 Young-Ho Ha
Young-Ho Ha Changkyun Kim
Changkyun Kim Kyung Choi2
Kyung Choi2 Joo-Hwan Kim
Joo-Hwan Kim- 1Department of Life Science, Gachon University, Seongnam, South Korea
- 2Korea National Arboretum, Pocheon, South Korea
Tribe Forsythieae (Oleaceae), containing two genera (Abeliophyllum and Forsythia) and 13 species, is economically important plants used as ornamentals and in traditional medicine. This tribe species occur primarily in mountainous regions of Eurasia with the highest species diversity in East Asia. Here, we examine 11 complete chloroplast genome and nuclear cycloidea2 (cyc2) DNA sequences of 10 Forsythia species and Abeliophyllum distichum using Illumina platform to provide the phylogeny and biogeographic history of the tribe. The chloroplast genomes of the 11 Forsythieae species are highly conserved, except for a deletion of about 400 bp in the accD–psaI region detected only in Abeliophyllum. Within Forsythieae species, analysis of repetitive sequences revealed a total of 51 repeats comprising 26 forward repeats, 22 palindromic repeats, and 3 reverse repeats. Of those, 19 repeats were common and 32 were unique to one or more Forsythieae species. Our phylogenetic analyses supported the monophyly of Forsythia and its sister group is Abeliophyllum using the concatenated dataset of 78 chloroplast genes. Within Forsythia, Forsythia likiangensis and F. giraldiana were basal lineages followed by F. europaea; the three species are characterized by minutely serrate or entire leaf margins. The remaining species, which are distributed in East Asia, formed two major clades. One clade included F. ovata, F. velutina, and F. japonica; they are morphologically supported by broadly ovate leaves. Another clade of F. suspensa, F. saxatilis, F. viridissima, and F. koreana characterized by lanceolate leaves (except F. suspensa which have broad ovate leaves). Although cyc2 phylogeny is largely congruent to chloroplast genome phylogeny, we find the discordance between two phylogenies in the position of F. ovata suggesting that introgression of the chloroplast genome from one species into the nuclear background of another by interspecific hybridization in East Asian Forsythia species. Molecular dating and biogeographic reconstructions suggest an origin of the Forsythieae species in East China in the Miocene. Distribution patterns in Forsythia indicated that the species were radially differentiated from East China, and the speciation of the European F. europaea was the result of both vicariance and dispersal in the late Miocene to Pliocene.
Introduction
Forsythieae H. Taylor ex L. Johnson (Oleaceae) is comprised of two genera, Forsythia Vahl and Abeliophyllum Nakai (Angiosperm Phylogeny Group [APG], 2016). Abeliophyllum is a monotypic and endemic genus in Korea characterized by samara-type fruits and white flowers (Kim, 2007). Forsythia consists of 13 locally derived endemic species restricted to certain geographical regions. They are deciduous shrubs characterized by opposite simple or rarely 3-parted to 3-foliolate petiolate leaves, and yellow flowers blooming in the early spring before leaves (Chang et al., 1996; Wu et al., 2010). Nowadays, many ornamental species of Forsythia have been created by horticultural scholars to improve their cold hardiness and enrich the colorful display of flowers (DeWolf and Hebb, 1971). Additionally, the fruits of Forsythia suspensa Vahl and F. viridissima Lindl. have been used in traditional herbal medicine in Korea and China (Xia et al., 2009; Ryuk et al., 2010). In light of the need to preserve native species and utilize their genetic information in various phylogenetic and conservational studies, we determined the chloroplast (cp) genome sequences of Forsythieae species.
Since Nakai (1919b) described Abeliophyllum as a new genus, taxonomical position of the genus within the Oleaceae was contentious and many studies have been performed to resolve their relationships (O’Mara, 1930; Sax and Abbe, 1932; Taylor, 1945; Johnson, 1957; Maekawa, 1962; Lee and Park, 1982a,b; Lim et al., 1989). Based on morphological and anatomical characteristics of the fruit (winged samara) and pollen aperture type (tricolporate), Abeliophyllum is most closely related to Fontanesia (Taylor, 1945; Johnson, 1957). In contrast, Abeliophyllum is most closely related to Forsythia according to the same chromosome number (x = 14) and karyotype pattern (Sax and Abbe, 1932; Tae et al., 2005). While recent molecular phylogenetic studies have verified Forsythia and Abeliophyllum are sister groups in Oleaceae (Lee et al., 2007; Kim and Kim, 2011), the relationships within Forsythia remain controversial. Using restriction fragment length polymorphism, Kim (1999) suggested that Forsythia species are divided into four groups according to their geographical distribution. Lee (2011), however, proposed the division of Forsythia species into two groups, the F. koreana complex and F. nakaii complex, based on morphological characters such as petiole length, darkness of the petals, floral tube and lobes, and sepals. In contrast, Kim and Kim (2011) proposed three Forsythia lineages using internal transcribed spacers (ITS) and plastid DNA trnL-F and matK gene sequences. Previous studies resolved different relationships (especially between F. suspensa, F. saxatilis Nakai, and F. viridissima) and did not fully resolve the complexes/clades because of insufficient supporting characters (Kim, 1999; Kim and Kim, 2011). These incongruous relationships reveal difficulties in recognizing morphologically ambiguous species.
In North Hemisphere, disjunctive pattern of plants has been intensively studied between East Asia and North America (Wen, 1999; Donoghue and Smith, 2004) since Asa Gray (Gray, 1846) first reported the phenomenon. In contrast, disjunction of species between East Asia and Europe has been less focused because the migration of plants between two regions could be due to continuous mountain chains toward east–west direction across Eurasia (Green, 1972b). Forsythia species are distributed in Eurasia, with high species diversity in East Asia—six species are distributed in China (F. giraldiana Lingelsh., F. likiangensis Ching & Feng ex P.Y.Pai, F. mandschurica Uyeki, F. mira M.C.Chang, F. suspensa, and F. viridissima), four in Korea (F. koreana Nakai, F. ovata Nakai, F. saxatilis, and F. velutina Nakai), and two in Japan (F. japonica Makino and F. togashii Hara) (Yamazaki and Noshiro, 1993; Chang et al., 1996; Lee, 2002). F. europaea Degen & Bald is disjunctively distributed in northern Albania and adjacent parts of the former Yugoslavia (Green, 1972a; Willis and Shaw, 1973; Mabberley, 1997). Although the distribution of Forsythia species shows a noticeably distinct pattern between East Asia and Europe, the biogeographical study by Kim (1999) is the only study that examined this pattern based on chloroplast (cp) DNA substitution rate. Additionally, age estimation of the Oleaceae was conducted at higher taxonomic level with only a few species representing Forsythia (Wikström et al., 2001; Bell et al., 2010). Therefore, biogeographic origin of the tribe Forsythieae and their biogeographic patterns of distribution between East Asia and Europe has been not addressed.
The cp genome, containing genes coding for photosynthesis, is generally maternally inherited in Angiosperm (Corriveau and Coleman, 1988). It has a conserved quadripartite structure consisted of two copies of inverted repeats (IRs), a large single copy (LSC) region, and a small single copy (SSC) region (Palmer, 1991; Raubeson and Jansen, 2005). In addition, the analyses using whole cp genome sequences were efficient and essential for verifying variations in phylogeny as a result of sequence divergence among plant species at genus and tribal levels (Huang et al., 2014; Feng et al., 2016; Givnish et al., 2016). Due to variations in the cp genome, more studies have focused on genomic events such as simple sequence repeats (SSRs) (Park et al., 2016), indels (Ye et al., 2014), expansions/contractions (Yang et al., 2013), and inversions (Lee et al., 2007; Kim et al., 2016). Moreover, complete cp genome sequences have been used extensively to resolve phylogenetic relationships (Moore et al., 2007), evaluate species identification (Wu et al., 2010; Nock et al., 2011), and reveal biogeographical history (Givnish et al., 2016).
Next-generation sequencing (NGS) technologies provide a cost-effective method by accessing extensive amounts of data that can provide insights into the phylogenetic relationship and biogeographic history of plants (Hörandl and Appelhans, 2015). This technology can be used to recover whole cp genomes, mitochondrial data, and numerous nuclear markers which can aid in resolving Forsythieae species. Here, we obtained complete cp genome sequences and nuclear single-copy gene, cycloidea2 (cyc2) that encodes a transcription factor involved in the evolution of corolla zygomorphy (Zhong and Kellogg, 2015) of Abeliophyllum distichum and 10 Forsythia species using NGS technology. Our aims are: (1) to examine global patterns of structural variations in the cp genome and repetitive sequences of Forsythieae; (2) to reconstruct the phylogenetic relationship based on cp genome protein coding sequences and nuclear cyc2; and (3) to infer the biogeographic origin of Forsythia and explain their disjunctive distribution pattern.
Materials and Methods
Taxon Sampling
Three datasets were designed for this study. In the first phylogenetic analysis using cp genome data, we included 11 species—Forsythia (10 species) and Abeliophyllum (1 species)—to encompass the major lineages of Forsythieae based on the previous study by Kim and Kim (2011). We also included two cp genome sequences from other genera in Oleaceae (Hesperelaea palmeri A.Gray [GenBank no., LN515489] and Olea europaea L. [GenBank no., GU228899]) as outgroups. For the second phylogenetic analyses using cyc2 sequences, in addition to 11 Forsythieae species (12 accessions), we included 14 species (21 accessions) from GenBank. To estimate the divergence time and biogeography, we analyzed Forsythieae species within a broad phylogenetic framework using the third dataset. This dataset included 72 species representing 25 genera of Oleaceae sensu Angiosperm Phylogeny Group [APG] (2016). The outgroup comprised 16 species from Verbenaceae (3 species), Byblidaceae (2), Carlemanniaceae (2), Plocospermataceae (1), Gelsemiaceae (1), Loganiaceae (1), Strychnaceae (2), and Rubiaceae (4) based on previous phylogenetic analyses (Wallander and Albert, 2000); their sequences were obtained from GenBank. The sampled taxa, localities, and voucher information are listed in Supplementary Table S1.
DNA Sequencing, Assembly, Annotation, and Comparative Analysis
Total genomic DNA was extracted from silica-dried plant material using a DNeasy Plant Mini Kit (Qiagen, CA, United States). Genomic DNA was sequenced using an Illumina MiSeq2000 sequencer. The obtained raw reads were trimmed using Geneious v.7.1.9 (Kearse et al., 2012) to remove regions with chance of error greater than 0.05% per base. The cp genome sequences of 11 species were mapped on the reference genome of O. europaea using the ‘Map to Reference’ option implemented in Geneious. The mapping was conducted at medium-low sensitivity, and assembled reads were then de novo assembled with zero mismatches and gaps allowed among the reads. The reads were then re-aligned to the resulting de novo contigs with zero mismatches and gaps and with 100 iterations. Gaps and mismatches among the reads were not allowed in final assembled contigs. The cp genomes were annotated with DOGMA (Wyman et al., 2004) to identify coding sequences and rRNAs. The tRNA sequences were also confirmed by tRNAscan-SE (Lowe and Eddy, 1997). Genome maps were constructed using the Web-based tool GenomeVx (Conant and Wolfe, 2008). Visualization of the alignment of chloroplast sequences was conducted with mVISTA1, in which O. europaea was used as a reference sequence. Repetitive sequences were identified using REPuter program (Kurtz et al., 2001) and three types of repeats (forward, palindrome, and reverse) were identified following the procedure of Ni et al. (2016). The minimum repeat size was set to eight, and duplicated sequences in the IR region were excluded. We scored 1 (present) or 0 (absent) in a binary matrix for each minimum repeat size (Supplementary Table S2). Afterward, monomorphic bands across all species were discarded. To illustrate the genetic relationships at species levels, we analyzed the matrix of the minimum repeat sizes with the unweighted pair group method with arithmetic averages (UPGMA) based on pairwise distances (p-distance) using PAUP v.4.0 (Swofford, 2003).
The availability of some cyc2 sequences in GenBank for Forsythia and Abeliophyllum allowed the comparison of a common marker from the nuclear genome. Row reads of the 11 Forsythieae species samples were assembled to de novo segments of cyc2 region using default parameters in Geneious. We also checked the cyc2 sequences from NGS by direct sequencing. The cyc2 was amplified and sequenced using the primers Olea-CYC-126F and Olea-CYC693R as described in Zhong and Kellogg (2015).
Phylogenetic Analyses
We derived phylogenies from the datasets of 78 coding genes of the cp genome and nuclear cyc2 using maximum likelihood (ML) and Bayesian inference (BI) methods. Multiple-sequence alignment was performed in MAFFT v.6 (Katoh and Standley, 2013) using the default alignment parameters. Gaps were treated as missing data. The ML analysis was conducted on the RAxML BlackBox online server (Stamatakis, 2014), which supports GTR-based models of nucleotide substitution. The ML search option and the gamma model of rate heterogeneity were used to find the best scoring tree after bootstrapping. The statistical support for the branches (BS) was calculated by rapid bootstrap analyses with 1000 replicates (Stamatakis, 2014).
The BI analysis was conducted using MrBayes v.3.12 (Huelsenbeck and Ronquist, 2001; Ronquist et al., 2012). The best models of molecular evolution for the cp genome dataset (GTR + I + G) and cyc2 (GTR + I) were evaluated in MrModeltest v.2.0 (Nylander, 2004). Four chains of Markov chain Monte Carlo (MCMC) were run simultaneously and sampled every 1000 generations for a total of 20 million generations. We plotted the log-likelihood scores of sample points against the generation time using Tracer v.1.5 (Rambaut and Drummond, 2014) to ensure that stationarity was achieved after the first 2,000,000 generations by checking whether the log-likelihood values of the sample points reached a stable equilibrium (Huelsenbeck and Ronquist, 2001). In addition, we used AWTY (Nylander et al., 2008) to compare split frequencies in different runs and to plot cumulative split frequencies to ensure that stationarity was reached. The first 5000 (25%) of sampled trees from each run were discarded as burn-in in Tracer v.1.5 (Rambaut and Drummond, 2014). The remaining trees were used to construct a 50% majority-rule consensus tree, and the proportion of trees that contained the clade was given as posterior probability (PP) on the consensus tree to estimate robustness of each clade.
Estimates of Divergence Times
We used BEAST v.1.5.22 (Drummond and Rambaut, 2007) to estimate divergence time based on the combined dataset of six cpDNA regions (matK, rbcL, ndhF, atpB, rps16, and trnL-F) (Supplementary Table S1). To generate input files for BEAST, the BEAUti interface was used in which a selected model (GTR + I + G) for the combined dataset was applied with a Yule speciation tree prior and an uncorrelated lognormal molecular clock model. Two runs of 100 million generations of MCMC chains were produced, sampling every 1000 generations. Convergence of the stationary distribution was checked by visual inspection of plotted posterior estimates in Tracer v.1.6 (Rambaut and Drummond, 2014). After discarding the first 10,000 trees as burn-in, the samples were summarized in the maximum clade credibility tree using TreeAnnotator v1.6.1 (Rambaut and Drummond, 2010) with a PP limit of 0.50 and summarizing mean node heights. Means and 95% higher posterior densities (HPDs) of age estimates are obtained from the combined outputs using Tracer. The results were visualized using FigTree v.1.3.1 (Rambaut and Drummond, 2012).
Calibrations of molecular phylogenetic trees are generally better when performed using multiple fossil records (Forest, 2009). Because there is no reliable fossil assigned to Forsythieae, we constrained the ages of four nodes from outgroups as follows: (C1) the crown age of Olea was constrained with a uniform distribution from 23 to 30 million years ago (mya) following Besnard et al. (2009) and Hong-Wa and Besnard (2013); (C2) the crown node of Fraxinus was constrained to 45 (±1.5) mya with a normal distribution based on the macrofossil of the genus described from the middle Eocene (Call and Dilcher, 1992); (C3) the crown age of Byblis was constrained to 45 (±1.5) mya with a normal distribution based on the fossil from the middle Eocene (Conran and Christophel, 2004); and (C4) the stem age of Oleaceae was constrained to 101.6 (±3.4) mya with a normal distribution following Roalson and Roberts (2016).
Ancestral Area Reconstruction
The biogeographic data for the Forsythieae species were obtained from herbarium specimens and the literature (Chang et al., 1996). The distribution range of Forsythieae was divided into five regions: (A) Korea-Japan, (B) Central China, (C) East China, (D) Sikang-Yuennan, and (E) Western Europe following Takhtajan’s (1986) classification. The ancestral area reconstruction and the estimation of the spatial patterns of geographic diversification within Forsythieae were inferred using the Bayesian binary method (BBM) implemented in Reconstruct Ancestral State in Phylogenies (RASP) v.3.0 (Yu et al., 2015). The BBM was selected for its tendency to suggest single distribution areas for ancestral nodes compared to others and capability of providing precise and reliable results (Kim et al., 2015; Müller et al., 2015; Ito et al., 2017). For the biogeographic analysis, we used 100,000 trees obtained from the BEAST MCMC output after removing outgroups. The BBM was run with the fixed state frequencies model (Jukes–Cantor) with equal among-site rate variation for 50,000 generations, 10 chains each, and 2 parallel runs. The consensus tree used to map the ancestral distribution on each node was obtained with the Compute Condense option in RASP from stored trees. The maximum number of ancestral areas was set to five.
Results
Features of CpDNA Genomes in Forsythieae
The length of the complete cp genome sequences of 11 Forsythieae species ranged from 156,009 bp (A. distichum) to 156,397 bp (F. japonica) (Figure 1 and Table 1). The mean genome coverages of the 11 cp sequences ranged from 80 (F. velutina) to 1830 (F. giraldiana). The GC contents of all Forsythieae species were identical (37.8%). Similar to other angiosperms, the Forsythieae cp genomes showed a typical quadripartite structure, consisting of a pair of IRs (25,682–25,711 bp) separated by the LSC (86,772–87,075 bp) and SSC (17,827–17,859 bp) regions (Table 1). The cp genomes were found to encode an identical set of 132 predicted functional genes, of which 114 genes were unique and 18 were duplicated in the IR regions. The 114 unique genes comprised 80 protein coding genes, 30 transfer RNA genes, and 4 ribosomal RNA genes (Table 2). The 18 duplicated genes in the IR region consisted of 7 coding genes (rpl12, rpl23, rps7, rps12, ndhB, ycf2, ycf15), 7 transfer RNA genes (trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG, trnN-GUU), and 4 ribosomal RNA genes (rrn4.5, rrn5, rrn16, rrn23) (Table 2). Fourteen genes, rpl2, rpl16, rpoC1, trnA-UGC, trnI-GAU, trnK-UUU, trnL-CAA, trnL-UAA, trnV-UAC, ycf3, petB, petD, atpF, ndhA, ndhB, and clpP, contained one intron, while two genes (clpP, ycf3) contained two introns. Partial ycf1 was located at the boundary between IRa/SSC, leading to incomplete duplication of the gene within IRs. The rps12 gene was trans-spliced, with the 5′ end of the transcript for rps12 exon 1 were located in the LSC and the 3′ ends of exon 2 and exon 3 were duplicated in the IR regions. We found variation in indel events among Forsythieae species (Table 3); the smallest indel was 20 bp in length. The indels were specific and shared within the tribe Forsythieae. Specifically, a deletion of 416 bp in accD–psaI intergenic sequence (IGS) region was identified only in A. distichum, five indels were shared between F. giraldiana and F. likiangensis, and a deletion of 28 bp in ndhF–rpl32 IGS region was observed in three species, F. ovata, F. velutina, and F. japonica.
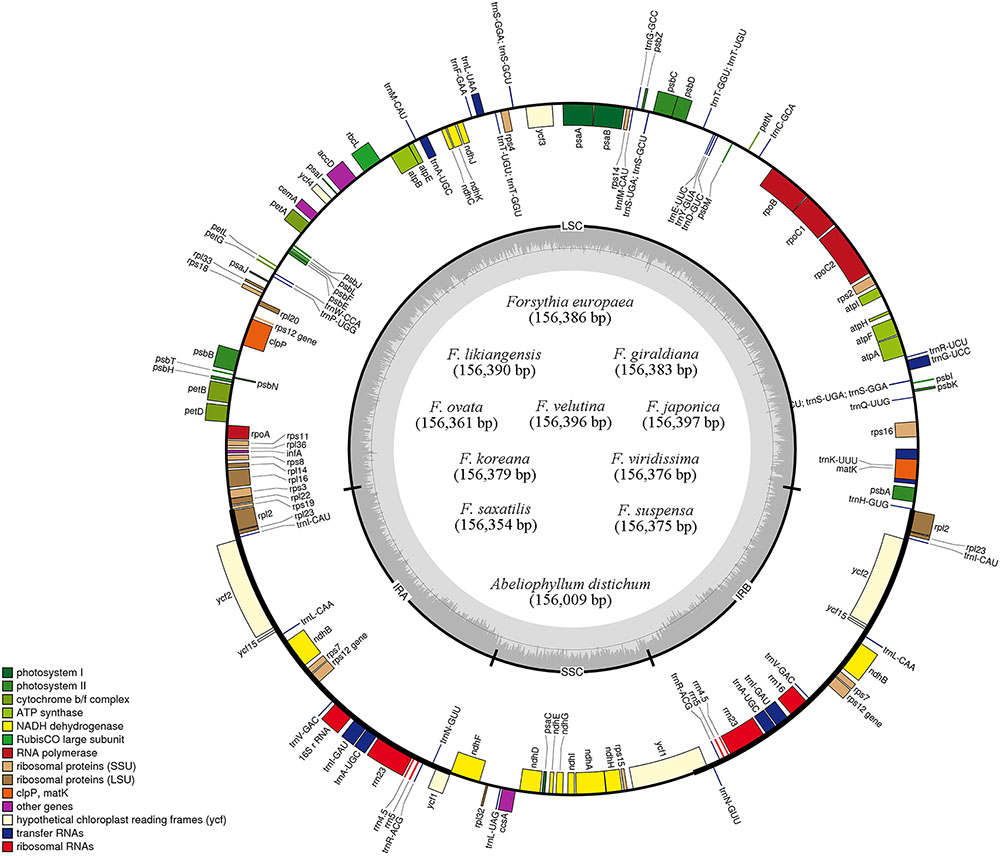
FIGURE 1. Gene maps of 11 Forsythieae chloroplast genomes. Genes drawn outside of the map circle are transcribed clockwise, while those drawn inside are transcribed counterclockwise. Genes belonging to different functional groups were color-coded. The darker gray in the inner circle corresponds to GC while the lighter gray corresponds to AT content.
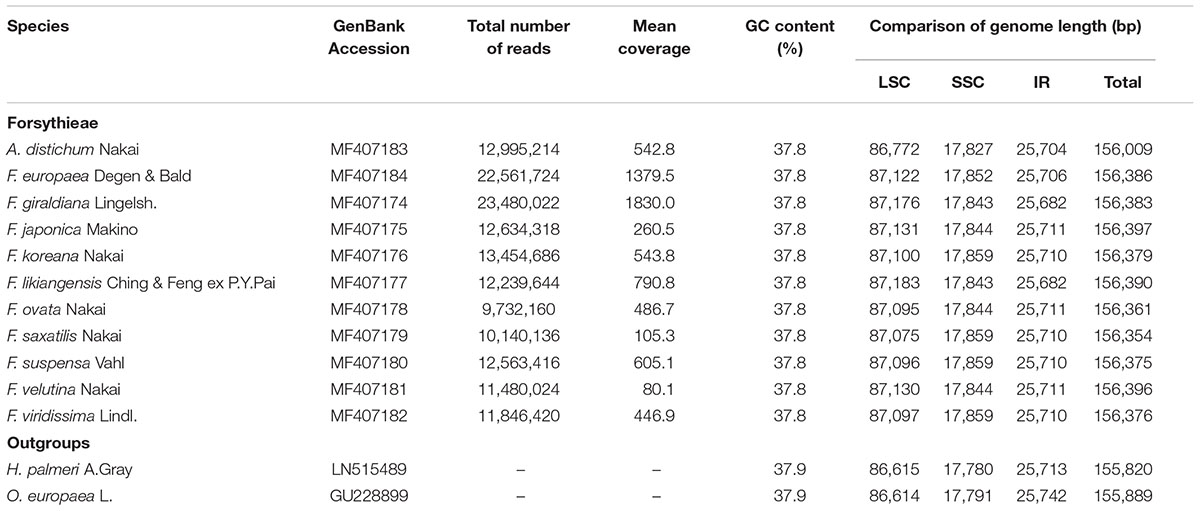
TABLE 1. Summary of the chloroplast genome sequences used in this study.

TABLE 2. List of genes encoded in chloroplast genomes of Forsythieae.
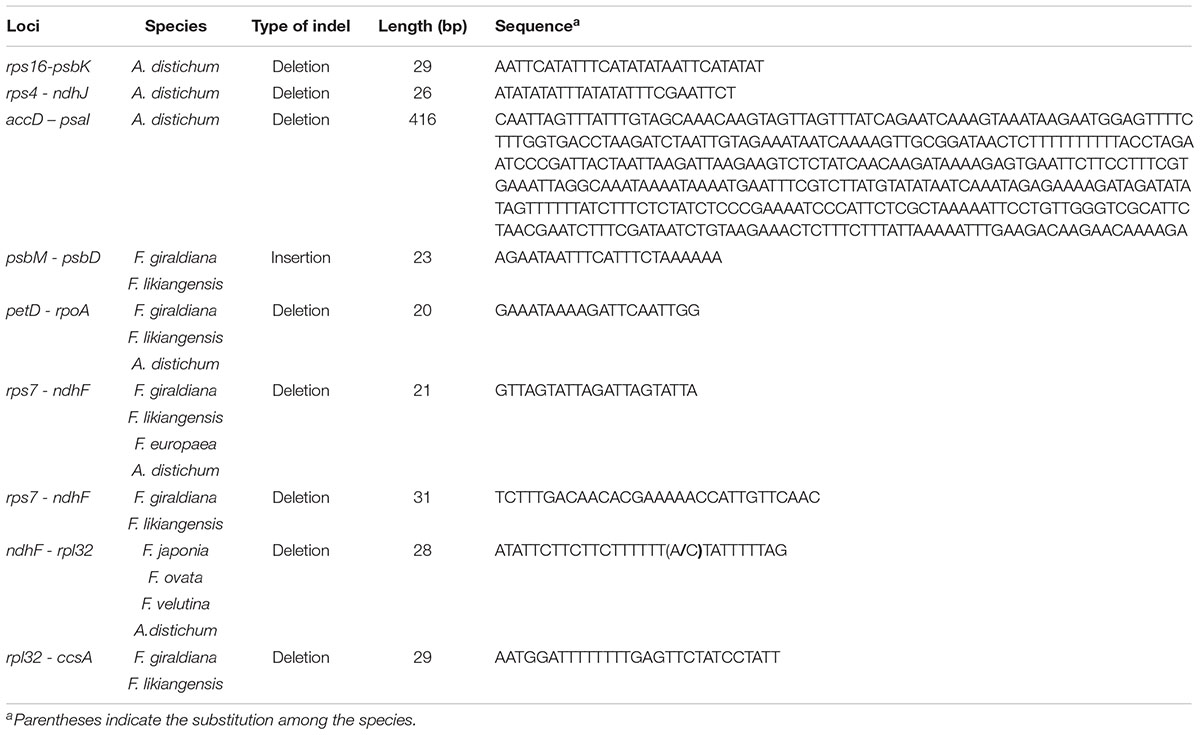
TABLE 3. Indels (Insertion/Deletion) identified in Forsythieae.
Sequences were plotted to check their identity using the mVISTA tool by aligning the 11 Forsythieae cp genomes with the annotation of O. europaea as a reference (Figure 2). The whole aligned sequences indicate that the Forsythieae cp genomes are rather conservative although some divergent regions were formed between these genomes. Moreover, similar to other plant species, the coding region is more conservative than the non-coding counterpart.
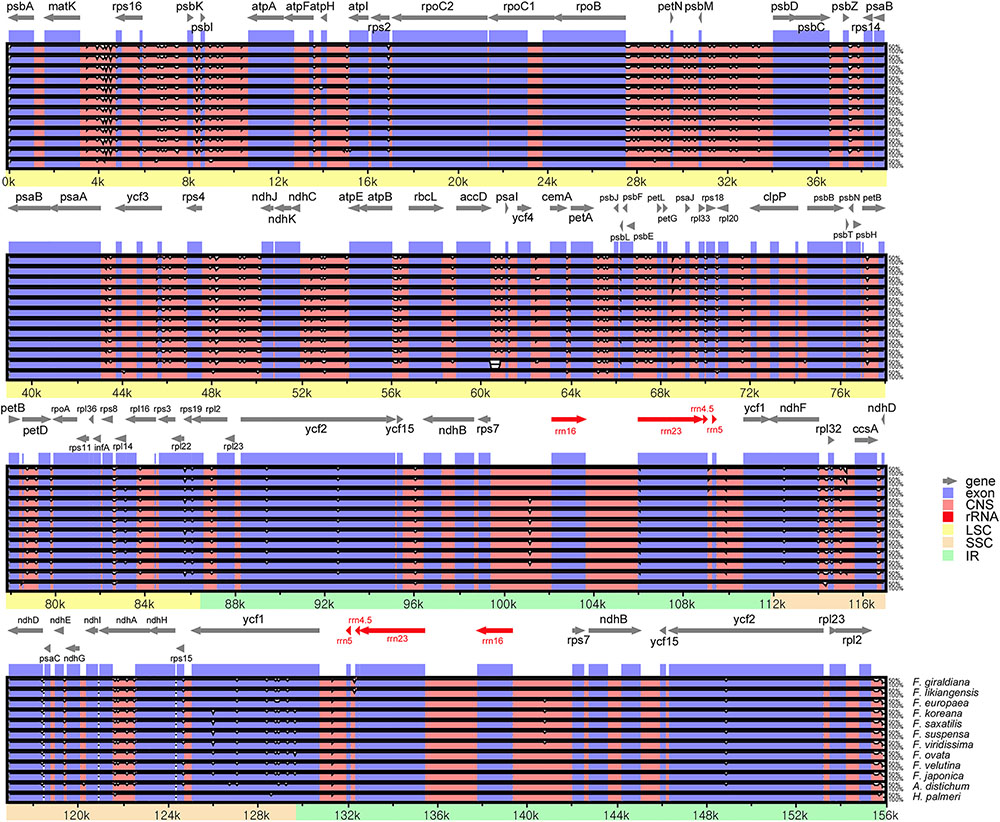
FIGURE 2. Sequence alignments of the Forsythieae chloroplast genomes using mVISTA program. VISTA-based identity plots showing sequence identity between 11 sequences of Forsythieae using Olea europaea (GenBank no., GU228899) as a reference. Black arrow shows the inverted repeats (IRs) in the chloroplast genomes.
Repetitive Sequences
Within the 11 cp genomes of the Forsythieae, we identified 51 repeat sequences, whose length varied from 17 to 41 bp. These repeat sequences comprised 26 forward (51%), 22 palindromic (41.1%), and 3 reverse (5.9%) sequences (Figure 3A). Among 11 Forsythieae species, the total number of repeats varied from 23 (F. viridissima and F. saxatilis) to 28 (A. distichum) (Figure 3B). Out of 51 repeats, 19 (37%) were commonly observed in Forsythieae and 32 were unique to one or more cp genomes (Supplementary Table S2). Eight repeats were specific to A. distichum. Similarly, a group of 13 repeats was shared by three species (F. giraldiana, F. likiangensis, and F. europaea). F. ovata, F. velutina, and F. japonica shared seven repeats; two of them can be used as potential markers for identifying these three species.
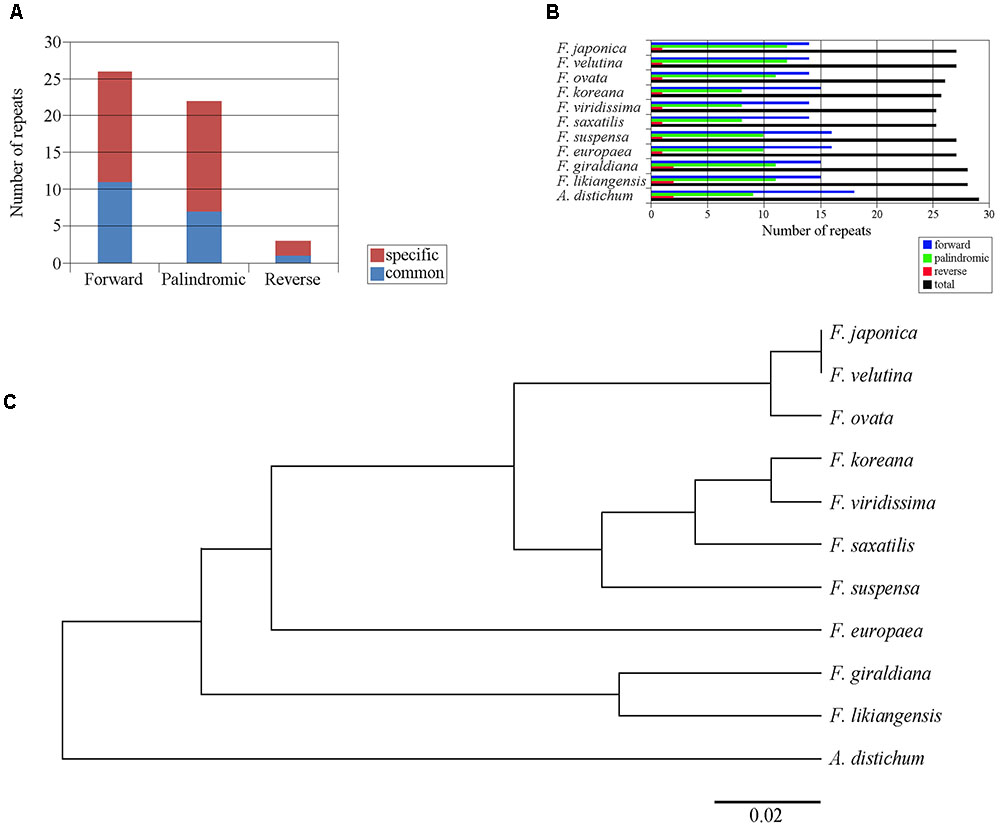
FIGURE 3. Repeat structure analyses in the 11 Forsythieae chloroplast genomes. (A) Histogram showing total number of the three repeat types in the Forsythieae. (B) Frequency of the three repeat types in each Forsythieae chloroplast genomes. (C) UPGMA tree based on the presence or absence of repeat structure.
Using the dataset of repeat sequences, the UPGMA dendrogram of the 11 Forsythieae species revealed four defined groups: (1) A. distichum, (2) F. likiangensis and F. giraldiana, (3) F. europaea, and (4) the remaining species, subdivided into two clusters (a) F. suspensa, F. saxatilis, F. viridissima, and F. koreana and (b) F. ovata, F. japonica, and F. velutina (Figure 3C).
Phylogenetic Analyses
The cp genome dataset included 68,965 aligned nucleotides from 13 species and 78 protein coding genes, 1,684 (2.4%) of which were variable. The BI tree was identical in topology to the ML tree (Figure 4; ML tree not shown). All clades were strongly supported (BS = 100; PP = 1.00), except for F. viridissima and F. koreana (BS = 72; PP = 0.94). Abeliophyllum was sister to the Forsythia species. Two species (F. likiangensis and F. giraldiana; clade I) occupied the most basal position followed by the clade with F. europaea, which was sister to the rest of the Forsythieae species (clade II). The clade II species were divided into two subclades: (1) F. suspensa, F. saxatilis, F. viridissima, and F. koreana (subclade a) and (2) F. ovata, F. velutina, and F. japonica (subclade b).
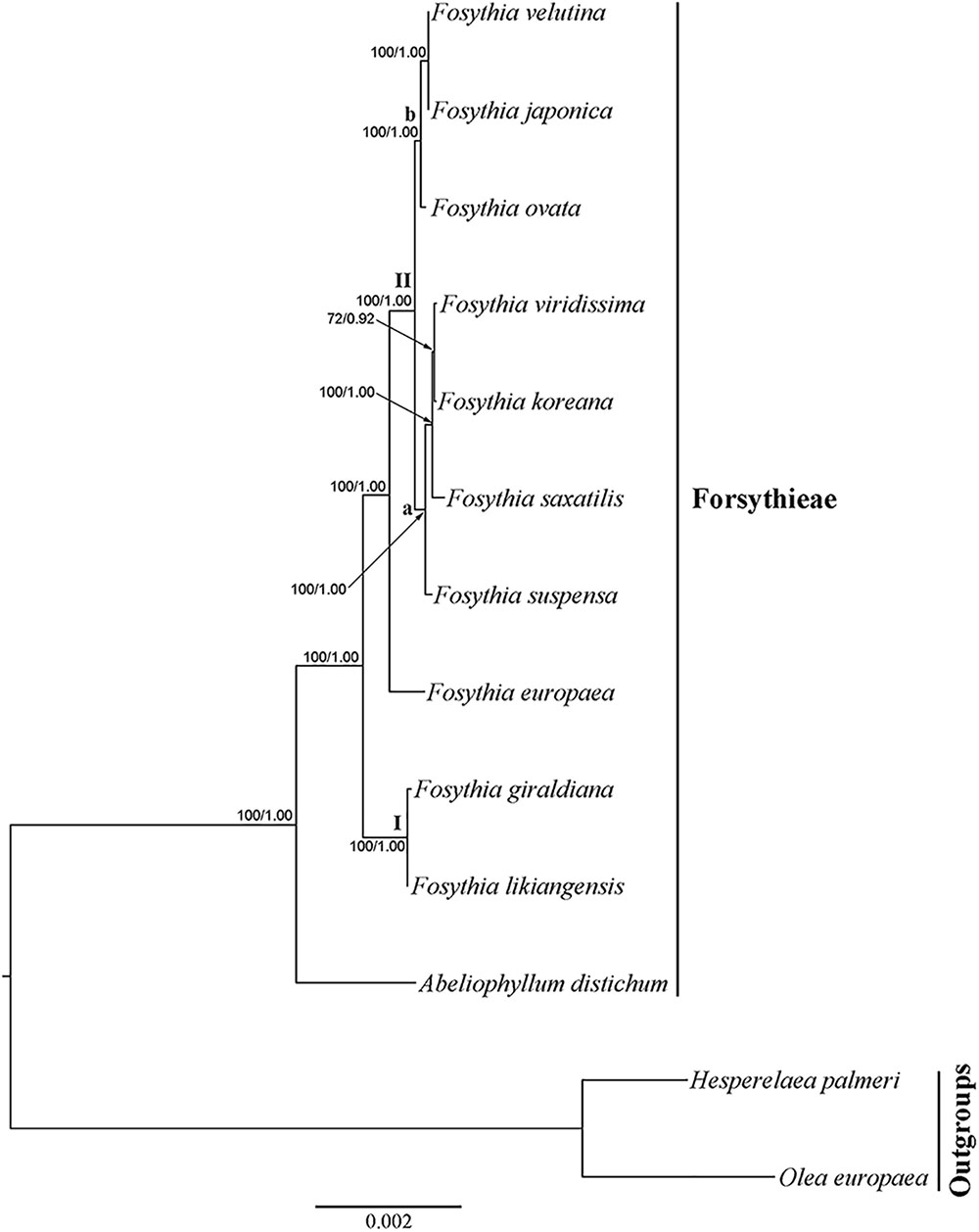
FIGURE 4. Phylogenetic tree resulting from Bayesian inference analysis of chloroplast 78 protein coding genes. Number above the branches indicate support values (maximum likelihood (BS)/Bayesian posterior probability (PP).
The aligned cyc2 data matrix for 11 Forsythieae species (14 accessions) and 13 outgroup species (19) consisted of 582 nucleotides, 285 (50.0%) of which were variable. Within the ingroup, there were 40 (6.9%) variable sites. The ML tree (not shown) was identical in topology to the BI tree (Figure 5). Two conflicts were found in the positions of F. ovata and F. viridissima between cp genome and cyc2 phylogenies. F. ovata was sister to F. velutina-F. japonica in the cp genome results (BS = 100, PP = 1.00; Figure 4), but was formed a clade with F. viridissima–F. saxatilis–F. koreana–F. suspensa in the cyc2 phylogeny (moderately supported; BS = 82%, PP = 0.99; Figure 5). F. viridissima was sister to F. koreana in the cp genome tree (weakly supported; BS = 72, PP = 0.92), but was sister to F. saxatilis in the cyc2 phylogeny (BS = 99, PP = 1.00). However, the topology of the cyc2 tree is largely similar to that of cp genome tree in highly supported relationships.
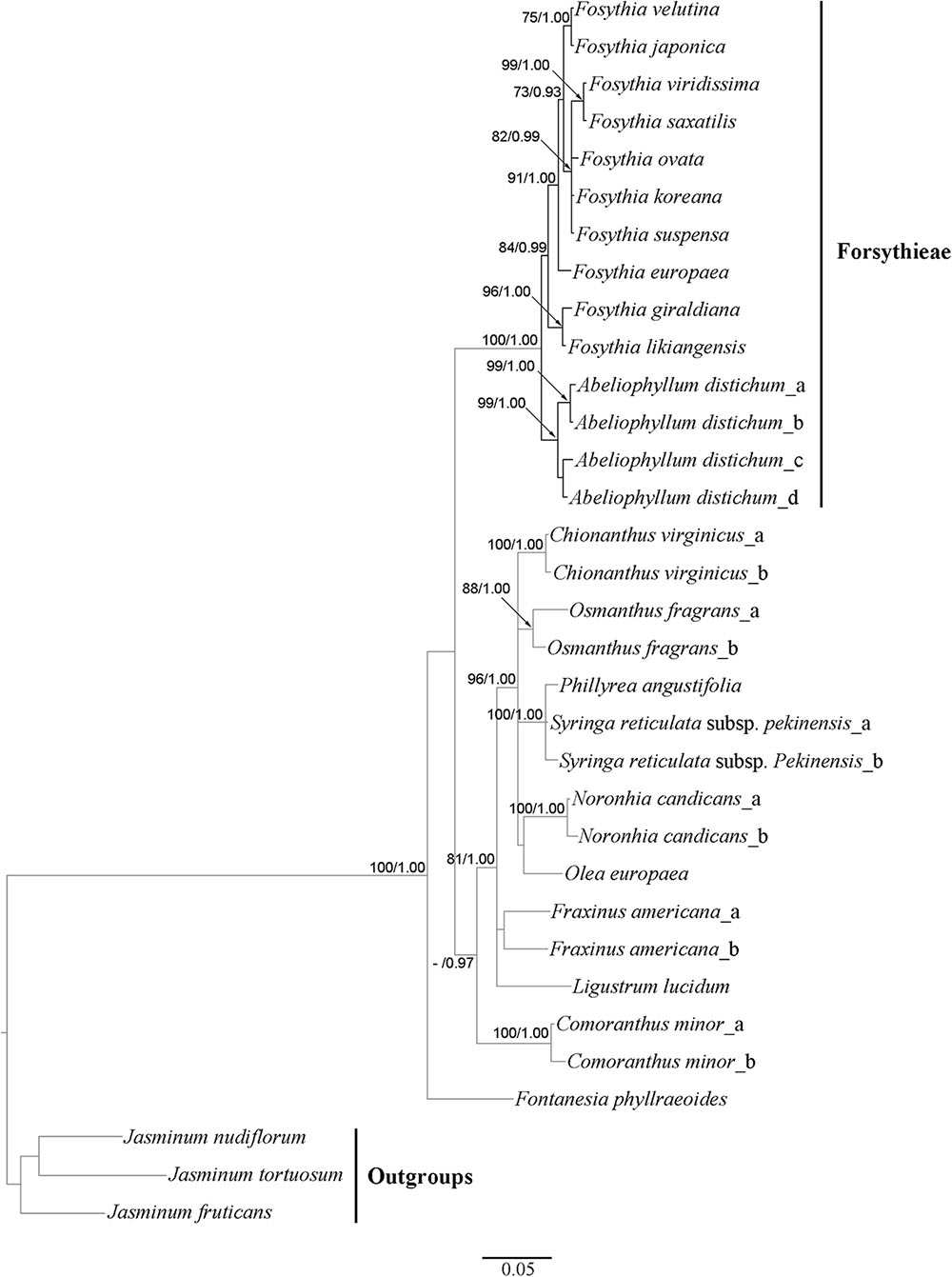
FIGURE 5. Phylogenetic tree resulting from Bayesian inference analysis of nuclear cyc2 gene. Number above the branches indicate support values (maximum likelihood (BS)/Bayesian posterior probability (PP); a dash (–) indicates BP < 70%.
Divergence Times Analyses
The mean divergence age estimates and 95% HPDs for nodes of interest based on the BEAST analysis of the combined dataset of six cpDNA sequences are presented in Figure 6 and Table 4. The age of the crown node of Forsythieae was estimated at 16.6 mya (95% HPD = 5.0–33.6 mya; node 1) in the Miocene. The age of the most recent common ancestor of Forsythia was estimated at 7.1 mya (95% HPD = 2.6–12.9 mya; node 2) in the late Miocene. Within the genus, the age estimate of the crown node for F. likiangensis and F. giraldiana (clade I) was dated to be 0.3 mya (95% HPD = 1.2 × 10-5–1.2 mya; node 5) in the Pleistocene. The divergence time between Europe (F. europaea) and East Asia (F. ovata, F. velutina, F. japonica, F. suspensa, F. saxatilis, F. viridissima, and F. koreana; clade II) was estimated at 5.2 mya (95% HPD = 1.8–9.7 mya; node 3) in the late Miocene/Pliocene interface. The crown age of clade II was dated at 2.5 mya (95% HPD = 0.7–4.8 mya; node 4) in the Pliocene/Pleistocene interface. The age of the subclades was estimated to be the Pleistocene (subclade a, 0.9 mya, 95% HPD = 0.02–2.2 mya [node 6]; subclade b, 1.2 mya, 95% HPD = 0.2–2.5 mya [node 7]).
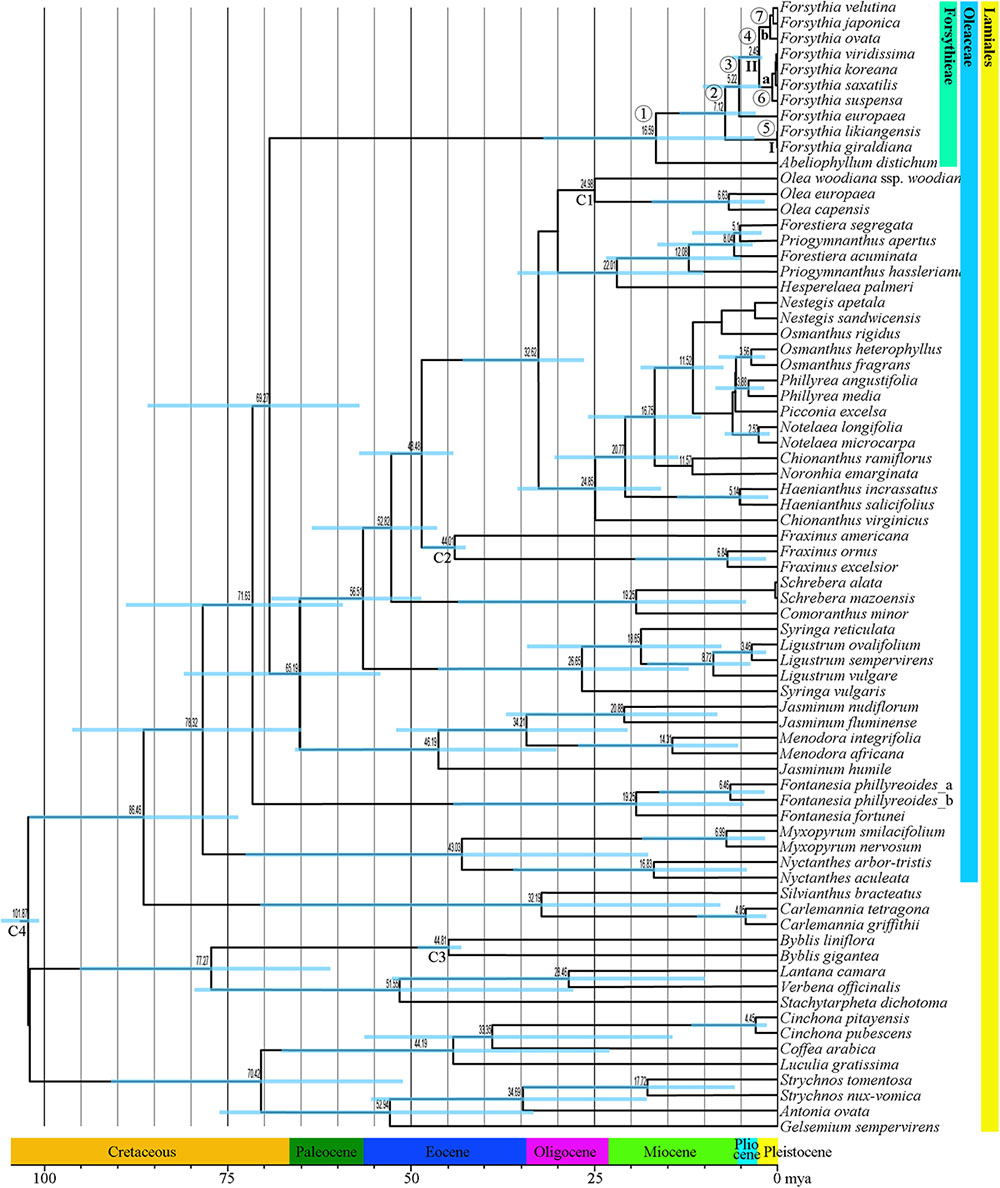
FIGURE 6. Chronogram showing divergence times estimated in BEAST based on six cpDNA sequence data (matK, rbcL, ndhF, atpB, rps16, and trnL-F). The divergence times are shown near each node. Blue bars represent 95% high posterior density for the estimated mean dates. The clades (I and II) and subclades (a and b) correspond to those in Figure 4. Nodes labeled C1–C4 are calibration points used in the analysis (for more details, see section “Materials and Methods”). Numbers 1–7 indicate nodes of interest (for details, see Table 4).

TABLE 4. Posterior age distributions of major nodes of Forsythieae with results of ancestral area reconstruction using BBM analysis.
Ancestral Area Reconstruction
The summary of the ancestral ranges at the nodes of interest inferred by BBM are presented in Figure 7 and Table 4. The BBM reconstruction suggests that Forsythieae (node 1) and Forsythia (node 2) originated in East China (C) with 82 and 84% marginal probabilities, respectively. Similar results were obtained for node 3 consisted of European (F. europaea) and East Asian Forsythia species, node 4 consisted of the clade II species, and node 5 which included SW Chinese F. likiangensis and Central and East Chinese F. giraldiana (clade I). The BBM analyses indicated two possible ranges, Central + East China (BC) and Korea and Japan + East China (AC), as the ancestral areas for the subclades (nodes 6 and 7); the occurrence of these ranges was 41 and 87%, respectively.
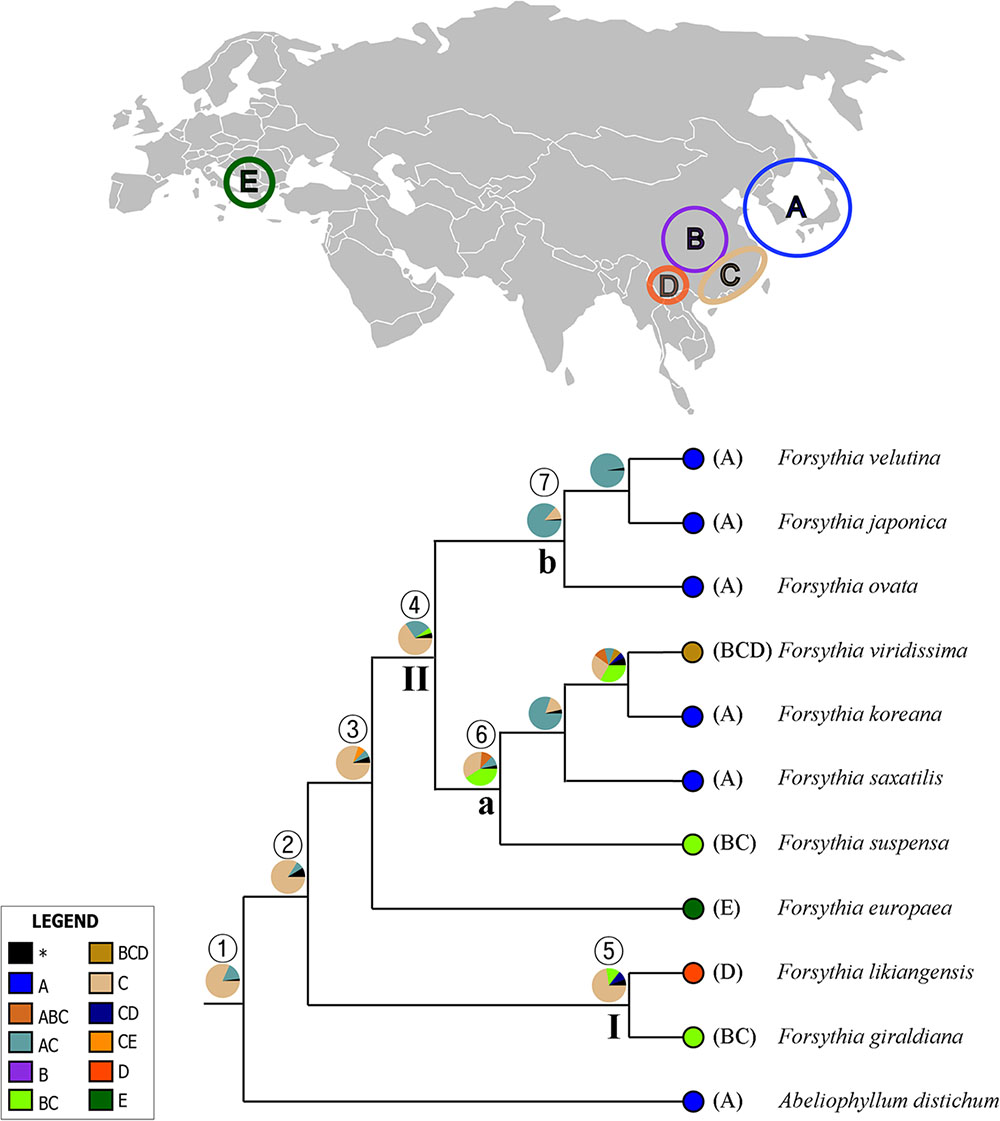
FIGURE 7. Summary of the Bayesian Binary Method (BBM) model of ancestral area reconstruction in Forsythieae based on a reduced BEAST combined-gene chronogram. The BBM ancestral area reconstructions with the highest likelihood are shown as pies for each clade of Forsythieae. Biogeographic regions used in BBM: A, Korea and Japan; B, Central China; C, East China; D, Sikang-Yuennan; E, West Europe. The clades (I and II) and subclades (a and b) correspond to those in Figure 4. Numbers 1–7 indicate nodes of interest (for details, see Table 4).
Discussion
Characteristics of Chloroplast Genomes in Forsythieae
In this study, we report the first complete cp genome sequences of 11 species from Abeliophyllum and Forsythia (Forsythieae) (Figure 1 and Table 1). These cpDNAs exhibited a highly conserved pattern of gene content and order in comparison to the previously reported cp genomes of O. europaea and H. palmeri (Oleaceae). The LSC/IR and IR/SSC junctions were similar to those of a typical eudicot cp genome, but the length between junctions varied. Among the well-preserved junctions, Forsythia is divided into four groups. In other words, Forsythia species tend to have similar expansions/contractions within the group caused by intramolecular recombination between two short directly repeated sequences (Kim and Lee, 2004). Although the gene content and their order were highly conserved, we identified nine indels in the IGS region of the cp genomes of the 11 examined the Forsythieae species (Table 3). Repetitive sequences are patterns of DNA fragments that occur in multiple copies in the genome (Mehrotra and Goyal, 2014). They play a crucial role in higher plants due to their effectiveness in structural rearrangement and the expansion and contraction of chloroplast genome (SanMiguel et al., 1996). For example, the hairpin structure, created by the modification of the palindromic repeat sequence, affects the replication mechanism (Kurtz et al., 2001). Recently, the variation in repetitive sequences and SSRs has been used extensively in comparative genomic studies and species identification (Ni et al., 2016; Zhang et al., 2016). In the present study, the indels were not only unique, but they were also shared among the species of Forsythieae (Figure 2, Table 3 and Supplementary Table S2), suggesting their value as molecular markers for species identification in the tribe. The UPGMA tree based on the presence/absence of the repetitive sequences recognized four clusters, which were congruent with the results of the phylogenetic analysis of the Forsythieae (Figure 3C).
Phylogenetic Relationship within Forsythieae
Our phylogenetic analyses indicate that the sister group of Abeliophyllum with strong support (BS = 100%; Figures 4, 5) is Forsythia. This result is consistent with previous studies of the molecular phylogenetic relationships of Oleaceae based on limited sampling of Forsythia (Kim, 1999; Lee et al., 2007; Kim and Kim, 2011). The sister relationship between Abeliophyllum and Forsythia is also supported by morphological and cytological evidence: (1) morphology of the corolla tube (Nakai, 1920), (2) unique form of polymorphism: heterostylous flowers (Ryu et al., 1976), and (3) same basic chromosomal number (x = 14; Taylor, 1945; Maekawa, 1962).
Our cp genome and nuclear phylogenetic analyses produced conflicting results regarding species relationships in Forsythia (Figures 4, 5). Two striking differences between the two topologies are in the positions of F. ovata and F. viridissima. For example, in the cp genome tree, F. ovata is sister to F. japonica–F. velutina (BS = 100; Figure 4), the pattern also found by Kim and Kim (2011) using nrITS and two cpDNA regions. In contrast, the nuclear cyc2 topology clustered F. ovata–(F. viridissima–F. saxatilis)–F. suspensa–F. koreana as a single moderately supported clade (BS = 82; Figure 5). Discordance between cp genome and nuclear phylogenies is common in plants (e.g., Soltis and Kuzoff, 1995; Deng et al., 2015). A possible explanation for the conflict has invoked introgression of cp genome from one species into the nuclear background of another (or vice versa) by interspecific hybridization, in which case the incongruent tree topologies represent the different histories of cp and nuclear genomes. Interspecific hybridization in Forsythia is not surprising by showing the heterostyly, with anther and stigma located at different height in the flowers (pin and thrum types; Rosati et al., 2007).
Within Forsythia, F. giraldiana and F. likiangensis, which are distributed in north-central and southern regions of China, respectively (Chang et al., 1996), formed a basal and strongly supported (BS = 100) clade (Figure 4). This relationship is also supported by similar morphological characteristics (ovate to elliptic leaf blade and entire leaf margin), and similar indels and repeat sequences in the cp genome analyses (Supplementary Table S2). This study is the first to conduct phylogenetic analysis of F. likiangensis, a species closely related to F. giraldiana.
In previous studies, the position of F. europaea within Forsythia was contentious. Based on RFLP and nuclear and plastid DNA sequence data, this species was placed as sister to F. giraldiana (Kim, 1999; Kim and Kim, 2011), whereas the RAPD analysis resolved it as sister to F. viridissima (Tae et al., 2005). Additionally, similar to F. giraldiana and F. likiangensis, F. europaea has oblong leaves with entire margins. In our analyses, F. europaea did not group with either F. giraldiana or F. likiangensis, but it was sister to the other two subclades comprised of seven Forsythia species (Figure 4). This point is reinforced by our cyc2 phylogeny (Figure 5). This discrepancy between our and previous studies might have resulted from the addition of F. likiangensis in the data matrix and/or the use of the whole sequences of protein coding genes from the cp genome.
The remaining seven species, distributed in East Asia, were divided into two subclades in cp genome phylogeny (Figure 4). The first clade includes F. ovata, F. japonica, and F. velutina, and the monophyly of this group was supported by both morphological and molecular data (Lee, 1984; Kim and Kim, 2004; Lee, 2011). The second clade consists of F. viridissima (Southern China), F. koreana (Korea), F. saxatilis (Korea), and F. suspensa (widely distributed in China). In this clade, F. suspensa, the first described species in Forsythia, was sister to the remaining species; it is defined by apomorphic hollow stem and 3-parted to 3-foliolate leaf blade. However, the relationship of F. viridissima, F. koreana, and F. saxatilis presented herein did not corroborate the results of previous studies (Kim, 1999; Kim and Kim, 2011). F. saxatilis is characterized by ovate to lanceolate leaves and originally Nakai (1919a) treated it as variety saxatilis in F. japonica. Therefore, it is necessary to evaluate whether leaf shape is a key character in classifying the species within the genus Forsythia and conduct the taxonomic treatment of F. saxatilis.
Historical Biogeography
Our molecular dating showed that the tribe Forsythieae originated in East China (Figure 7) and differentiated into two genera (Forsythia and Abeliophyllum) during the Miocene (16.6 mya, 95% HPD = 5.0–33.6 mya) (Figure 6). Due to the influence of the mid-Miocene climatic optimum (17–15 mya), the temperature in the Miocene was 4–5°C higher than it is today (You et al., 2009). Climate change offered an opportunity for accelerated establishment of the new lineage (Hinsinger et al., 2013). Therefore, Forsythieae were probably divided into different lineages during the period of climate change in the middle Miocene. The two genera of Forsythieae are clearly distinguished by their fruits (i.e., winged compressed indehiscent fruits in Abeliophyllum and dried capsule in Forsythia). The functional significance of samaras is to produce lift and drag to counter the forces of gravity, thereby reducing the falling speed of the fruit and increasing the distance, it may be dispersed by winds (Augspurger, 1986). Thus, the samara fruit may have been a key innovation for Abeliophyllum, triggering diversification and dispersal and conserved during its divergence and evolution.
Within Forsythia, F. europaea, a species restricted to areas in Europe (Green, 1972a; Willis and Shaw, 1973; Mabberley, 1997), originated in East China and along with its sister groups separated from the common ancestor at 5.2 mya (95% HPD = 1.8–9.7 mya) in the late Miocene–Pliocene interface (Figure 6). Based on low cpDNA substitution rate, Kim (1999) suggested relatively recent long-distance dispersal event at 0.46 mya (the Pleistocene) to explain the disjunction between Europe and East China. However, it is well known that age estimates based on a substitution rate from inter-species comparisons can be biased by heterogeneity in rates of molecular evolution (Pulquério and Nichols, 2006). This problem can be alleviated by analyzing multiple gene loci simultaneously and by using multiple calibration points (Yang and Yoder, 2003). In this study, we examined six cpDNA regions (matK, rbcL, ndhF, atpB, rps16, and trnL-F) across the broad sampling of Oleaceae and outgroups to address the divergence time and biogeographic origin of Forsythia.
Several factors, including the presence of the Turgai Strait, the Quaternary ice period, Qinghai-Tibetan Plateau (QTP) uplift, and climate change, in the Pliocene have been suggested as explanations for disjunct distribution of species between East Asia and Europe. In the Paleocene and early Oligocene, the Turgai Strait was a barrier that extended from the Arctic Ocean to the Tethys Seaway, separating the two regions (Legendre and Hartenberger, 1992); with its collapse the migration of the species was possible (Carlson et al., 2012). However, our divergence time between European and East Asian species is too recent to support this hypothesis (Figure 6 and Table 4). Southern mountain chains stretching east to west between Asia and Europe provided possible paths for migration of plant species; however, the onset of the glacial period and the advancement of glaciers could have caused the extinction of species in the Pleistocene (Green, 1972b). This suggests that the Quaternary glaciers interrupted the migration route of Forsythia, isolating its populations into two areas, Europe and Asia. However, our results indicate that the two lineages split before the Pleistocene and therefore do not support this hypothesis. The uplift of the QTP in the Miocene prevented the dispersal of species between Europe and Asia (Harrison and Copeland, 1992; Axelrod et al., 1998; Sun et al., 2001; Zhang et al., 2006). In the case of Forsythia, the divergence time between European and East Asian species was estimated to 5.3 mya, thus supporting the hypothesis that vicariance due to the QTP uplift contributed to the disjunction in Forsythia. Finally, climate fluctuations during the Pliocene (Fiz-Palacios et al., 2010; Tu et al., 2010) probably influenced this disjunction as the rapid raise of the QTP resulted in drying and desertification of the regions around the QTP (Zhisheng et al., 2001; Xie et al., 2014). Similar to our results, climate change and glaciations were the main factors affecting the distribution of Scabiosa (Carlson et al., 2012). Thus, our results support the vicariance hypothesis and identify it as the main factor for the differentiation of Forsythia between Europe and Asia approximately 5.2 mya. The best hypothetical scenario to explain the distribution of F. europaea is the dispersal in the late-Miocene to Pliocene, followed by vicariance during the Pliocene climate fluctuations that caused the disjunction between European and East Asian populations. Seven species of Forsythia in East Asia (China, Korea, and Japan) diverged after the Pleistocene. However, it is difficult to interpret the distribution patterns of these species based on the current data and further phylogeographic studies at the population level should be carried out to understand the evolutionary history of these Forsythia species.
Author Contributions
J-HK conceived and designed the experiments. Y-HH and KC collected the plant materials. Y-HH, CK, and J-HK performed the experiments and analyzed the data. Y-HH and CK wrote the draft and J-HK revised the draft. All authors agreed on the contents of the paper and declared that no competing interests exist.
Funding
This work was supported by grants from Scientific Research (No. KNA1-1-13, 14-1 to J-HK) of Korea National Arboretum.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer WW and handling Editor declared their shared affiliation.
Acknowledgments
The authors would like to thank Dr. Dong-Kap Kim and Dr. Hyuk-Jin Kim for kindly providing leaf material of F. koreana and F. suspensa from KNA and Lucy Reed (KEW Botanical Garden) for supplying leaf material of F. giraldiana, F. likiangensis, and F. europaea. They are grateful to Dr. Sang-Chul Kim for the help with data analysis and Do Hoang Dang Khoa for the valuable comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00099/full#supplementary-material
Footnotes
References
Angiosperm Phylogeny Group [APG] (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Augspurger, C. K. (1986). Morphology and dispersal potential of wind-dispersed diaspores of neotropical trees. Am. J. Bot. 73, 353–363.
Axelrod, D., Al Shehbaz, I., and Raven, P. (1998). “History of the modern flora of China. Floristic characteristics and diversity of east Asian plants,” in Proceedings of the 1st International Symposium of Floristic Characteristics and Diversity of East Asian Plants, (Beijing: Higher Education Press).
Bell, C. D., Soltis, D. E., and Soltis, P. S. (2010). The age and diversification of the angiosperms re-revisited. Am. J. Bot. 97, 1296–1303. doi: 10.3732/ajb.0900346
Besnard, G., DeCasas, R. R., Christin, P.-A., and Vargas, P. (2009). Phylogenetics of Olea (Oleaceae) based on plastid and nuclear ribosomal DNA sequences: tertiary climatic shifts and lineage differentiation times. Ann. Bot. 104, 143–160. doi: 10.1093/aob/mcp105
Call, V. B., and Dilcher, D. L. (1992). Investigations of angiosperms from the Eocene of southeastern North America: samaras of Fraxinus wilcoxiana Berry. Rev. Palaeobot. Palynol. 74, 249–266. doi: 10.1016/0034-6667(92)90010-E
Carlson, S. E., Linder, H. P., and Donoghue, M. J. (2012). The historical biogeography of Scabiosa (Dipsacaceae): implications for Old World plant disjunctions. J. Biogeogr. 39, 1086–1100. doi: 10.1111/j.1365-2699.2011.02669.x
Chang, M.-C., Qiu, L., and Green, P. (1996). “Oleaceae,” in Flora of China, Vol. 15, eds Z. Y. Wu and P. H. Raven (Beijing: Science Press), 272–319.
Conant, G. C., and Wolfe, K. H. (2008). GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics 24, 861–862. doi: 10.1093/bioinformatics/btm598
Conran, J. G., and Christophel, D. C. (2004). A fossil Byblidaceae seed from Eocene South Australia. Int. J. Plant Sci. 165, 691–694. doi: 10.1086/386555
Corriveau, J. L., and Coleman, A. W. (1988). Rapid screening method to detect potential biparental inheritance of plastid DNA and results for over 200 angiosperm species. Am. J. Bot. 75, 1443–1458. doi: 10.2307/2444695
Deng, T., Nie, Z.-L., Drew, B. T., Volis, S., Kim, C., Siang, C.-L., et al. (2015). Does the arcto-tertiary biogeographic hypothesis explain the disjunct distribution of Northern hemisphere herbaceous plants? The case of Meehania (Lamiaceae). PLOS ONE 10:e0117171. doi: 10.1371/journal.pone.0117171
Donoghue, M. J., and Smith, S. A. (2004). Patterns in the assembly of temperate forests around the Northern Hemisphere. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 1633–1644. doi: 10.1098/rstb.2004.1538
Drummond, A. J., and Rambaut, A. (2007). BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Feng, X., Liu, J., and Gong, X. (2016). Species delimitation of the Cycas segmentifida complex (Cycadaceae) resolved by phylogenetic and distance analyses of molecular data. Front. Plant Sci. 7:134. doi: 10.3389/fpls.2016.00134
Fiz-Palacios, O., Vargas, P., Vila, R., Papadopulos, A. S., and Aldasoro, J. J. (2010). The uneven phylogeny and biogeography of Erodium (Geraniaceae): radiations in the Mediterranean and recent recurrent intercontinental colonization. Ann. Bot. 106, 871–884. doi: 10.1093/aob/mcq184
Forest, F. (2009). Calibrating the tree of life: fossils, molecules and evolutionary timescales. Ann. Bot. 104, 789–794. doi: 10.1093/aob/mcp192
Givnish, T. J., Zuluaga, A., Marques, I., Lam, V. K., Gomez, M. S., Iles, W. J., et al. (2016). Phylogenomics and historical biogeography of the monocot order Liliales: out of Australia and through Antarctica. Cladistics 32, 581–605. doi: 10.1111/cla.12153
Gray, A. (1846). Analogy between the flora of Japan and that of the United States. Am. J. Sci. Arts 2, 135–136.
Green, P. (1972a). “Forsythia,” in Flora Europaea 3, eds T. G. Tutin, V. H. Heywood, N. A. Burges, D. M. Moore, D. H. Valentine, S. M. Walters, et al. (Cambridge: Cambridge University Press), 53.
Green, P. (1972b). Osmanthus decorus and disjunct Asiatic-European distributions in the Oleaceae. Kew Bull. 26, 487–490. doi: 10.2307/4120313
Harrison, T. M., and Copeland, P. (1992). Raising tibet. Science 255, 1663–1670. doi: 10.1126/science.255.5052.1663
Hinsinger, D. D., Basak, J., Gaudeul, M., Cruaud, C., Bertolino, P., Frascaria-Lacoste, N., et al. (2013). The phylogeny and biogeographic history of ashes (Fraxinus, Oleaceae) highlight the roles of migration and vicariance in the diversification of temperate trees. PLOS ONE 8:e80431. doi: 10.1371/journal.pone.0080431
Hong-Wa, C., and Besnard, G. (2013). Intricate patterns of phylogenetic relationships in the olive family as inferred from multi-locus plastid and nuclear DNA sequence analyses: a close-up on Chionanthus and Noronhia (Oleaceae). Mol. Phylogenet. Evol. 67, 367–378. doi: 10.1016/j.ympev.2013.02.003
Hörandl, E., and Appelhans, M. S. (2015). Next-Generation Sequencing in Plant Systematics. Koenigstein: Koeltz Scientific Books.
Huang, H., Shi, C., Liu, Y., Mao, S.-Y., and Gao, L.-Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Huelsenbeck, J. P., and Ronquist, F. (2001). MrBayes: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Ito, Y., Tanaka, N., Albach, D. C., Barfod, A. S., Oxelman, B., and Muasya, A. M. (2017). Molecular phylogeny of the cosmopolitan aquatic plant genus Limosella (Scrophulariaceae) with a particular focus on the origin of the Australasian L. curdieana. J. Plant Res. 130, 107–116. doi: 10.1007/s10265-016-0872-6
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, C., Deng, T., Wen, J., Nie, Z.-L., and Sun, H. (2015). Systematics, biogeography, and character evolution of Deutzia (Hydrangeaceae) inferred from nuclear and chloroplast DNA sequences. Mol. Phylogenet. Evol. 87, 91–104. doi: 10.1016/j.ympev.2015.03.002
Kim, D.-K., and Kim, J.-H. (2004). Numerucal taxonomy of tribe Forsythieae (Oleaceae) in Korea. Korean J. Plant Taxon. 34, 189–203.
Kim, D.-K., and Kim, J.-H. (2011). Molecular phylogeny of tribe Forsythieae (Oleaceae) based on nuclear ribosomal DNA internal transcribed spacers and plastid DNA trnL-F and matK gene sequences. J. Plant Res. 124, 339–347. doi: 10.1007/s10265-010-0383-9
Kim, K.-J. (1999). Molecular phylogeny of Forsythia (Oleaceae) based on chloroplast DNA variation. Plant Syst. Evol. 218, 113–123. doi: 10.1007/BF01087039
Kim, K.-J. (2007). “Abeliophyllum nakai,” in The Genera of Vascular Plants of Korea, ed. C.-W. Park (Seoul: Academy Publishing Co.), 849–851.
Kim, K.-J., and Lee, H.-L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kim, S.-C., Kim, J. S., and Kim, J.-H. (2016). Insight into infrageneric circumscription through complete chloroplast genome sequences of two Trillium species. AoB Plants 8:plw015. doi: 10.1093/aobpla/plw015
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lee, H.-L., Jansen, R. K., Chumley, T. W., and Kim, K.-J. (2007). Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 24, 1161–1180. doi: 10.1093/molbev/msm036
Lee, S., and Park, E. (1982b). A cladistic analysis of the Korean Oleaceae. Korean J. Bot. 25, 57–64.
Lee, S.-T. (1984). A systematic study of Korean Forsythia species. Korean J. Plant Taxon. 14, 87–107.
Lee, S. T. (2011). Palynological contributions to the taxonomy of family Oleaceae, with special empahsis on genus Forsythia (tribe Forsytheae). Korean J. Plant Taxon. 41, 175–181.
Lee, S.-T., and Park, E.-J. (1982a). A palynotaxonomic study of the Korean Oleaceae. Korean J. Plant Taxon. 12, 1–11.
Legendre, S., and Hartenberger, J.-L. (1992). “The evolution of mammalian faunas in Europe during the Eocene and Oligocene,” in Eocene-Oligocene Climatic and Biotic Evolution, eds D. R. Prothero and W. A. Berggren (Princeton, NJ: Princeton University Press), 516–528.
Lim, J. H., Ko, Y. T., Lee, D. H., and Kim, S. Y. (1989). Clonorchiasis: sonographic findings in 59 proved cases. Am. J. Roentgenol. 152, 761–764. doi: 10.2214/ajr.152.4.761
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.0955
Mabberley, D. J. (1997). The Plant-Book. A Portable Dictionary of the Vascular Plants. Cambridge: Cambridge University Press.
Maekawa, F. (1962). Major polyploidy with special reference to the phylogeny of Oleaceae. J. Jap. Bot. 37, 25–27.
Mehrotra, S., and Goyal, V. (2014). Repetitive sequences in plant nuclear DNA: types, distribution, evolution and function. Genomics Proteomics Bioinformatics 12, 164–171. doi: 10.1016/j.gpb.2014.07.003
Moore, M. J., Bell, C. D., Soltis, P. S., and Soltis, D. E. (2007). Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. U.S.A. 104, 19363–19368. doi: 10.1073/pnas.0708072104
Müller, S., Salomo, K., Salazar, J., Naumann, J., Jaramillo, M. A., Neinhuis, C., et al. (2015). Intercontinental long-distance dispersal of Canellaceae from the New to the Old World revealed by a nuclear single copy gene and chloroplast loci. Mol. Phylogenet. Evol. 84, 205–219. doi: 10.1016/j.ympev.2014.12.010
Nakai, T. (1919b). Genus novum Oleacearum in Corea media inventum. Bot. Mag. Tokyo 33, 153–154. doi: 10.15281/jplantres1887.33.392_153
Ni, L., Zhao, Z., Xu, H., Chen, S., and Dorje, G. (2016). The complete chloroplast genome of Gentiana straminea (Gentianaceae), an endemic species to the Sino-Himalayan subregion. Gene 577, 281–288. doi: 10.1016/j.gene.2015.12.005
Nock, C. J., Waters, D. L., Edwards, M. A., Bowen, S. G., Rice, N., Cordeiro, G. M., et al. (2011). Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 9, 328–333. doi: 10.1111/j.1467-7652.2010.00558.x
Nylander, J. A. A. (2004). MrModeltest 2.0. Evolutionary Biology Centre. Uppsala: Uppsala University.
Nylander, J. A. A., Wilgenbusch, J. C., Warren, D. L., and Swofford, D. L. (2008). AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics 24, 581–583. doi: 10.1093/bioinformatics/btm388
Palmer, J. D. (1991). “Plastid chromosomes: structure and evolution,” in Cell Culture and Somatic Cell Genetics of Plants, eds L. Bogorad, and I. Vasil (San Diego, CA: Academic Press), 5–53. doi: 10.1016/B978-0-12-715007-9.50009-8
Park, H., Kim, C., Lee, Y.-M., and Kim, J.-H. (2016). Development of chloroplast microsatellite markers for the endangered Maianthemum bicolor (Asparagaceae sl). Appl. Plant Sci. 4:apps.1600032. doi: 10.3732/apps.1600032
Pulquério, M. J. F., and Nichols, R. A. (2006). Dates from the molecular clock: how wrong can we be? Trends Ecol. Evol. 22, 180–184.
Rambaut, A., and Drummond, A. (2010). TreeAnnotator v1.6.1 in Computer Program and Documentation Distributed by the Author. Available at: http://beast.bio.ed.ac.uk
Rambaut, A., and Drummond, A. (2012). FigTree v1.4.0 in Computer Program and Documentation Distributed by the Author. Available at: http://tree.bio.ed.ac.uk/software/figtree/
Rambaut, A., and Drummond, A. (2014). Tracer v1.6 in Computer Program and Documentation Distributed by the Author. Available at: http://tree.bio.ed.ac.uk/software/tracer
Raubeson, L. A., and Jansen, R. K. (2005). “Chloroplast genomes of plants,” in Plant Diversity and Evolution of Plants: Genotypic and Phenotypic Variation in Higher Plants, ed. R. Henry (Wallingford: CABI Publishing), 45–68.
Roalson, E. H., and Roberts, W. R. (2016). Distinct processes drive diversification in different clades of Gesneriaceae. Syst. Biol. 65, 662–684. doi: 10.1093/sysbio/syw012
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rosati, C., Cadic, A., Duron, M., and Simoneau, P. (2007). “Forsythia,” in Transgenic Crops IV. Biotechnology in Agriculture and Forestry, Vol. 61, eds E. C. Pua and M. R. Davey (Berlin: Springer), 299–318.
Ryu, T. Y., Yeam, D. Y., Kim, Y. J., and Kim, S. J. (1976). Studies on dimorphism and heterostyle-incompatability of Abeliophyllum distichum. Seoul Nat. Coll. Agric. Bull. 1, 113–120.
Ryuk, J. A., Choi, G. Y., Kim, Y. H., Lee, H. W., Lee, M. Y., Choi, J. E., et al. (2010). Application of genetic marker and real-time polymerase chain reaction for discrimination between Forsythia viridissima and Forsythia suspensa. Biol. Pharm. Bull. 33, 1133–1137. doi: 10.1248/bpb.33.1133
SanMiguel, P., Tikhonov, A., Jin, Y.-K., and Motchoulskaia, N. (1996). Nested retrotransposons in the intergenic regions of the maize genome. Science 274, 765–768. doi: 10.1126/science.274.5288.765
Sax, K., and Abbe, E. C. (1932). Chromosome numbers and the anatomy of the secondary xylem in the Oleaceae. J. Arnold Arbor. 13, 37–48.
Soltis, D. E., and Kuzoff, R. K. (1995). Discordance between nuclear and chloroplast phylogenies in the Heuchera group (Saxifragaceae). Evolution 49, 727–742. doi: 10.1111/j.1558-5646.1995.tb02309.x
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun, H., McLewin, W., and Fay, M. F. (2001). Molecular phylogeny of Helleborus (Ranunculaceae), with an emphasis on the East Asian-Mediterranean disjunction. Taxon 50, 1001–1018. doi: 10.2307/1224717
Swofford, D. L. (2003). PAUP∗: Phylogenetic Analysis using Parsimony and Other Methods. Version 4. Sunderland, MA: Sinauer Associates.
Tae, K.-H., Kim, D.-K., and Kim, J.-H. (2005). Genetic variations and phylogenetic relationships of tribe Forsythieae (Oleaceae) based on RAPD analysis. Plant Resour. 8, 135–144.
Taylor, H. (1945). Cyto-taxonomy and phylogeny of the Oleaceae. Brittonia 5, 337–367. doi: 10.2307/2804889
Tu, T., Volis, S., Dillon, M. O., Sun, H., and Wen, J. (2010). Dispersals of Hyoscyameae and Mandragoreae (Solanaceae) from the New World to Eurasia in the early Miocene and their biogeographic diversification within Eurasia. Mol. Phylogenet. Evol. 57, 1226–1237. doi: 10.1016/j.ympev.2010.09.007
Wallander, E., and Albert, V. A. (2000). Phylogeny and classification of Oleaceae based on rps16 and trnL-F sequence data. Am. J. Bot. 87, 1827–1841. doi: 10.2307/2656836
Wen, J. (1999). Evolution of eastern Asian and eastern North American disjunct distributions in flowering plants. Annu. Rev. Ecol. Syst. 30, 421–455. doi: 10.1016/j.ympev.2012.05.003
Wikström, N., Savolainen, V., and Chase, M. W. (2001). Evolution of the angiosperms: calibrating the family tree. Proc. R. Soc. Lond. B Biol. Sci. 268, 2211–2220. doi: 10.1098/rspb.2001.1782
Willis, J. C., and Shaw, H. K. A. (1973). A Dictionary of the Flowering Plants and Ferns. Cambridge: Cambridge University Press.
Wu, F.-H., Chan, M.-T., Liao, D.-C., Hsu, C.-T., Lee, Y.-W., Daniell, H., et al. (2010). Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 10:68. doi: 10.1186/1471-2229-10-68
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xia, Y., Yang, B., Wang, Q., Liang, J., Wei, Y., Yu, H., et al. (2009). Quantitative analysis and chromatographic fingerprinting for the quality evaluation of Forsythia suspensa extract by HPLC coupled with photodiode array detector. J. Sep. Sci. 32, 4113–4125. doi: 10.1002/jssc.200900488
Xie, L., Yang, Z.-Y., Wen, J., Li, D.-Z., and Yi, T.-S. (2014). Biogeographic history of Pistacia (Anacardiaceae), emphasizing the evolution of the Madrean-Tethyan and the eastern Asian-Tethyan disjunctions. Mol. Phylogenet. Evol. 77, 136–146. doi: 10.1016/j.ympev.2014.04.006
Yamazaki, T., and Noshiro, S. (1993). “Oleaceae,” in Flora of Japan, eds K. Iwatsuki, T. Yamazaki, D. E. Boufford, and H. Ohba (Tokyo: Kodansha Ltd.), 122–135.
Yang, J.-B., Tang, M., Li, H.-T., Zhang, Z.-R., and Li, D.-Z. (2013). Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 13:84. doi: 10.1186/1471-2148-13-84
Yang, Z., and Yoder, A. D. (2003). Comparison of likelihood and Bayesian methods for estimating divergence times using multiple gene loci and calibration points, with application to a radiation of cute-looking mouse lemur species. Syst. Biol. 52, 705–716. doi: 10.1080/10635150390235557
Ye, C.-Y., Lin, Z., Li, G., Wang, Y.-Y., Qiu, J., Fu, F., et al. (2014). Echinochloa chloroplast genomes: insights into the evolution and taxonomic identification of two weedy species. PLOS ONE 9:e113657. doi: 10.1371/journal.pone.0113657
You, Y., Huber, M., Müller, R., Poulsen, C., and Ribbe, J. (2009). Simulation of the middle Miocene climate optimum. Geophys. Res. Lett. 36:L04702. doi: 10.1029/2008GL036571
Yu, Y., Harris, A. J., and He, X. J. (2015). RASP (reconstruct ancestral state in phylogenies): a tool for historical biogeography. Mol. Phylogenet. Evol. 87, 46–49. doi: 10.1016/j.ympev.2015.03.008
Zhang, Y., Du, L., Liu, A., Chen, J., Wu, L., Hu, W., et al. (2016). The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7:306. doi: 10.3389/fpls.2016.00306
Zhang, Z., Fan, L., Yang, J., Hao, X., and Gu, Z. (2006). Alkaloid polymorphism and ITS sequence variation in the Spiraea japonica complex (Rosaceae) in China: traces of the biological effects of the Himalaya-Tibet Plateau uplift. Am. J. Bot. 93, 762–769. doi: 10.3732/ajb.93.5.762
Zhisheng, A., Kutzbach, J. E., Prell, W. L., and Porter, S. C. (2001). Evolution of Asian monsoons and phased uplift of the Himalaya–Tibetan plateau since Late Miocene times. Nature 411, 62–66. doi: 10.1038/35075035
Keywords: Abeliophyllum, biogeographic origin, chloroplast genome, Forsythia, Forsythieae, molecular dating, phylogenetic relationship
Citation: Ha Y-H, Kim C, Choi K and Kim J-H (2018) Molecular Phylogeny and Dating of Forsythieae (Oleaceae) Provide Insight into the Miocene History of Eurasian Temperate Shrubs. Front. Plant Sci. 9:99. doi: 10.3389/fpls.2018.00099
Received: 31 July 2017; Accepted: 18 January 2018;
Published: 05 February 2018.
Edited by:
Tian Tang, Sun Yat-sen University, ChinaReviewed by:
Wei Wu, Sun Yat-sen University, ChinaJinfeng Chen, University of California, Riverside, United States
Copyright © 2018 Ha, Kim, Choi and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joo-Hwan Kim, kimjh2009@gachon.ac.kr
†These authors have contributed equally to this work.