 Sylvie Tordjman1,2*
Sylvie Tordjman1,2* Eszter Somogyi1
Eszter Somogyi1 Nathalie Coulon1Solenn Kermarrec1,2
Nathalie Coulon1Solenn Kermarrec1,2 David Cohen3Guillaume Bronsard4Olivier Bonnot1Catherine Weismann-Arcache5
David Cohen3Guillaume Bronsard4Olivier Bonnot1Catherine Weismann-Arcache5 Michel Botbol1,6Bertrand Lauth7Vincent Ginchat3Pierre Roubertoux8
Michel Botbol1,6Bertrand Lauth7Vincent Ginchat3Pierre Roubertoux8 Marianne Barburoth1Viviane Kovess9
Marianne Barburoth1Viviane Kovess9 Marie-Maude Geoffray10
Marie-Maude Geoffray10 Jean Xavier3
Jean Xavier3
- 1Laboratoire Psychologie de la Perception, Université Paris Descartes, CNRS UMR 8158, Paris, France
- 2Pôle Hospitalo-Universitaire de Psychiatrie de l’Enfant et de l’Adolescent, Université de Rennes 1, Centre Hospitalier Guillaume Régnier, Rennes, France
- 3Department of Child and Adolescent Psychiatry, AP-HP, GH Pitié-Salpétrière, CNRS FRE 2987, University Pierre and Marie Curie, Paris, France
- 4Laboratoire de Santé Publique (EA3279), School of Medicine of La Timone, Marseille, France
- 5Laboratoire Psychologie et Neurosciences de la Cognition et de l’Affectivité, Université de Rouen, Mont Saint Aignan, France
- 6Service Hospitalo-Universitaire de Psychiatrie de l’Enfant et de l’Adolescent, Université de Bretagne Occidentale, CHU de Brest, Brest, France
- 7Department of Child and Adolescent Psychiatry, Landspitali University Hospital, University of Iceland, Reykjavik, Iceland
- 8Laboratoire de Génétique Médicale, Génomique Fonctionnelle, INSERM U 910, Université d’Aix-Marseille 2, Marseille, France
- 9Department of Epidemiology and Biostatistics, EHESP School for Public Health, EA 4057 University Paris Descartes, Paris, France
- 10Service Universitaire de Psychiatrie de l’Enfant et de l’Adolescent Hospitalier Le Vinatier, Bron, France
Several studies support currently the hypothesis that autism etiology is based on a polygenic and epistatic model. However, despite advances in epidemiological, molecular and clinical genetics, the genetic risk factors remain difficult to identify, with the exception of a few chromosomal disorders and several single gene disorders associated with an increased risk for autism. Furthermore, several studies suggest a role of environmental factors in autism spectrum disorders (ASD). First, arguments for a genetic contribution to autism, based on updated family and twin studies, are examined. Second, a review of possible prenatal, perinatal, and postnatal environmental risk factors for ASD are presented. Then, the hypotheses are discussed concerning the underlying mechanisms related to a role of environmental factors in the development of ASD in association with genetic factors. In particular, epigenetics as a candidate biological mechanism for gene × environment interactions is considered and the possible role of epigenetic mechanisms reported in genetic disorders associated with ASD is discussed. Furthermore, the example of in utero exposure to valproate provides a good illustration of epigenetic mechanisms involved in ASD and innovative therapeutic strategies. Epigenetic remodeling by environmental factors opens new perspectives for a better understanding, prevention, and early therapeutic intervention of ASD.
Introduction
Biological research in autism has attempted to improve our understanding of the neurobiological mechanisms possibly involved in autistic disorder (AD); studies have been conducted in domains as diverse as genetics, neurochemistry, neuropharmacology, neuroendocrinology, neuroanatomy, brain imaging, and neuroimmunology. For example, structural and functional imaging and neuropathological techniques applied to autism spectrum disorders (ASD) brains have revealed developmental macroscopic and microscopic abnormalities suggesting neuroinflammation in frontal cortex and cerebellar regions, including cytokine production and activation of microglia and astrocytes (1). Studies stress increasingly that AD cannot be summed up or explained by a single biological factor, but rather by a multifactorial etiology. A multidisciplinary biological approach allows us to compare different fields and methodological processes, thus to understand better the neurobiology of autism. However, in spite of the numerous studies conducted on AD during the last decades, it appears that no etiological model, no biological or behavioral marker, and no specific psychopathological process have been clearly identified (negative or contradictory results, associations not replicated). Although the genetic factors and the mode of transmission of AD are not yet fully determined, the underlying genetic architecture, such as known chromosomal rearrangements or single gene disorders, are being identified through, for instance, more and more routine chromosome microarray analysis (CMA) (2). Thus, more than 200 autism susceptibility genes have been identified to date, and complex patterns of inheritance, such as oligogenic heterozygosity, appear to contribute to the etiopathogenesis of autism. Similarly, cytogenetic abnormalities have been reported for almost every chromosome [for a review, see Ref. (3–7) and http://projects.tcag.ca/autism/]. Because of the lack of conclusive results and concensus, it is probably more appropriate to use the concept of syndrome to characterize autism. Autism is defined in the ICD-10 and DSM-5 as a delay or abnormal functioning with onset prior 3 years in social communication, and manifestation of restricted, repetitive and stereotyped patterns of behavior, interests, and activities.
Several authors support the hypothesis that the mechanism underlying autism etiology is most likely polygenic and potentially epistatic, and that environmental factors may interact with genetic factors to increase risk (8, 9). Arguments for an environmental contribution to AD come from the growing number of studies on environmental factors in ASD, but also from the current lack of conclusive results on an etiopathological genetic model of autism. It seems important to reframe autism in a multifactorial context. Autism could be considered as a psychopathological organization that would result from the effects of diverse biological factors and/or psychological factors, including genetic factors, environmental factors, and gene × environment interactions. The environmental factors could be post- or prenatal (psychosocial environment but also cytoplasmic and uterine environment, with placental exchanges and hormonal effects).
First, we will examine arguments for a genetic contribution to AD based on updated family and twin studies. Then, after reviewing the possible prenatal, perinatal, and postnatal environmental risk factors for AD, we will discuss the hypotheses concerning the underlying mechanisms related to their role in the development of AD in association with genetic factors. In particular, the possible role of epigenetic mechanisms reported in genetic disorders associated with autism will be considered.
Genetic Architecture of Autism Risk
Several recent literature reviews underline the important role of genetics in the etiology of AD (10–13). Much of the data come from family and twin studies. The concordance rate among monozygotic (MZ) twins ranges on average from 60 to 90%, and from 0 to 20% among dizygotic (DZ) twins. These rates depend on the diagnosis and on the subtype of autism considered. In addition, they are not sufficient to explain by themselves the autistic syndrome. Autism could be considered as a multifactorial hereditary disorder, in other words a disorder that depends on numerous genes (polygenic heredity) and environmental factors. Although genetic studies have identified hundreds of genes associated with ASD, the exact number remains unknown (10, 14). The wide phenotypic variability of autism may reflect the interaction between genes and environment but also the interaction of multiple genes within an individual’s genome and the existence of distinct genes and gene combinations among those affected.
Family Studies
The prevalence of autism in the general population has been estimated in various ways that depend mainly on sampling methods and diagnostic criteria, as noted already many years ago in the report by Agence Nationale pour le Développement de l’Evaluation Médicale (ANDEM) (15). Thus, the prevalence of autism varies according to the diagnostic criteria of Kanner, DSM-III, and DSM-IV classifications: from 1 to 5/10,000 according to Kanner or DSM-III criteria up to 20/10,000 according to DSM-IV-TR criteria (16). Prevalence of parent-reported diagnosis of ASD among 3- to 17-year-old children in the USA reaches the very high rate of 1/91 (17). Similar results are expected using the DSM-5 criteria given that the diagnosis of autism is only based on ASD in this classification (according to DSM-5 criteria, ASD includes two main domains of autistic behavioral impairments: social communication impairments and stereotyped behaviors or interests). Broadening of the diagnostic criteria for autism and better recognition of the autism behavioral phenotype may explain this rising prevalence, but a true increase in incidence cannot be ruled out [Autism and Developmental Disabilities Monitoring Network, 2008; (18)]. However, Fisch (19, 20) showed clearly that this rising prevalence is related to the use of different diagnostic criteria. He concluded by saying “There is no autism epidemic but a research epidemic on autism.” Still, the reasons of such an increased interest in autism remain to be understood (for example, a better organization of association of parents, more funding contributing to an increase in the number of researchers and studies in autism, a growing interest for social communication impairments in a society promoting social communication networks).
It is noteworthy that there is a male prevalence in autism [about three to four times higher in males than in females (21)], which might also fit with greater social communication difficulties observed in males compared to females with typical development. Studies on the prevalence of autism in families with autistic children show a higher rate than in the general population. The concordance rate for siblings of individuals with autism of unknown cause ranges from 5 to 10% and approaches 35% in families with two or more affected children (22–25). Taken together, the rates of AD in siblings of children with autism are on average 50–150 times higher than the rate of autism in the general population, which suggests that autism has a family feature (family meaning here environmental as much as genetic). Carlier and Roubertoux (26) emphasized that in evaluating the risk, the degree of genetic proximity and the degree of environmental similarity were correlated. Only two studies have attempted to assess the presence of parental pathology at the same time as sibling pathology in the families of autistic individuals. The first was the Utah epidemiological survey (1989) and the second was Gillberg et al. (27) study. Ritvo et al. (25) reported that of the 214 parents seen in the Utah survey, 7 were autistic, the majority being fathers. In the epidemiologically based, case–control study by Gillberg et al. (27) four fathers of the 33 autistic probands were considered to have Asperger’s syndrome. This gives an overall prevalence of autism in parents between the two studies of 2.3%. As underscored by Todd and Hudziak (28) the presence of affected father–son pairs is not compatible with simple X-linked transmission.
Twin Studies
In the field of genetic research on AD, which compares MZ with DZ twins, three interesting results can be presented (see Table 1 for a summary of the results from the updated studies):
• In each study, the concordance rate for MZ twins is higher than for DZ twins.
• The concordance rate of AD in MZ twins is incomplete, suggesting a contribution of environmental factors. Hallmayer et al. (9) underline in their twin studies the involvement of both genetic and environmental factors in the development of ASD.
• In the Folstein and Rutter (29), Bailey et al. (30), and Hallmayer et al. (9) studies, concordance rates vary according to the diagnosis: the concordance rates are higher for the broader autism phenotype than for AD (full diagnostic criteria).
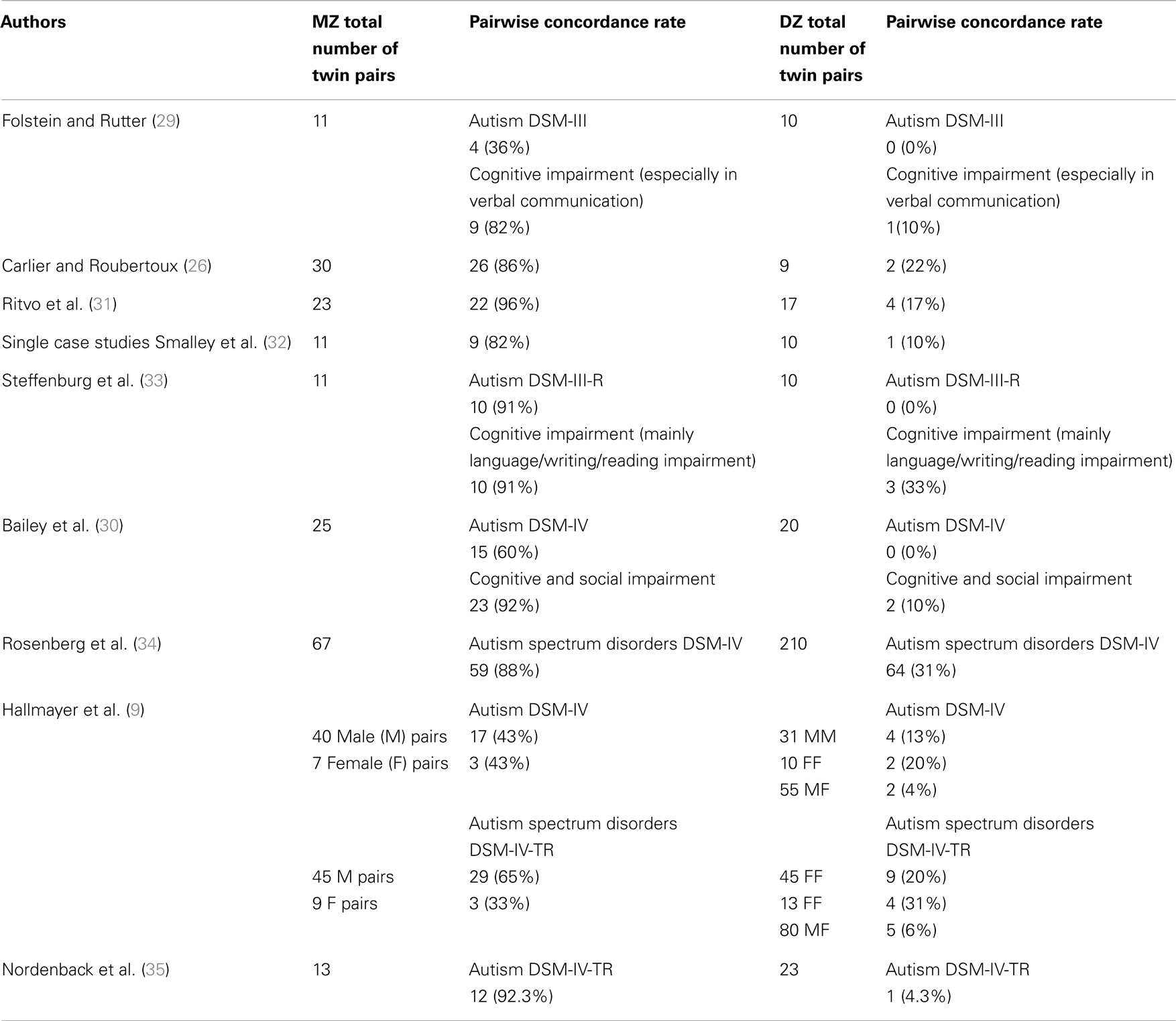
Table 1. Pairwise concordance rates for autism in monozygotic twins (MZ) and dizygotic twins (DZ).
These results point to a possible etiological heterogeneity of autism. The etiology could be different according to the subtype of autism considered, a subtype that could be clinical as much as biological. This may help us to better understand why none of the genetics inheritance models proposed for autism, including the polygenic model, can fully explain the autism phenotype in the family and twin studies presented above. One of the current issues in the field of genetic research on AD is to work on different subtypes in order to identify the relevant genes. There are three main approaches to identifying genetic hotspots or chromosomal regions likely to contain relevant genes: (1) cytogenetic studies, (2) whole genome screens, and (3) evaluation of a priori selected candidate genes known to affect brain development or possibly involved in the pathogenesis of autism.
Genome-wide association studies (GWAS) examine associations between disease and genetic variants such as single-nucleotide polymorphisms (SNPs) or copy number variations (CNVs). Genetic variants can be either inherited or caused (which is often the case) by de novo mutations. CNVs and SNPs have both been reported to play a major role in autism incidence (36–41). Common SNPs acting additively have been reported as a major source of risk for ASD (42) with heritability exceeding 60% for ASD individuals from multiplex families and approximately 40% for simplex families. CNVs, including insertions, deletions, and repeated sequences, can be highly disruptive to developmentally regulated genes. Several CNV studies (36, 43, 44) identified also structural changes in DNA, which contribute to the risk for ASD. Recent findings suggest the possibility that not only single, but also aggregate molecular genetic risk factors, linked in particular to alterations in calcium-channel signaling, are shared between autism and four other psychiatric disorders (schizophrenia, attention-deficit hyperactivity disorder, bipolar disorder, and major depressive disorder) (45, 46). However, the mechanisms underlying the role of these mutations in the development of ASD phenotypes remain to be ascertained. More generally, children with neurodevelopmental problems, including ASD, are often affected in more than one area of functioning of mental health to the extent that hierarchies of mutually excluding categorical diagnoses have to be considered as conflicting with scientific evidence (47). It suggests, according to Anckarsäter (48), that genetic susceptibilities behind mental health problems have to be sought both in relation to specific problem types and to general dysfunction, using multivariate analyses with measures of all types of mental disorders.
Concerning candidate genes, several of them have been studied at chromosome regions 7q22–q33 or 15q11–q13, and variant alleles of the serotonin transporter gene at 17q11–q12 are more frequent in individuals with autism [see Ref. (12), for a review]. Linkage data from genome screens and animal models suggest also a possible role of the oxytocin receptor gene at 3p25–p26 (49). Interestingly, the majority of the genes reported to be associated with autism is involved in various physiological processes, such as chromatin remodeling, metabolism, translation, and synaptogenesis. These genes may converge into pathways affecting distinct neuronal functions such as synaptic homeostasis. Such a genetic basis of synaptic and neuronal signaling dysfunction in ASD has been confirmed by recent findings (50) demonstrating differences in transcriptome organization between autistic and normal brain through gene co-expression network analysis.
Finally, it should be highlighted that the polygenic model does not exclude a role of environment. It is noteworthy that heritability (h2) is defined as h2 = GV/(GV + EV) where GV is the cumulative genetic variance and EV, the environmental variance (51). The possible prenatal, perinatal, and postnatal environmental risk factors for ASD are presented below.
Prenatal, Perinatal, and Postnatal Environmental Risk Factors for ASD
The prenatal factors associated with autism risk in the meta-analysis provided by Gardener et al. (52) were advanced paternal and maternal age at birth, gestational diabetes, gestational bleeding, multiple birth, being first born compared to being third or after, and maternal birth abroad. In fact, several recent studies suggest that parental immigration, especially maternal immigration but also paternal immigration, is a risk factor for ASD (53–59). This association between migration and autism is more particularly observed in male children of immigrant parents living in urban areas compared to rural areas (60). In addition, concerning the prenatal risk factors for AD, a rare consistent association with AD is in utero exposure to two known teratogenic medications, thalidomide, and valproate (valproate is a broad-spectrum anticonvulsant drug used in seizures, bipolar disorder, or migraine headache), or the abortifactant misoprostol (7, 61–63). Thus, children exposed to valproate in utero were seven times more likely to develop autism than those not exposed to antiepileptic drugs (61, 62). A large population-based cohort study of all children born alive in Denmark from 1996 to 2006 was conducted on 655,615 children, including 508 prenatally exposed to valproate and 5437 identified with autism spectrum disorder (2067 with AD). Children of women who used a high valproate dose (>750 mg/day) or a low valproate dose (<750 mg/day) early (first trimester) or later in pregnancy had significantly a higher risk of ASD and AD compared with children of women who did not use valproate (even after adjusting for maternal epilepsy and parental psychiatric history, or restricting the analysis to children without congenital malformations), whereas this increased risk was not observed for other antiepileptic drugs used as monotherapy. The mechanisms of action of valproate will be developed later in the next section. Furthermore, prenatal exposure to folic acid (known to decrease the risk of neural tube defects) has also been associated with the risk of autism (64). As folate and folic acid are essential for basic cellular processes (including DNA replication as well as DNA, RNA, and protein methylation), it could not be excluded that, depending on timing and dose, such nutritional supplements might also have adverse effects. Also, the time period at which folic acid was added to the diet of women of childbearing age coincides with the apparent onset of a continuous increase in the prevalence of autism. However, a recent well-controlled epidemiological study (65) disconfirms this claim and reports, as underlined by Berry et al. (66) or Vahabzadeh and McDougle (67), a lower incidence of AD in children whose mothers received prenatal folic acid supplementation around the time of conception (64/61 042 or 0.10%) than in children whose mothers did not take folic acid (50/24 134 or 0.21%). Similarly, in children from countries without folic acid supplementation, autism has been linked to two polymorphisms of the methylenetetrahydrofolate reductase gene (MTHFR), which is essential for DNA biosynthesis and the epigenetic process of DNA methylation (68). Although such findings do not establish a causal relation between folic acid use and a lower incidence of AD, they do provide an impetus for further study. Finally, some authors (69, 70) have suggested a low but possible risk of neurological problems and imprinting disorders (such as Beckwith–Wiedemann syndrome and Angelman syndrome which are genetic disorders associated with autism) in children conceived by in vitro fertilization (IVF). However, earlier investigations on possible links between assisted reproductive technologies (ART) and autism have shown inconsistent results (71, 72). More recent epidemiological studies involving larger populations show that IVF is not associated with autism (73), but rather with a small increased risk of intellectual disability (74). According to Sandin et al. (74), the risk for ASD was significantly higher following intra-cytoplasmic spermatozoid injection (ICSI) using surgically extracted sperm and fresh embryos compared to IVF without ICSI with fresh embryo transfer.
During the perinatal period, Guinchat et al. (75) pointed to three factors associated positively with the development of AD: prematurity (the risk for autism increased with the severity of preterm birth), abnormal presentation in general and breech presentation in particular, and planned cesarean section. However, the role of cesarean section as an independent risk factor for AD needs to be clarified given that breech presentation is a common cause of first cesarean delivery. Thus, Bilder et al. (76) reported that after correction for breech presentation, the observed association between cesarean section and AD lost statistical significance. Other studies grouped cesarean section in one specific variable [for a review, see Ref. (75, 77, 78)], but cesarean section emerged as an independent risk factor only in the Hultman et al. (79) study. During the neonatal period, conditions potentially related to hypoxia, such as umbilical-cord complications, low 5-min Apgar score, being small for gestational age, low birth weight (especially when <1500 g), fetal distress, or meconium aspiration, as well as birth injury or trauma, summer birth, feeding difficulties, neonatal anemia, ABO or Rh incompatibility, and hyperbilirubinemia were significantly (P < 0.05) associated with autism [for a review, see Ref. (8, 75)]. Thus, Maimburg et al. (80, 81) and Buchmayer et al. (82) published large population-based studies associating hyperbilirubinemia with independent risks for AD that might be related to the potential toxicity of hyperbilirubinemia on basal ganglia and cerebellum. It is noteworthy that prematurity might be a variable masking the effect of hyperbilirubinemia. Interestingly, summer season [summer birth was significantly associated with an elevated risk of autism, RR: 1.14, P = 0.02; (8)] corresponds to the longest days of the year. It strengthens the hypothesis developed by several authors [for a review, see Ref. (83–85)] of a possible role of a deficit in melatonin in the development of ASD (the production of melatonin is powerfully suppressed by light acting through the retino-hypothalamic tract). Finally, the perinatal factors with the strongest evidence against a role in autism risk included anesthesia use during delivery, assisted vaginal delivery, post-term birth, high birth weight, or head circumference (8).
Effects of exposure to air pollution during pregnancy in the first year of life deserve particular attention, especially because they might be mediated by epigenetic mechanisms as in valproate exposure. Epidemiological studies (86, 87) reported associations between autism and air pollution at the birth and early life residences. Thus, residential proximity to freeways in California within 309 m during the third trimester of pregnancy and at birth was found associated with a risk of ASD about twofold higher (88). Studies in animal models (rodents) and humans described developmental effects of air pollution following prenatal and early life exposure, such as altered neuronal differentiation, impaired cognitive functions, and white matter abnormalities (89–91). Given the male prevalence observed in autism, it is noteworthy that adult male mice but not females, showed increased depression-like responses and low resilience to stress in the tail suspension test following prenatal exposure to urban freeway nanoparticulate matter. In this line, Volk et al. (92) found that exposure during pregnancy and the first year of life to traffic-related air pollution was associated with autism (DSM-IV and ICD-10 criteria based on the ADI-R and ADOS scales). Children residing in homes with the highest levels of modeled air pollution (>31.8 ppb) were three times as likely to have autism compared to children residing in homes with the lowest levels of exposure (<9.7 ppb). An increasing probability of autism was seen with increasing air pollution (nitrogen dioxide and particulate matter less than 2.5 and 10 μm in diameter: PM2.5 and PM10) with a plateau reached at a threshold above 25–30 ppb. Associations were reported for each trimester of pregnancy but the smallest magnitude of the effects was observed for the first trimester. Neurodevelopmental effects of prenatal and/or early life exposure to polycyclic aromatic hydrocarbons may be mediated by epigenetic effects (93). However, the results could also be affected by unmeasured confounding factors associated with both autism and exposure to traffic-related air pollution.
Furthermore, maternal depression (prenatal but also postnatal depression given that it is in fact very difficult to dissociate depression during pregnancy from perinatal/postnatal depression) raises an interesting issue with regard to risk factors for ASD. Interestingly, common genetic factors contributing to depression and autism have been reported (45, 46). A study (94) conducted on 4429 cases of ASD (1828 with and 2601 without intellectual disability; antidepressant use during pregnancy for 1679 cases) showed that a history of depression during pregnancy but not paternal depression was associated with an approximately 60% increase in risk of ASD in offspring (raw odds ratio 1.61, 95% confidence interval 1.17–2.23, P = 0.004), particularly without intellectual disability (adjusted odds ratio 1.86, 95% confidence interval 1.25–2.77, P = 0.002), and more precisely for mothers reporting antidepressant use during pregnancy but independently of the type of antidepressant (adjusted odds ratio when depression with antidepressant use 3.34, 1.50–7.47, P = 0.003; adjusted odds ratio in case of depression without antidepressant use 1.06, 0.68–1.66, not significant). These results are in line with the Croen et al. (95) study reporting association between use of selective reuptake inhibitor (SSRI) antidepressants during pregnancy and ASD in offspring. However, antidepressant use during pregnancy explained only 0.6% of the 4429 cases of ASD in the Rai et al. (94) study. The authors conclude that assuming causality, antidepressant use during pregnancy is unlikely to have contributed significantly toward the observed prevalence of ASD as it explained less than 1% of the cases. In summary, the Croen et al. (95) study suggests an effect of antidepressant use during pregnancy but, as supported by the Rai et al. (94) study, a risk for ASD in severe maternal depression during pregnancy cannot be excluded or reduced to the effect of antidepressant use. Further studies are requested to test fully these hypotheses through a scientific approach and methodology. It is noteworthy that to avoid the possibility of reverse causality reported by Daniels et al. (96), only diagnoses of maternal depression recorded before birth were considered by Rai et al. (94). Also, two meta-analyses (52, 97) underlined the lack of studies with psychiatric diagnosis of parents before the birth of children with autism, suggesting that depression during pregnancy may be underestimated. Finally, given the continuity between prenatal and postnatal depression, an effect of maternal postnatal depression on early infant development cannot be ruled out.
It is difficult to establish if these prenatal, perinatal, and neonatal factors are independent environmental risk factors for ASD involved in cause–effect relationship or are associated with ASD but result themselves from other factors that still need to be identified. Thus, maternal immigration is probably not an independent environmental risk factor for ASD but might be related to other factors such as prenatal and/or postnatal maternal depression. Similarly, several potential risk factors for ASD that occur during the neonatal period, such as low Apgar scores or birth weight, shortened gestational age or even breech presentation, may be the consequence of other risk factors during the prenatal and perinatal periods, such as prematurity. Furthermore, the possible effects mentioned previously of prematurity or severe maternal depression on the risk for ASD could be related, for example, to a dysfunction in brain development (e.g., prenatal depression is associated with a modified biological profile involving, in particular, the serotoninergic system and the hypothalamo-pituitary adrenal axis) and/or a deficit in very early social interaction and sensory stimulation. Finally, it is noteworthy that in Gardener et al. (8) meta-analysis, overall, preterm birth was not associated with the risk of autism, although there was significant heterogeneity across studies.
Concerning psychosocial factors occurring during the postnatal period, such as a severe deficit in very early social interaction, AD has been reported in children institutionally exposed to profound early social deprivation (98–100). However, several issues have to be raised. First, if the pattern of autistic-like behavior found by Rutter et al. (99) at 4 years of age in the sample of children from Romanian institutions was indistinguishable from that seen in typical children with autism, by age 6 years this “quasi-autistic” pattern had diminished and a number of atypical features were observed: the children displayed more flexibility in communication and a substantial social approach up to, for some of them, an indiscriminately overfriendliness that is usually associated with disinhibited attachment. Furthermore, the follow-up of 28 children with “quasi-autism” from Romanian institutions, using the autism diagnostic interview-revised [ADI-R; (101)] showed that by age 11–12 years, just over 1 out of 10 children who experienced profound institutional deprivation still displayed a quasi-autistic pattern but a quarter of the children lost their autistic-like features, and dishinibited attachment with poor peer relationships was also observed in over half of these children (100). It is noteworthy that most of the children with quasi-autism had an IQ in the normal range. Finally, methodological issues can be raised concerning the Rutter et al. follow-up that did not study the relationship between the presence of autistic-like features and the duration of the profound institutional deprivation or the age of the children when they experienced this social deprivation.
Concerning neurophysiological factors occurring during the postnatal period, and more specifically severe sensory deprivation, several studies report a high risk for AD in congenitally severely visually impaired children. Thus, AD was observed in 10 out of 24 (42%) congenitally blind children (102) and autism was also reported in 30 out of 157 (19%) children with varying ophthalmological problems (103). In addition, only 31% of Swedish children with AD (n = 45) were found to have normal visual acuity (104). Similarly, several studies have reported abnormally high rates of AD in hearing impaired individuals [from 1.7 up to 7%; (105, 106)]. Inversely, hearing problems, including hearing loss, were described in 10% of Swedish children and adolescents with autism (n = 199) (107). Furthermore, very high rates of ASD have been observed in congenital conditions (up to 68% in CHARGE syndrome, 45% in Möbius sequence, 42% in oculo-auriculo-vertebral spectrum) involving multiple sensory deficits such as impaired vision associated with reduced hearing. These study groups and case reports suggest that severe multimodal sensory deprivation is a good candidate risk factor for AD. Genetic disorders such as CHARGE syndrome support the hypothesis that genetic factors could lead to multiple simultaneous sensory deficit occurring at a critical period of development that, in turn, would play a role in the pathogenesis of AD. Also, genetic disorders such as CHARGE syndrome support the hypothesis of common genetic risk factors for ASD given that CHARGE syndrome is due to mutations in CHD7, which is a homolog of CHD8, one of the most recurrently affected genes in autism cohorts (108). Taken together, these studies suggest that impairment in cross-modal sensory perception (impaired vision associated with reduced hearing) contributes more to the development of AD than impaired vision alone, which appears to be a more important risk factor for AD than reduced hearing alone. This is in line with a model, which postulates that AD would be related to impairment in the developmental sequence involving cross-modal sensory perception, body-image construction, sense of body self, self/non-self differentiation and in turn development of social communication (109). However, it is noteworthy that CHARGE syndrome, Möbius sequence, and oculo-auriculo-vertebral spectrum are associated with extended developmental defects that might be involved in the risk for ASD. Thus, CHARGE syndrome includes multiple malformations (see Table 2), Möbius sequence is associated with brainstem hypoplasia (110), and cerebral abnormalities are observed in oculo-auriculo-vertebral spectrum (111). Finally, another limitation is related to the assessment of ASD in congenital conditions due to the difficulty to use current autism diagnostic instruments in individuals with intellectual disability, cranial nerve palsies (most commonly affecting the facial nerve), and sensory deficits (patients often being severely visually impaired or blind, hearing impaired or deaf).
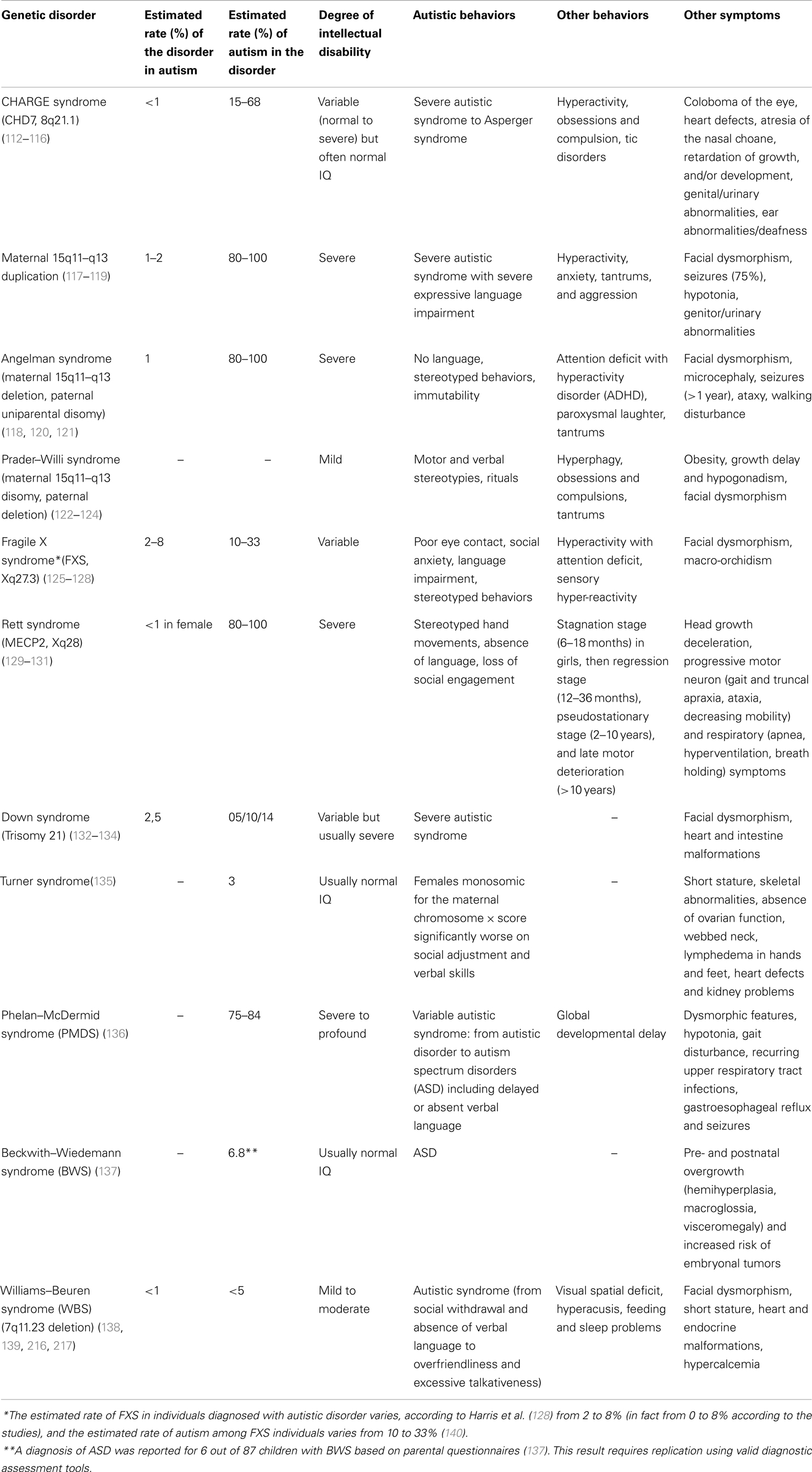
Table 2. Genetic disorders with epigenetic mechanisms associated with autistic syndrome.
In conclusion of this descriptive part on the prenatal, perinatal, and postnatal risk factors, it appears that no specific factor has been identified and no individual factor has been consistently validated as an independent environmental risk factor for ASD. This suggests that future research on environmental risk factors for AD, rather than focusing on a single factor, should study a combination of factors through an integrated approach including gene × environment interactions and conduct multivariate analyses.
Hypotheses on the Role of Environmental Factors in Association with Genetic Factors
First, “the epiphenomenon hypothesis” argues for a primary role of the genetic susceptibility to autism and proposes that genetic factors increase the risk for both autism and the associated prenatal, perinatal, and postnatal complications. This hypothesis is supported by Glasson et al. (141).
A second hypothesis, “the heterogeneity hypothesis,” proposes that the contribution of genetic and/or environmental factors varies according to the cases. AD might be due mainly to genetic factors in some cases [for example, neonatal congenital malformations are significantly associated with an increased risk for autism; (8)] or mainly to environmental factors in other cases (for example, in utero exposure to valproate) with possible cumulative effects mediated by different environmental factors and/or genetic factors (for example, see the CHARGE syndrome in Table 2). Some authors (142–144) suggest that the greater the genetic contribution, the more dysmorphic signs, and cognitive impairment are present. Thus, children with AD showing a higher number of minor physical anomalies have lower IQ and are more at risk for genetic variations (142). The finding that unbalanced chromosome abnormalities are found predominantly in children with autism who are dysmorphic (142, 143, 145), strengthens this hypothesis. It is noteworthy that large chromosomal abnormalities are more often found in children with dysmorphic features, whereas smaller CNVs involving the same region and de novo single-nucleotide variants of major effect are found often in individuals without dysmorphic features. However, there is no evidence that the role of environment is more important in the case of absence of de novo event, as common variants were shown to contribute substantially to autism risk (42). The debate does not focus anymore on a possible contribution of genetic factors to the risk for autism but on the magnitude of this contribution. Contemporary research efforts are moving away from the search for a condition-specific genetic factor to embrace a more cumulative model based on elevated risk as a function of smaller gene point mutations. Interestingly, the hypothesis, mentioned previously, related to genes altering the synaptic homeostasis leads to the perspective of possible autism phenotype reversals (146) and raises issues concerning a possible role of environmental factors associated with genetic factors. Indeed, it has been shown in mouse models of autism that certain neuronal defects can be reversed in the mature mouse brain, either by restoring the gene function, decreasing mRNA translation, or modulating the balance between excitation and inhibition. The early signs of autism are in fact still largely unknown, which hints that during this premorbid period, there might be a discrete window for reversing the pathological process. This window of development could correspond to early critical periods when brain development is particularly sensitive to experience and when brain plasticity, involving sensory systems but also motor functions and cognition, is possible (147). After these critical periods, the level of plasticity is reduced due to the development of myelin and perineuronal networks that drastically prune neuronal outgrowth in the mature brain and lead to functional modifications or fine-tuning in the excitation–inhibition balance (148).
Finally, another hypothesis is to consider the genetic pre-disposition to AD as resulting from different chromosomal or gene variations and to propose that environmental factors associated with these genetic factors would modify the phenotypic expression of AD and lead to a similar clinical phenotype. The different genetic disorders associated with autistic syndrome might share the same environmental factors that could contribute to the expression of behavioral autistic impairments. For example, oxidative stress might be a candidate mechanism linking genetic and environmental factors in genetic disorders associated with autism (149–151). Thus, regarding the possible molecular mechanisms that might be shared in autism and fragile X syndrome (FXS), it has been proposed that increased oxidative stress in the brain might be a common factor. The loss of FMR1 expression in FXS leads to the absence of fragile X mental retardation protein (FMRP), which is primarily involved in binding mRNAs. The absence of FMRP, as shown in FMR1-knockout mice, leads to increased oxidative stress (152–154). It can be hypothesized that any chromosomal or gene variations leading to increased oxidative stress would contribute to the expression of a behavioral autistic phenotype. Inversely, heterogeneity of environmental factors may lead to clinical subgroups with different cognitive–behavioral phenotypes. Lacaria et al. (155) developed a mouse model for Potocki–Lupski syndrome (PTLS) (PTLS is a genetic disorder associated with autism and caused by a 3.7-Mb duplication in 17p11.2), and rearing this animal model in an enriched environment (enriched cages were larger and contained enrichment items such as increased number of mice per cage to enhance social behavior) mitigated the autistic-like abnormal behavioral phenotypes, suggesting a role for gene–environment interactions in the determination of CNV-mediated autism severity. The authors suggest a potential link between the behavioral benefits of environmental enrichment and the underlying changes observed in their study for the serotoninergic and dopaminergic pathways. This is particularly interesting with regard to the abnormalities of the serotoninergic and dopaminergic systems reported in autism [for a review, see Nakamura et al. (156)]. Furthermore, epigenetics is a good illustration of the effects of environmental factors on gene expression. Epigenetics can be viewed as a candidate biological mechanism that is a part of the more general hypothesis of “gene × environment interactions.” Environmental factors/events can occur during early life and be involved in the regulation of neural development associated with synaptic plasticity even at later developmental stages (157). This hypothesis states that environmental factors interact with genetic factors that would not or less influence development otherwise. Gene × environment studies are needed to test this hypothesis and raise the issue of power. The issue of power refers to the possibility to detect an effect based on sample size. Indeed, interaction effects, such as gene × environment ones, require large samples and high power in order to detect the effects as these effects are often small but perhaps truly present and worth integrating in models. Furthermore, interaction effects are known to be difficult to observe because measurement errors or random noise associated with the variables involved in the interaction cumulate, thus “hiding” interaction effects unless they are very strong. The use of large samples reduces the tendency for noise in the data to be an issue, as errors tend to cancel each other out in a large sample, thereby increasing the ability to see an interaction effect.
More precisely, epigenetics refers to functionally relevant modifications to the genome that influence gene expression without involving a change in nucleotide sequence. Epigenetic modifications include DNA methylation and various modifications (e.g., methylation, acetylation) of histone proteins that are complexed with DNA to form the chromatin. Epigenetic modifications of histone proteins are generally transient and reversible, whereas epigenetic modifications of DNA are usually more stable. It is noteworthy that environmental events involved in epigenetic mechanisms may be, as underlined by Bagot and Meaney (157), internal or external to the organism (e.g., changes in the availability of glucose, electrical impulses, or social interaction and maternal care). Thus, maternal care such as pup licking/grooming from the mother over the first week of postnatal life provides tactile stimulation for the neonate, which increases hippocampal glucocorticoid receptor gene expression through epigenetic modifications of DNA and decreases HPA responses to stress (158, 159). Inversely, manipulations imposed on the mother that decrease pup licking/grooming such as chronic stressors, are associated with decreased hippocampal glucocorticoid receptor expression, increased hypothalamic expression of CRF, and enhanced behavioral and HPA responses to stress (160, 161). According to Bagot and Meaney (157), these findings suggest that maternal care can stably affect gene expression that in turn mediates the expression of individual differences in behavioral and neuroendocrine responses to stress in adulthood. Futhermore, epigenetic modifications of DNA are stable and can also be transmitted across generations. Indeed, early-life postnatal adversity in animal models can persistently affect behavior across generations and DNA methylation in the germline. Thus, chronic and unpredictable early maternal separation from day 1 to 14 induces in mice depressive-like behaviors and DNA methylation changes in the separated male pups (this finding was not observed in female pups confirming previous findings that maternal separation and prenatal stress have a negative influence primarily in males [162]), but also in female offspring of males subjected to maternal separation, despite the fact that these males were reared normally (163). The authors (163) showed that chronic and unpredicatble maternal separation alters DNA methylation in the promoter of several genes in the germline of the separated males and these changes are transmitted across several generations through a complex and sex-specific mode (transmission occurs through males by epigenetic germline inheritance and affects the offspring in a sex-dependent manner).
Environmental events occurring during early development can activate cellular signaling pathways associated with synaptic plasticity even at later stages in development. The critical period could be during fetal development [as suggested by in utero exposure to valproate; (164)] but also during early-postnatal development [as suggested by studies in animal models concerning the first weeks of postnatal life (157)]. In animal studies, autistic behaviors have been observed following administration of valproate during prenatal life or during weaning (stereotyped behaviors, social interaction deficit, self-injurious behaviors, enhanced anxiety as well as impaired cognitive, motor, and attention development) (165–167). In addition, Rodier’s group exposed rat embryos to valproate at the period of neural tube closure and it led to a reduction of the vermal posterior lobe (168). This study, showing that early chemical exposure can provoke late developmental cerebellar anomalies is of interest but the face validity and construct or etiological validity of this model of autism are questionable considering that animal behaviors have not been studied and cerebellar vermian anomalies reported in individuals with autism are controversial. In fact, several authors have proposed exposure to valproate as a possible neurobehavioral model of autism (169–172). However, as underlined in our review on animal models relevant to autism and schizophrenia (173), the main interest of animal models is not to validate a specific categorical model of autism, but rather to study behavioral and neurobiological mechanisms possibly involved in ASD through a multidimensional approach. Concerning its mechanisms of action, valproate has known epigenetic effects. Phiel et al. (174), using cell lines, showed that valproate at therapeutic concentrations inhibits histone deacetylases; this inhibition correlates with increased expression of multiple genes. Milutinovic et al. (175) reported that valproate affects not only histone modification and gene expression, but also DNA de-methylation in human embryonic kidney cell lines, linking epigenetic modifications of both histones and DNA to gene expression.
Oh et al. (176) studied effects of other environmental factors on DNA methylation, using a mouse model of maternal adversity – based on a deficit in the maternal 5-HT1A receptor [reduced binding of 5-HT1A has been found in peri/postpartum depression, a condition that can represent early life adversity for the offspring (177)], which causes innate anxiety, increased stress reactivity, and impaired vocal communication in the offspring – and genome-wide DNA methylation analyses. One rationale for this study was that adverse gestational and postpartum maternal environment might be a contributing factor in the development of autism based on autistic behaviors following prenatal exposure to valproate (as mentioned above), mental disorders associated with parental adversities during early-postnatal life (178), and also effects of deficit in maternal care on increased stress responses in the offspring (157). The authors found that adverse maternal environment induced DNA methylation changes in the offspring and the affected genes encode proteins involved in synapse formation and function (reduced expression of cell adhesion and neurotransmitter receptor genes was observed). They conclude that the differential methylation expression of a large number of pre- and post-synaptic cell adhesion molecules and neurotransmitter receptor genes all involved in neuronal excitability as well as anxiety, suggests a wide-ranging and permanent epigenetic effect of the adverse maternal environment on synaptic plasticity and neuronal excitability. It is noteworthy that mutations in some of these genes such as genes encoding cadherins and neurexin–neuroligins in human, which were differentially methylated following exposure to adverse maternal environment in mice, have been associated with ASD (179–182). A limitation of this animal model is that it does not offer a model of autism, but rather a model of 5-HT1A receptor-deficient maternal environment. However, studying the effect of 5-HT1A receptor-deficient maternal environment on DNA methylation as well as on anxiety-like behavior and impaired vocal communication in the offspring is of interest in autism, especially given central and peripheral alterations in serotonin in autism (156, 183).
Concerning epigenetic histone modifications possibly involved in autism, epigenetic anomalies in histone methylation patterns may contribute to the cerebellum neuroanatomical alterations, especially concerning Purkinje cells, observed in some individuals with ASD [for a review of cerebellar abnormalities reported in autism, see Ref. (184) and (185)]. The Purkinje cell maturation in the cerebellum is signaled by a normal downregulation of the engrailed-2 (EN-2) gene during late prenatal and early-postnatal development (186). James et al. (187) conducted a study on post-mortem cerebellar samples from 13 individuals with autism and found histone methylation modifications in the EN-2 promoter associated with increased EN-2 gene expression and EN-2 protein levels. These results suggest a postnatal persistence of EN-2 overexpression in some individuals with autism that may contribute to autism cerebellar abnormalities.
Another possible epigenetic mechanism that may underlie autism is RNA editing, a neurodevelopmentally regulated post-transcriptional mechanism responsible for producing mRNA molecules with sequence information not specifically encoded in the genome. More particularly, adenosine-to-inosine (A-to-I) RNA editing fine-tunes synaptic function (strength and duration) in response to environmental stimuli, affecting the transmission of all sensory stimuli to the CNS. A recent study (188) surveyed A-to-I RNA editing in autistic brains and found differential editing patterns and a dysfunctional form of the adenosine deaminase enzyme involved in RNA editing in post-mortem cerebella from individuals with autism.
Epigenetic mechanisms have been also reported in various genetic disorders associated with autism, including maternal 15q11–q13 duplication and several syndromes such as Fragile X, Rett, Down, Turner, Phelan-McDermid, Beckwith–Wiedemann, Williams–Beuren, CHARGE, Angelman, or Prader–Willi syndrome (see Tables 2 and 3). It is noteworthy that among the most commonly recurrent cytogenetic abnormalities associated with autism are duplications of sequences in a region on the proximal part of the long arm of chromosome 15, specifically the interval 15q11–q13. The behavioral phenotypes associated with 15q11–q13 defects show a parent-of-origin specific effect on phenotypic expression. More specifically, it is the maternally derived duplications that convey a high risk of autism (189–191). Similarly, paternal-specific deletion of multiple imprinted, paternally expressed genes on the 15q11–q13 region results in Prader–Willi syndrome, whereas maternal deletion of a single, imprinted, maternally expressed gene encoding a ubiquitin-protein ligase (UBE3A) on this same region gives rise to the Angelman syndrome phenotype. Phenotypic comparisons between Prader–Willi syndrome, Angelman syndrome, and maternal 15q11–q13 duplication reveal commonalities possibly related to a shared genetic basis. These phenotype overlaps concern common areas of cognitive impairment (or, more rarely, superior performance in discrete cognitive domains), motor stereotypies, motor coordination, seizures, language impairment, or behavioral manifestations such as compulsions or tantrums (see Table 2).

Table 3. Epigenetic mechanisms and potential therapeutic targets in genetic disorders associated with autistic syndrome.
Given the number of genetic disorders associated with epigenetic etiologies comorbid with autism, it can be suggested that epigenetic mechanisms involving gene × environment interactions might be a common pathway for many cases of ASD. Furthermore, as underlined by Grafodatskaya et al. (164), new molecular technologies allowing the identification of critical epigenetic determinants open interesting perspectives, including therapeutic ones. It might serve in the development of innovative therapeutic strategies, as already applied with the treatment by the histone deactylase inhibitor valproate.
Conclusion
Considering that environmental factors can modify the expression of genes and the potential role of epigenetic mechanisms in the development of ASD, it appears necessary to study in concert the genetic factors and the environmental factors in autism. Despite recent studies on environmental risk factors for ASD, no single and major environmental factor has been identified, suggesting that further research should study a combination of factors through an integrated approach including gene × environment interactions. This integrated clinico-biological approach takes into account the interactions between the genetic factors and the postnatal or prenatal environmental factors (psychosocial environment but also cytoplasmic and uterine environment with placental exchanges and hormonal effects). An allelic form can be present in the genotype without expressing itself if it is inhibited by signals mediating environmental, epigenetic, or genetic contributions. It is therefore important not to focus on the “genes of autism,” which implies determinism, but to study instead the effects of the genome integrated with the effects of the environment with possible plasticity. Epigenetic remodeling by environmental factors opens new perspectives for a better understanding, prevention and early therapeutic intervention of ASD. Finally, autism could be considered as the result of several genetic and environmental factors. Given this multifactorial etiology, it is probably through multivariate analyses and a multidisciplinary approach including the participation of biologists as well as clinicians that advances will be made.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vargas DL, Nascimbene C, Krishan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol (2005) 57:67–81. doi: 10.1002/ana.20315
2. Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics (2010) 125(4):e727–35. doi:10.1542/peds.2009-1684
3. Reddy KS. Cytogenetic abnormalities and fragile-X syndrome in autism spectrum disorders. BMC Med Genet (2005) 6:3. doi:10.1186/1471-2350-6-3
4. Vorstman JA, Staal WG, van Daalen E, van Engeland H, Hochstenbach PF, Franke L. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry (2006) 11:18–28. doi:10.1038/sj.mp.4001757
5. Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet (2008) 82(2):477–88. doi:10.1016/j.ajhg.2007.12.009
6. Lintas C, Persico AM. Autistic phenotypes and genetic testing: state-of-the-art for the clinical geneticist. J Med Genet (2009) 46(1):1–8. doi:10.1136/jmg.2008.060871
7. Miles JH. Autism spectrum disorders-a genetics review. Genet Med (2011) 13(4):278–94. doi:10.1097/GIM.0b013e3181ff67ba
8. Gardener H, Spiegelman D, Buka SL. Perinatal and neonatal risk factors for autism: a comprehensive meta-analysis. Pediatrics (2011) 128(2):344–55. doi:10.1542/peds.2010-1036
9. Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry (2011) 68(11):1095–102. doi:10.1001/archgenpsychiatry.2011.76
10. Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet (2008) 9:341–55. doi:10.1038/nrg2346
11. Bill BR, Geschwind DH. Genetic advances in autism: heterogeneity and convergence on shared pathways. Curr Opin Genet Dev (2009) 19:271–8. doi:10.1016/j.gde.2009.04.004
13. State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci (2011) 14(12):1499–506. doi:10.1038/nn.2924
14. Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev (2012) 22:229–37. doi:10.1016/j.gde.2012.03.002
16. Fombonne E. Epidemiology of pervasive developmental disorders. Pediatr Res (2009) 65(6):591–8. doi:10.1203/PDR.0b013e31819e7203
17. Kogan MD, Blumberg SJ, Schieve LA, Boyle CA, Perrin JM, Ghandour RM, et al. Prevalence of parent-reported diagnosis of autism spectrum disorder among children in the US, 2007. Pediatrics (2009) 124(5):1395–403. doi:10.1542/peds.2009-1522
18. Hertz-Picciotto I, Delwiche L. The rise in autism and the role of age at diagnosis. Epidemiology (2009) 20(1):84–90. doi:10.1097/EDE.0b013e3181902d15
19. Fisch GS. Nosology and epidemiology in autism: classification counts. Am J Med Genet C Semin Med Genet (2012) 160C:91–103. doi:10.1002/ajmg.c.31325
20. Fisch GS. Autism and epistemology III: child development, behavioral stability, and reliability of measurement. Am J Med Genet A (2012) 158A:969–79. doi:10.1002/ajmg.a.35269
21. Tordjman S, Gutknecht L, Carlier M, Spitz E, Antoine C, Slama F, et al. Role of the serotonin transporter gene in the behavioral expression of autism. Mol Psychiatry (2001) 6:434–9. doi:10.1038/sj.mp.4000873
22. Landa RJ. Diagnosis of autism spectrum disorders in the first 3 years of life. Nat Clin Pract Neurol (2008) 4(3):138–47. doi:10.1038/ncpneuro0731
23. Lauritsen MB, Pedersen CB, Mortensen PB. Effects of familial risk factors and place of birth on the risk of autism: a nationwide register-based study. J Child Psychol Psychiatry (2005) 46(9):963–71.
24. Selkirk CG, McCarthy VP, Lian F, Schimmenti L, Leroy BS. Parents’ perceptions of autism spectrum disorder etiology and recurrence risk and effects of their perceptions on family planning: recommendations for genetic counselors. J Genet Couns (2010) 18:507–19. doi:10.1007/s10897-009-9233-0
25. Ritvo ER, Freeman BJ, Pingree C, Mason-Brothers A, Jorde L, Jenson WR, et al. The UCLA-University of Utah epidemiologic survey of autism: prevalence. Am J Psychiatry (1989) 146(2):194–9.
26. Carlier M, Roubertoux PL. Psychopathologie et génétique. Mode d’emploi. In: Widlöcher D, editor. Traité de Psychopathologie. Paris: PUF (1994). p. 586–613.
27. Gillberg C, Gillberg IC, Steffenburg S. Siblings and parents of children with autism: a controlled population-based study. Dev Med Child Neurol (1992) 34(5):389–98. doi:10.1111/j.1469-8749.1992.tb11450.x
29. Folstein SE, Rutter ML. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry (1977) 18(4):297–321. doi:10.1111/j.1469-7610.1977.tb00443.x
30. Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med (1995) 25:63–77. doi:10.1017/S0033291700028099
31. Ritvo ER, Spence MA, Freeman BJ, Mason-Brothers A, Mo A, Marazita ML. Evidence for autosomal recessive inheritance in 46 families with multiple incidences of autism. Am J Psychiatry (1985) 142(2):187–92.
32. Smalley SL, Asarnow RF, Spence MA. Autism and genetics. A decade of research. Arch Gen Psychiatry (1988) 45(10):953–61.
33. Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobson G, et al. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry (1989) 30:405–16.
34. Rosenberg RE, Law JK, Yenokyan G, McGready J, Kaufmann WE, Law PA. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med (2009) 163(10):907–14. doi:10.1001/archpediatrics.2009.98
35. Nordenbæk C, Jørgensen M, Kyvik KO, Bilenberg N. A Danish population-based twin study on autism spectrum disorders. Eur Child Adolesc Psychiatry (2014) 23(1):35–43. doi:10.1007/s00787-013-0419-5
36. Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science (2007) 316:445–9. doi:10.1126/science.1138659
37. Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron (2012) 74(2):285–99. doi:10.1016/j.neuron.2012.04.009
38. Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature (2012) 485(7397):242–5. doi:10.1038/nature11011
39. O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature (2012) 485(7397):246–50. doi:10.1038/nature10989
40. Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature (2012) 485:237–41. doi:10.1038/nature10945
41. Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron (2013) 77:259–73. doi:10.1016/j.neuron.2012.11.002
42. Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ, et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism (2012) 3(1):9. doi:10.1186/2040-2392-3-9
43. Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature (2010) 466:368–72. doi:10.1038/nature09146
44. Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron (2011) 70:863–85. doi:10.1016/j.neuron.2011.05.002
45. Cross-Disorder Group of the Psychiatric Genomics Consortium. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet (2013) 45(9):984–94. doi:10.1038/ng.2711
46. Smoller JW, Craddock N, Kendler K, Lee PH, Neale BM, Nurnberger JI, et al. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet (2013) 381(9875):1371–9. doi:10.1016/S0140-6736(12)62129-1
47. Anckarsäter H, Lundstrom S, Kollberg L, Kerekes N, Palm C, Carlstrom E, et al. The child and adolescent twin study in Sweden (CATSS). Twin Res Hum Genet (2011) 14(6):495–508. doi:10.1375/twin.14.6.495
48. Anckarsäter H. Beyond categorical diagnostics in psychiatry: scientific and medicolegal implications. Int J Law Psychiatry (2010) 33:59–65. doi:10.1016/j.ijlp.2009.12.001
49. Schanen NC. Epigenetics of autism spectrum disorders. Hum Mol Genet (2006) 15(Suppl 2):R138–50. doi:10.1093/hmg/ddl213
50. Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature (2011) 474:380–4. doi:10.1038/nature10110
51. Hegmann JP, Possidente B. Estimating genetic correlations from inbred strains. Behav Genet (1981) 11:103–14. doi:10.1007/BF01065621
52. Gardener H, Spiegelman D, Buka SL. Prenatal risk factors for autism: comprehensive meta-analysis. Br J Psychiatry (2009) 196:416–7. doi:10.1192/bjp.bp.108.051672
53. Barnevik-Olsson M, Gillberg C, Fernell E. Prevalence of autism in children of Somali origin living in Stockholm: brief report of an at-risk population. Dev Med Child Neurol (2008) 50(8):598–601. doi:10.1111/j.1469-8749.2010.03812.x
54. Fountain C, Bearman P. Risk as social context: immigration policy and autism in California. Sociol Forum (Randolph N J) (2011) 26(2):215–40. doi:10.1111/j.1573-7861.2011.01238.x
55. Haglund NG, Källén KB. Risk factors for autism and Asperger syndrome. Perinatal factors and migration. Autism (2011) 15(2):163–83. doi:10.1177/1362361309353614
56. Keen DV, Reid FD. Armone, D. Autism, ethnicity and maternal immigration. Br J Psychiatry (2012) 196(4):274–81. doi:10.1192/bjp.bp.109.065490
57. Magnusson C, Rai D, Goodman A, Lundberg M, Idring S, Svensson A, et al. Migration and autism spectrum disorder: population-based study. Br J Psychiatry (2012) 201:109–15. doi:10.1192/bjp.bp.111.095125
58. Schieve LA, Boulet SL, Blumberg SJ, Kogan MD, Yeargin-Allsopp M, Boyle CA, et al. Association between parental nativity and autism spectrum disorder among US-born non-Hispanic white and Hispanic children, 2007 National Survey of Children’s Health. Disabil Health J (2012) 5(1):18–25. doi:10.1016/j.dhjo.2011.09.001
59. Lord C. Fetal and sociocultural environments and autism. Am J Psychiatry (2013) 170(4):355–8. doi:10.1176/appi.ajp.2013.13010078
60. Gillberg IC, Gillberg C. Autism in immigrants: a population-based study from Swedish rural and urban areas. J Intellect Disabil Res (1996) 40(1):24–31. doi:10.1111/j.1365-2788.1996.tb00599.x
61. Bromley RL, Mawer G, Clayton-Smith J, Baker GA. Autism spectrum disorders following in utero exposure to antiepileptic drugs. Neurology (2008) 71:1923–4. doi:10.1212/01.wnl.0000339399.64213.1a
62. Bromley RL, Mawer GE, Briggs M, Cheyne C, Clayton-Smith J, García-Fiñana M, et al. The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. J Neurol Neurosurg Psychiatry (2013) 84(6):637–43. doi:10.1136/jnnp-2012-304270
63. Newschaffer CJ, Croen LA, Daniels J, Giarelli E, Grether JK, Levy SE, et al. The epidemiology of autism spectrum disorders. Annu Rev Public Health (2007) 28:235–58.
64. Rogers EJ. Has enhanced folate status during pregnancy altered natural selection and possibly Autism prevalence? A closer look at a possible link. Med Hypotheses (2008) 71:406–10. doi:10.1016/j.mehy.2008.04.013
65. Surén P, Roth C, Bresnahan M, Haugen M, Hornig M, Hirtz D, et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA (2013) 309(6):570–7. doi:10.1001/jama.2012.155925
66. Berry RJ, Crider KS, Yeargin-Allsopp M. Periconceptional folic acid and risk of autism spectrum disorders. JAMA (2013) 309(6):611–3. doi:10.1001/jama.2013.198
67. Vahabzadeh A, McDougle CJ. Maternal folic acid supplementation and risk of autism. JAMA (2013) 309(21):2208. doi:10.1001/jama.2013.4876
68. Pu D, Shen Y, Wu J. Association between MTHFR gene polymorphisms and the risk of autism spectrum disorders: a meta-analysis. Autism Res (2013) 6(5):384–92. doi:10.1002/aur.1300
69. Stromberg B, Dahlquist G, Ericson A, Finnstrom O, Koster M, Stjernqvist K. Neurological sequelae in children born after in-vitro fertilisation: a population-based study. Lancet (2002) 359:461–5. doi:10.1016/S0140-6736(02)07674-2
70. Odom LN, Segars J. Imprinting disorders and assisted reproductive technology. Curr Opin Endocrinol Diabetes Obes (2010) 17(6):517–22.
71. Hvidtjørn D, Grove J, Schendel D, Schieve LA, Svaerke C, Ernst E, et al. Risk of autism spectrum disorders in children born after assisted conception: a population-based follow-up study. J Epidemiol Community Health (2011) 65(6):497–502. doi:10.1136/jech.2009.093823
72. Zachor DA, Ben Itzchak E. Assisted reproductive technology and risk for autism spectrum disorder. Res Dev Disabil (2011) 32(6):2950–6. doi:10.1016/j.ridd.2011.05.007
73. Lehti V, Brown AS, Gissler M, Rihko M, Suominen A, Sourander A. Autism spectrum disorders in IVF children: a national case-control study in Finland. Hum Reprod (2013) 28(3):812–8. doi:10.1093/humrep/des430
74. Sandin S, Nygren KG, Iliadou A, Hultman CM, Reichenberg A. Autism and mental retardation among offspring born after in vitro fertilization. JAMA (2013) 310(1):75–84. doi:10.1001/jama.2013.7222
75. Guinchat V, Thorsen P, Laurent C, Cans C, Bodeau N, Cohen D. Pre-peri-, and neonatal risk factors for autism. Acta Obstet Gynecol Scand (2012) 91(3):287–300. doi:10.1111/j.1600-0412.2011.01325.x
76. Bilder D, Pinborough-Zimmerman J, Miller J, McMahon W. Prenatal, perinatal, and neonatal factors associated with autism spectrum disorders. Pediatrics (2009) 123(5):1293–300. doi:10.1542/peds.2008-0927
77. Mason-Brothers A, Ritvo ER, Pingree C, Petersen PB, Jenson WR, McMahon WM, et al. The UCLA-University of Utah epidemiologic survey of autism: prenatal, perinatal, and postnatal factors. Pediatrics (1990) 86(4):514–9.
78. Brimacombe M, Ming X, Lamendola M. Prenatal and birth complications in autism. Matern Child Health J (2007) 11(1):73–9. doi:10.1007/s10995-006-0142-7
79. Hultman CM, Sparen P, Cnattingius S. Perinatal risk factors for infantile autism. Epidemiology (2002) 13(4):417–23. doi:10.1097/00001648-200207000-00009
80. Maimburg RD, Vaeth M, Schendel DE, Bech BH, Olsen J, Thorsen P. Neonatal jaundice: a risk factor for infantile autism? Paediatr Perinat Epidemiol (2008) 22(6):562–8. doi:10.1111/j.1365-3016.2008.00973.x
81. Maimburg RD, Bech BH, Vaeth M, Moller-Madsen B, Olsen J. Neonatal jaundice, autism, and other disorders of psychological development. Pediatrics (2010) 126(5):872–8. doi:10.1542/peds.2010-0052
82. Buchmayer S, Johansson S, Johansson A, Hultman CM, Sparen P, Cnattingius S. Can association between preterm birth and autism be explained by maternal or neonatal morbidity? Pediatrics (2009) 124(5):e817–25. doi:10.1542/peds.2008-3582
83. Tordjman S, Pichard N, Anderson GM, Touitou Y. Nocturnal urinary excretion of melatonin in children and adolescents with autistic disorder. Biol Psychiatry (2005) 57:134–8. doi:10.1016/j.biopsych.2004.11.003
84. Tordjman S, Anderson GM, Bellissant E, Botbol M, Charbuy H, Camus F, et al. Day and nighttime excretion of 6-sulphatoxymelatonin in adolescents and young adults with autistic disorder. Psychoneuroendocrinology (2012) 37(12):1990–7. doi:10.1016/j.psyneuen.2012.04.013
85. Tordjman S, Najjar I, Bellissant E, Anderson GM, Barburoth M, Cohen D, et al. Advances in the research of melatonin in autism spectrum disorders: literature review and new perspectives. Int J Mol Sci (2013) 14:20508–42. doi:10.3390/ijms141020508
86. Windham GC, Zhang L, Gunier R, Croen LA, Grether JK. Autism spectrum disorders in relation to distribution of hazardous air pollutants in the San Francisco bay area. Environ Health Perspect (2006) 114(9):1438–44. doi:10.1289/ehp.9120
87. Kalkbrenner AE, Daniels JL, Chen JC, Poole C, Emch M, Morrissey J. Perinatal exposure to hazardous air pollutants and autism spectrum disorders at age 8. Epidemiology (2010) 21(5):631–41. doi:10.1097/EDE.0b013e3181e65d76
88. Volk HE, Hertz-Picciotto I, Delwiche L, Lurmann F, McConnell R. Residential proximity to freeways and autism in the CHARGE study. Environ Health Perspect (2011) 119(6):873–7. doi:10.1289/ehp.1002835
89. Currie J, Neidell M, Schmieder JF. Air pollution and infant health: lessons from New Jersey. J Health Econ (2009) 28(3):688–703. doi:10.1016/j.jhealeco.2009.02.001
90. Hansen CA, Barnett AG, Pritchard G. The effect of ambient air pollution during early pregnancy on fetal ultrasonic measurements during mid-pregnancy. Environ Health Perspect (2008) 116(3):362–9. doi:10.1289/ehp.10720
91. Davis DA, Bortolato M, Godar SC, Sander TK, Iwata N, Pakbin P, et al. Prenatal exposure to urban air nanoparticles in mice causes altered neuronal differentiation and depression-like responses. PLoS One (2013) 8(5):e64128. doi:10.1371/journal.pone.0064128
92. Volk HE, Lurmann F, Penfold B, Hertz-Picciotto I, McConnel R. Traffic-related air pollution, particulate matter, and autism. JAMA Psychiatry (2013) 70(1):71–7. doi:10.1001/jamapsychiatry.2013.266
93. Perera FP, Whyatt R, Hoepner L, Wang S, Camann D, Rauh V. Prenatal air-borne polycyclic aromatic hydrocarbon exposure and child IQ at age 5 years. Pediatrics (2009) 124(2):e195–202. doi:10.1542/peds.2008-3506
94. Rai D, Lee BK, Dalman C, Golding J, Lewis G, Magnusson C. Parental depression, maternal antidepressant use during pregnancy, and risk of autism spectrum disorders: population based case-control study. BMJ (2013) 346:f2059. doi:10.1136/bmj.f2059
95. Croen LA, Grether JK, Yoshiba CK, Odouli R, Hendrick V. Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch Gen Psychiatry (2011) 68:1104–12. doi:10.1001/archgenpsychiatry.2011.73
96. Daniels K, Lewin S, Practice Policy Group. Translating research into maternal health care policy: a qualitative case study of the use of evidence in policies for the treatment of eclampsia and pre-eclampsia in South Africa. Health Res Policy Syst (2008) 6:12. doi:10.1186/1478-4505-6-12
97. Yirmiya N, Shaked M. Psychiatric disorders in parents of children with autism: a meta-analysis. J Child Psychol Psychiatry (2005) 46(1):69–83. doi:10.1111/j.1469-7610.2004.00334.x
98. Hoksbergen R, ter Laak J, Rijk K, van Dijkum C, Stoutjesdijk F. Post-institutional autistic syndrome in Romanian adoptees. J Autism Dev Disord (2005) 35:615–23. doi:10.1007/s10803-005-0005-x
99. Rutter M, Andersen-Wood L, Beckett C, Bredenkamp D, Castle J, Groothues C, et al. Quasi-autistic patterns following severe early global privation. English and Romanian Adoptees (ERA) study team. J Child Psychol Psychiatry (1999) 40(4):537–49.
100. Rutter M, Kreppner J, Croft C, Murrin M, Colvert E, Beckett C, et al. Early adolescent outcomes of institutionally deprived and non-deprived adoptees. III. Quasi-autism. J Child Psychol Psychiatry (2007) 48:1200–7. doi:10.1111/j.1469-7610.2006.01688.x
101. Lord C. Diagnostic instruments in autism spectrum disorders 2nd ed. In: Cohen DJ, Volkmar FR, editors. Handbook of Autism and Pervasive Developmental Disorders. New York: John Wiley and Sons Inc (1997). p. 460–83.
102. Brown R, Hobson RP, Lee A, Stevenson J. Are there “autistic-like” features in congenitally blind children? J Child Psychol Psychiatry (1997) 38(6):693–703. doi:10.1111/j.1469-7610.1997.tb01696.x
103. Mukaddes NM, Kilincaslan A, Kucukyazici G, Sevketoglu T, Tuncer S. Autism in visually impaired individuals. Psychiatry Clin Neurosci (2007) 61(1):39–44.
104. Steffenburg S. Neuropsychiatric assessment of children with autism: a population-based study. Dev Med Child Neurol (1991) 33(6):495–511. doi:10.1111/j.1469-8749.1991.tb14915.x
105. Donaldson AI, Heavner KS, Zwolan TA. Measuring progress in children with autism spectrum disorder who have cochlear implants. Arch Otolaryngol (2004) 130:666–71. doi:10.1001/archotol.130.5.666
106. Daneshi A, Hassanzadeh S. Cochlear implantation in prelingually deaf with additional disability. J Laryngol Otol (2007) 121(7):635–8.
107. Rosenhall U, Nordin V, Sandström M, Ahlsen G, Gillberg C. Autism and hearing loss. J Autism Dev Disord (1999) 29(5):349–57. doi:10.1023/A:1023022709710
108. O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science (2012) 338(6114):1619–22. doi:10.1126/science.1227764
109. Tordjman S, Maillhes AS. Developmental disorder in body image occurring in early infancy: a common dimension shared by schizophrenia and autism? Neuropsychiatr Enfance Adolesc (2009) 57(1):6–13. doi:10.1016/j.neurenf.2008.09.005
110. Johansson M, Wentz E, Fernell E, Strömland K, Miller MT, Gillberg C. Autistic spectrum disorders in Moëbius sequence: a comprehensive study of 25 cases. Dev Med Child Neurol (2001) 43:338–45. doi:10.1017/S0012162201000627
111. Johansson M, Billstedt E, Danilesson S, Strömland K, Miller M, Granström G. Autism spectrum disorders and underlying brain mechanisms in the oculoauriculovertebral spectrum. Dev Med Child Neurol (2007) 49:280–8. doi:10.1111/j.1469-8749.2007.00280.x
112. Hartshorne TS, Grialou TL, Parker KR. Autistic-like behavior in CHARGE syndrome. Am J Med Genet A (2005) 133A:257–61. doi:10.1002/ajmg.a.30541
113. Johansson M, Råstam M, Billstedt E, Danielsson S, Strömland K, Miller M, et al. Autism spectrum disorders and underlying brain pathology in CHARGE association. Dev Med Child Neurol (2006) 48:40–50. doi:10.1017/S0012162206000090
114. Smith IM, Nichols SL, Issekutz K, Blake K, Canadian Paediatric Surveillance Program. Behavioral profiles and symptoms of autism in CHARGE syndrome: preliminary Canadian epidemiological data. Am J Med Genet A (2005) 133A:248–56. doi:10.1002/ajmg.a.30544
115. Blake KD, Prasad C. CHARGE syndrome. Orphanet J Rare Dis (2006) 1:34. doi:10.1186/1750-1172-1-34
116. Johansson M, Gillberg C, Råstam M. Autism spectrum conditions in individuals with Möbius sequence, CHARGE syndrome and oculo-auriculo-vertebral spectrum: diagnostic aspects. Res Dev Disabil (2010) 31:9–24. doi:10.1016/j.ridd.2009.07.011
117. Sutcliffe JS, Nurmi EL, Lombroso PJ. Genetics of childhood disorders: XLVII. Autism, part 6: duplication and inherited susceptibility of chromosome 15q11-q13 genes in autism. J Am Acad Child Adolesc Psychiatry (2003) 42(2):253–6. doi:10.1097/00004583-200302000-00021
118. Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci U S A (2010) 107(1):17668–73. doi:10.1073/pnas.1004487107
119. Rangasamy S, D’Mello SR, Narayanan V. Epigenetics, autism spectrum, and neurodevelopmental disorders. Neurotherapeutics (2013) 10(4):742–56. doi:10.1007/s13311-013-0227-0
120. Steffenburg S, Gillberg CL, Seffenburg U, Kyllerman M. Autism in Angelman syndrome: a population-based study. Pediatr Neurol (1996) 14:131–6. doi:10.1016/0887-8994(96)00011-2
121. Peters SU, Beaudet AL, Madduri N, Bacino CA. Autism in Angelman syndrome: implications for autism research. Clin Genet (2004) 66(6):530–6.
122. Veltman MW, Thompson RJ, Roberts SE, Thomas NS, Whittington J, Bolton PF. Prader-Willi syndrome – a study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur Child Adolesc Psychiatry (2004) 13(1):42–50. doi:10.1007/s00787-004-0354-6
123. Descheemaeker MJ, Govers V, Vermeulen P, Fryns JP. Pervasive developmental disorders in Prader-Willi syndrome: the Leuven experience in 59 subjects and controls. Am J Med Genet A (2006) 140(11):1136–42. doi:10.1002/ajmg.a.31235
124. Hogart A, Wu D, LaSalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis (2010) 38(2):181–91. doi:10.1016/j.nbd.2008.08.011
125. Kau AS, Tierney E, Bukelis I, Stump MH, Kates WR, Trescher WH, et al. Social behavior profile in young males with fragile X syndrome: characteristics and specificity. Am J Med Genet A (2004) 126A(1):9–17. doi:10.1002/ajmg.a.20218
126. Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci (2006) 9(10):1221–5. doi:10.1038/nn1765
127. Loesch DZ, Bui QM, Dissanayake C, Clifford S, Gould E, Bulhak-Paterson D, et al. Molecular and cognitive predictors of the continuum of autistic behaviours in fragile X. Neurosci Biobehav Rev (2007) 31(3):315–26.
128. Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, et al. Autism profiles of males with fragile X syndrome. Am J Ment Retard (2008) 113(6):427–38. doi:10.1352/2008.113:427-438
129. Meloni I, Bruttini M, Longo I, Mari F, Rizzolio F, D’Adamo P, et al. A mutation in the Rett syndrome gene, MECP2, causes X-linked mental retardation and progressive spasticity in males. Am J Hum Genet (2000) 67(4):982–5.
130. Smeets E, Terhal P, Casaer P, Peters A, Midro A, Schollen E, et al. Rett syndrome in females with CTS hot spot deletions: a disorder profile. Am J Med Genet A (2005) 132A:117–20. doi:10.1002/ajmg.a.30410
131. Young DJ, Bebbington A, Anderson A, Ravine D, Ellaway C, Kulkarni A, et al. The diagnosis of autism in a female: could it be Rett syndrome? Eur J Pediatr (2008) 167:661–9. doi:10.1007/s00431-007-0569-x
132. Bregmen JD, Volkmar FR. Autistic social dysfunction and Down syndrome. J Am Acad Child Adolesc Psychiatry (1988) 27:440–1. doi:10.1097/00004583-198807000-00011
133. Ghaziuddin M. Autism in Down syndrome: family history correlates. J Intellect Disabil Res (1997) 41:87–91. doi:10.1111/j.1365-2788.1997.tb00681.x
134. Moss J, Howlin P. Autism spectrum disorders in genetic syndromes: implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J Intellect Disabil Res (2009) 53(10):852–73. doi:10.1111/j.1365-2788.2009.01197.x
135. Skuse DH, James RS, Bishop DVM, Coppin B, Dalton P, Aamodt-Leeper G, et al. Evidence from Turner’s syndrome of an imprinted X-linked locus affecting cognitive function. Nature (1997) 387:705–8. doi:10.1038/42706
136. Soorya L, Kolevzon A, Zweifach J, Lim T, Dobry Y, Schwartz L, et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol Autism (2013) 4(1):18. doi:10.1186/2040-2392-4-18
137. Kent L, Bowdin S, Kirby GA, Cooper WN, Maher ER. Beckwith Wiedemann syndrome: a behavioral phenotype-genotype study. Am J Med Genet B Neuropsychiatr Genet (2008) 147B(7):1295–7. doi:10.1002/ajmg.b.30729
138. Gagliardi C, Bonaglia MC, Selicorni A, Borgatti R, Giorda R. Unusual cognitive and behavioural profile in a Williams syndrome patient with atypical 7q11.23 deletion. J Med Genet (2003) 40:526–30. doi:10.1136/jmg.40.7.526
139. Edelmann L, Prosnitz A, Pardo S, Bhatt J, Cohen N, Lauriat T, et al. An atypical deletion of the Williams-Beuren syndrome interval implicates genes associated with defective visuospatial processing and autism. J Med Genet (2007) 44(2):136–43. doi:10.1136/jmg.2006.044537
140. Rogers SJ, Wehner DE, Hagerman R. The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Beh Pediatrics (2001) 22:409–17.
141. Glasson EJ, Bower C, Petterson B, de Klerk N, Chaney G, Hallmayer JF. Perinatal factors and the development of autism: a population study. Arch Gen Psychiatry (2004) 61(6):618–27. doi:10.1001/archpsyc.61.6.618
142. Miles JH, Hillman RE. Value of a clinical morphology examination in autism. Am J Med Genet (2000) 91(4):245–53.
143. Miles JH, Takahashi TN, Bagby S, Sahota PK, Vaslow DF, Wang CH, et al. Essential versus complex autism: definition of fundamental prognostic subtypes. Am J Med Genet A (2005) 135(2):171–80.
144. Cohen D, Pichard N, Tordjman S, Baumann C, Burglen L, Excoffier E, et al. Specific genetic disorders and autism: clinical contribution towards their identification (Review). J Autism Dev Disord (2005) 35(1):103–16.
145. Jacquemont ML, Sanlaville D, Redon R, Raoul O, Cormier-Daire V, Lyonnet S, et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet (2006) 43(11):843–9. doi:10.1136/jmg.2006.043166
146. Delorme R, Ey E, Toro R, Leboyer M, Gillberg C, Bourgeron T. Progress toward treatments for synaptic defects in autism. Nat Med (2013) 19(6):685–94. doi:10.1038/nm.3193
147. Kuhl PK. Brain mechanisms in early language acquisition. Neuron (2010) 67(5):713–27. doi:10.1016/j.neuron.2010.08.038
148. Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci (2005) 6:877–88. doi:10.1038/nrn1787
149. Damodaran LP, Arumugam G. Urinary oxidative stress markers in children with autism. Redox Rep (2011) 16(5):216–22. doi:10.1179/1351000211Y.0000000012
150. McGinnis WR. Oxidative stress in autism. Altern Ther Health Med (2004) 10(6):22–36; quiz 37, 92.
151. Ming X, Stein TP, Brimacombe M, Johnson WG, Lambert GH, Wagner GC. Increased excretion of a lipid peroxidation biomarker in autism. Prostaglandins Leukot Essent Fatty Acids (2005) 73(5):379–84. doi:10.1016/j.plefa.2005.06.002
152. Bechara EG, Didiot MC, Melko M, Davidovic L, Bensaid M, Martin P, et al. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol (2009) 7(1):e1000016. doi:10.1371/journal.pbio.1000016
153. de Diego-Otero Y, Romero-Zerbo Y, El Bekay R, Decara J, Sanchez L, Rodriguez-de Fonseca F, et al. Alpha-tocopherol protects against oxidative stress in the fragile X knockout mouse: an experimental therapeutic approach for the Fmr1 deficiency. Neuropsychopharmacology (2009) 34(4):1011–26. doi:10.1038/npp.2008.152
154. el Bekay R, Romero-Zerbo Y, Decara J, Sanchez-Salido L, Del Arco-Herrera I, Rodriguez-de Fonseca F, et al. Enhanced markers of oxidative stress, altered antioxidants and NADPH-oxidase activation in brains from fragile X mental retardation 1-deficient mice, a pathological model for fragile X syndrome. Eur J Neurosci (2007) 26(11):3169–80. doi:10.1111/j.1460-9568.2007.05939.x
155. Lacaria M, Spencer C, Gu W, Paylor R, Lupski JR. Enriched rearing improves behavioral responses of an animal model for CNV-based autistic-like traits. Hum Mol Genet (2012) 21(14):3083–96. doi:10.1093/hmg/dds124
156. Nakamura K, Sekine Y, Ouchi Y, Tsujii M, Yoshikawa E, Futatsubashi M, et al. Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch Gen Psychiatry (2010) 67(1):59–68. doi:10.1001/archgenpsychiatry.2009.137
157. Bagot RC, Meaney MJ. Epigenetics and the biological basis of gene X interactions. J Am Acad Child Adolesc Psychiatry (2010) 49(8):752–71. doi:10.1016/j.jaac.2010.06.001
158. Gonzalez A, Lovic V, Ward GR, Wainwright PE, Fleming AS. Intergenerational effects of complete maternal deprivation and replacement stimulation on maternal behaviour and emotionality in female rats. Dev Psychobiol (2001) 38(1):11–32. doi:10.1002/1098-2302(2001)38:1<11::AID-DEV2>3.0.CO;2-B
159. Jutapakdeegul N, Casalotti SO, Govitrapong P, Kotchabhakdi N. Postnatal touch stimulation acutely alters corticosterone levels and glucocorticoid receptor gene expression in the neonatal rat. Dev Neurosci (2003) 25(1):26–33. doi:10.1159/000071465
160. Champagne FA, Meaney MJ. Stress during gestation alters postpartum maternal care and the development of the offspring in a rodent model. Biol Psychiatry (2006) 59(12):1227–35. doi:10.1016/j.biopsych.2005.10.016
161. Fenoglio KA, Brunson KL, Avishai-Eliner S, Stone BA, Kapadia BJ, Baram TZ. Enduring, handling-evoked enhancement of hippocampal memory function and glucocorticoid receptor expression involves activation of the corticotropin-releasing factor type 1 receptor. Endocrinology (2005) 146(9):4090–6. doi:10.1210/en.2004-1285
162. Graignic-Philippe R, Dayan J, Chokron S, Jacquet AY, Tordjman S. Effects of prenatal stress on fetal and child development: a critical literature review. Neurosci Biobehav Rev (2014) 43:137–62. doi:10.1016/j.neubiorev.2014.03.022
163. Franklin TB, Russig H, Weiss IC, Gräff J, Linder N, Michalon A, et al. Epigenetic transmission of the impact of early stress across generations. Biol Psychiat (2010) 68:408–15.
164. Grafodatskaya D, Chung B, Szatmari P, Weksberg R. Autism spectrum disorders and epigenetics. J Am Acad Child Adolsec Psychiatry (2010) 49(8):794–809. doi:10.1016/j.jaac.2010.05.005
165. Chapman JB, Cutler MG. Effects of sodium valproate on development and social behaviour in the Mongolian gerbil. Neurotoxicol Teratol (1989) 11:193–8. doi:10.1016/0892-0362(89)90058-5
166. Voorhees CV. Behavioral teratogenicity of valporic acid: selective effects on behavior after prenatal exposure to rats. Psychopharmacology (1987) 92:173–9. doi:10.1007/BF00177911
167. Wu Y, Wang L. The effects of antiepileptic drugs on spatial learning and hippocampal protein kinase C (in immature rats). Brain Dev (2002) 24:82–7. doi:10.1016/S0387-7604(02)00012-8
168. Ingram JL, Peckham SM, Tisdale B, Rodier PM. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol Teratol (2000) 22:319–24. doi:10.1016/S0892-0362(99)00083-5
169. Markram K, Rinaldi T, La Mendola D, Sandi C, Markram H. Abnormal fear conditioning and amygdala processing in an animal model of autism. Neuropsychopharmacology (2008) 33(4):901–12.
170. Wagner GC, Reuhl KR, Cheh M, McRae P, Halladay AK. A new neurobehavioral model of autism in mice: pre- and postnatal exposure to sodium valproate. J Autism Dev Disord (2006) 36(6):779–93. doi:10.1007/s10803-006-0117-y
171. Schneider T, Przewlocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology (2005) 30(1):80–9. doi:10.1038/sj.npp.1300518
172. Schneider T, Turczak J, Przewłocki R. Environmental enrichment reverses behavioral alterations in rats prenatally exposed to valproic acid: issues for a therapeutic approach in autism. Neuropsychopharmacology (2006) 31(1):36–46.
173. Tordjman S, Drapier D, Bonnot O, Graignic R, Fortes S, Cohen D, et al. Animal models relevant to schizophrenia and autism: validity and limitations. Behav Genet (2007) 37(1):61–78. doi:10.1007/s10519-006-9120-5
174. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsivant, mood stabilizer, and teratogen. J Biol Chem (2001) 276:36734–41. doi:10.1074/jbc.M101287200
175. Milutinovic S, D’Alessio AC, Detich N, Szyf M. Valproate Induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis (2007) 28(3):560–71.
176. Oh JE, Chambwe N, Klein S, Gal J, Andrews S, Gleason G, et al. Differential gene body methylation and reduced expression of cell adhesion and neurotransmitter receptor genes in adverse maternal environment. Transl Psychiatry (2013) 3:e218. doi:10.1038/tp.2012.130
177. Gleason G, Zupan B, Toth M. Maternal genetic mutations as gestational and early life influences in producing psychiatric disease-like phenotypes in mice. Front Psychiatry (2011) 2:25. doi:10.3389/fpsyt.2011.00025
178. Green JG, McLaughlin KA, Berglund PA, Gruber MJ, Sampson NA, Zaslavsky AM, et al. Childhood adversities and adult psychiatric disorders in the national comorbidity survey replication I: associations with first onset of DSM-IV disorders. Arch Gen Psychiatry (2010) 67(2):113–23. doi:10.1001/archgenpsychiatry.2009.186
179. Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet (2007) 39:319–28. doi:10.1038/ng1985
180. Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet (2008) 82:199–207. doi:10.1016/j.ajhg.2007.09.011
181. Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature (2009) 459:528–33. doi:10.1038/nature07999
182. Wisniowiecka-Kowalnik B, Nesteruk M, Peters SU, Xia Z, Cooper ML, Savage S, et al. Intragenic rearrangements in NRXN1 in three families with autism spectrum disorder, developmental delay, and speech delay. Am J Med Genet B Neuropsychiatr Genet (2010) 153B:983–93. doi:10.1002/ajmg.b.31064
183. Anderson GM, Gutknecht L, Cohen DJ, Brailly-Tabard S, Cohen JH, Ferrari P, et al. Serotonin transporter promoter variants in autism: functional effects and relationship to platelet hyperserotonemia. Mol Psychiatry (2002) 7(8):831–6.
184. Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci (2008) 31:137–45. doi:10.1016/j.tins.2007.12.005
185. Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum (2008) 7:406–16. doi:10.1007/s12311-008-0043-y
186. Benayed R, Choi J, Matteson PG, Gharani N, Kamdar S, Brzustowicz LM, et al. Autism-associated haplotype affects the regulation of the homeobox gene, ENGRAILED 2. Biol Psychiatry (2009) 66(10):911–7. doi:10.1016/j.biopsych.2009.05.027
187. James SJ, Shpyleva S, Melnyk S, Pavliv O, Pogribny IP. Complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl Psychiatry (2013) 3:e232. doi:10.1038/tp.2013.8
188. Eran A, Li JB, Vatalaro K, McCarthy J, Rahimov F, Collins C, et al. Comparative RNA editing in autistic and neurotypical cerebella. Mol Psychiatry (2013) 18(9):1041–8. doi:10.1038/mp.2012.118
189. Cook EH, Courchesne RY, Cox NJ, Lord C, Gonen D, Guter SJ, et al. Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers. Am J Hum Genet (1998) 62(5):1077–83. doi:10.1086/301832
190. Schroer RJ, Phelan MC, Michaelis RC, Crawford EC, Skinner SA, Cuccaro M, et al. Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet (1998) 76(4):327–36. doi:10.1002/(SICI)1096-8628(19980401)76:43.0.CO;2-M
191. Bolton PF, Dennis NR, Browne CE, Thomas NS, Veltman MW, Thompson RJ, et al. The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am J Med Genet (2001) 105(8):675–85. doi:10.1002/ajmg.1551
192. Patten SA, Jacobs-McDaniels NL, Zaouter C, Drapeau P, Albertson RC, Moldovan F. Role of Chd7 in zebrafish: a model for CHARGE syndrome. PLoS One (2012) 7(2):e31650. doi:10.1371/journal.pone.0031650
193. Huang HS, Allen JA, Mabb AM. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature (2011) 481:185–9. doi:10.1038/nature10726
194. Mabb AM, Judson MC, Zylka MJ, Philpot BD. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci (2011) 34(6):293–303. doi:10.1016/j.tins.2011.04.001
195. Meng L, Person RE, Beaudet AL. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet (2012) 21(13):3001–12. doi:10.1093/hmg/dds130
196. Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet A (2008) 146:2041–52. doi:10.1002/ajmg.a.32364
197. Buiting K. Prader-Willi and Angelman syndrome. Am J Med Genet C Semin Med Genet (2010) 154C:365–76. doi:10.1002/ajmg.c.30273
198. Cassidy SB, Schwartz S, Miller H, Driscoll DJ. Prader-Willi syndrome. Genet Med (2012) 14:10–26. doi:10.1038/gim.0b013e31822bead0
199. Dölen G, Carpenter RL, Ocain TD, Bear MF. Mechanism-based approaches to treating fragile X. Pharmacol Ther (2010) 127:78–93. doi:10.1016/j.pharmthera.2010.02.008
200. McLennan Y, Polussa J, Tassone F, Hagerman R. Fragile X syndrome. Curr Genomics (2011) 12(3):216–24. doi:10.2174/138920211795677886
201. Bar-Nur O, Caspi I, Benvenisty N. Molecular analysis of FMR 1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J Mol Cell Biol (2012) 4(3):180–3. doi:10.1093/jmcb/mjs007
202. Hagerman R, Lauterborn J, Au J, Berry-Kravis E. Fragile X syndrome and targeted treatment trials. Results Probl Cell Differ (2012) 54:297–335. doi:10.1007/978-3-642-21649-7_17
203. Tabolacci E, Chiurazzi P. Epigenetics, fragile X syndrome and transcriptional therapy. Am J Med Genet A (2013) 161(11):2797–808. doi:10.1002/ajmg.a.36264
204. Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science (2003) 302(5646):890–3.
205. Gadalla K, Bailey ME, Cobb SR. MeCP2 and Rett syndrome: reversibility and potential avenues for therapy. Biochem J (2011) 14:439. doi:10.1042/BJ20110648
206. Bahi-Buisson N, Bienvenu T. CDKL5-related disorders: from clinical description to molecular genetics. Mol Syndromol (2012) 2(3–5):137–52.
207. Garg SK, Lioy DT, Cheval H. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci (2013) 33:13612–20. doi:10.1523/JNEUROSCI.1854-13.2013
208. Jiang J, Jing Y, Cost GJ, Chiang JC, Kolpa HJ, Cotton AM, et al. Translating dosage compensation to trisomy 21. Nature (2013) 500(7462):296–300. doi:10.1038/nature12394
209. Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature (2013) 503(7475):267–71. doi:10.1038/nature12618
210. Phelan K, McDermid HE. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol Syndromol (2012) 2(3–5):186–201.
211. Peca J, Feliciano C, Ting JT, Wang W, Wells ME, Venkatraman TN, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature (2011) 472(7344):437–42. doi:10.1038/nature09965
212. DeBaun MR, Niemitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet (2003) 72:156–60. doi:10.1086/346031
213. Smith AC, Choufani S, Ferreira JC, Weksberg R. Growth regulation, imprinted genes, and chromosome 11p15.5. Pediatr Res (2007) 61:43–7. doi:10.1203/pdr.0b013e3180457660
214. Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet (2010) 18(1):8–14. doi:10.1038/ejhg.2009.106
215. Makeyev AV, Enkhmandakh B, Hong SH, Joshi P, Shin DG, Bayarsaihan D. Diversity and complexity in chromatin recognition by TFII-I transcription factors in pluripotent embryonic stem cells and embryonic tissues. PLoS One (2012) 7(9):e44443. doi:10.1371/journal.pone.0044443
216. Tordjman S, Anderson GM, Botbol M, Toutain A, Sarda P, Carlier M, et al. Autistic disorder in patients with Williams-Beuren syndrome: a reconsideration of the Williams-Beuren syndrome phenotype. PLoS One (2012) 7(3):e30778. doi:10.1371/journal.pone.0030778
Keywords: autistic spectrum disorders, environment, gene × environment interactions, epigenetics, multifactorial, multidisciplinary
Citation: Tordjman S, Somogyi E, Coulon N, Kermarrec S, Cohen D, Bronsard G, Bonnot O, Weismann-Arcache C, Botbol M, Lauth B, Ginchat V, Roubertoux P, Barburoth M, Kovess V, Geoffray M-M and Xavier J (2014) Gene × environment interactions in autism spectrum disorders: role of epigenetic mechanisms. Front. Psychiatry 5:53. doi: 10.3389/fpsyt.2014.00053
Received: 16 August 2013; Accepted: 02 May 2014;
Published online: 04 August 2014.
Edited by:
Bruno Etain, Assistance Publique des Hôpitaux de Paris et INSERM, FranceReviewed by:
Bernhard J. Mitterauer, Volitronics-Institute for Basic Research Psychopathology and Brain Philosophy, AustriaPauline Jeanne Chaste, University of Pittsburgh, USA
Copyright: © 2014 Tordjman, Somogyi, Coulon, Kermarrec, Cohen, Bronsard, Bonnot, Weismann-Arcache, Botbol, Lauth, Ginchat, Roubertoux, Barburoth, Kovess, Geoffray and Xavier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvie Tordjman, Professor in Child Psychiatry, Chef de Pôle Hospitalo-Universitaire de Psychiatrie de l’Enfant et de l’Adolescent, 154 rue de Châtillon, 35 200 Rennes, France e-mail: s.tordjman@yahoo.fr