
- 1 Virginia Institute for Psychiatric and Behavioral Genetics, Virginia Commonwealth University, Richmond, VA, USA
- 2 Department of Psychology, Neuroscience and Behaviour, McMaster University, Hamilton, ON, Canada
- 3 Department of Psychiatry, Virginia Commonwealth University, Richmond, VA, USA
Some evolutionary researchers have argued that current diagnostic criteria for major depressive disorder (MDD) may not accurately distinguish true instances of disorder from a normal, adaptive stress response. According to disorder advocates, neurochemicals like the monoamine neurotransmitters (serotonin, norepinephrine, and dopamine) are dysregulated in major depression. Monoamines are normally under homeostatic control, so the monoamine disorder hypothesis implies a breakdown in homeostatic mechanisms. In contrast, adaptationist hypotheses propose that homeostatic mechanisms are properly functioning in most patients meeting current criteria for MDD. If the homeostatic mechanisms regulating monoamines are functioning properly in these patients, then oppositional tolerance should develop with prolonged antidepressant medication (ADM) therapy. Oppositional tolerance refers to the forces that develop when a homeostatic mechanism has been subject to prolonged pharmacological perturbation that attempt to bring the system back to equilibrium. When pharmacological intervention is discontinued, the oppositional forces cause monoamine levels to overshoot their equilibrium levels. Since depressive symptoms are under monoaminergic control, this overshoot should cause a resurgence of depressive symptoms that is proportional to the perturbational effect of the ADM. We test this prediction by conducting a meta-analysis of ADM discontinuation studies. We find that the risk of relapse after ADM discontinuation is positively associated with the degree to which ADMs enhance serotonin and norepinephrine in prefrontal cortex, after controlling for covariates. The results are consistent with oppositional tolerance, and provide no evidence of malfunction in the monoaminergic regulatory mechanisms in patients meeting current diagnostic criteria for MDD. We discuss the evolutionary and clinical implications of our findings.
Introduction
Depression is an affective state of negative mood and low arousal. According to the current text revision version of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR), about of depression constitutes an episode of major depressive disorder (MDD) if it is denoted by at least five of nine symptoms: (1) depressed mood; (2) anhedonia; (3) significant weight loss (or gain), or a decrease (or increase) in appetite; (4) insomnia (or hypersomnia); (5) psychomotor retardation (or agitation); (6) fatigue or loss of energy; (7) feelings of worthlessness or guilt; (8) diminished ability to concentrate; and (9) recurrent thoughts of death (not just fear of dying), or suicidal thoughts or actions. One of the symptoms must be either depressed mood or anhedonia. Additionally, the episode must last for at least 2 weeks, and it must cause substantial distress or impairment in an important domain of functioning. Finally, the episode must not be better accounted for by bereavement (which assumes that the episode is not disordered if it causes significant impairment for up to 2 months), and must not be caused by a substance or a medical condition (American Psychiatric Association, 2000).
Millions of people are affected by depression each year. Approximately 6.6% of adults, or about 13 million of the adult US population, are estimated to have had episodes that meet current criteria for MDD within a year’s time (Kessler et al., 2007). Based on a single interview, the lifetime prevalence of MDD is estimated to be 16.2%, or 33 million of the adult population (Kessler et al., 2007). However, lifetime estimates from single interviews are conservative because interviewees vary greatly in their current age and they have difficulty recalling prior episodes of depressive symptoms (Wells and Horwood, 2004; Kessler et al., 2005; Kruijshaar et al., 2005; Moffitt et al., 2009). Longitudinal studies of community samples in which participants are repeatedly interviewed yield higher estimates of the lifetime prevalence of MDD, often exceeding 40% (Wells and Horwood, 2004; Kruijshaar et al., 2005; Moffitt et al., 2009).
Most episodes that meet diagnostic criteria for MDD are associated with stressors (e.g., bereavement, marital difficulties, interpersonal conflict, financial difficulties, health issues). For instance, in a longitudinal study of a large community sample of twins of both sexes, 88.1% of diagnosed episodes of MDD were associated with a stressor of some sort (Keller et al., 2007). Only 11.9% of episodes appeared to be endogenous (depression in the absence of an environmental trigger). However, even many of the apparently endogenous episodes could have reflected a reluctance to disclose stressors of a sensitive nature (Leff et al., 1970).
There are two broadly divergent approaches to the relationship between stressors and depression. In some way, the relationship must be mediated by the brain, but the neurological causes of depression are generally acknowledged to be unknown (Berton and Nestler, 2006). Many researchers, certain that depressive symptoms are maladaptive, search for evidence of stress-induced malfunction in the brain (for recent reviews, see Krishnan and Nestler, 2008; Savitz and Drevets, 2009). These researchers attempt to identify the neurological mechanisms responsible for the chronic nature of depression and its association with a variety of medical conditions (McEwen, 1998, 2007), the structural changes in the brain associated with depression and their effects on cognition (Sapolsky, 1996, 2000, 2001; Duman, 2004; Duman and Monteggia, 2006; Savitz and Drevets, 2009), and the neurochemical pathways of depression, including the mechanisms by which antidepressant medications (ADMs) reduce symptoms (for a review, see Krishnan and Nestler, 2008). An integrating theme in this research is that the neurochemistry of depression is disordered, although the precise neurochemicals that are dysregulated are the subject of much debate and research. Monoamine neurotransmitters, glucocorticoids, neurotrophins, and cytokines are the major chemicals thought to be involved in depression (Krishnan and Nestler, 2008). Regardless of the role of other chemicals, there seems to be consensus that the monoamine neurotransmitters – particularly serotonin (5-hydroxytryptamine or 5-HT), norepinephrine (NE), and, to a lesser extent, dopamine (DA) – are directly or indirectly involved in the biochemical pathways to depression (Krishnan and Nestler, 2008). All ADMs, for instance, act on monoamines through a variety of mechanisms (see below). Moreover, research on rodents in which monoaminergic receptors have been knocked out, or monoamine transmission has been disabled, has provided strong experimental evidence that monoamines play a role in depressive symptoms and in the antidepressant response, particularly 5-HT and NE (Heisler et al., 1998; Mayorga et al., 2001; Cryan et al., 2004; Dziedzicka-Wasylewska et al., 2006).
Other researchers argue that depressive symptoms might be better described by adaptive mechanisms that respond to stressors and threats (Price et al., 1994; Nesse, 2000, 2004; Watson and Andrews, 2002; Allen and Badcock, 2003, 2006; Hagen, 2003; Gilbert, 2006; Horwitz and Wakefield, 2007; Andrews and Thomson Jr., 2009; Nesse and Ellsworth, 2009; Price, 2009). These hypotheses do not propose that all episodes of depression are adaptive since all evolved adaptations in the body are susceptible to malfunction and disorder at some rate. Rather, the issue is whether the symptoms currently used to diagnose depressive disorder accurately distinguish the disordered state from normal, adaptive processes (Spitzer and Wakefield, 1999; Horwitz and Wakefield, 2007; Wakefield et al., 2007, 2010). Some adaptationist hypotheses restrict their application to symptoms and episodes that do not meet diagnostic thresholds, but such restrictions are intended to exclude true instances of disorder. Consequently, these hypotheses are not falsified in any rigorous sense by evidence that current diagnostic criteria are inaccurate. In any event, evolutionarily oriented researchers tend to focus on the ecological mapping of the symptoms of depression (and the cognitive and behavioral outputs) to the stressors that trigger episodes (Gilbert and Allan, 1998; Hagen, 1999, 2002; Badcock and Allen, 2003; Gilbert et al., 2004; Keller and Nesse, 2004, 2005, 2006; Andrews et al., 2007; Wrosch and Miller, 2009), largely treating the brain as a black box whose internal structure and operation are unknown.
Both approaches agree that the body has evolved adaptations for responding to stress. They both believe that, in general, environmental stressors trigger mechanisms in the body that regulate multiple body systems so that cognition and behavior can be adaptively modulated to meet the environmental challenge. They also agree that these evolved stress response mechanisms can malfunction. They primarily differ in what constitutes evidence of depressive disorder and the causes of depressive disorder.
Most articulations of the monoamine disorder hypothesis propose that forebrain levels of monoamine neurotransmitters are depleted in depressive disorder – particularly serotonin (5-HT), norepinephrine (NE) and, to a lesser extent, dopamine (DA). However, some have suggested that monoaminergic transmission may be enhanced in depression (Sapolsky, 2004). The precise direction of association between depression and forebrain monoamine levels is not crucial for the present paper. It is sufficient to say that the neurochemical disorder hypothesis proposes that monoamines, particularly 5-HT and NA, are perturbed in some way.
But monoaminergic perturbation, per se, is not evidence of disorder. The most prominent definition of disorder is based in evolutionary theory (Spitzer, 2007), and it argues that at heart of every disorder is an evolved adaptation that is malfunctioning (Wakefield, 1992). Under this definition, the claim that depression is a disorder of too much or too little of a neurotransmitter is ultimately a claim that the evolved mechanisms that control neurotransmitter levels are malfunctioning.
Monoamine neurotransmitter levels are normally under homeostatic control (Best et al., 2010). Homeostasis involves the regulation of an important substance or physiological parameter within a narrow range around an equilibrium. Minimally, homeostatic mechanisms have a sensor for determining how far the parameter deviates from the equilibrium and feedback mechanisms for bringing the parameter back to equilibrium (Woods, 2009). For instance, the homeostatic control of core body temperature involves neuronal sensors in the preoptic anterior hypothalamus that connect with various efferent pathways to exert feedback and keep temperature at equilibrium (Romanovsky, 2007). Additionally, many homeostatic mechanisms can raise or lower the equilibrium in response to environmental contingencies. Thus, the body often responds to an infection by raising the core body temperature equilibrium – otherwise known as fever (Romanovsky et al., 2005). Feedback mechanisms then maintain core body temperature around this elevated equilibrium. Because some infections last for weeks or months, fever can last for extended periods of time.
If depression is, in whole or in part, a disorder in the homeostatic mechanisms that regulate monoamine transmission, then it could involve malfunction in: (1) the sensors that monitor monoamine levels (2) the feedback mechanisms that maintain monoamine levels at the equilibrium; or (3) the mechanisms that alter the equilibrium point.
Although most adaptationist research treats the brain as a black box, a common (if unstated) implication of all adaptationist hypotheses for depression is that the relevant neurochemistry is altered to change cognition and behavior in ways that adapt the organism to the environmental contingencies that triggered the episode. An adaptationist perspective on the chronic nature of depression suggests that, like fever, monoamines may be under the control of a homeostatic mechanism that maintains forebrain levels at an altered equilibrium for an extended period of time, presumably because the nature of the stressor requires it (see, e.g., Andrews and Thomson Jr., 2009).
The disruption of a mechanism by pharmacological intervention can reveal whether it is under homeostatic control (Young and Goudie, 1995). Acute exposure to opiates inhibits the firing of noradrenergic neurons in the locus coeruleus by suppressing cyclic adenosine 3′,5′-monophosphate (cAMP) levels (Nestler and Aghajanian, 1997). However, with long-term (chronic) exposure, firing rates return to normal because a negative feedback mechanism upregulates the cAMP pathway so that it counteracts the drug’s effect and returns the system to the equilibrium (Nestler and Aghajanian, 1997). The neurological changes brought about by homeostatic mechanisms that counteract the disrupting effects of chronic drug exposure are referred to as oppositional tolerance (Young and Goudie, 1995; Fava and Offidani, 2011). These counteractive forces can cause the system to overshoot the equilibrium when the drug is abruptly discontinued (Young and Goudie, 1995; Fava and Offidani, 2011). Thus, when an opioid receptor antagonist is used to abruptly block an opiate, the upregulated cAMP pathway causes firing rates to exceed the normal rate (Nestler and Aghajanian, 1997).
Some researchers have called for greater research on the neurological responses of homeostatic mechanisms to chronic psychotropic drug use, including ADMs (Hyman and Nestler, 1996). Others have specifically hypothesized that oppositional tolerance to ADMs may develop in depression (Fava and Offidani, 2011). Whether the monoamines thought to cause major depression are under homeostatic control has important implications for debates about whether most episodes of MDD are instances of a properly functioning adaptation or true instances of disorder. Homeostatic mechanisms are classic examples of adaptations. Survival and reproduction require the internal environment of the organism to be regulated (Hochochka and Somero, 2002; Woods, 2009). The machinery needed to exert negative feedback to maintain physiological conditions within a narrow band around an equilibrium are often complex and difficult to explain by any process other than natural selection. Consequently, evidence that monoamine neurotransmitters were under homeostatic control in patients diagnosed with MDD would suggest that those mechanisms were not malfunctioning.
In this paper, we focus on a straightforward implication of the hypothesis that depression neurochemistry is under homeostatic control: ADM-induced perturbations to monoamine neurotransmitter levels should trigger oppositional tolerance. Previous research provides some support for oppositional tolerance to ADMs in patients diagnosed with MDD. It is not uncommon for depressive symptoms to reemerge after remission while patients are on maintenance ADM therapy (Byrne and Rothschild, 1998), which is consistent with tolerance (Fava and Offidani, 2011). Other support comes from a meta-analysis of studies examining the risk of relapse among patients with remitted symptoms after ADM treatment had been discontinued (Viguera et al., 1998). The authors found that the risk of relapse was positively associated with the duration of ADM treatment. While this relationship was not statistically significant, it became marginally significant in a re-analysis that included an additional study (Baldessarini et al., 2002). Such evidence suggests oppositional tolerance because longer treatment periods provide homeostatic mechanisms with more time to make neurological changes that oppose pharmacological perturbation (Fava and Offidani, 2011). However, several subsequent meta-analyses have failed to find any evidence that treatment duration affected the risk of relapse after discontinuation (Geddes et al., 2003; Kaymaz et al., 2008; Glue et al., 2010). At present, evidence of oppositional tolerance in depression is equivocal.
Antidepressant medications vary widely in the degree to which they perturb monoamine levels in the brain (see below). Consequently, perturbational differences in ADMs could affect the degree of oppositional tolerance. This is analogous to the negative feedback forces that springs exert when they are displaced from their equilibrium position. As one pulls a spring from its equilibrium position, the spring exerts an oppositional force that attempts to bring the spring back to equilibrium; the more further one displaces the spring from its equilibrium position, the greater the oppositional force that the spring produces. Similarly, ADMs with greater perturbational effects should trigger stronger oppositional forces that attempt to bring monoamine levels back to equilibrium. The buildup of oppositional tolerance under ADM treatment could then cause the system to overshoot its equilibrium upon discontinuation, and the degree of overshoot should be proportional to the perturbational effect of the ADM.
We hypothesize that monoamine levels are under homeostatic control in patients diagnosed with MDD, and that ADM treatment will cause oppositional tolerance. There are two implications for the present paper. First, the buildup of oppositional tolerance during ADM treatment will attempt to bring monoamine levels back to equilibrium, which will cause them to overshoot the equilibrium after ADM treatment is discontinued. Second, the degree to which monoamine levels overshoot the equilibrium will be positively related to the perturbational effect of the ADM. We do not directly test these implications because we lack relevant perturbational data in humans. Rather, we test two predictions that follow from them.
Since depressive symptoms are under monoaminergic control, ADM treatment tends to decrease depressive symptoms. However, oppositional tolerance during ADM treatment should build pressure for increasing symptoms (Fava and Offidani, 2011). After ADM treatment is discontinued, the overshoot of monoamines will cause an increase in symptoms that could exceed diagnostic thresholds for a relapse or a recurrence of MDD (hereafter, just relapse). Since oppositional tolerance will be absent in patients who resolve their episodes without ADM treatment, we predict that the risk of relapse among patients who discontinue ADM treatment will be higher than the risk of relapse among patients who remit without ADM treatment. Second, and more importantly, we predict that the risk of relapse will be positively related to the perturbational effect of the ADM.
We conduct a meta-analysis to test these predictions. Other meta-analyses of ADM discontinuation studies have been conducted (Viguera et al., 1998; Geddes et al., 2003; Kaymaz et al., 2008; Glue et al., 2010), but they have not been specifically designed to address these questions. Moreover, our meta-analysis offers an important methodological improvement. ADM discontinuation studies vary widely in the criteria that are used to define a relapse. Some studies use a threshold score on a single assessment instrument, while others may use a complex combination of different instruments assessed over multiple time periods. These definitional criteria can have a dramatic affect on the relapse rate (Montgomery and Dunbar, 1993; Keller et al., 1998). Studies that use more stringent relapse criteria make it more difficult for an increase in symptoms to qualify as a relapse, resulting in lower relapse rates (Montgomery and Dunbar, 1993). Prior meta-analyses have not attempted to control for the variability in the stringency of relapse criteria in discontinuation studies, possibly because it is difficult to objectively compare relapses defined by different instruments. For each study, we construct a variable (called stringency) that can be objectively calculated as the number of assessment hurdles that have to be passed before an increase in symptoms will be defined as a relapse. In general, studies with more assessment hurdles should make it more difficult for an increase in symptoms to qualify as a relapse. If so, our stringency variable should be a negative predictor of the relapse rate.
Before describing the design of our meta-analysis in detail, we discuss how the major ADM classes differ in the degree to which they perturb monoamine neurotransmitter levels.
Perturbational Effects of ADMs
Monoaminergic neurons originate in various areas of the midbrain and project to forebrain regions. After monoamines are released into the synapse, they are eventually taken back into the presynaptic neuron by molecules called transporters. Once they are taken back inside the cell, monoamines are broken down by two enzymes – monoamine oxidase A (MAO-A), which breaks down 5-HT, NE, and DA, and monoamine oxidase B (MAO-B), which breaks down DA, but not 5-HT or NE.
All ADMs have multiple pharmacological effects by binding to various receptors, transporters, and enzymes. This could make it difficult to classify them in terms of their perturbational impact. However, one relatively clear measure of perturbational impact is by whether cortical levels of the monoamine neurotransmitters – 5-HT, NE, and DA – are actually affected. When cortical levels of a neurotransmitter are influenced by an ADM, the drug exerts perturbational influence through all the receptors, transporters, and enzymes to which the neurotransmitter binds.
The perturbational effects of ADMs on monoamine forebrain levels have not been directly measured in humans because it requires invasive techniques. However, microdialysis studies of rodents have examined the perturbational effects of ADMs in various brain regions. Since there is regional variability in the effects of ADMs, we look at microdialysis studies assessing the in vivo effects of ADMs on rodent medial prefrontal cortex (mPFC) monoamine levels. We choose the mPFC because it plays an important role in behavioral depression in rodents (Amat et al., 2005, 2006, 2010; Baratta et al., 2009). In human depression, the ventrolateral prefrontal cortex (VLPFC) is thought to be important (Drevets, 1998; Andrews and Thomson Jr., 2009), which may seem incongruent with the rodent literature. However, the rodent brain is organized differently than the human brain, and the rodent mPFC is a likely structural and functional homolog to the human VLPFC (Uylings and Vaneden, 1990; Kesner, 2000; Uylings et al., 2003). The perturbational effects of ADMs on rodent mPFC monoamine levels may correspond to their perturbational effects in human brain regions implicated in depression, such as the VLPFC. Unless otherwise indicated, we rely on microdialysis studies in which a single dose in the 10–15 mg/kg range was systemically administered (either by intraperitoneal injection or oral administration). This dose range is commonly used and approximates clinically relevant doses (Koch et al., 2003). From these studies, we extracted the average percent increase of each monoamine (5-HT, NE, and DA) above baseline in the mPFC (see Table 1).
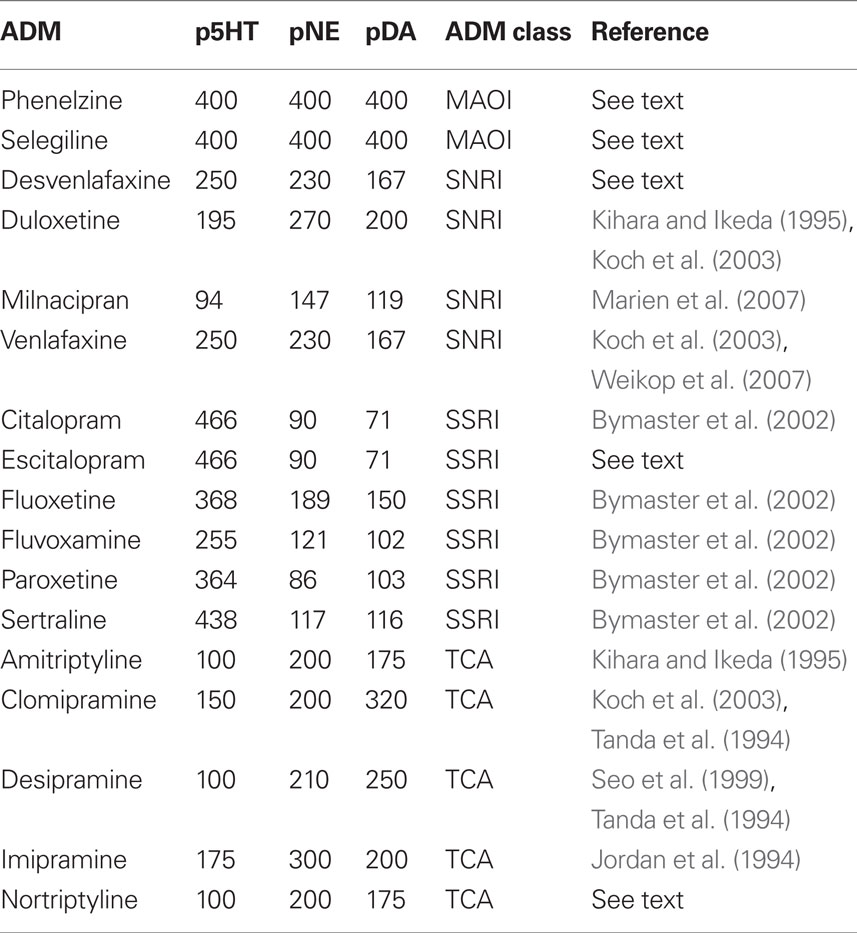
Table 1. Perturbational effects of antidepressants on mPFC levels of monoamine neurotransmitters (expressed as percent over baseline).
Microdialysis studies of mPFC monoamine levels can be found for most of the major standalone ADMs used in discontinuation studies. However, we lack mPFC microdialysis data for the two monoamine oxidase inhibitors (MAOIs) – phenelzine and selegiline – that have been used in discontinuation studies. Phenelzine is a dual MAO-A/B inhibitor, and so is selegiline at the doses used in the discontinuation study (Stahl, 2008). As noted above, the inhibition of MAO-A primarily increases 5-HT and NE. Although MAO-B does not metabolize 5-HT or NE, it does metabolize β-phenylethylamine, which is related to the release of catecholamines, including 5-HT and NE (Kitaichi et al., 2010). The inhibition of MAO-B can therefore indirectly increase 5-HT and NE as well as directly increase DA (Kitaichi et al., 2010).
Of the ADMs, dual MAO-A/B inhibitors exert the most powerful effects on monoamine levels. Due to differences in regional effects of ADMs, many ADMs do not increase monoamine levels when averaged across the whole rodent brain, yet dual MAO-A/B inhibitors are powerful enough to do so (Campbell and Marshall, 1974; Fuller et al., 1974; Renard et al., 2004; Bano et al., 2010). In one study, a single dose of phenelzine increased whole brain levels of 5-HT by 240%, NE by 150%, and DA by 170% (Parent et al., 2000).
In mPFC, dual MAO-A/B inhibitors tend to have greater effect than the whole brain average. The inhibition of MAO-A by itself increases mPFC levels of 5-HT and NE by 200–300% (Finberg et al., 1993; Bel and Artigas, 1995), but the simultaneous inhibition of MAO-A and MAO-B causes increases of at least 400% (Bel and Artigas, 1995; Takahashi et al., 2000; Kitaichi et al., 2010). We therefore conservatively assume that phenelzine and selegiline (at dual inhibition doses) increase mPFC monoamine levels by 400% above baseline (Table 1). However, since we do not have precise mPFC measurements for phenelzine and selegiline, for our analyses we capped the maximum perturbational impact of ADMs at 400%. We label the perturbational effect of a drug for each monoamine as follows: p5HT (for serotonin), pNE (for norepinephrine), and pDA (for dopamine).
The perturbational effects of other ADMs generally correspond to the class to which they belong, but there are exceptions and, more importantly, differences in degree. The selective serotonin reuptake inhibitors (SSRIs) selectively bind to the serotonin transporter, and so they tend to increase cortical levels of serotonin. The effects of citalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline on mPFC monoamine levels were assessed in a single study (Bymaster et al., 2002). Fluoxetine is unique among the SSRIs in that it also increases NE and, to a lesser extent, DA in the mPFC (Bymaster et al., 2002; Stahl, 2008). The mechanism is not by reuptake blockade, but by antagonism of 5-HT2C receptors (Stahl, 2008). While we lack microdialysis data for escitalopram in the 10–15 mg/kg range, escitalopram is the active S-enantiomer of citalopram (Stahl, 2008), so we use the same perturbational values as for citalopram.
The serotonin norepinephrine reuptake inhibitors (SNRIs) selectively bind to both the 5-HT and NE transporters, although, as a class, they tend to have greater affinity for the 5-HT transporter than the NE transporter. ADMs that inhibit NE reuptake also tend to increase prefrontal levels of DA to some degree because there are few DA transporter molecules in the prefrontal cortex, so DA has a tendency to diffuse widely from the synapse (Stahl, 2008). DA eventually gets taken back into neurons by the NE transporter, which has a higher affinity for DA than NE (Stahl, 2008). We relied on several studies (Kihara and Ikeda, 1995; Koch et al., 2003; Marien et al., 2007; Weikop et al., 2007) to get data on the mPFC monoamine effects of the SNRIs used in discontinuation studies – desvenlafaxine, duloxetine, milnacipran, and venlafaxine. Venlafaxine does not have much effect on NE at 15 mg/kg, but it has a larger effect at 40 mg/kg (Koch et al., 2003). The discontinuation studies of venlafaxine use doses that are much higher than the recommended dose, so we use the data for 40 mg/kg. We lacked microdialysis data for desvenlafaxine, but it is the active metabolite of venlafaxine (Stahl, 2008). In the discontinuation study of desvenlafaxine, it was also administered at doses that are much higher than the recommended dose, so we use the same perturbational values as for venlafaxine.
The tricyclic antidepressants (TCAs) also bind to the 5-HT and NE transporters, although, as a class, they tend to have greater affinity for the NE transporter. We relied on several studies (Jordan et al., 1994; Tanda et al., 1994; Kihara and Ikeda, 1995; Koch et al., 2003; Bongiovanni et al., 2008) to get data on the mPFC monoamine effects of the TCAs that have been used in discontinuation studies – amitriptyline, clomipramine, desipramine, imipramine, and nortriptyline. We lacked microdialysis data for nortriptyline, but since it is the active metabolite of amitriptyline, we assigned it the same perturbational values.
Study Overview
To summarize, we conduct a meta-analysis to test two predictions of the hypothesis that monoamine levels are under homeostatic control in patients diagnosed with MDD. First, patients who remit with ADM treatment will have a higher risk of relapse after treatment stops than patients who remit without ADM treatment. Second, the risk of relapse after ADM treatment is discontinued will be positively related to the perturbational effects of the ADM on prefrontal monoamine levels.
Two types of studies are relevant to these predictions. In ADM extension studies, patients diagnosed with MDD are initially randomly assigned to ADM or placebo during the treatment phase, and then remitters in both groups are followed in an extension phase in which they continue to receive the same treatment. Since our first prediction relates to the risk of relapse after remission without ADM treatment, we only consider the Placebo–Placebo arms of extension studies. We put placebo in its own class (PBO) and assign to placebo treatment a perturbational score of 100 (no effect on baseline levels) for each of the monoamines.
In ADM discontinuation studies, all patients diagnosed with MDD go through an initial ADM treatment phase and then remitters go through a discontinuation phase in which they are randomly assigned, under double-blind conditions, to either continued treatment (Drug–Drug) or placebo (Drug–Placebo). Since our second prediction deals with the risk of relapse after ADM discontinuation, we only consider the Drug–Placebo arms of discontinuation studies.
In our meta-analysis, we include all the randomized, double-blind, placebo-controlled ADM extension studies we can find since patients in the Placebo–Placebo arms of these studies have not received any pharmacological treatment. However, we limit analysis to ADM discontinuation studies in which patients receive chronic treatment of a single ADM from one of the major standalone classes – the MAOIs, the TCAs, the SNRIs, or the SSRIs. As a practical matter, microdialysis data on the mPFC monoaminergic effects of ADMs from outside these classes is limited.
Study Selection
Design of relevant studies
Studies included in our meta-analysis had to meet several design criteria. First, patients must have had a primary diagnosis of an episode of unipolar major depression (MDD, recurrent or chronic, with or without dysthymia). A variety of diagnostic methods were allowed. Additionally, most published studies excluded depressed patients who had a diagnosis of bipolar disorder or concurrent alcohol or substance abuse. We therefore excluded studies specifically focusing on these special populations. We did not exclude studies in which patients were diagnosed with other comorbid conditions, such as anxiety disorder or diabetes (Lustman et al., 2006) provided they met all other criteria.
Second, each study had to have an initial period of ADM treatment followed by randomization to continued treatment with ADM or placebo under double-blind conditions. We allowed studies with multiple treatment phases, provided ADM treatment was continuous from the beginning of the study until discontinuation.
Third, we excluded any study that did not involve at least one of the major ADM classes – MAOI, TCA, SNRI, or SSRI.
Fourth, we excluded any study in which multiple ADMs from different classes were simultaneously administered to the same individuals because we were interested in isolating the effects of individual drug classes. We did not allow the sequential administration of different ADMs, because they have different perturbational effects.
Fifth, we excluded any study in which patients also received psychotherapy because it has been shown to influence relapse rates after discontinuation.(Hollon et al., 2006).
Sixth, to avoid double-counting, we excluded any report that was a re-analysis of data already included in the analysis.
We included part of a study in which patients were given fluoxetine for 12 weeks and then randomized to one of four branches (Reimherr et al., 1998): (1) placebo for the duration of the study; (2) continued treatment with fluoxetine for 14 weeks before assignment to placebo; (3) continued treatment with fluoxetine for an additional 36 weeks followed by placebo; and (4) continued treatment with fluoxetine for the duration of the study. We include the relapse results from only the first assessment period (12 weeks after randomization), because this was the only assessment in which all subjects in the placebo group had been treated with fluoxetine for the same period of time.
We excluded another study (Eric, 1991) because it was a conference abstract that provided a preliminary report of an ongoing study. Consequently, important information was missing (e.g., average age, proportion of patients who were women), and it appeared to be a preliminary report of a later published study (Montgomery and Dunbar, 1993).
Search strategy
Our search strategy involved two steps. First, we used four meta-analyses of discontinuation studies (Viguera et al., 1998; Geddes et al., 2003; Kaymaz et al., 2008; Glue et al., 2010) and a recent meta-analysis of extension studies (Zimmerman et al., 2007) that used rigorous search criteria as baseline sources. Second, we conducted searches of the Web of Science database covering 1965 until October 8, 2010. To search for discontinuation studies, we used the keywords “antidepress* and placebo and (relapse or recurren*) and (discontinu* or mainten* or continua*).” Then, we conducted similar searches substituting specific drugs for “antidepress*” (e.g., “sertraline,” “phenelzine,” “amitriptyline,” etc.). To search for extension studies, we used the keywords “antidepress* and placebo and (relapse or recurren*) and (mainten* or continua*) and extension,” The flow diagram for the search procedure is in Figure 1.
Figure 1. Flow diagram of the search strategy.
Variables
From each study, we extracted information on a number of variables that were either directly relevant to our hypotheses, or were important covariates to control for.
Demographic variables (age, sex)
We coded each study for the average age of patients (variable name = age), and for the proportion of patients who were women (variable name = sex), who were randomized to placebo. Two studies did not report the average age or sex composition of the patients randomized to placebo (Doogan and Caillard, 1992; Montgomery et al., 1993), so we took the demographic characteristics of all those who were subject to randomization instead (Doogan and Caillard, 1992; Montgomery et al., 1992). Five studies did not report any information on the age of the sample, and two of these studies did not provide any information on the sex composition of the sample (see Table A1 in Appendix).
Depression history (dephx)
In principle, people with greater histories of depression could be at a greater risk of relapse (Kendler et al., 2000). We therefore coded each study for the minimum number of prior episodes required for entry into the study (variable name = dephx). Where not reported, we assumed no restriction.
Treatment resistance (txresist)
In principle, patients who are more resistant to pharmacological treatment could be at greater risk of relapse (Rush et al., 2006). We therefore coded each study for whether treatment resistant patients were targeted. Studies were coded as involving treatment resistant patients if they included hospitalized patients, or targeted patients with chronic or atypical depression (variable name = txresist).
Drug variables (class, half-life)
Characteristics of ADMs other than their perturbational effects on mPFC monoamine levels could, in principle, be predictors of the risk of relapse. The elimination half-life of the drug is thought to be pharmacologically important, so we included it in some of our analyses (variable name = half-life) (see Table 2). We do not use the half-lives of phenelzine or selegiline as they irreversibly bind to monoamine oxidase, so effects may last for 2 weeks after discontinuation (Lemke et al., 2010). We took all half-life values from Sadock and Sadock (2005), using the value of the parent compound or its longest acting active metabolite. Where a range in half-life was reported, we took the midpoint.
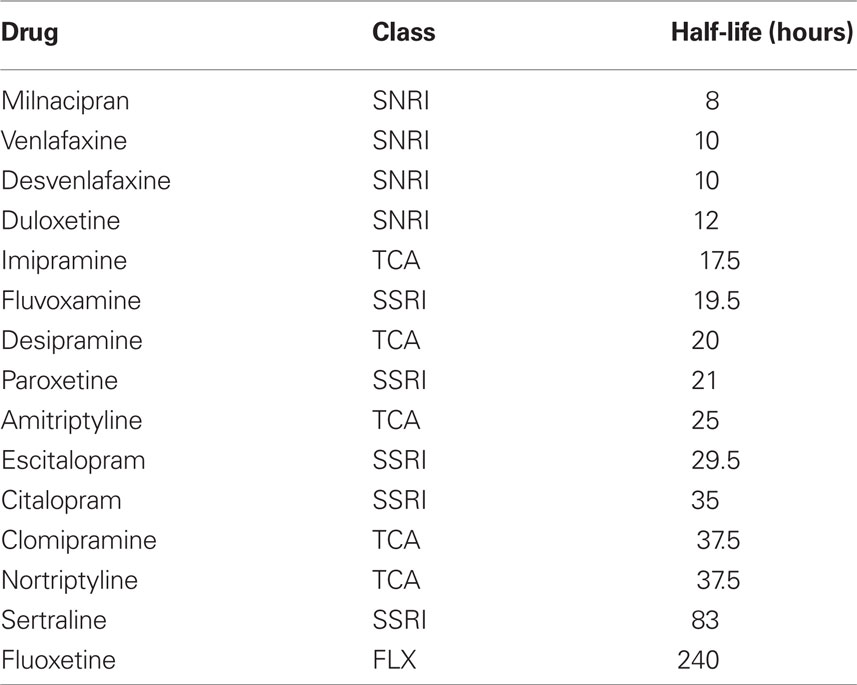
Table 2. Half-lives of different antidepressant drugs.
Treatment duration (txdur)
The duration of acute treatment with an ADM before transition to placebo could potentially be an important predictor of the risk of relapse, as discussed in the introduction. Ideally, each study would have a clearly defined treatment period, which is the time from the onset of ADM initiation to randomization to placebo (variable name = txdur). Unfortunately, patients were subjected to treatment for varying time periods in three of the MAOI studies (Davidson and Raft, 1984; Harrison et al., 1986; Stewart et al., 1997) and four of the TCA studies (Mindham et al., 1973; Coppen et al., 1978; Cook et al., 1986). For these studies, we used the average treatment period or the midpoint in the range. For one TCA study, the only information available was that acute treatment persisted for 6 weeks after remission of the index episode (Coppen et al., 1978). Since there is a therapeutic delay of several weeks for all ADMs, we assumed a treatment period of 10 weeks. Time periods are usually reported in weeks. We convert this time to 30-day months. Often, time periods of 24 weeks are referred to as being equal to 6 months, which only makes sense if months are considered 4 week periods. For this reason, when time periods are reported in months, we assume that each month is equal to 4 weeks.
Dropout rate during treatment (droprate)
Patients may drop out of discontinuation and extension studies, most commonly because they fail to remit or because they have adverse effects from the treatment. This could be an important covariate influencing the relapse rate. In principle, the people who drop out could be more vulnerable to relapsing, leaving patients who remain in the sample more resistant to relapse. We calculated the proportion of patients who dropped out of each study during the treatment phase as the number of patients who dropped out divided by the total number of patients who entered the study (variable name = droprate). Ten studies lacked information on dropout rate (see Table A1 in Appendix).
Relapse risk period (riskper)
The studies also varied in the period of time after randomization to placebo during which patients were to be monitored for relapse. This could be an important predictor of relapse, with longer monitoring periods associated with a greater risk of relapse. As with treatment duration, this is often reported in weeks. We convert such time periods into 30-day months. When reported in months, we assumed that each month was equivalent to 4 weeks.
Stringency of criteria for defining depressive relapse (stringency)
We do not distinguish between patients who experienced a recurrence (onset of a new episode) from those who relapsed back into their index episode. For consistency, we refer to all lapses of remission as relapses, though some may be recurrences.
As discussed in the introduction, the stringency of criteria used to define a relapse (hereafter, stringency) should be an important negative predictor of the relapse rate. Commonly used tools for assessing depressive symptoms included the Hamilton Rating Scale for Depression (HRSD; Hamilton, 1960), the Clinical Global Impression-Improvement (CGI-I) or –Severity (CGI-S) scales (Guy, 1976), the Montgomery–Asberg Depression Research Scale (Montgomery and Asberg, 1979), DSM-III or DSM-IV criteria, the Beck Depression Inventory (Beck and Beamesderfer, 1974), or clinical judgment. Studies could vary on the number of these assessment tools and the cutoff scores used to diagnose a relapse. Additionally, studies varied on the duration of time that symptoms must persist.
Taking into account the stringency of relapse criteria is difficult as the variety of qualitatively different criteria prohibits easy comparison. However, there are strong a priori reasons to believe that stringency must be an important variable that affects relapse rates. Simply put, the more stringent the criteria that a study uses to define a relapse, the more difficult it will be for patients to meet the criteria, and overall relapse rates should be lower.
We operationalize stringency as the number of assessment hurdles that must be passed for an episode to be deemed a relapse. This has the drawback of ignoring cutoff differences. Thus, a study that defines relapse as requiring a HRSD > 18 would be given the same stringency score as a study that requires a HRSD > 12. However, there are very few studies that could be directly compared solely on the basis of differences in cutoff scores, so it is an impractical metric.
The advantage of using the number of assessment hurdles is that it is an objective way for us to compare the studies, and it leads to a natural way of taking into account differences in the requirements for duration of symptoms. For instance, if a study defines a relapse as requiring a HRSD > 15 and a CGI-S > 3 for 2 weeks in a row, then we assign it a stringency rating of four (one HRSD assessment and one CGI-S assessment must be passed each of the 2 weeks). When a study uses a number of criteria paths by which a patient can be designated as relapsed, we give priority to the path with the shortest duration requirement, then to the path with the fewest number of assessments that must be passed.
Two of the authors, Paul W. Andrews and Lisa J. Halberstadt, independently rated each study for the stringency of the relapse criteria. Agreement between the two ratings was very high: kappa = 0.782, weighted kappa = 0.903, and tetrachoric correlation = 0.986. Differences between the two ratings were then reconciled.
Nearly all the studies had stringency scores ranging from 1 to 4. However, two studies used a complex combination of assessments and symptom duration requirements (Keller et al., 1998; Amsterdam and Bodkin, 2006). Using our strict definition for the number of assessment hurdles that must be passed, one study (Amsterdam and Bodkin, 2006) would receive a rating of 8 (2 for the number of weeks that DSM-IV criteria must be satisfied, 2 for the number of weeks that HRSD > 13, 2 for the number of weeks that patients must have CGI-S ≥ 3, and 2 for the number of weeks that the CGI-S score must be elevated by 2 points above the baseline score). The other study (Keller et al., 1998) would receive a rating of 11 (4 for the number of weeks that DSM-IV criteria must be satisfied, 2 for the number of weeks that patients must have a CGI-S ≥ 4, 2 for the number of weeks that a patient must have a CGI-I ≥ 3, 2 for the number of weeks that there must be a 4 point increase in HRSD score over that of the maintenance phase, and 1 for confirmation by a senior investigator). These studies are clearly outliers. Moreover, retaining these scores could be misleading if some of the additional hurdles were redundant and had little impact on relapse rates. We therefore considered a range of stringency scores of 5–8 for these studies, with the minimum value reflecting the fact that their relapse definitions were more stringent than those of other studies. All reported results are based on stringency scores of 5. Unless otherwise stated, results do not change significantly with different stringency scores.
Relapse rate (relrate)
We calculated the relapse rate (variable = relrate) as the proportion of patients who had been randomized to placebo who satisfied relapse criteria over the relapse risk period. It was not uncommon for the authors of a study to report in the text of the paper a relapse number that was lower than the number that could be derived from the Kaplan–Meier survival curve (Montgomery et al., 1993; Robert and Montgomery, 1995; Hochstrasser et al., 2001; Rickels et al., 2010). This occurred because the study authors were, for some reason, analyzing relapse disparities between the Drug–Drug and Drug–Placebo groups at a time point before the end of the relapse risk period. This may not have been an unreasonable thing to do for those studies, but for the purposes of this analysis it is important to understand precisely how many relapses occurred over a specific interval. In such situations, we took the relapse number from the Kaplan–Meier survival curves for the relapse risk period that the authors reported in their study.
Imputation of Missing Values
As discussed above, some studies did not provide information about the average age, sex composition, or the dropout rate during acute treatment. We assumed that this data were missing at random, which means that the missing data may depend on variables that are known, but not on unknown variables. Under such circumstances, it is reasonable to use multiple regression to impute the missing data based on the data for which information is available (Little and Rubin, 1989), which we did using IVEware for SAS (Raghunathan et al., 2002). We used the following variables to impute values: year in which the report was published, txresist, dephx, stringency, the specific antidepressant, ADM class, the perturbational scores for each of the monoamines in mPFC, relapse rate, txdur, and riskper.
Often, a missing data point represents a person about whom a piece of information is missing. In this case, imputation involves using regression to predict the unknown information based on all the known information in the entire sample plus a stochastic element to represent the fact that the missing information cannot be predicted with certainty. However, in our situation, a missing data point is not an individual but a study in which a summary statistic – the average age, sex composition, or dropout rate – is missing. We want to impute the missing summary statistics (rather than the missing data of an individual) based on the summary statistics from all the studies that were available. However, imputing with a stochastic component would introduce far more variability into the summary statistic than actually existed. For this reason, we imputed without stochasticity. Due to the non-stochasticity, the estimates for analyses in which imputed data were used are slightly more precise (their SE are smaller) than they should be. However, since age, sex, and dropout rate were not significant predictors of the relapse rate, this problem would not apply to our best-fitting model.
Statistical Analysis
We conducted weighted least squares regression analyses using PROC REG in SAS on both the non-imputed and the imputed datasets. Since the results were not significantly different, we only report the results from the imputed data. All studies were weighted by the inverse of the variance for the proportion of patients with a relapse during the discontinuation phase. To avoid over-fitting problems that can arise from having more variables than studies, we constructed a best-fitting model. We did this by examining how the removal of each variable influenced the adjusted R2, keeping all the other variables in the model. We then eliminated the one variable that improved the fit the most. This procedure was repeated with the remaining variables until all model variables were significant or their elimination could not improve the fit. In all reported models, the results have been examined for, and are unaffected by, influential data points. No reported models showed signs of over-fitting.
Results
Forty-six papers met inclusion criteria, constituting 49 samples and 3,454 patients (Klerman et al., 1974; Coppen et al., 1978; Stein et al., 1980; van Praag and de Haan, 1980; Davidson and Raft, 1984; Harrison et al., 1986; Montgomery et al., 1988, 1993, 1998, 2004; Georgotas et al., 1989; Robinson et al., 1991; Doogan and Caillard, 1992; Claghorn and Feighner, 1993; Montgomery and Dunbar, 1993; Anton et al., 1994; Robert and Montgomery, 1995; Bremner and Smith, 1996; Entsuah et al., 1996; Kocsis et al., 1996, 2007; Stewart et al., 1997; Keller et al., 1998; Reimherr et al., 1998; Terra and Montgomery, 1998; Reynolds et al., 1999; Alexopoulos et al., 2000; Rouillon et al., 2000; Schmidt et al., 2000; Gilaberte et al., 2001; Hochstrasser et al., 2001; Klysner et al., 2002; Wilson et al., 2003; Detke et al., 2004; Simon et al., 2004; Amsterdam and Bodkin, 2006; Kamijima et al., 2006; Lustman et al., 2006; McGrath et al., 2006; Perahia et al., 2006, 2009; Gorwood et al., 2007; Cheung et al., 2008; Dobson et al., 2008; Emslie et al., 2008; Rickels et al., 2010). The information extracted from each study is listed in Table A1 in the Appendix. In Figure 2, we provide a forest plot of the relapse rates of individual studies, grouped by the particular ADM used, and ADMs in the same class are organized alphabetically.
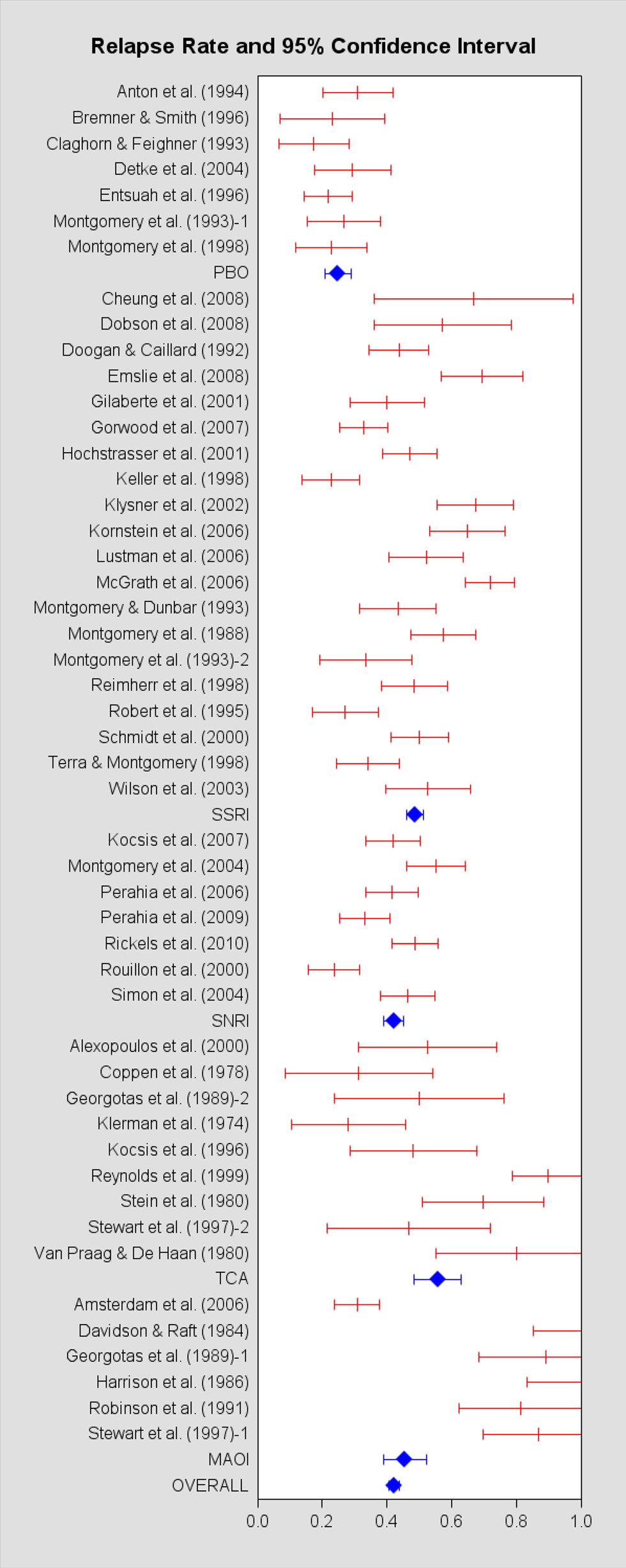
Figure 2. Forest plot of the relapse rate and 95% confidence interval for each study meeting inclulsion criteria.
Table 3 provides basic summary statistics. The PBO studies had the unadjusted relapse rate, which is consistent with our first prediction. However, this is also consistent with the possibility that the PBO studies tended to include patients with a lower risk of relapse (e.g., patients were less likely to have a prior history of depression, or they were more resistant to pharmacological treatment). Moreover, the PBO studies had the highest dropout rate during acute treatment, while the ADM classes had similar dropout rates. The dropout rate pattern suggests a potential selection bias problem for the PBO group, so we include the dropout rate as a covariate in all our regression analyses, unless otherwise specified. The SSRI studies had the second highest raw relapse rate, after the TCAs, while the MAOIs had the second lowest raw relapse rate of the ADM classes.
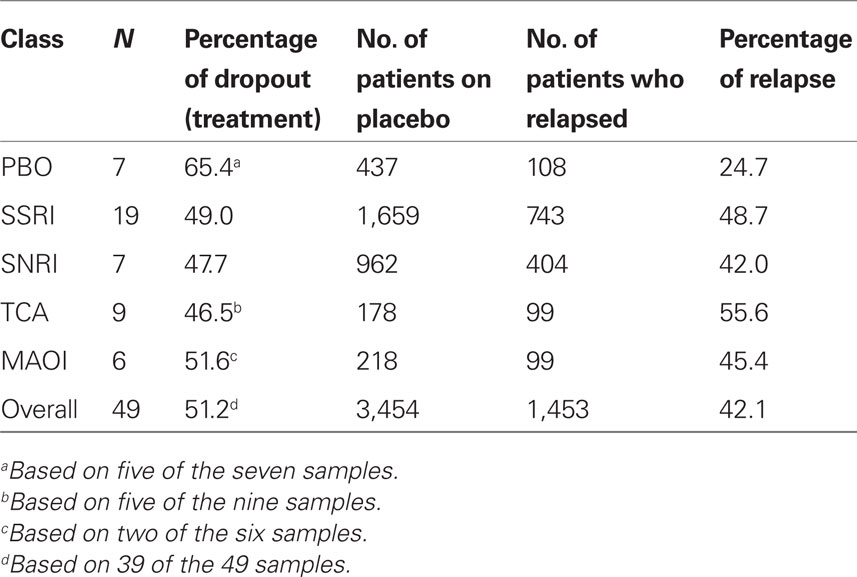
Table 3. The number of samples (N), the dropout rate during the treatment phase, and the relapse rate after randomization to placebo for each ADM class.
Correlations
For the ADM discontinuation studies, we examined the bivariate correlations between the continuous and count variables (Table 4). The perturbational effects of NE and DA are both significantly correlated with the relapse rate, but the correlation between pNE and pDA approaches unity, indicating a collinearity problem. The perturbational effect of 5-HT is not significantly correlated with the relapse rate, but it is related to other variables that could be potential confounds (risk period, treatment duration). Partially consistent with our prediction, there is a marginally significant, negative correlation between the stringency variable and the relapse rate. The stringency variable also shows a marginally significant relationship between the risk period and treatment duration, suggesting that they could be potential confounds for each other. Finally, the risk period and treatment duration are significantly correlated with each other, suggesting that they could also be potential confounds for each other.
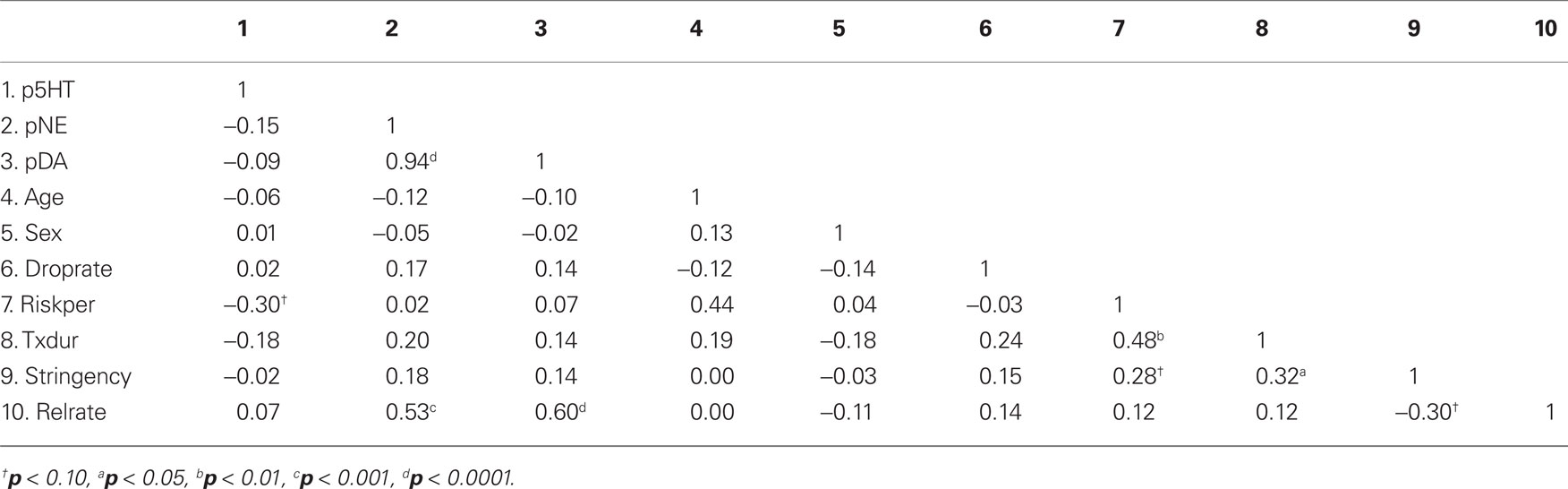
Table 4. The bivariate correlations between the continuous and count variables.
Predictors of the Dropout Rate
To formally test whether there is a potential selection bias problem in the PBO group due to a higher dropout rate during acute treatment, we regressed the dropout rate on the ADM classes and tested for a formal contrast of the ADM classes against the PBO class. We included as covariates the average age, the proportion of patients who were female, the duration of acute treatment, whether treatment resistant patients were targeted in the study, and the minimum number of depressive episodes required for entry. The contrast was significant, p = 0.003, indicating that the ADM classes had lower dropout rates than the PBO group. Additionally, the duration of acute treatment was a positive predictor of dropout rate, β =+0.023, SE = 0.010, p = 0.03. Subsequent analysis showed that it was driven by the ADM classes (not the PBO class), suggesting that longer drug treatment caused higher dropout, possibly due to adverse drug effects. In short, these results suggested that there could be a potential selection bias problem in the PBO studies.
Does the Dropout Rate Predict the Relapse Rate?
Next, we regressed the relapse rate on the dropout rate to see whether the potential selection bias problem was affecting the relapse rate. By itself, dropout rate was not a significant predictor, β = −0.213, SE = 0.225, p = 0.35. We then added covariates (age, sex, txresist, txdur, dephx), but the dropout rate was still not significant, β = −0.262, SE = 0.257, p = 0.32.
Even though we had no evidence that the dropout rate – by itself or in conjunction with covariates – was a predictor of the relapse rate, we included it as a covariate in our subsequent analyses unless otherwise indicated. We also included the interaction of dropout rate with a binary treatment variable (PBO or ADM), but it was not significant in any of the analyses, so we do not report it.
Does the Stringency Variable Negatively Predict the Relapse Rate?
The correlational results suggested that the stringency of relapse criteria could be an important negative predictor of the relapse rate. To give a visual presentation of this, we graphed the relapse rate as a function of the stringency variable for each class (see Figure 3). Within each class, the relapse rates tend to decline with stringency, suggesting that stringency is an important covariate. Moreover, the MAOIs have the highest relapse rates at each level of stringency. It is only for a stringency score of 5 or greater that the MAOI relapse rate drops off substantially. This is actually due to a single study (Amsterdam and Bodkin, 2006) with the second highest score on the stringency variable in our meta-analysis (stringency score = 8). Comparison of this figure with Table 3 suggests that, consistent with our second prediction, the ADMs with the greatest perturbational effects may actually have the highest relapse rates, but these can be obscured if one does not take the stringency of relapse criteria into account. To help illustrate this point further, we separated out the fluoxetine (FLX) studies from the other SSRIs. As discussed above, fluoxetine is unique among the SSRIs in that it causes NE and DA to increase in the mPFC through a mechanism other than reuptake blockade, which makes it substantially more perturbational than other SSRIs. Consistent with our second prediction, the fluoxetine studies tend to have higher relapse rates than the other SSRIs. In fact, the relapse rates of the fluoxetine studies seem to be more comparable to that of the SNRI and TCA studies – other ADM classes that influence NE and DA.
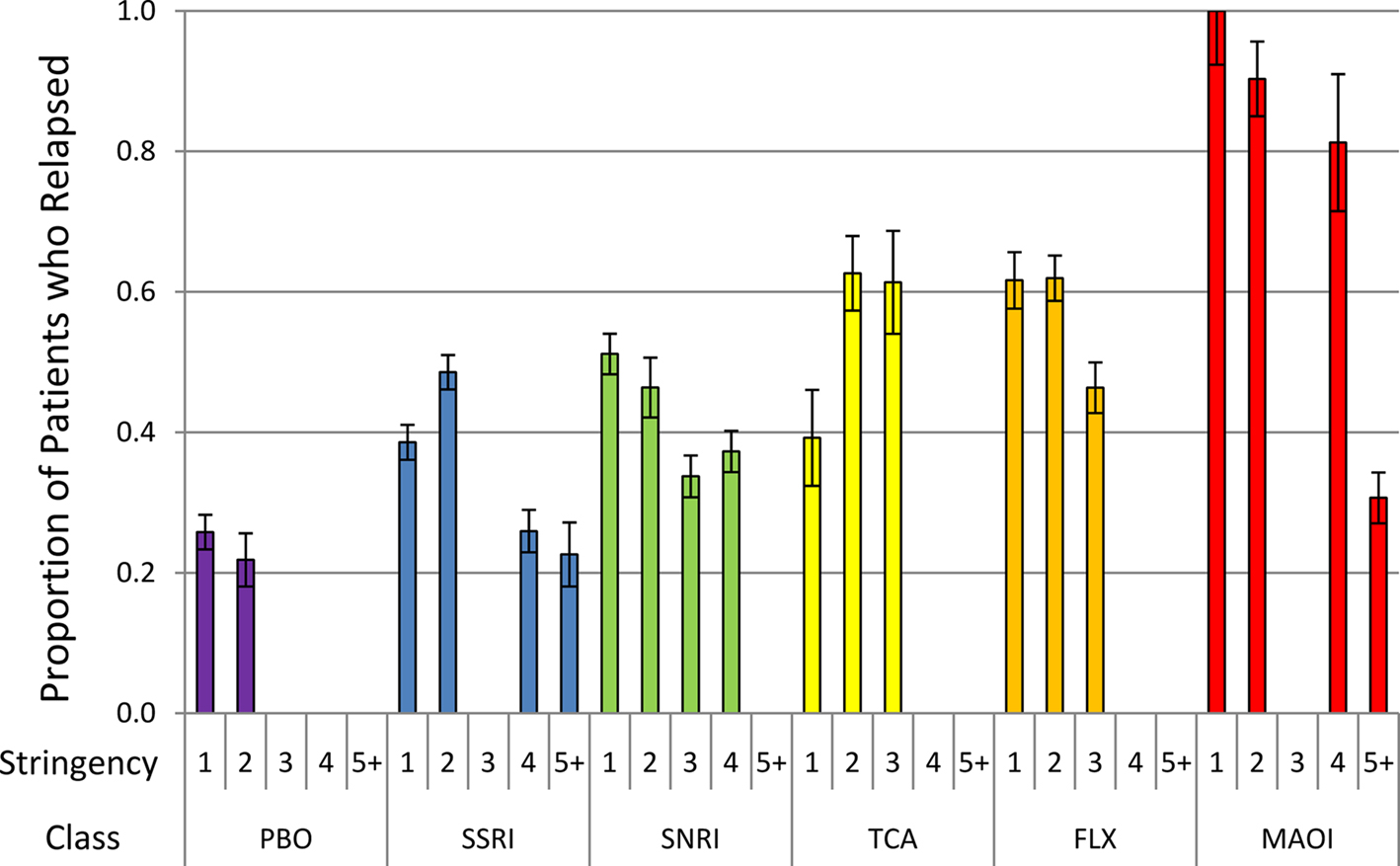
Figure 3. Unadjusted proportions (with SE bars) of patients with a relapse after discontinuation (to placebo) (y-axis) as a function of the stringency of the definition of relapse (x-axis) for each ADM class.
To formally test our prediction, we regressed the relapse rate on the stringency variable. The stringency variable, by itself, is only a marginally significant negative predictor of the relapse rate when all 49 samples are analyzed, β = −0.035, SE = 0.018, p = 0.06. However, it reaches significance when only the 42 ADM discontinuation samples are analyzed, β = −0.057, SE = 0.017, p = 0.002. It also reaches significance in the entire sample when covariates (age, sex, txresist, dephx, droprate, txdur, riskper) are included, β = −0.050, SE = 0.019, p = 0.01.
Does the PBO Group Have a Lower Relapse Rate than the ADM Classes?
Our first prediction was that the PBO group would have a lower relapse rate than that of the ADM studies, and this was supported by the summary statistic results. To formally test this, we constructed a binary treatment variable (0 = PBO, 1 = the ADM classes). We then regressed the relapse rate on this treatment variable. The treatment variable was significant, β = 0.277, SE = 0.067, p = 0.0001, indicating that the PBO group had a significantly lower relapse rate than the ADM classes. Inclusion of the stringency variable only increased the significance of the results. As discussed above, the lower relapse rate of the PBO group could be due to a selection bias problem in which more relapse–vulnerable patients dropped out during acute treatment, or because more patients in the PBO studies were more vulnerable to relapse at the outset. To formally test this, we included as covariates the minimum number of depressive episodes required for entry into the study, whether treatment resistant patients were targeted, and the dropout rate during the acute treatment phase. None of the sample characteristic variables were significant, while the treatment variable remained significant, β = 0.342, SE = 0.080, p = 0.0001. The inclusion of other covariates also did not influence the significance of this variable.
Do the Perturbational Properties of ADMS Predict the Relapse Rate?
Our second prediction was that the perturbational properties of ADMs on mPFC monoamine levels would be positive predictors of the relapse rate. To formally test this, we regressed the relapse rate on the perturbational effects on 5-HT and NE. (Since the perturbational effects on NE and DA are highly collinear, we do not analyze them at the same time.) We included as covariates average age, the proportion of women in the sample, the minimum number of prior episodes required for entry, whether treatment resistant patients were targeted, the dropout rate during acute treatment, the duration of acute treatment, the length of the risk period, and the stringency of relapse criteria. We report the estimates of this model in Table 5, Model I. The perturbational effects of ADMs on 5-HT and NE were both significant positive predictors of the relapse rate, as was stringency and the duration of the risk period.
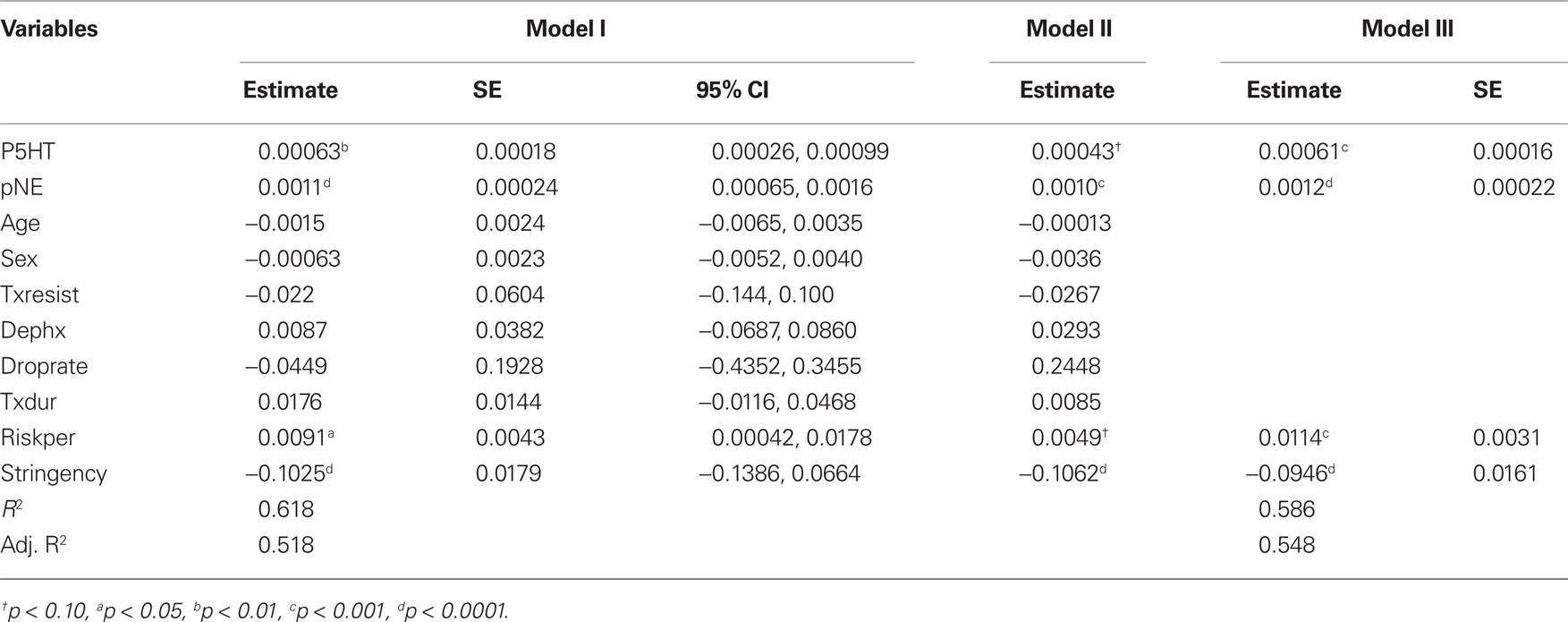
Table 5. Parameter estimates and SE from the weighted least squares analyses (see text).
Next, we excluded the PBO studies and re-ran the analyses, the results of which are in Table 5, Model II. Comparing Model II with Model I shows that the estimates for Model II are well within the 95% confidence limits of the estimates for Model I. Thus, including the PBO studies does not fundamentally change the parameter estimates – they only increase the power.
Then, we included the PBO studies and excluded the MAOI studies, to determine how the parameter estimates were affected by the MAOI studies. The estimates for this model (not shown) were also well within the 95% confidence limits of the Model I estimates. Additionally, the perturbational variables for 5-HT and NE, and the risk period and stringency variables, were all statistically significant. Thus, the inclusion of the MAOI studies also does not fundamentally change the parameter estimates.
Using the entire sample, we report the best-fitting model in Table 5, Model III. Only the perturbational variables for 5-HT and NE, the stringency variable, and the risk period remained in the model, but they still explained nearly 60% of the variance.
Finally, we re-ran each of these analyses using the perturbational variable for DA in place of NE (results not shown). They all fit nearly as well, as might be expected from their collinearity.
Discussion
We found robust support for our predictions. The risk of relapse after ADM discontinuation was higher than the risk of relapse after remission on placebo. Moreover, the risk of relapse was after ADM discontinuation was positively predicted by the degree to which ADMs perturbed 5-HT and NE levels in the rodent mPFC. The risk of relapse was also positively predicted by the degree to which ADMs perturbed DA levels in the mPFC. Given the high degree of collinearity between the perturbational effects of ADMs on NE and DA, we prefer the 5-HT/NE model over the 5-HT/DA model because the evidence for the involvement of NE in depression is stronger.
While the results are robust, their implications depend on their interpretation, so we first discuss several potential confounds. Is it possible, for instance, that the results do not reflect the risk of relapse, but rather withdrawal symptoms? The discontinuation of ADM can cause withdrawal symptoms (also known as discontinuation syndrome) that are unrelated to relapse, but may sometimes be mistaken for relapse (Haddad, 1997). Such mistakes are more likely with untrained physicians (Haddad, 1997), so it should be less of a problem in ADM discontinuation studies where trained psychiatrists are employed to evaluate putative cases of relapse. Regardless, drugs with longer half-lives are less likely to show discontinuation syndrome (Michelson et al., 2000; Fava, 2006). If discontinuation syndrome were being mistaken for relapse, then drugs with longer half-lives should have lower relapse rates. To test this possibility, we added half-life to the best-fitting model and eliminated the perturbational variables (results not shown). The resultant model did not fit as well, and, in fact, half-life was a significant positive predictor of relapse rate. This is because drugs with stronger perturbational effects on 5-HT tend to have longer half-lives. We therefore have no evidence that withdrawal symptoms were mislabeled as relapses.
There is some evidence that ADM treatment resistant patients may have higher relapse rates (Rush et al., 2006). Is it possible that studies using ADMs with stronger perturbational properties had higher relapse rates, not due to the pharmacological properties of the drugs used, but because they also had higher proportions of treatment resistant patients participating in them? There are two ways this could happen. First, studies using stronger ADMs could have been more likely to target treatment resistant patients at the outset. Second, the characteristics of patient samples in discontinuation studies may change during treatment due to dropouts caused by the treatment, causing a potential selection bias problem.
A related issue is whether the PBO class (denoted by the Placebo–Placebo arms of the extension studies) was fundamentally different from the other classes (denoted by the Drug–Placebo arms of the ADM discontinuation studies). For instance, perhaps the PBO studies targeted fundamentally different samples at the outset, or perhaps they had higher dropout during the acute treatment phase so that patient populations were substantially different by the beginning of the extension phase. Consistent with this, the PBO studies did have a higher dropout rate and a lower relapse rate than the ADMs as a group. Of course, this result might also be expected if the PBO studies were quantitatively different only because they were on one end of the perturbational continuum.
To deal with these issues in our analyses, we included two variables as covariates that captured differences in the initial patient populations: whether treatment resistant patients were being targeted, and the minimum number of depressive episodes required for entry into the study. Neither variable was significant in any of our analyses, and their inclusion did not change the statistical significance of any result. We also included the dropout rate during acute treatment because the PBO studies had significantly higher dropout rates, and the dropout rate in the ADM discontinuation studies was positively associated with the duration of treatment. Both findings suggested that the dropout rate during treatment might be a proxy for the degree of selection bias at discontinuation. If selection bias problems were driving the lower relapse rate in the PBO studies or the relationship between relapse rate and perturbational variables, then dropout rate during the treatment phase could predict relapse, either as a main effect or in interaction with binary treatment status (Placebo vs. Drug). Neither term was significant in any of our analyses, nor did they affect the significance of any result.
In any event, the strongest reason for inferring that the PBO studies were not qualitatively different from the other studies is that excluding them did not significantly change the parameter estimates of any variables, including the perturbational variables. This suggests that the PBO studies simply anchor one end of the perturbational continuum and increase the power of our analyses. (Similarly, even though we had to make assumptions about the prefrontal perturbational effects of phenelzine and selegiline based on evidence from other dual MAO-A/B inhibitors, our results did not change significantly if we excluded the MAOI studies.)
Another potential confound is that we are only using published studies in our meta-analysis. It has been shown, for instance, that there is a publication bias of studies dealing with the efficacy of ADMs during acute treatment (Turner et al., 2008). For publication bias to explain our results, the likelihood of publication would have to be related to the perturbational effects of ADMs. This seems to us unlikely, but we cannot rule out this possibility.
Other limitations of this meta-analysis highlight difficulties with the ADM literature on which it is based. To address the question of how ADM discontinuation affects relapse rates, we were forced to rely on between-study relapse rates that may be biased by a number of other between-study differences, such as how relapse is defined. Prior meta-analyses have not dealt directly with this issue, despite evidence that definitional criteria affect relapse rates (Montgomery and Dunbar, 1993; Keller et al., 1998). We constructed a stringency variable that measured the number of assessment hurdles that had to be passed before an increase in symptoms was deemed to be a relapse. As expected, this variable was a significant negative predictor of the relapse rate, and it generally improved the explanatory power of other variables in our analyses (results not shown), suggesting that the failure to take this variable into account can obscure findings. Nevertheless, our findings are necessarily correlational.
Is Behavioral Depression in Rodents Homologous to Clinical Depression in Humans?
We could not use data on how ADMs perturb monoamine levels in humans. Such data do not exist because they require highly invasive techniques. Consequently, we used evidence on the perturbational effects in rat mPFC, which has been found to play a special role in processing information about stressors and triggering depressive symptoms in response to repeated, uncontrollable shock (Amat et al., 2005). ADMs have dramatically different effects on monoamine levels in different brain regions (Dawson and Li, 2003; Valentini et al., 2004, 2005), so the gambit was that the perturbational effects in the rodent mPFC would predict the risk of relapse after ADM discontinuation in humans. This need not have been true.
Indeed, there are ongoing debates about whether behavioral measures of depression in rodents are good models of human depression (Nestler et al., 2002; Anisman and Matheson, 2005; Kalueff et al., 2007). These debates are, at least implicitly, about whether rodent models are homologous to depression in humans. Consequently, arguments tend to focus on whether the models induce behavioral symptoms in rodents that map onto depressive symptoms in humans, and whether the symptoms are alleviated by ADM treatment.
Our results show that the degree to which an ADM perturbs monoamines in the mPFC predicts the risk of depressive relapse in humans after discontinuation of the ADM. In other words, the region that predicts behavioral depression in rats also predicts depression in humans, which provides further evidence that rodent and human depression are homologous. Moreover, these results suggest that the rodent mPFC may be homologous to a region in the human brain that plays a similar role in clinical depression. One possible candidate is the VLPFC, of which the mPFC has been argued to be a structural and functional homolog (Kesner, 2000). The VLPFC has been implicated in clinical and subclinical depression and may play an important role in depressive rumination (Drevets, 1998; Andrews and Thomson Jr., 2009).
Are the Symptoms Currently Used to Diagnose MDD Better Characterized by Disorder or Adaptationist Hypotheses?
Our results are also relevant to debates about whether the symptoms used to diagnose MDD are better characterized by disorder or adaptationist hypotheses. Of course, we cannot definitively answer this question. Our results are most relevant to claims that depressive episodes that meet diagnostic criteria for MDD are monoaminergic disorders.
If every disorder is to be understood as involving a malfunction in some adaptation (Wakefield, 1992), then mapping out the evolved structure and operation of adaptations can facilitate the identification of disorder by revealing the precise points where malfunction can occur. As discussed above, homeostatic mechanisms have sensor and feedback components, and some also have components for altering the equilibrium level of the parameter. To produce oppositional tolerance, the sensor component must be able to discern perturbations from equilibrium so that the feedback component can produce an oppositional force proportional to the degree of perturbation.
Our results support the conclusion that prolonged ADM use triggers oppositional tolerance in patients meeting diagnostic criteria for MDD. As predicted by an oppositional tolerance model, the risk of relapse was higher after ADM discontinuation than the unmedicated risk of relapse. Also, the risk of relapse after ADM discontinuation was proportional to the perturbational effect of the ADM on rodent mPFC. This result is analogous to the oppositional force produced by a spring, which is proportional to the deviation from the equilibrium position. Our results therefore provide no evidence of malfunction in either the sensor or feedback components of monoaminergic homeostatic mechanisms in patients with episodes that satisfy diagnostic criteria for MDD.
However, it is possible that there could be some malfunction in the component that alters equilibrium monoamine levels. Demonstrating malfunction in this component requires more than merely showing that the equilibrium point is altered, since that is the function of this component. In other words, the claim that depression is a disorder in which monoamine levels are too high or too low is not simply supported by evidence that monoamine levels are altered since this could merely be the consequence of adaptive changes in the equilibrium levels in response to stressors. Undoubtedly, the equilibrium altering component must malfunction at some rate, but it is not clear that it is malfunctioning as frequently as current diagnostic criteria suggest. Understanding when it is malfunctioning will require greater research into the structure and operation of this component in response to stress. In any event, our results provide no positive evidence of malfunction in the homeostatic mechanisms regulating monoamine levels in people that meet diagnostic criteria for MDD.
Of course, our results do not speak to other possible malfunctions in patients who meet criteria for MDD. However, there is currently no evidence that the mechanisms regulating other neurochemicals are malfunctioning in patients diagnosed with MDD. The conceptual basis for considering an episode of depression to be a mental disorder is that it causes significant distress or impairment (American Psychiatric Association, 2000). However, it is clear that the body has evolved adaptations for causing significant pain and impairment.
For instance, organisms with infection can be incapacitated for days, weeks, or months, yet such incapacitation is usually considered to be an adaptive response to infection (Dantzer, 2001). Since mounting an effective immune response requires massive amounts of energy (Lochmiller and Deerenberg, 2000; Wolowczuk et al., 2008), social, sexual, locomotor, or cognitive activity could draw on limited energetic resources and interfere with the immune response. Consequently, the social, sexual, locomotor, and cognitive incapacity exhibited by sick organisms reflects an adaptive downregulation of systems that could otherwise interfere with mounting an effective immune response. Consistent with this, there is growing evidence that pharmacological disruption of fever, which is the best studied sickness behavior, prolongs infection and, for serious infections, may lower survivorship (Brandts et al., 1997; Hasday et al., 2000; Schulman et al., 2005).
In short, we found no positive evidence to support, and some positive evidence to contradict, the claim that most patients diagnosed with MDD have a monoaminergic disorder. Nor is there any evidence that the current diagnostic reliance on distress and impairment accurately identifies depressive disorder. Consequently, our results are just as consistent with adaptationist hypotheses, although they do not speak to the issue of what function depression may have evolved to serve.
Clinical Implications
Our results have several important clinical implications.
Modest efficacy of ADMs during acute therapy
Hypotheses that depression is caused by a dysregulation in monoamine levels were developed based on drugs that lower symptoms and had monoaminergic properties, such as blockade of the serotonin transporter or inhibition of the MAO enzymes (Valenstein, 1998). However, these drugs also have limited efficacy during acute treatment (Kirsch et al., 2008; Fournier et al., 2010), which is one reason why researchers are investigating the role of other neurochemicals in depression (Krishnan and Nestler, 2008). However, even if monoamines are the neurochemical cause of depression, the limited efficacy of ADMs could be explained by the development of oppositional tolerance.
Maintenance therapy
It is commonly thought that ADM treatment should continue until the index episode resolves, else relapse will recur. For this reason, maintenance ADM therapy is often recommended for 4–12 months or more after the acute management of the index episode (Geddes et al., 2003). Nevertheless, there are troubling empirical problems with these guidelines. For instance, in naturalistic studies, unmedicated patients have much shorter episodes, and better long-term prospects, than medicated patients (Coryell et al., 1995; Goldberg et al., 1998; Posternak et al., 2006). Several of these studies have found that the average duration of an untreated episode of major depression is 12–13 weeks (Coryell et al., 1995; Posternak et al., 2006). Since acute ADM management of major depression minimally requires several weeks to reduce symptoms, the duration of untreated episodes is much shorter than the recommended duration of ADM therapy. This suggests that ADM therapy may delay resolution of depressive episodes.
Current maintenance therapy guidelines are based primarily on prior meta-analyses that have found that the relapse rates in the Drug–Drug arms of ADM discontinuation studies are consistently lower than in the Drug–Placebo arms (Viguera et al., 1998; Geddes et al., 2003; Kaymaz et al., 2008; Glue et al., 2010). This is often interpreted as evidence that ADM therapy prevents depressive relapse. However, our results suggest that oppositional tolerance to the ADM contributes to the high risk of relapse in the Drug–Placebo arms of these studies, which undermines the rationale for maintenance therapy. Moreover, if continuous ADM therapy were needed until the index episode resolved, then ADM discontinuation studies with longer periods of continuous treatment should have lower relapse rates. This prediction has not been supported in any prior meta-analysis of ADM discontinuation studies. We also did not find treatment duration to have a protective effect on the risk of relapse. Rather, we found a non-significant trend for longer periods of continuous ADM treatment to be associated with a greater risk of relapse. Maintenance therapy guidelines should be re-examined.
Dependency on ADMs
Drugs that promote the risk of relapse or withdrawal upon discontinuation can cause dependence on the drug to prevent the return of symptoms (Chouinard, 2004). Consequently, such drugs must be managed carefully and patients must provide informed consent for their use (Bursztajn and Brodsky, 1998). ADMs are sometimes prescribed to people with alcohol or illicit drug dependencies (Petrakis et al., 2003), because the use of such substances to medicate feelings of anxiety and depression is thought to play a role in the dependency (Tomlinson et al., 2006). Ironically, the use of ADMs to help people wean off such substances might merely replace one dependency with another.
Treating depression with multiple psychotropic drugs
Since many patients on antidepressants alone do not achieve full remission (Rush et al., 2006), possibly due to oppositional tolerance, atypical antipsychotic drugs, and other agents are increasingly prescribed to enhance the efficacy of ADMs. Our results suggest that the concurrent use of multiple drugs could cause greater monoaminergic perturbations, possibly further increasing the risk of relapse after they are discontinued.
Conflict of Interest Statement
The funding source had no role in the study design, the collection, analysis, or interpretation of data, the writing of the paper, or the decision to submit the paper for publication. Paul W. Andrews had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Paul W. Andrews, Michael C. Neale, Lisa J. Halberstadt, and Charles O. Gardner declare no conflicts of interest. Susan G. Kornstein has served as a consultant for Bristol-Myers Squibb, Forest Laboratories, Lilly, Pfizer, Rexahn, Dey Pharma, PGxHealth, Takeda, and Wyeth. She has received research support from National Institute of Mental Health, Bristol-Myers Squibb, Lilly, Forest Laboratories, Wyeth, Novartis, Boehringer-Ingelheim, Pfizer, Otsuka, and Takeda. She has received book royalties from Guilford Press.
Acknowledgments
Paul W. Andrews and Michael C. Neale were supported by NIH grant DA-018673 (PI: Michael C. Neale). We thank Andy Thomson for commenting on prior versions of the manuscript.
References
Alexopoulos, G. S., Meyers, B. S., Young, R. C., Kalayam, B., Kakuma, T., Gabrielle, M., Sirey, J. A., and Hull, J. (2000). Executive dysfunction and long-term outcomes of geriatric depression. Arch. Gen. Psychiatry 57, 285–290.
Allen, N. B., and Badcock, P. B. T. (2003). The social risk hypothesis of depressed mood: evolutionary, psychosocial, and neurobiological perspectives. Psychol. Bull. 129, 887–913.
Allen, N. B., and Badcock, P. B. T. (2006). Darwinian models of depression: a review of evolutionary accounts of mood and mood disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 30, 815–826.
Amat, J., Aleksejev, R. M., Paul, E., Watkins, L. R., and Maier, S. F. (2010). Behavioral control over shock blocks behavioral and neurochemical effects of later social defeat. Neuroscience 165, 1031–1038.
Amat, J., Baratta, M. V., Paul, E., Bland, S. T., Watkins, L. R., and Maier, S. F. (2005). Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nat. Neurosci. 8, 365–371.
Amat, J., Paul, E., Zarza, C., Watkins, L. R., and Maier, S. F. (2006). Previous experience with behavioral control over stress blocks the behavioral and dorsal raphe nucleus activating effects of later uncontrollable stress: role of the ventral medial prefrontal cortex. J. Neurosci. 26, 13264–13272.
American Psychiatric Association (APA). (2000). (DSM-IV-TR) Diagnostic and Statistical Manual of Mental Disorders, 4th Edn. Washington, DC: American Psychiatric Press, Inc.
Amsterdam, J. D., and Bodkin, J. A. (2006). Selegiline transdermal system in the prevention of relapse of major depressive disorder: a 52-week, double-blind, placebo-substitution, parallel-group clinical trial. J. Clin. Psychopharmacol. 26, 579–586.
Andrews, P. W., Aggen, S. H., Miller, G. F., Radi, C., Dencoff, J. E., and Neale, M. C. (2007). The functional design of depression’s influence on attention: a preliminary test of alternative control-process mechanisms. Evol. Psychol. 5, 584–604.
Andrews, P. W., and Thomson, J. A. Jr. (2009). The bright side of being blue: depression as an adaptation for analyzing complex problems. Psychol. Rev. 116, 620–654.
Anisman, H., and Matheson, K. (2005). Stress, depression, and anhedonia: caveats concerning animal models. Neurosci. Biobehav. Rev. 29, 525–546.
Anton, S. F., Robinson, D. S., Roberts, D. L., Kensler, T. T., English, P. A., and Archibald, D. G. (1994). Long-term treatment of depression with nefazodone. Psychopharmacol. Bull. 30, 165–169.
Badcock, P. B. T., and Allen, N. B. (2003). Adaptive social reasoning in depressed mood and depressive vulnerability. Cogn. Emot. 17, 647–670.
Baldessarini, R. J., Ghaemi, S. N., and Viguera, A. C. (2002). Tolerance in antidepressant treatment. Psychother. Psychosom. 71, 177–179.
Bano, S., Gitay, M., Ara, I., and Badawy, A. (2010). Acute effects of serotonergic antidepressants on tryptophan metabolism and corticosterone levels in rats. Pak. J. Pharm. Sci. 23, 266–272.
Baratta, M. V., Zarza, C. M., Gomez, D. M., Campeau, S., Watkins, L. R., and Maier, S. F. (2009). Selective activation of dorsal raphe nucleus-projecting neurons in the ventral medial prefrontal cortex by controllable stress. Eur. J. Neurosci. 30, 1111–1116.
Beck, A. T., and Beamesderfer, A. (1974). “Assessment of depression: the depression inventory,” in Modern Problems in Pharmacopsychiatry, ed. P. Pichot (Basel: Karger), 151–169.
Bel, N., and Artigas, F. (1995). In vivo evidence for the reversible action of the monoamine oxidase inhibitor brofaromine on 5-hydroxytryptamine release in rat brain. Naunyn Schmiedebergs Arch. Pharmacol. 351, 475–482.
Berton, O., and Nestler, E. J. (2006). New approaches to antidepressant drug discovery: beyond monoamines. Nat. Rev. Neurosci. 7, 137–151.
Best, J., Nijhout, H. F., and Reed, M. (2010). Serotonin synthesis, release and reuptake in terminals: a mathematical model. Theor. Biol. Med. Model. doi: 10.1186/1742-4682-7-34.
Bongiovanni, R., Newbould, E., and Jaskiw, G. E. (2008). Tyrosine depletion lowers dopamine synthesis and desipramine-induced prefrontal cortex catecholamine levels. Brain Res. 1190, 39–48.
Brandts, C. H., Ndjave, M., Graninger, W., and Kremsner, P. G. (1997). Effect of paracetamol on parasite clearance time in Plasmodium falciparum malaria. Lancet 350, 704–709.
Bremner, J. D., and Smith, W. T. (1996). Org 3770 vs amitriptyline in the continuation treatment of depression: a placebo controlled trial. Eur. J. Psychiatry 10, 5–15.
Bursztajn, H. J., and Brodsky, A. (1998). Ethical and legal dimensions of benzodiazepine prescription: a commentary. Psychiatr. Ann. 28, 121–128.
Bymaster, F. P., Zhang, W., Carter, P. A., Shaw, J., Chernet, E., Phebus, L., Wong, D. T., and Perry, K. W. (2002). Fluoxetine, but not other selective serotonin uptake inhibitors, increases norepinephrine and dopamine extracellular levels in prefrontal cortex. Psychopharmacology (Berl.) 160, 353–361.
Byrne, S. E., and Rothschild, A. J. (1998). Loss of antidepressant efficacy during maintenance therapy: possible mechanisms and treatments. J. Clin. Psychiatry 59, 279–288.
Campbell, I., and Marshall, E. (1974). Effects of chronic regimes of phenelzine, tranylcypromine and imipramine on rat brain noradrenaline and 5-hydroxytryptamine. J. Pharmacol. 5, 14–15.
Cheung, A., Kusumakar, V., Kutcher, S., Dubo, E., Garland, J., Weiss, M., Kiss, A., and Levitt, A. (2008). Maintenance study for adolescent depression. J. Child Adolesc. Psychopharmacol. 18, 389–394.
Chouinard, G. (2004). Issues in the clinical use of benzodiazepines: potency, withdrawal, and rebound. J. Clin. Psychiatry 65, 7–12.
Claghorn, J. L., and Feighner, J. P. (1993). A double-blind comparison of paroxetine with imipramine in the long-term treatment of depression. J. Clin. Psychopharmacol. 13, S23–S27.
Cook, B. L., Helms, P. M., Smith, R. E., and Tsai, M. (1986). Unipolar depression in the elderly: reoccurrence on discontinuation on tricyclic antidepressants. J. Affect. Disord. 10, 91–94.
Coppen, A., Ghose, K., Montgomery, S., Rao, V. A. R., Bailey, J., and Jorgensen, A. (1978). Continuation therapy with amitriptyline in depression. Br. J. Psychiatry 133, 28–33.
Coryell, W., Endicott, J., Winokur, G., Akiskal, H., Solomon, D., Leon, A., Mueller, T., and Shea, T. (1995). Characteristics and significance of untreated major depressive disorder. Am. J. Psychiatry 152, 1124–1129.
Cryan, J. F., O’Leary, O. F., Jin, S. H., Friedland, J. C., Ouyang, M., Hirsch, B. R., Page, M. E., Dalvi, A., Thomas, S. A., and Lucki, I. (2004). Norepinephrine-deficient mice lack responses to antidepressant drugs, including selective serotonin reuptake inhibitors. Proc. Natl. Acad. Sci. U.S.A. 101, 8186–8191.
Dantzer, R. (2001). Cytokine-induced sickness behavior: where do we stand? Brain Behav. Immun. 15, 7–24.
Davidson, J., and Raft, D. (1984). Use of phenelzine in continuation therapy. Neuropsychobiology 11, 191–194.
Dawson, L. A., and Li, P. (2003). Effects of 5-HT6 receptor blockade on the neurochemical outcome of antidepressant treatment in the frontal cortex of the rat. J. Neural Transm. 110, 577–590.
Detke, M. J., Wiltse, C. G., Mallinckrodt, C. H., McNamara, R. K., Demitrack, M. A., and Bitter, I. (2004). Duloxetine in the acute and long-term treatment of major depressive disorder: a placebo- and paroxetine-controlled trial. Eur. Neuropsychopharmacol. 14, 457–470.
Dobson, K. S., Hollon, S. D., Dimidjian, S., Schmaling, K. B., Kohlenberg, R. J., Gallop, R. J., Rizvi, S. L., Gollan, J. K., Dunner, D. L., and Jacobson, N. S. (2008). Randomized trial of behavioral activation, cognitive therapy, and antidepressant medication in the prevention of relapse and recurrence in major depression. J. Consult. Clin. Psychol. 76, 468–477.
Doogan, D. P., and Caillard, V. (1992). Sertraline in the prevention of depression. Br. J. Psychiatry 160, 217–222.
Drevets, W. C. (1998). Functional neuroimaging studies of depression: the anatomy of melancholia. Annu. Rev. Med. 49, 341.
Duman, R. S., and Monteggia, L. M. (2006). A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127.
Dziedzicka-Wasylewska, M., Faron-Gorecka, A., Kusmider, M., Drozdowska, E., Rogoz, Z., Siwanowicz, J., Caron, M. G., and Bönisch, H. (2006). Effect of antidepressant drugs in mice lacking the norepinephrine transporter. Neuropsychopharmacology 31, 2424–2432.
Emslie, G. J., Kennard, B. D., Mayes, T. L., Nightingale-Teresi, J., Carmody, T., Hughes, C. W., Rush, A. J., Tao, R., and Rintelmann, J. W. (2008). Fluoxetine versus placebo in preventing relapse of major depression in children and adolescents. Am. J. Psychiatry 165, 459–467.
Entsuah, A. R., Rudolph, R. L., Hackett, D., and Miska, S. (1996). Efficacy of venlafaxine and placebo during long-term treatment of depression: a pooled analysis of relapse rates. Int. Clin. Psychopharmacol. 11, 137–145.
Eric, L. (1991). A prospective, double-blind, comparative, multicentre study of paroxetine and placebo in preventing recurrent major depressive episodes. Biol. Psychiatry 29, 254S–255S.
Fava, G. A., and Offidani, E. (2011). The mechanisms of tolerance in antidepressant action. Prog. Neuropsychopharmacol. Biol. Psychiatry. doi: 10.1016/j.pnpbp.2010.07.026. [Epub ahead of print].
Fava, M. (2006). Prospective studies of adverse events related to antidepressant discontinuation. J. Clin. Psychiatry 67, 14–21.
Finberg, J. P. M., Pacak, K., Kopin, I. J., and Goldstein, D. S. (1993). Chronic inhibition of monoamine oxidase type A increases noradrenaline release in rat frontal cortex. Naunyn Schmiedebergs Arch. Pharmacol. 347, 500–505.
Fournier, J. C., DeRubeis, R. J., Hollon, S. D., Dimidjian, S., Amsterdam, J. D., Shelton, R. C., and Fawcett, J. (2010). Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA 303, 47–53.
Fuller, R. W., Perry, K. W., and Molloy, B. B. (1974). Effect of an uptake inhibitor on serotonin metabolism in rat brain: studies with 3-para-trifluoromethylphenoxy)-n-methyl-3-phenylpropylamine (Lilly 110140). Life Sci. 15, 1161–1171.
Geddes, J. R., Carney, S. M., Davies, C., Furukawa, T. A., Kupfer, D. J., Frank, E., and Goodwin, G. M. (2003). Relapse prevention with antidepressant drug treatment in depressive disorders: a systematic review. Lancet 361, 653–661.
Georgotas, A., Mccue, R. E., and Cooper, T. B. (1989). A placebo-controlled comparison of nortriptyline and phenelzine in maintenance therapy of elderly depressed-patients. Arch. Gen. Psychiatry 46, 783–786.
Gilaberte, I., Montejo, A. L., de la Gandara, J., Perez-Sola, V., Bernardo, M., Massana, J., Martin-Santos, R., Santiso, A., Noguera, R., Casais, L., Perez-Camo, V., Arias, M., and Judge, R., and Fluoxetine Long-Term Study Group. (2001). Fluoxetine in the prevention of depressive recurrences: a double-blind study. J. Clin. Psychopharmacol. 21, 417–424.
Gilbert, P., and Allan, S. (1998). The role of defeat and entrapment (arrested flight) in depression: an exploration of an evolutionary view. Psychol. Med. 28, 585–598.
Gilbert, P., Gilbert, J., and Irons, C. (2004). Life events, entrapments and arrested anger in depression. J. Affect. Disord. 79, 149–160.
Glue, P., Donovan, M. R., Kolluri, S., and Emir, B. (2010). Meta-analysis of relapse prevention antidepressant trials in depressive disorders. Aust. N. Z. J. Psychiatry 44, 697–705.
Goldberg, D., Privett, M., Ustun, B., Simon, G., and Linden, M. (1998). The effects of detection and treatment on the outcome of major depression in primary care: a naturalistic study in 15 cities. Br. J. Gen. Pract. 48, 1840–1844.
Gorwood, P., Weiller, E., Lemming, O., and Katona, C. (2007). Escitalopram prevents relapse in older patients with major depressive disorder. Am. J. Geriatr. Psychiatry 15, 581–593.
Guy, W. (1976). ECDEU Assessment Manual for Psychopharmacology-Revised (DHEW publication no. ADM 76-338). Rockville, MD: US Department of Health, Education, and Welfare.
Haddad, P. (1997). Newer antidepressants and the discontinuation syndrome. J. Clin. Psychiatry 58, 17–22.
Hagen, E. H. (2003). “The bargaining model of depression,” in The Genetic and Cultural Evolution of Cooperation, ed. P. Hammerstein (Cambridge, MA: MIT Press), 95–123.
Harrison, W., Rabkin, J., Stewart, J. W., McGrath, P. J., Tricamo, E., and Quitkin, F. (1986). Phenelzine for chronic depression: a study of continuation treatment. J. Clin. Psychiatry 47, 346–349.
Hasday, J. D., Fairchild, K. D., and Shanholtz, C. (2000). The role of fever in the infected host. Microbes Infect. 2, 1891–1904.
Heisler, L. K., Chu, H. M., Brennan, T. J., Danao, J. A., Bajwa, P., Parsons, L. H., and Tecott, L. H. (1998). Elevated anxiety and antidepressant-like responses in serotonin 5-HT1A receptor mutant mice. Proc. Natl. Acad. Sci. U.S.A. 95, 15049–15054.
Hochochka, P. W., and Somero, G. N. (2002). Biochemical Adaptation: Mechanism and Process in Physiological Evolution. New York, NY: Oxford University Press.
Hochstrasser, B., Isaksen, P. M., Koponen, H., Lauritzen, L., Mahnert, F. A., Rouillon, F., Wade, A. G., Andersen, M., Pedersen, S. F., Swart, J. C., and Nil, R. (2001). Prophylactic effect of citalopram in unipolar, recurrent depression – Placebo-controlled study of maintenance therapy. Br. J. Psychiatry 178, 304–310.
Hollon, S. D., Stewart, M. O., and Strunk, D. (2006). Enduring effects for cognitive behavior therapy in the treatment of depression and anxiety. Annu. Rev. Psychol. 57, 285–315.
Horwitz, A. V., and Wakefield, J. C. (2007). The Loss of Sadness: How Psychiatry Transformed Normal Sorrow into Depressive Disorder. New York, NY: Oxford University Press.
Hyman, S. E., and Nestler, E. J. (1996). Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am. J. Psychiatry 153, 151–162.
Jordan, S., Kramer, G. L., Zukas, P. K., Moeller, M., and Petty, F. (1994). In vivo biogenic amine efflux in medial prefrontal cortex with imipramine, fluoxetine, and fluvoxamine. Synapse 18, 294–297.
Kalueff, A. V., Wheaton, M., and Murphy, D. L. (2007). What’s wrong with my mouse model? Advances and strategies in animal modeling of anxiety and depression. Behav. Brain Res. 179, 1–18.
Kamijima, K., Burt, T., Cohen, G., Arano, I., and Hamasaki, T. (2006). A placebo-controlled, randomized withdrawal study of sertraline for major depressive disorder in Japan. Int. Clin. Psychopharmacol. 21, 1–9.
Kaymaz, N., van Os, J., Loonen, A. J. M., and Nolen, W. A. (2008). Evidence that patients with single versus recurrent depressive episodes are differentially sensitive to treatment discontinuation: a meta-analysis of placebo-controlled randomized trials. J. Clin. Psychiatry 69, 1423–1436.
Keller, M. B., Kocsis, J. H., Thase, M. E., Gelenberg, A. J., Rush, A. J., Koran, L., Schatzberg, A., Russell, J., Hirschfeld, R., Klein, D., McCullough, J. P., Fawcett, J. A., Kornstein, S., LaVange, L., and Harrison, W. (1998). Maintenance phase efficacy of sertraline for chronic depression - A randomized controlled trial. JAMA 280, 1665–1672.
Keller, M. C., Neale, M. C., and Kendler, K. S. (2007). Association of different adverse life events with distinct patterns of depressive symptoms. Am. J. Psychiatry 164, 1521–1529.
Keller, M. C., and Nesse, R. M. (2004). Subtypes of low mood provide evidence for its adaptive significance. J. Affect. Disord. 78, S77–S78.
Keller, M. C., and Nesse, R. M. (2005). Is low mood an adaptation? Evidence for subtypes with symptoms that match precipitants. J. Affect. Disord. 86, 27–35.
Keller, M. C., and Nesse, R. M. (2006). The evolutionary significance of depressive symptoms: different adverse situations lead to different depressive symptom patterns. J. Pers. Soc. Psychol. 91, 316–330.
Kendler, K. S., Thornton, L. M., and Gardner, C. O. (2000). Stressful life events and previous episodes in the etiology of major depression in women: an evaluation of the “kindling” hypothesis. Am. J. Psychiatry 157, 1243–1251.
Kesner, R. P. (2000). Subregional analysis of mnemonic functions of the prefrontal cortex in the rat. Psychobiology 28, 219–228.
Kessler, R. C., Berglund, P., Demler, O., Jin, R., and Walters, E. E. (2005). Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the national comorbidity survey replication. Arch. Gen. Psychiatry 62, 593–602.
Kessler, R. C., Merikangas, K. R., and Wang, P. S. (2007). Prevalence, comorbidity, and service utilization for mood disorders in the United States at the beginning of the twenty-first century. Annu. Rev. Clin. Psychol. 3, 137–158.
Kihara, T., and Ikeda, M. (1995). Effects of duloxetine, a new serotonin and norepinephrine uptake inhibitor, on extracellular monoamine levels in rat frontal cortex. J. Pharmacol. Exp. Ther. 272, 177–183.
Kirsch, I., Deacon, B. J., Huedo-Medina, T. B., Scoboria, A., Moore, T. J., and Johnson, B. T. (2008). Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med. 5, 260–268. doi: 10.1371/journal.pmed.0050045
Kitaichi, Y., Inoue, T., Nakagawa, S., Boku, S., Izumi, T., and Koyama, T. (2010). Combined treatment with MAO-A inhibitor and MAO-B inhibitor increases extracellular noradrenaline levels more than MAO-A inhibitor alone through increases in beta-phenylethylamine. Eur. J. Pharmacol. 637, 77–82.
Klerman, G. L., Dimascio, A., Weissman, M., Prusoff, B., and Paykel, E. S. (1974). Treatment of depression by drugs psychotherapy. Am. J. Psychiatry 131, 186–191.
Klysner, R., Bent-Hansen, J., Hansen, H. L., Lunde, M., Pleidrup, E., Poulsen, D. L., Andersen, M., and Petersen, H. E. (2002). Efficacy of citalopram in the prevention of recurrent depression in elderly patients: placebo-controlled study of maintenance therapy. Br. J. Psychiatry 181, 29–35.
Koch, S., Hemrick-Luecke, S. K., Thompson, L. K., Evans, D. C., Threlkeld, P. G., Nelson, D. L., Perry, K. W., and Bymaster, F. P. (2003). Comparison of effects of dual transporter inhibitors on monoamine transporters and extracellular levels in rats. Neuropharmacology 45, 935–944.
Kocsis, J. H., Friedman, R. A., Markowitz, J. C., Leon, A. C., Miller, N. L., Gniwesch, L., and Parides, M. (1996). Maintenance therapy for chronic depression – A controlled clinical trial of desipramine. Arch. Gen. Psychiatry 53, 769–774.
Kocsis, J. H., Thase, M. E., Trivedi, M. H., Shelton, R. C., Kornstein, S. G., Nemeroff, C. B., Friedman, E. S., Gelenberg, A. J., Dunner, D. L., Hirschfeld, R. M., Rothschild, A. J., Ferguson, J. M., Schatzberg, A. F., Zajecka, J. M., Pedersen, R. D., Yan, B., Ahmed, S., Musgnung, J., Ninan, P. T., and Keller, M. B. (2007). Prevention of recurrent episodes of depression with venlafaxine ER in a 1-year maintenance phase from the PREVENT study. J. Clin. Psychiatry 68, 1014–1023.
Krishnan, V., and Nestler, E. J. (2008). The molecular neurobiology of depression. Nature 455, 894–902.
Kruijshaar, M. E., Barendregt, J., Vos, T., de Graaf, R., Spijker, J., and Andrews, G. (2005). Lifetime prevalence estimates of major depression: an indirect estimation method and a quantification of recall bias. Eur. J. Epidemiol. 20, 103–111.
Leff, M. J., Roatch, J. F., and Bunney, W. E. (1970). Environmental factors preceding onset of severe depressions. Psychiatry 33, 293–311.
Lemke, T. L., Williams, D. A., Roche, V. F., and Zito, S. W. (2010). Foye’s Principles of Medicinal Chemistry. Baltimore, MD: Lippincott Williams & Wilkins.
Little, R. J. A., and Rubin, D. B. (1989). The analysis of social science data with missing values. Sociol. Methods Res. 18, 292–326.
Lochmiller, R. L., and Deerenberg, C. (2000). Trade-offs in evolutionary immunology: just what is the cost of immunity? Oikos 88, 87–98.
Lustman, P. J., Clouse, R. E., Nix, B. D., Freedland, K. E., Rubin, E. H., Mcgill, J. B., Williams, M. M., Gelenberg, A. J., Ciechanowski, P. S., and Hirsch, I. B. (2006). Sertraline for prevention of depression recurrence in diabetes mellitus - A randomized, double-blind, placebo-controlled trial. Arch. Gen. Psychiatry 63, 521–529.
Marien, M., Floutard, J., and Ladure, P. (2007). Effect of milnacipran on extracellular levels of noradrenaline, dopamine and serotonin (5-HT) in the medial prefrontal cortex of freely moving rats, as measured by intracerebral microdialysis. Fundam. Clin. Pharmacol. 21, 386.
Mayorga, A. J., Dalvi, A., Page, M. E., Zimov-Levinson, S., Hen, R., and Lucki, I. (2001). Antidepressant-like behavioral effects in 5-hydroxytryptamine(1A) and 5-hydroxytryptamine(1B) receptor mutant. J. Pharmacol. Exp. Ther. 298, 1101–1107.
McEwen, B. S. (1998). Protective and damaging effects of stress mediators. N. Engl. J. Med. 338, 171–179.
McEwen, B. S. (2007). Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol. Rev. 87, 873–904.
McGrath, P. J., Stewart, J. W., Quitkin, F. M., Ying, C., Alpert, J. E., Nierenberg, A. A., Fava, M., Cheng, J., and Petkova, E. (2006). Predictors of relapse in a prospective study of fluoxetine treatment of major depression. Am. J. Psychiatry 163, 1542–1548.
Michelson, D., Fava, M., Amsterdam, J., Apter, J., Londborg, P., Tamura, R., and Tepner, R. G. (2000). Interruption of selective serotonin reuptake inhibitor treatment: double-blind, placebo-controlled trial. Br. J. Psychiatry 176, 363–368.
Mindham, R. H. S., Howland, C., and Shepherd, M. (1973). Evaluation of continuation therapy with tricyclic antidepressants in depressive illness. Psychol. Med. 3, 5–17.
Moffitt, T. E., Caspi, A., Taylor, A., Kokaua, J., Milne, B. J., Polanczyk, G., and Poulton, R. (2009). How common are mental disorders? Evidence that lifetime prevalence rates are doubled by prospective versus retrospective ascertainment. Psychol. Med. 40, 899–909.
Montgomery, S. A., and Asberg, M. (1979). A new depression scale designed to be sensitive to change. Br. J. Psychiatry 134, 382–389.
Montgomery, S. A., Dufour, H., Brion, S., Gailledreau, J., Laqueille, X., Ferrey, G., Moron, P., Parant-Lucena, N., Singer, L., Danion, J. M., Beuzen, J. M., and Pierredoin, M. A. (1988). The prophylactic efficacy of fluoxetine in unipolar depression. Br. J. Psychiatry 153 (Suppl. 3), 69–76.
Montgomery, S. A., and Dunbar, G. (1993). Paroxetine is better than placebo in relapse prevention and the prophylaxis of recurrent depression. Int. Clin. Psychopharmacol. 8, 189–195.
Montgomery, S. A., Entsuah, R., Hackett, D., Kunz, N. R., and Rudolph, R. L. (2004). Venlafaxine versus placebo in the preventive treatment of recurrent major depression. J. Clin. Psychiatry 65, 328–336.
Montgomery, S. A., Rasmussen, J. G. C., Lyby, K., Connor, P., and Tanghoj, P. (1992). Dose-response relationship of citalopram 20 mg, citalopram 40 mg and placebo in the treatment of moderate and severe depression. Int. Clin. Psychopharmacol. 6, 65–70.
Montgomery, S. A., Rasmussen, J. G. C., and Tanghoj, P. (1993). A 24 week study of 20 mg citalopram, 40 mg citalopram, and placebo in the prevention of relapse of major depression. Int. Clin. Psychopharmacol. 8, 181–188.
Montgomery, S. A., Reimitz, P. E., and Zivkov, M. (1998). Mirtazapine versus amitriptyline in the long-term treatment of depression: a double-blind placebo-controlled study. Int. Clin. Psychopharmacol. 13, 63–73.
Nesse, R. M. (2004). Natural selection and the elusiveness of happiness. Philos. Trans. R. Soc. Lond. BBiol. Sci. 359, 1333–1347.
Nesse, R. M., and Ellsworth, P. C. (2009). Evolution, emotions, and emotional disorders. Am. Psychologist 64, 129–139.
Nestler, E. J., and Aghajanian, G. K. (1997). Molecular and cellular basis of addiction. Science 278, 58–63.
Nestler, E. J., Gould, E., Manji, H., Bucan, M., Duman, R. S., Gershenfeld, H. K., Hen, R., Koester, S., Lederhendler, I., Meaney, M., Robbins, T., Winsky, L., and Zalcman, S. (2002). Preclinical models: status of basic research in depression. Biol. Psychiatry 52, 503–528.
Parent, M. B., Habib, M. K., and Baker, G. B. (2000). Time-dependent changes in brain monoamine oxidase activity and in brain levels of monoamines and amino acids following acute administration of the antidepressant/antipanic drug phenelzine. Biochem. Pharmacol. 59, 1253–1263.
Perahia, D. G., Gilaberte, I., Wang, F. J., Wiltse, C. G., Huckins, S. A., Clemens, J. W., Montgomery, S. A., Montejo, A. L., and Detke, M. J. (2006). Duloxetine in the prevention of relapse of major depressive disorder – Double-blind placebo-controlled study. Br. J. Psychiatry 188, 346–353.
Perahia, D. G. S., Maina, G., Thase, M. E., Spann, M. E., Wang, F. J., Walker, D. J., and Detke, M. J. (2009). Duloxetine in the prevention of depressive recurrences: a randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 70, 706–716.
Petrakis, I. L., Leslie, D., and Rosenheck, R. (2003). The use of antidepressants in alcohol-dependent veterans. J. Clin. Psychiatry 64, 865–870.
Posternak, M. A., Solomon, D. A., Leon, A. C., Mueller, T. I., Shea, M. T., Endicott, J., and Keller, M. B. (2006). The naturalistic course of unipolar major depression in the absence of somatic therapy. J. Nervous Ment. Dis. 194, 324–329.
Price, J., Sloman, L., Gardner, R., Gilbert, P., and Rohde, P. (1994). The social competition hypothesis of depression. Br. J. Psychiatry 164, 309–315.
Raghunathan, T. E., Solenberger, P. W., and Van Hoewyk, J. (2002). IVEware: Imputation and Variance Estimation Software User Guide [Computer software]. Ann Arbor, MI: The Regents of the University of Michigan.
Reimherr, F. W., Amsterdam, J. D., Quitkin, F. M., Rosenbaum, J. F., Fava, M., Zajecka, J., Beasley, C. M. Jr., Michelson, D., Roback, P., and Sundell, K. (1998). Optimal length of continuation therapy in depression: a prospective assessment during long-term fluoxetine treatment. Am. J. Psychiatry 155, 1247–1253.
Renard, C. E., Dailly, E., Nic Dhonnchadha, B. A., Hascoet, M., and Bourin, M. (2004). Is dopamine a limiting factor of the antidepressant-like effect in the mouse forced swimming test? Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 1255–1259.
Reynolds, C. F., Frank, E., Perel, J. M., Imber, S. D., Cornes, C., Miller, M. D., Mazumdar, S., Houck, P. R., Dew, M. A., Stack, J. A., Pollock, B. G., and Kupfer, D. J. (1999). Nortriptyline and interpersonal psychotherapy as maintenance therapies for recurrent major depression – A randomized controlled trial in patients older than 59 years. JAMA 281, 39–45.
Rickels, K., Montgomery, S. A., Tourian, K. A., Guelfi, J. D., Pitrosky, B., Padmanabhan, S. K., Germain, J. M., Leurent, C., and Brisard, C. (2010). Desvenlafaxine for the prevention of relapse in major depressive disorder: results of a randomized trial. J. Clin. Psychopharmacol. 30, 18–24.
Robert, P., and Montgomery, S. A. (1995). Citalopram in doses of 20-60 mg Is effective in depression relapse prevention: a placebo-controlled 6-month study. Int. Clin. Psychopharmacol. 10, 29–35.
Robinson, D. S., Lerfald, S. C., Bennett, B., Laux, D., Devereaux, E., Kayser, A., Corcella, J., and Albright, D. (1991). Continuation and maintenance treatment of major depression with the monoamine oxidase inhibitor Phenelzine: a double-blind placebo controlled discontinuation study. Psychopharmacol. Bull. 27, 31–39.
Romanovsky, A. A. (2007). Thermoregulation: some concepts have changed. Functional architecture of the thermoregulatory system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R37–R46.
Romanovsky, A. A., Almeida, M. C., Aronoff, D. M., Ivanov, A. I., Konsman, J. P., Steiner, A. A., and Turek, V. F. (2005). Fever and hypothermia in systemic inflammation: recent discoveries and revisions. Front. Biosci. 10, 2193–2216.
Rouillon, F., Berdeaux, G., Bisserbe, J. C., Warner, B., Mesbah, M., Smadja, C., and Chwalow, J. (2000). Prevention of recurrent depressive episodes with milnacipran: consequences on quality of life. J. Affect. Disord. 58, 171–180.
Rush, A. J., Trivedi, M. H., Wisniewski, S. R., Nierenberg, A. A., Stewart, J. W., Warden, D., Niederehe, G., Thase, M. E., Lavori, P. W., Lebowitz, B. D., McGrath, P. J., Rosenbaum, J. F., Sackeim, H. A., Kupfer, D. J., Luther, J., and Fava, M. (2006). Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am. J. Psychiatry 163, 1905–1917.
Sadock, B. J., and Sadock, V. A. (2005). Kaplan & Sadock’s Comprehensive Textbook of Psychiatry, 8th Edn. Philadelphia, PA: Lippincott Williams & Wilkins.
Sapolsky, R. M. (2000). The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death. Biol. Psychiatry 48, 755–765.
Sapolsky, R. M. (2001). Depression, antidepressants, and the shrinking hippocampus. Proc. Natl. Acad. Sci. U.S.A. 98, 12320–12322.
Savitz, J. B., and Drevets, W. C. (2009). Imaging phenotypes of major depressive disorder: genetic correlates. Neuroscience 164, 300–330.
Schmidt, M. E., Fava, M., Robinson, J. M., and Judge, R. (2000). The efficacy and safety of a new enteric-coated formulation of fluoxetine given once weekly during the continuation treatment of major depressive disorder. J. Clin. Psychiatry 61, 851–857.
Schulman, C. I., Namias, N., Doherty, J., Manning, R. J., Li, P., Elhaddad, A., Lasko, D., Amortegui, J., Dy, C. J., Dlugasch, L., Baracco, G., and Cohn, S. M. (2005). The effect of antipyretic therapy upon outcomes in critically ill patients: a randomized, prospective study. Surg. Infect. (Larchmt) 6, 369–375.
Seo, D. O., Shin, C. Y., Lee, C. J., Dailey, J. W., Reith, M. E. A., Jobe, P. C., and Ko, K. H. (1999). Effect of alterations in extracellular norepinephrine on adrenoceptors: a microdialysis study in freely moving rats. Eur. J. Pharmacol. 365, 39–46.
Simon, J. S., Aguiar, L. M., Kunz, N. R., and Lei, D. (2004). Extended-release venlafaxine in relapse prevention for patients with major depressive disorder. J. Psychiatr. Res. 38, 249–257.
Spitzer, R. L., and Wakefield, J. C. (1999). DSM-IV diagnostic criterion for clinical significance: does it help solve the false positives problem? Am. J. Psychiatry 156, 1856–1864.
Stahl, S. (2008). Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications, 3rd Edn. Cambridge: Cambridge University Press.
Stein, M. K., Rickels, K., and Weise, C. C. (1980). Maintenance therapy with amitriptyline: controlled trial. Am. J. Psychiatry 137, 370–371.
Stewart, J. W., Tricamo, E., McGrath, P. J., and Quitkin, F. M. (1997). Prophylactic efficacy of phenelzine and imipramine in chronic atypical depression: likelihood of recurrence on discontinuation after 6 months’ remission. Am. J. Psychiatry 154, 31–36.
Takahashi, H., Takada, Y., Nagai, N., Urano, T., and Takada, A. (2000). Serotonergic neurons projecting to hippocampus activate locomotion. Brain Res. 869, 194–202.
Tanda, G., Carboni, E., Frau, R., and Dichiara, G. (1994). Increase of extracellular dopamine in the prefrontal cortex: a trait of drugs with antidepressant potential. Psychopharmacology (Berl.) 115, 285–288.
Terra, J. L., and Montgomery, S. A. (1998). Fluvoxamine prevents recurrence of depression: results of a long-term, double-blind, placebo-controlled study. Int. Clin. Psychopharmacol. 13, 55–62.
Tomlinson, K. L., Tate, S. R., Anderson, K. G., McCarthy, D. M., and Brown, S. A. (2006). An examination of self-medication and rebound effects: psychiatric symptomatology before and after alcohol or drug relapse. Addict. Behav. 31, 461–474.
Turner, E. H., Matthews, A. M., Linardatos, E., Tell, R. A., and Rosenthal, R. (2008). Selective publication of antidepressant trials and its influence on apparent efficacy. N. Engl. J. Med. 358, 252–260.
Uylings, H. B. M., Groenewegen, H. J., and Kolb, B. (2003). Do rats have a prefrontal cortex? Behav. Brain Res. 146, 3–17.
Uylings, H. B. M., and Vaneden, C. G. (1990). Qualitative and quantitative comparison of the prefrontal cortex in rat and in primates, including humans. Prog. Brain Res. 85, 31–62.
Valenstein, E. S. (1998). Blaming the Brain: The Truth About Drugs and Mental Health. New York, NY: The Free Press.
Valentini, V., Cacciapaglia, F., Frau, R., and Di Chiara, G. (2005). Selective serotonin reuptake blockade increases extracellular dopamine in noradrenaline-rich isocortical but not prefrontal areas: dependence on serotonin-1A receptors and independence from noradrenergic innervation. J. Neurochem. 93, 371–382.
Valentini, V., Frau, R., and Di Chiara, G. (2004). Noradrenaline transporter blockers raise extracellular dopamine in medial prefrontal but not parietal and occipital cortex: differences with mianserin and clozapine. J. Neurochem. 88, 917–927.
van Praag, H., and de Haan, S. (1980). Depression vulnerability and 5-hydroxytryptophan prophylaxis. Psychiatry Res 3, 75–83.
Viguera, A. C., Baldessarini, R. J., and Friedberg, J. (1998). Discontinuing antidepressant treatment in major depression. Harv. Rev. Psychiatry 5, 293–306.
Wakefield, J. C. (1992). The concept of mental disorder: on the boundary between biological facts and social values. Am. Psychologist 47, 373–388.
Wakefield, J. C., Schmitz, M. F., and Baer, J. C. (2010). Does the DSM-IV clinical significance criterion for major depression reduce false positives? Evidence from the National Comorbidity Survey replication. Am. J. Psychiatry 167, 298–304.
Wakefield, J. C., Schmitz, M. F., First, M. B., and Horwitz, A. V. (2007). Extending the bereavement exclusion for major depression to other losses - Evidence from the National Comorbidity Survey. Arch. Gen. Psychiatry 64, 433–440.
Watson, P. J., and Andrews, P. W. (2002). Toward a revised evolutionary adaptationist analysis of depression: the social navigation hypothesis. J. Affect. Disord. 72, 1–14.
Weikop, P., Kehr, J., and Scheel-Kruger, J. (2007). Reciprocal effects of combined administration of serotonin, noradrenaline and dopamine reuptake inhibitors on serotonin and dopamine levels in the rat prefrontal cortex: the role of 5-HT1A receptors. J. Psychopharmacol. 21, 795–804.
Wells, J. E., and Horwood, L. J. (2004). How accurate is recall of key symptoms of depression? A comparison of recall and longitudinal reports. Psychol. Med. 34, 1001–1011.
Wilson, K. C. M., Mottram, P. G., Ashworth, L., and Abou-Saleh, M. T. (2003). Older community residents with depression: long-term treatment with sertraline – Randomised, double-blind, placebo-controlled study. Br. J. Psychiatry 182, 492–497.
Wolowczuk, I., Verwaerde, C., Viltart, O., Delanoye, A., Delacre, M., Pot, B., and Grangette, C. (2008). Feeding our immune system: impact on metabolism. Clin. Dev. Immunol. 1–19.
Woods, A. H. (2009). “Evolution of homeostatic physiological systems,” in Phenotypic Plasticity of Insects, eds D. Whitman and T. N. Ananthakrishnan (Plymouth: Science Publishers), 655–674.
Wrosch, C., and Miller, G. E. (2009). Depressive symptoms can be useful: self-regulatory and emotional benefits of dysphoric mood in adolescence. J. Pers. Soc. Psychol. 96, 1181–1190.
Young, A. M., and Goudie, A. J. (1995). “Adaptive processes regulating tolerance to behavioral effects of drugs,” in Psychopharmacology: 4th Generation of Progress, eds F. E. Bloom and D. J. Kupfer (New York, NY: Raven Press), 733–742.
Keywords: antidepressant medication, homeostasis, major depression, meta-analysis, oppositional tolerance, relapse, discontinuation
Citation: Andrews PW, Kornstein SG, Halberstadt LJ, Gardner CO and Neale MC (2011) Blue again: perturbational effects of antidepressants suggest monoaminergic homeostasis in major depression. Front. Psychology 2:159. doi: 10.3389/fpsyg.2011.00159
Received: 10 May 2011;
Paper pending published: 14 June 2011;
Accepted: 23 June 2011;
Published online: 07 July 2011.
Edited by:
Debra Lieberman, University of Miami, USACopyright: © 2011 Andrews, Kornstein, Halberstadt, Gardner and Neale. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Paul W. Andrews, Department of Psychology, Neuroscience and Behaviour, McMaster University, 1280 Main Street West, Hamilton, ON, Canada L8S 4K1. e-mail: pandrews@mcmaster.ca