (Galitski et al., 1999; Wu et al., 2010). However, if endoreduplication alone were responsible for the differential expression of genes in LGOoe sepals, we would expect substantial overlap with the genes expressed in mature trichomes, another highly endoreduplicated cell type. In fact, we observed modest overlap between LGOoe genes and mature trichome genes, suggesting that endoreduplication alone is not a sufficient mechanism to explain all of the gene expression in LGOoe sepals, although it may explain some transcriptomic effects. Endoreduplication in LGOoe sepals may regulate the expression of genes indirectly in that endoreduplication causes the formation of giant cells, which in turn means there are fewer cells available to enter the stomatal patterning pathway. The expression of stomatal patterning genes FAMA, ERECTA-LIKE2 (ERL2), and STOMATAL CARPENTER1 (SCAP1) is significantly downregulated in LGOoe atml1-3 sepals, which have very few stomata, relative to lgo-2 sepals, which have many stomata. However, ATML1 is also known to promote stomatal development (Peterson et al., 2013; Takada et al., 2013), so this may be due to a combination of factors. Second, because LGO regulates the cell cycle, and because E2F has been implicated in LGO-dependent transcriptional activation of defense-response genes in cpr5 plants, one might expect that LGO would regulate transcription in sepals via E2F transcription factors. However, the genes in our gene set have essentially no overlap with E2F target genes (Vandepoele et al., 2005). They also have a significantly lower frequency (compared to the genome as a whole) of genes with an E2F binding motif in their 500-nt 5′ flanks, suggesting that direct mediation of transcription by E2Fs is unlikely. In our analysis, RNA-seq was performed on RNA isolated from mature sepals after division has ceased; the lack of cell cycle genes in our LGOoe-driven gene sets is thus not unreasonable. It is likely that transcriptional effects of LGO regulating E2F were present at earlier stages of development. Although we do not find evidence of E2F targets in our dataset, it is possible that the changes in gene expression caused by LGOoe occur indirectly through the cell cycle via CDKs regulating other transcription factors and RBR. CDKA;1 has been shown to phosphorylate HEAT SHOCK FACTOR1 (HSF1) modulating the DNA binding activity in vitro (Reindl et al., 1997) and we found that genes involved in response to heat were upregulated by LGOoe in an ATML1-dependent manner. Third, previous results show that LGO can interact with the bZIP69 transcription factor and the chromatin remodeler BRAHMA (Van Leene et al., 2010), suggesting that LGO could play a more direct role in regulating transcription. In tomato, a set of protein–protein interactions was identified via yeast two-hybrid that included a SIAMESE-related CKI (SIP4), two 14-3-3 proteins, the SPAK kinase, the SPGB bZIP transcription factor, and the phosphatidylethanolamine binding protein SELF PRUNING (Pnueli et al., 2001). In plants, this family of phosphatidylethanolamine binding proteins includes many important developmental regulators including FLOWERING LOCUS T (FT) and TERMINAL FLOWER (TFL), which control flowering time and inflorescence meristem determinacy respectively (Alvarez et al., 1992; Corbesier et al., 2007). Notably, phosphatidylethanolamine binding [GO:0008429] is significantly overrepresented among genes upregulated by LGO overexpression that do not depend on ATML1 (Table 3); for instance, the genes FLOWERING LOCUS T (FT) and BROTHER OF FT AND TFL (BFT) are significantly more strongly expressed in LGOoe atml-3 than in atml1-3 sepals (Supplementary File S01). Further investigation of possible transcriptional complexes upon which LGO might act directly remains for future research.
LGO overexpression throughout the epidermis not only produces cells that cannot be phenotypically distinguished from giant cells (Figure 1), but also induces the expression of defense response genes. Transcriptional responses to the environment can be highly cell-type specific (Dinneny et al., 2008). As the epidermis is the first line of defense against pathogens, it would not be surprising to find that epidermal cell types have specific defensive roles mediated by upregulation of epidermis-specific batteries of genes. For example, the myrosinases that cleave glucosinolates into toxic and reactive compounds upon tissue disruption (i.e., herbivore predation) are expressed specifically in guard cells and myrosin idioblast cells (Thangstad et al., 2004; Li and Sack, 2014), which would biologically complement upregulation of glucosinolate biosynthesis in giant cells.
In conclusion, our findings support the hypothesis that LGO itself can directly or indirectly drive sepal epidermal cells to specific states of gene expression; in particular, they are consistent with recent findings showing LGO to be necessary for transcriptional activation of defense genes in Arabidopsis.
Materials and Methods
Plants
Plants were grown in pots of Lambert General Purpose Mix LM111 soil in Percival growth chambers in continuous light at 22°C to minimize circadian effects on the transcriptome. The Columbia-0 (Col-0) accession of Arabidopsis was used as wild type (Col_WT). The lgo-2 (SALK_033905) and atml1-3 (SALK_033408) alleles have been described previously (Roeder et al., 2010, 2012). Plants overexpressing LGO in the epidermis [LGOoe, i.e., plants carrying the transgene ATML1p::LGO, encoded by the plasmid pAR178 as described previously (Roeder et al., 2010)] were generated by Agrobacterium-mediated transformation of Col-0 wild type plants with pAR178 and selection of a line segregating 3:1 resistant:sensitive phenotypes to the herbicide Basta. The LGOoe atml1-3 plants were generated by crossing, genotyping for the atml1-3 mutant, and assessing homozygosity of LGOoe based on phenotypic segregation in subsequent generations.
Scanning Electron Microscopy (SEM)
Scanning electron microscopy was performed as described in Roeder et al. (2003). Stage 12 flowers were fixed in FAA (3.7% formaldehyde, 50% ethanol, 5% acetic acid) for 4 h with 15 min vacuum infiltration. Samples were dehydrated through an ethanol series (50, 60, 60, 60, 90, 95, 100%) and critical-point dried with a BAL-TEK CPD030. Sepals were dissected and mounted on stubs, sputter coated with platinum palladium and imaged with a Tescan Mira 3 FESEM scanning electron microscope in the Cornell Center for Materials Research.
RNA Isolation
For each genotype (wild type Col-0, lgo-2, atml1-3, LGOoe, and LGOoe atml1-3), three biological replicate samples were analyzed. The genotypes within one replicate were grown simultaneously together in a flat. Replicates 1, 2, and 3 were grown at different times in different flats. For each sample, 250 stage 12 sepals were dissected and immediately frozen in liquid nitrogen; sepals were staged according to Smyth et al. (1990). Sepals were dissected in the growth chamber where the plants were growing to minimize transcriptional changes during the dissection process. Sepals were dissected between 5:00 PM and 8:20 PM to minimize circadian effects on the transcriptome. RNA was purified using the QIAGEN RNeasy Plant Mini kit (Qiagen Cat# 74904) according to the manufacturer’s instructions. RNA was treated with DNAse I amplification grade (Invitrogen Cat# 18068015) according to manufacturer’s instructions before library preparation.
qPCR Measurement of LGO Expression
For each genotype (LGOeo, wild type Col, and lgo-2), three biological replicates were analyzed. For each sample, RNA was isolated from three pooled inflorescences including flowers up to stage 12 using the QIAGEN RNeasy Plant Mini kit (Qiagen Cat# 74904) according to the manufacturer’s instructions. One microgram of RNA for each sample was treated with DNAse I amplification grade (Invitrogen Cat# 18068015) according to manufacturer’s instructions. First strand cDNA was synthesized with oligo dT using Superscript II (Invitrogen Cat#18064014). Real-time PCR was performed using SYBR Green Master Mix (Roche Cat# 4707516001) on a Roche LightCycler 440 system with three technical replicates. LGO was quantified using primers oHM63 (5′-AGA ACA CAA GAT TCC CGC CG-3′) and oHH64 (5′-ACG GAG GAG AAG AAA CGG TC-3′). ROC1 (AT4G38740) was used as a reference gene to normalize gene expression with primers cyclo-F (5′-CGA TAA GAC TCC CAG GAC TGC CGA-3′) and cyclo-R (5′-TCG GCT TTC CAG ATG ATG ATC CAA CC-3′). qPCR results were analyzed by the ΔΔCt method. Statistics were calculated on ΔCt values using the t-test.
Generation of RNA-seq Data
RNA-seq library preparation was performed as in Kumar et al. (2012) with modifications for individual sample preparation. Briefly, mRNA was isolated with Dynabeads oligo dT beads (Invitrogen Cat# 61006) and fragmented for 5 min at 70°C with the PCR block lid at 105°C using Fragmentation buffer (Ambion Cat# AM8740). First strand cDNA was synthesized with random hexamers (Invitrogen Cat# 48190011) using Superscript II (Invitrogen Cat#18064014) and the second strand was synthesized with DNA Pol I (Fermentas Cat #EP0041) and RNAse H (Invitrogen Cat# 18021071). End repair was conducted with NEBNext End repair enzyme Mix (NEB Cat# E6050S) and Klenow DNA polymerase (NEB Cat# M0210S), A tailing with Klenow 3′ to 5′ exonuclease (Fermentas Cat# EP0421), and ligation of adapters containing 3 bp in read barcodes with Mighty Mix Ligase (Clonetech TAK6023). The library was size-selected using Agencourt AmPure Beads (Beckman Coulter A63880). PCR enrichment was conducted with primers PE 1.0 (5′-AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC* T-3′) and PE 2.0 (5′-CAA GCA GAA GAC GGC ATA CGA GAT CGG TCT CGG CAT TCC TGC TGA ACC GCT CTT CCG ATC* T-3′) for 15 cycles using Phusion polymerase (NEB). The entire library was run on a 1.2% agarose gel and size-selected (about 200–500 bp) to remove adapter dimers. Libraries for each replicate were pooled and sequenced on two lanes using rapid run settings on an Illumina HiSeq 2000 to generate single-end 51-nt reads at the Cornell University Biological Resource Center Genomics Facility.
Quality-Filtering and Barcoding RNA-seq Reads
We quality-filtered our raw 51-nt RNA-seq reads in two steps. First, we used a custom Perl script (quality_trim_fastq.pl, provided in Supplementary File S16) to remove reads that failed CHASTITY testing, to trim the 51st nucleotide off of our raw reads, and to censor any aberrant reads under 50 nt in length; the arguments used were “quality_trim_fastq.pl -i - [i.e., zcat input stream] -q 33 -u 50 -o [output FastQ file].” Second, we used Trimmomatic 0.33 (Bolger et al., 2014) to remove adapter sequences, to trim off unreliable residues, and to censor any reads that such trimming reduced to ≤49 nt; the arguments used were “java -jar trimmomatic SE -threads 7 -phred33 [input FastQ file] [output FastQ file] ILLUMINACLIP:[adapter sequences FastA file]:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50”. Adapter sequences provided to Trimmomatic are listed in Supplementary File S17. Third, having stringently quality-filtered our reads, we split them via barcodes into discrete RNA-seq data sets with fastx_barcode_splitter.pl from FastX-Toolkit 0.0.14123); the line command format and arguments used were “cat [input file] | fastx_barcode_splitter.pl –bcfile [barcode file] –bol –mismatches 0 –prefix [specific data output prefix] –suffix “.fq” ”. Barcodes provided to fastx_barcode_splitter.pl are listed in Supplementary File S18. Fourth, having sorted the reads by barcodes, we trimmed off their 5′-most four nucleotides in silico with fastx_trimmer from FastX-Toolkit 0.0.14; the arguments used were “-f 5 -i [input reads] -o [output reads].” We trimmed off the 5′-most four nucleotides in order to remove not only the 3-nt barcodes but also a fourth non-cDNA residue added during the construction of Illumina RNA-seq libraries. After this quality-filtering, we obtained between 7,615,242 and 33,257,356 reads per replicate (48,448,184 to 82,408,319 reads per genotype; Supplementary File S19). We used the final reads (quality-filtered, barcode-sorted, and barcode-trimmed) for all further RNA-seq analyses, and archived them in the NCBI Sequence Read Archive database (SRA4).
Analysis of RNA-seq Data
To enable mapping of RNA-seq reads to Arabidopsis genes, we constructed a transcript index with RSEM 1.2.21 (Li and Dewey, 2011). This index primarily contained 27,416 protein-coding sequences from the TAIR10 release of the Arabidopsis genome database (Berardini et al., 2015). As negative control sequences, it also included Arabidopsis ribosomal ncRNAs, a GFP transgenic coding sequence, and the paired-end adapter sequences used in our RNA-seq libraries. Sources for all of these sequences are given in Supplementary File S20. We computed RNA-seq expression values for Arabidopsis genes by mapping quality-filtered, barcode-split and -trimmed read sets to our transcript index with bowtie2 version 2.2.5 (Langmead and Salzberg, 2012), SAMTools 1.2 (Li et al., 2009), and rsem-calculate-expression in RSEM 1.2.21. For each RNA-seq data set, the numbers of reads mapped to a unique site in the transcriptome, mapped to two or more sites, or that were unmapped are listed in Supplementary File S19. The arguments used for RSEM included “rsem-calculate-expression –bowtie2 –calc-pme –phred33-quals –calc-ci –ci-credibility-level 0.99 –fragment-length-mean [mean insert size in nt] –fragment-length-sd [standard deviation of insert sizes in nt].” Mean insert sizes for each RNA-seq library were determined by reading the library’s peak insert size from its Bioanalyzer report, and subtracting 127 nt to account for linker sequences; the standard deviation for each library’s insert size was estimated visually from its Bioanalyzer report by taking the average of size changes that reduced the density of the library inserts by half. Mean sizes and standard deviations for RNA-seq libraries are given in Supplementary File S19. For each gene, from the RSEM results for each RNA-seq read set, we extracted the following data: posterior mean estimate of TPM, a normalized measurement of gene expression; the minimum TPM (minTPM) in the 99% confidence interval of expression values computed by RSEM; and the integer portion of the posterior mean estimate of reads mapped to that gene (rounding off decimal fractions in RSEM’s estimate). All of these gene expression data are provided in Supplementary File S01. All read sets mapped to Arabidopsis genes at high frequencies (83.6–87.2% per replicate), with 46.4–48.8% of reads per replicate mapping to unique sites in the Arabidopsis transcriptome (Supplementary File S19).
To determine whether a given Arabidopsis gene was actually expressed in a given RNA-seq data set (i.e., if its measured expression was above background noise), we compared the minTPM for that gene to the nominal expression value (in TPM, not minTPM) for the coding sequence of transgenic GFP. In reality, none of the sources of RNA-seq data in our work had any GFP transgene, let alone an active one. Therefore, the expression level computed by RSEM for GFP in a given RNA-seq data set was a reasonable empirical measurement of background noise. Thus, if the minTPM computed for an actual Arabidopsis gene in a given RNA-seq data set was greater than the TPM computed for GFP, we classified that gene as actually being expressed. We detected expression of 20,241 genes in wild-type sepals (73.8% of all genes), and expression of 1,358 more genes (5.0%) in sepals of other genotypes (Supplementary File S21), with expression levels per gene ranging from ∼92,000 to 0.07 TPM.
To determine which changes of gene expression between batches or genotypes were statistically significant, we analyzed integral readcounts per gene with DESeq2 version 1.10.1 (Love et al., 2014). We determined significant changes of gene expression between genotypes while controlling for the effects of batches, and also determined significant changes of gene expression between batches (replicates) while controlling for the effects of genotypes. Increases or decreases of gene expression were considered significant if they had a Benjamini–Hochberg-adjusted p-value of ≤0.1, i.e., if after correction for multiple hypothesis testing they had a collective false discovery rate (FDR) of ≤0.1 (Noble, 2009). The specific R commands used to generate DESeq2 values for these comparisons are given in Supplementary File S22. All instances of significantly changed gene expression are listed in Supplementary File S01. To check these results, we also analyzed readcounts per gene with the exactTest function of edgeR version 3.12.1 (Robinson et al., 2010). This gave sets of genes with significantly changed gene expression that were generally smaller than those sets predicted with DESeq2; at the same time, genes predicted by edgeR were almost always also predicted by DESeq2 (Supplementary File S03). The specific R commands used to generate edgeR values for these comparisons are given in Supplementary File S23. Because the edgeR predictions were essentially a subset of the DESeq2 predictions, we used the DESeq2 predictions for all subsequent analyses.
A heatmap of global gene activity (Figure 2A) was drawn with pheatmap 1.0.8 in R5 with the arguments “mat, scale = “row”, show_rownames = FALSE, cluster_cols = FALSE, clustering_method = “ward.D2”, width = 6, height = 6, clustering_distance_rows = “correlation”, color = colorRampPalette (c(“darkturquoise”, “black”, “yellow”))(150)”, using expression data in TPM for the set of 1,341 genes with significant differences in sepal expression between genotypes. The expression values of genes (in rows, with their conditions in column) were centered and scaled, so that relative rather than absolute expression levels would be displayed. These normalized expression values were grouped using an updated version of Ward’s minimum variance method, which is designed to find compact, spherical clusters6, with distances between the gene expression profiles being determined by their Pearson correlation. A principal component analysis (PCA) of global gene activity (Figure 2B) was generated with DESeq2, from the same readcount data used to produce statistical significances for changes in gene expression. Venn diagrams were drawn in Photoshop CC 2015 based on the data for significantly changed gene expression in Supplementary File S01. Heat maps for expression levels of individual genes (Tables 2 and 6) were created in Microsoft Excel using conditional formatting for the TPM values.
Gene Annotations
To provide informative descriptions for the genes listed in Supplementary File S01, we obtained annotations from the following sources. Gene aliases were downloaded and extracted from ftp://ftp.arabidopsis.org/home/tair/TAIR_Public_Releases/TAIR_Data_20140331/gene_aliases_20140331.txt. Gene descriptions (including short descriptions, summaries, and computational descriptions of gene function) were downloaded and extracted from ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR10_genome_release/gene_description_20131231.txt.gz. InterPro protein domains encoded by genes were downloaded and extracted from http://www.arabidopsis.org/download_files/Proteins/Domains/TAIR10_all.domains. Gene aliases, gene descriptions, and protein domains were from the TAIR10 release of the Arabidopsis genome database (Berardini et al., 2015). Lists of genes encoding transcription factors, and the specific class of transcription factor so encoded, were extracted from families_data.tbl (which itself was downloaded and extracted from http://arabidopsis.med.ohio-state.edu/Downloads/AtTFDB.zip) and downloaded and extracted from http://planttfdb.cbi.pku.edu.cn/download/gene_model_family/Ath, being merged into a single annotation set; these two sets of transcription factor annotation data were, respectively, from the databases AtTFDB (Yilmaz et al., 2010) and PlantTFDB (Jin et al., 2014).
Identifying Previously Published Sets of Genes
We extracted groups of genes from the following previously published analyses, because of their importance to understanding LGOoe-driven gene activity in sepals: 710 genes that are significantly more strongly expressed in cpr5 than in cpr5 sim lgo plants (i.e., genes upregulated by cpr5 in a SIM- and LGO-dependent fashion) (Wang et al., 2014), of which 698 could be identified with protein-coding genes in TAIR10; 181 genes that are likely targets of the transcription factor E2F in Arabidopsis (Vandepoele et al., 2005), of which 180 could be identified with protein-coding genes in TAIR10; 1,148 genes with trichome-specific expression, as observed by Jakoby et al. (2008), of which 1,143 could be identified with protein-coding genes in TAIR10; 802 genes with trichome-specific expression, as observed by Marks et al. (2009), of which 788 could be identified with protein-coding genes in TAIR10; and 264 protein-coding genes in TAIR10 that were observed, by Jégu et al. (2013) via ChIP-seq, to be bound by KRP5. Our lists of these gene sets are given in Supplementary File S24.
Genes that were significantly more strongly expressed in cpr5 than in cpr5 sim lgo were extracted from the microarray data of Wang et al. (2014)7 via the NCBI’s GEO2R portal8. We used GEO2R to compare GSM991297, GSM991298, and GS991299 (cpr5_rep1, cpr5_rep2, and cpr5_rep3) to GSM991303, GSM991304, and GSM991305 (cpr5 sim smr1_rep1, cpr5 sim smr1_rep2, and cpr5 sim smr1_rep3); we selected “Submitter supplied” platform annotations, but otherwise ran GEO2R with default settings. The resulting data were exported from NCBI as tab-delimited data tables, and filtered for genes that had any upregulation at all (i.e., any positive value for the logarithm of their expression ratios) with an adjusted p-value of ≤0.001.
Likely E2F target genes in Arabidopsis were extracted from Supplementary Table S3 of Vandepoele et al. (2005).
Trichome-specific genes were extracted from Supplementary Table S1A of Jakoby et al. (2008) by selecting all genes with a “FC mT/mLwoT” ratio of ≥1 that were also annotated as “trichome-specific” with an asterisk in the table. These genes were defined by Jakoby et al. (2008) as genes with both of the following two traits: they were up-regulated in mature trichomes relative to leaves without trichomes; and they were not expressed in leaves without trichomes.
Trichome-specific genes were extracted from Supplemental Table S1 of Marks et al. (2009) by the following method, derived from the text of Marks et al. (2009). The raw gene expression for wild-type mature trichomes, for mutant gl3-sst sim trichomes (with genetic blocks against the differentiation of mature trichomes), and for wild-type shoots were all compared against one another. Any gene that was expressed in wild-type mature trichomes, and that exhibited either ≥3-fold lower expression or no expression at all in either mutant gl3-sst sim trichomes or wild-type shoots, was then selected as trichome-specific.
Gene Ontology Term Analysis
We used func_hyper and func_hyper_refin in FUNC 0.4.7 (Prüfer et al., 2007) to identify non-redundant GO terms that were statistically overrepresented among groups of protein-coding genes. These groups primarily consisted of gene sets that we determined, in this work, to be significantly up- or down-regulated in responses to changes in genotype or batch (Supplementary File S01). However, we also analyzed GO terms associated with previously published groups of genes of relevance to this work (see above).
A key feature of FUNC is that it can use the GO term hierarchy to discount p-values for broad, high-level GO terms if these values arise solely from more specific, low-level GO terms annotating a subset of the genes annotated by a high-level term. The GO term hierarchy was downloaded from the GO consortium at http://archive.geneontology.org/full/2015-12-01/go_201512-termdb-tables.tar.gz. GO terms for protein-coding genes in TAIR10 were extracted from the GO annotation file http://geneontology.org/gene-associations (released on 2/1/2016), and reformatted into FUNC-usable input files with the custom Perl scripts build_func_boolean.pl and qual2func_table_arath.pl (Supplementary File S16). As noted above, each group of genes from our sepal expression data was selected by having an adjusted p-value of ≤0.1 and by sharing either increased or decreased expression in response to a given change of conditions. The GO terms associated with each group of genes were then tested against the GO terms associated with the total list of protein-coding genes in the TAIR10 release of the Arabidopsis genome database9. We first ran func_hyper with the arguments “-i [FUNC-formatted gene set file] -t go_201512-termdb-tables -o [output directory for FUNC analysis]”, which gave us an initial set of GO terms statistically overrepresented among the gene set by comparison to all genes in the genome, based on the hypergeometric statistical distribution for binary associated comparisons. We then refined these results, in order to select only the most specific GO terms that actually had statistical enrichment among differentially expressed genes while discarding more broad parent GO terms that derived all their significance from more specific child terms, by running func-refin (via the automatically generated script refin-YEAR-MM-DD.sh) with the arguments “0.01 [pvalue] 0.01 [pvalue-after-refinement] 1 [minimum number of genes in a subgroup for refinement].” All of the final, refined GO terms for gene groups from our sepal expression data are summarized in Supplementary File S05; final, refined GO terms for gene groups from previously published data are summarized in Supplementary File S15.
Gene ontology hierarchical graphs were made with AgriGO using Singular Enrichment Analysis (SEA) for Arabidopsis thaliana genemodel (TAIR910) with default parameters (Du et al., 2010).
Bar graphs for GO fractions of genes (Figures 4A,B) were generated using the func_hyper results prior to refinement, which provided gene counts. Specific terms were chosen to illustrate trends. The fraction of genes in the genome associated with a given GO term was calculated by dividing #genes_in_node by #genes_in_root_node. The fraction of genes from each gene set associated with a given GO term was calculated by dividing #genes_with_variable = 1_in_node by #genes_with_variable = 1_in_root_node. Bar graphs were created in Microsoft Excel. Terms that were significantly enriched (raw_p_overrepresentation_of_variable = 1 < 0.01) were marked with an asterisk, and terms that were significantly depleted (raw_p_underrepresentation_of_variable = 1 < 0.01), were marked with a dagger (†).
Non-coding DNA Motif Prediction and Analysis
We extracted non-coding DNA motifs from the 500-nt 5′-flanking sequences of Arabidopsis protein-coding genes (in TAIR10_upstream_500_translation_start_20101028.fa, obtained from ftp://ftp.arabidopsis.org/home/tair/Sequences/blast_datasets/TAIR10_blastsets/upstream_sequences/TAIR10_upstream_500_translation_start_20101028) with MEME 4.11.1 (Bailey et al., 2015), using the arguments “-evt 0.1 -dna -mod zoops -nmotifs 20 -minw 6 -maxw 12 -bfile TAIR10_500nt_trans_markov1-revcomp-maxsize 10000000”. Before running MEME, we generated the Markov-1 background file TAIR10_500nt_trans_markov1 with the argument “-m 1”. To produce this background, we extracted and used an all-contigs derivative of TAIR10_upstream_500_translation_start_20101028.fa in which non-ACGT residues were censored and their adjacent contigs were split into separate sequences; this was done in order to build the background model with a standard nucleotide alphabet (ACGT). MEME searches were performed on defined sets of genes, rather than all 27,416 protein-coding genes at once. Most of these sets were subsets of the 1,341 genes for which we observed statistically significant changes of gene activity between genotypes; the subsets were selected either by shared GO terms (e.g., response to heat [GO:0009408]) or by shared genotypic changes under which expression changed significantly (e.g., LGOoe atml1-3 versus atml1-3, upregulated). In addition, we searched for motifs in a number of previous published gene sets of possible relevance to sepal biology (e.g., E2F target genes and cpr5-upregulated, SIM/LGO-dependent genes).
For all motifs discovered by MEME, we used TOMTOM from MEME 4.11.1 (Tanaka et al., 2011) with the argument “-bfile TAIR10_500nt_trans_markov1” to check them for similarity to previously identified motifs in the following motif databases distributed with MEME: ARABD, CIS-BP, EUKARYOTE, FLY, HUMAN, JASPAR, MALARIA, MOUSE, TFBSshape, WORM, and YEAST. The motif databases (updated on Decermber 8, 2015) were obtained from http://meme-suite.org/meme-software/Databases/motifs/motif_databases.12.11.tgz. We used FIMO from MEME 4.11.1 (Grant et al., 2011) with the arguments “–bgfile TAIR10_500nt_trans_markov1 –max-stored-scores 10000000 –thresh 1e-4” to identify genomewide hits for all MEME motifs; the search was done against the all-contigs derivative of TAIR10_upstream_500_translation_start_20101028.fa, in order to run FIMO against DNA sequences with standard nucleotide alphabets (ACGT). We tested three p-value thresholds for motif hits (10-4, 10-5, and 10-6) with three sets of genes for which motif prediction gave well-defined positive results, and for which we could thus tune FIMO thresholds empirically when trying to redetect an original gene set in a genomewide motif search: glucosinolate genes (which identified a MYB binding site), gene upregulated in LGOoe versus LGOoe atml1-3 (which identified an HSF binding site), and known E2F target genes (which identified an E2F binding site). We found that, for all three of these controls, p ≤ 10-5 appeared to give the best balance between sensitivity and specificity in rediscovering known positive genes. We thus selected this as our FIMO threshold in all genomewide motif searches. The overall results (Supplementary File S14) were summarized with custom Perl scripts.
The statistical significance of overlaps between gene sets was evaluated by the two-sided exact binomial test at the 0.99 confidence level (i.e., with a 99% confidence interval), as implemented in the stats package of R (binom.test; R version 2.15.1; 2012-06-22).
Availability of Data and Material
Seeds are available from the Arabidopsis Biological Resource Center (ABRC) with the following accession numbers: atml1-3, CS68906; lgo-2, CS69160; ATML1p::LGO, CS69161; and ATML1p::LGO atml1-3, CS69162.
Sequence files for quality-filtered, barcode-sorted, and barcode-trimmed RNA-seq reads have been deposited in the NCBI Sequence Read Archive (SRA11) under the accession numbers SRR3179593 (Col_WT, replicate 1), SRR3179623 (Col_WT, replicate 2), SRR3179642 (Col_WT, replicate 3), SRR3179988 (atml1-3, replicate 1), SRR3180064 (atml1-3, replicate 2), SRR3180073 (atml1-3, replicate 3), SRR3180080 (lgo-2, replicate 1), SRR3180088 (lgo-2, replicate 2), SRR3180318 (lgo-2, replicate 3), SRR3180447 (LGOoe [ATML1p::LGO], replicate 1), SRR3180481 (LGOoe [ATML1p::LGO], replicate 2), SRR3180517 (LGOoe [ATML1p::LGO], replicate 3), SRR3180541 (LGOoe [ATML1p::LGO] atml1-3, replicate 1), SRR3180542 (LGOoe [ATML1p::LGO] atml1-3, replicate 2), and SRR3180543 (LGOoe [ATML1p::LGO] atml1-3, replicate 3).
Author Contributions
ES: bioinformatic analysis, writing the manuscript. AR: design of the experiment, isolation of RNA and creation of sequencing libraries, writing the manuscript.
Funding
This work was funded by the NSF Plant, Fungal, and Microbial Developmental Mechanisms grant IOS-1256733 and a 2015 President’s Council of Cornell Women Affinito-Stewart Grant. This work made use of the Cornell Center for Materials Research Shared Facilities which are supported through the NSF MRSEC program (DMR-1120296). The funding bodies had no role in the design of the study, interpretation of the data, or writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Lilan Hong, Jian Hua, Heather Meyer, Dana Robinson, Michael Scanlon, and Mingyuan Zhu for comments on the manuscript. We thank Endia Blunt, Heather Meyer, and Mingyuan Zhu for assistance with experiments. We thank Margaret Frank for instruction in RNA-seq library and heatmap preparation.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01744/full#supplementary-material
Abbreviations
ATML1, Arabidopsis thaliana MERISTEM LAYER1, AT4G21750; CDK, cyclin-dependent kinase; CKI, cyclin-dependent kinase inhibitor; CPR5, CONSTITUTUVE EXPRESSOR OF PATHOGENESIS RELATED GENES5 AT5G64930; GO, Gene ontology; HD-ZIP IV, homeodomain leucine zipper class IV; KRP, KIP-RELATED PROTEIN; LGO, LOSS OF GIANT CELLS FROM ORGANS, AT3G10525; LGOoe, overexpression of LGO throughout the epidermis (ATML1p::LGO); PR, pathogenesis related; SIM, SIAMESE (AT5G04470); SMR1, SIAMESE RELATED1, also known as LGO (AT3G10525); TPM, transcripts per million.
Footnotes
- ^http://hannonlab.cshl.edu/fastx_toolkit/index.html
- ^https://github.com/agordon/fastx_toolkit/releases/download/0.0.14/fastx_toolkit-0.0.14.tar.bz2
- ^https://github.com/agordon/libgtextutils/releases/download/0.7/libgtextutils-0.7.tar.gz
- ^http://www.ncbi.nlm.nih.gov/sra
- ^https://cran.r-project.org/web/packages/pheatmap/index.html
- ^https://stat.ethz.ch/R-manual/R-patched/library/stats/html/hclust.html
- ^http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc$=$GSE40322
- ^http://www.ncbi.nlm.nih.gov/geo/geo2r/?acc$=$GSE40322
- ^https://www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/TAIR10_cds_20101214_updated
- ^http://bioinfo.cau.edu.cn/agriGO
- ^http://www.ncbi.nlm.nih.gov/sra
References
Abe, M., Katsumata, H., Komeda, Y., and Takahashi, T. (2003). Regulation of shoot epidermal cell differentiation by a pair of homeodomain proteins in Arabidopsis. Development 130, 635–643. doi: 10.1242/dev.00292
Acosta, I. F., and Farmer, E. E. (2010). Jasmonates. Arabidopsis Book 8:e0129. doi: 10.1199/tab.0129
Alvarez, J., Guli, C. L., Yu, X. H., and Smyth, D. R. (1992). Terminal flower: a gene affecting inflorescence development in Arabidopsis thaliana. Plant J. 2, 103–116. doi: 10.1111/j.1365-313X.1992.00103.x
Bailey, T. L., Johnson, J., Grant, C. E., and Noble, W. S. (2015). The MEME Suite. Nucleic Acids Res. 43, W39–W49. doi: 10.1093/nar/gkv416
Bao, Z., and Hua, J. (2015). Linking the cell cycle with innate immunity in Arabidopsis. Mol. Plant 8, 980–982. doi: 10.1016/j.molp.2015.03.013
Bao, Z., Yang, H., and Hua, J. (2013). Perturbation of cell cycle regulation triggers plant immune response via activation of disease resistance genes. Proc. Natl. Acad. Sci. U.S.A. 110, 2407–2412. doi: 10.1073/pnas.1217024110
Berardini, T. Z., Reiser, L., Li, D., Mezheritsky, Y., Muller, R., Strait, E., et al. (2015). The Arabidopsis information resource: making and mining the “gold standard” annotated reference plant genome. Genesis 53, 474–485. doi: 10.1002/dvg.22877
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bramsiepe, J., Wester, K., Weinl, C., Roodbarkelari, F., Kasili, R., Larkin, J. C., et al. (2010). Endoreplication controls cell fate maintenance. PLoS Genet. 6:e1000996. doi: 10.1371/journal.pgen.1000996
Breuer, C., Ishida, T., and Sugimoto, K. (2010). Developmental control of endocycles and cell growth in plants. Curr. Opin. Plant Biol. 13, 654–660. doi: 10.1016/j.pbi.2010.10.006
Burow, M., Rice, M., Hause, B., Gershenzon, J., and Wittstock, U. (2007). Cell- and tissue-specific localization and regulation of the epithiospecifier protein in Arabidopsis thaliana. Plant Mol. Biol. 64, 173–185. doi: 10.1007/s11103-007-9143-1
Celenza, J. L., Quiel, J. A., Smolen, G. A., Merrikh, H., Silvestro, A. R., Normanly, J., et al. (2005). The Arabidopsis ATR1 Myb transcription factor controls indolic glucosinolate homeostasis. Plant Physiol. 137, 253–262. doi: 10.1104/pp.104.054395
Che, F. S., Nakajima, Y., Tanaka, N., Iwano, M., Yoshida, T., Takayama, S., et al. (2000). Flagellin from an incompatible strain of Pseudomonas avenae induces a resistance response in cultured rice cells. J. Biol. Chem. 275, 32347–32356. doi: 10.1074/jbc.M004796200
Churchman, M. L., Brown, M. L., Kato, N., Kirik, V., Hülskamp, M., Inzé, D., et al. (2006). SIAMESE, a plant-specific cell cycle regulator, controls endoreplication onset in Arabidopsis thaliana. Plant Cell 18, 3145–3157. doi: 10.1105/tpc.106.044834
Clay, N. K., Adio, A. M., Denoux, C., Jander, G., and Ausubel, F. M. (2009). Glucosinolate metabolites required for an Arabidopsis innate immune response. Science 323, 95–101. doi: 10.1126/science.1164627
Corbesier, L., Vincent, C., Jang, S., Fornara, F., Fan, Q., Searle, I., et al. (2007). FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 316, 1030–1033. doi: 10.1126/science.1140035
Cruz-Ramírez, A., Díaz-Triviño, S., Blilou, I., Grieneisen, V. A., Sozzani, R., Zamioudis, C., et al. (2012). A bistable circuit involving SCARECROW- RETINOBLASTOMA integrates cues to inform asymmetric stem cell division. Cell 150, 1002–1015. doi: 10.1016/j.cell.2012.07.017
Desvoyes, B., de Mendoza, A., Ruiz-Trillo, I., and Gutierrez, C. (2014). Novel roles of plant RETINOBLASTOMA-RELATED (RBR) protein in cell proliferation and asymmetric cell division. J. Exp. Bot. 65, 2657–2666. doi: 10.1093/jxb/ert411
Desvoyes, B., Ramirez-Parra, E., Xie, Q., Chua, N. H., and Gutierrez, C. (2006). Cell type-specific role of the retinoblastoma/E2F pathway during Arabidopsis leaf development. Plant Physiol. 140, 67–80. doi: 10.1104/pp.105.071027
Dinneny, J. R., Long, T. A., Wang, J. Y., Jung, J. W., Mace, D., Pointer, S., et al. (2008). Cell identity mediates the response of Arabidopsis roots to abiotic stress. Science 320, 942–945. doi: 10.1126/science.1153795
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70. doi: 10.1093/nar/gkq310
Franco-Zorrilla, J. M., López-Vidriero, I., Carrasco, J. L., Godoy, M., Vera, P., and Solano, R. (2014). DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc. Natl. Acad. Sci. U.S.A. 111, 2367–2372. doi: 10.1073/pnas.1316278111
Frerigmann, H., and Gigolashvili, T. (2014). MYB34, MYB51, and MYB122 distinctly regulate indolic glucosinolate biosynthesis in Arabidopsis thaliana. Mol. Plant 7, 814–828. doi: 10.1093/mp/ssu004
Frerigmann, H., Piślewska-Bednarek, M., Sánchez-Vallet, A., Molina, A., Glawischnig, E., Gigolashvili, T., et al. (2016). Regulation of pathogen triggered tryptophan metabolism in Arabidopsis thaliana by MYB transcription factors and indole glucosinolate conversion products. Mol. Plant 9, 682–695. doi: 10.1016/j.molp.2016.01.006
Galitski, T., Saldanha, A. J., Styles, C. A., Lander, E. S., and Fink, G. R. (1999). Ploidy regulation of gene expression. Science 285, 251–254. doi: 10.1126/science.285.5425.251
Gao, Q.-M., Venugopal, S., Navarre, D., and Kachroo, A. (2011). Low oleic acid-derived repression of jasmonic acid-inducible defense responses requires the WRKY50 and WRKY51 proteins. Plant Physiol. 155, 464–476. doi: 10.1104/pp.110.166876
Gigolashvili, T., Berger, B., Mock, H.-P., Müller, C., Weisshaar, B., and Flügge, U.-I. (2007). The transcription factor HIG1/MYB51 regulates indolic glucosinolate biosynthesis in Arabidopsis thaliana. Plant J. 50, 886–901. doi: 10.1111/j.1365-313X.2007.03099.x
Grant, C. E., Bailey, T. L., and Noble, W. S. (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. doi: 10.1093/bioinformatics/btr064
Hamdoun, S., Zhang, C., Gill, M., Kumar, N., Churchman, M., Larkin, J. C., et al. (2015). Differential roles of two homologous cyclin-dependent kinase inhibitor genes in regulating cell cycle and innate immunity in Arabidopsis. Plant Physiol. 170, 515–527. doi: 10.1104/pp.15.01466
Hardwick, L., Ali, F. R., Azzarelli, R., and Philpott, A. (2015). Cell cycle regulation of proliferation versus differentiation in the central nervous system. Cell Tissue Res. 359, 187–200. doi: 10.1007/s00441-014-1895-8
Haudry, A., Platts, A. E., Vello, E., Hoen, D. R., Leclercq, M., Williamson, R. J., et al. (2013). An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat. Genet. 45, 891–898. doi: 10.1038/ng.2684
Hervieux, N., Dumond, M., Sapala, A., Routier-Kierzkowska, A.-L., Kierzkowski, D., Roeder, A. H. K., et al. (2016). A mechanical feedback restricts sepal growth and shape in Arabidopsis. Curr. Biol. 26, 1019–1028. doi: 10.1016/j.cub.2016.03.004
Hong, L., Dumond, M., Tsugawa, S., Sapala, A., Routier-Kierzkowska, A.-L., Zhou, Y., et al. (2016). Variable cell growth yields reproducible organ development through spatiotemporal averaging. Dev. Cell 38, 15–32. doi: 10.1016/j.devcel.2016.06.016
Hu, J., Barlet, X., Deslandes, L., Hirsch, J., Feng, D. X., Somssich, I., et al. (2008). Transcriptional responses of Arabidopsis thaliana during wilt disease caused by the soil-borne phytopathogenic bacterium, Ralstonia solanacearum. PLoS ONE 3:e2589. doi: 10.1371/journal.pone.0002589
Jakoby, M. J., Falkenhan, D., Mader, M. T., Brininstool, G., Wischnitzki, E., Platz, N., et al. (2008). Transcriptional profiling of mature Arabidopsis trichomes reveals that NOECK encodes the MIXTA-like transcriptional regulator MYB106. Plant Physiol. 148, 1583–1602. doi: 10.1104/pp.108.126979
Jegu, T., Latrasse, D., Delarue, M., Mazubert, C., Bourge, M., Hudik, E., et al. (2013). Multiple functions of Kip-Related Protein5 connect endoreduplication and cell elongation. Plant Physiol. 161, 1694–1705. doi: 10.1104/pp.112.212357
Jiang, Y., and Deyholos, M. K. (2008). Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 69, 91–105. doi: 10.1007/s11103-008-9408-3
Jin, J., Zhang, H., Kong, L., Gao, G., and Luo, J. (2014). PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187. doi: 10.1093/nar/gkt1016
Jolma, A., Kivioja, T., Toivonen, J., Cheng, L., Wei, G., Enge, M., et al. (2010). Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 20, 861–873. doi: 10.1101/gr.100552.109
Kopriva, S., Calderwood, A., Weckopp, S. C., and Koprivova, A. (2015). Plant sulfur and big data. Plant Sci. 241, 1–10. doi: 10.1016/j.plantsci.2015.09.014
Koroleva, O. A., Davies, A., Deeken, R., Thorpe, M. R., Tomos, A. D., and Hedrich, R. (2000). Identification of a new glucosinolate-rich cell type in Arabidopsis flower stalk. Plant Physiol. 124, 599–608. doi: 10.1104/pp.124.2.599
Kulakovskiy, I. V., Vorontsov, I. E., Yevshin, I. S., Soboleva, A. V., Kasianov, A. S., Ashoor, H., et al. (2016). HOCOMOCO: expansion and enhancement of the collection of transcription factor binding sites models. Nucleic Acids Res. 44, D116–D125. doi: 10.1093/nar/gkv1249
Kumar, N., Harashima, H., Kalve, S., Bramsiepe, J., Wang, K., Sizani, B. L., et al. (2015). Functional conservation in the SIAMESE-RELATED family of cyclin-dependent kinase inhibitors in land plants. Plant Cell 27, 3065–3080. doi: 10.1105/tpc.15.00489
Kumar, R., Ichihashi, Y., Kimura, S., Chitwood, D. H., Headland, L. R., Peng, J., et al. (2012). A high-throughput method for Illumina RNA-Seq library preparation. Front. Plant Sci. 3:202. doi: 10.3389/fpls.2012.00202
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lawrence, M. S., Stojanov, P., Mermel, C. H., and Robinson, J. T. (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. doi: 10.1038/nature12912
Lee, H. O., Davidson, J. M., and Duronio, R. J. (2009). Endoreplication: polyploidy with purpose. Genes Dev. 23, 2461–2477. doi: 10.1101/gad.1829209
Leon, J., Rojo, E., and Sanchez-Serrano, J. J. (2001). Wound signalling in plants. J. Exp. Bot. 52, 1–9. doi: 10.1093/jexbot/52.354.1
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, H., Collado, M., Villasante, A., Matheu, A., Lynch, C. J., Cañamero, M., et al. (2012). p27Kip1 directly represses Sox2 during embryonic stem cell differentiation. Cell Stem Cell 11, 845–852. doi: 10.1016/j.stem.2012.09.014
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, M., and Sack, F. D. (2014). Myrosin idioblast cell fate and development are regulated by the Arabidopsis transcription factor FAMA, the auxin pathway, and vesicular trafficking. Plant Cell 26, 4053–4066. doi: 10.1105/tpc.114.129726
Li, S., Fu, Q., Chen, L., Huang, W., and Yu, D. (2011). Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta 233, 1237–1252. doi: 10.1007/s00425-011-1375-2
Lim, S., and Kaldis, P. (2013). Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140, 3079–3093. doi: 10.1242/dev.091744
Liu, S., Kracher, B., Ziegler, J., Birkenbihl, R. P., and Somssich, I. E. (2015). Negative regulation of ABA signaling by WRKY33 is critical for Arabidopsis immunity towards Botrytis cinerea 2100. Elife 4:e07295. doi: 10.7554/eLife.07295.001
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. doi: 10.1186/s13059-014-0550-8
Lu, P., Porat, R., Nadeau, J. A., and O’Neill, S. D. (1996). Identification of a meristem L1 layer-specific gene in Arabidopsis that is expressed during embryonic pattern formation and defines a new class of homeobox genes. Plant Cell 8, 2155–2168. doi: 10.1105/tpc.8.12.2155
Luna, E., Pastor, V., Robert, J., Flors, V., Mauch-Mani, B., and Ton, J. (2011). Callose deposition: a multifaceted plant defense response. Mol. Plant Microbe Interact. 24, 183–193. doi: 10.1094/MPMI-07-10-0149
Marks, M. D., Wenger, J. P., Gilding, E., Jilk, R., and Dixon, R. A. (2009). Transcriptome analysis of Arabidopsis wild-type and gl3-sst sim trichomes identifies four additional genes required for trichome development. Mol. Plant 2, 803–822. doi: 10.1093/mp/ssp037
Matos, J. L., Lau, O. S., Hachez, C., Cruz-Ramirez, A., Scheres, B., and Bergmann, D. C. (2014). Irreversible fate commitment in the Arabidopsis stomatal lineage requires a FAMA and RETINOBLASTOMA-RELATED module. Elife 3:e03271. doi: 10.7554/eLife.03271
Melaragno, J. E., Mehrotra, B., and Coleman, A. W. (1993). Relationship between endopolyploidy and cell size in epidermal tissue of Arabidopsis. Plant Cell 5, 1661–1668. doi: 10.1105/tpc.5.11.1661
Minami, E., Kuchitsu, K., He, D.-Y., Kouchi, H., Midoh, N., Ohtsuki, Y., et al. (1996). Two novel genes rapidly and transiently activated in suspension-cultured rice cells by treatment with N-Acetylchitoheptaose, a biotic elicitor for phytoalexin production. Plant Cell Physiol. 37, 563–567. doi: 10.1093/oxfordjournals.pcp.a028981
Murray, S. L., Ingle, R. A., Petersen, L. N., and Denby, K. J. (2007). Basal resistance against Pseudomonas syringae in Arabidopsis involves WRKY53 and a protein with homology to a nematode resistance protein. Mol. Plant Microbe Interact. 20, 1431–1438. doi: 10.1094/MPMI-20-11-1431
Nakamura, M., Katsumata, H., Abe, M., Yabe, N., Komeda, Y., Yamamoto, K. T., et al. (2006). Characterization of the class IV homeodomain-Leucine Zipper gene family in Arabidopsis. Plant Physiol. 141, 1363–1375. doi: 10.1104/pp.106.077388
Noble, W. S. (2009). How does multiple testing correction work? Nat. Biotechnol. 27, 1135–1137. doi: 10.1038/nbt1209-1135
Pandey, S. P., and Somssich, I. E. (2009). The role of WRKY transcription factors in plant immunity. Plant Physiol. 150, 1648–1655. doi: 10.1104/pp.109.138990
Peres, A., Churchman, M. L., Hariharan, S., Himanen, K., Verkest, A., Vandepoele, K., et al. (2007). Novel plant-specific cyclin-dependent kinase inhibitors induced by biotic and abiotic stresses. J. Biol. Chem. 282, 25588–25596. doi: 10.1074/jbc.M703326200
Peterson, K. M., Shyu, C., Burr, C. A., Horst, R. J., Kanaoka, M. M., Omae, M., et al. (2013). Arabidopsis homeodomain-leucine zipper IV proteins promote stomatal development and ectopically induce stomata beyond the epidermis. Development 140, 1924–1935. doi: 10.1242/dev.090209
Pieterse, C. M. J., Leon-Reyes, A., Van der Ent, S., and Van Wees, S. C. M. (2009). Networking by small-molecule hormones in plant immunity. Nat. Chem. Biol. 5, 308–316. doi: 10.1038/nchembio.164
Pippa, R., Espinosa, L., Gundem, G., and Garcia-Escudero, R. (2012). p27Kip1 represses transcription by direct interaction with p130/E2F4 at the promoters of target genes. Oncogene 31, 4207–4220. doi: 10.1038/onc.2011.582
Pnueli, L., Gutfinger, T., Hareven, D., Ben-Naim, O., Ron, N., Adir, N., et al. (2001). Tomato SP-interacting proteins define a conserved signaling system that regulates shoot architecture and flowering. Plant Cell 13, 2687–2702. doi: 10.1105/tpc.010293
Prüfer, K., Muetzel, B., Do, H.-H., Weiss, G., Khaitovich, P., Rahm, E., et al. (2007). FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics 8:41. doi: 10.1186/1471-2105-8-41
Ratzka, A., Vogel, H., Kliebenstein, D. J., Mitchell-Olds, T., and Kroymann, J. (2002). Disarming the mustard oil bomb. Proc. Natl. Acad. Sci. U.S.A. 99, 11223–11228. doi: 10.1073/pnas.172112899
Reindl, A., Schoffl, F., Schell, J., Koncz, C., and Bako, L. (1997). Phosphorylation by a cyclin-dependent kinase modulates DNA binding of the Arabidopsis heat-shock transcription factor HSF1 in vitro. Plant Physiol. 115, 93–100. doi: 10.1104/pp.115.1.93
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Roeder, A. H. K., Chickarmane, V., Cunha, A., Obara, B., Manjunath, B. S., and Meyerowitz, E. M. (2010). Variability in the control of cell division underlies sepal epidermal patterning in Arabidopsis thaliana. PLoS Biol. 8:e1000367. doi: 10.1371/journal.pbio.1000367
Roeder, A. H. K., Cunha, A., Ohno, C. K., and Meyerowitz, E. M. (2012). Cell cycle regulates cell type in the Arabidopsis sepal. Development 139, 4416–4427. doi: 10.1242/dev.082925
Roeder, A. H. K., Ferrándiz, C., and Yanofsky, M. F. (2003). The role of the REPLUMLESS homeodomain protein in patterning the Arabidopsis fruit. Curr. Biol. 13, 1630–1635. doi: 10.1016/j.cub.2003.08.027
Sessions, A. A., Weigel, D. D., and Yanofsky, M. F. M. (1999). The Arabidopsis thaliana MERISTEM LAYER 1 promoter specifies epidermal expression in meristems and young primordia. Plant J. 20, 259–263. doi: 10.1046/j.1365-313x.1999.00594.x
Shibuya, N., and Minami, E. (2001). Oligosaccharide signalling for defence responses in plant. Physiol. Mol. Plant Pathol. 59, 223–233. doi: 10.1006/pmpp.2001.0364
Smyth, D. R., Bowman, J. L., and Meyerowitz, E. M. (1990). Early flower development in Arabidopsis. Plant Cell 2, 755–767. doi: 10.1105/tpc.2.8.755
Suzuki, N., Miller, G., Morales, J., Shulaev, V., Torres, M. A., and Mittler, R. (2011). Respiratory burst oxidases: the engines of ROS signaling. Curr. Opin. Plant Biol. 14, 691–699. doi: 10.1016/j.pbi.2011.07.014
Takada, S., Takada, N., and Yoshida, A. (2013). ATML1 promotes epidermal cell differentiation in Arabidopsis shoots. Development 140, 1919–1923. doi: 10.1242/dev.094417
Tanaka, E., Bailey, T., Grant, C. E., Noble, W. S., and Keich, U. (2011). Improved similarity scores for comparing motifs. Bioinformatics 27, 1603–1609. doi: 10.1093/bioinformatics/btr257
Thangstad, O. P., Gilde, B., Chadchawan, S., Seem, M., Husebye, H., Bradley, D., et al. (2004). Cell specific, cross-species expression of myrosinases in Brassica napus, Arabidopsis thaliana and Nicotiana tabacum. Plant Mol. Biol. 54, 597–611. doi: 10.1023/B:PLAN.0000038272.99590.10
Torres, M. A., Jones, J. D. G., and Dangl, J. L. (2006). Reactive oxygen species signaling in response to pathogens. Plant Physiol. 141, 373–378. doi: 10.1104/pp.106.079467
Ullah, Z., Lee, C. Y., Lilly, M. A., and DePamphilis, M. L. (2009). Developmentally programmed endoreduplication in animals. Cell Cycle 8, 1501–1509. doi: 10.4161/cc.8.10.8325
Van Leene, J., Hollunder, J., Eeckhout, D., Persiau, G., Van De Slijke, E., Stals, H., et al. (2010). Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Mol. Syst. Biol. 6, 1–12. doi: 10.1038/msb.2010.53
Van Loon, L. C., Rep, M., and Pieterse, C. (2006). Significance of inducible defense-related proteins in infected plants. Annu. Rev. Phytopathol. 44, 135–162. doi: 10.1146/annurev.phyto.44.070505.143425
Vandepoele, K., Vlieghe, K., Florquin, K., Hennig, L., Beemster, G., Gruissem, W., et al. (2005). Genome-wide identification of potential plant E2F target genes. Plant Physiol. 139, 316–328. doi: 10.1104/pp.105.066290
Wang, S., Gu, Y., Zebell, S. G., Anderson, L. K., Wang, W., Mohan, R., et al. (2014). A noncanonical role for the CKI-RB-E2F cell-cycle signaling pathway in plant effector-triggered immunity. Cell Host Microbe 16, 787–794. doi: 10.1016/j.chom.2014.10.005
Wittstock, U., and Halkier, B. A. (2002). Glucosinolate research in the Arabidopsis era. Trends Plant Sci. 7, 263–270. doi: 10.1016/S1360-1385(02)02273-2
Wu, C.-Y., Rolfe, P. A., Gifford, D. K., and Fink, G. R. (2010). Control of transcription by cell size. PLoS Biol. 8:e1000523. doi: 10.1371/journal.pbio.1000523.s014
Xing, D. H., Lai, Z. B., Zheng, Z. Y., Vinod, K. M., and Fan, B. F. (2008). Stress-and pathogen-induced Arabidopsis WRKY48 is a transcriptional activator that represses plant basal defense. Mol. Plant 1, 459–470. doi: 10.1093/mp/ssn020
Yang, K.-Z., Jiang, M., Wang, M., Xue, S., Zhu, L.-L., Wang, H.-Z., et al. (2015). Phosphorylation of serine 186 of bHLH transcription factor SPEECHLESS promotes stomatal development in Arabidopsis. Mol. Plant 8, 783–795. doi: 10.1016/j.molp.2014.12.014
Yilmaz, A., Mejia-Guerra, M. K., Kurz, K., Liang, X., Welch, L., and Grotewold, E. (2010). AGRIS: the Arabidopsis gene regulatory information server, an update. Nucleic Acids Res. 39, D1118–D1122. doi: 10.1093/nar/gkq1120
Zheng, Z., Mosher, S. L., Fan, B., Klessig, D. F., and Chen, Z. (2007). Functional analysis of Arabidopsis WRKY25 transcription factor in plant defense against Pseudomonas syringae. BMC Plant Biol. 7:2. doi: 10.1186/1471-2229-7-2
Zheng, Z., Qamar, S. A., Chen, Z., and Mengiste, T. (2006). Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 48, 592–605. doi: 10.1111/j.1365-313X.2006.02901.x
Keywords: loss of giant cells from organs (lgo), siamese related1 (smr1), endoreduplication, endoreplication, Arabidopsis thaliana, RNA-seq, sepal, transcriptome
Citation: Schwarz EM and Roeder AHK (2016) Transcriptomic Effects of the Cell Cycle Regulator LGO in Arabidopsis Sepals. Front. Plant Sci. 7:1744. doi: 10.3389/fpls.2016.01744
Received: 16 July 2016; Accepted: 04 November 2016;
Published: 22 November 2016.
Edited by:
Keqiang Wu, National Taiwan University, TaiwanReviewed by:
Junxian He, The Chinese University of Hong Kong, Hong KongJohn Larkin, Louisiana State University, USA
Copyright © 2016 Schwarz and Roeder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrienne H. K. Roeder, ahr75@cornell.edu