Commentary: The Impact of Neuroimmune Alterations in Autism Spectrum Disorder
 Carmem Gottfried
Carmem Gottfried Victorio Bambini-Junior
Victorio Bambini-Junior Fiona Francis
Fiona Francis Rudimar Riesgo
Rudimar Riesgo Wilson Savino
Wilson Savino- 1Translational Research Group in Autism Spectrum Disorder (GETTEA), Federal University of Rio Grande do Sul, Porto Alegre, Brazil
- 2Research Group in Neuroglial Plasticity, Department of Biochemistry, Federal University of Rio Grande do Sul, Porto Alegre, Brazil
- 3Laboratory on Thymus Research, Oswaldo Cruz Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil
- 4Sorbonne Université, Université Pierre et Marie Curie, Paris, France
- 5INSERM UMR-S 839, Paris, France
- 6Institut du Fer à Moulin, Paris, France
- 7Child Neurology Unit, Clinical Hospital of Porto Alegre, Federal University of Rio Grande do Sul, Porto Alegre, Brazil
Autism spectrum disorder (ASD) involves a complex interplay of both genetic and environmental risk factors, with immune alterations and synaptic connection deficiency in early life. In the past decade, studies of ASD have substantially increased, in both humans and animal models. Immunological imbalance (including autoimmunity) has been proposed as a major etiological component in ASD, taking into account increased levels of pro-inflammatory cytokines observed in postmortem brain from patients, as well as autoantibody production. Also, epidemiological studies have established a correlation of ASD with family history of autoimmune diseases; associations with major histocompatibility complex haplotypes and abnormal levels of immunological markers in the blood. Moreover, the use of animal models to study ASD is providing increasing information on the relationship between the immune system and the pathophysiology of ASD. Herein, we will discuss the accumulating literature for ASD, giving special attention to the relevant aspects of factors that may be related to the neuroimmune interface in the development of ASD, including changes in neuroplasticity.
History of ASD Studies
The first use of the term “autistic” was in 1911, by the Swiss psychiatrist Eugen Bleuler (1857–1939), referring to the limitation of human relationships and the loss of contact with reality presented by patients with schizophrenia (1). The term was then adopted by the Austrian pediatrician Hans Asperger (1906–1980) working at the University Children’s Hospital-Vienna, referring to “autistic psychopaths.” Asperger was investigating a form of autism spectrum disorder (ASD) now known as Asperger syndrome and not widely recognized as a separate diagnosis until 1981. In 1943, the Austrian-American psychiatrist Leo Kanner (1894–1981) used the term “autistic disturbances of affective contact” to describe 11 children with behavior marked by difficulties in establishing affective and interpersonal contact (2). He reported a form distinct from other diseases, such as schizophrenia, and that seemed to affect patients from the beginning of their lives. In the following year, Hans Asperger described cases exhibiting some characteristics similar to autism, which included difficulty in social communication, but without cognitive loss. For further reading, see Ref. (3), in which Asperger’s 1944 manuscript was translated.
In 1980, the term “autism” was first inserted in the third edition of Diagnostic and Statistical Manual of Mental Disorders (DSM-III). In 1994, the fourth edition of the DSM included new criteria due to the need to identify subgroups of individuals with autism, for both practical purposes and research, considering the subdivisions: typical autism, pervasive developmental disorder not otherwise specified (PDD-NOS), and Asperger syndrome (4).
In the fifth edition, DSM considered instead of three domains of autism symptoms (social impairment, language/communication impairment, and repetitive/restricted behaviors), only two categories: (1) social communication impairment and (2) restricted interest/repetitive behaviors. Also, the new classification eliminated the previously separate subcategories into the broad term ASD (5–7). To simplify reading, the term “autism” or “ASD” will be used throughout the text representing the entire spectrum.
As a developmental disorder, ASD includes different degrees of severity and males are more affected than females by a ratio 5:1 approximately (8).
The number of cases in children increased by 23% between 2006 and 2008, reaching 1:88 children under 8 years diagnosed with any of the spectrum subtypes, and increased by 78% when the 2008 data were compared with the data for 2002. The overall prevalence of ASD for 2010 in the United States of America was 1:68 children aged 8 years and there is a clear growth in the number of identified cases (8). This can be due to advances in the knowledge of the symptoms associated with improvement in diagnostic criteria, as well as increase of environmental risk factors (drugs, pollutants, stress, etc.), especially during pregnancy, which may be related to changes in lifestyle of the society (8). In any case, this high prevalence indicates that the subject requires emergency measures due to the high economic, social, and family cost. The Autism Speaks organization estimates in the USA that the current costs of ASD reach US$137 billion per year, a number that has increased more than threefold since 2006.
Clinical Approach and Molecular Phenotypes
There are two complementary issues in the clinical approach for autism. The first is the general management, including diagnosis and evaluation of the intensity level of eventual core behavioral symptoms (9). The second considers treatment options, such as psychopharmacotherapy and different types of non-medical treatments. It is important to consider that ASD symptoms usually change during the patient’s lifetime, and therefore, it is crucial for clinicians to be aware of age-related differences. Future perspectives in the treatment of ASD will probably include immunomodulation, stem cell therapy, and other approaches, after careful randomized controlled trials attesting the corresponding efficiency of these various strategies.
Although a number of definitions and improvements have been made in ASD, the etiological aspects remain unclear. The growing number of publications, especially in the last decade, leaves no doubt of the multifactorial aspect of the spectrum and indicates a complex interplay between genetic/environmental factors and the immune system, including stimulation of immune cells, generation of autoantibodies, cytokine/chemokine imbalance, and increased permeability of the blood–brain barrier (BBB) favoring leukocyte migration into the brain tissue (10).
In addition to clinical knowledge related to ASD, intense efforts have been directed toward identifying genes that specifically cause or increase the risk of developing autism, through both large genome-wide association studies and investigation of new candidate genes (11–16). It is estimated that 400–1000 genes may be related to ASD and large-scale studies in ASD and respective families have allowed the identification of candidate genes that may be related to the development of this disorder. Single-gene polymorphisms have been associated with ASD (17, 18), including those affecting contactin-associated protein like 2 (CNTNAP2); SH3 and multiple ankyrin repeat domains 3 (SHANK3); neuroligin 3 (NLGN3/4); copy-number variations and chromosomal abnormalities, such as the 15q11–q13 duplication and 16p11.2 deletions or duplications.
Other ASD candidate genes include forkhead box P2 (FOXP2); suppression of tumorigenicity 7 (ST7); IMP2 inner mitochondrial membrane peptidase-like (IMMP2L); reelin (RELN) at 7q22–q33, gamma-amino butyric acid (GABA)A receptor subunit; and ubiquitin-protein ligase E3A (UBE3A) on chromosome 15q11–q13 (19).
Table 1 illustrates polymorphisms with correlation to gut–brain axis abnormalities. The communication between these two systems needs to be further studied in order to identify possible key elements involved in ASD symptomatology. We mention a few examples as follows. The protein MET is a pleiotropic tyrosine-kinase receptor that functions in both brain development and gastrointestinal repair. An important functional variant (rs1858830 allele “C”) of the gene encoding this protein has been demonstrated to be strongly associated with autism, as seen in a group of patients that also presented gastrointestinal disturbances (20) and altered immune response (21). Also, it was demonstrated that MET protein levels were significantly decreased in the cerebral cortex from ASD cases, compared to healthy controls (22), suggesting a dysregulation of signaling that may contribute to altered neural circuit formation and function. As the MET receptor plays important function in regulating immune responsiveness (21), it is conceivable that it has a simultaneous influence upon both brain development and gastrointestinal function.
TABLE 1
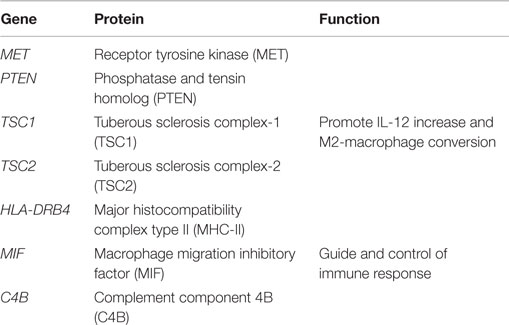
Table 1. Selected genes altered in ASD, correlated with immune function.
Overall, these data indicate molecular phenotypes, genetic risk factors, and gastrointestinal abnormalities, with the gut–brain axis. This hypothesis emerges from the observation that MET expression is decreased in temporal cortex from postmortem ASD brains and that the endogenous MET ligand, hepatocyte growth factor (HGF) is decreased in the gastrointestinal tract from ASD patients (17).
In a second vein, specific haplotypes related to the polymorphism of the SLC6A4 serotonin transporter (SERT) gene correlate with hyperfunctioning of serotonin transporter SERT in brain, in circulating platelets, and in enterocytes (17), further indicating interconnections between genetic risk factors for autism and gastrointestinal abnormalities. The SLC64A gene is found on chromosome 17q11–12 and encodes one of the SERT genes. The 5-hydroxytryptamine-transporter length polymorphism (5HTTLPR) of the SLC64A gene has been considered to be associated with abnormalities seen in serotonin transporter binding in ASD (17, 23, 24). Serotonin receptors have also been found in the gut mucous layer (25), indicating possible implications in ASD since drugs that alter serotonin levels are taken orally.
In future studies, it will be important to improve the understanding of the relationships between genetic variation and phenotype. In fact, the wide diversity of core features in ASD and a varied occurrence of comorbidities make diagnostic procedure and clinical management of the patient more difficult, presenting a complex range of brain alterations with important changes in the frontal cortex.
It should be pointed out that, in addition to genetic alterations, environmental risk factors (such as infections, and drug use) during key periods of embryonic/fetal development may be associated with triggering ASD (26). It was demonstrated that modeling a situation of maternal infection (by maternal immune activation, MIA) in mice leads to permanent immune dysregulation in the progeny animals, together with autistic-like symptoms.
Cortical Connectivity Dysfunction in ASD
Although a consensus concerning structural and functional abnormalities in ASD remains difficult, a number of studies on these topics bring together important data, as shown in Table 2. Several abnormalities have been identified, which may have a relationship with neuroimmune changes during development. These include subtle defects in cortical architecture, aggravated perhaps by perturbed critical period activity-dependent remodeling of the network. Such changes lead to white matter defects and connectivity problems, which can, in some cases, be linked to behavioral abnormalities, as discussed below.
TABLE 2
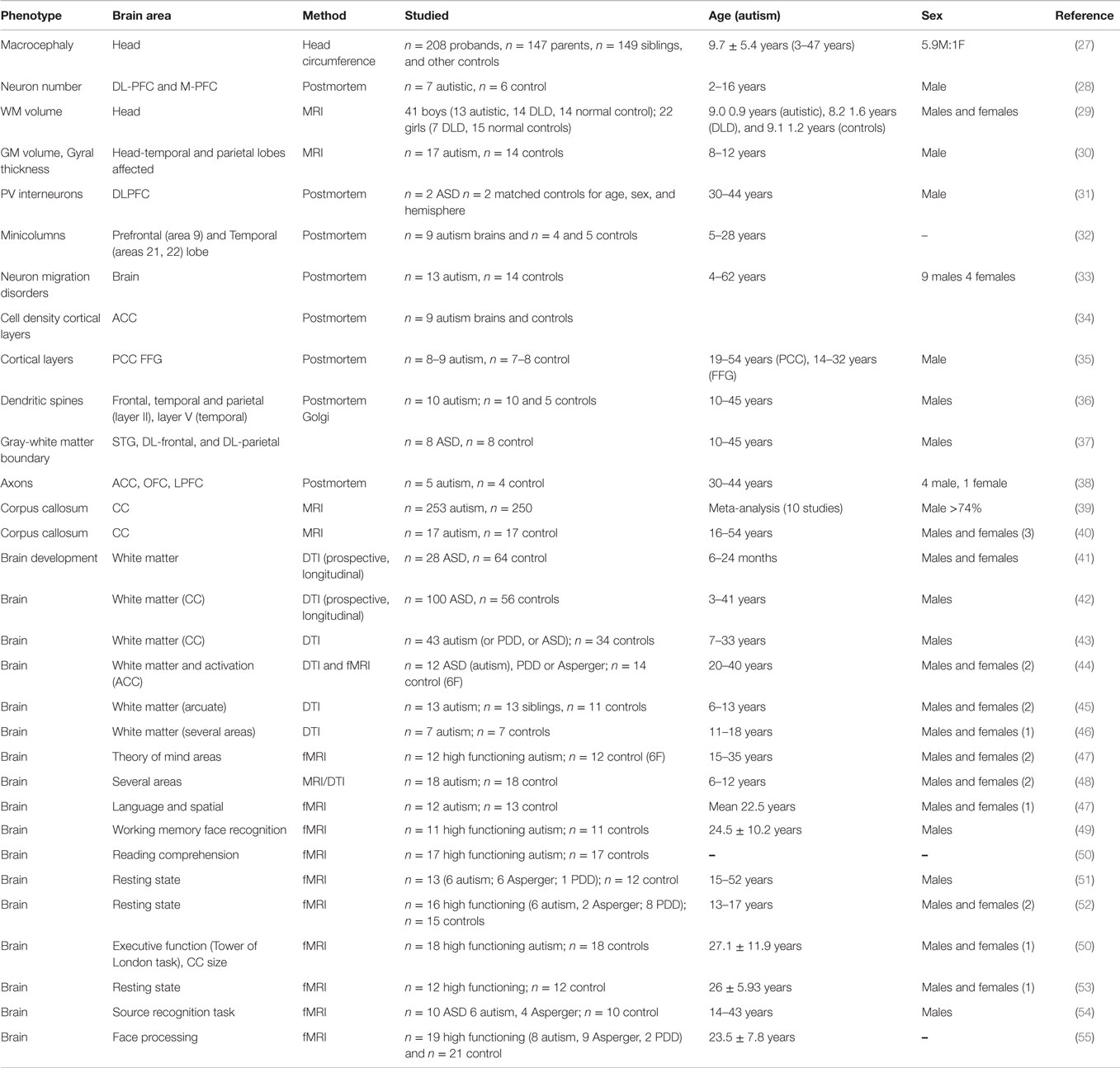
Table 2. Anatomical studies of brains from individual with ASD.
As previously mentioned, structural abnormalities are likely to be developmental in origin but may have diverse causes. Yet, before entering into this issue, it seemed worthwhile to describe normal cortical development (Box 1), also described in Ref. (56).
Box 1. General features of normal cortical development.
During early steps of cortical development, stem cells and progenitor cells divide in zones close to the cerebral ventricles before giving rise to neurons, which migrate long distances to reach the developing cortical plate. Future principal and inhibitory neurons are derived mainly from dorsal and ventral telencephalic regions, respectively. Critically timed neuronal activity is essential for circuit development, both intrinsic activity and sensory derived, affecting synaptogenesis and remodeling. Synaptic pruning removes unused and unwanted connections to refine the synaptic patterns. Timing is critical and activity-dependent processes contribute to spine turnover and maturation (57). Excitatory synapses are generally formed first, followed by inhibitory synapses. The temporal regulation of synaptogenesis is likely to be highly regulated for a correct excitatory: inhibitory balance. Myelination of mature neurons is another critical process ensuring correct functional connectivity in a timely fashion between neurons. In the primate, it has been shown that cortical areas take different amounts of time to form (58).
Environmental risk factors acting during cortical development (in utero effects related to maternal infections, stress, other agents, such as pharmaceutics, alcohol and drugs of abuse, and postnatal experience-dependent activities), can, hence, have heterogeneous influences on the formation of cortical areas. For example, maternal autoimmunity, infection during pregnancy, maternal age and obesity, gestational diabetes, and the presence of maternal MET variant rs1858830 “C” allele have been associated with the triggering of ASD through maternal immune activation, which could manifests as changes in the maternal peripheral cytokine milieu, generation of immunoglobulin G (IgG) and autoantibodies reactive to fetal brain proteins in different areas, such as frontal cortex (59).
Increased brain size (“macrocephaly”) in the first years of life is now firmly associated with ASD (60, 61). This may have its origins in increased numbers of neurons in some brain areas (as the prefrontal) compared to normal individuals (28), or more prominently in increased cell size (soma, axonal tracts, and dendrites), or in combination of both, in localized regions. Accordingly, differences are observed in ASD patients in gray or white matter volumes, both identified in MRI studies (62, 63). These increased volumes in ASD are associated with aberrant connectivity, which may combine over and under-connectivity. As mentioned below, structural and functional data revealed a connectivity disturbance, affecting frontal, fronto-temporal, fronto-limbic, fronto-parietal, and inter-hemispheric connections (31, 64).
Concerning potential gray matter abnormalities during childhood, in postmortem studies, 79 and 29% more neurons were identified in dorsolateral and medial prefrontal regions, respectively, in ASD (28). Furthermore, subtle defects in the radial organization and local densities of neurons (“minicolumns”) in different cortical areas, including the frontal cortex have been identified in brains from both ASD children and adults (65, 66), reviewed by Zikopoulos and Barbas (31). Occasionally, nodular subependymal heterotopia has been identified in ASD (33), suggesting local progenitor or neuronal migration abnormalities, although this may be rare. In the adult, increased numbers of neurons are not obvious (31), although minicolumn changes, with subtle abnormal lamination have been identified occasionally in certain areas (35, 67).
Increased dendritic spine densities have also been observed notably in layer 2 of lateral prefrontal association areas (36). Parvalbumin positive interneurons have been shown in postmortem studies to be less numerous in dorsolateral prefrontal regions (31). Blurred white and gray matter boundaries are also regularly observed in ASD (37, 65), and this has been suggested in other situations (e.g., schizophrenia) to be due to an excess of interstitial neurons in the white matter, which may have their origin in the subplate (68). Overall, several neuronal density and distribution alterations, localized to certain areas, are associated with ASD.
A number of changes in brain seem to be related to late prenatal or early postnatal periods. Transiently enlarged white matter volumes (related to abnormal axonal tracts) have been documented in ASD infants exhibiting enlarged head circumferences. White matter volumes in these individuals then increase less slowly during childhood compared to control individuals [reviewed by Cassina et al. (69)]. Axonal tracts have been studied by confocal and electron microscopy in postmortem tissue (38), showing fewer large axons in the deep white matter of the anterior cingulate cortex (likely representing long-range cortico-cortical connections), a higher proportion of branched axons of medium caliber, and a significantly increased density of thinner branched, axons in the superficial white matter (likely connecting nearby areas). Other neuroimaging studies have shown reductions in the strength of long-distance connections, e.g., sensory input to prefrontal cortex and inter-hemispheric connections (40, 43, 70–72). Such defects would be expected to have quite severe network effects.
Travers et al. (73) (and references therein) summarize and compare 48 peer-reviewed diffusion tensor imaging (DTI) studies. Preliminary findings suggest that developmental trajectories of fractional anisotropy in ASD infants are also different from controls, and may mimic the accelerated brain volume phenotype (41, 73). Despite small sample sizes, the corpus callosum was found in several DTI studies to be reduced in volume [Ref. (43), see Figure 3 of Ref. (73)], and in one study the authors further found this result to be specific to patients who did not have macrocephaly (61). Interestingly and vice versa, callosal agenesis is also associated with autism-like symptoms. Concerning microstructure, fractional anisotropy was found reduced in anterior regions or across the length of the corpus callosum in multiple studies (42, 43, 73). This may be due to reduced myelination or larger axon diameter or reduced density. In some studies, this finding was associated with reduced performance IQ and reduced callosal volume (43). Differences were observed in ASD patients, concerning the cingulum bundles, which are primary inter-hemispheric-association pathways associated with executive function, connecting the medial cingulate cortex with temporal lobe structures, such as the hippocampus, consistent macrostructural, and reduced fractional anisotropy (44). Relatively concordant results of decreased fractional anisotropy were obtained at the beginning of the arcuate fasciculus (although heterogeneous results were obtained for the whole tract) in the region of the temporo-parietal junction and superior temporal gray matter (45, 46, 73). This latter region is associated with social perception, and gray matter structure and functional connectivity differences have also previously been identified (47). For the moment, relationships between DTI measures and ASD symptoms remain only preliminary and future work with defined patient groups will deepen these correlations [see also Ref. (44)].
Studies using DTI also show differences in the cerebellar fibers that connect to various brain regions, demonstrating altered cerebellar feedback projections in ASD (74). In addition, neuropathological studies have also reported a decrease in Purkinje cell density in the cerebellum of ASD patients indicating that this abnormality may contribute to selected clinical features of the autism phenotype (75).
Functional magnetic resonance imaging (fMRI) studies are being used to assess synchronous activated and deactivated brain regions during cognitive tasks and in resting states in ASD patients [reviewed by Rathinam et al. (76)]. It appears that the most consistent functional results refers to a decreased connectivity between frontal and more posterior brain regions (in high-functioning patients), performing a variety of tasks. These include task integrating language comprehension (frontal) and spatial processing (parietal) (77); in working memory tasks related to face recognition [involving frontal executive and occipito-temporal fusiform gyrus regions (49)], and in reading comprehension requiring language comprehension and working memory (50). Similarly, frontal-posterior under-connectivity has also been found in studies of patients at rest, revealing hence spontaneous brain activity connections (51, 52, 78). These resting state studies suggest that abnormal connectivity may already exist in patient brains, not specifically related to different tasks, and perhaps indicating a structural basis for some differences, as suggested above. There is, however, some heterogeneity in other fMRI results, since some tasks in some patients have also shown frontal-posterior over-connectivity (79), fronto-frontal, or posterio-posterior over-connectivity in the resting state (53). Analyzing connections with other brain regions, e.g., subcortico-cortico, has also in several cases revealed over-connectivity [e.g., in task-independent tasks, Ref. (72) and references therein], or under-connectivity (55). A variety of other brain regions have been analyzed contributing to the variability of the results obtained [Ref. (80) and references therein]. In addition, transcranial ultrasonography may be a useful screening technique for children at potential risk of ASD, providing rapid, non-invasive evaluation of extra-axial fluid and cortical lesions (81). Further work, potentially involving new methods, may help to clarify under- or over-connectivity in different brain regions.
Whether or not these changes are related to neuroimmune interactions is a completely open field of investigation. In particular, it should be helpful to perform correlation studies between the above described changes with specific immune activation states, such as infections.
Crosstalk between the CNS and the Immune System in ASD
The crosstalk involving the immune and nervous systems encompasses a complex and intricate pathway of signals with extensive communication between them in health and disease (82, 83). Cytokines and chemokines modulate brain function, as well as systemic and CNS responses to infection, injury, and inflammation (84). In fact, cytokines, such as TNF-α, IL-1β, IL-6, and TGF-β family, are able to modulate neuronal activity (85) and IL-6 promotes oligodendrocyte survival (86).
Pro-inflammatory cytokines, including interleukin (IL)-1, IL-6, IL-12, interferon-γ (IFN-γ), and tumor necrosis factor α (TNF-α), are involved in CNS pleiotropic effects during neurodevelopment (87) and have been extensively studied in patients with ASD. In a pioneer work indicating immune dysfunction in ASD (88), cell-mediated immune response was assessed in vitro by phytohemagglutinin (PHA) stimulation in lymphocyte cultures from 12 children with ASD and 13 control subjects: the ASD against neural antigens, produced by the mother during pregnancy (89–92), and that may induce changes in neural development and plasticity in the developing embryo/fetus.
Anti-double-stranded DNA antibodies and anti-nuclear antibodies were measured in the sera of 100 autistic children, aged between 4 and 11 years, in comparison to 100 healthy-matched children (93). In this study, the authors found increased levels of anti-double-strand DNA (34%) or anti-nuclear antibodies (25%) in ASD children. Furthermore, meta-analysis of data reported in patients with ASD clearly revealed alterations in different cytokines, both in plasma and in brain, as seen in Table 3.
TABLE 3
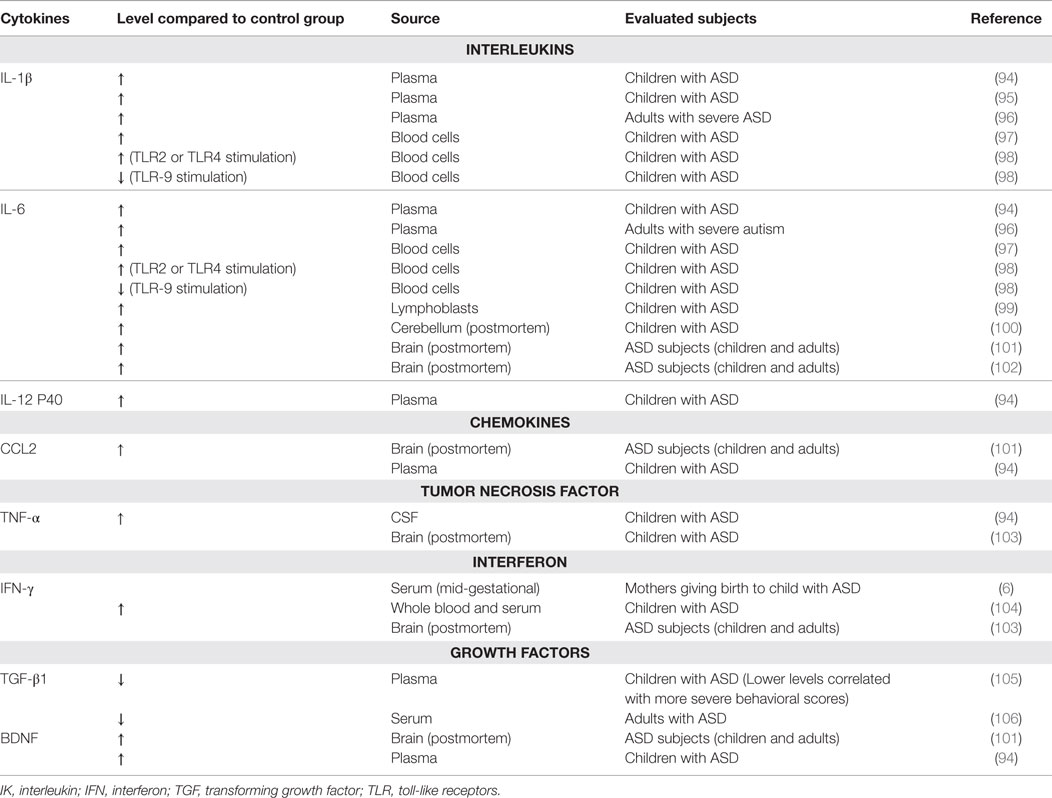
Table 3. Altered cytokines in autism spectrum disorder (ASD).
Also, although ASD patients present reduced amounts of total IgM and IgG immunoglobulins contents, they exhibit increased levels of antibodies against various proteins expressed in the nervous tissue, e.g., serotonin receptors, myelin basic protein, heat shock protein, and glial fibrillary acidic protein (GFAP) (107, 108). Recently, the presence of autoantibodies against human neuronal progenitor cells (NPCs) was assayed in sera from children with ASD (109). Immunoreactivity against multiple NPC proteins of molecular sizes ranging from 55 to 210 kDa was found in the ASD group, significantly differing from control individuals. This is in keeping with the fact that in the mouse model of autism following maternal immune activation triggered by poly(I:C)-injection, offspring exhibited a reduction of 50% in the numbers of regulatory T lymphocytes (CD4+Foxp3+CD25+) in the spleen (110), indicating a dysfunction in the regulation of the immune response in autism.
As mentioned above, studies in animal models indicate that maternal immune activation leads to autistic-like behavioral patterns in the offspring (111, 112). In addition to B and T cell abnormalities, changes in the innate immune response have been reported. Using in vitro experiments, it was demonstrated that ASD individuals have a reduced capacity of natural killer (NK) cells to kill K562 target cells (an immortalized myelogenous leukemia cell line) (113). Thus, it is likely that an aberrant group showed impaired lymphocyte PHA-induced proliferation when compared to control subjects.
In the following years, the hypothesis of autoimmunity involving the CNS was postulated as a key issue in the pathogenesis of autism and various clinical studies indicated a link between dysfunctional immune activity and ASD, including maternal immune abnormalities during early pregnancy (10, 114) and increased incidence of familial autoimmunity (115). Additionally, autoimmunity triggered by viral or bacterial infections has been considered as risk factor to ASD development (87, 116, 117). It has also been demonstrated in humans that family history of autoimmune disorders is more common in families of children with ASD (118). In addition, immune-mediated disorders during pregnancy, such as allergy and psoriasis, are more frequent in mothers of children with ASD compared with mothers of children with typical development (119).
Yet, the biological mechanism(s) of maternal immune dysfunction that could be involved in triggering ASD remain(s) unclear. One possibility involves the generation of antibodies activity of these components of innate immunity may also contribute to atypical immune activity seen in patients with ASD.
Moreover, increased numbers of circulating monocytes, important precursors for macrophages, dendritic, and microglial cells, have been observed in the blood and in the postmortem brain tissue from ASD individuals, associated with the presence of perivascular macrophages (101, 120). Furthermore, analysis of cytokine serum levels in children with ASD revealed a representative profile of myeloid cell activation, with increased production of IL-14, IL-12p40, TNF-α, IL-1β, and IL-6 (94–97, 121). Also, increased level of TNF-α was found in cerebrospinal fluid of children with ASD (122).
In respect to caspases, a group of cysteinyl aspartate-specific proteases involved in apoptosis and several other cell functions, it has been shown that the activation of some members of the caspase family contributes to the differentiation of monocytes into macrophages, in the absence of cell death (123). Interestingly, the mRNA levels for caspases 1–5, 7, and 12 were significantly increased in ASD patients as compared to healthy subjects, suggesting a role of the caspase pathway in ASD clinical outcome and as potential diagnostic and/or as therapeutic tools (124). These studies will hopefully provide new insights in the mechanisms of caspase activation and abnormal differentiation of monocytes into macrophages in ASD.
Considering that monocytes are key elements for the immune response, these alterations may result in long-term immune alterations in ASD children, with adverse neuroimmune interactions, ultimately contributing to the ASD pathophysiology. Also, it was found increased expression levels of pro-inflammatory cytokines TNF-alpha and IL-6, and decreased Bcl2 expression in lymphoblasts (99) and decreased levels of TGF-β in plasma (105) and in serum (106) of autistic subjects.
Moreover, considering that increased levels of anti- and pro-inflammatory cytokines have been observed in ASD individuals (6), it is conceivable that cytokines are also involved in the pathophysiology of ASD.
Taking into account, the environmental in utero influence in triggering ASD changes in oxidative stress responses may also correlate with activation of the hypothalamic–pituitary–adrenal (HPA) axis. Upon activation, the hypothalamus secretes corticotropin-releasing hormone (CRH), stimulating the anterior pituitary gland to secrete adrenocorticotropic hormone (ACTH), which in turn stimulates the cortex of the adrenal glands to release glucocorticoid, which plays an important role in adaptive responses (125), including immunosuppression. This response can signal to the organism under stressful events, such as environmental adverse factors in utero. In fact, patients with ASD present elevated blood levels of nitric oxide (NO), nitrites, and nitrates (126). These molecules might increase the permeability of BBB and intestinal permeability, as commonly found in autism (127). Furthermore, ASD patients have diminished antioxidant systems in plasma, including decreased amounts of glutathione (GSH), vitamins (A, C, and E), and antioxidant enzymes (superoxide dismutase and glutathione peroxidase) (128–130). The increase in oxidative stress can potentially induce dysfunction in the immune system, plasticity and function of the thymus and stimulate neuroinflammatory infiltrates. Potentially, this set of dysfunctions may be associated with the behavioral abnormalities, gastrointestinal disorders, and sleep disturbances present in autism. In an animal model of ASD induced by prenatal exposure to valproic acid (VPA), a reduced thymus size was observed in the VPA group, compared to the control animals (131), indicating that T-cell development can also be affected in autism, and may be at the origin of both T and B cell dysfunctions seen in ASD, including neuroinflammation.
One important point is that, although the well accepted fact that the CNS undergoes constant immune surveillance that takes place within the meningeal compartment (132), the real mechanism(s) that guide(s) the entrance and exit of immune cells from the CNS remains to be demonstrated. Recently, an interesting investigation revealed the presence of structures with functional lymphatic vessels lining the dural sinuses, in a place difficult to visualize and actually so far ignored. These structures present characteristics of lymphatic endothelial cells and are able to carry both fluid and immune cells from and into the cerebrospinal fluid. Importantly, these structures are connected to the deep cervical lymph nodes (132). From this view, it is clear that a new and important window of investigation starts, in the search for possible link(s) connecting triggering of ASD to immune system impairment and vice versa.
Evidence for Neuroimmune Interactions in ASD
The intercommunication between the brain and blood systems is followed by integrative exchanges, and the BBB permeability is variable, depending on the vessel type (artery, capillary, or vein) (8). During development, neurons, astrocytes, oligodendrocytes, and microglia intercommunicate in a paracrine/autocrine manner (133), withstand endocrine and immune systems influences, particularly during pregnancy, which can impair functions of the nervous system. Microglial cells in turn act as surveillance systems, with the capacity to respond phenotypically with varying degrees of activation to fluctuations in microenvironment stimuli or to transient or chronic damage, reaching the phagocytic state in the event of cell death (134). These cells also present dynamic movements or projections able to detect irregularities in neural microenvironments, in both intra and extracellular milieu and can increase in number by proliferation or through the entrance of macrophages into the brain (135).
In this vein, it was demonstrated by analysis of postmortem brain tissue that individuals with autism have an increased number of activated microglial cells (136).
Figure 1 illustrates alterations found in both blood and postmortem brains of patients with ASD, including blood/brain cell activation, autoantibody production, and alterations in levels of different molecules that can modify cell signaling, brain response, and BBB permeability. The associated neuroinflammatory process does support the hypothesis of neuroimmune interactions in the pathogenesis of ASD.
FIGURE 1
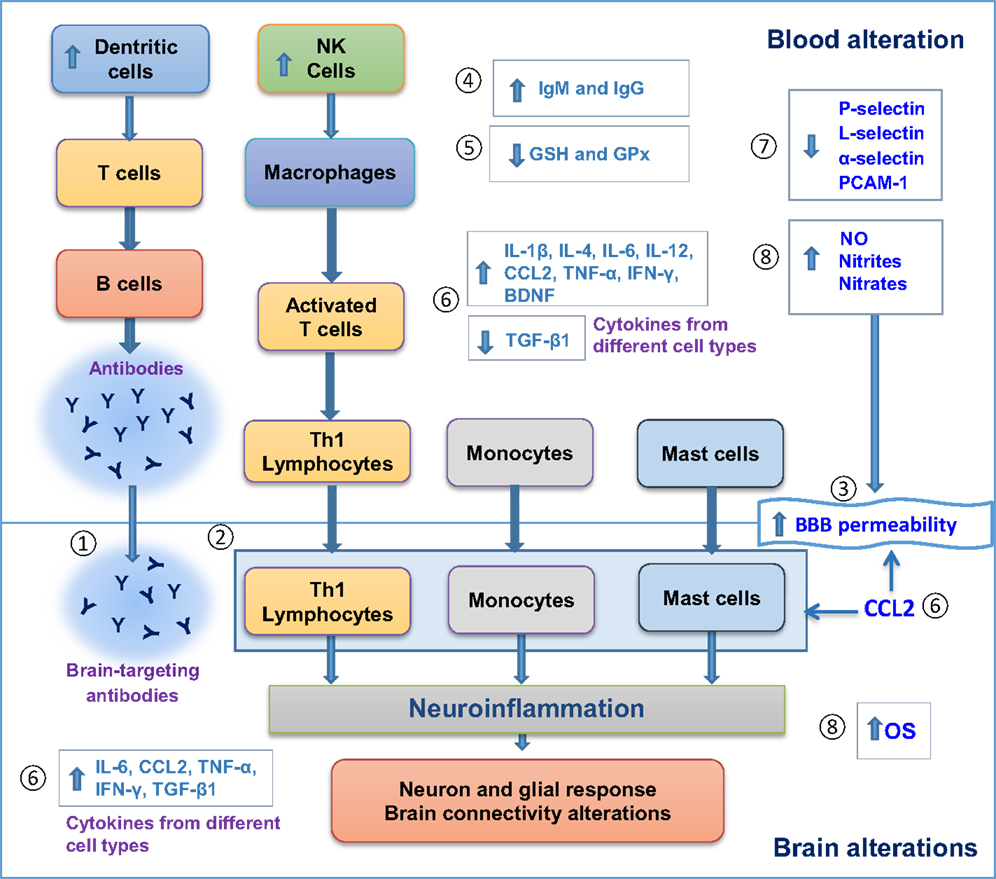
Figure 1. Evidence for neuroimmune interactions in autism spectrum disorder (ASD). Blood and postmortem brain alterations in individuals with ASD. (1) Antibody production in blood against brain antigens. (2) Brain cell infiltration of Th1 lymphocytes, monocytes and mast cells. (3) Increase in blood brain barrier (BBB) permeability. (4) Increase in IgG and IgM levels. (5) Less antioxidant defenses. (6) Changes in cytokine levels. (7) Decrease in cell adhesion molecules, such as Selectins and PCAM-1. 8. Increase in oxidative stress. All these alterations can promote neuroinflammation, followed by neuron–glial response and brain connectivity dysfunction that ultimately can influence behavioral features in ASD. GSH, glutathione; GPx, glutathione peroxidase; NO, nitric oxide; Th, T-helper; OS, oxidative stress; CCL2, C–C motif chemokine 2.
The analysis of postmortem brains from ASD individuals indicates changes in synaptic organization, dendritic arborization, neurotransmission (i.e., GABAergic, serotonergic, and glutamatergic pathways), and glial cells. Accordingly, recent studies suggested an important role for astrocytes and microglial cells in ASD, with alterations in GFAP expression (137), and increases of pro-inflammatory cytokines (6).
Molecules secreted by the brain’s immune system may influence neurodevelopment. As already mentioned above, individuals with autism have a marked neuroinflammation, with microglial activation and increased NO, as well as production of chemokines and pro-inflammatory cytokines (6, 101). There is evidence that an increase of TNF-α is associated with stereotypic behaviors similar to those found in individuals with autism (138). Moreover, soluble cytokine receptors that are normally present in blood can regulate peripheral cytokine and lymphoid activity (139–141). Further elucidation and characterization of the molecular pathways that mediate soluble cytokine receptor signaling in ASD will promote new strategies for therapeutic interventions.
In addition, as demonstrated in Table 1, the genes MET, PTEN, TSC1, and TSC2, for example, encode proteins related to the phosphoinositide3-kinase (PI3K) pathway, which plays an important role in suppressing the production of the pro-inflammatory cytokine IL-12 (142). MET is important to the developing brain, particularly to the neocortex and cerebellum in two regions frequently compromised in autism (22). Also, alteration in this gene can be correlated with increased immune response, involving cytokine expression (21) and regulation by small RNAs (miRNA) (143), which are presently known to be associated with the immunological response, such as lymphocytic phenotypes or key points during hematopoiesis (144). In ASD, various miRNA are altered in the blood, providing new clues in the search for new molecular targets in the study of autism (145–147). One is miR-132, altered in both autism and schizophrenia, and that can participate in brain plasticity, connectivity, and regulation of immune responses (145).
Another area implicated in autism is the cerebellum, and immunological studies indicate increased levels of IL-6 in the cerebellum of ASD subjects, stimulating the formation of granule cell excitatory synapses, without affecting inhibitory synapses (100).
A relevant point to be considered in the neuroimmune interactions occurring in autism is the fact that the intestinal mucosa of children with autism has a higher frequency of TNF-α+ T cells and lower frequency of IL-10+ T cells (148, 149). These studies indicate that such lymphocytes assume a pro-inflammatory profile, which corroborates with the increased levels of pro-inflammatory cytokines found in plasma and brain of patients with ASD.
Another important issue is the strong association between autism and allergic response involving mast cells, which correlates with various cellular processes, including allergic reactions enteric nervous system (ENS) (87, 150).
Increased plasma levels of IgG4 in children with ASD were also observed (151). These changes may be linked to changes in BBB permeability and also may influence neural plasticity and function, resulting in impairment in social interaction, communication, and behavior (87).
It is also important to consider cell adhesion molecules (CAM), which are present in endothelial cells, promoting a direct and selective interaction between blood cells and the cerebral endothelium (152). It is well known that CAMs play an important role in mediating the passage of T cells through endothelial barriers (153). These data indicate that the modulation of immune cell entry into the brain from patients with autism might also be a potential therapeutic target.
Working with the animal model of ASD induced by prenatal exposure of VPA, we recently demonstrated that the treatment of pregnant females with the antioxidant and anti-inflammatory resveratrol (RSV), before and after VPA exposure, prevented all behavioral impairments observed in the offspring (154). This is a naturally occurring phytochemical that was detected in 1963 in the dried roots of Polygonum cuspidatum (Itadori tea) and has been proposed as a pharmacological tool for neuroprotection against neuronal injury, including age-associated chronic diseases (155), ischemic brain damage (156), and cerebral models of stroke (157). For a systematic review and recommendations on the use of RSV, read (158).
Since similar alterations are also observed in the animal model induced by VPA (131, 159–161) and RSV exerts anti-inflammatory effects (158), future studies will be relevant to evaluate the influence of RSV in the immune system, particular in the ASD context. There is evidence for RSV use to establish immunological tolerance during treatment of autoimmune diseases that ablate or suppress the immune system. Specifically, RSV effect on tolerance is likely to be in the induction of Foxp3+ T cells and IL-10 expression, which are critical to development of T cells that are protective against autoimmune diseases, such as multiple sclerosis (162). In addition, the administration of RSV to mice developing experimental autoimmune encephalomyelitis – an animal model of human multiple sclerosis – increases expression of IL-10 and Foxp3 in T cells, the animal model of multiple sclerosis (163). In order to advance the knowledge related ASD development, it is important to also evaluate intracellular targets of VPA and RSV to clarify molecules and pathways affected by both. In this respect, we anticipate that further understanding of these molecular targets will be relevant to both therapeutic and etiological aspects of ASD. Similarly, such studies will hopefully help us to understand ASD-related epigenetic modulation and developmental alterations implicated in the neural and behavioral impairments.
In Table 4, we have summarized outcomes, breakthroughs, or major findings in animal models, relating to ASD and immune system activation. In the case of animal models of maternal immune activation, there is a cascade of inflammatory responses that are dependent on the pathogenic agent and can potentiate immune responses in offspring in a strain-dependent manner (111). It is hypothesized that pro-inflammatory cytokines, brain-reactive antibodies, and endocrine mediators, such as corticotropin-releasing factor and glucocorticoids participate in the etiology of autoimmunity-associated behavioral syndrome (164). Also, neonatal rat infection with Borna disease virus results in abnormalities of early development and increase in locomotor activity; stereotypies and brain expression of mRNA for IL-1α, IL1-β, IL-6, TNF-α, and TNF-β (165).
TABLE 4
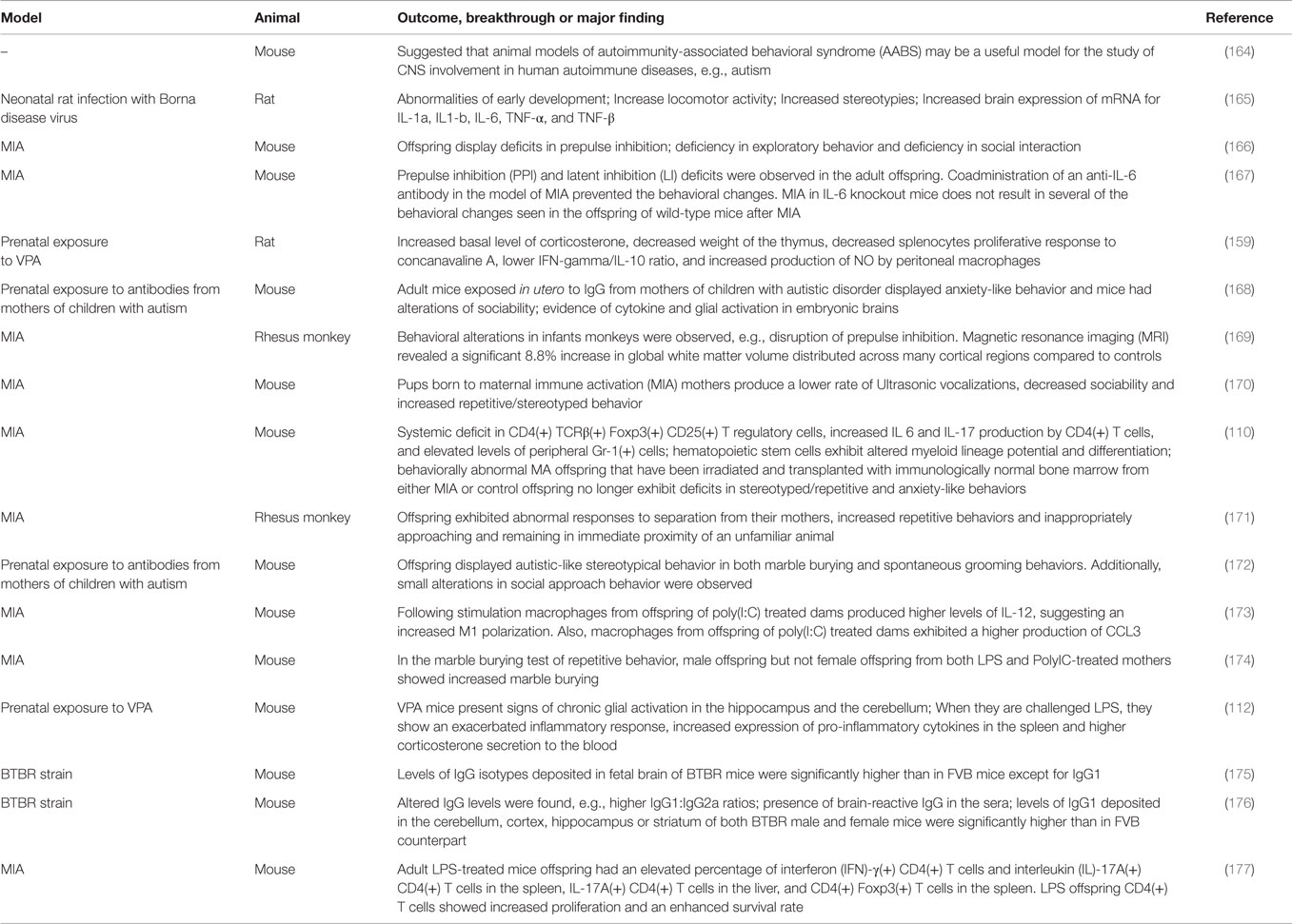
Table 4. Selected findings in animal models related to ASD and immune system.
Animal models of maternal infection have also been used to study behavioral impairments and brain alterations, such as maternal influenza infection (166), maternal immune activation (110, 167, 169–171, 173, 174, 177, 178), and prenatal exposure to antibodies (168, 172). In addition, the inbred BTBR T + tf/J (BTBR) mouse strain has been used as an animal model of core behavioral deficits in autism. BTBR mice exhibit repetitive behaviors and deficits in sociability and communication, presenting higher IgG1:IgG2a ratios and increased levels of IgG1 in brain (175, 176).
Summary and Outlook
Since the first descriptions of autism, 70 years of investigation have passed, with great efforts mainly in the last decade, bringing important information and knowledge on the mechanisms underlying ASD. Nevertheless, even with these advances, the etiology of ASD remains largely unknown and we are still searching for specific clinical marker(s) able to improve early diagnosis. We work on the hypothesis that integrating maternal–embryo systems will contribute to the understanding of ASD. One possibility, which was summarized here, concerns the hypothesis of neuroimmune interactions being involved in triggering ASD development, as schematically depicted in Figure 2. The presence of environmental risk factors during critical periods of embryonic/fetal development may influence the immune system in the mother, promoting localized or systemic inflammatory responses with the release of cytokines and hormonal molecules, which in turn, via neuroimmunomodulatory responses and crosstalk between circulatory and neural systems, may impair circuitry development, neuronal plasticity, and neuroglial function in the embryo/fetus. As immunological factors interfere with neural development since the embryonic period, and considering that inflammation or immune response may arise due to abnormal environmental interactions in utero, a better understanding of the neuroimmune changes that may underlie the pathogenesis or pathophysiology of ASD will hopefully have a large impact on the development of new clinical and therapeutic strategies to better deal with ASD.
FIGURE 2
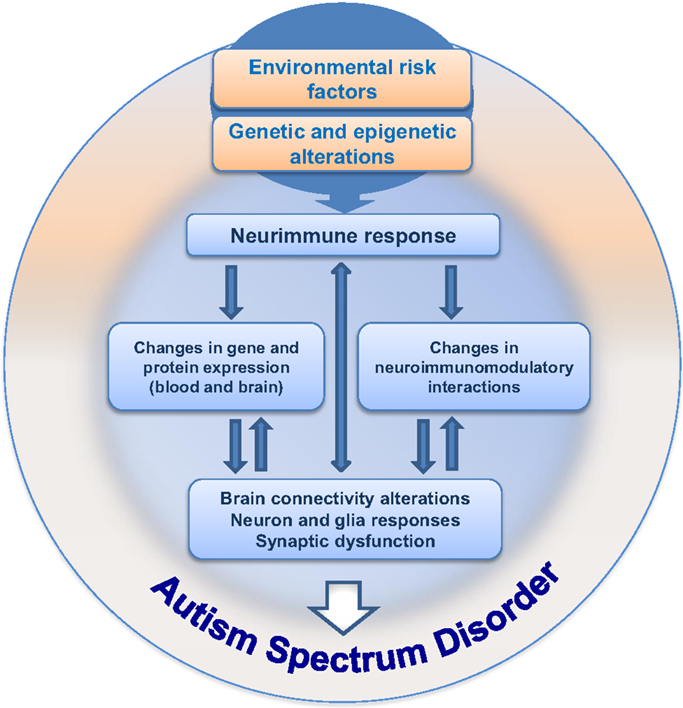
Figure 2. Hypothesis for neuroimmune interactions in triggering the development of ASD. This hypothesis considers the presence of environmental risk factors during pregnancy, followed by immunoneuroendocrine response from the mother to the developing embryo/fetus. The risk factors (such as VPA) would influence central and peripheral neural responses in the context of a crosstalk with the immune system, followed by gradual changes in neural plasticity and function, resulting in behavioral impairment during development, ultimately leading to ASD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
CG and RR: National Council of Technological and Scientific Development (CNPq), Coordination for the Improvement of Higher Education Personnel (CAPES), and Clinical Hospital of Porto Alegre (FIPE-HCPA). FF thanks other members of the lab for their contribution to discussions on this subject. Financial support for FF’s lab was received from INSERM and UPMC and the Région Ile-de-France. FF is supported by the CNRS, and is associated with the BioPsy Labex project and the Ecole des Neurosciences de Paris Ile-de-France network. VB-J and WS: CNPq, CAPES, FAPERJ (The funding research agency of the Estado do Rio de Janeiro), Fiocruz, and Mercosur Fund for Convergence (FOCEM), Mercosur.
References
1. Ashok AH, Baugh J, Yeragani VK. Paul Eugen Bleuler and the origin of the term schizophrenia. Indian J Psychiatry (2012) 54:95–6. doi:10.4103/0019-5545.94660
4. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523(7560):337–41. doi:10.1038/nature14432
5. Giovannoni G, Miller RF, Heales SJ, Land JM, Harrison MJ, Thompson EJ. Elevated cerebrospinal fluid and serum nitrate and nitrite levels in patients with central nervous system complications of HIV-1 infection: a correlation with blood-brain-barrier dysfunction. J Neurol Sci (1998) 156:53–8. doi:10.1016/S0022-510X(98)00021-5
6. Goines PE, Ashwood P. Cytokine dysregulation in autism spectrum disorders (ASD): possible role of the environment. Neurotoxicol Teratol (2013) 36:67–81. doi:10.1016/j.ntt.2012.07.006
8. Forrester JV, Xu H, Lambe T, Cornall R. Immune privilege or privileged immunity? Mucosal Immunol (2008) 1:372–81. doi:10.1038/mi.2008.27
9. Riesgo RS, Gottfried C, Becker M. Clinical Approach in Autism: Management and Treatment, In Recent Advances in Autism Spectrum Disorders. Rijeka, Croatia: InTech (2013).
10. Verkhratsky A, Rodriguez JJ, Parpura V. Neuroglia in ageing and disease. Cell Tissue Res (2014) 357:493–503. doi:10.1007/s00441-014-1814-z
11. Weiss LA. Autism genetics: emerging data from genome-wide copy-number and single nucleotide polymorphism scans. Expert Rev Mol Diagn (2009) 9:795–803. doi:10.1586/erm.09.59
12. Wall DP, Esteban FJ, Deluca TF, Huyck M, Monaghan T, Velez de Mendizabal N, et al. Comparative analysis of neurological disorders focuses genome-wide search for autism genes. Genomics (2009) 93:120–9. doi:10.1016/j.ygeno.2008.09.015
13. Matuszek G, Talebizadeh Z. Autism genetic database (AGD): a comprehensive database including autism susceptibility gene-CNVs integrated with known noncoding RNAs and fragile sites. BMC Med Genet (2009) 10:102. doi:10.1186/1471-2350-10-102
14. Miles JH. Autism spectrum disorders – a genetics review. Genet Med (2011) 13:278–94. doi:10.1097/GIM.0b013e3181ff67ba
15. Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron (2013) 77:259–73. doi:10.1016/j.neuron.2012.11.002
16. Moreno-De-Luca D, Sanders SJ, Willsey AJ, Mulle JG, Lowe JK, Geschwind DH, et al. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry (2012) 18:1090–5. doi:10.1038/mp.2012.138
17. Hsiao EY. Gastrointestinal issues in autism spectrum disorder. Harv Rev Psychiatry (2014) 22:104–11. doi:10.1097/HRP.0000000000000029
18. Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet (2013) 93:249–63. doi:10.1016/j.ajhg.2013.06.012
19. Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatrics (2004) 113:e472–86. doi:10.1542/peds.113.5.e472
20. Campbell DB, Buie TM, Winter H, Bauman M, Sutcliffe JS, Perrin JM, et al. Distinct genetic risk based on association of MET in families with co-occurring autism and gastrointestinal conditions. Pediatrics (2009) 123:1018–24. doi:10.1542/peds.2008-0819
21. Heuer L, Braunschweig D, Ashwood P, Van de Water J, Campbell DB. Association of a MET genetic variant with autism-associated maternal autoantibodies to fetal brain proteins and cytokine expression. Transl Psychiatry (2011) 1:e48. doi:10.1038/tp.2011.48
22. Campbell DB, D’Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt P, et al. Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann Neurol (2007) 62:243–50. doi:10.1002/ana.21180
23. Sakurai T, Reichert J, Hoffman EJ, Cai G, Jones HB, Faham M, et al. A large-scale screen for coding variants in SERT/SLC6A4 in autism spectrum disorders. Autism Res (2008) 1:251–7. doi:10.1002/aur.30
24. Coutinho AM, Sousa I, Martins M, Correia C, Morgadinho T, Bento C, et al. Evidence for epistasis between SLC6A4 and ITGB3 in autism etiology and in the determination of platelet serotonin levels. Hum Genet (2007) 121:243–56. doi:10.1007/s00439-006-0301-3
25. Kunze A, Lengacher S, Dirren E, Aebischer P, Magistretti PJ, Renaud P. Astrocyte-neuron co-culture on microchips based on the model of SOD mutation to mimic ALS. Integr Biol (Camb) (2013) 5:964–75. doi:10.1039/c3ib40022k
26. Kim YS, Leventhal BL. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol Psychiatry (2014) 77:66–74. doi:10.1016/j.biopsych.2014.11.001
27. Lainhart JE, Bigler ED, Bocian M, Coon H, Dinh E, Dawson G, et al. Head circumference and height in autism: a study by the collaborative program of excellence in autism. Am J Med Genet A (2006) 140(21):2257–74. doi:10.1002/ajmg.a.31465
28. Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, et al. Neuron number and size in prefrontal cortex of children with autism. JAMA (2011) 306:2001–10. doi:10.1001/jama.2011.1638
29. Herbert MR, Ziegler DA, Makris N, Filipek PA, Kemper TL, Normandin JJ, et al. Localization of white matter volume increase in autism and developmental language disorder. Ann Neurol (2004) 55(4):530–40. doi:10.1002/ana.20032
30. Hardan AY, Muddasani S, Vemulapalli M, Keshavan MS, Minshew NJ. An MRI study of increased cortical thickness in autism. Am J Psychiatry (2006) 163(7):1290–2. doi:10.1176/ajp.2006.163.7.1290
31. Zikopoulos B, Barbas H. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front Hum Neurosci (2013) 7:609. doi:10.3389/fnhum.2013.00609
32. Casanova MF, Buxhoeveden DP, Switala AE, Roy E. Minicolumnar pathology in autism. Neurology (2002) 58(3):428–32. doi:10.1212/WNL.58.3.428
33. Wegiel J, Kuchna I, Nowicki K, Imaki H, Marchi E, Ma SY, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol (2010) 119:755–70. doi:10.1007/s00401-010-0655-4
34. Simms ML, Kemper TL, Timbie CM, Bauman ML, Blatt GJ. The anterior cingulate cortex in autism: heterogeneity of qualitative and quantitative cytoarchitectonic features suggests possible subgroups. Acta Neuropathol (2009) 118(5):673–84. doi:10.1007/s00401-009-0568-2
35. Oblak AL, Rosene DL, Kemper TL, Bauman ML, Blatt GJ. Altered posterior cingulate cortical cyctoarchitecture, but normal density of neurons and interneurons in the posterior cingulate cortex and fusiform gyrus in autism. Autism Res (2011) 4:200–11. doi:10.1002/aur.188
36. Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res (2009) 1309:83–94. doi:10.1016/j.brainres.2009.09.120
37. Avino TA, Hutsler JJ. Abnormal cell patterning at the cortical gray-white matter boundary in autism spectrum disorders. Brain Res (2010) 1360:138–46. doi:10.1016/j.brainres.2010.08.091
38. Zikopoulos B, Barbas H. Changes in prefrontal axons may disrupt the network in autism. J Neurosci (2010) 30:14595–609. doi:10.1523/JNEUROSCI.2257-10.2010
39. Frazier TW, Hardan AY. A meta-analysis of the corpus callosum in autism. Biol Psychiatry (2009) 66(10):935–41. doi:10.1016/j.biopsych.2009.07.022
40. Casanova MF, El-Baz A, Elnakib A, Switala AE, Williams EL, Williams DL, et al. Quantitative analysis of the shape of the corpus callosum in patients with autism and comparison individuals. Autism (2011) 15:223–38. doi:10.1177/1362361310386506
41. Wolff JJ, Gu H, Gerig G, Elison JT, Styner M, Gouttard S, et al. Differences in white matter fiber tract development present from 6 to 24 months in infants with autism. Am J Psychiatry (2012) 169:589–600. doi:10.1176/appi.ajp.2011.11091447
42. Travers BG, Tromp do PM, Adluru N, Lange N, Destiche D, Ennis C, et al. Atypical development of white matter microstructure of the corpus callosum in males with autism: a longitudinal investigation. Mol Autism (2015) 6:15. doi:10.1186/s13229-015-0001-8
43. Alexander AL, Lee JE, Lazar M, Boudos R, DuBray MB, Oakes TR, et al. Diffusion tensor imaging of the corpus callosum in autism. Neuroimage (2007) 34:61–73. doi:10.1016/j.neuroimage.2006.08.032
44. Thakkar KN, Polli FE, Joseph RM, Tuch DS, Hadjikhani N, Barton JJ, et al. Response monitoring, repetitive behaviour and anterior cingulate abnormalities in autism spectrum disorders (ASD). Brain (2008) 131:2464–78. doi:10.1093/brain/awn099
45. Barnea-Goraly N, Lotspeich LJ, Reiss AL. Similar white matter aberrations in children with autism and their unaffected siblings: a diffusion tensor imaging study using tract-based spatial statistics. Arch Gen Psychiatry (2010) 67:1052–60. doi:10.1001/archgenpsychiatry.2010.123
46. Noriuchi M, Kikuchi Y, Yoshiura T, Kira R, Shigeto H, Hara T, et al. Altered white matter fractional anisotropy and social impairment in children with autism spectrum disorder. Brain Res (2010) 1362:141–9. doi:10.1016/j.brainres.2010.09.051
47. Kana RK, Keller TA, Cherkassky VL, Minshew NJ, Just MA. Atypical frontal-posterior synchronization of theory of mind regions in autism during mental state attribution. Soc Neurosci (2009) 4:135–52. doi:10.1080/17470910802198510
48. Poustka L, Jennen-Steinmetz C, Henze R, Vomstein K, Haffner J, Sieltjes B. Fronto-temporal disconnectivity and symptom severity in children with autism spectrum disorder. World J Biol Psychiatry (2012) 13(4):269–80. doi:10.3109/15622975.2011.591824
49. Koshino H, Kana RK, Keller TA, Cherkassky VL, Minshew NJ, Just MA. fMRI investigation of working memory for faces in autism: visual coding and underconnectivity with frontal areas. Cereb Cortex (2008) 18:289–300. doi:10.1093/cercor/bhm054
50. Just MA, Cherkassky VL, Keller TA, Kana RK, Minshew NJ. Functional and anatomical cortical underconnectivity in autism: evidence from an FMRI study of an executive function task and corpus callosum morphometry. Cereb Cortex (2007) 17:951–61. doi:10.1093/cercor/bhl006
51. Kennedy DP, Courchesne E. Functional abnormalities of the default network during self- and other-reflection in autism. Soc Cogn Affect Neurosci (2008) 3:177–90. doi:10.1093/scan/nsn011
52. Weng SJ, Wiggins JL, Peltier SJ, Carrasco M, Risi S, Lord C, et al. Alterations of resting state functional connectivity in the default network in adolescents with autism spectrum disorders. Brain Res (2013) 1313:202–14. doi:10.1016/j.brainres.2009.11.057
53. Monk CS, Peltier SJ, Wiggins JL, Weng SJ, Carrasco M, Risi S, et al. Abnormalities of intrinsic functional connectivity in autism spectrum disorders. Neuroimage (2009) 47:764–72. doi:10.1016/j.neuroimage.2009.04.069
54. Noonan SK, Haist F, Muller RA. Aberrant functional connectivity in autism: evidence from low-frequency BOLD signal fluctuations. Brain Res (2009) 1262:48–63. doi:10.1016/j.brainres.2008.12.076
55. Kleinhans NM, Richards T, Sterling L, Stegbauer KC, Mahurin R, Johnson LC, et al. Abnormal functional connectivity in autism spectrum disorders during face processing. Brain (2008) 131:1000–12. doi:10.1093/brain/awm334
57. Doll CA, Broadie K. Impaired activity-dependent neural circuit assembly and refinement in autism spectrum disorder genetic models. Front Cell Neurosci (2012) 8:30. doi:10.3389/fncel.2014.00030
58. Ning ZY, Zhao DM, Liu HX, Yang JM, Han CX, Cui YL, et al. Altered expression of the prion gene in rat astrocyte and neuron cultures treated with prion peptide 106-126. Cell Mol Neurobiol (2005) 25:1171–83. doi:10.1007/s10571-005-8357-5
59. Estes ML, McAllister AK. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat Rev Neurosci (2015) 16:469–86. doi:10.1038/nrn3978
60. Hazlett HC, Poe MD, Gerig G, Styner M, Chappell C, Smith RG, et al. Early brain overgrowth in autism associated with an increase in cortical surface area before age 2 years. Arch Gen Psychiatry (2011) 68:467–76. doi:10.1001/archgenpsychiatry.2011.39
61. Kilian S, Brown WS, Hallam BJ, McMahon W, Lu J, Johnson M, et al. Regional callosal morphology in autism and macrocephaly. Dev Neuropsychol (2008) 33:74–99. doi:10.1080/87565640701729821
62. Potts MB, Siu JJ, Price JD, Salinas RD, Cho MJ, Ramos AD, et al. Analysis of Mll1 deficiency identifies neurogenic transcriptional modules and Brn4 as a factor for direct astrocyte-to-neuron reprogramming. Neurosurgery (2014) 75:472–82. doi:10.1227/NEU.0000000000000452
63. Chamak B, Fellous A, Autillo-Touati A, Barbin G, Prochiantz A. Are neuronotrophic neuron-astrocyte interactions regionally specified? Ann N Y Acad Sci (1987) 495:528–36. doi:10.1111/j.1749-6632.1987.tb23698.x
64. Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol (2007) 17:103–11. doi:10.1016/j.conb.2007.01.009
65. Pitanga BP, Silva VD, Souza CS, Junqueira HA, Fragomeni BO, Nascimento RP, et al. Assessment of neurotoxicity of monocrotaline, an alkaloid extracted from Crotalaria retusa in astrocyte/neuron co-culture system. Neurotoxicology (2011) 32:776–84. doi:10.1016/j.neuro.2011.07.002
66. Casanova MF. Neuropathological and genetic findings in autism: the significance of a putative minicolumnopathy. Neuroscientist (2006) 12:435–41. doi:10.1177/1073858406290375
67. Zungun C, Yilmaz FM, Tutkun E, Yilmaz H, Uysal S. Assessment of serum S100B and neuron specific enolase levels to evaluate the neurotoxic effects of organic solvent exposure. Clin Toxicol (Phila) (2013) 51:748–51. doi:10.3109/15563650.2013.820831
68. Zhou BY, Liu Y, Kim B, Xiao Y, He JJ. Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol Cell Neurosci (2004) 27:296–305. doi:10.1016/j.mcn.2004.07.003
69. Cassina P, Pehar M, Vargas MR, Castellanos R, Barbeito AG, Estevez AG, et al. Astrocyte activation by fibroblast growth factor-1 and motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem (2005) 93:38–46. doi:10.1111/j.1471-4159.2004.02984.x
70. Aschner M, Bennett BA. Astrocyte and neuron coculturing method. Methods Mol Med (1999) 22:133–44. doi:10.1385/0-89603-612-X:133
71. Yang CZ, Zhao R, Dong Y, Chen XQ, Yu AC. Astrocyte and neuron intone through glutamate. Neurochem Res (2008) 33:2480–6. doi:10.1007/s11064-008-9758-x
72. Schipul SE, Keller TA, Just MA. Inter-regional brain communication and its disturbance in autism. Front Syst Neurosci (2011) 5:10. doi:10.3389/fnsys.2011.00010
73. Travers BG, Adluru N, Ennis C, Tromp do PM, Destiche D, Doran S, et al. Diffusion tensor imaging in autism spectrum disorder: a review. Autism Res (2012) 5:289–313. doi:10.1002/aur.1243
74. Catani M, Jones DK, Daly E, Embiricos N, Deeley Q, Pugliese L, et al. Altered cerebellar feedback projections in Asperger syndrome. Neuroimage (2008) 41:1184–91. doi:10.1016/j.neuroimage.2008.03.041
75. Skefos J, Cummings C, Enzer K, Holiday J, Weed K, Levy E, et al. Regional alterations in purkinje cell density in patients with autism. PLoS One (2014) 9:e81255. doi:10.1371/journal.pone.0081255
76. Rathinam ML, Watts LT, Stark AA, Mahimainathan L, Stewart J, Schenker S, et al. Astrocyte control of fetal cortical neuron glutathione homeostasis: up-regulation by ethanol. J Neurochem (2006) 96:1289–300. doi:10.1111/j.1471-4159.2006.03674.x
77. Blackburn D, Sargsyan S, Monk PN, Shaw PJ. Astrocyte function and role in motor neuron disease: a future therapeutic target? Glia (2009) 57:1251–64. doi:10.1002/glia.20848
78. Amiri M, Hosseinmardi N, Bahrami F, Janahmadi M. Astrocyte-neuron interaction as a mechanism responsible for generation of neural synchrony: a study based on modeling and experiments. J Comput Neurosci (2013) 34:489–504. doi:10.1007/s10827-012-0432-6
79. Liu QY, Schaffner AE, Chang YH, Barker JL. Astrocyte-conditioned saline supports embryonic rat hippocampal neuron differentiation in short-term cultures. J Neurosci Methods (1998) 86:71–7. doi:10.1016/S0165-0270(98)00146-0
80. Agarwal A, Bergles DE. Astrocyte morphology is controlled by neuron-derived FGF. Neuron (2014) 83:255–7. doi:10.1016/j.neuron.2014.07.005
81. Bradstreet JJ, Pacini S, Ruggiero M. A new methodology of viewing extra-axial fluid and cortical abnormalities in children with autism via transcranial ultrasonography. Front Hum Neurosci (2014) 7:934. doi:10.3389/fnhum.2013.00934
82. Marchetti B. Cross-talk signals in the CNS: role of neurotrophic and hormonal factors, adhesion molecules and intercellular signaling agents in luteinizing hormone-releasing hormone (LHRH)-astroglial interactive network. Front Biosci (1997) 2:d88–125.
83. Kerschensteiner M, Meinl E, Hohlfeld R. Neuro-immune crosstalk in CNS diseases. Results Probl Cell Differ (2009) 51:197–216. doi:10.1007/400_2009_6
84. Besedovsky HO, del Rey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev (1996) 17:64–102. doi:10.1210/edrv-17-1-64
85. Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun (2011) 26:383–92. doi:10.1016/j.bbi.2011.08.007
86. Vitkovic L, Konsman JP, Bockaert J, Dantzer R, Homburger V, Jacque C. Cytokine signals propagate through the brain. Mol Psychiatry (2000) 5:604–15. doi:10.1038/sj.mp.4000813
87. Theoharides TC, Stewart JM, Panagiotidou S, Melamed I. Mast cells, brain inflammation and autism. Eur J Pharmacol (2015). doi:10.1016/j.ejphar.2015.03.086
88. Stubbs EG, Crawford ML. Depressed lymphocyte responsiveness in autistic children. J Autism Child Schizophr (1977) 7:49–55. doi:10.1007/BF01531114
89. Wills S, Cabanlit M, Bennett J, Ashwood P, Amaral D, Van de Water J. Autoantibodies in autism spectrum disorders (ASD). Ann N Y Acad Sci (2007) 1107:79–91. doi:10.1196/annals.1381.009
90. Zimmerman AW, Connors SL, Matteson KJ, Lee LC, Singer HS, Castaneda JA, et al. Maternal antibrain antibodies in autism. Brain Behav Immun (2007) 21:351–7. doi:10.1016/j.bbi.2006.08.005
91. Braunschweig D, Van de Water J. Maternal autoantibodies in autism. Arch Neurol (2012) 69:693–9. doi:10.1001/archneurol.2011.2506
92. Gupta S, Samra D, Agrawal S. Adaptive and innate immune responses in autism: rationale for therapeutic use of intravenous immunoglobulin. J Clin Immunol (2010) 30(Suppl 1):S90–6. doi:10.1007/s10875-010-9402-9
93. Mostafa GA, El-Sherif DF, Al-Ayadhi LY. Systemic auto-antibodies in children with autism. J Neuroimmunol (2014) 272:94–8. doi:10.1016/j.jneuroim.2014.04.011
94. Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun (2011) 25:40–5. doi:10.1016/j.bbi.2010.08.003
95. Suzuki K, Matsuzaki H, Iwata K, Kameno Y, Shimmura C, Kawai S, et al. Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PLoS One (2011) 6:e20470. doi:10.1371/journal.pone.0020470
96. Emanuele E, Orsi P, Boso M, Broglia D, Brondino N, Barale F, et al. Low-grade endotoxemia in patients with severe autism. Neurosci Lett (2010) 471:162–5. doi:10.1016/j.neulet.2010.01.033
97. Jyonouchi H, Sun S, Le H. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. J Neuroimmunol (2001) 120:170–9. doi:10.1016/S0165-5728(01)00421-0
98. Enstrom AM, Onore CE, Van de Water JA, Ashwood P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav Immun (2009) 24(1):64–71. doi:10.1016/j.bbi.2009.08.001
99. Malik M, Sheikh AM, Wen G, Spivack W, Brown WT, Li X. Expression of inflammatory cytokines, Bcl2 and cathepsin D are altered in lymphoblasts of autistic subjects. Immunobiology (2011) 216:80–5. doi:10.1016/j.imbio.2010.03.001
100. Wei H, Zou H, Sheikh AM, Malik M, Dobkin C, Brown WT, et al. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflammation (2011) 8:52. doi:10.1186/1742-2094-8-52
101. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol (2005) 57:67–81. doi:10.1002/ana.20315
102. Xu N, Li X, Zhong Y. Inflammatory cytokines: potential biomarkers of immunologic dysfunction in autism spectrum disorders. Mediators Inflamm (2015) 2015:531518. doi:10.1155/2015/531518
103. Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, et al. Elevated immune response in the brain of autistic patients. J Neuroimmunol (2009) 207(1–2):111–6. doi:10.1016/j.jneuroim.2008.12.002
104. Croonenberghs J, Bosmans E, Deboutte D, Kenis G, Maes M. Activation of the inflammatory response system in autism. Neuropsychobiology (2002) 45(1):1–6. doi:10.1159/000048665
105. Ashwood P, Enstrom A, Krakowiak P, Hertz-Picciotto I, Hansen RL, Croen LA, et al. Decreased transforming growth factor beta1 in autism: a potential link between immune dysregulation and impairment in clinical behavioral outcomes. J Neuroimmunol (2008) 204:149–53. doi:10.1016/j.jneuroim.2008.07.006
106. Okada K, Hashimoto K, Iwata Y, Nakamura K, Tsujii M, Tsuchiya KJ, et al. Decreased serum levels of transforming growth factor-beta1 in patients with autism. Prog Neuropsychopharmacol Biol Psychiatry (2007) 31:187–90. doi:10.1016/j.pnpbp.2006.08.020
107. Brimberg L, Sadiq A, Gregersen PK, Diamond B. Brain-reactive IgG correlates with autoimmunity in mothers of a child with an autism spectrum disorder. Mol Psychiatry (2013) 18:1171–7. doi:10.1038/mp.2013.101
108. Parker-Athill EC, Tan J. Maternal immune activation and autism spectrum disorder: interleukin-6 signaling as a key mechanistic pathway. Neurosignals (2010) 18:113–28. doi:10.1159/000319828
109. Mazur-Kolecka B, Cohen IL, Gonzalez M, Jenkins EC, Kaczmarski W, Brown WT, et al. Autoantibodies against neuronal progenitors in sera from children with autism. Brain Dev (2014) 36:322–9. doi:10.1016/j.braindev.2013.04.015
110. Hsiao EY, McBride SW, Chow J, Mazmanian SK, Patterson PH. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc Natl Acad Sci U S A (2012) 109:12776–81. doi:10.1073/pnas.1202556109
111. Schwartzer JJ, Careaga M, Onore CE, Rushakoff JA, Berman RF, Ashwood P. Maternal immune activation and strain specific interactions in the development of autism-like behaviors in mice. Transl Psychiatry (2013) 3:e240. doi:10.1038/tp.2013.16
112. Lucchina L, Depino AM. Altered peripheral and central inflammatory responses in a mouse model of autism. Autism Res (2014) 7:273–89. doi:10.1002/aur.1338
113. Enstrom AM, Lit L, Onore CE, Gregg JP, Hansen RL, Pessah IN, et al. Altered gene expression and function of peripheral blood natural killer cells in children with autism. Brain Behav Immun (2009) 23:124–33. doi:10.1016/j.bbi.2008.08.001
114. Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med (2011) 17:389–94. doi:10.1016/j.molmed.2011.03.001
115. Atladottir HO, Pedersen MG, Thorsen P, Mortensen PB, Deleuran B, Eaton WW, et al. Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics (2009) 124:687–94. doi:10.1542/peds.2008-2445
116. Warrington AE, Bieber AJ, Van Keulen V, Ciric B, Pease LR, Rodriguez M. Neuron-binding human monoclonal antibodies support central nervous system neurite extension. J Neuropathol Exp Neurol (2004) 63:461–73.
117. Baslow MH, Dyakin VV, Nowak KL, Hungund BL, Guilfoyle DN. 2-PMPA, a NAAG peptidase inhibitor, attenuates magnetic resonance BOLD signals in brain of anesthetized mice: evidence of a link between neuron NAAG release and hyperemia. J Mol Neurosci (2005) 26:1–15. doi:10.1385/JMN:26:1:001
118. Enstrom AM, Van de Water JA, Ashwood P. Autoimmunity in autism. Curr Opin Investig Drugs (2009) 10:463–73.
119. Lyall K, Ashwood P, Van de Water J, Hertz-Picciotto I. Maternal immune-mediated conditions, autism spectrum disorders, and developmental delay. J Autism Dev Disord (2013) 44:1546–55. doi:10.1007/s10803-013-2017-2
120. Sweeten TL, Posey DJ, McDougle CJ. High blood monocyte counts and neopterin levels in children with autistic disorder. Am J Psychiatry (2003) 160:1691–3. doi:10.1176/appi.ajp.160.9.1691
121. Enstrom AM, Onore CE, Van de Water JA, Ashwood P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav Immun (2009) 24:64–71. doi:10.1016/j.bbi.2009.08.001
122. Chez MG, Dowling T, Patel PB, Khanna P, Kominsky M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr Neurol (2007) 36:361–5. doi:10.1016/j.pediatrneurol.2007.01.012
123. Sordet O, Rebe C, Plenchette S, Zermati Y, Hermine O, Vainchenker W, et al. Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood (2002) 100:4446–53. doi:10.1182/blood-2002-06-1778
124. Siniscalco D, Sapone A, Giordano C, Cirillo A, de Novellis V, de Magistris L, et al. The expression of caspases is enhanced in peripheral blood mononuclear cells of autism spectrum disorder patients. J Autism Dev Disord (2012) 42:1403–10. doi:10.1007/s10803-011-1373-z
125. Goncharova ND. Stress responsiveness of the hypothalamic-pituitary-adrenal axis: age-related features of the vasopressinergic regulation. Front Endocrinol (2013) 4:26. doi:10.3389/fendo.2013.00026
126. Tostes MH, Teixeira HC, Gattaz WF, Brandao MA, Raposo NR. Altered neurotrophin, neuropeptide, cytokines and nitric oxide levels in autism. Pharmacopsychiatry (2012) 45:241–3. doi:10.1055/s-0032-1301914
127. Sweeten TL, Posey DJ, Shankar S, McDougle CJ. High nitric oxide production in autistic disorder: a possible role for interferon-gamma. Biol Psychiatry (2004) 55:434–7. doi:10.1016/j.biopsych.2003.09.001
128. Ghanizadeh A, Akhondzadeh S, Hormozi M, Makarem A, Abotorabi-Zarchi M, Firoozabadi A. Glutathione-related factors and oxidative stress in autism, a review. Curr Med Chem (2012) 19:4000–5. doi:10.2174/092986712802002572
129. Frustaci A, Neri M, Cesario A, Adams JB, Domenici E, Dalla Bernardina B, et al. Oxidative stress-related biomarkers in autism: systematic review and meta-analyses. Free Radic Biol Med (2012) 52:2128–41. doi:10.1016/j.freeradbiomed.2012.03.011
130. Meguid NA, Dardir AA, Abdel-Raouf ER, Hashish A. Evaluation of oxidative stress in autism: defective antioxidant enzymes and increased lipid peroxidation. Biol Trace Elem Res (2010) 143:58–65. doi:10.1007/s12011-010-8840-9
131. Schneider T, Przewlocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology (2005) 30:80–9. doi:10.1038/sj.npp.1300518
132. Rao SP, Sikdar SK. Acute treatment with 17beta-estradiol attenuates astrocyte-astrocyte and astrocyte-neuron communication. Glia (2007) 55:1680–9. doi:10.1002/glia.20564
133. Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Front Neuroendocrinol (2012) 33:267–86. doi:10.1016/j.yfrne.2012.08.006
134. Sierra A, Tremblay ME, Wake H. Never-resting microglia: physiological roles in the healthy brain and pathological implications. Front Cell Neurosci (2014) 8:240. doi:10.3389/fncel.2014.00240
135. Sierra A, Beccari S, Diaz-Aparicio I, Encinas JM, Comeau S, Tremblay ME. Surveillance, phagocytosis, and inflammation: how never-resting microglia influence adult hippocampal neurogenesis. Neural Plast (2014) 2014:610343. doi:10.1155/2014/610343
136. Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry (2010) 68:368–76. doi:10.1016/j.biopsych.2010.05.024
137. Tetreault NA, Hakeem AY, Jiang S, Williams BA, Allman E, Wold BJ, et al. Microglia in the cerebral cortex in autism. J Autism Dev Disord (2012) 42:2569–84. doi:10.1007/s10803-012-1513-0
138. Patel AS, Zalcman SS. Interleukin-2 treatment induces an acquired behavioral response pattern (repetitive stereotyped movements) mediated by dopamine D1 and D2 receptors. Int Neuropsychiatr Dis J (2014) 2:175–85. doi:10.9734/INDJ/2014/7284
139. Fernandez-Botran R. Soluble cytokine receptors: novel immunotherapeutic agents. Expert Opin Investig Drugs (2000) 9:497–514. doi:10.1517/13543784.9.3.497
140. Fernandez-Botran R. Soluble cytokine receptors: their role in immunoregulation. FASEB J (1991) 5:2567–74.
142. Knowles MA, Platt FM, Ross RL, Hurst CD. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev (2009) 28:305–16. doi:10.1007/s10555-009-9198-3
143. Brighenti M. MicroRNA and MET in lung cancer. Ann Transl Med (2015) 3:68. doi:10.3978/j.issn.2305-5839.2015.01.26
144. Wang L, He L, Zhang R, Liu X, Ren Y, Liu Z, et al. Regulation of T lymphocyte activation by microRNA-21. Mol Immunol (2014) 59:163–71. doi:10.1016/j.molimm.2014.02.004
145. Mellios N, Sur M. The emerging role of microRNAs in schizophrenia and autism spectrum disorders. Front Psychiatry (2012) 3:39. doi:10.3389/fpsyt.2012.00039
146. Mundalil Vasu M, Anitha A, Thanseem I, Suzuki K, Yamada K, Takahashi T, et al. Serum microRNA profiles in children with autism. Mol Autism (2014) 5:40. doi:10.1186/2040-2392-5-40
147. Olde Loohuis NF, Kole K, Glennon JC, Karel P, Van der Borg G, Van Gemert Y, et al. Elevated microRNA-181c and microRNA-30d levels in the enlarged amygdala of the valproic acid rat model of autism. Neurobiol Dis (2015) 80:42–53. doi:10.1016/j.nbd.2015.05.006
148. Ashwood P, Wakefield AJ. Immune activation of peripheral blood and mucosal CD3+ lymphocyte cytokine profiles in children with autism and gastrointestinal symptoms. J Neuroimmunol (2006) 173:126–34. doi:10.1016/j.jneuroim.2005.12.007
149. Ashwood P, Anthony A, Torrente F, Wakefield AJ. Spontaneous mucosal lymphocyte cytokine profiles in children with autism and gastrointestinal symptoms: mucosal immune activation and reduced counter regulatory interleukin-10. J Clin Immunol (2004) 24:664–73. doi:10.1007/s10875-004-6241-6
150. Theoharides TC, Angelidou A, Alysandratos KD, Zhang B, Asadi S, Francis K, et al. Mast cell activation and autism. Biochim Biophys Acta (2012) 1822:34–41. doi:10.1016/j.bbadis.2010.12.017
151. Enstrom A, Krakowiak P, Onore C, Pessah IN, Hertz-Picciotto I, Hansen RL, et al. Increased IgG4 levels in children with autism disorder. Brain Behav Immun (2009) 23:389–95. doi:10.1016/j.bbi.2008.12.005
152. Coisne C, Dehouck L, Faveeuw C, Delplace Y, Miller F, Landry C, et al. Mouse syngenic in vitro blood-brain barrier model: a new tool to examine inflammatory events in cerebral endothelium. Lab Invest (2005) 85:734–46. doi:10.1038/labinvest.3700281
153. Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol (2008) 9:137–45. doi:10.1038/ni1551
154. Bambini-Junior V, Zanatta G, Della Flora Nunes G, Mueller de Melo G, Michels M, Fontes-Dutra M, et al. Resveratrol prevents social deficits in animal model of autism induced by valproic acid. Neurosci Lett (2014) 583:176–81. doi:10.1016/j.neulet.2014.09.039
155. Harikumar KB, Aggarwal BB. Resveratrol: a multitargeted agent for age-associated chronic diseases. Cell Cycle (2008) 7:1020–35. doi:10.4161/cc.7.8.5740
156. Dong W, Li N, Gao D, Zhen H, Zhang X, Li F. Resveratrol attenuates ischemic brain damage in the delayed phase after stroke and induces messenger RNA and protein express for angiogenic factors. J Vasc Surg (2008) 48(3):709–14. doi:10.1016/j.jvs.2008.04.007
157. Sinha K, Chaudhary G, Gupta YK. Protective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life Sci (2002) 71(6):655–65. doi:10.1016/S0024-3205(02)01691-0
158. Vang O, Ahmad N, Baile CA, Baur JA, Brown K, Csiszar A, et al. What is new for an old molecule? Systematic review and recommendations on the use of resveratrol. PLoS One (2011) 6:e19881. doi:10.1371/journal.pone.0019881
159. Schneider T, Roman A, Basta-Kaim A, Kubera M, Budziszewska B, Schneider K, et al. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology (2008) 33:728–40. doi:10.1016/j.psyneuen.2008.02.011
160. Bambini-Junior V, Nunes GD, Schneider T, Gottfried C. Comment on “oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring”. Science (2014) 346:176. doi:10.1126/science.1255679
161. Bambini-Junior V, Rodrigues L, Behr GA, Moreira JC, Riesgo R, Gottfried C. Animal model of autism induced by prenatal exposure to valproate: behavioral changes and liver parameters. Brain Res (2011) 1408:8–16. doi:10.1016/j.brainres.2011.06.015
162. Petro TM. Regulatory role of resveratrol on Th17 in autoimmune disease. Int Immunopharmacol (2011) 11:310–8. doi:10.1016/j.intimp.2010.07.011
163. Singh NP, Hegde VL, Hofseth LJ, Nagarkatti M, Nagarkatti P. Resveratrol (trans-3,5,4’-trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol Pharmacol (2007) 72:1508–21. doi:10.1124/mol.107.038984
164. Sakic B, Szechtman H, Denburg JA. Neurobehavioral alterations in autoimmune mice. Neurosci Biobehav Rev (1997) 21:327–40. doi:10.1016/S0149-7634(96)00018-8
165. Hornig M, Weissenbock H, Horscroft N, Lipkin WI. An infection-based model of neurodevelopmental damage. Proc Natl Acad Sci U S A (1999) 96:12102–7. doi:10.1073/pnas.96.21.12102
166. Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci (2003) 23:297–302.
167. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci (2007) 27:10695–702. doi:10.1523/JNEUROSCI.2178-07.2007
168. Singer HS, Morris C, Gause C, Pollard M, Zimmerman AW, Pletnikov M. Prenatal exposure to antibodies from mothers of children with autism produces neurobehavioral alterations: a pregnant dam mouse model. J Neuroimmunol (2009) 211:39–48. doi:10.1016/j.jneuroim.2009.03.011
169. Willette AA, Lubach GR, Knickmeyer RC, Short SJ, Styner M, Gilmore JH, et al. Brain enlargement and increased behavioral and cytokine reactivity in infant monkeys following acute prenatal endotoxemia. Behav Brain Res (2011) 219:108–15. doi:10.1016/j.bbr.2010.12.023
170. Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun (2012) 26:607–16. doi:10.1016/j.bbi.2012.01.011
171. Bauman MD, Iosif AM, Smith SE, Bregere C, Amaral DG, Patterson PH. Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biol Psychiatry (2014) 75:332–41. doi:10.1016/j.biopsych.2013.06.025
172. Camacho J, Jones K, Miller E, Ariza J, Noctor S, Van de Water J, et al. Embryonic intraventricular exposure to autism-specific maternal autoantibodies produces alterations in autistic-like stereotypical behaviors in offspring mice. Behav Brain Res (2014) 266:46–51. doi:10.1016/j.bbr.2014.02.045
173. Onore CE, Schwartzer JJ, Careaga M, Berman RF, Ashwood P. Maternal immune activation leads to activated inflammatory macrophages in offspring. Brain Behav Immun (2014) 38:220–6. doi:10.1016/j.bbi.2014.02.007
174. Xuan IC, Hampson DR. Gender-dependent effects of maternal immune activation on the behavior of mouse offspring. PLoS One (2014) 9:e104433. doi:10.1371/journal.pone.0104433
175. Hwang SR, Kim CY, Shin KM, Jo JH, Kim HA, Heo Y. Altered expression levels of neurodevelopmental proteins in fetal brains of BTBR T+tf/J mice with autism-like behavioral characteristics. J Toxicol Environ Health A (2015) 78:516–23. doi:10.1080/15287394.2015.1010466
176. Kim SN, Jo GH, Kim HA, Heo Y. Aberrant IgG isotype generation in mice with abnormal behaviors. J Immunotoxicol (2015):1–5. doi:10.3109/1547691X.2015.1014581
177. Luan R, Cheng H, Li L, Zhao Q, Liu H, Wu Z, et al. Maternal lipopolysaccharide exposure promotes immunological functional changes in adult offspring CD4 T cells. Am J Reprod Immunol (2015) 73:522–35. doi:10.1111/aji.12364
Keywords: autism, neuroimmune interactions, environmental risk factors, rodent models, valproic acid
Citation: Gottfried C, Bambini-Junior V, Francis F, Riesgo R and Savino W (2015) The impact of neuroimmune alterations in autism spectrum disorder. Front. Psychiatry 6:121. doi: 10.3389/fpsyt.2015.00121
Received: 06 July 2015; Accepted: 17 August 2015;
Published: 09 September 2015
Edited by:
Rochelle S. Cohen, University of Illinois at Chicago, USAReviewed by:
Dario Siniscalco, Second University of Naples, ItalyHongen Wei, Shanxi Medical University, China
Copyright: © 2015 Gottfried, Bambini-Junior, Francis, Riesgo and Savino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmem Gottfried, Ramiro Barcelos, 2600-Anexo, Porto Alegre, Rio Grande do Sul 90035003, Brazil, cgottfried@ufrgs.br;
Wilson Savino, Laboratório de Pesquisa sobre o Timo, Instituto Oswaldo Cruz, Fundação Oswaldo Cruz, Ave. Brasil 4365, Manguinhos, Rio de Janeiro 21045-900, Brazil, savino.w@gmail.com