 Hongwei Wang1,2
Hongwei Wang1,2 Feng Qin3*
Feng Qin3*- 1Agricultural College, Yangtze University, Jingzhou, China
- 2Hubei Collaborative Innovation Center for Grain Industry, Yangtze University, Jingzhou, China
- 3College of Biological Sciences, China Agricultural University, Beijing, China
Crops are often cultivated in regions where they will face environmental adversities; resulting in substantial yield loss which can ultimately lead to food and societal problems. Thus, significant efforts have been made to breed stress tolerant cultivars in an attempt to minimize these problems and to produce more stability with respect to crop yields across broad geographies. Since stress tolerance is a complex and multi-genic trait, advancements with classical breeding approaches have been challenging. On the other hand, molecular breeding, which is based on transgenics, marker-assisted selection and genome editing technologies; holds great promise to enable farmers to better cope with these challenges. However, identification of the key genetic components underlying the trait is critical and will serve as the foundation for future crop genetic improvement. Recently, genome-wide association studies have made significant contributions to facilitate the discovery of natural variation contributing to stress tolerance in crops. From these studies, the identified loci can serve as targets for genomic selection or editing to enable the molecular design of new cultivars. Here, we summarize research progress on this issue and focus on the genetic basis of drought tolerance as revealed by genome-wide association studies and quantitative trait loci mapping. Although many favorable loci have been identified, elucidation of their molecular mechanisms contributing to increased stress tolerance still remains a challenge. Thus, continuous efforts are still required to functionally dissect this complex trait through comprehensive approaches, such as system biological studies. It is expected that proper application of the acquired knowledge will enable the development of stress tolerant cultivars; allowing agricultural production to become more sustainable under dynamic environmental conditions.
Introduction
In order to meet the demands of the ever-growing human population, it is predicted that world food production will need to double by the year 2050 (Tilman et al., 2002). Unfortunately, crop production is facing severe potential threats from changes within our global climate (Battisti and Naylor, 2009; Boyer et al., 2013). For example, when crops suffer under abiotic stress conditions such as drought, salinity, and temperature extremes, primary losses of crop productivity might occur worldwide with an average yield loss of >50% for major crops (Boyer, 1982). Drought stress dramatically reduces agricultural harvests, resulting in widespread risk of food insecurity and social problems. Thus, given the unpredictable nature of drought and climate variability over the years, breeding crops that are tolerant to abiotic stresses, especially to drought, is one of the most important approaches to maintain or increase crop production (Gill and Tuteja, 2010; Tester and Langridge, 2010).
Due to the complex genetic basis of stress tolerance, classical breeding strategies often fail to meet the needs for stabilizing yield under variable conditions (Tester and Langridge, 2010), although these strategies have significantly impacted the yield potential of crops during the past century, without preexisting knowledge of the exact genetic factors controlling the trait (Collins et al., 2008). On the other hand, molecular breeding approaches, which include transgenic, genome-wide marker-assisted selection and genome editing technologies, are more accurate and rely on clear genetic information of the favorable alleles contributing to the trait. As such, transgenic approaches can insert or create novel alleles beyond those that are available within naturally occurring populations. However, there are limitations for the broad utility of these products due to the requirements for passing environmental safety and regulatory approvals prior to their acceptance for cultivation. In contrast, marker-assisted selection (MAS), which uses DNA markers to track specific chromosomal regions carrying natural favorable alleles in sequential runs of crossing and selection, is free from artificial genetic modification and is widely considered to be natural and safe. Although this approach is time-consuming, the products generated from these breeding strategies have a greatly accelerated path to market. By leveraging the knowledge of biochemistry and genetics, the emerging genome editing technology can efficiently introduce specific mutations into target loci referring and can be considered as transgenic-free. This technology enables direct functional studies of genes of interest in the crop context and provides a suite of new methodologies for crop improvement (Voytas and Gao, 2014; Gao, 2015).
Due to their sessile nature, crops have evolved sophisticated mechanisms to adapt to environmental changes. As a result, it is likely that the functional mechanisms underlying environmental stress responses in plants are probably more advanced and prominent than in animals. The question of how plants survive various environmental stresses is one of the most attractive topics to plant biologists and agronomists. As a result, there is tremendous interest in and demand for enhancing the stress tolerance of crops through biotechnology with the knowledge of how plants react and resist to drought stress. Thus, this review describes advances in natural genetic variations detected for drought tolerance, which may offer genomic selection targets for molecular breeding to accelerate drought-resistance breeding and sheds light into the mechanism of how plants react to stress.
Qunatitative Trait Loci (QTL) Analysis for Drought Tolerance
The mechanism of how plants respond to drought is sophisticated, which includes processes such as the regulation of genetic and metabolic pathways on molecular, physiological and population levels (Chaves et al., 2003; Izanloo et al., 2008; Xu et al., 2009). Overall, the adaptation to drought can be split into different categories including: ‘drought avoidance’ by escaping the water deficit, owing to inherent developmental distinctions such as early/late flowering time and robust root systems (Geber and Dawson, 1990); ‘water preservation’ of specialized anatomic leaf structures (Schulze, 1986; Jackson et al., 2000) and advanced leaf senescence (Hoffmann and Merila, 1999; Chaves et al., 2003; Sherrard et al., 2009; Maherali et al., 2010); and ‘cellular drought tolerance’ to dehydration (Bartoli et al., 1999). The last aspect is often inferred as ‘drought tolerance’ by researchers specialized in plant abiotic stress response. With respect to the cellular drought tolerance, when soil water potential is decreased, stress related signal transduction networks involving ABA dependent or non-ABA dependent pathways will be stimulated and transduced after these stress signals are perceived by plant cells. Transcription factors, which are master regulators of stress-responsive gene expression, are then subsequently activated. The expression of stress-responsive genes will result in biochemical and physiological processes, including hormone biosynthesis and transport, adjusted osmotic status, photosynthesis, stomatal regulation and detoxification in plant cells. Thus, stress induced morphology changes, including root growth enhancement and leaf area reduction, will result in acquired tolerance and survival strategies (Figure 1). All these categories, which can enhance plant survival and growth under the stress, are referred as drought resistance.
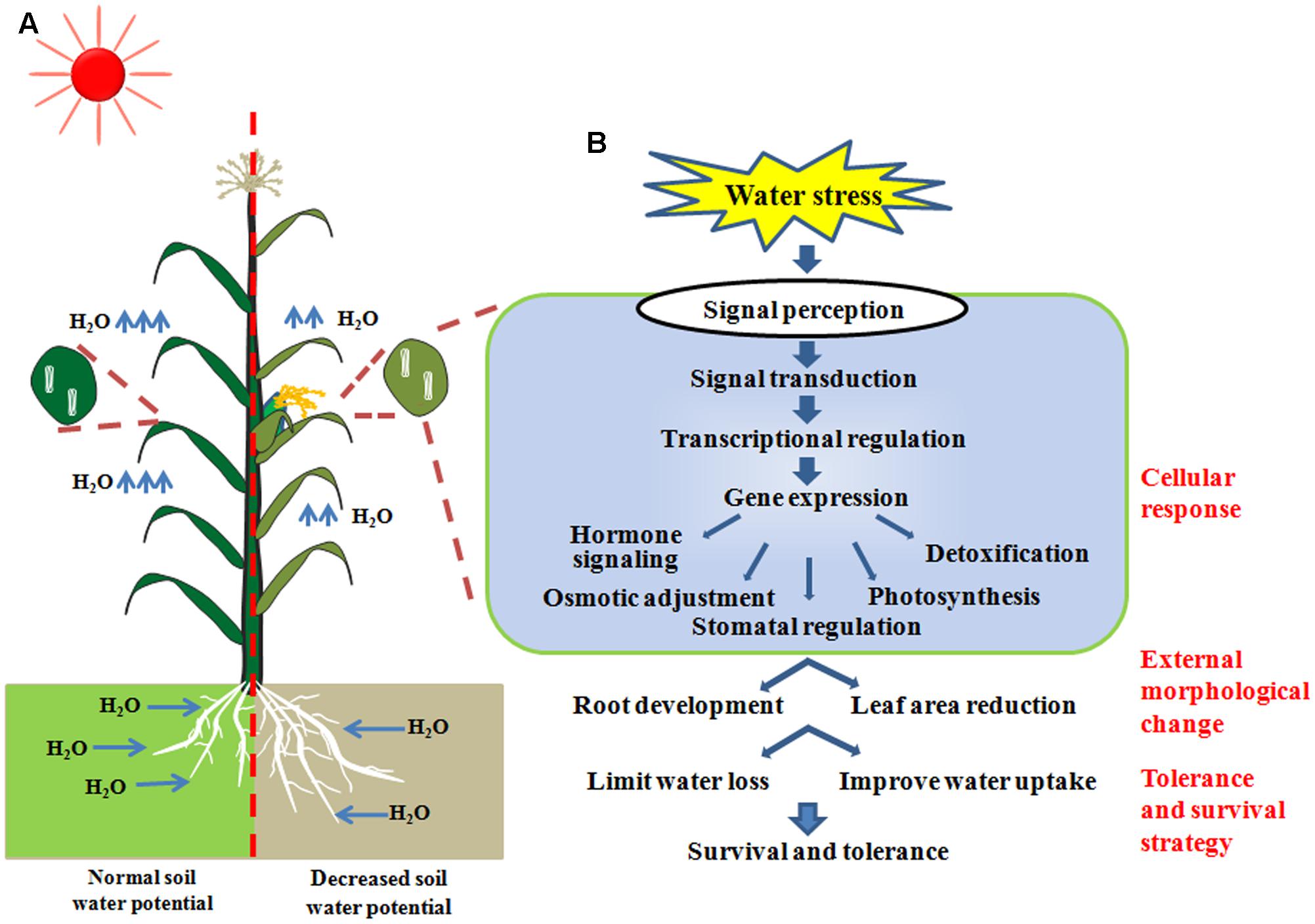
FIGURE 1. (A) The physiological and morphological adaptations of plants grown under water-deficit conditions. (B) Cellular response to water stress, including signaling transduction and physiological changes to achieve survival strategies.
In order to dissect the genetic basis of plant drought resistance, forward genetics strategies have been utilized to identify QTLs underlying the trait. However, due to the complexity and poor heritability of the crop yield trait, individual trait components are more frequently identified and characterized due to their better heritability in replicated experiments (Meyer et al., 2007; Riedelsheimer et al., 2012). For example, the trait components regarding maize drought resistance are usually dissected into (1) seedling survival rate under severe water deficits (Liu et al., 2013; Mao et al., 2015; Wang et al., 2016); (2) yield loss which is often indexed by ear length, kernel number per row, and hundred-kernel weight (Lu et al., 2006; Vikram et al., 2012; Yadaw et al., 2013); (3) anthesis and silking interval (ASI), which is an important indicative trait for maize drought tolerance. Drought stress often delays maize ear silk emergence and elongation, but not tassel development and pollen shedding; which leads to a significantly prolonged ASI. Thus, it dramatically reduces pollination efficiency and yields (Lebreton et al., 1995; Ribaut et al., 1996; Tuberosa et al., 2002a; Welcker et al., 2007; Lu et al., 2010). When identifying QTLs for grain yield and ASI across populations and under water stressed and well-watered environments, 68 QTLs were discovered with each physical interval of the QTL consisting of approximately five candidate genes (Semagn et al., 2013).
In addition, ABA is regarded as a stress hormone which can be significantly produced under drought and stimulates the expression of a large number of stress responsive genes and triggers stomatal closure (Bray, 1993; Close, 1996; And and Bartels, 2003). Thus, many studies have focused on the identification of QTLs that could enhance the concentration of ABA under stress in an attempt to better understand drought tolerance (Lebreton et al., 1995; Sanguineti et al., 1999; Landi et al., 2005; Rahman et al., 2011; Iehisa et al., 2014; Barakat et al., 2015). Specially, Landi et al. (2005) found a major maize QTL for L-ABA in bin 2.04 which indicated that the QTL not only enhances yield but also affects root architecture under drought conditions.
In the mean time, root achitechure (e.g., seminal root number; nodal root number; root pulling force; primary root diameter, length and weight; and adventitious seminal root weight) also play an important role in the process of drought resistance (Guingo et al., 1998; Tuberosa et al., 2002b; Gowda et al., 2011; Messmer et al., 2011; Sebastian et al., 2016). Tuberosa and Salvi (2002) reported that a deep root system with higher root density increased the survivability of plants under drought. Furthermore, four QTLs for nodal root angle (qRA), three for root dry weight, two for shoot dry weight, and three for plant leaf area were reported in sorghum; of which nodal root angle QTL presented new opportunities for improving drought adaptation mechanisms (Mace et al., 2012). Specially, introducing DEEPER ROOTING 1 (DRO1) into a shallow-rooting rice cultivar by backcrossing enabled the resulting line to avoid drought via increased root depth; which ultimately resulted in maintenance of high yield performance under drought conditions relative to the recipient cultivar (Uga et al., 2013). The 1 bp deletion in the open reading frame may be the causative site responsible for the differentiating root morphology between IR64 and KP, two rice varieties which differ in root architecture. The discovered allelic variation of DRO1 not only reveals the possible mechanism of how root morphology is modulated, but also suggests that the control of root architecture will contribute to the drought avoidance of crops. Over the last two decades, many studies have focused on the aforementioned drought resistance trait components in crops (Table 1). Thus, our knowledge pertaining to the genetic loci controlling drought resistance has increased substantially.
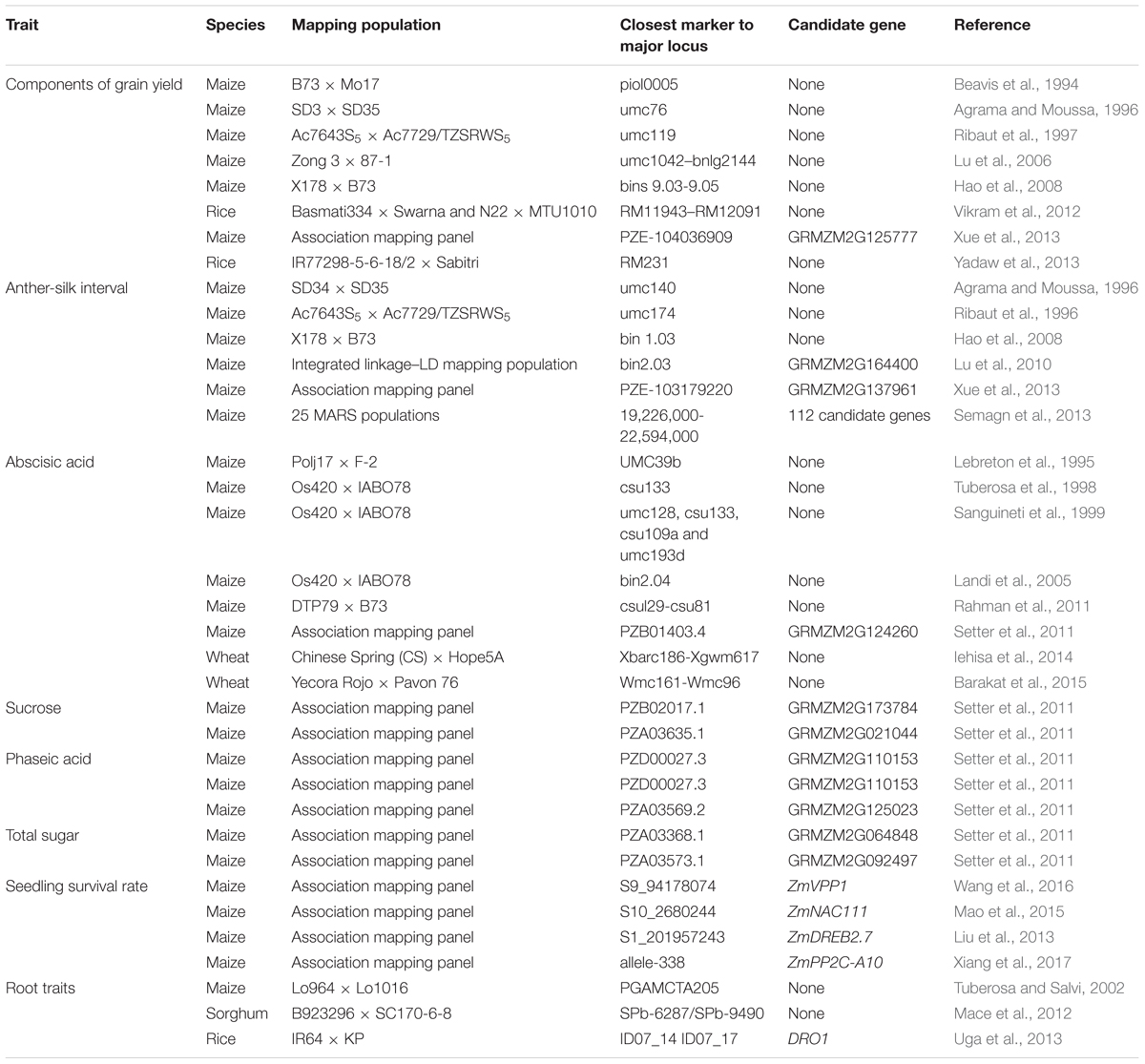
TABLE 1. Genetic dissction for drought resistance and related traits.
The Development of Population Design, Genotyping, and Variance Components Estimation Approaches for Genome-Wide Association Study (GWAS)
In plants, linkage mapping and genome-wide association study (GWAS) are the two major adopted methods to identify the QTLs for complex traits. The fundamental basis for linkage mapping and association mapping is genetic recombination. Linkage mapping exploits the functional polymorphisms and adjacent markers within families or pedigrees with known ancestry, whereas, association mapping exploits historical and evolutionary recombinations at a natural population level. Populations adopted for linkage mapping are usually derived from a bi-parental cross with a clear ancestry, while a collection of cultivars with unobserved ancestry are often adopted for association mapping (Cockram et al., 2010; Zhao et al., 2011; Hung et al., 2012; Riedelsheimer et al., 2012; Zhang et al., 2015; Zhou et al., 2015). Thus, biased association may occur due to the population structure and imbalanced familial relatedness among cultivars. In order to break the population structure and improve the statistic power for detecting rare variations, the creation and usage of newly designed populations in plants has emerged. In 2008, nested association mapping (NAM) and multi-parent advanced generation inter-cross (MAGIC) were used (Cavanagh et al., 2008; Yu et al., 2008) as methodologies to enable the functional identification of loci of interest. The NAM approach benefits from the historical and recent recombination events with clear population structure, whereas, MAGIC can examine the effect of loci unbiased due to the balanced contributions from all founders. In addition to these approaches, an additional population design is the random open-parents association mapping (ROAM) population which improves genetic resolution and statistical power for detecting rare variations (Pan et al., 2016; Xiao et al., 2016). Collectively, the recently designed populations have demonstrated their capacity to reveal the genetic components underlying complicated traits such as heterosis (Huang et al., 2016), leaf architecture (Tian et al., 2011) and flowering time (Huang et al., 2011).
However, no matter which mapping population is chosen for a study, a large number of molecular markers, which record the level of genetic diversity between two parental lines or amongst different cultivars, are needed for the mapping of QTLs. For example, it was estimated that more than 10 million markers were required for an efficient GWAS in maize, not taking into account of epigenetic variations (Myles et al., 2009). In brief, the development of molecular marker methodologies has undergone three major stages. The earliest type of molecular markers, such as restriction fragment length polymorphisms (Botstein et al., 1980), random amplified polymorphism DNA (Williams et al., 1990), sequence characterized amplified regions (Paran and Michelmore, 1993), cleaved amplified polymorphic sequences (Konieczny and Ausubel, 1993), simple sequence repeats (Litt and Luty, 1989) and amplified fragment length polymorphisms (Vos et al., 1995) rely on DNA hybridization or polymerase chain reaction (PCR) techniques; which are either too expensive or inefficient. As genomic and expression sequence tag information began to accumulate (Adams et al., 1991), the primary molecular markers tools changed from DNA fragment lengths to the single nucleotide polymorphism (SNP) level (Wang et al., 1998); since SNP consists of the largest amount of variations in different genomes (Rafalski, 2002; Zhu et al., 2003). However, SNP detection methods, such as Taqman and molecular beacons, were still time-consuming and expensive (Tapp et al., 2000). Lately, a high-throughput sequencing technology by massively parallel sequencing and image recognition, such as Roche454 FLX Titanium (Thudi et al., 2012), HiSeq2500 (Bentley et al., 2008), Ion Torrent PGM (Rothberg et al., 2011), could obtain more than 100 million nucleotide sequences that could offer millions of SNP markers at a comparatively low cost for a mapping population (Shendure and Ji, 2008; Edwards and Batley, 2010). This is especially true for genotyping-by-sequencing (GBS), which focuses on sequencing the ends of DNA restriction fragments rather than the whole genome. As a result, GBS provides scientists an alternative genotyping method to quickly identify the genomic variations from an unprecedented number of samples; especially for species with large genomes (Altshuler et al., 2000; Narum et al., 2013). Due to the advantages of its efficiency, speed, simplicity and cost-effectiveness (Davey et al., 2011); GBS is an ideal marker acquisition technology (Poland and Rife, 2012). Notably, the GBS method was used for QTL mapping and genome diversity studies (Fu and Peterson, 2011; Deschamps et al., 2012; Fu Y. B. et al., 2013; Lu et al., 2013). For example, 140 million SNP and 200 thousand Insertion/Deletion (InDel) markers amongst 5000 recombinant maize inbred lines (Gore et al., 2009), 680 thousand SNP markers in 2815 maize inbred lines (Romay et al., 2013), 200 thousand SNP markers between wild and cultivated soybeans (Lam et al., 2010) were identified by GBS. Thus, high-throughput sequencing technology undoubtedly provides a revolutionary tool for offering a large amount of whole genome-coverage genetic markers for gene mapping either through linkage and/or association analyses to dissect the genetic loci underlying any interested traits in crops.
In addition to the sufficient amount of molecular markers, the fast and accurate variance component estimation method is also one of the prerequisites to perform a GWAS. In the early stage of GWAS, the general linear model was employed which included genome control (Henderson, 1975), family test (Abecasis et al., 2000) and structural correlation (Pritchard et al., 2000) to control possible false positive associations. Later, Yu et al. (2006) proposed a method based on a mixed linear model (MLM) for better control of the population structure and the imbalance correlation among various materials; of which the population structure (Pritchard et al., 2000) and the familial relatedness among the different genotypes were treated as fixed effects and random effects, respectively. However, no matter which statistical method is applied, a large amount of samples is needed to obtain sufficient statistical power (Balding, 2006). This is especially important to detect the associations of a complex trait to loci with small effects (Buckler et al., 2009). Since the computational burden is proportional to the cube of the sample number fitted as random effects, massive computational time is demanded by the MLM. Especially, when associations are analyzed for a large number of markers with the phenotypic information from thousands of individuals, heavy computational burden occurs. In order to solve this problem, the sire model was first used in animal association studies to enhance the computational speed by reducing random effects (Henderson, 1975; Thompson, 1979; Quaas and Pollak, 1980; Pollak and Quaas, 1983). After that, genome-wide rapid association analysis using mixed model and regression (GRAMMER) built in GenABLE software was developed to approximately estimate the random effects via a two-step residuals approach. It estimates the residuals from the linear mixed model by removing the marker effects first, and then the residuals are treated as a phenotype for each marker under a standard linear regression model (Aulchenko et al., 2007). This method significantly reduced the computational time for each marker association calculation. Unlike the approximate estimation, efficient mixed-model association (EMMA) matrix is a method for an accurate estimation of variance component, including the genetic variance and residual variance, which can speed up the iterative process (Kang et al., 2008). However, for a GWAS of a population consisting of a few thousand individuals and a half million SNPs, it is probable that several years of central processing unit time would be necessary to complete the computing when using the EMMA method (Kang et al., 2010; Zhang et al., 2010). Population parameters previously determined (P3D) (Zhang et al., 2010) and EMMA eXpedited (EMMAX) (Kang et al., 2010) were the other two complex approximate estimation methods to reduce the computational processing. The two calculation methods made it possible for a personal desktop computer to perform GWAS; which adopts 1000 samples with 500 thousand markers or more. Although in most cases, the approximate variance estimation method would generate a nearly identical estimation as compared to the accurate method (Kang et al., 2010; Zhang et al., 2010). The accuracy of approximate estimation would be unknown in the absence of accurate estimation for variance components. Thus, factored spectrally transformed linear mixed models (FaST-LMM) (Lippert et al., 2011) and genome-wide efficient mixed-model association (GEMMA) (Zhou and Stephens, 2012) were developed. These approaches had advantages to directly estimate variance components including fixed effects in MLM and reduce the calculation burden for the single maker analysis. Benefitting from continuous efforts, with the massively reduced computational time and effective statistics method, the GWAS strategy became computationally practical the enable the genetic dissection for a trait of interest.
GWAS Facilitates QTL Cloning for Drought Tolerance
Although several QTLs for drought resistance have been identified through traditional linkage analysis, to date, no QTLs responsible for drought resistance have been cloned; despite the reports of their mapping information (Hao et al., 2009; Messmer et al., 2009; Almeida et al., 2013; Semagn et al., 2013). In order to clone a stress resistant QTL through traditional segregation and linkage analysis, not only do the different recombinants in the identified QTL region need to be screened, but the resistance of their progenies also need to be phenotyped and compared to determine which genetic region co-segregates with the resistance. In order to narrow down the QTL region to candidate genes, it usually takes several rounds of the fine mapping step; which makes QTL cloning time-consuming and laborious. As a result, the identification of the genetic components underlying drought resistance has been considered and proven to be very challenging. Nevertheless, owing to the advantage of a rapid life cycle and less space requirements for the cultivation of the model plant compared with crops; significant achievements have been made in Arabidopsis thaliana to study the molecular mechanisms for drought tolerance. Although, the genetic basis of quantitative variation for drought resistance in crops remains poorly understood, a complex gene network involving various kinds of genes has been conceived (Li et al., 2005; Qin et al., 2011; Osakabe et al., 2014).
Based on linkage disequilibrium (LD), the GWAS method enabled researchers to greatly increase the knowledge base of the genetic architecture and it also facilitated the cloning of genes underlying complex traits in crops. For example, the genetic basis of heterosis (Huang et al., 2016) and some important agronomic traits (Cockram et al., 2010; Tian et al., 2011; Sukumaran et al., 2014; Zhang et al., 2015; Si et al., 2016) were investigated through GWAS in a large amount of collected or constructed populations. However, it was estimated that for self-pollinating species, such as rice and soybean, the LD decay of cultivated rice had a long-range from about 100 kb to over 200 kb (McNally et al., 2009; Huang et al., 2010); whereas those of soybean landraces and improved cultivars were approximately 83 and 133 kb, respectively (Zhou et al., 2015). Whereas, for maize, which is an out-pollination species, the LD decay was estimated to be ≤2 kb among 26 founder genotypes of a nested association mapping population (Gore et al., 2009); whereas the LD decay was ∼500 bp in a maize natural variation population (Fu J. J. et al., 2013). Therefore, the rapid LD decay in the maize genome with above 1 million high-quality SNPs (Fu J. J. et al., 2013; Li et al., 2013) could make the resolution of GWAS reach the single gene level (Fu J. J. et al., 2013; Li et al., 2013; Wang et al., 2016). As a result, GWAS facilitated the gene cloning of ZmCCT which controls maize photoperiod sensitivity (Hung et al., 2012; Yang et al., 2013) and the investigatigation of evolutionary ZmWAK locus which controls the resistance to Sporisorium reilianum, a soil-borne fungus causing head smut disease in maize (Zuo et al., 2015).
Drought resistance is a complex and intrinsic trait, and the improvement of plant drought resistance through molecular breeding strategy is an ever growing hotspot for both basic and applied research areas. By leveraging knowledge gained from model plants, plant drought tolerance has been enhanced by manipulating the expression of drought-responsive genes (Li et al., 2005; Qin et al., 2011; Osakabe et al., 2014). In crops, however, limited allelic variation underlying drought tolerance has been identified. Recently, GWAS has contributed to the identification of favorable alleles for drought tolerance, especially in maize. Setter et al. (2011) reported that a candidate gene association analysis identified loci involved in the accumulation of carbohydrates and ABA metabolites under drought; of which eight candidate SNPs were confirmed to be significantly associated with the tolerance. Later, each genotype of this association panel was crossed with a common tester (CML312) and the generated F1 plants were phenotyped for nine traits under well-watered and water-stressed conditions in seven environments. As a result of this analysis, an additional 42 associated SNPs were identified (Xue et al., 2013). Liu et al. (2013) analyzed all the functional (Dehydration Responsive Element Binding protein) DREB genes in maize and examined their associations with the natural variation in drought tolerance among 368 maize varieties collected from tropical/subtropical and temperate regions (Fu J. J. et al., 2013; Li et al., 2013). A significant association of the natural variation in ZmDREB2.7 gene with drought tolerance was detected which locates in the gene promoter region and most likely enables an early induction of stress responsive gene expression. Thirunavukkarasu et al. (2014) reported that by using 240 accessions of subtropical maize with a high density marker set, 61 significant SNPs were identified under water stress condition; 48% of which were stress tolerance related genes. Moreover, the genetic basis of drought resistance in Hordeum vulgare, Cicer arietinum, A. thaliana, Medicago truncatula were also investigated by GWAS (Thudi et al., 2014; Verslues et al., 2014; Kang et al., 2015; Wehner et al., 2015). Recently, combined data from 15 bi-parental populations of maize were developed under the Water-Efficient Maize for Africa (WEMA) project to identify drought resistance genes by GWAS. The study identified several hundred genetic variants that are associated with plant height and flowering time under various water deficit conditions (Wallace et al., 2016). By adopting a global maize diversity panel (Yang et al., 2010; Fu J. J. et al., 2013; Li et al., 2013), Wang et al. (2016) reported a GWAS of maize drought resistance at the seedling stage and identified 83 genetic variants, which were resolved to 42 candidate genes. The peak GWAS signal overlapped with a previously reported drought related QTL9.3 (Semagn et al., 2013), and is directly located in the ZmVPP1 gene on Chromosome 9 which encodes a vacuolar-type H+-pyrophosphatase (H+-PPase). The ZmVPP1 gene is a homolog of AVP1 which encodes an Arabidopsis H+-PPase. The AVP1 gene was shown to confer drought and salt resistance when over-expressed in Arabidopsis (Gaxiola et al., 2001). Yeast and E. coli H+-pyrophosphatases can establish a proton gradient across the vacuolar membrane by means of pyrophosphate (PPi) hydrolysis (Maeshima, 2000; Rea and Poole, 2003) and acidify the vacuole (Lin et al., 2012). Other studies also found that transgenic plants expressing the H+-PPase from Arabidopsis or other species developed larger root systems and were more resistant to salt, drought stress, and phosphorous deficiency (Gaxiola et al., 2001; Park et al., 2005; Zhao et al., 2006; Li et al., 2008; Lv et al., 2008; Bao et al., 2009; Pasapula et al., 2011; Pei et al., 2012; Schilling et al., 2014). The GWAS study demonstrated that the natural variation in ZmVPP1, had the most significant impact to the drought resistance of young seedlings on a genome-wide scale. In the tolerant allele of ZmVPP1 containing three MYB recognition sequences (WAACCA, W: A or T), a 366-bp insertion was found as the causative variation that conferred the drought inducible expression of ZmVPP1 (Wang et al., 2016). Moreover, an additional natural variation in ZmNAC111 located on maize Chromosome 10, which encodes a NAM, ATAF, CUC2 (NAC) type transcription factor, was also found to contribute to the tolerance. ZmNAC111 belongs to a plant-specific transcription factor superfamily which is comprised of members that participate in various biological processes, including plant development, stress response, leaf senescence, and ion re-location (Uauy et al., 2006; Jeong et al., 2010; Nakashima et al., 2012; Liang et al., 2014). An 82-bp miniature inverted-repeat transposable element, forming a stem-loop structure, inserts in the gene promoter. It represses the gene expression most likely through RNA-directed DNA and histone methylation (Mao et al., 2015). Further transgenic data suggested that the biological function of ZmNAC111 and ZmVPP1 were different and are most likely functionally involved in the reduction of water loss under water deficit or the enhancement of water uptake and leaf photosynthesis (Mao et al., 2015; Wang et al., 2016). Notably, in Arabidopsis it is found that clade A protein phosphatase 2C genes (PP2C-As) negatively function in ABA signaling and plant drought response (Ma et al., 2009; Park et al., 2009). In order to find the natural variations of ZmPP2C-As that are directly associated with maize drought tolerance, a candidate gene association analysis was conducted. Among this gene family, ZmPP2C-A10 was found to be tightly associated with drought tolerance. Furthermore, a causal natural variation of this gene was identified, lacking an endoplasmic reticulum stress response element (ERSE) in the gene promoter due to a 14-bp deletion, could lead to increased plant drought tolerance (Xiang et al., 2017).
Furthermore, the adoption of global natural varieties which originate from different regions and periods of time, GWAS can infer the trend of evolution and/or the artificial selection of an important trait. Crops have been subjected to cultivation and extensive selection for grain productivity and quality to meet with human demand. For example, the ZmWAK locus, which conferred resistance to head smut, was lost from the teosinte wild ancestry during the domestication of maize (Zuo et al., 2015). Thus, a GWAS population consisting of the ancestral varieties of a species and those from adverse environments would be of great value to identify genetic loci contributing to the stress tolerance or help to understand the domestication history of an important trait by utilization of a superior allele (Hung et al., 2012; Yang et al., 2013; Zhou et al., 2015).
Network Analysis Offers A New Tool to Reveal the Mechanism for Drought Tolerance
Despite the fact that stress responsive genes are often studied in a one by one manner to undercover their roles in stress tolerance, coordinated expression of related genes has also been identified. For example, microarray and RNA gel blot analyses confirmed that a drought-inducible transcription factor (DREB2A) can regulate the expression of many stress-responsive genes (Sakuma et al., 2006). Findings such as this support the strategy for enhancing stress tolerance by manipulating a single regulatory gene and systematically altering the expression of a large number of genes involved in the drought-responsive network. Despite a large number of genes that are involved in the co-expression network, based on the integrative large scale data acquisition and analysis, systems biology offers a new and integrated tool to dissect the network. Sulpice et al. (2010) found that by profiling maximum catalytic activities of 37 enzymes from central metabolism, a matrix was generated to investigate species-wide connectivity among metabolites, enzymes, and biomass. The results showed that biomass can be predicted by two independent integrative metabolic biomarkers that could result in the preferential investment in photosynthetic machinery and optimization of carbon use. The genetic loci controlling differential gene expression in maize kernels were investigated through GWAS, which revealed a comprehensive gene regulatory network consisting of 31 zein and 16 key kernel genes (Fu J. J. et al., 2013). A comprehensive study of maize kernel metabolites, in relation to genetic variations and gene expressions, identified biomarkers associated with kernel weight which may facilitate the genetic improvement of maize (Wen et al., 2014). Undoubtedly, stress resistance consists of a combination of dynamic networks that contain many correlated genes. The potential for the expression of a specific gene to enhance stress tolerance or not depends on multiple factors such as its position, branch point, direction, and redundant reactions in the network. Recently, network analysis has provided a framework for understanding and modulating plant responses to salt stress and abscisic acid application, through chromatin immuno-precipitation sequencing. Genome-wide targets of 21 ABA-related transcription factors were identified and a comprehensive regulatory network was constructed. Moreover, a new family of transcriptional regulators was discovered to be functionally involved in ABA and salt responsiveness, and shown to modulate plant tolerance to osmotic stress in Arabidopsis (Song et al., 2016). Taken together, knowledge derived from network analyses will provide us with new strategies to cope with the ongoing challenges of drought stress.
Stacking or Combining Favorable Alleles to Improve Drought Tolerance
The key to meet the ever-growing demand for crops yield, and to relieve the threat on crop productivity imposed by environmental stress, is to cultivate high-yield and high-resistance varieties. As a result, it is very important that breeders can have access to tools and technologies that will enable them to improve their selection efficiency. Molecular breeding, which includes MAS, genome editing, genome-wide selection and transgenics, is based on the knowledge of QTL/gene mapping and cloning and is considered as an innovative approach for precision breeding in the 21st century to meet growing demand for food sources (Collard and Mackill, 2008; Voytas and Gao, 2014; Gao, 2015). A favorable allele from wild soybean was used to increase seed protein content in soybean by MAS (Sebolt et al., 2000). Merchuk-Ovnat et al. (2016) generated a near isogenic line (NIL-7A-B-2) by introgressing a drought-related QTL on chromosome 7A from wild emmer wheat into a cultivated wheat variety; resulting in enhanced grain yield and biomass production across environments. Notably, by using the (CRISPR)-Cas9 genome editing technology (Cong et al., 2013; Mali et al., 2013), targeted mutations were successfully introduced into the three homoeoalleles encoding MILDEW-RESISTANCE LOCUS (MLO) proteins; which conferred heritable resistance to powdery mildew in hexaploid bread wheat (Wang et al., 2014). The reported natural allele variation of DRO1 (Uga et al., 2013), ZmDREB2.7 (Liu et al., 2013), ZmNAC111 (Mao et al., 2015), and ZmVPP1 (Wang et al., 2016) shed novel insight into how natural variation factors into improving crop drought tolerance or resistance. Moreover, the cumulative effects of multiple quantitative resistance loci could be exploited to produce high tolerance (Miedaner et al., 2006; Richardson et al., 2006). Thus, it is plausible that several favorable alleles, not restricted to ZmDREB2.7, ZmNAC111, and ZmVPP1, can be stacked to improve drought tolerance in maize and other crops by marker-assisted selection or genome editing technology.
Perspectives
Global climate change threatens crop production worldwide. Unexpected changes in weather patterns, such as high temperature and drought have dramatically affected crop yield which in turn could result in food and societal crises. It is plausible that molecular breeding strategies may speed up the traditional breeding processes to increase stress resistance in cultivars; enabling them to better cope with the changing environment. Along with the development of high-throughput DNA sequencing technology, whole genome covered markers can be produced more cost-effectively with unprecedented increases of accuracy. Meanwhile, the advances of statistical analyses for quantitative genetics provide new methodologies to dissect the genetic basis of complex traits. It can be anticipated that systematic network analyses, consisting of genomics, transcriptomics, proteomics, metabolomics, and phenomics, will provide integrative information for increase our understanding of the balance between stress response and grain yield and quality. Undoubtedly, this acquired knowledge will be of great value to put us in a better position to enable the precise molecular design of new cultivars with desired stress resistance and yields.
Author Contributions
HW wrote the manuscript. FQ designed and revised the manuscript.
Funding
This work is supported by National Key Research and Development Plan of China (2016YFD0100605) and the National Natural Science Foundation of China (31625022).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Drs. Shengxue Liu and Xianglan Wang for their discussions and inputs to this work.
References
Abecasis, G. R., Cardon, L. R., and Cookson, W. O. (2000). A general test of association for quantitative traits in nuclear families. Am. J. Hum. Genet. 66, 279–292. doi: 10.1086/302698
Adams, M. D., Kelley, J. M., Gocayne, J. D., Dubnick, M., Polymeropoulos, M. H., Xiao, H., et al. (1991). Complementary DNA sequencing: expressed sequence tags and human genome project. Science 252, 1651–1656. doi: 10.1126/science.2047873
Agrama, H. A. S., and Moussa, M. E. (1996). Mapping QTLs in breeding for drought tolerance in maize (Zea mays L.). Euphytica 91, 89–97. doi: 10.1007/bf00035278
Almeida, G. D., Makumbi, D., Magorokosho, C., Nair, S., Borem, A., Ribaut, J. M., et al. (2013). QTL mapping in three tropical maize populations reveals a set of constitutive and adaptive genomic regions for drought tolerance. Theor. Appl. Genet. 126, 583–600. doi: 10.1007/s00122-012-2003-7
Altshuler, D., Pollara, V. J., Cowles, C. R., Van, Etten WJ, Baldwin, J., Linton, L., et al. (2000). An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 407, 513–516.
And, J. I., and Bartels, D. (2003). The molecular basis of dehydration tolerance in plants. Annu. Rev. Plant Biol. 47, 377–403. doi: 10.1146/annurev.arplant.47.1.377
Aulchenko, Y. S., Ripke, S., Isaacs, A., and van Duijn, C. M. (2007). GenABEL: an R library for genome-wide association analysis. Bioinformatics 23, 1294–1296. doi: 10.1093/bioinformatics/btm108
Balding, D. J. (2006). A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 7, 781–791. doi: 10.1038/nrg1916
Bao, A. K., Wang, S. M., Wu, G. Q., Xi, J. J., Zhang, J. L., and Wang, C. M. (2009). Overexpression of the Arabidopsis H+-PPase enhanced resistance to salt and drought stress in transgenic alfalfa (Medicago sativa L.). Plant Sci. 176, 232–240. doi: 10.1016/j.plantsci.2008.10.009
Barakat, M. N., Saleh, M. S., Al-Doss, A. A., Moustafa, K. A., Elshafei, A. A., Zakri, A. M., et al. (2015). Mapping of QTLs associated with abscisic acid and water stress in wheat. Biol. Plant. 59, 291–297. doi: 10.1007/s10535-015-0499-9
Bartoli, C. G., Simontacchi, M., Tambussi, E., Beltrano, J., Montaldi, E., and Puntarulo, S. (1999). Drought and watering-dependent oxidative stress: effect on antioxidant content in Triticum aestivum L. leaves. J. Exp. Bot. 50, 375–385. doi: 10.1093/jxb/50.332.375
Battisti, D. S., and Naylor, R. L. (2009). Historical warnings of future food insecurity with unprecedented seasonal heat. Science 323, 240–244. doi: 10.1126/science.1164363
Beavis, W. D., Smith, O. S., Grant, D., and Fincher, R. R. (1994). Identification of quantitative trait loci using a small sample of topcrossed and F4 progeny from maize. Crop Sci. 34, 882–896.
Bentley, D. R., Balasubramanian, S., Swerdlow, H. P., Smith, G. P., Milton, J., Brown, C. G., et al. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59. doi: 10.1038/nature07517
Botstein, D., White, R. L., Skolnick, M., and Davis, R. W. (1980). Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32, 314–331.
Boyer, J. S. (1982). Plant productivity and environment. Science 218, 443–448. doi: 10.1126/science.218.4571.443
Boyer, J. S., Byrne, P., Cassman, K. G., Cooper, M., Delmer, D., Greene, T., et al. (2013). The U.S. drought of 2012 in perspective: a call to action. Glob. Food Sec. 2, 139–143. doi: 10.1016/j.gfs.2013.08.002
Bray, E. A. (1993). Molecular responses to water deficit. Plant Physiol. 103, 1035–1040. doi: 10.1104/pp.103.4.1035
Buckler, E. S., Holland, J. B., Bradbury, P. J., Acharya, C. B., Brown, P. J., Browne, C., et al. (2009). The genetic architecture of maize flowering time. Science 325, 714–718. doi: 10.1126/science.1174276
Cavanagh, C., Morell, M., Mackay, I., and Powell, W. (2008). From mutations to MAGIC: resources for gene discovery, validation and delivery in crop plants. Curr. Opin. Plant Biol. 11, 215–221. doi: 10.1016/j.pbi.2008.01.002
Chaves, M. M., Maroco, J. P., and Pereira, J. S. (2003). Understanding plant response to drought-from genes to the whole plant. Funct. Plant Biol. 30, 239–264. doi: 10.1071/FP02076
Close, T. J. (1996). Dehydrins: emergence of a biochemical role of a family of plant dehydration proteins. Physiol. Plant. 97, 795–803. doi: 10.1111/j.1399-3054.1996.tb00546.x
Cockram, J., White, J., Zuluaga, D. L., Smith, D. J., Comadran, J., Macaulay, M., et al. (2010). Genome-wide association mapping to candidate polymorphism resolution in the unsequenced barley genome. Proc. Natl. Acad. Sci. U.S.A. 107, 21611–21616. doi: 10.1073/pnas.1010179107
Collard, B. C. Y., and Mackill, D. J. (2008). Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B 363, 557–572. doi: 10.1098/rstb.2007.2170
Collins, N. C., Tardieu, F., and Tuberosa, R. (2008). Quantitative trait loci and crop performance under abiotic stress: where do we stand? Plant Physiol. 147, 469–486. doi: 10.1104/pp.108.118117
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Catchen, J. M., and Blaxter, M. L. (2011). Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 12, 499–510. doi: 10.1038/nrg3012
Deschamps, S., Llaca, V., and May, G. D. (2012). Genotyping-by-sequencing in plants. Biology (Basel) 1, 460–483. doi: 10.3390/biology1030460
Edwards, D., and Batley, J. (2010). Plant genome sequencing: applications for crop improvement. Plant Biotechnol. J. 8, 2–9. doi: 10.1111/j.1467-7652.2009.00459.x
Fu, J. J., Cheng, Y. B., Linghu, J. J., Yang, X. H., Kang, L., Zhang, Z. X., et al. (2013). RNA sequencing reveals the complex regulatory network in the maize kernel. Nat. Commun. 4:2832. doi: 10.1038/ncomms3832
Fu, Y. B., Cheng, B. F., and Peterson, G. W. (2013). Genetic diversity analysis of yellow mustard (Sinapis alba L.) germplasm based on genotyping by sequencing. Genet. Resour. Crop Evol. 61, 579–594. doi: 10.1007/s10722-013-0058-1
Fu, Y. B., and Peterson, G. W. (2011). Genetic diversity analysis with 454 pyrosequencing and genomic reduction confirmed the eastern and western division in the cultivated barley gene pool. Plant Genome J. 4, 226–237. doi: 10.3835/plantgenome2011.08.0022
Gao, C. (2015). Genome editing in crops: from bench to field. Natl. Sci. Rev. 2, 13–15. doi: 10.1093/nsr/nwu054
Gaxiola, R. A., Li, J., Undurraga, S., Dang, L. M., Allen, G. J., Alper, S. L., et al. (2001). Drought- and salt-tolerant plants result from overexpression of the AVP1 H+-pump. Proc. Natl. Acad. Sci. U.S.A. 98, 11444–11449. doi: 10.1073/pnas.191389398
Geber, M. A., and Dawson, T. E. (1990). Genetic variation in and covariation between leaf gas exchange, morphology and development in Polygonum arenastrum, an annual plant. Oecologia 85, 153–158. doi: 10.1007/BF00319396
Gill, S. S., and Tuteja, N. (2010). Polyamines and abiotic stress tolerance in plants. Plant Signal. Behav. 5, 26–33. doi: 10.4161/psb.5.1.10291
Gore, M. A., Chia, J. M., Elshire, R. J., Sun, Q., Ersoz, E. S., Hurwitz, B. L., et al. (2009). A first-generation haplotype map of maize. Science 326, 1115–1117. doi: 10.1126/science.1177837
Gowda, V. R. P., Henry, A., Yamauchi, A., Shashidhar, H. E., and Serraj, R. (2011). Root biology and genetic improvement for drought avoidance in rice. Field Crops Res. 122, 1–13. doi: 10.1016/j.fcr.2011.03.001
Guingo, E., Hebert, Y., and Charcosset, A. (1998). Genetic analysis of root traits in maize. Agronomie 18, 225–235. doi: 10.1051/agro:19980305
Hao, Z., Li, X., Liu, X., Xie, C., Li, M., Zhang, D., et al. (2009). Meta-analysis of constitutive and adaptive QTL for drought tolerance in maize. Euphytica 174, 165–177. doi: 10.1007/s10681-009-0091-5
Hao, Z. F., Li, X. H., Xie, C. X., Li, M. S., Zhang, D. G., Bai, L., et al. (2008). Two consensus quantitative trait loci clusters controlling anthesis-silking interval, ear setting and grain yield might be related with drought tolerance in maize. Ann. Appl. Biol. 153, 73–83. doi: 10.1111/j.1744-7348.2008.00239.x
Henderson, C. R. (1975). Comparison of alternative sire evaluation methods. J. Anim. Sci. 41, 760–770. doi: 10.2527/jas1975.413760x
Hoffmann, A. A., and Merila, J. (1999). Heritable variation and evolution under favourable and unfavourable conditions. Trends Ecol. Evol. 14, 96–101. doi: 10.1016/S0169-5347(99)01595-5
Huang, X., Paulo, M. J., Boer, M., Effgen, S., Keizer, P., Koornneef, M., et al. (2011). Analysis of natural allelic variation in Arabidopsis using a multiparent recombinant inbred line population. Proc. Natl. Acad. Sci. U.S.A. 108, 4488–4493. doi: 10.1073/pnas.1100465108
Huang, X., Wei, X., Sang, T., Zhao, Q., Feng, Q., Zhao, Y., et al. (2010). Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 42, 961–967. doi: 10.1038/ng.695
Huang, X., Yang, S., Gong, J., Zhao, Q., Feng, Q., Zhan, Q., et al. (2016). Genomic architecture of heterosis for yield traits in rice. Nature 537, 629–633. doi: 10.1038/nature19760
Hung, H. Y., Shannon, L. M., Tian, F., Bradbury, P. J., Chen, C., Flint-Garcia, S. A., et al. (2012). ZmCCT and the genetic basis of day-length adaptation underlying the postdomestication spread of maize. Proc. Natl. Acad. Sci. U.S.A. 109, E1913–E1921. doi: 10.1073/pnas.1203189109
Iehisa, J. C. M., Matsuura, T., Mori, I. C., Yokota, H., Kobayashi, F., and Takumi, S. (2014). Identification of quantitative trait loci for abscisic acid responsiveness in the D-genome of hexaploid wheat. J. Plant Physiol. 171, 830–841. doi: 10.1016/j.jplph.2014.02.003
Izanloo, A., Condon, A. G., Langridge, P., Tester, M., and Schnurbusch, T. (2008). Different mechanisms of adaptation to cyclic water stress in two South Australian bread wheat cultivars. J. Exp. Bot. 59, 3327–3346. doi: 10.1093/jxb/ern199
Jackson, R. B., Sperry, J. S., and Dawson, T. E. (2000). Root water uptake and transport: using physiological processes in global predictions. Trends Plant Sci. 5, 482–488. doi: 10.1016/S1360-1385(00)01766-0
Jeong, J. S., Kim, Y. S., Baek, K. H., Jung, H., Ha, S., Choi, Y. D., et al. (2010). Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiol. 153, 185–197. doi: 10.1104/pp.110.154773
Kang, H. M., Sul, J. H., Service, S. K., Zaitlen, N. A., Kong, S. Y., Freimer, N. B., et al. (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354. doi: 10.1038/ng.548
Kang, H. M., Zaitlen, N. A., Wade, C. M., Kirby, A., Heckerman, D., Daly, M. J., et al. (2008). Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723. doi: 10.1534/genetics.107.080101
Kang, Y., Sakiroglu, M., Krom, N., Stantongeddes, J., Wang, M., Lee, Y., et al. (2015). Genome-wide association of drought-related and biomass traits with HapMap SNPs in Medicago truncatula. Plant Cell Environ. 38, 1997–2011. doi: 10.1111/pce.12520
Konieczny, A., and Ausubel, F. M. (1993). A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J. 4, 403–410. doi: 10.1046/j.1365-313X.1993.04020403.x
Lam, H. M., Xu, X., Liu, X., Chen, W., Yang, G., Wong, F. L., et al. (2010). Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–1059. doi: 10.1038/ng.715
Landi, P., Sanguineti, M. C., Salvi, S., Giuliani, S., Bellotti, M., Maccaferri, M., et al. (2005). Validation and characterization of a major QTL affecting leaf ABA concentration in maize. Mol. Breed. 15, 291–303. doi: 10.1007/s11032-004-7604-7
Lebreton, C., Lazicjancic, V., Steed, A., Pekic, S., and Quarrie, S. A. (1995). Identification of QTL for drought responses in maize and their use in testing causal relationships between traits. J. Exp. Bot. 46, 853–865. doi: 10.1093/jxb/46.7.853
Li, B., Wei, A., Song, C., Li, N., and Zhang, J. (2008). Heterologous expression of the TsVP gene improves the drought resistance of maize. Plant Biotechnol. J. 6, 146–159. doi: 10.1111/j.1467-7652.2007.00301.x
Li, H., Peng, Z., Yang, X., Wang, W., Fu, J., Wang, J., et al. (2013). Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43–50. doi: 10.1038/ng.2484
Li, J., Yang, H., Peer, W. A., Richter, G., Blakeslee, J., Bandyopadhyay, A., et al. (2005). Arabidopsis H+-PPase AVP1 regulates auxin-mediated organ development. Science 310, 121–125. doi: 10.1126/science.1115711
Liang, C., Wang, Y., Zhu, Y., Tang, J., Hu, B., Liu, L., et al. (2014). OsNAP connects abscisic acid and leaf senescence by fine-tuning abscisic acid biosynthesis and directly targeting senescence-associated genes in rice. Proc. Natl. Acad. Sci. U.S.A. 111, 10013–10018. doi: 10.1073/pnas.1321568111
Lin, S. M., Tsai, J. Y., Hsiao, C. D., Huang, Y. T., Chiu, C. L., Liu, M. H., et al. (2012). Crystal structure of a membrane-embedded H+-translocating pyrophosphatase. Nature 484, 399–403. doi: 10.1038/nature10963
Lippert, C., Listgarten, J., Liu, Y., Kadie, C. M., Davidson, R. I., and Heckerman, D. (2011). FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–835. doi: 10.1038/nmeth.1681
Litt, M., and Luty, J. A. (1989). A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am. J. Hum. Genet 44, 397–401.
Liu, S., Wang, X., Wang, H., Xin, H., Yang, X., Yan, J., et al. (2013). Genome-wide analysis of ZmDREB genes and their association with natural variation in drought tolerance at seedling stage of Zea mays L. PLoS Genet. 9:e1003790. doi: 10.1371/journal.pgen.1003790
Lu, F., Lipka, A. E., Glaubitz, J., Elshire, R., Cherney, J. H., Casler, M. D., et al. (2013). Switchgrass genomic diversity, ploidy, and evolution: novel insights from a network-based SNP discovery protocol. PLoS Genet. 9:e1003215. doi: 10.1371/journal.pgen.1003215
Lu, G., Tang, J., Yan, J., Ma, X., Li, J., Chen, S., et al. (2006). Quantitative trait loci mapping of maize yield and its components under different water treatments at flowering time. J. Integr. Plant Biol. 48, 1233–1243. doi: 10.1111/j.1744-7909.2006.00289.x
Lu, Y., Zhang, S., Shah, T., Xie, C., Hao, Z., Li, X., et al. (2010). Joint linkage-linkage disequilibrium mapping is a powerful approach to detecting quantitative trait loci underlying drought tolerance in maize. Proc. Natl. Acad. Sci. U.S.A. 107, 19585–19590. doi: 10.1073/pnas.1006105107
Lv, S., Zhang, K., Gao, Q., Lian, L., Song, Y., and Zhang, J. (2008). Overexpression of an H+-PPase gene from Thellungiella halophila in cotton enhances salt tolerance and improves growth and photosynthetic performance. Plant Cell Physiol. 49, 1150–1164. doi: 10.1093/pcp/pcn090
Ma, Y., Szostkiewicz, I., Korte, A., Moes, D., Yang, Y., Christmann, A., et al. (2009). Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 324, 1064–1068. doi: 10.1126/science.1172408
Mace, E. S., Singh, V., Van Oosterom, E. J., Hammer, G. L., Hunt, C. H., and Jordan, D. R. (2012). QTL for nodal root angle in sorghum (Sorghum bicolor L. Moench) co-locate with QTL for traits associated with drought adaptation. Theor. Appl. Genet. 124, 97–109. doi: 10.1007/s00122-011-1690-9
Maeshima, M. (2000). Vacuolar H+-pyrophosphatase. Biochim. Biophys. Acta 1465, 37–51. doi: 10.1016/S0005-2736(00)00130-9
Maherali, H., Caruso, C. M., Sherrard, M. E., and Latta, R. G. (2010). Adaptive value and costs of physiological plasticity to soil moisture limitation in recombinant inbred lines of Avena barbata. Am. Nat. 175, 211–224. doi: 10.1086/649598
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. doi: 10.1126/science.1232033
Mao, H., Wang, H., Liu, S., Li, Z., Yang, X., Yan, J., et al. (2015). A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat. Commun. 6:8326. doi: 10.1038/ncomms9326
McNally, K. L., Childs, K. L., Bohnert, R., Davidson, R. M., Zhao, K., Ulat, V. J., et al. (2009). Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl. Acad. Sci. U.S.A. 106, 12273–12278. doi: 10.1073/pnas.0900992106
Merchuk-Ovnat, L., Fahima, T., Krugman, T., and Saranga, Y. (2016). Ancestral QTL alleles from wild emmer wheat improve grain yield, biomass and photosynthesis across enviroinments in modern wheat. Plant Sci. 251, 23–34. doi: 10.1016/j.plantsci.2016.05.003
Messmer, R., Fracheboud, Y., Banziger, M., Stamp, P., and Ribaut, J. (2011). Drought stress and tropical maize: QTLs for leaf greenness, plant senescence, and root capacitance. Field Crops Res. 124, 93–103. doi: 10.1016/j.fcr.2011.06.010
Messmer, R., Fracheboud, Y., Banziger, M., Vargas, M., Stamp, P., and Ribaut, J. M. (2009). Drought stress and tropical maize: QTL-by-environment interactions and stability of QTLs across environments for yield components and secondary traits. Theor. Appl. Genet. 119, 913–930. doi: 10.1007/s00122-009-1099-x
Meyer, R. C., Steinfath, M., Lisec, J., Becher, M., Wituckawall, H., Torjek, O., et al. (2007). The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 104, 4759–4764. doi: 10.1073/pnas.0609709104
Miedaner, T., Wilde, F., Steiner, B., Buerstmayr, H., Korzun, V., and Ebmeyer, E. (2006). Stacking quantitative trait loci (QTL) for Fusarium head blight resistance from non-adapted sources in an European elite spring wheat background and assessing their effects on deoxynivalenol (DON) content and disease severity. Theor. Appl. Genet. 112, 562–569. doi: 10.1007/s00122-005-0163-4
Myles, S., Peiffer, J., Brown, P. J., Ersoz, E. S., Zhang, Z., Costich, D. E., et al. (2009). Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21, 2194–2202. doi: 10.1105/tpc.109.068437
Nakashima, K., Takasaki, H., Mizoi, J., Shinozaki, K., and Yamaguchishinozaki, K. (2012). NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 1819, 97–103. doi: 10.1016/j.bbagrm.2011.10.005
Narum, S. R., Buerkle, C. A., Davey, J. W., Miller, M. R., and Hohenlohe, P. A. (2013). Genotyping-by-sequencing in ecological and conservation genomics. Mol. Ecol. 22, 2841–2847. doi: 10.1111/mec.12350
Osakabe, Y., Yamaguchi-Shinozaki, K., Shinozaki, K., and Tran, L. S. (2014). ABA control of plant macroelement membrane transport systems in response to water deficit and high salinity. New Phytol. 202, 35–49. doi: 10.1111/nph.12613
Pan, Q., Li, L., Yang, X., Tong, H., Xu, S., Li, Z., et al. (2016). Genome-wide recombination dynamics are associated with phenotypic variation in maize. New Phytol. 210, 1083–1094. doi: 10.1111/nph.13810
Paran, I., and Michelmore, R. W. (1993). Development of reliable PCR-based markers linked to downy mildew resistance genes in lettuce. Theor. Appl. Genet. 85, 985–993. doi: 10.1007/bf00215038
Park, S., Li, J., Pittman, J. K., Berkowitz, G. A., Yang, H., Undurraga, S., et al. (2005). Up-regulation of a H+-pyrophosphatase (H+-PPase) as a strategy to engineer drought-resistant crop plants. Proc. Natl. Acad. Sci. U.S.A. 102, 18830–18835. doi: 10.1073/pnas.0509512102
Park, S. Y., Fung, P., Nishimura, N., Jensen, D. R., Fujii, H., Zhao, Y., et al. (2009). Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 324, 1068–1071. doi: 10.1126/science.1173041
Pasapula, V., Shen, G., Kuppu, S., Paez-Valencia, J., Mendoza, M., Hou, P., et al. (2011). Expression of an Arabidopsis vacuolar H+-pyrophosphatase gene (AVP1) in cotton improves drought- and salt tolerance and increases fibre yield in the field conditions. Plant Biotechnol. J. 9, 88–99. doi: 10.1111/j.1467-7652.2010.00535.x
Pei, L., Wang, J., Li, K., Li, Y., Li, B., Gao, F., et al. (2012). Overexpression of Thellungiella halophila H+-pyrophosphatase gene improves low phosphate tolerance in maize. PLoS ONE 7:e43501. doi: 10.1371/journal.pone.0043501
Poland, J. A., and Rife, T. W. (2012). Genotyping-by-sequencing for plant breeding and genetics. Plant Genome J. 5:92. doi: 10.3835/plantgenome2012.05.0005
Pollak, E. J., and Quaas, R. L. (1983). Definition of group effects in sire evaluation models. J. Dairy Sci. 66, 1503–1509. doi: 10.3168/jds.S0022-0302(83)81965-1
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1111/j.1471-8286.2007.01758.x
Qin, F., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2011). Achievements and challenges in understanding plant abiotic stress responses and tolerance. Plant Cell Physiol. 52, 1569–1582. doi: 10.1093/pcp/pcr106
Quaas, R. L., and Pollak, E. J. (1980). Mixed model methodology for farm and ranch beef cattle testing programs. J. Anim. Sci. 51, 1277–1287. doi: 10.1038/nrg1916
Rafalski, A. (2002). Applications of single nucleotide polymorphisms in crop genetics. Curr. Opin. Plant Biol. 5, 94–100. doi: 10.1016/S1369-5266(02)00240-6
Rahman, H., Pekic, S., Lazicjancic, V., Quarrie, S., Shah, S. M. A., Pervez, A., et al. (2011). Molecular mapping of quantitative trait loci for drought tolerance in maize plants. Gen. Mol. Res. 10, 889–901. doi: 10.4238/vol10-2gmr1139
Rea, A. P., and Poole, R. J. (2003). Vacuolar H+-translocating pyrophosphatase. Annu. Rev. Plant Biol. 44, 157–180. doi: 10.1146/annurev.pp.44.060193.001105
Ribaut, J. M., Hoisington, D. A., Deutsch, J., Jiang, C., and Gonzalezdeleon, D. (1996). Identification of quantitative trait loci under drought conditions in tropical maize. 1. Flowering parameters and the anthesis.silking interval. Theor. Appl. Genet. 92, 905–914. doi: 10.1007/BF00221905
Ribaut, J.-M., Jiang, C., Gonzalez-de-Leon, D., Edmeades, G. O., and Hoisington, D. A. (1997). Identification of quantitative trait loci under drought conditions in tropical maize.2. Yield components and marker-assisted selection strategies. Theor. Appl. Genet. 94, 887–896. doi: 10.1007/s001220050492
Richardson, K., Vales, M. I., Kling, J. G., Mundt, C. C., and Hayes, P. M. (2006). Pyramiding and dissecting disease resistance QTL to barley stripe rust. Theor. Appl. Genet. 113, 485–495. doi: 10.1007/s00122-006-0314-2
Riedelsheimer, C., Lisec, J., Czedik-Eysenberg, A., Sulpice, R., Flis, A., Grieder, C., et al. (2012). Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl. Acad. Sci. U.S.A. 109, 8872–8877. doi: 10.1073/pnas.1120813109
Romay, M. C., Millard, M. J., Glaubitz, J. C., Peiffer, J. A., Swarts, K. L., Casstevens, T. M., et al. (2013). Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol. 14:R55. doi: 10.1186/gb-2013-14-6-r55
Rothberg, J. M., Hinz, W., Rearick, T. M., Schultz, J., Mileski, W., Davey, M., et al. (2011). An integrated semiconductor device enabling non-optical genome sequencing. Nature 475, 348–352. doi: 10.1038/nature10242
Sakuma, Y., Maruyama, K., Osakabe, Y., Qin, F., Seki, M., Shinozaki, K., et al. (2006). Functional analysis of an Arabidopsis transcription factor, DREB2A, involved in drought-responsive gene expression. Plant Cell 18, 1292–1309. doi: 10.1105/tpc.105.035881
Sanguineti, M. C., Tuberosa, R., Landi, P., Salvi, S., Maccaferri, M., Casarini, E., et al. (1999). QTL analysis of drought-related traits and grain yield in relation to genetic variation for leaf abscisic acid concentration in field-grown maize. J. Exp. Bot. 50, 1289–1297. doi: 10.1093/jxb/50.337.1289
Schilling, R. K., Marschner, P., Shavrukov, Y., Berger, B., Tester, M., Roy, S. J., et al. (2014). Expression of the Arabidopsis vacuolar H+-pyrophosphatase gene (AVP1) improves the shoot biomass of transgenic barley and increases grain yield in a saline field. Plant Biotechnol. J. 12, 378–386. doi: 10.1111/pbi.12145
Schulze, E. D. (1986). Carbon dioxide and water vapor exchange in response to drought in the atmosphere and the soil. Annu. Rev. Plant Physiol. 37, 247–274. doi: 10.1146/annurev.pp.37.060186.001335
Sebastian, J., Yee, M. C., Goudinho Viana, W., Rellan-Alvarez, R., Feldman, M., Priest, H. D., et al. (2016). Grasses suppress shoot-borne roots to conserve water during drought. Proc. Natl. Acad. Sci. U.S.A. 113, 8861–8866. doi: 10.1073/pnas.1604021113
Sebolt, A., Shoemaker, R. C., and Diers, B. W. (2000). Analysis of a quantitative trait locus allele from wild soybean that increases seed protein concentration in soybean. Crop Sci. 40, 1438–1444. doi: 10.2135/cropsci2000.4051438x
Semagn, K., Beyene, Y., Warburton, M. L., Tarekegne, A., Mugo, S., Meisel, B., et al. (2013). Meta-analyses of QTL for grain yield and anthesis silking interval in 18 maize populations evaluated under water-stressed and well-watered environments. BMC Genomics 14:313. doi: 10.1186/1471-2164-14-313
Setter, T. L., Yan, J., Warburton, M., Ribaut, J. M., Xu, Y., Sawkins, M., et al. (2011). Genetic association mapping identifies single nucleotide polymorphisms in genes that affect abscisic acid levels in maize floral tissues during drought. J. Exp. Bot. 62, 701–716. doi: 10.1093/jxb/erq308
Shendure, J., and Ji, H. (2008). Next-generation DNA sequencing. Nat. Biotechnol. 26, 1135–1145. doi: 10.1038/nbt1486
Sherrard, M. E., Maherali, H., and Latta, R. G. (2009). Water stress alters the genetic architecture of functional traits associated with drought adaptation in Avena barbata. Evolution 63, 702–715. doi: 10.1111/j.1558-5646.2008.00580.x
Si, L., Chen, J., Huang, X., Gong, H., Luo, J., Hou, Q., et al. (2016). OsSPL13 controls grain size in cultivated rice. Nat. Genet. 48, 447–456. doi: 10.1038/ng.3518
Song, L., Huang, S. C., Wise, A., Castanon, R., Nery, J. R., Chen, H., et al. (2016). A transcription factor hierarchy defines an environmental stress response network. Science 354:aag1550 doi: 10.1126/science.aag1550
Sukumaran, S., Dreisigacker, S., Lopes, M. S., Chavez, P., and Reynolds, M. P. (2014). Genome-wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theor. Appl. Genet. 128, 353–363. doi: 10.1007/s00122-014-2435-3
Sulpice, R., Trenkamp, S., Steinfath, M., Usadel, B., Gibon, Y., Witucka-Wall, H., et al. (2010). Network analysis of enzyme activities and metabolite levels and their relationship to biomass in a large panel of Arabidopsis accessions. Plant Cell 22, 2872–2893. doi: 10.1105/tpc.110.076653
Tapp, I., Malmberg, L., Rennel, E., Wik, M., and Syvanen, A. C. (2000). Homogeneous scoring of single-nucleotide polymorphisms: comparison of the 5′-nuclease TaqMan assay and Molecular Beacon probes. Biotechniques 28, 732–738.
Tester, M., and Langridge, P. (2010). Breeding technologies to increase crop production in a changing world. Science 327, 818–822. doi: 10.1126/science.1183700
Thirunavukkarasu, N., Hossain, F., Arora, K., Sharma, R., Shiriga, K., Mittal, S., et al. (2014). Functional mechanisms of drought tolerance in subtropical maize (Zea mays L.) identified using genome-wide association mapping. BMC Genomics 15:1182. doi: 10.1186/1471-2164-15-1182
Thudi, M., Li, Y., Jackson, S. A., May, G. D., and Varshney, R. K. (2012). Current state-of-art of sequencing technologies for plant genomics research. Brief Funct. Genomics 11, 3–11. doi: 10.1093/bfgp/elr045
Thudi, M., Upadhyaya, H. D., Rathore, A., Gaur, P. M., Krishnamurthy, L., Roorkiwal, M., et al. (2014). Genetic dissection of drought and heat tolerance in chickpea through genome-wide and candidate gene-based association mapping approaches. PLoS ONE 9:e96758. doi: 10.1371/journal.pone.0096758
Tian, F., Bradbury, P. J., Brown, P. J., Hung, H., Sun, Q., Flint-Garcia, S., et al. (2011). Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 43, 159–162. doi: 10.1038/ng.746
Tilman, D., Cassman, K. G., Matson, P. A., Naylor, R., and Polasky, S. (2002). Agricultural sustainability and intensive production practices. Nature 418, 671–677. doi: 10.1038/nature01014
Tuberosa, R., and Salvi, S. (2002). “QTL for agronomic traits in maize production,” in Handbook of Maize: Its Biology, eds J. L. Bennetzen and S. C. Hake (Dordrecht: Springer), 501–542. doi: 10.1007/978-0-387-79418-1_26
Tuberosa, R., Salvi, S., Sanguineti, M. C., Landi, P., Maccaferri, M., and Conti, S. (2002a). Mapping QTLs regulating morpho-physiological traits and yield: case studies, shortcomings and perspectives in drought-stressed maize. Ann. Bot. 89, 941–963. doi: 10.1093/aob/mcf134
Tuberosa, R., Sanguineti, M. C., Landi, P., Giuliani, M. M., Salvi, S., and Conti, S. (2002b). Identification of QTLs for root characteristics in maize grown in hydroponics and analysis of their overlap with QTLs for grain yield in the field at two water regimes. Plant Mol. Biol. 48, 697–712. doi: 10.1023/A:1014897607670
Tuberosa, R., Sanguineti, M. C., Landi, P., Salvi, S., Casarini, E., and Conti, S. (1998). RFLP mapping of quantitative trait loci controlling abscisic acid concentration in leaves of drought-stressed maize (Zea mays L.). Theor. Appl. Genet. 97, 744–755. doi: 10.1007/s001220050951
Uauy, C., Distelfeld, A., Fahima, T., Blechl, A., and Dubcovsky, J. (2006). A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 314, 1298–1301.
Uga, Y., Sugimoto, K., Ogawa, S., Rane, J., Ishitani, M., Hara, N., et al. (2013). Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat. Genet. 45, 1097–1102. doi: 10.1038/ng.2725
Verslues, P. E., Lasky, J. R., Juenger, T. E., Liu, T. W., and Kumar, M. N. (2014). Genome-wide association mapping combined with reverse genetics identifies new effectors of low water potential-induced proline accumulation in Arabidopsis. Plant Physiol. 164, 144–159. doi: 10.1104/pp.113.224014
Vikram, P., Swamy, B. P. M., Dixit, S., Ahmed, H., Cruz, M. T. S., Singh, A. K., et al. (2012). Bulk segregant analysis: “An effective approach for mapping consistent-effect drought grain yield QTLs in rice”. Field Crops Res. 134, 185–192. doi: 10.1016/j.fcr.2012.05.012
Vos, P., Hogers, R., Bleeker, M., Reijans, M., van de Lee, T., Hornes, M., et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 23, 4407–4414. doi: 10.1093/nar/23.21.4407
Voytas, D. F., and Gao, C. (2014). Precision genome engineering and agriculture: opportunities and regulatory challenges. PLoS Biol. 12:e1001877. doi: 10.1371/journal.pbio.1001877
Wallace, J. G., Zhang, X., Beyene, Y., Semagn, K., Olsen, M., Prasanna, B. M., et al. (2016). Genome-wide association for plant height and flowering time across 15 tropical maize populations under managed drought stress and well-watered conditions in sub-saharan africa. Crop Sci. 56, 2365–2378. doi: 10.2135/cropsci2015.10.0632
Wang, D. G., Fan, J. B., Siao, C. J., Berno, A., Young, P., Sapolsky, R., et al. (1998). Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 280, 1077–1082. doi: 10.1126/science.280.5366.1077
Wang, X., Wang, H., Liu, S., Ferjani, A., Li, J., Yan, J., et al. (2016). Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 48, 1233–1241. doi: 10.1038/ng.3636
Wang, Y., Cheng, X., Shan, Q., Zhang, Y., Liu, J., Gao, C., et al. (2014). Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat. Biotechnol. 32, 947–951. doi: 10.1038/nbt.2969
Wehner, G. G., Balko, C. C., Enders, M. M., Humbeck, K. K., and Ordon, F. F. (2015). Identification of genomic regions involved in tolerance to drought stress and drought stress induced leaf senescence in juvenile barley. BMC Plant Biol. 15:125. doi: 10.1186/s12870-015-0524-3
Welcker, C., Boussuge, B., Bencivenni, C., Ribaut, J., and Tardieu, F. (2007). Are source and sink strengths genetically linked in maize plants subjected to water deficit? A QTL study of the responses of leaf growth and of Anthesis-Silking Interval to water deficit. J. Exp. Bot. 58, 339–349. doi: 10.1093/jxb/erl227
Wen, W., Li, D., Li, X., Gao, Y., Li, W., Li, H., et al. (2014). Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 5:3438. doi: 10.1038/ncomms4438
Williams, J. G., Kubelik, A. R., Livak, K. J., Rafalski, J. A., and Tingey, S. V. (1990). DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 18, 6531–6535. doi: 10.1093/nar/18.22.6531
Xiang, Y., Sun, X., Gao, S., Qin, F., and Dai, M. (2017). Deletion of an endoplasmic reticulum stress response element in a ZmPP2C-A gene facilitates drought tolerance of maize seedlings. Mol. Plant 10, 456–469. doi: 10.1016/j.molp.2016.10.003
Xiao, Y., Tong, H., Yang, X., Xu, S., Pan, Q., Qiao, F., et al. (2016). Genome-wide dissection of the maize ear genetic architecture using multiple populations. New Phytol. 210, 1095–1106. doi: 10.1111/nph.13814
Xu, Z. Z., Zhou, G. S., and Shimizu, H. (2009). Effects of soil drought with nocturnal warming on leaf stomatal traits and mesophyll cell ultrastructure of a perennial grass. Crop Sci. 19, 1843–1851. doi: 10.2135/cropsci2008.12.0725
Xue, Y., Warburton, M. L., Sawkins, M., Zhang, X., Setter, T., Xu, Y., et al. (2013). Genome-wide association analysis for nine agronomic traits in maize under well-watered and water-stressed conditions. Theor. Appl. Genet. 126, 2587–2596. doi: 10.1007/s00122-013-2158-x
Yadaw, R. B., Dixit, S., Raman, A., Mishra, K. K., Vikram, P., Swamy, B. P. M., et al. (2013). A QTL for high grain yield under lowland drought in the background of popular rice variety Sabitri from Nepal. Field Crops Res. 144, 281–287. doi: 10.1016/j.fcr.2013.01.019
Yang, Q., Li, Z., Li, W., Ku, L., Wang, C., Ye, J., et al. (2013). CACTA-like transposable element in ZmCCT attenuated photoperiod sensitivity and accelerated the postdomestication spread of maize. Proc. Natl. Acad. Sci. U.S.A. 110, 16969–16974. doi: 10.1073/pnas.1310949110
Yang, X., Yan, J., Shah, T., Warburton, M. L., Li, Q., Li, L., et al. (2010). Genetic analysis and characterization of a new maize association mapping panel for quantitative trait loci dissection. Theor. Appl. Genet. 121, 417–431. doi: 10.1007/s00122-010-1320-y
Yu, J., Holland, J. B., McMullen, M. D., and Buckler, E. S. (2008). Genetic design and statistical power of nested association mapping in maize. Genetics 178, 539–551. doi: 10.1534/genetics.107.074245
Yu, J., Pressoir, G., Briggs, W. H., Vroh Bi, I., Yamasaki, M., Doebley, J. F., et al. (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. doi: 10.1038/ng1702
Zhang, Y., He, J., Wang, Y., Xing, G., Zhao, J., Li, Y., et al. (2015). Establishment of a 100-seed weight quantitative trait locus-allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes. J. Exp. Bot. 66, 6311–6325. doi: 10.1093/jxb/erv342
Zhang, Z., Ersoz, E., Lai, C. Q., Todhunter, R. J., Tiwari, H. K., Gore, M. A., et al. (2010). Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 42, 355–360. doi: 10.1038/ng.546
Zhao, F. Y., Zhang, X. J., Li, P. H., Zhao, Y. X., and Zhang, H. (2006). Co-expression of the Suaeda salsa SsNHX1 and Arabidopsis AVP1 confer greater salt tolerance to transgenic rice than the single SsNHX1. Mol. Breed. 17, 341–353. doi: 10.1007/s11032-006-9005-6
Zhao, K., Tung, C. W., Eizenga, G. C., Wright, M. H., Ali, M. L., Price, A. H., et al. (2011). Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2:467. doi: 10.1038/ncomms1467
Zhou, X., and Stephens, M. (2012). Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824. doi: 10.1038/ng.2310
Zhou, Z., Jiang, Y., Wang, Z., Gou, Z., Lyu, J., Li, W., et al. (2015). Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 33, 408–414. doi: 10.1038/nbt.3096
Zhu, Y. L., Song, Q. J., Hyten, D. L., Van Tassell, C. P., Matukumalli, L. K., Grimm, D. R., et al. (2003). Single-nucleotide polymorphisms in soybean. Genetics 163, 1123–1134.
Keywords: GWAS, drought tolerance, gene cloning, crops, natural variation
Citation: Wang H and Qin F (2017) Genome-Wide Association Study Reveals Natural Variations Contributing to Drought Resistance in Crops. Front. Plant Sci. 8:1110. doi: 10.3389/fpls.2017.01110
Received: 14 April 2017; Accepted: 08 June 2017;
Published: 30 June 2017.
Edited by:
Lam-Son Tran, RIKEN, JapanReviewed by:
Mingqiu Dai, Huazhong Agricultural University, ChinaBaichen Wang, Institute of Botany (CAS), China
Copyright © 2017 Wang and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Qin, qinfeng@cau.edu.cn