 Jeri D. Barak1,2*
Jeri D. Barak1,2* Taca Vancheva1,3
Taca Vancheva1,3 Pierre Lefeuvre4
Pierre Lefeuvre4 Jeffrey B. Jones5
Jeffrey B. Jones5 Sujan Timilsina5
Sujan Timilsina5 Gerald V. Minsavage5
Gerald V. Minsavage5 Gary E. Vallad6
Gary E. Vallad6 Ralf Koebnik1*
Ralf Koebnik1*- 1UMR Interactions – Plantes – Microorganismes – Environnement, IRD-Cirad-Université Montpellier, Montpellier, France
- 2Department of Plant Pathology, University of Wisconsin, Madison, WI, USA
- 3Faculty of Biology, Sofia University St. Kliment Ohridski, Sofia, Bulgaria
- 4Pôle de Protection des Plantes, UMR Peuplements Végétaux et Bioagresseurs en Milieu Tropical, Cirad-Université de la Réunion, Saint-Pierre, Ile de la Réunion, France
- 5Department of Plant Pathology, University of Florida, Gainsville, FL, USA
- 6Gulf Coast Research and Education Center, University of Florida, Wimauma, FL, USA
Multiple species of Xanthomonas cause bacterial spot of tomato (BST) and pepper. We sequenced five Xanthomonas euvesicatoria strains isolated from three continents (Africa, Asia, and South America) to provide a set of representative genomes with temporal and geographic diversity. LMG strains 667, 905, 909, and 933 were pathogenic on tomato and pepper, except LMG 918 elicited a hypersensitive reaction (HR) on tomato. Furthermore, LMG 667, 909, and 918 elicited a HR on Early Cal Wonder 30R containing Bs3. We examined pectolytic activity and starch hydrolysis, two tests which are useful in differentiating X. euvesicatoria from X. perforans, both causal agents of BST. LMG strains 905, 909, 918, and 933 were nonpectolytic while only LMG 918 was amylolytic. These results suggest that LMG 918 is atypical of X. euvesicatoria. Sequence analysis of all the publicly available X. euvesicatoria and X. perforans strains comparing seven housekeeping genes identified seven haplotypes with few polymorphisms. Whole genome comparison by average nucleotide identity (ANI) resulted in values of >99% among the LMG strains 667, 905, 909, 918, and 933 and X. euvesicatoria strains and >99.6% among the LMG strains and a subset of X. perforans strains. These results suggest that X. euvesicatoria and X. perforans should be considered a single species. ANI values between strains of X. euvesicatoria, X. perforans, X. allii, X. alfalfa subsp. citrumelonis, X. dieffenbachiae, and a recently described pathogen of rose were >97.8% suggesting these pathogens should be a single species and recognized as X. euvesicatoria. Analysis of the newly sequenced X. euvesicatoria strains revealed interesting findings among the type 3 (T3) effectors, relatively ancient stepwise erosion of some T3 effectors, additional X. euvesicatoria-specific T3 effectors among the causal agents of BST, orthologs of avrBs3 and avrBs4, and T3 effectors shared among xanthomonads pathogenic against various hosts. The results from this study supports the finding that T3 effector repertoire and host range are fundamental for the study of host—microbe interaction but of little relevance to bacterial speciation.
Introduction
The genus Xanthomonas includes numerous phytopathogenic bacteria. While the physiological characteristics of Xanthomonas are quite homogeneous, biological diversity is evident in that the phytopathogenic xanthomonads cause disease on more than 400 hosts, ranging across 11 monocotyledonous, and 57 dicotyledonous families (Leyns et al., 1984). Although the genus Xanthomonas infects a wide variety of hosts that inhabit the full spectrum of ecological niches, individual strains usually have a narrow host range (Jacques et al., 2016). Historically, phytopathogenic bacteria nomenclature has been based on their host range. Xanthomonas that caused the same symptomology on the same host range were grouped into an infrasub-specific division, pathovar (Dye et al., 1980). However, Xanthomonas phylogeny based on nucleic acid analysis has begun to upend the rationale for phytobacterial systematics to be based on host range.
Classification of species within the genus Xanthomonas underwent major revision based on nucleic acid analysis. A comprehensive DNA-DNA hybridization study resulted in the recognition of 20 species (Vauterin et al., 1995). Subsequently, three additional species, X. euvesicatoria, X. perforans, and X. gardneri, that all cause bacterial spot of tomato (BST) were designated based on DNA-DNA hybridization (Jones et al., 2004). Using DNA-DNA hybridizations, repetitive element palindromic (Rep)-PCR, and amplified fragment length polymorphism (AFLP) genomic fingerprints the major phytopathogenic species of Xanthomonas were divided into six groups designated 9.1 to 9.6 (Rademaker et al., 2000, 2005; Ah-You et al., 2009). Collectively, these analyses confirmed nucleic acid distinctions among the causal agents of BST. X. euvesicatoria and X. perforans were placed in group 9.2 and X. vesicatoria with distinct rep-PCR fingerprints matched no other examined strains and was left outside any group. Each group was designated a distinct species, usually one for each group; however, within some groups, historical species nomenclature has retained, such as group 9.2 recognized as X. euvesicatoria, also includes X. perforans, X. dieffenbachiae, X. alfalfae, and several pathovars of X. axonopodis.
Further nucleic acid examination continues to erode phytobacterial systematics based on host range. Mutilocus sequencing analysis (MLSA) based on a very limited number of bacterial spot causing strains hypothesized that (i) X. euvesicatoria and X. perforans, and (ii) X. gardneri and X. cynarae likely are synonyms (Young et al., 2008). Using MLSA, average nucleotide identity (ANI), and DNA-DNA hybridizations Constantin et al. also concluded that X. perforans should be considered X. euvesicatoria (Constantin et al., 2016). More importantly, results from all of these nucleic acid analyses with an extensive collection of X. dieffenbachiae strains isolated from three distinct hosts support that these strains belong in four bacterial species, X. euvesicatoria, X. citri, X. phaseoli, and X. axonopodis independent of host range.
Xanthomonas phylogeny is not driven by host range and therefore its systematics should also be independent of the historical constraints commonly imposed on phytopathogenic bacteria. Evidence to support this supposition already exists in the case of the causal agents of bacterial spot of tomato and/or pepper. Bacterial spot is caused by four distinct species: X. euvesicatoria, X. vesicatoria, X. perforans, and X. gardneri (Jones et al., 2000). Among the four species, X. euvesicatoria and X. gardneri strains infect both tomato and pepper, X. perforans strains until recently only cause disease in tomato (Schwartz et al., 2015), and X. vesicatoria strains primarily infect tomato. Interestingly, a recent phylogenomic analysis of these four species included a X. perforans isolated from symptomatic pepper (Schwartz et al., 2015). The authors concluded that host range was determined by type 3 effector repertoire and to an extent AvrBsT limited it to tomato. Although this study included 67 genomes of X. euvesicatoria, X. perforans, and X. gardneri, collectively, they were all isolated from symptomatic tissue collected in the United States, a narrow geographical range when one considers that X. euvesicatoria has a worldwide distribution (Jones et al., 2005) and X. perforans and X. gardneri strains increasingly have been isolated in Canada (Cuppels et al., 2006), South America, and regions bordering the Indian Ocean (Bouzar et al., 1996, 1999; Hamza et al., 2010). Although this recent trove of genomes of X. euvesicatoria, X. perforans, and X. gardneri is a useful set to examine questions of pathogen population structure and recent pathogenicity factor changes among some of the causal agents of bacterial spot of pepper and tomato, the available sequenced genomes remain temporally and geographically biased.
In this study we sequenced five Xanthomonas euvesicatoria strains isolated from three continents (Africa, Asia, and South America) to provide a set of representative genomes for further comparative analyses with the available sequenced strains isolated from the United States, the Balkan Peninsula, and Italy. Strains were isolated from either symptomatic Capsicum or Lycopersicon when recorded. We broadly analyzed nucleic acids and gene content of the strains we sequenced as well as all the available X. euvesicatoria and X. perforans sequenced strains. By ANI, we examined the phylogeny of X. euvesicatoria, X. perforans, X. allii, X. alfalfa subsp. citrumelonis, and X. dieffenbachiae, members of Rademaker group 9.2. By comparing multiple members of Rademaker group 9.2, we provide a unique, integrated phylogeny of X. euvesicatoria independent of host range. We also provide evidence based on genomic sequencing of a xanthomonad isolated from rose that should be placed in X. euvesicatoria as a new pathovar. With the genome sequences of these geographically and temporally diverse set of X. euvesicatoria LMG strains, as well as the X. euvesicatoria pv. rosa strain, T3 effector evolution has been examined and sets a foundation for future hypothesis-driven research.
Results/Methods/Discussion
Phenotypic Evaluation
Five strains of X. euvesicatoria were selected from the Belgium Co-ordinated Collection of Micro-organisms/LMG (http://bccm.belspo.be/about-us/bccm-lmg) for inquiry to expand our understanding about the causal agent of bacterial spot of tomato and pepper. Strains LMG 918 and LMG 933 were isolated from Capsicum frutescens from India in 1957 and Brazil, respectively. Strain LMG 909 was isolated from Capsicum sp. from the Ivory Coast in 1979. Strain LMG 667 was isolated from Lycopersicon esculentum, origin unknown and strain LMG 905 was isolated in India, host unknown. Since these strains were isolated from various hosts, we performed pathogenicity tests on tomato and pepper by infiltration using X. euvesicatoria strain 85-10 as a positive control. Overnight cultures were grown using in nutrient broth, cells were pelleted by centrifugation, and pellets were resuspended in water. Plant leaves were infiltrated by needleless syringe containing a water-bacterial suspension of 108 CFU/ml or water as a control. All the strains caused typical bacterial spot lesions on tomato (Bonny Best) and pepper (Early Cal Wonder, ECW), except LMG 918 which was only pathogenic on pepper. Race analysis on pepper with ECW-30R which contains Bs3 showed LMG 667, 909, and 918 elicited a hypersensitive reaction (HR) and thus contain avrBs3. LMG strains 667, 905, 909, and 933, but not 918, elicited a HR on ECW-20R which contains Bs2. Race analysis on tomato with Hawaii 7998 which has resistance that interacts with avrRxv resulted in a HR for LMG strains 667, 905, 909, and 933. These results suggest differences in the functional type 3 effectors among the LMG strains which we examined following genome sequencing.
Historically, BST pathogens, X. euvesicatoria and X. perforans, have been differentiated biochemically by pectate utilization and starch hydrolysis (Stall et al., 1994). In general, X. euvesicatoria strains are neither pectolytic nor amylolytic. All the strains, LMG 667, 905, 909, 918, and 933, failed to cause a depression around a bacterial colony on CVP medium by 48 h suggesting they are nonpectolytic. Surprisingly, LMG 918 was amylolytic as it and the positive control, X. perforans 485, displayed copious growth and a turbid halo around each colony grown for 48 h on nutrient agar supplemented with 1.5% soluble starch following flooding of plates with Lugol's iodine solution. The other LMG strains were nonamylolytic. These results suggest that LMG strains 667, 905, 909, and 933 react similar to X. euvesicatoria strains. In contrast, LMG 918 is different than the typical X. euvesicatoria or X. perforans strains, since X. perforans strains are strongly pectolytic and amylolytic (Jones et al., 2004). Neither these biochemical tests nor pathogenicity tests can confirm the species of all the LMG strains tested.
Genome Sequencing and Annotation
The genomes of the five LMG strains, 667, 905, 909, 918, and 933, were sequenced using the Illumina Hi-Seq2500 platform (Fasteris SA, Switzerland). The shotgun sequencing yielded between 1,917,527 and 3,384,562 100-bp paired-end reads (474–966 Mb), with insert sizes ranging from of 250 bp to 1.5 kb (Table 1). Draft genome sequences were assembled using the Edena algorithm v3.131028 (Hernandez et al., 2014), yielding between 375 and 498 contigs ≥200 bp (N50 between 19,253 and 29,903 bp) with 65–165 × coverage. Contigs were annotated with GeneMarkS + release 2.9 (revision 452131) (Borodovsky and Lomsadze, 2014), as implemented in the NCBI Prokaryotic Genome Annotation Pipeline (http://www.ncbi.nlm.nih.gov/genome/annotationprok/), which predicted between 4539 and 4879 genes per genome. These whole genome shotgun projects have been deposited at DDBJ/EMBL/GenBank under the accession no. JTEH00000000 to JTEL00000000 and the raw sequence reads are accession nos. SRR4712532, SRR4713555, SRR4714146, SRR4714148, and SRR4714149.
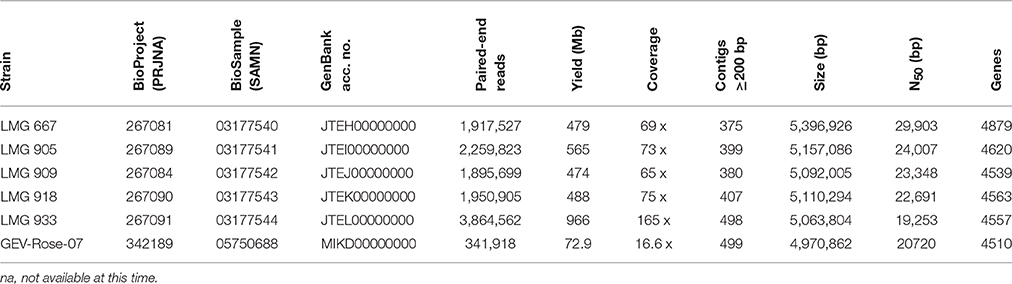
Table 1. Whole-genome sequence details.
The genome of GEV-Rose-07 strain was sequenced using Illumina Miseq platform (Interdisciplinary Center for Biotechnology Research, University of Florida). The sequences yielded 341,918 reads of average 241-bp paired-end reads (72.86 Mb). Draft genome was assembled using CLC Genomics Workbench v5, yielding 499 contigs ≥ 500 bp (N50 = 20,720 bp) with 16.6 × coverage. The assembled sequence was annotated using IMG/JGI platform, with the gene prediction underway. The genome has been deposited at GenBank under the accession number MIKD00000000 and the raw sequence read is accession no. SRR4457940.
Hydrolytic Enzymes Related to Taxonomy
To gain an understanding of the biochemical differentiation among LMG 918 and other X. euvesicatoria strains, we examined the genome sequences for amylases using the Carbohydrate-Active enZymes Database (CAZy; www.cazy.org). We found five putative polysaccharide lyases from three families in X. euvesicatoria 85-10, X. perforans 4P1S2, and the LMG strains 667, 905, 909, 918, and 933. Three CAZy families, GH13, GH14, and GH57, contain amylases. Amylases from the GH13 family were identified in X. euvesicatoria strain 85-10. Of the seven genes that were found, five encode cytoplasmic proteins with regions of similarity to alpha-amylases (annotated as putative alpha-amylase family protein, putative trehalose synthase, maltooligosyltrehalose synthase, alpha-glucosidase, and sucrose hydrolase). Two genes encode secreted proteins, annotated as putative alpha-amylase and cyclomaltodextrin glucanotransferase precursor. Interestingly, the putative alpha-amylase in X. euvesicatoria strain 85-10 contains a frameshift. A comparison of the X. euvesicatoria and X. perforans genome sequences revealed that all but three X. euvesicatoria strains but none of the X. perforans strains contain this frameshift (XCV0850/XCV0849). These results suggest that X. euvesicatoria strains LMG 918, Xe 259, and Xe 315 could be amylolytic which was confirmed in this study for LMG 918.
Comparison of Genome Sequences
Since phenotypic evaluation separated X. euvesicatoria LMG 918 from the other LMG strains and previously described X. perforans and X. euvesicatoria strains, we executed an extensive analysis of the sequenced genomes of both species. We compared portions of seven housekeeping genes (4722 bp in total), atpD, dnaK, efp, glnA, gyrB, lepA, and rpoD, from all publicly available genomes of X. euvesicatoria and X. perforans, as well as the strains sequenced in this study. Polymorphisms were rare. In total, seven haplotypes were found among the 68 strains. A large X. euvesicatoria haplotype group (H1) consists of strains from each geographical region examined with a total of 31 strains (Table 2). Thus, sequences of these essential genes from four of the newly characterized X. euvesicatoria strains in this study were identical to the bulk of strains from the United States. X. euvesicatoria strains 259 and 315 were separated from H1 by one SNP. A large X. perforans group (H6) consists of 29 strains. The sequence of strain X. perforans 4P1S2 contains five “N” which introduce frameshifts in the genes dnaK and glnA; removal of the Ns results in a sequence identical to the other members of H6. X. perforans strain Xp17-12 has one SNP with respect to H6. X. perforans strains 4–20 and 5–6 are more similar to H1 than H6 while LMG 918, which lacks 7 bp in the atpD gene due to its split into two contigs, is as similar to H1 as H6. Recently, a core protein-coding genome phylogenetic analysis identified a division among these X. perforans strains separating them into three groups (Schwartz et al., 2015), which grouped strains Xp 17-2, Xp 4-20, and Xp 5-6 together. Furthermore, their own SNP analysis including 22,105 SNPs in the X. perforans genomes compared to the reference X. axonopodis pv. citri strain 306 grouped these strains (Xp 17-2, Xp 4-20, and Xp 5-6) together tightly with strains we separated into H6, e.g., Xp15-11, Xp11-2, and Xp18-15. In general, the strains analyzed from the United States are relatively young, with the oldest isolated from 1998 while the LMG strains we sequenced in this study are significantly older, when isolation dates are known, such as LMG 918 and LMG 909 were isolated in 1957 and 1979, respectively. Neither geographic nor temporal factors appear to influence divisions among these strains based on SNP analysis. Results from the comparison of these niche independent genes suggest that although a few polymorphisms exist among X. euvesicatoria and X. perforans strains, separating the strains into distinct species is poorly supported by examination of a broad collection of strains.
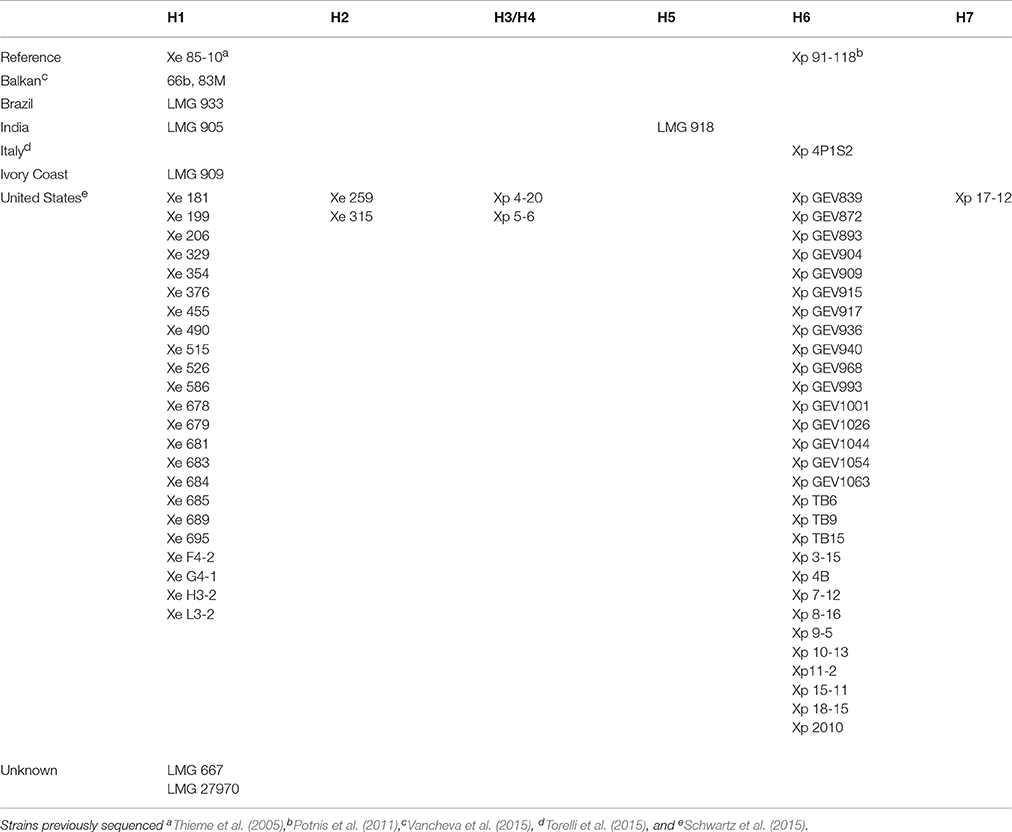
Table 2. SNP analysis of seven housekeeping genes, atpD, dnaK, efp, glnA, gyrB, lepA, and rpoD, separating all publicly available genomes of Xanthomonas euvesicatoria and X. perforans strains into haplotypes (H#).
To determine the validity of separating X. euvesicatoria and X. perforans into two species, ANI was calculated using JSpecies (Richter and Rosselló-Móra, 2009) for all the LMG strains sequenced in this study, X. euvesicatoria strains 66b and 83M, isolated from symptomatic Capsicum annuum from Bulgaria in 2012 and Macedonia in 2013, respectively (Vancheva et al., 2015), and X. euvesicatoria 85-10, the reference strain for genomics (Thieme et al., 2005). ANI values among all these X. euvesicatoria strains were >99.1% (Table 3). A BLAST-based comparison of a subset of X. perforans strains isolated in the United States and separated into three groups by Schwartz et al. based on ML analysis based on partitioned analysis by codon position, revealed ANI values between 99.62 and 99.7%. Neither SNP analysis, presence (or absence) of specific type 3 (T3) effectors, nor ANI support meaningful divisions within X. perforans. These results suggest that differences within X. perforans genomes are not relevant to bacterial infrasub-specific division phylogeny. We found ANI values between the same subset of X. perforans strains and X. euvesicatoria strains (85-10, 66b, 83M, and all the LMG strains described herein) were >98.1% (Table 3). Since ANI is considered the new standard for species definition, these results suggest that the strains sequenced in this study are X. euvesicatoria and that X. perforans strains should be considered X. euvesicatoria, similar to the findings of others (Young et al., 2008; Constantin et al., 2016).
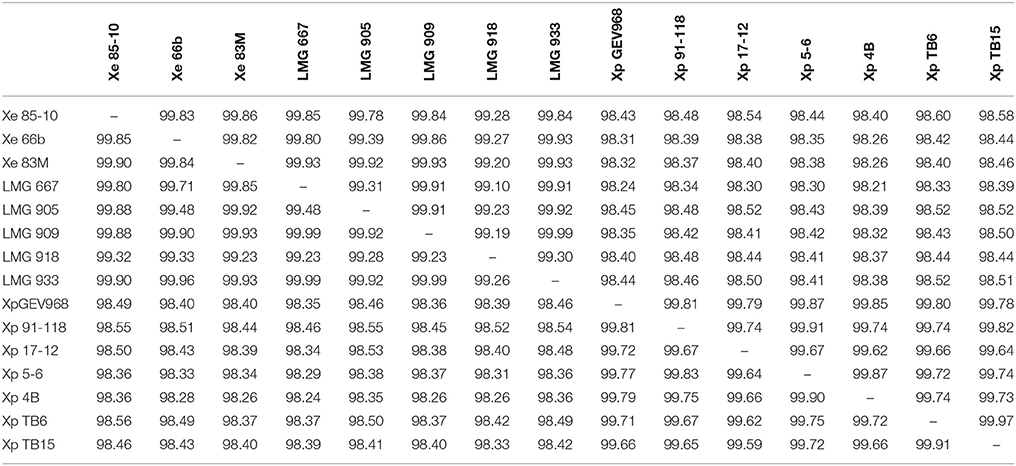
Table 3. Average nucleotide identity (ANI) values for a two-way comparison between two genomes of Xanthomonas euvesicatoria and/or X. perforans strains.
Since the ANI values between X. euvesicatoria and X. perforans strains were above the 95–96% transition zone, above which strains are considered to be a taxonomically prokaryotic species (Konstantinidis and Tiedje, 2005), we did a BLAST-based comparison of members of the Rademaker group 9.2, X. euvesicatoria, X. perforans, X. allii, X. alfalfae subsp. citrumelonis, and X. dieffenbachiae, as well as a recently described pathogen of rose (Huang et al., 2013). This comparison revealed ANI values between 99.11 and 97.83% among the compared genomes (Table 4). These results are significantly above the transition zone and suggest that members of Rademaker group 9.2 are a single species and should be recognized as X. euvesicatoria, regardless of host range.
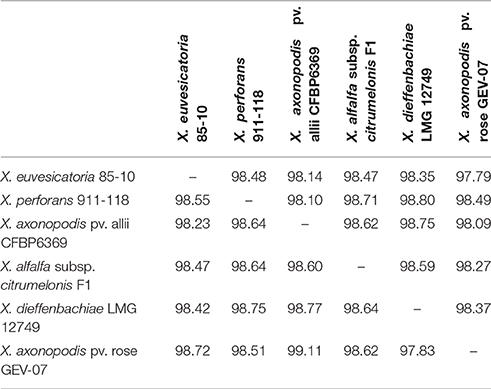
Table 4. Average nucleotide identity (ANI) values for a two-way comparison between two genomes of Xanthomonas species assigned to Rademaker group 9.2.
Our results add to the mounting evidence that Xanthomonas phylogeny is not driven by host range. Nonetheless, the systematics of phytobacterial pathogens which reflects host range is valuable to the scientific community as well as to a broader audience, such as regulators. Our phylogenetic analysis support designation of the recently described pathogen of rose as X. euvesicatoria, and yet we advocate for the use of pathovar rosa for those strains which share the same host range (Huang et al., 2013).
Type III Effectors
Pathogenicity on specific hosts by xanthomonads has been attributed to the presence or absence of specific T3 effectors (Schwartz et al., 2015). Previously, a group of T3 effectors were identified as a set core in the four Xanthomonas species which cause BST (Potnis et al., 2011). The LMG strains sequenced in this study possess each of these T3 effectors with the exception of XopAD (Table 5). The xopAD gene, which encodes a SKWP repeat protein, was found to be intact in X. perforans, the rose isolate, and LMG 918, but has several conserved internal stops in the other strains, suggesting that they all originate from a common ancestor. Similar findings were observed with xopC2, which shares the same inactivating frameshift mutations in all the LMG strains, the two strains from the Balkan Peninsula, and the X. euvesicatoria reference strain 85-10, and with xopAE, which has a conserved frameshift in strains 85-10, 83M, and all LMG strains except LMG 918. A stepwise erosion process of xopADΩ, xopC2Ω, and xopAEΩ is relatively ancient to the species as suggested by their G+C content for strain 85-10, 66.9, 60.9, and 63.7%, respectively (Figure 1). We speculate that LMG 918 and X. perforans strains 91-118 and 4P1S2 may have a shared ancestor separate from X. euvesicatoria strains 85-10, 66b, 83M, and LMG 667, 905, 909, and 933 that started to accumulate mutations in xopAD. The ancestor shared by X. euvesicatoria strains 85-10, 66b, 83M, and LMG 667, 905, 909, and 933 accumulated inactivating mutations in xopC2. Later, the lineage with the inactivated xopC2 incurred a frameshift mutation in xopAE. This scenario is an example of T3 effector repertoire evolution which may influence host specificity.
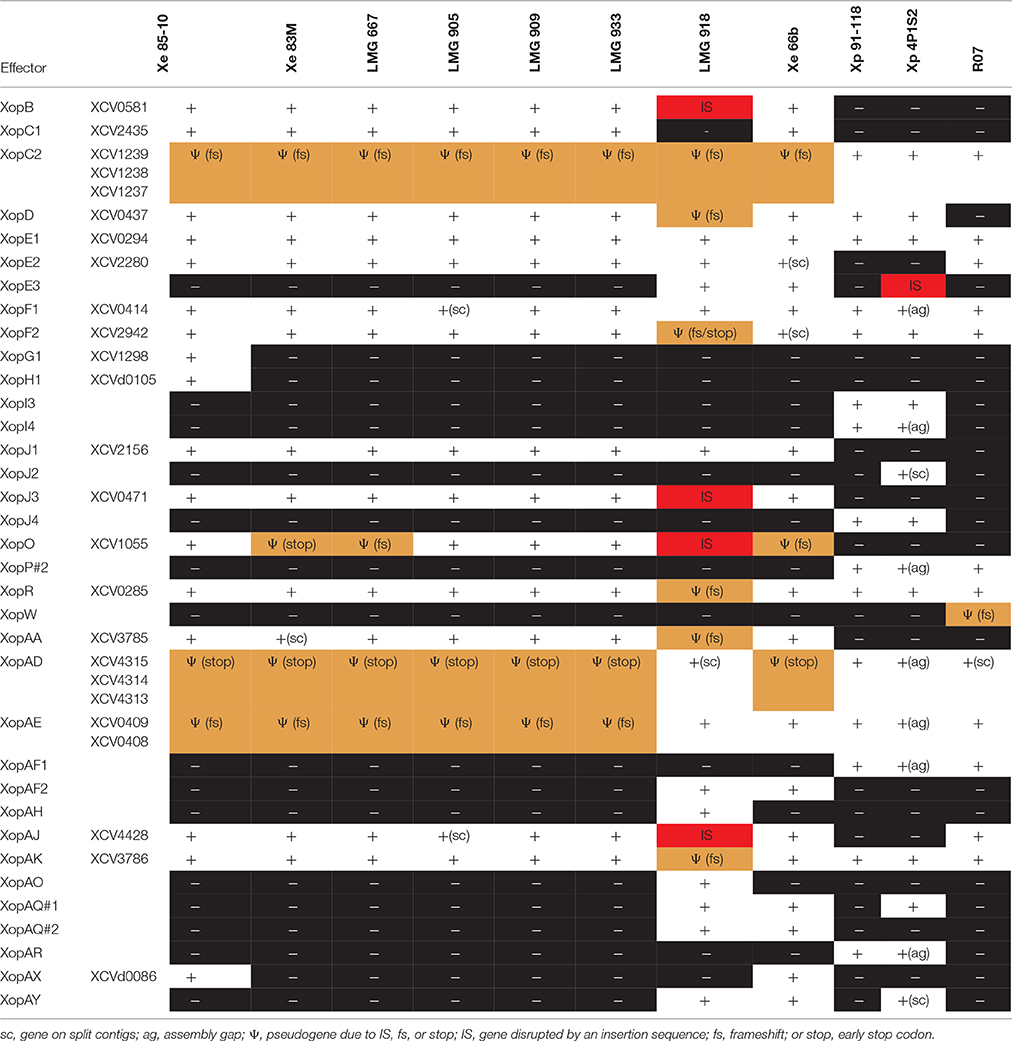
Table 5. Distribution of type 3 effectors not found in every Xanthomonas strain sequenced in this study.
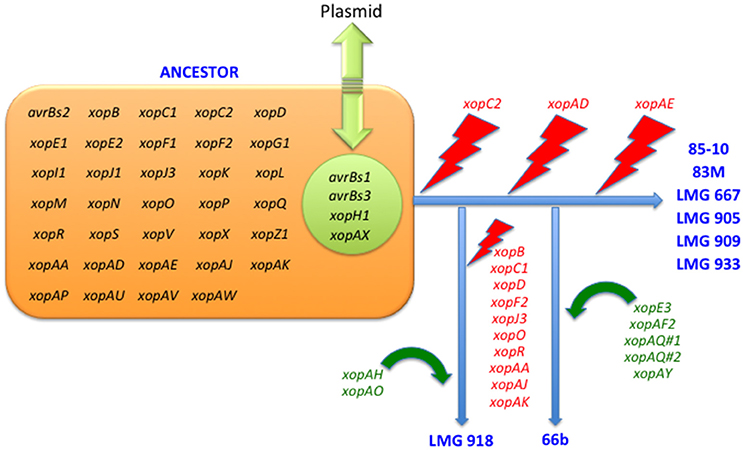
Figure 1. Erosion and acquisition of type 3 effectors among Xanthomonas euvesicatoria strains.
The stepwise erosion process of some T3 effector genes as described above appears to be compensated by additional, lineage-specific T3 effectors. Strains 66b and LMG 918 share five T3 effectors, which are not present in the other LMG strains, 83b, 85-10, or X. euvesicatoria pv. rosa (Table 5). However, it is widespread in field isolates of X. euvesicatoria from the United States (Schwartz et al., 2015), listed in Table 2. An inactivated variant, containing an IS element, was found in the genome of X. perforans 4P1S2. The low G+C content of this gene (59.0%) suggests a more recent acquisition. XopE3 belongs to the XopE class of T3 effectors and has close homologs in X. axonopodis, X. citri, X. fuscans, X. arboricola, X. cassavae, and X. campestris. Next to xopE3, facing each other, we found a homolog of xopAQ (G+C content of 56.6% with strain 85-10). A paralog of xopAQ with an even lower G+C content (53.8% with strain 85-10) is upstream of the T3 effector gene xopAY (G+C content of 52.6%). XopAY, which is related to HopW1 from Pseudomonas syringae (Lee et al., 2008), was first found in several X. translucens genomes but is also present in X. bromi, X. hyacinthi, and X. vasicola, all infecting monocots. The finding that this T3 effector is also present in strains 66b, LMG 918 and the X. perforans strain 4P1S2 suggests a function in dicotyledonous plants as well. XopAQ is related to a T3 effector (Rip6/Rip11) from Ralstonia solanacearum (Mukaihara et al., 2010). The two xopAQ paralogs have homologs in X. arboricola, X. citri, and X. gardneri, where it was first described in Xanthomonas (Potnis et al., 2011). XopAF2, another T3 effector specific to only X. euvesicatoria strains 66b and LMG 918, is related to the widespread HopAF1 effector from P. syringae, which suppresses plant immunity by targeting methionine recycling to block ethylene induction (Washington et al., 2016). Close homologs are currently only found in X. citri and X. fuscans, two species that were placed in Rademaker group 9, and in strains of X. arboricola.
Although we hypothesize that strains 66b and LMG 918 arose from separate lineages, they share T3 effectors absent in the reference strain 85-10 which we suppose shares a common ancestor with 66b. All three 66b/LMG 918-specific loci are syntenic in both strains. For the xopE3-xopAQ locus, we found a corresponding region in the reference strain 85-10, harboring XCV2439 and XCV2440 (hpaJ), in one of the two flanking sequences (2 kb). Similarly, for the xopAF2 locus, only one of the two flanking sequences has a counterpart in strain 85-10, encoding the two cointegrate resolution proteins S and T (XCV2438 and XCV2437). This close vicinity of the two loci is striking, and their vicinity to cointegrate resolution genes suggests a recombination-based acquisition mechanism, either from a plasmid or a transposon. Finally, the xopAQ-xopAY loci are also syntenic in strains 66b and LMG 918, and no homologous region is found in the reference strain 85-10. These data suggest that determination of ancestral lineage based on T3 effector repertoires may be futile whereas individual T3 effector lineage may be possible and informative to host—microbe interactions.
We also identified additional T3 effectors shared among all the LMG strains, X. euvesicatoria 85-10, and X. perforans 91-118 and 4P1S2, XopE1, XopI1, XopM, XopP#1, XopS, XopV, XopAP, XopAU, XopAV, and XopAW (Table 5). Surprisingly, all of these T3 effectors, as well as the ones identified as shared among BST pathogens previously (Potnis et al., 2011), were also found in X. euvesicatoria pv. rose. These T3 effectors must not be responsible for pathogenicity of tomato since X. euvesicatoria pv. rose was shown to be non-pathogenic to tomato (Huang et al., 2013). Taken together, these findings suggest that these T3 effectors are common among members of the Rademaker 9.2 clade and may be shared by a common ancestor but they are not involved in host determination.
Species-specific T3 effectors have also been previously identified for X. perforans (XopC2, XopJ4, XopAF, and XopAE) and X. euvesicatoria (AvrBs1, XopC1, XopJ1, XopJ3, XopO, XopAA, and XopAI) (Potnis et al., 2011). With the exception of LMG 918, in general, the LMG strains sequenced here have the classic X. euvesicatoria-specific T3 effectors previously identified, except AvrBs1. X. euvesicatoria strain 85-10 also includes XopH1, which like AvrBs1 is encoded next to each other on a plasmid, and XopG1, which is also unique to strain 85-10. The xopG1 (XCV1298) gene is located between insertion elements ISxac2 (XCV1297/XCV1296) and IS1477 (XCV1301/XCV1300) upstream of another 85-10-specific gene, XCV1299, which encodes a putative secreted protein.
Using the LMG strains 667, 905, 909, and 933 and X. euvesicatoria 85-10, 66B, and 83M sequences, we identified additional X. euvesicatoria-specific T3 effectors, XopB, XopD, XopE2, XopF1, XopF2, XopK, XopL, XopN, XopQ, XopR, XopX, XopAA, XopAJ, and XopAK. LMG 918 has a single X. euvesicatoria-specific T3 effector, XopC1 and a single X. perforans-specific effector, XopAE. Since we found that LMG 918 infects pepper, a reduced number of X. euvesicatoria-specific T3 effectors is surprising. None of the remaining LMG strains have any of the X. perforans-specific effectors and furthermore, they only shared T3 effectors with X. perforans strains 91-118 and 4P1S2 that were also shared with X. euvesicatoria strains. Previously XopF1 has been hypothesized to be a pathogenicity determinant of Xanthomonas in tomato. However, we identified XopF1 in X. euvesicatoria pv. rose and this strain was shown to not infect tomato (Huang et al., 2013). Collectively, these results emphasize the conclusion that T3 repertoires may be poor determinants of phylogeny and inconclusive for speciation.
A pathogen population shift in X. perforans has been observed in Florida from tomato race 3 to race 4 due to null mutations in the xopAE/avrXv3 gene. This shift has been noted as significant since the X. perforans reference strain 91-118, isolated in 1991, has XopAE while the X. euvesicatoria reference strain 85-10 does not have this T3 effector (Potnis et al., 2011). XopAE is a translational fusion of hpaG and hpaF. X. euvesicatoria strain 85-10, and X. euvesicatoria strains isolated in the United States before 1997, have separate hpaG and hpaF genes. We hypothesize that the LMG strains sequenced in this study would also have separate hpaG and hpaF genes and thus, lack XopAE. We found that only strain LMG 918 had an intact XopAE, similar to X. euvesicatoria 66b, X. perforans 91-118 and 4P1S2, and X. euvesicatoria pv. rose. This result is interesting since LMG 918 was isolated 40 years before XopAE appeared in the X. perforans population in the United States. All of the X. euvesicatoria strains isolated after 1997 from the United States also possess XopE3 (Schwartz et al., 2015); again, we only found XopE3 in LMG 918 and strain 66b from the Balkan Peninsula. The lag in appearance of XopE3 in the United States X. euvesicatoria population suggests a later introduction of X. euvesicatoria XopE3-containing strains into the United States or XopE3 arose multiple times in X. euvesicatoria.
A striking observation concerns the xopO gene, which suffered from mutational inactivation by at least four different events. The 211-codon gene has a frameshift at codon 14 in strain 66b, another frameshift at codon 73 in strain LMG 667, an early stop codon at codon 77 in strain 83M, and an IS element insertion with an 8-bp target site supplication at codon 136 in strain LMG 918. These multiple inactivation events that were retained suggest an advantage for getting rid of this protein, and it is tempting to speculate that XopO might be recognized as an avirulence factor by a resistance gene.
Targeting of T3 effectors to specific intracellular structures has been shown to affect the function. For instance, post-translational modification involving the covalent attachment of a lipid moiety (e.g., myristate or palmitate) has been shown to target proteins to the cytoplasmic membrane; this targeting is facilitated by a simple sequence motif at the N terminus of the polypeptide chain (Thieme et al., 2007). Prediction of such motifs in T3 effectors in the newly sequenced strains, using the CSS-Palm suite, reveals potential myristoylation/palmitoylation motifs for XopE1, XopE2, XopJ1, XopJ3, XopAF2, XopAH, XopAK, and XopAQ. Notably, we identified such a motif, MGNC, within the polypeptide chain of XopS (Schulze et al., 2012). Scrutinizing the 5′ region of the gene suggests that the corresponding ATG translational start codon is 15 nucleotides downstream of the annotated start codon in the X. euvesicatoria reference strain 85-10. This alternative start site would be accompanied by a well-defined Shine-Dalgarno sequence (GGAG) eight nucleotides upstream of the start codon. Another strongly predicted myrostoylation/palmitoylation motif, MGLC (preceded by a GGAG Shine-Dalgarno sequence six nucleotides upstream of the ATG start codon), is encoded 317 bp in front of the annotated ORF for XopB, only 29 bp downstream of a consensus PIP box-regulated promoter (see below). It will be interesting to analyze whether this candidate translational start site is functional, which would lead to the synthesis of a 24-amino acid peptide.
Predicted HrpX Regulons
Many, if not most, T3 effector genes are co-regulated with the hrp genes that encode the T3 secretion machinery (Roux et al., 2015). Moreover, additional virulence factors, such as cell-wall degrading enzymes, are co-regulated as well. All these genes are under control of a key regulatory protein of the AraC family, HrpX, which binds to a conserved sequence element in their promoter regions, the so-called PIP box (Koebnik et al., 2006). We therefore analyzed the six new genome sequences for the presence of the promoter motif TTCGB-N15-TTCGB-N30−32-TYNNNT (B represents C, G, or T; Y represents C or T). Using this conservative query, 24 putative PIP box-regulated promoters were found in the X. euvesicatoria strains and 18 were found in the X. euvesicatoria pv. rosa strain (Table 6). Most of the identified promoters corresponded to known HrpX-dependent genes, such as the hrp/hpa genes, genes for T3 effectors or cell-wall degrading enzymes (Noël et al., 2001; Jacobs et al., 2015). In addition, we found genes for three putative secreted proteins (XCV2568, XCV2729, and XCV4424) and a gene annotated to encode an anthranilate synthase component to be under control of HrpX, as previously experimentally confirmed for strain 85-10 (Noël et al., 2001; Koebnik et al., 2006). Two of these genes are not present in the rose isolate, neither did we find the T3 effector genes xopB, xopC1, xopJ1, and xopAA, which explains the smaller number of predicted PIP box-regulated promoters in this strain. We found only one predicted promoter that was oriented opposite to an annotated gene (XCV1852), a case that might represent a false positive. We would like to emphasize, however, that the hrpX regulon is certainly larger than the set of 18 to 24 genes discussed above. It is well known that mismatches to the PIP box consensus sequence can be tolerated for HrpX-dependent expression (Tsuge et al., 2005), as it was previously demonstrated by cDNA-AFLP and RT-PCR for the X. euvesicatoria genes XCV3407 and XCV3765 (Noël et al., 2001; Koebnik et al., 2006). Yet we prefer to use a conservative prediction approach because relaxing the stringency would increase the number of false positives.
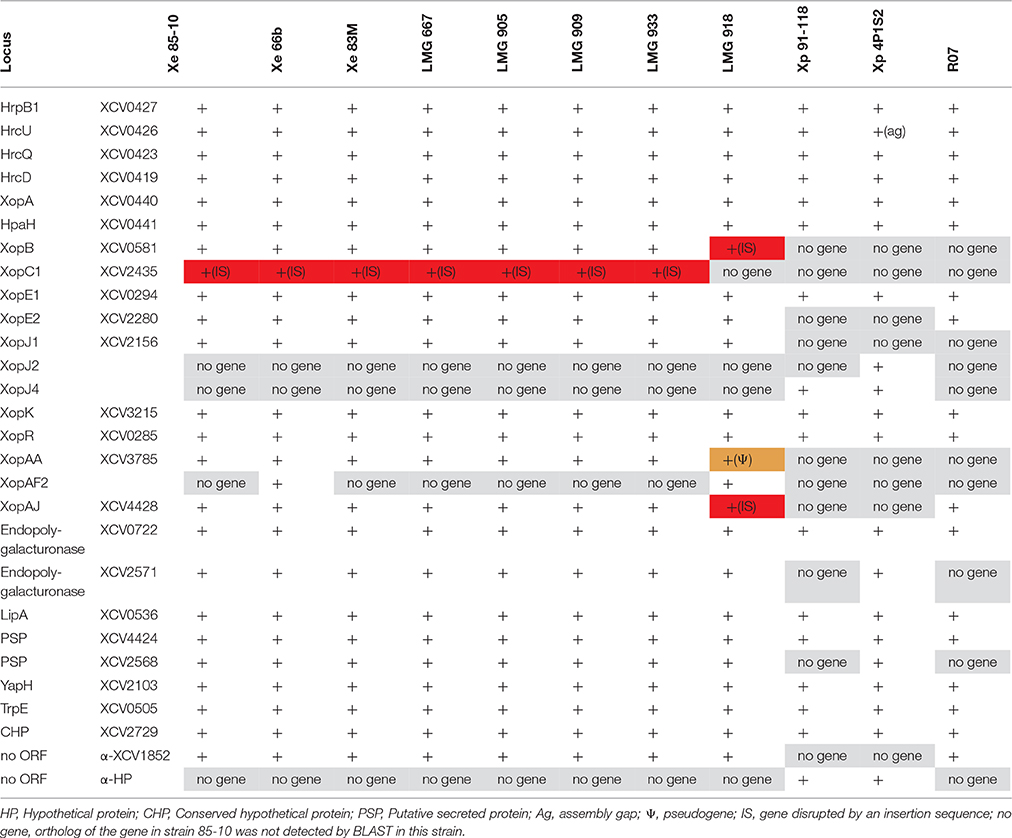
Table 6. Presence of consensus PIP-regulated promoter motifs in the Xanthomonas strains sequenced in this study.
ORFs for two T3 effectors were found downstream of a PIP box-regulated promoter, but are probably no longer under the control of HrpX due to the presence of IS elements. For example, in all the analyzed X. euvesicatoria genomes, the xopC1 ORF starts 1423 bp downstream of the predicted promoter, which suggests that the gene is not HrpX-dependently transcribed in any of the strains due to the polar effects of the IS element. In contrast, an IS element inserted directly behind the—10 promoter motif of xopB in LMG 918 likely disrupting gene transcription and/or regulation. These events enlarge the number of T3 effector genes that are in a process of functional erosion, in addition to the cases where IS elements insert into the coding sequence (e.g., xopJ3, xopO, and xopAJ in strain LMG 918). More experimental work is required to fully elucidate the hrpX regulon in the different strains, which might reveal that not only gene repertoires but also gene expression patterns contribute to host and tissue specificity of plant-pathogenic bacteria, such as Xanthomonas.
TAL effectors are among the best studied T3 effectors in Xanthomonas (Boch and Bonas, 2010). Upon import into the plant cell nucleus, they bind to the DNA in a sequence specific manner and induce transcription of eukaryotic genes in a way that TAL effectors can be considered as trans-kingdom remote controls for gene expression. Their modular structure, however, makes it nearly impossible to assemble TAL genes from short next generation sequencing reads. TBLASTN reads revealed the presence of TAL genes in the LMG strains 667, 909, and 918. Only two TAL effectors have been described and functionally characterized for X. euvesicatoria, AvrBs3, and AvrBs4. They are both encoded on plasmids and are extremely similar to each other. Yet, a small indel in the 3′ region of the avrBs4 gene allows to distinguish them. Based on this polymorphism we predict that LMG 667 and LMG 909 contain an ortholog of avrBs3 and LMG 918 contains an ortholog of avrBs4. We verified a functional copy of avrBs4 in LMG 918 as it caused HR on tomato and a functional copy of avrBs3 for LMG 667 and 909 which caused HR on pepper (ECW-30R). This conclusion is further supported by the observation that the upstream and downstream regions are syntenic between strains LMG 667 and 909 and the Macedonian strain 83M, which contains a functional avrBs3 gene that triggers HR on ECW-30R (Bs3) pepper plants but not on ECW plants. Similarly, the upstream region of LMG 918 is syntenic to the corresponding region in the Bulgarian strain 66b, which triggers an HR on the tomato cultivar Moneymaker (Bs4) but not in a Moneymaker line with the bs4 crossed in, and thus contains a functional avrBs4 gene. BLASTN analyses of the flanking regions of the TAL gene-containing contigs suggests that all X. euvesicatoria TAL effectors are encoded on plasmids, including the orthologs that we describe here for the LMG strains.
Conclusion
This study expands the publicly available genome sequences of X. euvesicatoria to include one from each continent where bacterial spot of tomato and pepper exists and from strains isolated in the 1950s and 1970s. Analysis of all the available sequences supports the conclusion that X. euvesicatoria and X. perforans are one bacterial species. Furthermore, a plethora of bioinformatic data as well as our own analyses supports the designation of all members of Rademaker group 9.2 as X. euvesicatoria. We offer direct evidence that X. euvesicatoria, X. perforans, X. axonopodis pv. allii, X. alfalfa subsp. citrumelonis, and X. dieffenbachiae belong to the same species, X. euvesicatoria. This species should also include the recently described pathogen of rose, herein designated X. euvesicatoria pv. rosa. Bioinformatic analysis of whole genomes alone for bacterial phylogeny should be relied upon instead of host range and T3 effectors.
Pathogenicity tests, race analysis, and bioinformatics analysis of T3 effectors are fundamental for the study of host—microbe interaction, but of little relevance to bacterial speciation. Relying on these tests or analyses for phylogeny of bacterial plant pathogens can confuse the concept of bacterial speciation which is now being built on whole genome sequencing. In this study, we inventoried the full repertoire of T3 effectors in sequenced strains of X. euvesicatoria. We describe relatively ancient stepwise erosion and acquisition of some T3 effectors. We identified orthologs of avrBs3 and avrBs4 highlighting a restriction to host expansion by this pathogen lineage.
Author Contributions
JB and RK conceived the study, analyzed the data, and wrote the manuscript. TV and PL participated in sequencing, assembly and annotation of the LMG strains. JJ, ST, and GV participated in sequencing and annotation of the rose pathogen strain. GM conducted the pathogenicity and biochemical tests. All authors have read and approved the final manuscript.
Funding
Work in RK's laboratory was supported by the French Agence National de la Recherche (grants ANR-2010-BLAN-1723 and ANR-2010-GENM-013). This work was partially funded by the United States Department of Agriculture, National Institute of Food and Agriculture (USDA-NIFA; grant 2011-670137-30166), and Food Research Institute at the University of Wisconsin—Madison to JB; funds supporting research efforts of GV, JJ, GM, and ST include Specialty Crop Program award #018015 from the Florida Department of Agriculture & Consumer Services, and Specialty Crop Research Initiative award #2015-51181-24312 from the USDA-NIFA. TV thanks the European Union Erasmus+ Program and Campus France for support.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Olivier Pruvost and Lionel Gagnevin, Cirad Saint-Pierre, La Réunion, for advice on representative strains for genome sequencing. We are grateful to Sébastien Cunnac and Lionel Moulin, IRD Montpellier, for advice and assistance on ANI analyses. We kindly acknowledge technical assistance of Claudine Boyer, Cirad Saint-Pierre, La Réunion, and Lucie Poulin, IRD Montpellier.
References
Ah-You, N., Gagnevin, L., Grimont, P. A., Brisse, S., Nesme, X., Chiroleu, F., et al. (2009). Polyphasic characterization of xanthomonads pathogenic to members of the Anacardiaceae and their relatedness to species of Xanthomonas. Int. J. Syst. Evol. Microbiol. 59, 306–318. doi: 10.1099/ijs.0.65453-0
Boch, J., and Bonas, U. (2010). Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu. Rev. Phytopathol. 48, 419–436. doi: 10.1146/annurev-phyto-080508-081936
Borodovsky, M., and Lomsadze, A. (2014). Gene identification in prokaryotic genomes, phages, metagenomes, and EST sequences with GeneMarkS suite. Curr. Protoc. Microbiol. 32, 1E.7.1–1E.7.17. doi: 10.1002/9780471729259.mc01e07s32
Bouzar, H., Jones, J. B., Somodi, G. C., Stall, R. E., Daouzli, N., Lambe, R. C., et al. (1996). Diversity of Xanthomonas campestris pv. vesicatoria in tomato and pepper fields of Mexico. Can. J. Plant Pathol. 18, 75–77. doi: 10.1080/07060669609500659
Bouzar, H., Jones, J. B., Stall, R. E., Louws, F. J., Schneider, M., Rademaker, J. L. W., et al. (1999). Multiphasic analysis of xanthomonads causing bacterial spot disease on tomato and pepper in the Caribbean and central america: evidence for common lineages within and between countries. Phytopathology 89, 328–335. doi: 10.1094/PHYTO.1999.89.4.328
Constantin, E. C., Cleenwerck, I., Maes, M., Baeyen, S., Van Malderghem, C., De Vos, P., et al. (2016). Genetic characterization of strains named as Xanthomonas axonopodis pv. dieffenbachiae leads to a taxonomic revision of the X. axonopodis species complex. Plant Pathol. 65, 792–806. doi: 10.1111/ppa.12461
Cuppels, D. A., Louws, F. J., and Ainsworth, T. (2006). Development and evaluation of PCR-based diagnostic assays for the bacterial speck and bacterial spot pathogens of tomato. Plant Dis. 90, 451–458. doi: 10.1094/PD-90-0451
Dye, D. W., Bradbury, J. F., Goto, M., Hayward, A. C., Lelliott, R. A., and Schroth, M. N. (1980). International standards for naming pathovars of phytopathgenic bacteria and a list of pathovar names and pathotype strains. Rev. Plant Pathol. 59, 153–168.
Hamza, A. A., Robène-Soustrade, I., Jouen, E., Gagnevin, L., Lefeuvre, P., Chiroleu, F., et al. (2010). Genetic and pathological diversity among xanthomonas strains responsible for bacterial spot on tomato and pepper in the southwest indian ocean region. Plant Dis. 94, 993–999. doi: 10.1094/PDIS-94-8-0993
Hernandez, D., Tewhey, R., Veyrieras, J. B., Farinelli, L., Østerås, M., François, P., et al. (2014). De novo finished 2.8 Mbp Staphylococcus aureus genome assembly from 100 bp short and long range paired-end reads. Bioinformatics 30, 40–49. doi: 10.1093/bioinformatics/btt590
Huang, C.-H., Vallad, G. E., Adkison, H., Summers, C., Margenthaler, E., Schneider, C., et al. (2013). A novel Xanthomonas sp. causes bacterial spot of rose (Rosa spp.). Plant Dis. 97, 1301–1307. doi: 10.1094/PDIS-09-12-0851-RE
Jacobs, J. M., Pesce, C., Lefeuvre, P., and Koebnik, R. (2015). Comparative genomics of a cannabis pathogen reveals insight into the evolution of pathogenicity in Xanthomonas. Front. Plant Sci. 6:431. doi: 10.3389/fpls.2015.00431
Jacques, M.-A., Arlat, M., Boulanger, A., Boureau, T., Carrère, S., Cesbron, S., et al. (2016). Using ecology, physiology, and genomics to understand host specificity in Xanthomonas. Annu. Rev. Phytopathol. 54, 163–187. doi: 10.1146/annurev-phyto-080615-100147
Jones, J. B., Bouzar, H., Stall, R. E., Almira, E. C., Roberts, P. D., Bowen, B. W., et al. (2000). Systematic analysis of xanthomonads (Xanthomonas spp.) associated with pepper and tomato lesions. Int. J. Syst. Evol. Microbiol. 50, 1211–1219. doi: 10.1099/00207713-50-3-1211
Jones, J. B., Lacy, G. H., Bouzar, H., Minsavage, G. V., Stall, R. E., Schaad, N. W., et al. (2005). Bacterial spot - worldwide distribution, importance and review. Acta Horticult. 695, 27–36. doi: 10.17660/ActaHortic.2005.695.1
Jones, J. B., Lacy, G. H., Bouzar, H., Stall, R. E., and Schaad, N. W. (2004). Reclassification of the Xanthomonads associated with bacterial spot disease of tomato and pepper. Syst. Appl. Microbiol. 27, 755–762. doi: 10.1078/0723202042369884
Koebnik, R., Krüger, A., Thieme, F., Urban, A., and Bonas, U. (2006). Specific binding of the Xanthomonas campestris pv. vesicatoria AraC-type transcriptional activator HrpX to plant-inducible promoter boxes. J. Bacteriol. 188, 7652–7660. doi: 10.1128/jb.00795-06
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Lee, M. W., Jelenska, J., and Greenberg, J. T. (2008). Arabidopsis proteins important for modulating defense responses to Pseudomonas syringae that secrete HopW1-1. Plant J. 54, 452–465. doi: 10.1111/j.1365-313X.2008.03439.x
Leyns, F., De Cleene, M., Swings, J.-G., and De Ley, J. (1984). The host range of the genus Xanthomonas. Bot. Rev. 50, 308–356. doi: 10.1007/BF02862635
Mukaihara, T., Tamura, N., and Iwabuchi, M. (2010). Genome-wide identification of a large repertoire of Ralstonia solanacearum type III effector proteins by a new functional screen. Mol. Plant Microbe Interact. 23, 251–262. doi: 10.1094/MPMI-23-3-0251
Noël, L., Thieme, F., Nennstiel, D., and Bonas, U. (2001). cDNA-AFLP analysis unravels a genome-wide hrpG-regulon in the plant pathogen Xanthomonas campestris pv. vesicatoria. Mol. Microbiol. 41, 1271–1281. doi: 10.1046/j.1365-2958.2001.02567.x
Potnis, N., Krasileva, K., Chow, V., Almeida, N., Patil, P., Ryan, R., et al. (2011). Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genomics 12:146. doi: 10.1186/1471-2164-12-146
Rademaker, J. L., Hoste, B., Louws, F. J., Kersters, K., Swings, J., Vauterin, L., et al. (2000). Comparison of AFLP and rep-PCR genomic fingerprinting with DNA-DNA homology studies: Xanthomonas as a model system. Int. J. Syst. Evol. Microbiol. 50, 665–677. doi: 10.1099/00207713-50-2-665
Rademaker, J. L., Louws, F. J., Schultz, M. H., Rossbach, U., Vauterin, L., Swings, J., et al. (2005). A comprehensive species to strain taxonomic framework for Xanthomonas. Phytopathology 95, 1098–1111. doi: 10.1094/PHYTO-95-1098
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Roux, B., Bolot, S., Guy, E., Denancé, N., Lautier, M., Jardinaud, F. M., et al. (2015). Genomics and transcriptomics of Xanthomonas campestris species challenge the concept of core type III effectome. BMC Genomics 16:975. doi: 10.1186/s12864-015-2190-0
Schulze, S., Kay, S., Büttner, D., Egler, M., Eschen-Lippold, L., Hause, G., et al. (2012). Analysis of new type III effectors from Xanthomonas uncovers XopB and XopS as suppressors of plant immunity. New Phytol. 195, 894–911. doi: 10.1111/j.1469-8137.2012.04210.x
Schwartz, A. R., Potnis, N., Timilsina, S., Wilson, M., Patane, J., Martins, J., et al. (2015). Phylogenomics of Xanthomonas field strains infecting pepper and tomato reveals diversity in effector repertoires and identifies determinants of host specificity. Front. Microbiol. 6:535. doi: 10.3389/fmicb.2015.00535
Stall, R. E. C., Beaulieu, D., Egel, N. C., Hodge, R. P., Leite, G. V., Minsavage, et al. (1994). Two genetically diverse groups of strains are included in Xanthomonas campestris pv. vesicatoria†. Int. J. Syst. Evol. Microbiol. 44, 47–53. doi: 10.1099/00207713-44-1-47
Thieme, F., Koebnik, R., Bekel, T., Berger, C., Boch, J., Büttner, D., et al. (2005). Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by the complete genome sequence. J. Bacteriol. 187, 7254–7266. doi: 10.1128/JB.187.21.7254-7266.2005
Thieme, F., Szczesny, R., Urban, A., Kirchner, O., Hause, G., and Bonas, U. (2007). New type III effectors from Xanthomonas campestris pv. vesicatoria trigger plant reactions dependent on a conserved n-myristoylation motif. Mol. Plant Microbe Interact. 20, 1250–1261. doi: 10.1094/MPMI-20-10-1250
Torelli, E., Aiello, D., Polizzi, G., Firrao, G., and Cirvilleri, G. (2015). Draft genome of a Xanthomonas perforans strain associated with pith necrosis. FEMS Microbiol. Lett. 362:1. doi: 10.1093/femsle/fnv001
Tsuge, S., Terashima, S., Furutani, A., Ochiai, H., Oku, T., Tsuno, K., et al. (2005). Effects on promoter activity of base substitutions in the cis-acting regulatory element of HrpXo regulons in Xanthomonas oryzae pv. oryzae. J. Bacteriol. 187, 2308–2314. doi: 10.1128/JB.187.7.2308-2314.2005
Vancheva, T., Lefeuvre, P., Bogatzevska, N., Moncheva, P., and Koebnik, R. (2015). Draft genome sequences of two Xanthomonas euvesicatoria strains from the balkan peninsula. Genome Announc. 3, e01528–e01514. doi: 10.1128/genomea.01528-14
Vauterin, L., Hoste, B., Kersters, K., and Swings, J. (1995). Reclassification of Xanthomonas. Int. J. Syst. Bacteriol. 45, 472–489. doi: 10.1099/00207713-45-3-472
Washington, E. J., Mukhtar, M. S., Finkel, O. M., Wan, L., Banfield, M. J., Kieber, J. J., et al. (2016). Pseudomonas syringae type III effector HopAF1 suppresses plant immunity by targeting methionine recycling to block ethylene induction. Proc. Natl. Acad. Sci. U.S.A. 113, E3577–E3586. doi: 10.1073/pnas.1606322113
Keywords: Xanthomonas euvesicatoria, Xanthomonas perforans, comparative genomics, taxonomy, type III effectors, PIP box, HrpX regulon, cell wall degrading enzymes
Citation: Barak JD, Vancheva T, Lefeuvre P, Jones JB, Timilsina S, Minsavage GV, Vallad GE and Koebnik R (2016) Whole-Genome Sequences of Xanthomonas euvesicatoria Strains Clarify Taxonomy and Reveal a Stepwise Erosion of Type 3 Effectors. Front. Plant Sci. 7:1805. doi: 10.3389/fpls.2016.01805
Received: 23 September 2016; Accepted: 15 November 2016;
Published: 09 December 2016.
Edited by:
Fabienne Vailleau, Centre Toulouse Midi-Pyrénées (INRA), FranceReviewed by:
David John Studholme, University of Exeter, UKFrederik Börnke, Leibniz Institute of Vegetable and Ornamental Crops (LG), Germany
Copyright © 2016 Barak, Vancheva, Lefeuvre, Jones, Timilsina, Minsavage, Vallad and Koebnik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeri D. Barak, jeri.barak@wisc.edu
Ralf Koebnik, koebnik@gmx.de