 Cedric Boeckx
Cedric Boeckx Antonio Benítez-Burraco
Antonio Benítez-Burraco- 1Catalan Institute for Advanced Studies and Research (ICREA), Barcelona, Spain
- 2Department of Linguistics, Universitat de Barcelona, Barcelona, Spain
- 3Department of Spanish Philology and its Didactics, University of Huelva, Huelva, Spain
This study builds on the hypothesis put forth in Boeckx and Benítez-Burraco (2014), according to which the developmental changes expressed at the levels of brain morphology and neural connectivity that resulted in a more globular braincase in our species were crucial to understand the origins of our language-ready brain. Specifically, this paper explores the links between two well-known ‘language-related’ genes like FOXP2 and ROBO1 implicated in vocal learning and the initial set of genes of interest put forth in Boeckx and Benítez-Burraco (2014), with RUNX2 as focal point. Relying on the existing literature, we uncover potential molecular links that could be of interest to future experimental inquiries into the biological foundations of language and the testing of our initial hypothesis. Our discussion could also be relevant for clinical linguistics and for the interpretation of results from paleogenomics.
Overview
A central goal of the biolinguistic enterprise (Di Sciullo and Boeckx, 2011) is to shed light on the genetic basis of a complex trait, characteristic of modern humans, which we dub the language-ready brain, understood as the set of neural structures that serve as a critical biological substrate for the acquisition and use of grammatical systems routinely called natural languages.
In this paper we would like to expand on the hypothesis we put forth in Boeckx and Benítez-Burraco (2014), taking into account considerations that we set aside when we formulated our original claim and constructed the initial set of genes we took to be of interest. This hypothesis, in a nutshell, amounted to claiming that the developmental changes expressed at the levels of brain morphology and neural connectivity that resulted in a more globular braincase in our species (‘globularity’) were crucial to understand a central aspect of the language-readiness of the modern human brain, viz. the ability to form complex, cross-modular thoughts. Using terminology common among linguists, we can say that this aspect of language pertained to the syntax-semantics interface. As we pointed out at the outset of our paper, this facet of our language-ready brain is distinct from another ability that is equally central to the acquisition and use of language: the ability to communicate these thoughts to conspecifics (“morpho-phonology” for linguists). In the technical literature (Berwick et al., 2013), this ability is said to be the responsibility of an ‘externalization’ component. The latter is by far the best studied aspect of our language-ready brain, as it can be related most directly to the vocal learning abilities manifested in other species, about which we are beginning to know a fair amount. The externalization component is also the one that most obviously breaks down in a number of language disorders. Thanks to this, it has been possible to begin to relate its functioning to some genes, most notably FOXP2 (Lai et al., 2001; Fisher and Marcus, 2006; Graham and Fisher, 2013).
Our aim here is to examine possible connections between the set of genes considered in Boeckx and Benítez-Burraco (2014) and what we know about the molecular substrate that allows humans to externalize the complex thoughts that our language-ready brain allows us to form. One particular reason to suspect that such connections are worth examining is that our initial investigation already revealed points of contact between our core set of genes (USF1, RUNX2, DLX1, DLX2, DLX5, DLX6, BMP2, BMP7, and DISP1) and FOXP2 and its partners. In addition, the anatomical structure we focused on in our 2014 paper, the thalamus, not only plays a crucial role in establishing and maintaining efficient cortico-cortical connections that we deemed necessary for cross-modularity (Boeckx and Benítez-Burraco, 2014: see Results and Discussion; Wang et al., 2010a; Buckner and Krienen, 2013), it also functions as a bridge between the cortex and the subcortical structures most frequently associated with the externalization component of language (the basal ganglia and the cerebellum). The fact that it has long been suspected that the FOXP2 network appears to play a critical role in the establishment and maintenance of these neural circuits (Vargha-Khadem et al., 2005; Fisher and Marcus, 2006; Kurt et al., 2012) suggest to us that it makes sense to look for potential connections. Thinking of Darwin’s (1871) concept of ‘correlated growth,’ we ought to expect changes at the level of the thalamus to affect the externalization component as much as the syntactic-semantic aspects of language that were our original focus, especially given the fact that unlike other species, our linguistic mode of communication appears to be able to express virtually all the thoughts we can construct. Thus, the present paper can be seen as an attempt to extend the scope of our initial study.
Much like we claimed that the roots of cross-modularity were quite ancient in our (2014) piece, we also think that the externalization component of the language-ready brain rests on solid evolutionary foundations. Although we believe that our nearest (extinct) relatives differed from us in certain ways that pertain not only to the syntax-semantics interface but also to the domain of externalization, we think that they were almost certainly capable of vocal learning, and had built a niche that favored communication and cooperation. Dunbar (1996), Deacon (1997), Mithen et al. (2006), Wrangham (2009), and Tomasello (2014), contain detailed arguments in favor of our communicative abilities and the contexts in which communication takes place having ancient roots: our (extinct) ancestors were highly social, cooperative, and undoubtedly displayed symbolic practices (rituals). They were big-brained, and displayed many anatomical and even molecular signs of vocal learning (Dediu and Levinson, 2013). In addition, they were skillful tool-users, and displayed some modern-like brain structures that people have long associated with aspects of language, such as a well-developed Broca’s area, and lateralization patterns reminiscent of ours (Frayer et al., 2010). All of these properties were unquestionably important for linguistic communication as we know it to ultimately develop. Thus we find ourselves in general agreement with the many researchers who treat Neanderthals as vocal learners, but we also side with scholars like Lieberman (2007), who has long insisted on modern speech abilities requiring an anatomy specific to modern humans. For us, this specific anatomy boils down to globularity, the outcome of a species-specific developmental trajectory that takes during the first year of life (Boeckx and Benítez-Burraco, 2014, and references therein), although some of its consequences only manifest themselves after a certain amount of maturation. The view we defend here is that globularity most likely entailed changes that led to some additional, possibly selected-for, adjustments within molecular pathways that were involved in vocal learning and were recruited to give rise to speech proper. In this respect, our stance fits well with the ‘musical protolanguage’ scenario first proposed by Darwin (1871) and recently refined by several authors (Mithen et al., 2006; Fitch, 2010). (Before proceeding, it may be worth mentioning that instead of speech, we should perhaps talk about sensori-motor abilities to capture the fact that the vocal channel is but one mode of linguistic externalization. Ultimately, signed languages must be integrated into our theorizing. Having said this, we will mostly discuss draw from the literature on vocal learning in what follows, if only because this is where progress in comparative cognition has been most obvious.)
The literature on vocal learning has been growing steadily over the last 10 years, and enables us to ground our proposal onto well-established anatomical and molecular findings. It is not our goal here to provide an exhaustive review of the literature, as several such reviews already exist. For example, Fitch and Jarvis (2013) provide numerous references in support of neural pathways necessary for vocal learning in birds, and by analogy in humans. One such pathway, the so-called posterior pathway, necessary to produce song/speech, provides a direct forebrain control of brainstem vocal motor neurons, lacking in vocal non-learners (Fitch, 2010). Another pathway, the so-called anterior pathway, is not necessary for producing songs, but is necessary for learning songs and for modifying already learned songs. This pathway consists of a loop linking basal ganglia (especially, the striatum), the thalamus, and (for humans) cortical structures (Miller and Buschman, 2007). Among the cortical structures, one finds the well-known fronto-temporal network connecting Broca’s and Wernicke’s areas (Hickok and Poeppel, 2007 and much subsequent work refining the classical Broca–Wernicke–Lichtheim–Geschwind model presented in all textbooks on this topic; in particular; Hickok et al., 2011; Hickok, 2012). Although traditionally taken to constitute ‘the language network,’ the fronto-temporal connections are best regarded implicated in aspects of the externalization component of language (see Boeckx et al., 2014, for review).
Our focus here will not be so much on these neuroanatomical findings, but rather on some of the most prominent molecular signatures of vocal learning identified in the literature. Besides FOXP2 and its partners (FOXP1, FOXP4, CNTNAP2, etc.), enriched gene regulation of the ROBO/SLIT family of genes has been identified (Fitch and Jarvis, 2013). Both FOXP2 and ROBO1 have been associated with language/speech disorders (specific language impairment and dyslexia, respectively; see below), and therefore strike us as valuable starting points in our quest. Recent studies, such as Kato et al. (2014), also use these genes as probes with the same confidence we want to use them here. We stress, though, that taking FOXP2 and ROBO1 as focal points does not mean that other “language-related” genes found in the literature are less important (see Benítez-Burraco, 2009, for a comprehensive survey). It is just that the potential functional links discussed below appear to us to be the most promising ones at this point, in large part because, as already mentioned above, these genes and their partners have been consistently related to brain areas and neural circuits that have been repeatedly implicated in the externalization of language. In part, our choice was also dictated by the fact that in most cases, and in contrast to FOXP2 and ROBO1, the genes related to language have not yet given rise to a solid body of knowledge on which we could rely. We certainly hope that future work will complement our perspective, which at this point is but a small corner of a much bigger puzzle.
Methods, Aims, and Limitations
As already stated, our goal in this paper is to expand the gene list that potentially fall under the scope of our initial hypothesis, and looking for potential functional connections with the initial gene set discussed in Boeckx and Benítez-Burraco (2014). To do so, we have extensively reviewed the literature on both the FOXP2 and ROBO1 interactomes and functional networks, as presently known, and examined connections with the network put forth in our (2014) paper.
Our modus operandi has been as follows:
-
We used PubMed1 to identify potential partners of the genes of interest in our (2014) paper, FOXP2 and ROBO1. We narrow our search by using key terms of interest for us, including (but not restricted to) “brain,” “cognition,” “language,” “syntax,” “semantics,” “phonology,” “speech,” or “vocal learning.”
-
We also searched the literature via PubMed looking for genes related to clinical conditions and symptoms of interest for us. Some of the key search terms used were: “language disorder,” cognitive disorder,” “intellectual disability,” “syntax deficit,” “semantic deficit,” “phonological deficit,” “speech deficit,” “dyslexia,” “schizophrenia,” “autism,” “autism spectrum disorder,” etc. When a gene of interest was identified, we looked for potential links with our core set of genes, refining the searching process by adding the name of genes of interest previously identified.
-
We also used PubMed to look for genes related to brain areas, circuits, neural processes, neurotransmitters, etc. of interest for us; specifically, the neurological considerations of Boeckx and Benítez-Burraco (2014) as well as those neural substrates highlighted in the vocal learning and FOXP2 literature. For this, we used search terms like “thalamus,” “thalamo-cortical connection,” “motor cortex,” “neurite outgrowth,” etc.
-
We did not systematically gather information beyond more than three connection levels. Thus, our limit was of the sort ‘ROBO1 is connected to X which is in turn connected to Y.’ We considered additional levels only if the identified genes were more closely connected to some other gene(s) of interest previously reviewed. For example, ‘ROBO1 is connected to X which is in turn connected to Y which is in turn connected to Z (but Z is connected to FOXP2).’
-
We explored potential connections with genes selected in anatomically modern humans (AMH; Pääbo’s 2014 list).
Because the amount of literature on single genes varies greatly, we did not necessarily discard any gene because the number of manuscripts was under a value we may have pre-selected.
In addition to PubMed, we also relied on the following databases, which we also used for our (2014) paper.
-
the microarray database of the Allen Brain Atlas2.
-
the Prenatal LMD Microarray search engine3.
-
the Developmental Transcriptome browser4 of the Allen Brain Atlas.
-
OMIM for the linguistic and cognitive deficits linked to the mutation of genes of interest5.
Additionally, we have exploited the information provided in Kuhlwilm et al. (2013), where 691 genes were found to be differentially expressed after RUNX2 transfection in neuroblastomic SH-SY5Y cells, and we have studied the overlap with the FOXP2 targets list provided in Konopka et al. (2009), who also relied on this cell line (see Table 1). [Other comprehensive lists of FOXP2 targets we also took into account are Spiteri et al. (2007) and Vernes et al. (2007).] We have also examined the overlap between the list provided in Kuhlwilm et al. (2013) and the list of differentially expressed genes in vocal learners in Wang (2011; see Table 2). Finally, we have explored links and predicted interactions generated by String 9.16. String 9.1 predicts direct/physical and indirect/functional associations between proteins that derive from four sources: genomic context, high-throughput experiments, conserved coexpression, and the knowledge previously gained from text mining (Szklarczyk et al., 2011). For each new candidate gene, we did an extensive literature survey to confirm its viability as a member of our network. The next section summarizes our results.

TABLE 1. Overlap between the FOXP2 targets list provided in Konopka et al. (2009) and the 691 genes found to be differentially expressed after RUNX2 transfection in neuroblastomic SHSY5Y cells in Kuhlwilm et al. (2013).

TABLE 2. Overlap between the list of differentially expressed genes in vocal learners in Wang (2011) and the list of genes found to be affected by RUNX2 overexpression in 10 different human cell lines in Kuhlwilm et al. (2013).
Results and Discussion
All the genes discussed in this section were selected as a result of the search method described in Section “Methods, Aims, and Limitations.” The information we provide about each gene pertains to the potential connections with the genes of interest in Boeckx and Benítez-Burraco (2014), with special emphasis on brain growth and neural interconnection. We also report on whether the gene is known as candidate for a clinical condition associated with a variety of linguistic, cognitive, and cranio-facial deficits (results from linkage or association analyses, GWAS, or the discovery of point mutations or chromosomal rearrangements affecting the gene function). We begin with ROBO genes and their partners (see The ROBO/SLIT Suite) (see Figure 1), then turn our attention to FOXP2 and its partners (see FOXP2 and Partners) (see Figure 2), and conclude this section by highlighting other related genes of interest (see Other Genes of Interest).
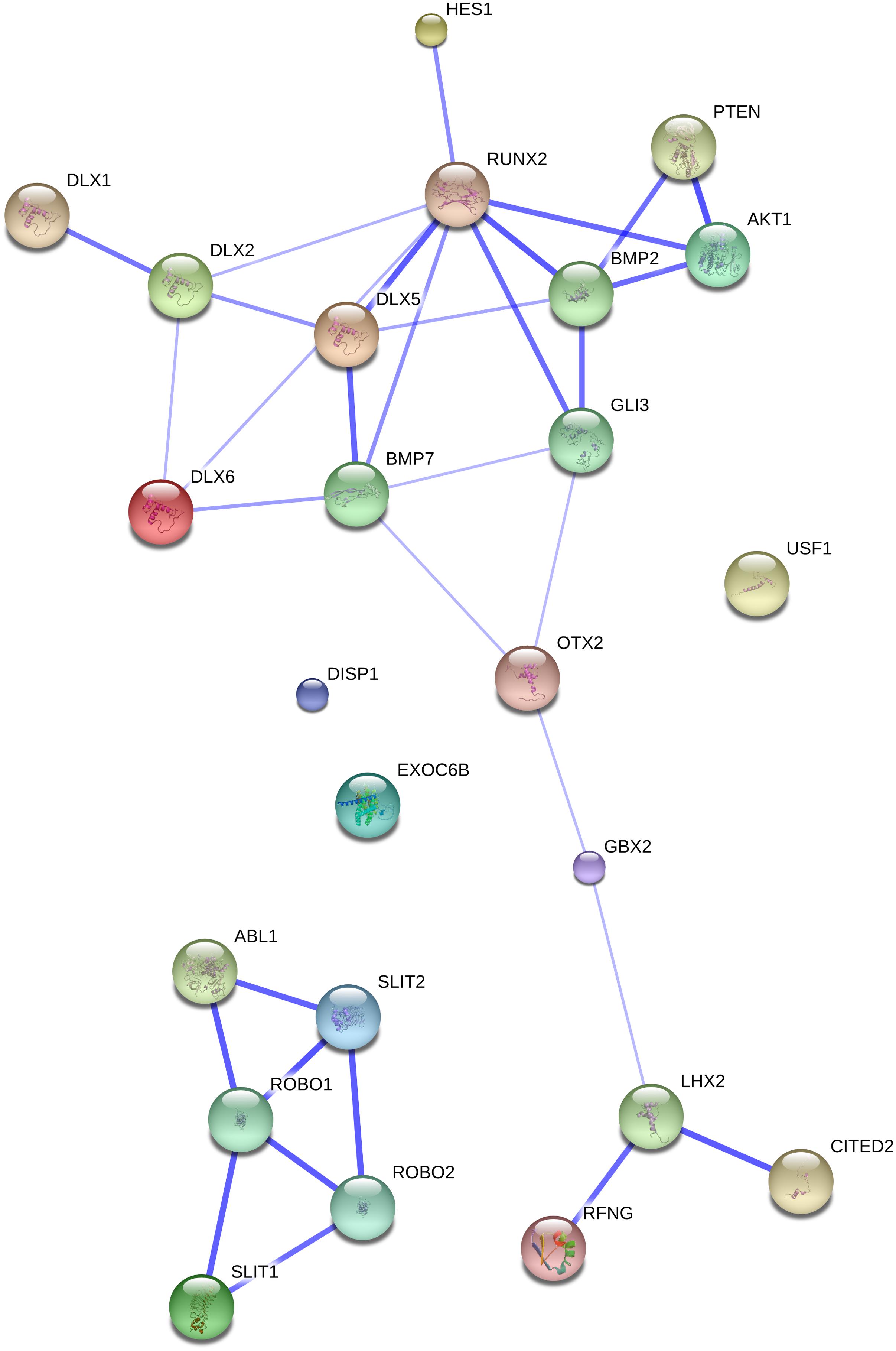
FIGURE 1. The ROBO/SLIT suite and the genes related to our initial gene set. The network was generated by String 9.1 with the proteins discussed in Section “The ROBO/SLIT Suite” plus the gene set related to globularity as advanced in Boeckx and Benítez-Burraco (2014). The medium confidence value was 0.0400. Nodes representing the proteins encompassing the network are colored randomly. In this confidence view, stronger associations between proteins are represented by thicker lines. The figure does not represent a fully connected graph, but readers are asked to bear in mind that String 9.1 predicts associations between proteins that derive from a limited set of databases. The material discussed in the main text lead us to suspect connections that String does not generate (although we wish to note that just adding a few genes, not discussed in this paper, yield a connected graph). It should be emphasized that the nature of String 9.1 is essentially predictive, and not explanatory. Although we have confirmed all the links we discuss here in the literature, they need to be confirmed at the brain level and in relation to language. Additionally, the diagram only represents the potential connectivity between the involved proteins, but this has to be mapped onto particular biochemical networks, signaling pathways, cellular properties, aspects of neuronal function, or cell-types of interest that can be confidently related to aspects of language development and function.
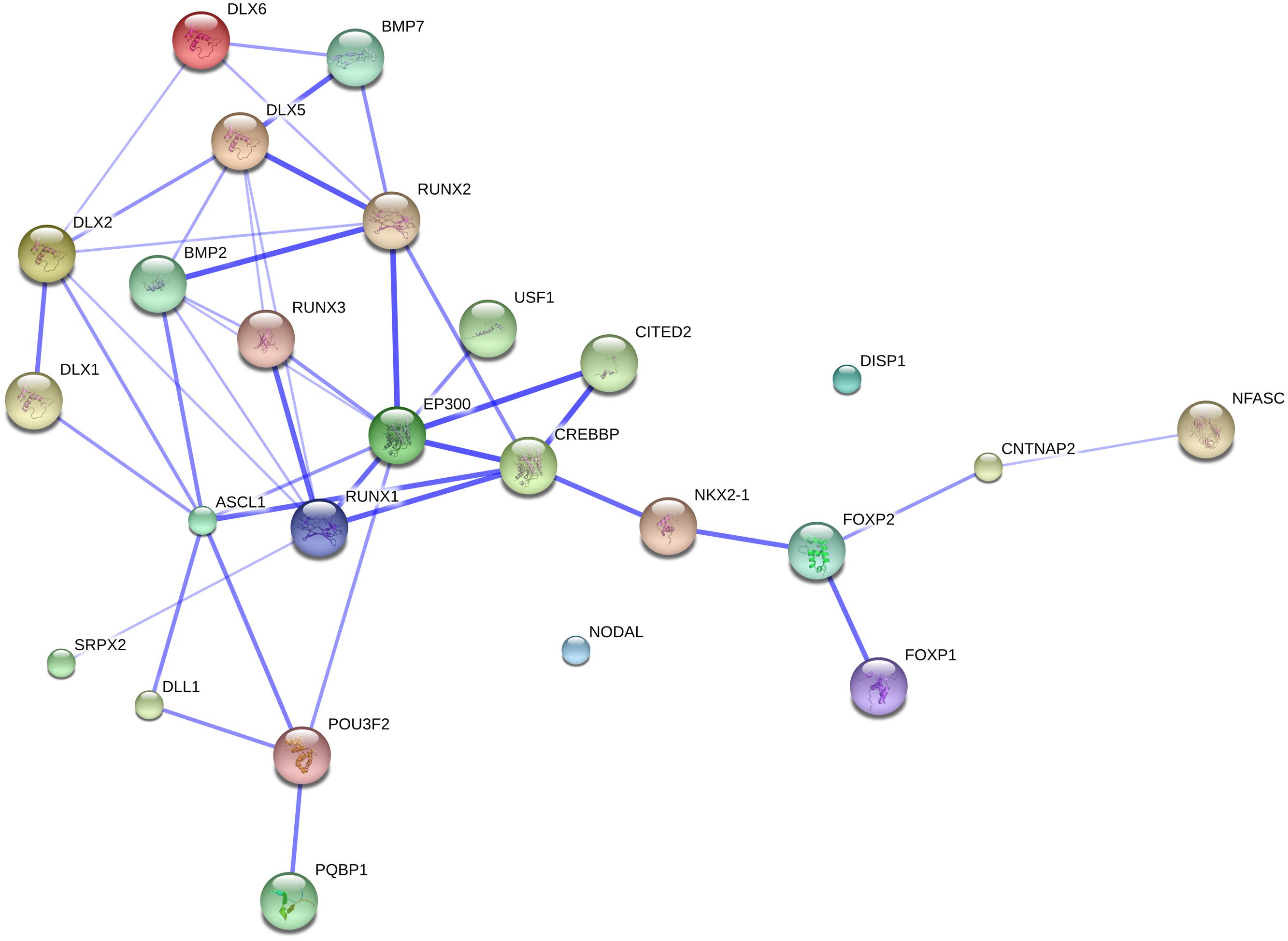
FIGURE 2. The FOXP2 suite and the genes related to globularity. The network was generated by String 9.1 with the proteins discussed in Section “FOXP2 and Partners” plus the gene set related to globularity as advanced in Boeckx and Benítez-Burraco (2014). The medium confidence value was 0.0400. The caveats noted for Figure 1 apply.
The ROBO/SLIT Suite
Members of the ROBO gene family, which includes ROBO1 and ROBO2, discussed here, play an important role to control the development of ascending or descending major axon tracts to or from the forebrain, and interneuron migration in the forebrain, through interactions with the ligands SLIT1, SLIT2, and SLIT3 (Andrews et al., 2006; López-Bendito et al., 2007; Dugan et al., 2011).
ROBO1
ROBO1 encodes an axon guidance receptor to navigate the growth of longitudinal axons (Devine and Key, 2008). Mutations in ROBO1 are associated with human dyslexia and speech sound disorder (Hannula-Jouppi et al., 2005; Mascheretti et al., 2014). As Wang (2011) notes, ROBOs/SLITs show differential expression in song nuclei of songbirds. The avian arcopallium contains large regions with parallels to the mammalian cortex. Particularly, the robust nucleus of the arcopallium (RA) projection neurons are proposed to be analogous to the layer V neurons of the facial motor cortex of mammals that sends direct projections to the brainstem vocal nucleus in humans (Jarvis, 2004), while the arcopallium surrounding the RA may be analogous to the layer V neurons of mammalian motor cortex (Feenders et al., 2008). Wang (2011) detected the expression of all ROBOs and SLITs in the RA and the surrounding arcopallium in the adult male zebra finch brain. As a comparison, three regions (layer V, entorhinal cortex and piriform cortex) of the adult rat cortex are found to express all five ROBOs/SLITs genes (Marillat et al., 2002). Of them, only layer V contains the descending motor pathway neurons. Thus, these findings seem to support the analogy between the RA surrounding arcopallium to the motor cortex (layer V) of mammals.
Additionally, Wang (2011) found specialized expression patterns of ROBO1, SLIT1, and ROBO2 (but not SLIT2 and SLIT3), in the RA of adult male zebra finch, relative to the surrounding arcopallium. Only ROBO1 is up-regulated in the RA, while ROBO2 and SLIT1 are down-regulated in this region. This difference between ROBO1 and ROBO2 in the RA is remarkable, since the two ROBOs typically act together for axon guidance in the forebrain (López-Bendito et al., 2007) and in rats, they are co-expressed in most telencephalic regions (Marillat et al., 2002). For birds, Wang (2011) suggests that ROBO1 is co-opted in the specialized vocal motor output nucleus of vocal learning birds, but admits that it remains to be seen whether it participates in forming the unique direct projections to the brainstem vocal nucleus. Wang (2011) also considers worth exploring the ROBO1 expression patterns in the analogous face motor cortex area in human and other mammalian vocal learners or non-learners. In this context, Wang (2011) observes that recent studies showed that a splice variant of ROBO1, called ROBO1a, is highly enriched in the temporal auditory neocortex and/or temporal association neocortex, while ROBO1b is enriched in the prefrontal neocortex where face motor cortex and Broca’s area develops (Johnson et al., 2009). Both ROBO1 and ROBO2 are specifically involved in thalamocortical axons (TCA) development (López-Bendito et al., 2007; Marcos-Mondéjar et al., 2012). TCAs represent the major input to the neocortex and modulate cognitive functions, consciousness, and alertness.
ROBO2
ROBO2 is a putative candidate for autism (Suda et al., 2011). Moreover, a polymorphism near the gene has been associated with expressive vocabulary growth in the normal population (St Pourcain et al., 2014). The locus for the gene is also a linkage region for dyslexia (Fisher et al., 2002) and speech-sound disorder and reading (Stein et al., 2004). ROBO2 is a functional partner of DLX1 and DLX2 too, two of the genes we highlighted in the context of our ‘globularity and language-readiness hypothesis.’ In particular, both Dlx1 and Dlx2 factors are required to promote tangential migration to the olfactory bulb via Robo2 (Long et al., 2007).
SLIT2
ROBO’s ligands SLITs are also worth considering in the context of our hypothesis. During human brain evolution, considerable enlargement of the association areas of the cortex is accompanied by a comparable enlargement of corresponding association thalamic nuclei in the diencephalon, but developmental mechanisms coordinating these expansions remain unknown. SLITs proteins have been claimed to play important roles in brain development before and after birth, and several pieces of evidence suggest that they may be involved in species-specific developmental patterns of the thalamus. Specifically, Slit2 has been hypothesized to act as a repellent for migrating sub-ventricular zone cells (Hu, 1999; Chen et al., 2001; Wu et al., 2001). The Slit/Robo guidance family also ensures the restraint of corticothalamic axons within the internal capsule, and upon reaching the diencephalon–telencephalon boundary, direct them dorsally toward the thalamus rather than crossing the midline (Bagri et al., 2002; López-Bendito et al., 2007; Braisted et al., 2009; Leyva-Díaz and López-Bendito, 2013). Slit1 and Slit2 are expressed in overlapping domains including the ganglionic eminences, prethalamus, hypothalamus, and the germinal zone of the thalamus (Bagri et al., 2002); while Robo1 and Robo2 are expressed in complementary patterns in the cortical plate, intermediate zone, and thalamus (López-Bendito et al., 2007).
Slit2 is also strongly expressed in the region of the cortical hem, a boundary region that has been shown to express transcripts of members of the patterning gene families Wnt, Bmp, and Msx, as well as Shh (see Grove et al., 1998), genes linked to our initial (2014) gene set of interest. Specifically, the midline repellent Slit2 orients migration of corridor neurons and thereby switches thalamic axons from an external to a mammalian-specific internal path. Bielle et al. (2011) reveal that subtle differences in the migration of conserved intermediate target neurons trigger large-scale changes in thalamic connectivity, and opens perspectives on Slit functions and the evolution of brain wiring. In particular, this suggests that SLIT2 may be involved in the human specific pattern of DLX-expressing interneuron migration from the ganglionic eminence into the thalamus (Letinic and Rakic, 2001), which is held to be responsible for the human-specific enlargement of higher-order thalamic nuclei like the mediodorsal nucleus or the pulvinar, structures highlighted by Boeckx and Benítez-Burraco (2014) in the context of globularity and language-readiness. It is also worth pointing out here that the promoter region of SLIT2 has been claimed to have been under significant positive selection in humans relative to other primates (Haygood et al., 2007).
SLIT1
Interestingly, another SLIT gene, SLIT1, an effector of ROBO1, serves a direct downstream target for the speech-language related gene FOXP2 (Vernes et al., 2007; Konopka et al., 2009). Moreover, three genes that encode proteins that are part of the centrosome assembly that interacts with SLIT1, necessary for proper neuron migration and neural process extension, show differential expressions among (mammalian) vocal learners (Wang, 2011): CKAP5, PCM1, and CEP192 (Higginbotham et al., 2006).
HES1
HES1 is functionally related to ROBO1. During neurogenesis the transcriptional activation of Hes1 is a key step for the Slit/Robo signaling pathway that modulates the transition between primary and intermediate progenitors (Borrell et al., 2012). Hes1 is also a direct interactor of runx2 (Suh et al., 2008), one of the most relevant genes discussed in Boeckx and Benítez-Burraco (2014) in the context of globularity. Hes1 is expressed in the ventral posterior part of the thalamus (Lein et al., 2007), and the HES1 pathway is related to language development and craniofacial development. Thus, the reduced expression of EXOC6B affects the expression of HES1 (Wen et al., 2013). In turn, EXOC6B haploinsufficiency has been related to intellectual disability, language delay, facial asymmetry, and vertebral and/or craniofacial abnormalities (Wen et al., 2013). Importantly, Hes1 is involved in the development of both GABAergic neurons, whose relevance we discussed in our (2014) paper, and of dopaminergic neurons, routinely mentioned in the literature on motor behavior and vocal learning in particular. According to Long et al. (2013), Hes1 silencing is able to promote bone marrow mesenchymal stem cells to differentiate into GABAergic neuron-like cells in vitro. In particular, it has been proposed that Hes1 plays a prominent role in regulating the location and density of mesencephalic dopaminergic neurons (Kameda et al., 2011). In Hes1 homozygous null mutant mice the lack of the gene results in disturbances in the inductive and repulsive activities of the isthmic organizer and leads to the failure of cranial neurulation due to the premature onset of neural differentiation.
GBX2 and its partners: LHX2, GLI3, and OTX2
Another gene of relevance that controls the expression of the Robo suite and is essential for TCA development is GBX2. In Gbx2-deficient mice TCAs are abolished. Specifically, thalamic axons are mostly misrouted to the ventral midbrain and dorsal midline of the diencephalon (Chatterjee et al., 2012). Gbx2 acts through three different mechanisms: first, Gbx2 and Lhx2 compete for binding to the Lmo3 promoter and exert opposing effects on its transcription; second, repressing Lmo3 by Gbx2 is essential for Lhx2 activity to induce Robo2; and third, Gbx2 represses Lhx9 transcription, which in turn induces Robo1.
Lhx2 affects the topographical sorting of axons by directly regulating the expression of Robo1 and Robo2 (Marcos-Mondéjar et al., 2012). Overexpression of Lhx2 gives rise to defective TCA guidance in vivo, while its conditional deletion in the thalamus alters projections from the medial geniculate nucleus and from the caudal ventrobasal nucleus (Marcos-Mondéjar et al., 2012). Lhx2 is also involved in thalamic development under the control of Shh, encoded by another member of our (2014) gene set. Specifically, low Shh signaling induces Lhx2 expression, as well as Gbx2 expression, in the caudal thalamus (Barth and Wilson, 1995; Hashimoto-Torii et al., 2003; Kiecker and Lumsden, 2004; Scholpp et al., 2006; Szabó et al., 2009; Vue et al., 2009), thus specifying thalamic neuronal subtype identities within this region (Scholpp and Lumsden, 2010). Additionally, Lhx2 (together with Lhx9) emerges as a crucial factor driving neurogenesis and maintaining the regional integrity of the caudal forebrain. Moreover, Lhx2-mediated neurogenesis seems to be involved in maintaining the integrity of cortex and regulating cortical size (Chou and O’Leary, 2013).
Interestingly, another of LHX2 interactors, namely, RFNG, also exhibits a fixed change (Y281H) in humans. Ectopic expression of Lhx2 induces Rfng expression in chicken (Rodríguez-Esteban et al., 1998). Importantly, rfng encodes an O-fucosylpeptide 3-β-N-acetylglucosaminyltransferase which negatively modulates Notch signaling in postmitotic neurons by inhibiting Hes1 expression in primary neurons (Mikami et al., 2001). In zebrafish rfng is required as well for wnt1 expression at hindbrain boundaries and contributes to the regulation of cell differentiation (Amoyel et al., 2005). Finally, LHX2 is a functional partner of CITED2 (Glenn and Maurer, 1999). As already mentioned in Boeckx and Benítez-Burraco (2014), CITED2 is an interesting gene for us, as it plays a key role in brain/skull development (as one of RUNX2 partners), in thalamus growth (as a partner of LHX2), and in the development of subcortical structures. We return to this gene in the next section, where we discuss its role as FOXP2 partner.
Going back to Gbx2, it is worth noting that Gbx2 expression is highly reduced in Gli3 mutants, in which the medial and intralaminar nuclei of the thalamus are specifically and severely affected (Haddad-Tóvolli et al., 2012). Like Lhx2, Gli3 interacts with Shh during thalamic development (Haddad-Tóvolli et al., 2012). Moreover, Gli3 regulates calvarial suture development by controlling Bmp-Smad signaling, which integrates a Dlx5/Runx2-II cascade (Tanimoto et al., 2012). Mutations in GLI3 have been found in people affected by Greig cephalopolysyndactyly syndrome, a condition in which craniosynostosis is an important feature (Debeer et al., 2003). Interestingly, most (∼98%) of Altaic Neanderthals and Denisovans had a different sequence in GLI3 compared to AMHs. While the latter retained the ancestral sequence, the former gained a non-synonymous change that appears to be mildly disruptive (Castellano et al., 2014).
Finally, Gbx2 interacts with Otx2 to determine the midbrain-hindbrain boundary in vertebrates. Moreover, both Gbx2 and Otx2 play a key role in the development of the cerebellum, a key brain component of vocal learning. Specifically, both genes are important during early neurogenesis and regulate the positioning and formation of the cerebellar primordium (Hashimoto and Hibi, 2012). In Boeckx and Benítez-Burraco (2014) we reviewed the link between OTX2 and language in connection with the FOXP2 interactome. Interestingly, a human-specific conserved deletion (hCONDEL) occurres downstream OTX2 (McLean et al., 2011). Of more direct interest is the fact that Gbx2 restricts Otx2 expression to forebrain and midbrain, competing with class III POU factors (Inoue et al., 2012). One of these factors is encoded by POU3F2, a target of FOXP2 (Konopka et al., 2012), to which we return below.
ABL1
ABL1 provides another link to ROBO1 that we find of interest in the context of our hypothesis. ABL1 encodes a cytoplasmic and nuclear tyrosine kinase that seems to play a prominent role in cell differentiation, division, and adhesion (see Matsumura and Toyoshima, 2012, for a recent review). Both ROBO1 and ABL1 show a differential expression profile in the hippocampus of schizophrenics (Benes et al., 2009). ABL1 is expressed in the thalamus too. In rats Abl1 levels are reduced specifically by safety learning but not fear conditioning, suggesting that Abl1 may be involved in the regulation and/or the activation of specific auditory networks within the thalamus (Habib et al., 2013). Moreover, high levels of ABL1 have been detected as well in the brains of people suffering from Parkinson (Hebron et al., 2013). Interestingly, Abl1 is also highly expressed in bone tissue in newborn mice and osteoblasts in the fetus. Mice homozygous for mutations in Abl1 display foreshortened crania (Schwartzberg et al., 1991) and are osteoporotic because of having dysfunctional osteoblasts (Li et al., 2000). Finally, ABL1 affects, via BMP signaling, the differentiation of cranial neural crest cells and the induction of myogenic cell proliferation in the cranial mesoderm during tongue development (Iwata et al., 2013). BMPs are among the genes we took to be central in our (2014) paper.
AKT1
AKT1 is another promising gene for us in the context of the ROBO suite, and the genes we discussed in our (2014) piece. Akt1 enhances transcriptional activity of Runx2 in mice (Fujita et al., 2004). AKT1 encodes a serine-threonine protein kinase. In the developing cerebellum AKT1 is a critical mediator of growth factor-induced neuronal survival (Dudek et al., 1997). In mice mutations affecting both Akt1 and Akt2 cause impairment of bone formation (Peng et al., 2003). In humans, mutations in AKT1 have been associated with the Proteus syndrome (Cohen, 2014), a condition similar to gigantism in which macrocephaly is a prominent symptom (Lindhurst et al., 2011). Moreover, AKT1 is a susceptibility gene for schizophrenia. According to Emamian et al. (2004) a significant association exists between schizophrenia and an AKT1 haplotype associated with lower AKT1 protein levels; moreover, a greater sensitivity to the sensorimotor gating-disruptive effect of amphetamine is conferred by AKT1 deficiency.
PTEN
Finally, PTEN is an effector of AKT1: in mice Pten is required for a robust depletion of nuclear phosphorylated Akt1 (Mistafa et al., 2010). PTEN is another candidate for Proteus syndrome (Lindhurst et al., 2011), but also for autism with macrocephaly (Buxbaum et al., 2007). Autism syndrome disorder subjects with PTEN mutations and reduced PTEN protein levels show strong reductions in working memory and processing speed, resulting from white-matter abnormalities (Frazier et al., 2014). Interestingly, in mice Pten is repressed by Hes1 as part of the Notch signaling pathway (Wong et al., 2012). Together with Hes1 (and other genes like Socs1 or Stat3), Pten comprisses a functional network that plays an important role in the control of the fate of ependymal stem progenitor cells of the spinal cord in response to motoneuron degeneration (Marcuzzo et al., 2014). Hes1, Notch1, Akt1, and Pten are functionally related (Palomero et al., 2008).
FOXP2 and Partners
Of all the language-related genes identified to date, none is as well characterized as FOXP2 (Graham and Fisher, 2013; French and Fisher, 2014). We begin this section by all too briefly summarizing some of the major facts about the gene. FOXP2 encodes a transcription factor that mostly works as a gene repressor (Shu et al., 2001). In the human brain it is expressed in several areas, including layer VI of the cortex, the thalamus, the cerebellum, and the basal ganglia (Ferland et al., 2003; Takahashi et al., 2003). The FOXP2 protein seemingly contributes to the development and function of cortico-thalamic-striatal circuits supporting motor planning, sequential tasks, and procedural learning (see Vargha-Khadem et al., 2005; Fisher and Scharff, 2009 for reviews). Recently, it has been suggested that the human variant may help achieve a faster transition between declarative and procedural learning systems (Schreiweis et al., 2014). Mutations in FOXP2 cause speech and language deficits (Vargha-Khadem et al., 1995; Watkins et al., 2002; MacDermot et al., 2005; Shriberg et al., 2006). However, oromotor and broad cognitive deficits have been claimed to exist as well in people bearing pathogenic mutations of FOXP2 (Vargha-Khadem et al., 1995; Watkins et al., 2002). In mice the knockout of Foxp2 (and the transformation with the human pathogenic variant of the gene) gives rise to structural and functional anomalies in the cerebellum (Shu et al., 2005; Groszer et al., 2008), increases long-term potentiation (LTP) in Purkinje cells, decreases long-term depression (LTD) in the striatum (Groszer et al., 2008), and impairs the ultrasonic vocalizations in the pups (Shu et al., 2005). In turn, the knockdown of FoxP2 in zebra finch mainly affects the area X of the song circuit (a structure homologous to the striatum), and decreases the accuracy of the imitative processes involved in song learning and shortens the critical period for song learning (Haesler et al., 2004).
In Boeckx and Benítez-Burraco (2014) we speculated about a potential link between FOXP2 and RUNX2 via SIRT1. We later found out that RUNX2 binds the FOXP2 promoter in human cells overexpressing RUNX2 (Kuhlwilm et al., 2013).
POU3F2
We begin our discussion of FOXP2 partners with POU3F2, which we briefly mentioned above. The POU3F2 protein binds a specific site within intron 8 of FOXP2 (Maricic et al., 2013). AMHs bear a derived allele of the binding site which is less efficient in activating transcription than the Neanderthal/Denisovan allele (Maricic et al., 2013). In mice the lack of the homopolymeric amino acid repeats that are characteristic of mammalian POU3F2 give rise to a decrease in the rate-limiting enzymes of dopamine and serotonin synthesis in various brain areas, and ultimately, to an impairment of pup retrieval behavior (Nasu et al., 2014). The POU3F2 factor also regulates the upper-layer neuronal migration and identity during the development of the neocortex (McEvilly et al., 2002; Sugitani et al., 2002). A region surrounding POU3F2 is a risk locus for bipolar disorder (Mühleisen et al., 2014). Moreover, sequence and copy number variations affecting POU3F2 have been found in patients with developmental and language delays, intellectual disability, schizophrenia, or autism spectrum disorders (Huang et al., 2005; Potkin et al., 2009; Lin et al., 2011). POU3F2 shows a frontal to temporal gradient patterning in the developing human neocortex and is associated with human accelerated conserved non-coding sequences (Miller et al., 2014). Additionally, POU3F2 interacts with ASCL1, a protein that regulates the development of GABAergic neurons (more on this gene below) and PQBP1 (Waragai et al., 1999), a protein involved in neurite growth and neuron projection (Wang et al., 2013a). PQBP1 has been linked to developmental delay and microcephaly (Li et al., 2013) and to intellectual disability (Wang et al., 2013a).
CNTNAP2
CNTNAP2 is a candidate for SLI and a FOXP2 target (Vernes et al., 2008). The gene has been also related to different forms of language delay and language impairment (Petrin et al., 2010; Sehested et al., 2010), to autism (Alarcón et al., 2008; Bakkaloglu et al., 2008), and to intellectual disability (Gregor et al., 2011). CNTNAP2 encodes a neurexin involved in synaptogenesis (Dean et al., 2003), which contributes to the establishment of interconnection patterns within the frontal lobe (Scott-Van Zeeland et al., 2010). In Drosophila Cntnap2 (known as NrxIV) functions in midline repulsive axon guidance in conjunction with Robo and Slit (NrxIV physically associates with Robo and Slit, and interactions between NrxIV and Slit are affected in NrxIV mutants; Banerjee et al., 2010). According to Banerjee et al. (2010), NrxIV is essential for proper Robo localization and identify Nrx IV as a novel interacting partner of the Slit/Robo signaling pathway. Interestingly, the human CNTNAP2 shows a fixed change (Ile345Val) compared to the Denisovan protein.
CNTNAP2 and a protein called NFASC are components of the nodal region of myelinated fibers (Brown et al., 2001). NFASC is a cell adhesion protein involved in mechanisms of neural plasticity such like neurite outgrowth or the formation of postsynaptic components and the organization of the axon initial segment and nodes of Ranvier during early development (Kriebel et al., 2012). Specifically, loss of the Nfasc factor in adult Purkinje neurons provokes loss of neuron activity and neuron disorganization, and eventually, ataxia (Buttermore et al., 2012). Interestingly, NFASC shows as well a fixed change (T987A) in AMHs compared to Neanderthals/Denisovans (Pääbo, 2014; Table S1).
The expression of CNTNAP2 in frontostriatal systems is interesting too in light of the pathology of SRPX2 mutations in the perisylvian cortex. SRPX2 is another of FOXP2 targets (Roll et al., 2010). Mutations in SRPX2 give rise to developmental verbal dyspraxia or to congenital bilateral perisylvian polymicrogyria (Roll et al., 2006).
FOXP1
The interaction between FOXP2 and FOXP1 is also worth considering in the present context. FOXP1 is a partner of FOXP2 (both proteins physically interact to form heterodimers; Li et al., 2004). Mutations affecting FOXP1 gives rise to intellectual disability, autism, and language impairment (Hamdan et al., 2010; Horn et al., 2010). In mice both Foxp1 and Foxp2 are highly expressed in the developing and mature basal ganglia. Foxp1 is also expressed in layers III-vof the cortex, the hippocampus, and thalamus (Ferland et al., 2003). Both Foxp1 and Foxp2 are expressed following neuronal migration, suggesting a role in postmigratory neuronal differentiation (Ferland et al., 2003).
Importantly for our hypothesis, FOXP1 is expressed along with RUNX1 and RUNX3 (two other members of the RUNX family, which are involved in the evolution of sophisticated sensory systems in higher vertebrates) in structures relevant to cortico-laryngeal connections (Inoue et al., 2008). As Inoue et al. (2008) review, RUNX3 has been shown to be upregulated in autism. The gene is also essential for the target-specific axon path finding of some dorsal root ganglion neurons, and it contributes to specify the termination pattern of sensory axons in the developing spinal cord (Inoue et al., 2008). It is thought to be essential not only for the functional glossopharyngeal system (swallowing), but to play a role in the forebrain as well, including language and social regions of brain (Inoue et al., 2008). Runx3 knockout mice show severe motor dis-coordination (Inoue et al., 2008).
As for RUNX1, it is synthesized in specific populations of somatic motor neurons in the spinal cord and in cholinergic branchial and visceral motoneurons in the hindbrain. Disruption of Runx1 in mice results in massive neuronal apoptosis (Inoue et al., 2008). Both Runx1 and Runx3 are acetylated by p300 (another gene of interest for us: see below), and these modifications are important for the control of transcriptional activity and protein stability (Jin et al., 2004; Yamaguchi et al., 2004).
NKX2-1
NKX2-1 is another gene potentially linking our initial gene set and the FOXP2 network. NKX2-1 plays a key role in the development of the basal ganglia. Hence, one of the two major subpopulations of GABAergic projection neurons in the basal ganglia (with descending projections to the subthalamus and substantia nigra) originates from progenitors expressing Nkx2-1 (Medina et al., 2014). Medina et al. (2014) also hypothesize that the novel expression of Nkx2-1 in the subpallium constitutes a major event in telencephalic evolution, and relate it to Shh expression and changes in the regulatory region of Nkx2-1 (SHH is a member of our initial gene set of interest). Moreover, the mutation of NKX2-1 gives rise to benign hereditary chorea, a clinical condition that not only involves hypotonia and chorea, but also learning difficulties in the adulthood (Gras et al., 2012). Interestingly, Foxp2 modulates Nkx2-1 DNA-binding and transcriptional activity, at least in the lung (Zhou et al., 2008; Yang et al., 2010a).
CITED2
In our previous work (Boeckx and Benítez-Burraco (2014)) we also highlighted that according to Prüfer et al. (2014) a highly disruptive intergenic change near CITED2 is 99% derived in AMHs and ancestral in both Altai Neanderthals and Denisovans. We also pointed out that this gene is highly expressed in the mediodorsal nucleus of the thalamus and that it is a regulatory target of FOXP2 (Vernes et al., 2011; Nelson et al., 2013). As we pointed out above, CITED2 is a functional partner of LHX2. Both Cited2 and Runx2 are regulated by Tgf (Luo et al., 2005). Moreover, CITED2 is also involved in the establishment of left-right axis (Preis et al., 2006). In mice Cited2 has proved to impact left-right patterning through interactions with the Bmp signaling and Nodal (Lopes Floro et al., 2011). BMPs are among the genes encompassing our core set of genes and NODAL is a robust candidate for the establishment of bilateral symmetry in the embryo (Zhou et al., 1993; Lowe et al., 1996; Gebbia et al., 1997). Interestingly, CITED2 may play some important role in craniofacial development too (Bhattacherjee et al., 2009). Additionally, Cited2 is a co-activator of Crebbp and Ep300, which play important roles in the initial development of the dorsal neural folds (Bhattacherjee et al., 2009).
CREBBP and EP300
Both CREBBP and EP300 are linked to our initial gene set of interest. In osteoblasts the CREB/CBP complex controls the RUNX2-mediated activation and expression of BMP2 (Shim et al., 2012). Interestingly, both EP300 and CREBBP are candidates for different subtypes of Rubinstein–Taybi syndrome, a complex condition characterized by mental and growth retardation and skeletal abnormalities (Viosca et al., 2010). Individuals with EP300 mutations have less severe mental impairment, but exhibit more severe microcephaly, and a greater degree of changes in facial bone structure (Hennekam, 2006). In this syndrome main abnormalities at the brain level occur in the rolandic area (Sener, 1995). In mice the knock-out of Crebbp is lethal, but when the gene is specifically deleted in postmitotic neurons of the forebrain, behavioral defects are greatly restricted and emerge mostly in the form of problems in object recognition memory (Valor et al., 2011). In turn, neonatal Crebbp(+/-) mice display perturbed vocalization behavior (Wang et al., 2010b).
Additionally, both EP300 and CREBBP are components linking our initial gene set to the FOXP2 and ROBO1 networks (see Figure 3). Their involvement in our network led us to search for other genes that are functionally connected to them. Thus, String 9.1 predicts confident links of CREBBP with ASCL1, CDKN1A, NCOA6, SIRT1, YAP1, RUNX1, and CTNNB1. In turn, EP300 is expected to be linked functionally to all those genes, but also to ETV4 and PIN1 (see Figure 3). Rather than going through all these genes, we decided to focus on only the most promising ones, for which we found robust links in the literature.
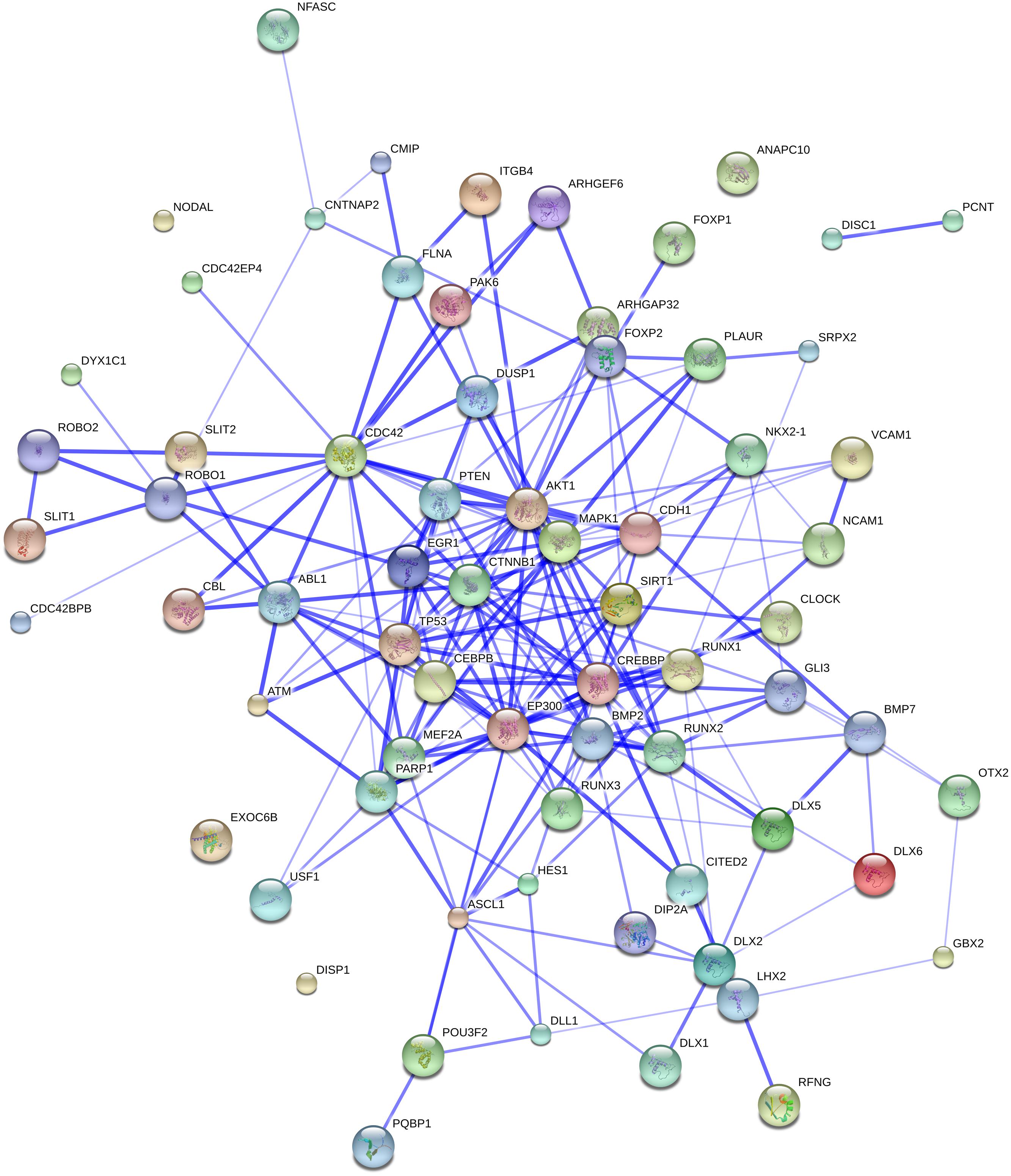
FIGURE 3. The whole set of genes discussed in this paper and our initial gene set related to globularity. The network was generated by String 9.1 with the proteins discussed in Sections “The ROBO/SLIT Suite,” “FOXP2 and Partners,” and “Other Genes of Interest” plus the gene set related to globularity as advanced in Boeckx and Benítez-Burraco (2014). The medium confidence value was 0.0400. The caveats noted for Figure 1 apply.
ASCL1
To begin with, ASCL1 is one of the proteins that regulate the DLX suite, a core component of our initial gene set in (2014). According to Letinic et al. (2002) 65% of neocortical GABAergic neurons in humans express the transcription factors ASCL1, DLX1, and DLX2 (the latter two are members of our initial gene set), and originate from ASCL1-expressing progenitors of the neocortical ventricular and subventricular zone of the dorsal forebrain (there exists a second lineage of neocortical GABAergic neurons, which express DLX1 and DLX2, but not ASCL1). Also according to Letinic et al. (2002), modifications in the expression pattern of transcription factors in the forebrain may underlie species-specific programs for the generation of neocortical local circuit neurons, with distinct lineages of cortical interneurons differentially affected in genetic and acquired diseases of the human brain. Moreover, over-expression of ASCL1 can improve GABAergic differentiation of bone marrow mesenchymal stem cells in vitro (Wang et al., 2013b). Together with Dlx1 and Dlx2 and the Foxp2 target Nkx2-1 discussed above, Ascl1 is involved in the development of the basal ganglia (Anderson et al., 1997; Casarosa et al., 1999). Additionally, ASCL1 is known to be involved in retinoic acid signaling: interestingly, both FOXP2 and retinoic acid strongly downregulate ASCL1 (Devanna et al., 2014). Finally, according to Wapinski et al. (2013) ASCL1 recruits POU3F2 during transdifferentiation of fibroblasts to neurons and likely other cell types.
DLL1
One partner of ASCL1 is DLL1 (see, for example, Nelson and Reh, 2008). DLL1 is linked to many of the genes involved in vocal learning (Wang, 2011). DLL1 encodes a member of the NOTCH signaling cascade (actually, DLL1 is a ligand of NOTCH1), which plays a central role in the regulation of neural proliferation and differentiation (Jung et al., 2011). An inverse correlation between oscillations in the Notch effectors Hes1, discussed above, and Dll1 controls neural growth and differentiation (Shimojo et al., 2008). Additionally, Dll1 may be related to the establishment of asymmetries during development. Hence, mutants for this gene exhibit defects in left-right asymmetry and do not express Nodal, the main left-sided determinant (Krebs et al., 2003).
Other Genes of Interest
Some other proteins may help to make more robust the links between our initial gene set and the two gene sets we have reviewed above (RUNX2, ROBO1, and FOXP2 networks, respectively). This is why we discuss them in the remainder of this section.
CDC42
Several lines of evidence converge onto CDC42 to make this gene an appealing one in the context of our hypothesis. According to the Human Brain Transcriptome (HBT) database CDC42 is highly expressed in the thalamus, with a peak around day 100th and then a slow drop until birth, a significant fact for our (2014) hypothesis, given that globularity is the result of key developmental events in the first year of life. Moreover, Cdc42 is required in pre-migratory progenitors of the medial ganglionic eminence in the ventricular zone for proper cortical interneuron migration (Katayama et al., 2013). Additionally, some partners of CDC42 are related to cognitive disorders. For example, a type of X-linked intellectual disability is caused by mutations in ARHGEF6 (and by the concomitant reduction in CDC42 activity), which result in structural anomalies in pyramidal neurons in the hippocampus, a reduction of early phase LTP, and an increase of LTD in some areas of this brain region (Ramakers et al., 2012). Similarly, the disruption of Pak5 and Pak6, which encode two effector proteins of Cdc42 gives rise to learning, memory and locomotion deficits in mice (Nekrasova et al., 2008). Additionally, Cdc42 activity is reduced in living growth cones by the inhibitory axon-guidance cue Slit2 (Myers et al., 2012). Ectopic expression of Slit2 on glioma cells attenuates cell migration and invasion through inhibition of Cdc42 activity in vitro. Moreover, cellular depletion of Robo1 prevents Slit2 inhibition of Cdc42 activity (Yiin et al., 2009).
CDC42 also allows for further connections between FOXP2 and RUNX2 networks. To begin with, FOXP2 regulates the expression of CDC42BPB, one effector of CDC42 (Spiteri et al., 2007). Moreover, during osteoclastogenesis CDC42 regulates FLNA function (Leung et al., 2010). FLNA encodes an actin-binding protein that regulates reorganization of the actin cytoskeleton and is required for neuronal migration to the cortex (Fox et al., 1998). Mutation in FLNA causes periventricular nodular heterotopias (Fox et al., 1998; Sheen et al., 2001). Interestingly, FLNA also binds CMIP (Fox et al., 1998), which is one strong candidate for SLI: it may modulate phonological short-term memory, which is a core deficit in SLI, dyslexia, and speech-sound disorder (Newbury et al., 2009). CMIP encodes one component anchoring the cellular membrane to the cytoskeleton, and seems to regulate neural migration and/or the assembly of synaptic complexes (Grimbert et al., 2003). FLNA interacts with ITGB4 (Travis et al., 2004), a protein that shows two fixed changes (T1689A and H1748R) in AMHs compared to Neanderthals/Denisovans (Pääbo, 2014; Table S1). Importantly, 1095 bp within the body of the gene CDC42EP4 (which encodes an effector of CDC42) are hypermethylated in humans compared to Denisovans (consequently, we should expect a lower expression of the gene in AMHs; Gokhman et al., 2014).
Another partner of CDC42, namely, ARHGAP32 bears a fixed change (E1489D) in humans compared to Denisovans (Meyer et al., 2012). ARHGAP32 encodes a receptor of NMDA which modulates Rho-GTPase activity, thus modulating dendritic spine morphology and strength, and promoting axon growth; moreover, the knockdown of Arhgap32 in mice results in impaired migration and axonal growth in the developing cerebellar cortex (Kannan et al., 2012). Interestingly, some polymorphisms within the human gene might increase susceptibility for schizophrenia and schizotypal personality traits (Ohi et al., 2012). ARHGAP32 promotes axon growth downstream of CDH1 (Kannan et al., 2012). This gene encodes a cadherin involved in the regulation of cell–cell adhesions, mobility and proliferation of epithelial cells. CDH1 also coordinates cortical neurogenesis and size, to the extent that the mutation of the gene gives rise to microcephaly (Delgado-Esteban et al., 2013). CDH1 seems to regulate neural connectivity as well, from axon and dendrite morphogenesis and growth to synapse differentiation and remodeling (Konishi et al., 2004; Huynh et al., 2009; Yang et al., 2010b). According to the HBT database the gene is expressed at high levels in the thalamus before birth. One of CDH1 partners, namely, ANAPC10 (Nourry et al., 2004) shows signals of a selective sweep in AMHs compared to Altai Neanderthals (Prüfer et al., 2014).
CBL
CBL encodes a negative regulator of several receptor protein tyrosine kinase signaling pathways (Joazeiro et al., 1999). Mutations in this gene cause Noonan syndrome-like disorder, a condition characterized by facial dysmorphism, a reduced growth, and variable cognitive deficits, among other symptoms (Martinelli et al., 2010). In mice activated Cdc42 prevents Cbl from catalyzing ubiquitination of specific receptors (Wu et al., 2003). Cbl interacts with some other of our genes of interest. Hence, Cbl phosphorylation depends on Abl1 (Miyoshi-Akiyama et al., 2001). Additionally, CBL belongs to the TRAIL pathway, associated with bone metabolism (Zhang et al., 2011). Moreover, Cbl ubiquitinates Notch1, triggering its degradation (Jehn et al., 2002). Finally, CBL is located in a region showing signal of a strong selective sweep (20-fold enrichment over random) in AMHs compared to Altai Neanderthals (Prüfer et al., 2014).
MEF2A
While discussing the role of SIRT1 in connection with FOXP2 and RUNX2 in our (2014) paper, we were led to consider MEF2A. The research we report on here led us back to this gene on numerous occasions. MEF2A has been recently implicated in differences between human and chimpanzee prefrontal cortex development (Liu et al., 2012). In addition, as reviewed in Pfenning (2012), MEF2A plays a significant role in songbirds. It turns out that MEF2A is linked to many of genes we have examined here. To begin with, MEF2A interacts with ASCL1 to modulate the expression of genes that are critical for neuronal differentiation (Mao and Nadal-Ginard, 1996; Gohlke et al., 2008). Additionally, EP300 interacts with MEF2A (He et al., 2011). Moreover, according to the HBT the gene is highly expressed in the thalamus. Finally, String 9.1 returns results where MEF2A is linked to ABL1 (an partner of ROBO1 that we have reviewed above) via ATM (a key controller of cell response to DNA damage and for genome stability that has been associated with ataxia telangiectasia, a condition involving cerebellar degeneration and dysarthria among other many symptoms, although not mental retardation; Ejima and Sasaki, 1998; Laake et al., 2000). According to String 9.1 ATM is also linked to PTEN and (indirectly) to FOXP1.
TP53
String 9.1 predicts TP53 to be linked to USF1, which we took to be central in our (2014) paper, but also to SIRT1, CDH1, ASPM (a well-known candidate for microcephaly), and PTEN. TP53 has been related to schizophrenia (Ni et al., 2005). Moreover, the expression level of the gene in humans is different compared to chimps/rhesus (Konopka et al., 2012). According to the HBT database the gene is expressed in the thalamus (this is the only structure where the gene is upregulated after birth). Additionally, there is a human-specific variant of the protein that bears an Arg in position 72, while Neanderthals/Denisovans exhibit a Pro (this is not a fixed change yet, since Arg72 frequencies range from 20 to 80% in AMHs, the lowest levels being observed among Sub-Saharan peoples; Paskulin et al., 2012).
CTNNB1
CTNNB1 is expected to be linked to many of the genes we are interested in, including RUNX2, EP300, CREBB4, SIRT1, BMP2, ROBO1, and CDC42 (prediction based on String 9.1 data). Specifically, Ctnnb1 binds the Runx2 promoter and upregulates Runx2 expression (Han et al., 2014). Additionally, the presence of an active Slit2/Robo1 signal blocks the translocation of Ctnnb1 into the cell nucleus (Chang et al., 2012). As mentioned in Boeckx and Benítez-Burraco (2014), CTNNB1 is a strong candidate for autism. Interestingly, it also interacts with PCDH11X/Y, a gene pair that underwent accelerated evolution in our lineage (Williams et al., 2006), and has been linked to cognitive disorders such as schizophrenia (Crow, 2013) and language acquisition delay (Speevak and Farrell, 2011).
EGR1
EGR1 is an immediate early gene that encodes a transcription factor involved in neuronal plasticity needed for consolidation of new memories (Veyrac et al., 2014). In the brain, signaling from synapses to the nucleus of neurons activated during learning tasks induce the expression of this kind of transcription factors, which mediates the gene programs needed for the stable functional and structural remodeling of the activated networks (this allows memory to be later reactivated upon recall). In mice Egr1 mutants are impaired in long-term (but not short-term) recognition memory (Bozon et al., 2003). Interestingly, in songbirds the expression of EGR1 is induced by singing in song nuclei, although the gene is also expressed in adjacent brain regions in response to non-vocal motor behaviors, quite contrary to DUSP1, which shows a motor-driven expression in the forebrain only in song nuclei and only in vocal learners (DUSP1 is up-regulated in sensory-input neurons of the thalamus and telencephalon; Horita et al., 2010, 2012). EGR1 functionally interacts with some of the genes we have examined, including PTEN (Kim et al., 2014) and AKT1 (Hernández et al., 2013). Similarly, EGR1 physically interacts with CBP and EP300 to modulate gene transcription (Silverman et al., 1998). Importantly, EGR1 is a target of both FOXP2 (Konopka et al., 2009) and RUNX2 (Kuhlwilm et al., 2013). Moreover, EGR1 downregulates PLAUR in osteosarcoma cell lines (Matsunoshita et al., 2011). PLAUR is a target of FOXP2 (Roll et al., 2010) and encodes an effector of SRPX2, another FOXP2 target (Royer-Zemmour et al., 2008). Finally, EGR1, acting as an effector of SOX9B and being regulated by RUNX3, modulates the BMP signaling needed for cranial cartilage development in zebrafish (Dalcq et al., 2012; BMPs are among the genes encompassing our core set of genes). According to the HBT database, EGR1 is expressed in the thalamus and the cortex: its expression level steadily increases until first year of life.
CEBPB
CEBPB encodes a transcription factor that binds – the promoter of EGR1 (Calella et al., 2007). Moreover, the protein CEBPB physically interacts with EGR1 to modulate gene transcription (Zhang et al., 2003). Compound Cebp knockout mice (i.e., for genes Cebpa and Cebpb) show defective differentiation of cortical dendrites (Calella et al., 2007). Transcriptional activation by CEBPB also involves the coactivators CBP and EP300 (Mink et al., 1997; Guo et al., 2001). Interestingly, CEBPB also plays a role in osteogenesis. Hence, Cebpb is a key modulator of Runx2 expression in bone formation, specifically during chondrocyte (Hirata et al., 2012) and osteoblast (Gutiérrez et al., 2002) formation. Deletion of Cebpb gives rise to suppressed differentiation of osteoblasts and delayed chondrocyte hypertrophy, thus postponing bone formation (Tominaga et al., 2008) Ultimately, CEBPB is claimed to be a candidate for cleidocranial dysplasia (Huang et al., 2014). CEBPB is also a target of FOXP2 (Konopka et al., 2009).
NCAM1
NCAM1 encodes a protein involved in cell-to-cell interactions that plays a key role in the development and differentiation of the brain (Fujita et al., 2000; Prag et al., 2002) The primary transcript of NCAM1 is modified both posttranscriptionally (alternative splicing of the mRNA generates three main protein isoforms and the ectodomain shedding of NCAM1 isoforms can produce an extracellular soluble neural cell adhesion molecule fragment) and posttranslationally (a residue of polysialic acid is added to the molecule) following developmental cues (Rutishauser and Goridis, 1986; Wang et al., 1998; Cox et al., 2009). Moreover, the gene is subject to epigenetic modifications that affect its splicing pattern (Schor et al., 2013). At the brain level NCAM1 plays a pivotal role in axonal and dendritic growth and synaptic plasticity, and ultimately, in cognition (Rønn et al., 2000; Hansen et al., 2008). Hence, Ncam1-deficient mice are impaired in working/episodic-like memory performance (Bisaz et al., 2013). Alterations in NCAM1 expression and/or proteolytic cleavage of the protein have been related to different neuropsychiatric conditions, including schizophrenia, bipolar disorder and Alzheimer’s disease (Atz et al., 2007) and may contribute to the cognitive dysfunctions observed in these diseases. Specifically, the amount of the NCAM1 extracellular proteolytic cleavage fragment has been reported to be increased in schizophrenics (Vawter et al., 2001). In mice when this fragment is overexpressed GABAergic innervation is impaired and the number of dendritic spines on pyramidal neurons in the prefrontal cortex becomes reduced. In turn this results in the impairment of long- and short-term potentiation in the prefrontal cortex, although synaptic plasticity is normal in the hippocampus (Brennaman et al., 2011). Interestingly, the absence of polysialic acid in the protein gives rise to misguidance of thalamocortical fibers and deficiencies of corticothalamic connections (Schiff et al., 2011). NCAM1 is functionally linked to some of the genes we considered. Hence, RUNX1 controls the expression of NCAM1 (Gattenloehner et al., 2007). Moreover, NCAM1 is a putative target of both RUNX2 (Kuhlwilm et al., 2013) and FOXP2 (Konopka et al., 2009).
VCAM1
NCAM1 interacts to VCAM1, one of the proteins showing a fixed change (D414G) in AMHs compared to Neanderthals/Denisovans (Pääbo, 2014; Table S1). VCAM1 is a cell surface glycoprotein involved in cell adhesion. In the adult forebrain, subventricular zone neurons arise from type B neural stem cells, which are anchored by specialized cells expressing high levels of VCAM1. Disruption of VCAM1 disturbs the architecture of the subventricular zone and increases neurogenesis in some areas (specifically, in the olfactory bulb; Kokovay et al., 2012). Interestingly, VCAM1 is upregulated by CLOCK (Gao et al., 2014), which interacts with RUNX2 (Reale et al., 2013), a link we reviewed in our paper (2014). According to Shimomura et al. (2013), Usf1, a member of our initial gene set, is able to compensate Clock mutations in mice, this ultimately suggesting that Usf1 is an important modulator of molecular and behavioral circadian rhythms in mammals. Also DUSP1 is upregulated by CLOCK (Doi et al., 2007).
MAPK1
MAPK1 is a potential hub linking many of our genes of interest. MAPK1 regulates the transcription of FOXP2 target PLAUR (Lengyel et al., 1996). Moreover, MAPK1 and DUSP1 physically interact at the brain level, since DUSP1 dephosphorylates MAPK1 (among other MAPK proteins; Choi et al., 2006; Lomonaco et al., 2008). Additionally, MAPK1 is a positive regulator of RUNX2 (Lee et al., 2011). Overall, MAPK1 seems to play an important role in osteogenesis. Specifically, inhibition of MAPK1 activity leads to significant decrease in BMP9-induced osteogenic differentiation and bone formation (Zhao et al., 2012). Moreover, BMP2 [also a member of our (2014) set] induces osteoblastic differentiation by a DUSP1–MAPK1 dependent mechanism (Ghayor et al., 2009). MAPK1 plays a key role in cognition and brain function too. The gene is required for neuronal cell fate determination. In mice deletion of Mapk1 results in a reduction in cortical thickness and mutant mice for Mapk1 exhibit important deficits in associative learning (Samuels et al., 2008). Additionally, a stimulus-dependent increase of Mapk1 signaling resulting from the ablation of Erk1 gives rise to a strong enhancement of striatum-dependent long-term memory, and ultimately, to a modification of the long-term adaptive changes underlying striatum-dependent behavioral plasticity (Mazzucchelli et al., 2002). In humans microdeletions on chromosome 22q11.2 encompassing MAPK1 give rise to microcephaly, impaired cognition, and developmental delay (Samuels et al., 2008). Actually, there exists a group of genetic disorders (including Costello syndrome, Leopard syndrome, and Noonan syndrome) that are caused by mutations in upstream elements of the MAPK signaling cascade. Among the distinctive symptoms, one finds craniofacial defects, developmental delay, and mental retardation (see Bentires-Alj et al., 2006, for a review). Similarly, the mutation of downstream elements in the MAPK cascade has been associated with mental retardation (Weeber and Sweatt, 2002).
‘Dyslexia’-related genes (beyond ROBO1)
In addition to ROBO1, other potential candidates for dyslexia (according to Poelmans et al., 2011) are linked to some of the genes we considered in the context of globularity in our (2014) article. Thus, DIP2A functions as a transcriptional co-activator of DLX2 and DLX5, and plays an important role in the development of the basal ganglia (Yu et al., 2001). Moreover, BMP2 blocks the binding of DIP2A to a protein called FRP (Tanaka et al., 2010), a member of the Wnt signaling pathway and a target of PAX6 during the regulation of axonal connectivity; in turn, Pax6 mediates the response of growing axons to Sfrp1 (Sebastián-Serrano et al., 2012), which functions as an enhancer of the Wnt/PCP signaling in dopamine cells and a regulator of Wnt/PCP-dependent functions in midbrain development (Kele et al., 2012).
Another potential candidate for dyslexia, PCNT, encodes pericentrin, a protein of the centrosome, which interacts with DISC1, the product of one robust candidate for schizophrenia (Miyoshi et al., 2004). DISC1 is a target of FOXP2 too (Walker et al., 2012). PCNT is cited by Green et al. (2010) among the 11 genes that show non-synonymous and non-fixed substitution changes in their coding sequences compared to Neanderthals. Moreover, the mutation of the gene gives rise to a condition called microcephalic osteodysplastic primordial dwarfism type II, characterized by different bone abnormalities and by a reduced brain size but usually with near-normal intelligence (Rauch et al., 2008).
Finally, among the genes that are differentially expressed among (mammalian) vocal learners (according to Wang, 2011), one finds PARP1 that regulates the dyslexia-susceptibility gene DYX1C1, important for neuronal migration in the developing cortex (Tapia-Páez et al., 2008).
Conclusion and Prospects
Boeckx and Benítez-Burraco (2014) put forth a hypothesis concerning the emergence of the language-ready brain that highlighted the potential role of a small set of genes such as RUNX2. Our original focus was on what one may want to call the syntax-semantics branch of our language faculty. The other branch, ‘the externalization component,’ crucial to convey the syntactically coded meanings of sentences, was left for future research. Our goal in this paper has been to attend to that aspect of language, and refine the gene set we originally put forward by taking into account what we know about vocal learning. In the vocal learning literature two neural components are often presented as critical: the direct cortico-laryngeal connection and the cortico-thalamo-basal ganglia pathway, and genes like ROBO1 and FOXP2 have been associated with these networks. The purpose of this paper has been to see if points of contact and potential functional links could be hypothesized between ROBO1, FOXP2, and their partners, and our initial gene set. Identifying such potential links would offer a much more comprehensive picture of our hypothesis liking globularity to language-readiness, opening up new ways to falsify it. Eventually, the genes we have advanced in this paper may be regarded as potential objects of inquiry for future research on the genetic underpinnings of language and of language disorders.
It stands to reason that as we learn more about each gene discussed here, the potential links between the various networks are likely to grow, so we are aware that we are just beginning to scratch the surface of a big puzzle. We are also aware of the limitations of attempts like ours to perform literature-based assembly of protein–protein and gene-regulatory networks. For this reason we think it fit to conclude this paper by stressing the need to test the robustness of each of the connections hypothesized here. Finding a transcription factor ChIP peak in the vicinity of a gene is not enough to conclude that the gene is regulated by the transcription factor. This interaction has to be demonstrated in vitro and/or in vivo. Likewise, finding that some protein has acquired a fixed coding change in AMHs compared to Neanderthals/Denisovans does not always entail that the change has impacted on protein function. This impact has to be demonstrated empirically. Similarly, although we have focused on the strongest links found in the literature, eventually these links (often binary) have to be properly evaluated in order to know if they are actually biologically significant and meaningful with respect to the phenotype of interest. We also acknowledge that the literature and the datasets we have relied on may be incomplete or biased because of the unavoidable focus on some genes as opposed to others. One should not forget that absence of evidence is not evidence of absence regarding protein–protein or DNA-protein interactions. It is also clear that the attested links for intensely studied proteins are always be more salient and more numerous than for less studied proteins. The same holds for disorders: some have been intensively examined, while others remain poorly described. So, we don’t want to exaggerate our results. Still, we hope that the current study can serve as a useful starting point for future investigations that regard our initial hypothesis as promising.
We would be delighted if the information we provided here help constrain the search space of future work.
To this end, we close with a few suggestions concerning how the expanded hypothesis presented here could be used and tested experimentally.
The most urgent task consists in showing that all the genes examined really interact in the human brain. In many cases evidence for this interaction comes from knockout experiments involving mice or from expression assays in cell lines. ChiP experiments are a good first approximation, but the results of these experiments need to be further refined. Additionally, we need to determine the exact anatomical and functional consequences of the mutation of all these genes in humans and/or of changes in their expression levels, with a special focus on the brain areas involved in language processing. We think that it would be extremely valuable to examine these consequences in non-human vocal learners, for which we can already rely on a fair amount of knowledge. Knockdown experiments could be conducted in vocal learners in order to test whether these enhanced networks centered on FOXP2 and ROBO1 and connected to RUNX2 really account for key aspects of externalization circuits. Finally, we need to check whether the fixed changes (or even the changes under selection) in the human proteins (compared to other hominins) had structural and functional consequences. This should also be complemented with additional examination of archaic genomes, with a special focus on changes in the regulatory regions of genes (promoters, enhancers, etc.) and in genes that do not encode proteins (e.g., ncRNAs). All of this will help us provide fuller accounts of how our species came to be language-ready.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The present work was made possible through a Marie Curie International Reintegration Grant from the European Union (PIRG- GA-2009-256413), and research funds from the Fundació Bosch i Gimpera [Cedric Boeckx].
Footnotes
- ^ http://www.ncbi.nlm.nih.gov/pubmed
- ^ http://human.brain-map.org/microarray/search
- ^ http://www.brainspan.org/lcm/search/index.html
- ^ http://www.brainspan.org/rnaseq/search/index.html
- ^ http://www.ncbi.nlm.nih.gov/omim/
- ^ http://string-db.org/
References
Alarcón, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M.,et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159. doi: 10.1016/j.ajhg.2007.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amoyel, M., Cheng, Y. C., Jiang, Y. J., and Wilkinson, D. G. (2005). Wnt1 regulates neurogenesis and mediates lateral inhibition of boundary cell specification in the zebrafish hindbrain. Development 132, 775–785. doi: 10.1242/dev.01616
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anderson, S. A., Qiu, M., Bulfone, A., Eisenstat, D. D., Meneses, J., Pedersen, R.,et al. (1997). Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron 19, 27–37. doi: 10.1016/S0896-6273(00)80345-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andrews, W, Liapi, A, Plachez, C, Camurri, L, Zhang, J, Mori, S., et al. (2006). Robo1 regulates the development of major axon tracts and interneuron migration in the forebrain. Development 133, 2243–2252. doi: 10.1242/dev.02379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Atz, M. E., Rollins, B., and Vawter, M. P. (2007). NCAM1 association study of bipolar disorder and schizophrenia: polymorphisms and alternatively spliced isoforms lead to similarities and differences. Psychiatr. Genet. 17, 55–67. doi: 10.1097/YPG.0b013e328012d850
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bagri, A., Marin, O, Plump, A. S., Mak, J., Pleasure, S. J., Rubenstein, L. R.,et al. (2002). Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron 33, 233–248. doi: 10.1016/S0896-6273(02)00561-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bakkaloglu, B., O’Roak, B. J., Louvi, A., Gupta, A. R., Abelson, J. F., Morgan, T. M.,et al. (2008). Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 82, 165–173. doi: 10.1016/j.ajhg.2007.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Banerjee, S., Blauth, K., Peters, K., Rogers, S. L., Fanning, A. S., and Bhat, M. A. (2010). Drosophila neurexin IV interacts with Roundabout and is required for repulsive midline axon guidance. J. Neurosci. 30, 5653–5667. doi: 10.1523/JNEUROSCI.6187-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barth, K. A., and Wilson, S. W. (1995). Expression of zebrafish nk2.2 is influenced by sonic hedgehog/vertebrate hedgehog-1 and demarcates a zone of neuronal differentiation in the embryonic forebrain. Development 121, 1755–1768.
Benes, F. M., Lim, B., and Subburaju, S. (2009). Site-specific regulation of cell cycle and DNA repair in post-mitotic GABA cells in schizophrenic versus bipolars. Proc. Natl. Acad. Sci. U.S.A. 106, 11731–11736. doi: 10.1073/pnas.0903066106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bentires-Alj, M., Kontaridis, M. I., and Neel, B. G. (2006). Stops along the RAS pathway in human genetic disease. Nat. Med. 12, 283–85. doi: 10.1038/nm0306-283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berwick, R. C., Friederici, A., Chomsky, N., and Bolhuis, J. J. (2013). Evolution, brain, and the nature of language. Trends Cogn. Sci. 17, 89–98. doi: 10.1016/j.tics.2012.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhattacherjee, V., Horn, K. H., Singh, S., Webb, C. L., Pisano, M. M., and Greene, R. M. (2009). CBP/p300 and associated transcriptional co-activators exhibit distinct expression patterns during murine craniofacial and neural tube development. Int. J. Dev. Biol. 53, 1097–1104. doi: 10.1387/ijdb.072489vb
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bielle, F., Marcos-Mondéjar, P., Keita, M., Mailhes, C., Verney, C., Nguyen Ba-Charvet, K.,et al. (2011). Slit2 activity in the migration of guidepost neurons shapes thalamic projections during development and evolution. Neuron 69, 1085–1098. doi: 10.1016/j.neuron.2011.02.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bisaz, R., Boadas-Vaello, P., Genoux, D., and Sandi, C. (2013). Age-related cognitive impairments in mice with a conditional ablation of the neural cell adhesion molecule. Learn. Mem. 20, 183–193. doi: 10.1101/lm.030064.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boeckx, C., and Benítez-Burraco, A. (2014). The shape of the language-ready brain. Front. Psychol. 5:282. doi: 10.3389/fpsyg.2014.00282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boeckx, C., Martínez-Álvarez, A., and Leivada, E. (2014). The functional neuroanatomy of serial order in language. J. Neurolinguist. 32, 1–15. doi: 10.1016/j.jneuroling.2014.07.001
Borrell, V., Cárdenas, A., Ciceri, G., Galcerán, J., Flames, N., Pla, R.,et al. (2012). Slit/Robo signaling modulates the proliferation of central nervous system progenitors. Neuron 76, 338–352. doi: 10.1016/j.neuron.2012.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bozon, B., Davis, S., and Laroche, S. (2003). A requirement for the immediate early gene zif268 in reconsolidation of recognition memory after retrieval. Neuron 40, 695–701. doi: 10.1016/S0896-6273(03)00674-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Braisted, J. E., Ringstedt, T., and O’Leary, D. D. M. (2009). Slits are chemorepellents endogenous to hypothalamus and steer thalamocortical axons into ventral telencephalon. Cereb. Cortex 19, i144–i151. doi: 10.1093/cercor/bhp035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brennaman, L. H., Kochlamazashvili, G., Stoenica, L., Nonneman, R. J., Moy, S. S., Schachner, M.,et al. (2011). Transgenic mice overexpressing the extracellular domain of NCAM are impaired in working memory and cortical plasticity. Neurobiol. Dis. 43, 372–378. doi: 10.1016/j.nbd.2011.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, A. A., Xu, T., Arroyo, E. J., Levinson, S. R., Brophy, P. J., Peles, E.,et al. (2001). Molecular organization of the nodal region is not altered in spontaneously diabetic BB-Wistar rats. J. Neurosci. Res. 65, 139–149. doi: 10.1002/jnr.1137
Buckner, R. L., and Krienen, F. M. (2013). The evolution of distributed association networks in the human brain. Trends Cogn. Sci. 17, 648–665. doi: 10.1016/j.tics.2013.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buttermore, E. D., Piochon, C., Wallace, M. L., Philpot, B. D., Hansel, C., and Bhat, M. A. (2012). Pinceau organization in the cerebellum requires distinct functions of neurofascin in Purkinje and basket neurons during postnatal development. J. Neurosci. 32, 4724–4742. doi: 10.1523/JNEUROSCI.5602-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buxbaum, J. D., Cai, G., Chaste, P., Nygren, G., Goldsmith, J., Reichert, J.,et al. (2007). Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 484–491. doi: 10.1002/ajmg.b.30493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calella, A. M., Nerlov, C., Lopez, R. G., Sciarretta, C., von Bohlen und Halbach, O., Bereshchenko, O.,et al. (2007). Neurotrophin/Trk receptor signaling mediates C/EBPα, -β and NeuroD recruitment to immediate-early gene promoters in neuronal cells and requires C/EBPs to induce immediate-early gene transcription. Neural Dev. 2:4. doi: 10.1186/1749-8104-2-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Casarosa, S., Fode, C., and Guillemot, F. (1999). Mash1 regulates neurogenesis in the ventral telencephalon. Development 126, 525–534.
Castellano, S., Parra, G., Sánchez-Quinto, F. A., Racimo, F., Kuhlwilm, M., Kircher, M.,et al. (2014). Patterns of coding variation in the complete exomes of three Neandertals. Proc. Natl. Acad. Sci. U.S.A. 111, 6666–6671. doi: 10.1073/pnas.1405138111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, P. H., Hwang-Verslues, W. W., Chang, Y. C., Chen, C. C., Hsiao, M., Jeng, Y. M.,et al. (2012). Activation of Robo1 signaling of breast cancer cells by Slit2 from stromal fibroblast restrains tumorigenesis via blocking PI3K/Akt/β-catenin pathway. Cancer Res. 72, 4652–4661. doi: 10.1158/0008-5472.CAN-12-0877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chatterjee, M., Li, K., Chen, L., Maisano, X., Guo, Q., Gan, L.,et al. (2012). Gbx2 regulates thalamocortical axon guidance by modifying the LIM and Robo codes. Development 139, 4633–4643. doi: 10.1242/dev.086991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, J.-H, Wen, L., Dupuis, S., Wu, J. Y., and Rao, Y. (2001). The N-terminal leucine-rich regions in slit are sufficient to repel olfactory bulb axons and subventricular zone neurons. J. Neurosci. 21, 1548–1556.
Choi, B. H., Hur, E. M., Lee, J. H., Jun, D. J., and Kim, K. T. (2006). Protein kinase Cδ-mediated proteasomal degradation of MAP kinase phosphatase-1 contributes to glutamate-induced neuronal cell death. J. Cell. Sci. 119, 1329–1340. doi: 10.1242/jcs.02837
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chou, S. J., and O’Leary, D. D. (2013). Role for Lhx2 in corticogenesis through regulation of progenitor differentiation. Mol. Cell. Neurosci. 56, 1–9. doi: 10.1016/j.mcn.2013.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cohen, M. M. Jr. (2014). Proteus syndrome review: molecular, clinical, and pathologic features. Clin. Genet. 85, 111–119. doi: 10.1111/cge.12266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cox, E. T., Brennaman, L. H., Gable, K. L., Hamer, R. M., Glantz, L. A., Lamantia, A. S.,et al. (2009). Developmental regulation of neural cell adhesion molecule in human prefrontal cortex. Neuroscience 162, 96–105. doi: 10.1016/j.neuroscience.2009.04.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crow, T. J. (2013). The XY gene hypothesis of psychosis: origins and current status. Am. J. Med. Genet. B Neuropsychiatr. Genet. 162, 800–824. doi: 10.1002/ajmg.b.32202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dalcq, J., Pasque, V., Ghaye, A., Larbuisson, A., Motte, P., Martial, J. A.,et al. (2012). RUNX3, EGR1 and SOX9B form a regulatory cascade required to modulate BMP-signaling during cranial cartilage development in zebrafish. PLoS ONE 7:e50140. doi: 10.1371/journal.pone.0050140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dean, C., Scholl, F. G., Choih, J., DeMaria, S., Berger, J., Isacoff, E.,et al. (2003). Neurexin mediates the assembly of presynaptic terminals. Nat. Neurosci. 6, 708–716. doi: 10.1038/nn1074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Debeer, P., Peeters, H., Driess, S., De Smet, L., Freese, K., Matthijs, G.,et al. (2003). Variable phenotype in Greig cephalopolysyndactyly syndrome: clinical and radiological findings in 4 independent families and 3 sporadic cases with identified GLI3 mutations. Am. J. Med. Genet. 120A, 49–58. doi: 10.1002/ajmg.a.20018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dediu, D., and Levinson, S. C. (2013). On the antiquity of language: the reinterpretation of Neandertal linguistic capacities and its consequences. Front. Psychol. 4:397. doi: 10.3389/fpsyg.2013.00397
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Delgado-Esteban, M., García-Higuera, I., Maestre, C., Moreno, S., and Almeida, A. (2013). APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nat. Commun. 4:2879. doi: 10.1038/ncomms3879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Devanna, P., Middelbeek, J., and Vernes, S. C. (2014). FOXP2 drives neuronal differentiation by interacting with retinoic acid signaling pathways. Front. Cell Neurosci. 8:305. doi: 10.3389/fncel.2014.00305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Devine, C. A., and Key, B. (2008). Robo-Slit interactions regulate longitudinal axon pathfinding in the embryonic vertebrate brain. Dev. Biol. 313, 371–383. doi: 10.1016/j.ydbio.2007.10.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Sciullo, A. M., and Boeckx, C. (eds.) (2011). The Biolinguistic Enterprise. Oxford: Oxford University Press.
Doi, M., Cho, S., Yujnovsky, I., Hirayama, J., Cermakian, N., Cato, A. C.,et al. (2007). Light-inducible and clock-controlled expression of MAP kinase phosphatase 1 in mouse central pacemaker neurons. J. Biol. Rhythms 22, 127–139. doi: 10.1177/0748730406298332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dudek, H., Datta, S. R., Franke, T. F., Birnbaum, M. J., Yao, R., Cooper, G. M.,et al. (1997). Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275, 661–665. doi: 10.1126/science.275.5300.661
Dugan, J. P., Stratton, A., Riley, H. P., Farmer, W. T., and Mastick, G. S. (2011). Midbrain dopaminergic axons are guided longitudinally through the diencephalon by Slit/Robo signals. Mol. Cell. Neurosci. 46, 347–356. doi: 10.1016/j.mcn.2010.11.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dunbar, R. (1996). Grooming, Gossip and the Evolution of Language. Cambridge: Harvard University Press.
Ejima, Y., and Sasaki, M. S. (1998). Mutations of the ATM gene detected in Japanese ataxia-telangiectasia patients: possible preponderance of the two founder mutations 4612del165 and 7883del5. Hum. Genet. 102, 403–408. doi: 10.1007/s004390050712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Emamian, E. S., Karayiorgou, M., and Gogos, J. A. (2004). Decreased phosphorylation of NMDA receptor type 1 at serine 897 in brains of patients with schizophrenia. J. Neurosci. 24, 1561–1564. doi: 10.1523/JNEUROSCI.4650-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feenders, G., Liedvogel, M., Rivas, M., Zapka, M., Horita, H., Hara, E.,et al. (2008). Molecular mapping of movement-associated areas in the avian brain: a motor theory for vocal learning origin. PLoS ONE 3:e1768. doi: 10.1371/journal.pone.0001768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferland, R. J., Cherry, T. J., Preware, P. O., Morrisey, E. E., and Walsh, C. A. (2003). Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J. Comp. Neurol. 460, 266–279. doi: 10.1002/cne.10654
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fisher, S. E., Francks, C., Marlow, A. J., MacPhie, I. L., Newbury, D. F., Cardon, L. R.,et al. (2002). Independent genome-wide scans identify a chromosome 18 quantitative-trait locus influencing dyslexia. Nat. Genet. 30, 86–91. doi: 10.1038/ng792
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fisher, S. E., and Marcus, G. F. (2006). The eloquent ape: genes, brains and the evolution of language. Nat. Rev. Genet. 7, 9–20. doi: 10.1038/nrg1747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fisher, S. E., and Scharff, C. (2009). FOXP2 as a molecular window into speech and language. Trends Genet. 25, 166–177. doi: 10.1016/j.tig.2009.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fitch, W. T. (2010). The Evolution of Language. Cambridge: Cambridge University doi: 10.1017/CBO9780511817779
Fitch, W. T., and Jarvis, E. D. (2013). “Birdsong and other animal models for human speech, song, and vocal learning,” in Language, Music,and the Brain, ed. M. Arbib (Harvard: MIT Press), 499–539.
Fox, J. W., Lamperti, E. D., Eksioglu, Y. Z., Hong, S. E., Feng, Y., Graham, D. A.,et al. (1998). Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21, 1315–1325. doi: 10.1016/S0896-6273(00)80651-0
Frayer, D. W., Lalueza-Fox, C., Fiore, I., Radovčić, J., and Bondioli, L. (2010). Right-handed Neandertals: vindija and beyond. J. Anthrop. Sci. 88, 113–127.
Frazier, T. W., Embacher, R., Tilot, A. K., Koenig, K., Mester, J., and Eng, C. (2014). Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol. Psychiatry doi: 10.1038/mp.2014.125 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
French, C. A., and Fisher, S. E. (2014). What can mice tell us about Foxp2 function? Curr. Opin. Neurobiol. 28C, 72–79. doi: 10.1016/j.conb.2014.07.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fujita, T., Azuma, Y., Fukuyama, R., Hattori, Y., Yoshida, C., Koida, M.,et al. (2004). Runx2 induces osteoblast and chondrocyte differentiation and enhances their migration by coupling with PI3K-Akt signaling. J. Cell Biol. 166, 85–95. doi: 10.1083/jcb.200401138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fujita, N., Saito, R., Watanabe, K., and Nagata, S. (2000). An essential role of the neuronal cell adhesion molecule contactin in development of the Xenopus primary sensory system. Dev. Biol. 221, 308–320. doi: 10.1006/dbio.2000.9692
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, Y., Meng, D., Sun, N., Zhu, Z., Zhao, R., Lu, C.,et al. (2014). Clock upregulates intercellular adhesion molecule-1 expression and promotes mononuclear cells adhesion to endothelial cells. Biochem. Biophys. Res. Commun. 443, 586–591. doi: 10.1016/j.bbrc.2013.12.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gattenloehner, S., Chuvpilo, S., Langebrake, C., Reinhardt, D., Müller-Hermelink, H. K., Serfling, E.,et al. (2007). Novel RUNX1 isoforms determine the fate of acute myeloid leukemia cells by controlling CD56 expression. Blood 110, 2027–2033. doi: 10.1182/blood-2007-02-074203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gebbia, M., Ferrero, G. B., Pilia, G., Bassi, M. T., Aylsworth, A. S., Penman-Splitt, M.,et al. (1997). X-linked situs abnormalities result from mutations in ZIC3. Nat. Genet. 17, 305–308. doi: 10.1038/ng1197-305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghayor, C., Ehrbar, M., San Miguel, B., Grätz, K. W., and Weber, F. E. (2009). cAMP enhances BMP2-signaling through PKA and MKP1-dependent mechanisms. Biochem. Biophys. Res. Commun. 381, 247–252. doi: 10.1016/j.bbrc.2009.02.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glenn, D. J., and Maurer, R. A. (1999). MRG1 binds to the LIM domain of Lhx2 and may function as a coactivator to stimulate glycoprotein hormone alpha-subunit gene expression. J. Biol. Chem. 274, 36159–36167. doi: 10.1074/jbc.274.51.36159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gohlke, J. M., Armant, O., Parham, F. M., Smith, M. V., Zimmer, C., Castro, D. S.,et al. (2008). Characterization of the proneural gene regulatory network during mouse telencephalon development. BMC Biol. 6:15. doi: 10.1186/1741-7007-6-15
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gokhman, D., Lavi, E., Prüfer, K., Fraga, M. F., Riancho, J. A., Kelso, J.,et al. (2014). Reconstructing the DNA methylation maps of the Neandertal and the Denisovan. Science 344, 523–527. doi: 10.1126/science.1250368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Graham, S. A., and Fisher, S. E. (2013). Decoding the genetics of speech and language. Curr. Opin. Neurobiol. 23, 43–51. doi: 10.1016/j.conb.2012.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gras, D., Jonard, L., Roze, E., Chantot-Bastaraud, S., Koht, J., Motte, J.,et al. (2012). Benign hereditary chorea: phenotype, prognosis, therapeutic outcome and long term follow-up in a large series with new mutations in the TITF1/NKX2-1 gene. J. Neurol. Neurosurg. Psychiatry 83, 956–962. doi: 10.1136/jnnp-2012-302505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Green, R. E., Krause, J., Briggs, A. W., Maricic, T., Stenzel, U., Kircher, M.,et al. (2010). A draft sequence of the Neandertal genome. Science 328, 710–722. doi: 10.1126/science.1188021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gregor, A., Albrecht, B., Bader, I., Bijlsma, E. K., Ekici, A. B., Engels, H.,et al. (2011). Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1. BMC Med. Genet. 12:106. doi: 10.1186/1471-2350-12-106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grimbert, P., Valanciute, A., Audard, V., Pawlak, A., Le Gouvelo, S., Lang, P.,et al. (2003). Truncation of C-mip (Tc-mip), a new proximal signaling protein, induces c-maf Th2 transcription factor and cytoskeleton reorganization. J. Exp. Med. 198, 797–807. doi: 10.1084/jem.20030566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Groszer, M., Keays, D. A., Deacon, R. M., de Bono, J. P., Prasad-Mulcare, S., Gaub, S.,et al. (2008). Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr. Biol. 18, 354–362. doi: 10.1016/j.cub.2008.01.060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grove, E. A., Tole, S., Limon, J., Yip, L., and Ragsdale, C. W. (1998). The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development 125, 2315–2325.
Guo, S., Cichy, S. B., He, X., Yang, Q., Ragland, M., Ghosh, A. K.,et al. (2001). Insulin suppresses transactivation by CAAT/enhancer-binding proteins beta (C/EBPβ). Signaling to p300/CREB-binding protein by protein kinase B disrupts interaction with the major activation domain of C/EBPβ. J. Biol. Chem. 276, 8516–8523. doi: 10.1074/jbc.M008542200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gutiérrez, S., Javed, A., Tennant, D. K., van Rees, M., Montecino, M., Stein, G. S.,et al. (2002). CCAAT/enhancer-binding proteins (C/EBP) β and δ activate osteocalcin gene transcription and synergize with Runx2 at the C/EBP element to regulate bone-specific expression. J. Biol. Chem. 277, 1316–1323. doi: 10.1074/jbc.M106611200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Habib, M. R., Ganea, D. A., Katz, I. K., and Lamprecht, R. (2013). ABL1 in thalamus is associated with safety but not fear learning. Front. Syst. Neurosci. 7:5. doi: 10.3389/fnsys.2013.00005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haddad-Tóvolli, R., Heide, M., Zhou, X., Blaess, S., and Álvarez-Bolado, G. (2012). Mouse thalamic differentiation: gli-dependent pattern and gli-independent prepattern. Front. Neurosci. 6:27. doi: 10.3389/fnins.2012.00027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haesler, S., Wada, K., Nshdejan, A., Morrisey, E. E., Lints, T., Jarvis, E. D.,et al. (2004). FoxP2 expression in avian vocal learners and non-learners. J. Neurosci. 24, 3164–3175. doi: 10.1523/JNEUROSCI.4369-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamdan, F. F., Daoud, H., Rochefort, D., Piton, A., Gauthier, J., Langlois, M.,et al. (2010). De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am. J. Hum. Genet. 87, 671–678. doi: 10.1016/j.ajhg.2010.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Han, N., Zheng, Y., Li, R., Li, X., Zhou, M., Niu, Y.,et al. (2014). β-catenin enhances odontoblastic differentiation of dental pulp cells through activation of Runx2. PLoS ONE 9:e88890. doi: 10.1371/journal.pone.0088890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hannula-Jouppi, K., Kaminen-Ahola, N., Taipale, M., Eklund, R., Nopola-Hemmi, J., Kääriäinen, H.,et al. (2005). The axon guidance receptor gene ROBO1 is a candidate gene for developmental dyslexia. PLoS Genetics 1:e50. doi: 10.1371/journal.pgen.0010050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hansen, S. M., Berezin, V., and Bock, E. (2008). Signaling mechanisms of neurite outgrowth induced by the cell adhesion molecules NCAM and N-cadherin. Cell Mol. Life Sci. 65, 3809–3821. doi: 10.1007/s00018-008-8290-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hashimoto, M., and Hibi, M. (2012). Development and evolution of cerebellar neural circuits. Dev. Growth Differ. 54, 373–389. doi: 10.1111/j.1440-169X.2012.01348.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hashimoto-Torii, K., Motoyama, J., Hui, C. C., Kuroiwa, A., Nakafuku, M., and Shimamura, K. (2003). Differential activities of Sonic hedgehog mediated by Gli transcription factors define distinct neuronal subtypes in the dorsal thalamus. Mech. Dev. 120, 1097–1111. doi: 10.1016/j.mod.2003.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haygood, R., Fedrigo, O., Hanson, B., Yokoyama, K. D., and Wray, G. A. (2007). Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nat. Genet. 39, 1140–1144. doi: 10.1038/ng2104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, J., Ye, J., Cai, Y., Riquelme, C., Liu, J. O., Liu, X.,et al. (2011). Structure of p300 bound to MEF2 on DNA reveals a mechanism of enhanceosome assembly. Nucleic Acids Res. 39, 4464–4474. doi: 10.1093/nar/gkr030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hebron, M. L., Lonskaya, I., and Moussa, C. E. (2013). Tyrosine kinase inhibition facilitates autophagic SNCA/α-synuclein clearance. Autophagy 9, 1249–1250. doi: 10.4161/auto.25368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hennekam, R. C. (2006). Rubinstein-Taybi syndrome. Eur. J. Hum. Genet. 14, 981–985. doi: 10.1038/sj.ejhg.5201594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hernández, V. J., Weng, J., Ly, P., Pompey, S., Dong, H., Mishra, L.,et al. (2013). Cavin-3 dictates the balance between ERK and Akt signaling. Elife 2:e00905. doi: 10.7554/eLife.00905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hickok, G. (2012). Computational neuroanatomy of speech production. Nat. Rev. Neurosci. 13, 135–145. doi: 10.1038/nrn3158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hickok, G., Houde, J., and Rong, F. (2011). Sensorimotor integration in speech processing: computational basis and neural organization. Neuron 69, 407–422. doi: 10.1016/j.neuron.2011.01.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hickok, G., and Poeppel, D. (2007). The cortical organization of speech processing. Nat. Rev. Neurosci. 8, 393–402. doi: 10.1038/nrn2113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Higginbotham, H., Tanaka, T., Brinkman, B. C., and Gleeson, J. G. (2006). GSK3β and PKCζ function in centrosome localization and process stabilization during Slit-mediated neuronal repolarization. Mol. Cell. Neurosci. 32, 118–132. doi: 10.1016/j.mcn.2006.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hirata, M., Kugimiya, F., Fukai, A., Saito, T., Yano, F., Ikeda, T.,et al. (2012). C/EBPβ and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2α as the inducer in chondrocytes. Hum. Mol. Genet. 21, 1111–1123. doi: 10.1093/hmg/ddr540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horita, H., Kobayashi, M., Liu, W. C., Oka, K., Jarvis, E. D., and Wada, K. (2012). Specialized motor-driven Dusp1 expression in the song systems of multiple lineages of vocal learning birds. PLoS ONE 7:e42173. doi: 10.1371/journal.pone.0042173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horita, H., Wada, K., Rivas, M. V., Hara, E., and Jarvis, E. D. (2010). The dusp1 immediate early gene is regulated by natural stimuli predominantly in sensory input neurons. J. Comp. Neurol. 518, 2873–901. doi: 10.1002/cne.22370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horn, D., Kapeller, J., Rivera-Brugués, N., Moog, U., Lorenz-Depiereux, B., Eck, S.,et al. (2010). Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum. Mutat. 31, E1851–E1860. doi: 10.1002/humu.21362
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, H. (1999). Chemorepulsion of neuronal migration by Slit2 in the developing mammalian forebrain. Neuron 23, 703–711. doi: 10.1016/S0896-6273(01)80029-5
Huang, B., Takahashi, K., Jennings, E. A., Pumtang-On, P., Kiso, H., Togo, Y.,et al. (2014). Prospective signs of cleidocranial dysplasia in Cebpb deficiency. J. Biomed. Sci. 21:44. doi: 10.1186/1423-0127-21-44
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, Y. T., Iwamoto, K., Kurosaki, T., Nasu, M., and Ueda, S. (2005). The neuronal POU transcription factor Brn-2 interacts with Jab1, a gene involved in the onset of neurodegenerative diseases. Neurosci. Lett. 382, 175–178. doi: 10.1016/j.neulet.2005.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huynh, M. A., Stegmüller, J., Litterman, N., and Bonni, A. (2009). Regulation of Cdh1-APC function in axon growth by Cdh1 phosphorylation. J. Neurosci. 29, 4322–4327. doi: 10.1523/JNEUROSCI.5329-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inoue, F., Kurokawa, D., Takahashi, M., and Aizawa, S. (2012). Gbx2 directly restricts Otx2 expression to forebrain and midbrain, competing with class III POU factors. Mol. Cell. Biol. 32, 2618–2627. doi: 10.1128/MCB.00083-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inoue, K. I., Shiga, T., and Ito, Y. (2008). Runx transcription factors in neuronal development. Neural Dev. 3, 8104–8103. doi: 10.1186/1749-8104-3-20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwata, J., Suzuki, A., Pelikan, R. C., Ho, T. V., and Chai, Y. (2013). Noncanonical transforming growth factor β (TGFβ) signaling in cranial neural crest cells causes tongue muscle developmental defects. J. Biol. Chem. 288, 29760–29770. doi: 10.1074/jbc.M113.493551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jarvis, E. D. (2004). Learned birdsong and the neurobiology of human language. Ann. N. Y. Acad. Sci. 1016, 749–777. doi: 10.1196/annals.1298.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jehn, B. M., Dittert, I., Beyer, S., von der, M. K., and Bielke, W. (2002). c-Cbl binding and ubiquitin-dependent lysosomal degradation of membrane-associated Notch1. J. Biol. Chem. 277, 8033–8040. doi: 10.1074/jbc.M108552200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jin, Y. H., Jeon, E. J., Li, Q. L., Lee, Y. H., Choi, J. K., Kim, W. J.,et al. (2004). Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J. Biol. Chem. 279, 29409–29417. doi: 10.1074/jbc.M313120200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joazeiro, C. A. P., Wing, S. S., Huang, H., Leverson, J. D., Hunter, T., and Liu., Y.-C. (1999). The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science 286, 309–312. doi: 10.1126/science.286.5438.309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johnson, M. B., Kawasawa, Y. I., Mason, C. E., Krsnik, Z., Coppola, G., Bogdanović, D.,et al. (2009). Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509. doi: 10.1016/j.neuron.2009.03.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jung, J., Mo, J. S., Kim, M. Y., Ann, E. J., Yoon, J. H., and Park, H. S. (2011). Regulation of Notch1 signaling by Delta-like ligand 1 intracellular domain through physical interaction. Mol. Cells 32, 161–165. doi: 10.1007/s10059-011-1046-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kameda, Y., Saitoh, T., and Fujimura, T. (2011). Hes1 regulates the number and anterior-posterior patterning of mesencephalic dopaminergic neurons at the mid/hindbrain boundary (isthmus). Dev. Biol. 358, 91–101. doi: 10.1016/j.ydbio.2011.07.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kannan, M., Lee, S. J., Schwedhelm-Domeyer, N., Nakazawa, T., and Stegmüller, J. (2012). p250GAP is a novel player in the Cdh1-APC/Smurf1 pathway of axon growth regulation. PLoS ONE 7:e50735. doi: 10.1371/journal.pone.0050735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Katayama, K., Imai, F., Campbell, K., Lang, R. A., Zheng, Y., and Yoshida, Y. (2013). RhoA and Cdc42 are required in pre-migratory progenitors of the medial ganglionic eminence ventricular zone for proper cortical interneuron migration. Development. 140, 3139–3145. doi: 10.1242/dev.092585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kato, M., Okanoya, K., Koike, T., Sasaki, E., Okano, H., Watanabe, S.,et al. (2014). Human speech- and reading-related genes display partially overlapping expression patterns in the marmoset brain. Brain Lang. 133, 26–38. doi: 10.1016/j.bandl.2014.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kele, J., Andersson, E. R., Villaescusa, J. C., Cajanek, L., Parish, C. L., Bonilla, S.,et al. (2012). SFRP1 and SFRP2 dose-dependently regulate midbrain dopamine neuron development in vivo and in embryonic stem cells. Stem Cells 30, 865–875. doi: 10.1002/stem.1049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiecker, C., and Lumsden, A. (2004). Hedgehog signaling from the ZLI regulates diencephalic regional identity. Nat. Neurosci. 7, 1242–1249. doi: 10.1038/nn1338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, J., Kang, H. S., Lee, Y. J., Lee, H. J., Yun, J., Shin, J. H.,et al. (2014). EGR1-dependent PTEN upregulation by 2-benzoyloxycinnamaldehyde attenuates cell invasion and EMT in colon cancer. Cancer Lett. 349, 35–44. doi: 10.1016/j.canlet.2014.03.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kokovay, E., Wang, Y., Kusek, G., Wurster, R., Lederman, P., Lowry, N.,et al. (2012). VCAM1 is essential to maintain the structure of the SVZ niche and acts as an environmental sensor to regulate SVZ lineage progression. Cell Stem Cell. 11, 220–230. doi: 10.1016/j.stem.2012.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konishi, Y., Stegmüller, J., Matsuda, T., Bonni, S., and Bonni, A. (2004). Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303, 1026–1030. doi: 10.1126/science.1093712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konopka, G., Bomar, J. M., Winden, K., Coppola, G., Jonsson, Z. O., Gao, F.,et al. (2009). Human-specific transcriptional regulation of CNS development genes by FOXP2. Nature 462, 213–217. doi: 10.1038/nature08549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konopka, G., Friedrich, T., Davis-Turak, J., Winden, K., Oldham, M. C., Gao, F.,et al. (2012). Human-specific transcriptional networks in the brain. Neuron 75, 601–617. doi: 10.1016/j.neuron.2012.05.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krebs, L. T., Iwai, N., Nonaka, S., Welsh, I. C., Lan, Y., Jiang, R.,et al. (2003). Notch signaling regulates left-right asymmetry determination by inducing Nodal expression. Genes Dev. 17, 1207–1212. doi: 10.1101/gad.1084703
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kriebel, M., Wuchter, J., Trinks, S., and Volkmer, H. (2012). Neurofascin: a switch between neuronal plasticity and stability. Int. J. Biochem. Cell. Biol. 44, 694–697. doi: 10.1016/j.biocel.2012.01.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuhlwilm, M., Davierwala, A., and Pääbo, S. (2013). Identification of putative target genes of the transcription factor RUNX2. PLoS ONE 8:e83218. doi: 10.1371/journal.pone.0083218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurt, S, Fisher, S. E., and Ehret, G. (2012). Foxp2 mutations impair auditory-motor association learning. PLoS ONE 7:e33130. doi: 10.1371/journal.pone.0033130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laake, K., Jansen, L., Hahnemann, J. M., Brondum-Nielsen, K., Lonnqvist, T., Kaariainen, H.,et al. (2000). Characterization of ATM mutations in 41 Nordic families with ataxia telangiectasia. Hum. Mutat. 16, 232–246. doi: 10.1002/1098-1004(200009)16:3<232::AID-HUMU6>3.0.CO;2-L
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lai, C. S., Fisher, S. E., Hurst, J. A., Vargha-Khadem, F., and Monaco, A. P. (2001). A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 413, 519–523. doi: 10.1038/35097076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, C. H., Huang, Y. L., Liao, J. F., and Chiou, W. F. (2011). Ugonin K promotes osteoblastic differentiation and mineralization by activation of p38 MAPK- and ERK-mediated expression of Runx2 and osterix. Eur. J. Pharmacol. 668, 383–389. doi: 10.1016/j.ejphar.2011.06.059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A.,et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. doi: 10.1038/nature05453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lengyel, E., Wang, H., Stepp, E., Juárez, J., Wang, Y., Doe, W.,et al. (1996). Requirement of an upstream AP-1 motif for the constitutive and phorbol ester-inducible expression of the urokinase-type plasminogen activator receptor gene. J. Biol. Chem. 271, 23176–23184. doi: 10.1074/jbc.271.38.23176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Letinic, K., and Rakic, P. (2001). Telencephalic origin of human thalamic GABAergic neurons. Nat. Neurosci. 4, 931–936. doi: 10.1038/nn0901-931
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Letinic, K., Zoncu, R., and Rakic, P. (2002). Origin of GABAergic neurons in the human neocortex. Nature 417, 645–649. doi: 10.1038/nature00779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leung, R., Wang, Y., Cuddy, K., Sun, C., Magalhaes, J., Grynpas, M.,et al. (2010). Filamin A regulates monocyte migration through Rho small GTPases during osteoclastogenesis. J. Bone Miner. Res. 25, 1077–1091. doi: 10.1359/jbmr.091114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leyva-Díaz, E., and López-Bendito, G. (2013). In and out from the cortex: development of major forebrain connections. Neuroscience 254C, 26–44. doi: 10.1016/j.neuroscience.2013.08.070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, B., Boast, S., de los Santos, K., Schieren, I., Quiroz, M., Teitelbaum, S. L.,et al. (2000). Mice deficient in Abl are osteoporotic and have defects in osteoblast maturation. Nat. Genet. 24, 304–308. doi: 10.1038/73542
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, S., Leshchyns’ka, I., Chernyshova, Y., Schachner, M., and Sytnyk, V. (2013). The neural cell adhesion molecule (NCAM) associates with and signals through p21-activated kinase 1 (Pak1). J. Neurosci. 33, 790–803. doi: 10.1523/JNEUROSCI.1238-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, S., Weidenfeld, J., and Morrisey, E. E. (2004). Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol. Cell. Biol. 24, 809–822. doi: 10.1128/MCB.24.2.809-822.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lieberman, P. (2007). The evolution of human speech; its anatomical and neural bases. Curr. Anthropol. 48, 39–66. doi: 10.1086/509092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, M., Pedrosa, E., Shah, A., Hrabovsky, A., Maqbool, S., Zheng, D.,et al. (2011). RNA-Seq of human neurons derived from iPS cells reveals candidate long non-coding RNAs involved in neurogenesis and neuropsychiatric disorders. PLoS ONE 6:e23356. doi: 10.1371/journal.pone.0023356.g001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lindhurst, M. J., Sapp, J. C., Teer, J. K., Johnston, J. J., Finn, E. M., Peters, K.,et al. (2011). A mosaic activating mutation in AKT1 associated with the Proteus syndrome. New. Eng. J. Med. 365, 611–619. doi: 10.1056/NEJMoa1104017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, X., Somel, M., Tang, L., Yan, Z., Jiang, X., Guo, S.,et al. (2012). Extension of cortical synaptic development distinguishes humans from chimpanzees and macaques. Genome Res. 22, 611–622. doi: 10.1101/gr.127324.111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lomonaco, S. L., Kahana, S., Blass, M., Brody, Y., Okhrimenko, H., Xiang, C.,et al. (2008). Phosphorylation of protein kinase Cdelta on distinct tyrosine residues induces sustained activation of Erk1/2 via down-regulation of MKP-1: role in the apoptotic effect of etoposide. J. Biol. Chem. 283, 17731–17739. doi: 10.1074/jbc.M801727200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Long, J. E., Garel, S., Álvarez-Dolado, M., Yoshikawa, K., Osumi, N., Álvarez-Buylla, A.,et al. (2007). Dlx-dependent and -independent regulation of olfactory bulb interneuron differentiation. J. Neurosci. 27, 3230–3243. doi: 10.1523/JNEUROSCI.5265-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Long, Q., Qiu, B., Wang, K., Yang, J., Jia, C., Xin, W.,et al. (2013). Genetically engineered bone marrow mesenchymal stem cells improve functional outcome in a rat model of epilepsy. Brain. Res. 1532, 1–13. doi: 10.1016/j.brainres.2013.07.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lopes Floro, K., Artap, S. T., Preis, J. I., Fatkin, D., Chapman, G., Furtado, M. B.,et al. (2011). Loss of Cited2 causes congenital heart disease by perturbing left-right patterning of the body axis. Hum. Mol. Genet. 20, 1097–1110. doi: 10.1093/hmg/ddq554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
López-Bendito, G., Flames, N., Ma, L., Fouquet, C., Di Meglio, T., Chedotal, A.,et al. (2007). Robo1 and Robo2 cooperate to control the guidance of major axonal tracts in the mammalian forebrain. J. Neurosci. 27, 3395–3407. doi: 10.1523/JNEUROSCI.4605-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lowe, L. A., Supp, D. M., Sampath, K., Yokoyama, T., Wright, C. V., Potter, S. S.,et al. (1996). Conserved left-right asymmetry of nodal expression and alterations in murine situs inversus. Nature 381, 158–161. doi: 10.1038/381158a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, X., Ding, L., Xu, J., and Chegini, N. (2005). Gene expression profiling of leiomyoma and myometrial smooth muscle cells in response to transforming growth factor-beta. Endocrinology 146, 1097–1118. doi: 10.1210/en.2004-1377
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
MacDermot, K. D., Bonora, E., Sykes, N., Coupe, A. M., Lai, C. S., Vernes, S. C.,et al. (2005). Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am. J. Hum. Genet. 76, 1074–1080. doi: 10.1086/430841
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mao, Z., and Nadal-Ginard, B. (1996). Functional and physical interactions between mammalian achaete-scute homolog 1 and myocyte enhancer factor 2A. J. Biol. Chem. 271, 14371–14375. doi: 10.1074/jbc.271.24.14371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marcos-Mondéjar, P., Peregrín, S., Li, J. Y., Carlsson, L., Tole, S., and López-Bendito, G. (2012). The lhx2 transcription factor controls thalamocortical axonal guidance by specific regulation of robo1 and robo2 receptors. J. Neurosci. 32, 4372–4385. doi: 10.1523/JNEUROSCI.5851-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marcuzzo, S., Kapetis, D., Mantegazza, R., Baggi, F., Bonanno, S., Barzago, C.,et al. (2014). Altered miRNA expression is associated with neuronal fate in G93A-SOD1 ependymal stem progenitor cells. Exp. Neurol. 253, 91–101. doi: 10.1016/j.expneurol.2013.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maricic, T., Günther, V., Georgiev, O., Gehre, S., Curlin, M., Schreiweis, C.,et al. (2013). A recent evolutionary change affects a regulatory element in the human FOXP2 gene. Mol. Biol. Evol. 30, 844–852. doi: 10.1093/molbev/mss271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marillat, V., Cases, O., Nguyenf Ba-Charvet, K. T., TessierLavigne, M., Sotelo, C., and Chédotal, A. (2002). Spatiotemporal expression patterns of slit and robo genes in the rat brain. J. Comp. Neurol. 442, 130–155. doi: 10.1002/cne.10068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martinelli, S., De Luca, A., Stellacci, E., Rossi, C., Checquolo, S., Lepri, F.,et al. (2010). Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am. J. Hum. Genet. 87, 250–257. doi: 10.1016/j.ajhg.2010.06.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mascheretti, S., Riva, V., Giorda, R., Beri, S., Lanzoni, L. F., Cellino, M. R.,et al. (2014). KIAA0319 and ROBO1: evidence on association with reading and pleiotropic effects on language and mathematics abilities in developmental dyslexia. J. Hum. Genet. 59, 189–197. doi: 10.1038/jhg.2013.141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsumura, S., and Toyoshima, F. (2012). ABL1 joins the cadre of spindle orientation machinery. Cell. Struct. Funct. 37, 81–87. doi: 10.1247/csf.12011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsunoshita, Y., Ijiri, K., Ishidou, Y., Nagano, S., Yamamoto, T., Nagao, H.,et al. (2011). Suppression of osteosarcoma cell invasion by chemotherapy is mediated by urokinase plasminogen activator activity via up-regulation of EGR1. PLoS ONE 6:e16234. doi: 10.1371/journal.pone.0016234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mazzucchelli, C., Vantaggiato, C., Ciamei, A., Fasano, S., Pakhotin, P., Krezel, W.,et al. (2002). Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34, 807–820. doi: 10.1016/S0896-6273(02)00716-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McEvilly, R. J., de Díaz, M. O., Schonemann, M. D., Hooshmand, F., and Rosenfeld, M. G. (2002). Transcriptional regulation of cortical neuron migration by POU domain factors. Science 295, 1528–1532. doi: 10.1126/science.1067132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McLean, C. Y., Reno, P. L., Pollen, A. A., Bassan, A. I., Capellini, T. D., Guenther, C.,et al. (2011). Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471, 216–219. doi: 10.1038/nature09774
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Medina, L., Abellán, A., Vicario, A., and Desfilis, E. (2014). Evolutionary and developmental contributions for understanding the organization of the Basal Ganglia. Brain Behav. Evol. 83, 112–125. doi: 10.1159/000357832
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meyer, M., Kircher, M., Gansauge, M. T., Li, H., Racimo, F., Mallick, S.,et al. (2012). A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226. doi: 10.1126/science.1224344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mikami, T., Ohnaka, Y., Nakamura, A., Kurosaka, A., and Itoh, N. (2001). Radical fringe negatively modulates Notch signaling in postmitotic neurons of the rat brain. Brain. Res. Mol. Brain. Res. 86, 138–144. doi: 10.1016/S0169-328X(00)00278-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, E. K., and Buschman, T. J. (2007). “Rules through recursion: how interactions between the frontal cortex and basal ganglia may build abstract, complex, rules from concrete, simple, ones,” in The Neuroscience of Rule-Guided Behavior, eds S. Bunge and J. Wallis (Oxford: Oxford University Press), 419–440.
Miller, J. A., Ding, S. L., Sunkin, S. M., Smith, K. A., Ng, L., Szafer, A.,et al. (2014). Transcriptional landscape of the prenatal human brain. Nature 508, 199–206. doi: 10.1038/nature13185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mink, S., Haenig, B., and Klempnauer, K. H. (1997). Interaction and functional collaboration of p300 and C/EBPβ. Mol. Cell. Biol. 17, 6609–6617.
Mistafa, O., Ghalali, A., Kadekar, S., Högberg, J., and Stenius, U. (2010). Purinergic receptor-mediated rapid depletion of nuclear phosphorylated Akt depends on pleckstrin homology domain leucine-rich repeat phosphatase, calcineurin, protein phosphatase 2A, and PTEN phosphatases. J. Biol. Chem. 285, 27900–27910. doi: 10.1074/jbc.M110.117093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mithen, S. J. (2006). The Singing Neanderthals: the Origins of Music, Language,Mind and Body. Cambridge: Harvard University Press.
Miyoshi, K., Asanuma, M., Miyazaki, I., Díaz-Corrales, F. J., Katayama, T., Tohyama, M.,et al. (2004). DISC1 localizes to the centrosome by binding to kendrin. Biochem. Biophys. Res. Commun. 317, 1195–1199. doi: 10.1016/j.bbrc.2004.03.163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyoshi-Akiyama, T., Alemán, L. M., Smith, J. M., Adler, C. E., and Mayer, B. J. (2001). Regulation of Cbl phosphorylation by the Abl tyrosine kinase and the Nck SH2/SH3 adaptor. Oncogene 20, 4058–4069. doi: 10.1038/sj.onc.1204528
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mühleisen, T. W., Leber, M., Schulze, T. G., Strohmaier, J., Degenhardt, F., Treutlein, J.,et al. (2014). Genome-wide association study reveals two new risk loci for bipolar disorder. Nat. Commun. 5:3339. doi: 10.1038/ncomms4339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Myers, J. P., Robles, E., Ducharme-Smith, A., and Gómez, T. M. (2012). Focal adhesion kinase modulates Cdc42 activity downstream of positive and negative axon guidance cues. J. Cell. Sci. 125, 2918–2929. doi: 10.1242/jcs.100107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nasu, M., Yada, S., Igarashi, A., Sutoo, D., Akiyama, K., Ito, M.,et al. (2014). Mammalian-specific sequences in Pou3f2 contribute to maternal behavior. Genome Biol. Evol. 6, 1145–1156. doi: 10.1093/gbe/evu072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nekrasova, T., Jobes, M. L., Ting, J. H., Wagner, G. C., and Minden, A. (2008). Targeted disruption of the Pak5 and Pak6 genes in mice leads to deficits in learning and locomotion. Dev. Biol. 322, 95–108. doi: 10.1016/j.ydbio.2008.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nelson, C. S., Fuller, C. K., Fordyce, P. M., Greninger, A. L., Li, H., and DeRisi, J. L. (2013). Microfluidic affinity and ChIP-seq analyses converge on a conserved FOXP2-binding motif in chimp and human, which enables the detection of evolutionarily novel targets. Nucleic Acids Res. 41, 5991–6004. doi: 10.1093/nar/gkt259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nelson, B. R., and Reh, T. A. (2008). Relationship between Delta-like and proneural bHLH genes during chick retinal development. Dev. Dyn. 237, 1565–1580. doi: 10.1002/dvdy.21550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Newbury, D. F., Winchester, L., Addis, L., Paracchini, S., Buckingham, L. L., Clark, A.,et al. (2009). CMIP and ATP2C2 modulate phonological short-term memory in language impairment. Am. J. Hum. Genet. 85, 264–272. doi: 10.1016/j.ajhg.2009.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ni, X., Trakalo, J., Valente, J., Azevedo, M. H., Pato, M. T., Pato, C. N.,et al. (2005). Human p53 tumor suppressor gene (TP53) and schizophrenia: case-control and family studies. Neurosci. Lett. 388, 173–178. doi: 10.1016/j.neulet.2005.06.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nourry, C., Maksumova, L., Pang, M., Liu, X., and Wang, T. (2004). Direct interaction between Smad3, APC10, CDH1 and HEF1 in proteasomal degradation of HEF1. BMC Cell Biol. 5:20. doi: 10.1186/1471-2121-5-20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ohi, K., Hashimoto, R., Nakazawa, T., Okada, T., Yasuda, Y., Yamamori, H.,et al. (2012). The p250GAP gene is associated with risk for schizophrenia and schizotypal personality traits. PLoS ONE 7:e35696. doi: 10.1371/journal.pone.0035696
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pääbo, S. (2014). The human condition-a molecular approach. Cell 157, 216–226. doi: 10.1016/j.cell.2013.12.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Palomero, T., Domínguez, M., and Ferrando, A. A. (2008). The role of the PTEN/AKT Pathway in NOTCH1-induced leukemia. Cell Cycle 7, 965–970. doi: 10.4161/cc.7.8.5753
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paskulin, D. D., Paixão-Côrtes, V. R., Hainaut, P., Bortolini, M. C., and Ashton-Prolla, P. (2012). The TP53 fertility network. Genet. Mol. Biol. 35, 939–946. doi: 10.1590/S1415-47572012000600008
Petrin, A. L., Giacheti, C. M., Maximino, L. P., Abramides, D. V., Zanchetta, S., Rossi, N. F.,et al. (2010). Identification of a microdeletion at the 7q33-q35 disrupting the CNTNAP2 gene in a Brazilian stuttering case. Am. J. Med. Genet. A 152A, 3164–3172. doi: 10.1002/ajmg.a.33749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfenning, A. R. (2012). A Computational Synthesis of Genes, Behavior, and Evolution Provides Insights into the Molecular Basis of Vocal Learning. Ph.D. thesis, Duke University, Durham.
Poelmans, G., Buitelaar, J. K., Pauls, D. L., and Franke, B. (2011). A theoretical molecular network for dyslexia: integrating available genetic findings. Mol. Psychiatry 16, 365–382. doi: 10.1038/mp.2010.105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Potkin, S. G., Turner, J. A., Guffanti, G., Lakatos, A., Fallon, J. H., Nguyen, D. D.,et al. (2009). A genome-wide association study of schizophrenia using brain activation as a quantitative phenotype. Schizophr. Bull. 35, 96–108. doi: 10.1093/schbul/sbn155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prag, S., Lepekhin, E. A., Kolkova, K., Hartmann-Petersen, R., Kawa, A., Walmod, P. S.,et al. (2002). NCAM regulates cell motility. J. Cell Sci. 115, 283–292.
Preis, J. I., Wise, N., Solloway, M. J., Harvey, R. P., Sparrow, D. B., and Dunwoodie, S. L. (2006). Generation of conditional Cited2 null alleles. Genesis 44, 579–583. doi: 10.1002/dvg.20251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prüfer, K., Racimo, F., Patterson, N., Jay, F., Sankararaman, S., Sawyer, S.,et al. (2014). The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49. doi: 10.1038/nature12886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramakers, G. J., Wolfer, D., Rosenberger, G., Kuchenbecker, K., Kreienkamp, H. J., Prange-Kiel, J.,et al. (2012). Dysregulation of Rho GTPases in the αPix/Arhgef6 mouse model of X-linked intellectual disability is paralleled by impaired structural and synaptic plasticity and cognitive deficits. Hum. Mol. Genet. 21, 268–286. doi: 10.1093/hmg/ddr457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rauch, A., Thiel, C. T., Schindler, D., Wick, U., Crow, Y. J., Ekici, A. B.,et al. (2008). Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science 319, 816–819. doi: 10.1126/science.1151174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reale, M. E., Webb, I. C., Wang, X., Baltazar, R. M., Coolen, L. M., and Lehman, M. N. (2013). The transcription factor Runx2 is under circadian control in the suprachiasmatic nucleus and functions in the control of rhythmic behavior. PLoS ONE 8:e54317. doi: 10.1371/journal.pone.0054317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodríguez-Esteban, C., Schwabe, J. W., Peña, J. D., Rincón-Limas, D. E., Magallón, J., Botas, J.,et al. (1998). Lhx2, a vertebrate homologue of apterous, regulates vertebrate limb outgrowth. Development 125, 3925–3934.
Roll, P., Rudolf, G., Pereira, S., Royer, B., Scheffer, I. E., Massacrier, A.,et al. (2006). SRPX2 mutations in disorders of language cortex and cognition. Hum. Mol. Genet. 15, 1195–1207. doi: 10.1093/hmg/ddl035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roll, P., Vernes, S. C., Bruneau, N., Cillario, J., Ponsole-Lenfant, M., Massacrier, A.,et al. (2010). Molecular networks implicated in speech-related disorders: FOXP2 regulates the SRPX2/uPAR complex. Hum. Mol. Genet. 19, 4848–4860. doi: 10.1093/hmg/ddq415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rønn, L. C., Berezin, V., and Bock, E. (2000). The neural cell adhesion molecule in synaptic plasticity and ageing. Int. J. Dev. Neurosci. 18, 193–199. doi: 10.1016/S0736-5748(99)00088-X
Royer-Zemmour, B., Ponsole-Lenfant, M., Gara, H., Roll, P., Lévêque, C., Massacrier, A.,et al. (2008). Epileptic and developmental disorders of the speech cortex: ligand/receptor interaction of wild-type and mutant SRPX2 with the plasminogen activator receptor uPAR. Hum. Mol. Genet. 17, 3617–3630. doi: 10.1093/hmg/ddn256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rutishauser, U., and Goridis, C. (1986). NCAM: the molecule and its genetics. Trends Genet. 2, 72–76. doi: 10.1016/0168-9525(86)90182-4
Samuels, I. S., Karlo, J. C., Faruzzi, A. N., Pickering, K., Herrup, K., Sweatt, J. D.,et al. (2008). Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J. Neurosci. 28, 6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schiff, M., Röckle, I., Burkhardt, H., Weinhold, B., and Hildebrandt, H. (2011). Thalamocortical pathfinding defects precede degeneration of the reticular thalamic nucleus in polysialic acid-deficient mice. J. Neurosci. 31, 1302–1312. doi: 10.1523/JNEUROSCI.5609-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scholpp, S., and Lumsden, A. (2010). Building a bridal chamber: development of the thalamus. Trends Neurosci. 33, 373–380. doi: 10.1016/j.tins.2010.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scholpp, S., Wolf, O., Brand, M., and Lumsden, A. (2006). Hedgehog signalling from the zona limitans intrathalamica orchestrates patterning of the zebrafish diencephalon. Development 133, 855–864. doi: 10.1242/dev.02248
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schor, I. E., Fiszbein, A., Petrillo, E., and Kornblihtt, A. R. (2013). Intragenic epigenetic changes modulate NCAM alternative splicing in neuronal differentiation. EMBO J. 32, 2264–2274. doi: 10.1038/emboj.2013.167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schreiweis, C., Bornschein, U., Burguière, E., Kerimoglu, C., Schreiter, S., Dannemann, M.,et al. (2014). Humanized Foxp2 accelerates learning by enhancing transitions from declarative to procedural performance. Proc. Natl. Acad. Sci. U.S.A. 111, 14253–14258. doi: 10.1073/pnas.1414542111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwartzberg, P. L., Stall, A. M., Hardin, J. D., Bowdish, K. S., Humaran, T., Boast, S.,et al. (1991). Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell 65, 1165–1175. doi: 10.1016/0092-8674(91)90012-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scott-Van Zeeland, A. A., Abrahams, B. S., Álvarez-Retuerto, A. I., Sonnenblick, L. I., Rudie, J. D., Ghahremani, D.,et al. (2010). Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci. Transl. Med. 2, 56ra80. doi: 10.1126/scitranslmed.3001344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sebastián-Serrano, A., Sandonis, A., Cardozo, M., Rodríguez-Tornos, F. M., Bovolenta, P., and Nieto, M. (2012). Pαx6 expression in postmitotic neurons mediates the growth of axons in response to SFRP1. PLoS ONE 7:e31590. doi: 10.1371/journal.pone.0031590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sehested, L. T., Møller, R. S., Bache, I., Andersen, N. B., Ullmann, R., Tommerup, N.,et al. (2010). Deletion of 7q34-q36.2 in two siblings with mental retardation, language delay, primary amenorrhea, and dysmorphic features. Am. J. Med. Genet. A 152A, 3115–31119. doi: 10.1002/ajmg.a.33476
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sener, R. N. (1995). Rubinstein-Taybi syndrome: cranial MR imaging findings. Comput. Med. Imaging. Graph. 19, 417–418. doi: 10.1016/0895-6111(95)00023-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sheen, V. L., Dixon, P. H., Fox, J. W., Hong, S. E., Kinton, L., Sisodiya, S. M.,et al. (2001). Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum. Mol. Genet. 10, 1775–1783. doi: 10.1093/hmg/10.17.1775
Shim, J. H., Greenblatt, M. B., Singh, A., Brady, N., Hu, D., Drapp, R.,et al. (2012). Administration of BMP2/7 in utero partially reverses Rubinstein-Taybi syndrome-like skeletal defects induced by Pdk1 or Cbp mutations in mice. J. Clin. Invest. 122, 91–106. doi: 10.1172/JCI59466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shimojo, H., Ohtsuka, T., and Kageyama, R. (2008). Oscillations in notch signaling regulate maintenance of neural progenitors. Neuron 58, 52–64. doi: 10.1016/j.neuron.2008.02.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shimomura, K., Kumar, V., Koike, N., Kim, T. K., Chong, J., Buhr, E. D.,et al. (2013). Usf1, a suppressor of the circadian Clock mutant, reveals the nature of the DNA-binding of the CLOCK:BMAL1 complex in mice. Elife 2:e00426. doi: 10.7554/eLife.00426
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shriberg, L. D., Ballard, K. J., Tomblin, J. B., Duffy, J. R., Odell, K. H., and Williams, C. A. (2006). Speech, prosody, and voice characteristics of a mother and daughter with a 7;13 translocation affecting FOXP2. J. Speech Lang. Hear. Res. 49, 500–525. doi: 10.1044/1092-4388(2006/038)
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shu, W., Cho, J. Y., Jiang, Y., Zhang, M., Weisz, D., Elder, G. A.,et al. (2005). Altered ultrasonic vocalization in mice with a disruption in the Foxp2 gene. Proc. Natl. Acad. Sci. U.S.A. 102, 9643–9648. doi: 10.1073/pnas.0503739102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shu, W., Yang, H., Zhang, L., Lu, M. M., and Morrisey, E. E. (2001). Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J. Biol. Chem. 276, 27488–27497. doi: 10.1074/jbc.M100636200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Silverman, E. S., Du, J., Williams, A. J., Wadgaonkar, R., Drazen, J. M., and Collins, T. (1998). cAMP-response-element-binding-protein-binding protein (CBP) and p300 are transcriptional co-activators of early growth response factor-1 (Egr-1). Biochem. J. 336, 183–189.
Speevak, M. D., and Farrell, S. A. (2011). Non-syndromic language delay in a child with disruption in the Protocadherin11X/Y gene pair. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156, 484–489. doi: 10.1002/ajmg.b.31186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spiteri, E., Konopka, G., Coppola, G., Bomar, J., Oldham, M., Ou, J.,et al. (2007). Identification of the transcriptional targets of FOXP2, a gene linked to speech and language, in developing human brain. Am. J. Hum. Genet. 81, 1144–1157. doi: 10.1086/522237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
St Pourcain, B., Cents, R. A., Whitehouse, A. J., Haworth, C. M., Davis, O. S., O’Reilly, P. F.,et al. (2014). Common variation near ROBO2 is associated with expressive vocabulary in infancy. Nat. Commun. 5:4831. doi: 10.1038/ncomms5831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stein, C. M., Schick, J. H., Gerry Taylor, H., Shriberg, L. D., Millard, C., Kundtz-Kluge, A.,et al. (2004). Pleiotropic effects of a chromosome 3 locus on speech-sound disorder and reading. Am. J. Hum. Genet. 74, 283–297. doi: 10.1086/381562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suda, S., Iwata, K., Shimmura, C., Kameno, Y., Anitha, A., Thanseem, I.,et al. (2011). Decreased expression of axon-guidance receptors in the anterior cingulate cortex in autism. Mol. Autism. 2:14. doi: 10.1186/2040-2392-2-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sugitani, M., Sugai, T., and Onoda, N. (2002). Postsynaptic activity of metabotropic glutamate receptors in the piriform cortex. Neuroreport 13, 1473–1476. doi: 10.1097/00001756-200208070-00025
Suh, J. H., Lee, H. W., Lee, J. W., and Kim, J. B. (2008). Hes1 stimulates transcriptional activity of Runx2 by increasing protein stabilization during osteoblast differentiation. Biochem. Biophys. Res. Commun. 367, 97–102. doi: 10.1016/j.bbrc.2007.12.100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szabó, N. E., Zhao, T., Zhou, X., and Álvarez-Bolado, G. (2009). The role of Sonic hedgehog of neural origin in thalamic differentiation in the mouse. J. Neurosci. 29, 2453–2466. doi: 10.1523/JNEUROSCI.4524-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szklarczyk, D., Franceschini, A., Kuhn, M., Simonovic, M., Roth, A., Mínguez, P.,et al. (2011). The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568. doi: 10.1093/nar/gkq973
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takahashi, K., Liu, F. C., Hirokawa, K., and Takahashi, H. (2003). Expression of Foxp2, a gene involved in speech and language, in the developing and adult striatum. J. Neurosci. Res. 73, 61–72. doi: 10.1002/jnr.10638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tanaka, M., Murakami, K., Ozaki, S., Imura, Y., Tong, X. P., Watanabe, T.,et al. (2010). DIP2 disco-interacting protein 2 homolog A (Drosophila) is a candidate receptor for follistatin-related protein/follistatin-like 1–analysis of their binding with TGF-β superfamily proteins. FEBS J. 277, 4278–4289. doi: 10.1111/j.1742-4658.2010.07816.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tanimoto, Y., Veistinen, L., Alakurtti, K., Takatalo, M., and Rice, D. P. (2012). Prevention of premature fusion of calvarial suture in GLI-Kruppel family member 3 (Gli3)-deficient mice by removing one allele of Runt-related transcription factor 2 (Runx2). J. Biol. Chem. 287, 21429–21438. doi: 10.1074/jbc.M112.362145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tapia-Páez, I., Tammimies, K., Massinen, S., Roy, A. L., and Kere, J. (2008). The complex of TFII-I, PARP1, and SFPQ proteins regulates the DYX1C1 gene implicated in neuronal migration and dyslexia. FASEB J. 22, 3001–3009. doi: 10.1096/fj.07-104455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tominaga, H., Maeda, S., Hayashi, M., Takeda, S., Akira, S., Komiya, S.,et al. (2008). CCAAT/enhancer-binding protein β promotes osteoblast differentiation by enhancing Runx2 activity with ATF4. Mol. Biol. Cell. 19, 5373–5386. doi: 10.1091/mbc.E08-03-0329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Travis, M. A., van der Flier, A., Kammerer, R. A., Mould, A. P., Sonnenberg, A., and Humphries, M. J. (2004). Interaction of filamin A with the integrin beta 7 cytoplasmic domain: role of alternative splicing and phosphorylation. FEBS Lett. 569, 185–190. doi: 10.1016/j.febslet.2004.04.099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Valor, L. M., Pulopulos, M. M., Jiménez-Minchán, M., Olivares, R., Lutz, B., and Barco, A. (2011). Ablation of CBP in forebrain principal neurons causes modest memory and transcriptional defects and a dramatic reduction of histone acetylation but does not affect cell viability. J. Neurosci. 31, 1652–1663. doi: 10.1523/JNEUROSCI.4737-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vargha-Khadem, F., Gadian, D. G., Copp, A., and Mishkin, M. (2005). FOXP2 and the neuroanatomy of speech and language. Nat. Rev. Neurosci. 6, 131–138. doi: 10.1038/nrn1605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vargha-Khadem, F., Watkins, K. E., Alcock, K. J., Fletcher, P., and Passingham, R. E. (1995). Praxic and nonverbal cognitive deficits in a large family with a genetically transmitted speech and language disorder. Proc. Natl. Acad. Sci. U.S.A. 92, 930–933. doi: 10.1073/pnas.92.3.930
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vawter, M. P., Usen, N., Thatcher, L., Ladenheim, B., Zhang, P., VanderPutten, D. M.,et al. (2001). Characterization of human cleaved N-CAM and association with schizophrenia. Exp. Neurol. 172, 29–46. doi: 10.1006/exnr.2001.7790
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vernes, S. C., Newbury, D. F., Abrahams, B. S., Winchester, L., Nicod, J., Groszer, M.,et al. (2008). A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 359, 2337–2345. doi: 10.1056/NEJMoa0802828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vernes, S. C., Oliver, P. L., Spiteri, E., Lockstone, H. E., Puliyadi, R., Taylor, J. M.,et al. (2011). Foxp2 regulates gene networks implicated in neurite outgrowth in the developing brain. PLoS Genet. 7:e1002145. doi: 10.1371/journal.pgen.1002145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vernes, S. C., Spiteri, E., Nicod, J., Groszer, M., Taylor, J. M., Davies, K. E.,et al. (2007). High-throughput analysis of promoter occupancy reveals direct neural targets of FOXP2, a gene mutated in speech and language disorders. Am. J. Hum. Genet. 81, 1232–1250. doi: 10.1086/522238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veyrac, A., Besnard, A., Caboche, J., Davis, S., and Laroche, S. (2014). The transcription factor Zif268/Egr1, brain plasticity, and memory. Prog. Mol. Biol. Transl. Sci. 122, 89–129. doi: 10.1016/B978-0-12-420170-5.00004-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Viosca, J., López-Atalaya, J. P., Olivares, R., Eckner, R., and Barco, A. (2010). Syndromic features and mild cognitive impairment in mice with genetic reduction on p300 activity: differential contribution of p300 and CBP to Rubinstein-Taybi syndrome etiology. Neurobiol. Dis. 37, 186–194. doi: 10.1016/j.nbd.2009.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vue, T. Y., Bluske, K., Alishahi, A., Yang, L. L., Koyano-Nakagawa, N., Novitch, B.,et al. (2009). Sonic hedgehog signaling controls thalamic progenitor identity and nuclei specification in mice. J. Neurosci. 29, 4484–4497. doi: 10.1523/JNEUROSCI.0656-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walker, R. M., Hill, A. E., Newman, A. C., Hamilton, G., Torrance, H. S., Anderson, S. M.,et al. (2012). The DISC1 promoter: characterization and regulation by FOXP2. Hum. Mol. Genet. 21, 2862–2872. doi: 10.1093/hmg/dds111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, B., Williams, H., Du, J. S., Terrett, J., and Kenwrick, S. (1998). Alternative splicing of human NrCAM in neural and nonneural tissues. Mol. Cell. Neurosci. 10, 287–295. doi: 10.1006/mcne.1997.0658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H. P., Spencer, D., Fellous, J. M., and Sejnowski, T. J. (2010a). Synchrony of thalamocortical inputs maximizes cortical reliability. Science 328, 106–109. doi: 10.1126/science.1183108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, J., Weaver, I. C., Gauthier-Fisher, A., Wang, H., He, L., Yeomans, J.,et al. (2010b). CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev. Cell 18, 114–125. doi: 10.1016/j.devcel.2009.10.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, R. (2011). Dissecting the Genetic Basis of Convergent Complex Traits Based on Molecular Homoplasy. Ph.D. thesis, Duke University, Durham.
Wang, Q., Moore, M. J., Adelmant, G., Marto, J. A., and Silver, P. A. (2013a). PQBP1, a factor linked to intellectual disability, affects alternative splicing associated with neurite outgrowth. Genes Dev. 27, 615–626. doi: 10.1101/gad.212308.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, K., Long, Q., Jia, C., Liu, Y., Yi, X., Yang, H.,et al. (2013b). Over-expression of Mash1 improves the GABAergic differentiation of bone marrow mesenchymal stem cells in vitro. Brain Res. Bull. 99, 84–94. doi: 10.1016/j.brainresbull.2013.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wapinski, O. L., Vierbuchen, T., Qu, K., Lee, Q. Y., Chanda, S., Fuentes, D. R.,et al. (2013). Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155, 621–635. doi: 10.1016/j.cell.2013.09.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waragai, M., Lammers, C. H., Takeuchi, S., Imafuku, I., Udagawa, Y., Kanazawa, I.,et al. (1999). PQBP-1, a novel polyglutamine tract-binding protein, inhibits transcription activation by Brn-2 and affects cell survival. Hum. Mol. Genet. 8, 977–87. doi: 10.1093/hmg/8.6.977
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watkins, K. E., Dronkers, N. F., and Vargha-Khadem, F. (2002). Behavioural analysis of an inherited speech and language disorder: comparison with acquired aphasia. Brain 125, 452–464. doi: 10.1093/brain/awf058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weeber, E. J., and Sweatt, J. D. (2002). Molecular neurobiology of human cognition. Neuron 33, 845–848. doi: 10.1016/S0896-6273(02)00634-7
Wen, J., Lopes, F., Soares, G., Farrell, S. A., Nelson, C., Qiao, Y.,et al. (2013). Phenotypic and functional consequences of haploinsufficiency of genes from exocyst and retinoic acid pathway due to a recurrent microdeletion of 2p13.2. Orphanet J Rare Dis. 8:100. doi: 10.1186/1750-1172-8-100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Williams, N. A., Close, J. P., Giouzeli, M., and Crow, T. J. (2006). Accelerated evolution of Protocadherin11X/Y: a candidate gene-pair for cerebral asymmetry and language. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141, 623–633. doi: 10.1002/ajmg.b.30357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong, G. W., Knowles, G. C., Mak, T. W., Ferrando, A. A., and Zúñiga-Pflücker, J. C. (2012). HES1 opposes a PTEN-dependent check on survival, differentiation, and proliferation of TCRβ-selected mouse thymocytes. Blood 120, 1439–1448. doi: 10.1182/blood-2011-12-395319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, J. Y., Feng, L., Park, H. T., Havlioglu, N., Wen, L., Tang, H.,et al. (2001). The neuronal repellent Slit inhibits leukocyte chemotaxis induced by chemotactic factors. Nature 410, 948–952. doi: 10.1038/35073616
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, W. J., Tu, S., and Cerione, R. A. (2003). Activated Cdc42 sequesters c-Cbl and prevents EGF receptor degradation. Cell 114, 715–725. doi: 10.1016/S0092-8674(03)00688-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamaguchi, Y., Kurokawa, M., Imai, Y., Izutsu, K., Asai, T., Ichikawa, M.,et al. (2004). AML1 is functionally regulated through p300-mediated acetylation on specific lysine residues. J. Biol. Chem. 279, 15630–15638. doi: 10.1074/jbc.M400355200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Z., Hikosaka, K., Sharkar, M. T., Tamakoshi, T., Chandra, A., Wang, B.,et al. (2010a). The mouse forkhead gene Foxp2 modulates expression of the lung genes. Life Sci. 87, 17–25. doi: 10.1016/j.lfs.2010.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Y., Kim, A. H., and Bonni, A. (2010b). The dynamic ubiquitin ligase duo: Cdh1-APC and Cdc20-APC regulate neuronal morphogenesis and connectivity. Curr. Opin. Neurobiol. 20, 92–99. doi: 10.1016/j.conb.2009.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yiin, J. J., Hu, B., Jarzynka, M. J., Feng, H., Liu, K. W., Wu, J. Y.,et al. (2009). Slit2 inhibits glioma cell invasion in the brain by suppression of Cdc42 activity. Neuro. Oncol. 11, 779–789. doi: 10.1215/15228517-2009-017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, G., Zerucha, T., Ekker, M., and Rubenstein, J. L. (2001). Evidence that GRIP, a PDZ-domain protein which is expressed in the embryonic forebrain, co-activates transcription with DLX homeodomain proteins. Brain Res. Dev. Brain Res. 130, 217–230. doi: 10.1016/S0165-3806(01)00239-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, F., Lin, M., Abidi, P., Thiel, G., and Liu, J. (2003). Specific interaction of Egr1 and c/EBPβ leads to the transcriptional activation of the human low density lipoprotein receptor gene. J. Biol. Chem. 278, 44246–44254. doi: 10.1074/jbc.M305564200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y. P., Liu, Y. Z., Guo, Y., Liu, X. G., Xu, X. H., Guo, Y. F.,et al. (2011). Pathway-based association analyses identified TRAIL pathway for osteoporotic fractures. PLoS ONE 6:e21835. doi: 10.1371/journal.pone.0021835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, Y., Song, T., Wang, W., Wang, J., He, J., Wu, N.,et al. (2012). P38 and ERK1/2 MAPKs act in opposition to regulate BMP9-induced osteogenic differentiation of mesenchymal progenitor cells. PLoS ONE 7:e43383. doi: 10.1371/journal.pone.0043383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, B., Zhong, Q., Minoo, P., Li, C., Ann, D. K., Frenkel, B.,et al. (2008). Foxp2 inhibits Nkx2.1-mediated transcription of SP-C via interactions with the Nkx2.1 homeodomain. Am. J. Respir. Cell Mol. Biol. 38, 750–758. doi: 10.1165/rcmb.2007-0350OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: globularity, language-ready brain, thalamus, cortex, basal ganglia, cerebellum, biolinguistics, vocal learning
Citation: Boeckx C and Benítez-Burraco A (2014) Globularity and language-readiness: generating new predictions by expanding the set of genes of interest. Front. Psychol. 5:1324. doi: 10.3389/fpsyg.2014.01324
Received: 24 July 2014; Accepted: 31 October 2014;
Published online: 25 November 2014.
Edited by:
Simon E. Fisher, Max Planck Institute for Psycholinguistics, NetherlandsReviewed by:
Genevieve Konopka, University of Texas Southwestern Medical Center, USASarah Anne Graham, Max Planck Institute for Psycholinguistics, Netherlands
Catherine Ann French, Champalimaud Centre for the Unknown, Portugal
Copyright © 2014 Boeckx and Benítez-Burraco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cedric Boeckx, Department of Linguistics, Universitat de Barcelona, Gran Via de les Corts Catalanes, 585, 08007 Barcelona, Spain e-mail: cedric.boeckx@ub.edu