 María I. Craig1*Maria F. Rojas2Claudia A. van der Ploeg3Valeria Olivera1Ariel E. Vagnozzi1Andrés M. Perez4Guido A. König5,6*
María I. Craig1*Maria F. Rojas2Claudia A. van der Ploeg3Valeria Olivera1Ariel E. Vagnozzi1Andrés M. Perez4Guido A. König5,6*
- 1Instituto de Virología, Centro de Investigación en Ciencias Veterinarias y Agronómicas, Instituto Nacional de Tecnología Agropecuaria (INTA)-Hurlingham, Buenos Aires, Argentina
- 2Laboratorio de Sanidad Aviar, Estación Experimental Agropecuaria, Instituto Nacional de Tecnología Agropecuaria (INTA)-Concepción del Uruguay, Entre Rios, Argentina
- 3Instituto Nacional de Producción de Biológicos, ANLIS “Dr. Carlos G. Malbrán”, Ciudad de Buenos Aires, Argentina
- 4Department of Veterinary Population Medicine, College of Veterinary Medicine, University of Minnesota, Saint Paul, MN, United States
- 5Instituto de Biotecnología, Centro de Investigación en Ciencias Veterinarias y Agronómicas INTA-Hurlingham, Buenos Aires, Argentina
- 6Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Buenos Aires, Argentina
Avian infectious laryngotracheitis (ILT) is a worldwide infectious disease that causes important economic losses in the poultry industry. Although it is known that ILT virus (ILTV) is present in Argentina, there is no information about the circulating strains. With the aim to characterize them, seven different genomic regions (thymidine kinase, glycoproteins D, G, B, C, and J, and infected cell polypeptide 4) were partially sequenced and compared between field samples. The gJ sequence resulted to be the most informative segment, it allowed the differentiation among field sample strains, and also, between wild and vaccine viruses. Specific changes in selected nucleotidic positions led to the definition of five distinct haplotypes. Tests for detection of clustering were run to test the null hypothesis that ILTV haplotypes were randomly distributed in time in Argentina and in space in the most densely populated poultry region of this country, Entre Rios. From this study, it was possible to identify a 46 km radius cluster in which higher proportions of haplotypes 4 and 5 were observed, next to a provincial route in Entre Rios and a significant decline of haplotype 5 between 2009 and 2011. Results here provide an update on the molecular epidemiology of ILT in Argentina, including data on specific genome segments that may be used for rapid characterization of the virus in the field. Ultimately, results will contribute to the surveillance of ILT in the country.
Introduction
Infectious laryngotracheitis (ILT) is an acute and highly contagious respiratory disease of chicken that causes far-reaching financial losses due to its high mortality rate and reduction in egg production. Chickens can be infected through the upper respiratory and ocular routes (1). The ILT etiological agent, generically referred to as ILT virus (ILTV) and also known as Gallid herpesvirus 1, is a member of the Herpesviridae family with a double-stranded DNA of approximately 155 kbp in size (2).
Traditionally, two types of ILTV attenuated vaccines have been widely used to control the disease, namely, chicken embryo-origin (CEO) vaccine, which is attenuated by serial passages in embryonated eggs (3), and a tissue culture-origin (TCO) vaccine, which is generated by multiple passages in tissue culture (4). Since 2006, use of the CEO vaccine has been banned in Argentina, and currently, TCO and two vector vaccines, which express the gB gene linked, are commercially available in the country.
The ILTV is globally distributed and several epidemiological studies have been conducted in different countries for detecting circulating ILTVs. Traditionally, differentiation of wild from vaccine strains has been based on the use of restriction length polymorphism (RFLP) profiles of the complete genome (5–8). Recently, however, amplification by polymerase chain reaction (PCR) combined with RFLP of different regions of the ILTV genome [timidine kinase, glycoprotein G, E, X, and C (gG, gE, gX, and gC) and infected cell polypeptide 4 (ICP4)] has been used to characterize strains, with varying degrees of success (9–12). Even in the absence of multiple ILTV serotypes, molecular characterizations is still epidemiologically useful to assess the virus dissemination process in a region.
Infectious laryngotracheitis virus occurs in Argentina sporadically and to our best knowledge, no evaluation of its prevalence and no characterization of field viruses have been conducted. Thus, considering the absence of information on ILTV strains circulating, the purpose of the study here was to assess the molecular epidemiology of ILTV, using sequences from the most variable regions of the genome corresponding to proteins with antigenic or functional relevance (TK, gD, gG, gB, gC, gJ, and ICP4). Results will help select the most variables ILTV genome regions, characterize field strains, and differentiate ILTV field and vaccine strains.
Materials and Methods
Clinical Samples
Field strains (n = 72) were obtained from the upper respiratory tract (tracheas or tracheal swabs) of broilers and layers showing mild to severe disease signs during different outbreaks between 2006 and 2013. The samples belong to 12 localities in Buenos Aires (there are nine samples with unknown location) and from 15 in Entre Rios province. Samples were spontaneously submitted to the diagnostic laboratory for disease testing and confirmation by producers that suspected the presence of the disease to one of two different INTA (Instituto Nacional de Tecnología Agropecuaria) avian virology laboratories, located in Buenos Aires and Entre Rios provinces.
Sample Collection and DNA Extraction
The DNA was extracted either from samples of tracheal mucosa or directly from tracheal swabs. Tracheal mucosa were scraped and homogenized in a mortar with sterile sand and PBS supplemented with penicillin 10,000 IU/ml, streptomycin 5,000 µg/ml, gentamicin sulfate 1,000 µg/ml, kanamycin sulfate 700 µg/ml, and amphotericin B 10 µg/ml (Sigma Chemical Co™, St. Louis, MO, USA); subsequently, centrifugation was performed to eliminate cell debris and sand. The supernatant was collected and stored at −70°C.
Tracheal swabs were obtained by a strong rubbing of the mucosal zone and were suspended in 2 ml of sterile PBS. DNA was extracted from the supernatants and reconstituted vaccines Laringo-Vac (CEO—Solvay Animal Health) and LT-IVAX (TCO—Schering Plough.) by QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA, USA), according to the manufacturer’s instructions.
Polymerase Chain Reaction
Primers for the gB and the gD were designed with Fast_PCR software version 3.3.64 (13), on the basis of sequences previously published in Genbank. The rest of the primers were obtained from published data (Table 1).
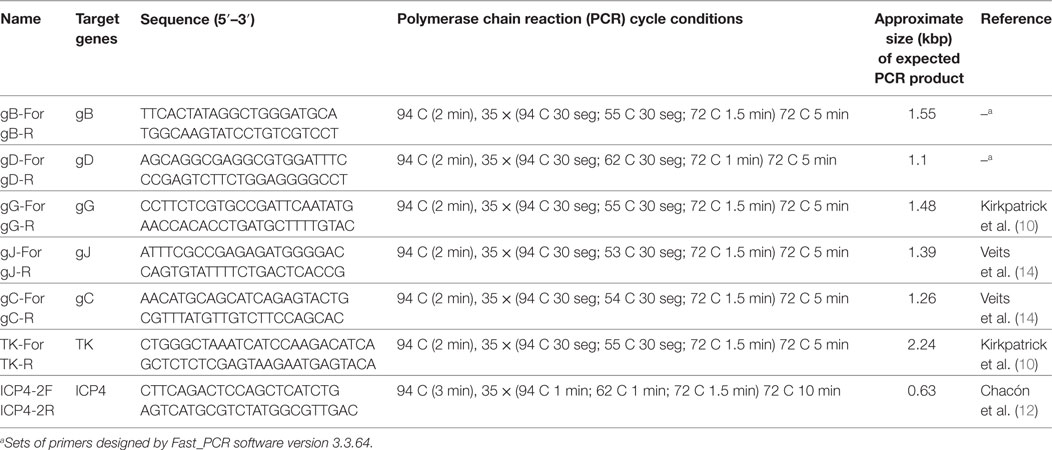
Table 1. Set of primers and cycling conditions for the amplification of different genomic regions of ILT virus.
DNA amplification was carried out in 50 µl reaction volume using 4 µl of extracted DNA, 5 µl of 10× reaction buffer, 1 µl of 10 mM dNTPs mix, 1 µl of each primer (5 µmol), and 0.25 µl of 5 U/μl of GoTaq DNA Polymerase (Promega). The cycling program for each amplification is described in Table 1.
Amplicon Purification and Sequence Analysis
The amplicons were purified by QIAquick PCR purification kit (Qiagen Inc., Valencia, CA, USA), according to the manufacturer’s instructions.
Sequences of each amplicon were directly sequenced with the Big Dye terminator kit TM (Applied Biosystems, Foster City, CA, USA) on an ABI 3130 or 3500 XL TM (Applied Biosystems, Foster City, CA, USA).
Sequences were edited using the BIOEDIT software (15). Sequences obtained here and reference strains sequences obtained from Genbank were included in a multiple alignment with CLUSTAL W program Version 1.8.3 (16).
Space and Time Cluster Analysis
The observed to expected case ratio (o/e), where the expected number of cases was computed under the null hypothesis of random distribution of cases, was estimated. A spatial multinomial scan statistic was implemented to assess whether there were areas in which the proportion of some haplotypes was significantly different to the rest of the study area. A temporal multinomial scan statistic was also carried out to assess whether there were periods when the proportion of some haplotypes was significantly different to the rest of the study period. Both analysis were performed in the SaTScan V.8.0 software (17). Clustering was assessed using a multivariate model and procedures described elsewhere (18, 19).
Results
Sample Distribution
Most (93%) of the 72 field samples were collected from the two provinces in which most of the poultry production is concentrated in Argentina, namely, Entre Rios (n = 39 samples, 54.2%) and Buenos Aires (n = 28 samples, 38.8%). Samples became from 20 flocks distributed in 12 localities in Buenos Aires and 32 flocks from 15 localities in Entre Rios province. Unfortunately, geolocalization from all Buenos Aires and 13 Entre Rios samples were not provided. The remaining 7% (n = 5) of the samples were collected in others provinces like Cordoba, Neuquén, Rio Negro, and Mendoza.
Selection of the Target Genomic Region
With the objective of selecting the most appropriate genomic sequence/s for epidemiological studies on circulating ILTV strains, many PCRs were performed on different genomic regions, including TK, ICP4, gC, gD, gB, gG, and gJ. Initially, the analysis was conducted on both types of attenuated vaccines (CEO and TCO) and 17 field samples.
Analysis of the amplicon sequences corresponding to TK and gC genes did not show differences among field samples or even between the vaccines.
Sequence alignment were compared to complete sequences available in the Genebank. Although the analysis of the ICP4 sequence allowed to clearly distinguishing between TCO and CEO vaccines, there were no differences between the CEO vaccine and field isolates. In contrast, gB showed two changes at positions 1043 (T/C) and 1931 (C/T), and those changes allowed distinguishing between TCO and CEO vaccines, and between vaccines and fields samples, although no differences were found among the latter (Table 2).
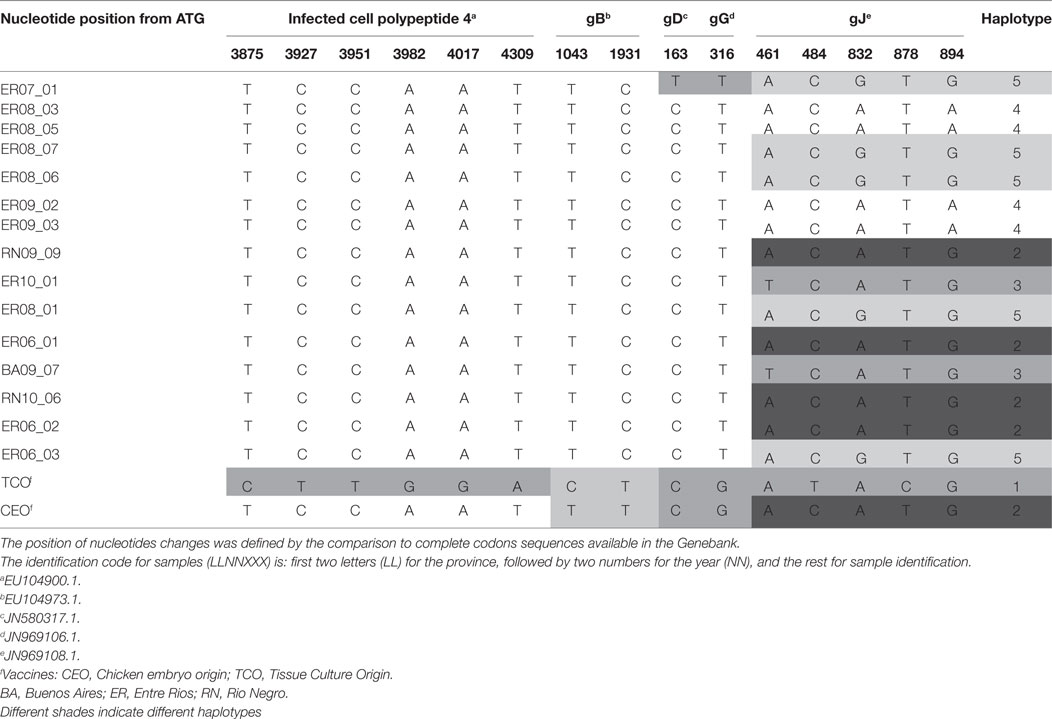
Table 2. Positions of nucleotide changes in amplicons of different genomic regions and defined haplotype according to gJ amplified sequence.
In gD and gG amplicons, only one position changed, 163 (C/T) and 316 (T/G), respectively. The nucleotide change detected in gD sequence was observed in only one field sample and the others showed no changes between them and the vaccines. The gG sequence was able to differentiate vaccines from field samples, but could not differentiate among field samples or between vaccines (Table 2).
Analysis of the amplified gJ region evidenced five changes, in positions 461 (A/T), 484 (C/T), 832 (G/A), 878 (T/C), and 894 (G/A), which permitted to differentiate between vaccines and among field strains. The combination of these nucleotide differences determined 5 haplotypes, numbered from 1 to 5 (Table 2).
Only gJ sequence was used to characterize the rest (n = 55) of the samples, in addition to the 17 samples initially assessed.
Glycoprotein J Sequence Analysis
Haplotypes 1, 2, and 3 were found in Buenos Aires, and haplotype 2 was also detected in other regions (Table 3).

Table 3. Presence of haplotypes in Buenos Aires and other provinces.
The five haplotypes were circulating in Entre Rios, the most densely populated poultry region and from where most (n = 39) samples were collected, and haplotypes 4 and 5 were found only in Entre Rios (Table 4). Interestingly, haplotypes 4 and 5 were mostly detected in different farms concentrated in an area of 6.600 km2 located in the Concepción del Uruguay and Colón counties, most of them were located near a road, referred to as provincial route 23 (Figure 1).
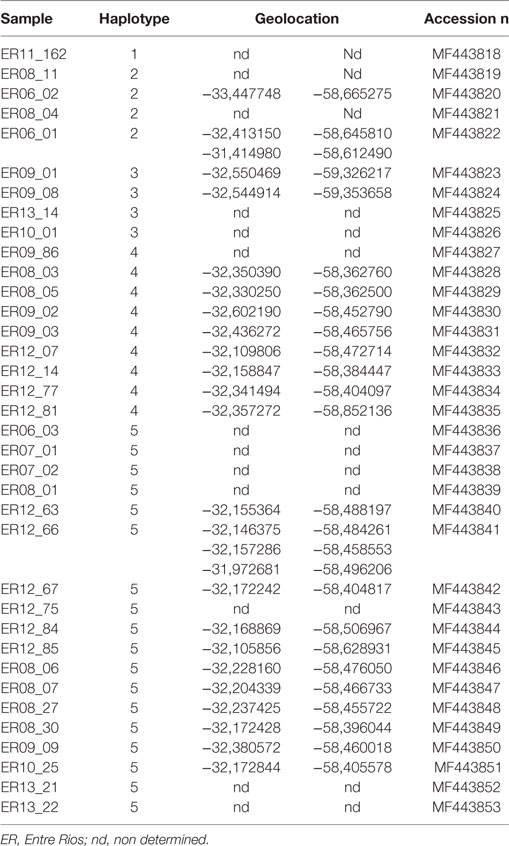
Table 4. Haplotypes and geolocation in Entre Rios province.
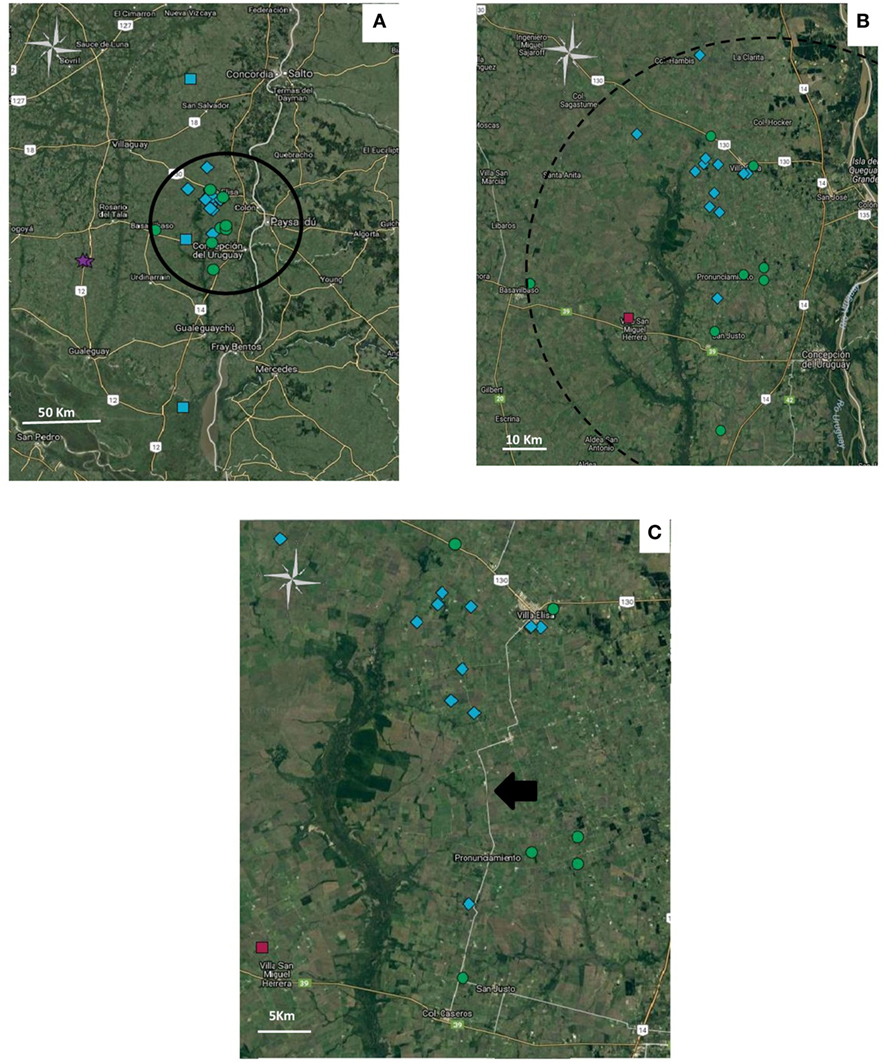
Figure 1. Geolocation of the haplotypes in Entre Rios province. Haplotypes: 2 ( ), 3 (
), 3 ( ), 4 (
), 4 ( ) y 5 (
) y 5 ( ). (A) Distribution of geolocated haplotypes in Entre Rios Province. Cluster is indicated as a circle. (B) Amplification of the zone where haplotype 5 and 4 are concentrated. (C) Distribution of haplotypes 4 and 5 along provincial route 23, pointed out with a black arrow. Figure was created with Google.
). (A) Distribution of geolocated haplotypes in Entre Rios Province. Cluster is indicated as a circle. (B) Amplification of the zone where haplotype 5 and 4 are concentrated. (C) Distribution of haplotypes 4 and 5 along provincial route 23, pointed out with a black arrow. Figure was created with Google.
The analysis of the deduced amino acid composition of gJ sequence showed differences among haplotypes. Nucleotides changes in haplotype 4 were synonymous respect to haplotype 2 (showing an identical amino acid composition); however, haplotype 1 showed changes in two positions, (W/R) 163 and (A/V) 293, whereas haplotype 3 and 5 showed differences in position, (I/N) 154 and (G/S) 278, respectively.
Cluster Analysis
The spatial multinomial scan was carried out only in the Entre Rios province, which concentrates most of the poultry production of the country and also most (n = 39) of samples collected, because there were too many missing locations from the rest of the study area. As 13/39 farm geolocations from the Entre Rios province were missing, the analysis was carried out on the remaining 26 samples collected. The temporal multinomial scan was carried out using the entire study area (72 samples).
In the analysis of spatial clustering centered in Entre Rios, only a cluster of 46 km radius was detected, in which a higher proportion of observed/expected cases of haplotype 4 and 5 cases (o/e = 2.22 and 2, 22, respectively) showed that the distribution of haplotypes was not randomized (Table 5).
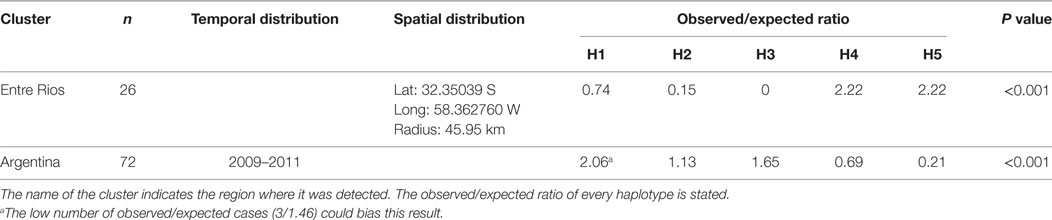
Table 5. Spatial clustering in the Entre Rios region and temporal clustering over the entire study area and period.
Results of the time cluster analysis suggested a remarkable decline of haplotype 5 detection during the second half of the study period (2009–2011). Overall distribution of haplotypes was significantly different (p < 0,001) for those 3 years compared to that expected under the null hypoteshsis of even distribution of haplotypes (o/e ratio of 2.06, 1.13, 1.65, 0.69, 0.21 for haplotype 1–5, respectively) (Table 5).
Discussion
Five haplotypes of circulating ILTV in Argentina were identified. Although the analysis by RFLP in different regions of the genome was successfully used by others to characterize ILTV strains (9–12), there is evidence that differentiation of field isolates from vaccine strains varies depending on the region of the world assessed (20). There are differences, not only in the regions of the genome analyzed but also in the number of these region used to characterized field strains and vaccines (20). Certainly, the analysis of several regions would be better to characterize the circulating strains as it would provide more informative sites and also have the potential to detect the presence of recombinants. Recently, a multi-allelic PCR-sequencing analysis proved to be an efficient tool in differentiating ILTV strains globally (21). However, we found that two of the assayed regions (ICP4 and gB) did not add more discriminative information than gJ alone, at least for Argentine strains. So, among the seven regions under study here, gJ resulted the most variable on region, determining 5 variants or haplotypes circulating in the country.
Haplotype 1 covers the TCO vaccine and few Argentine field strains. The comparison of these sequences and several sequences from the Genbank showed that nucleotide T in position 484 and nucleotide in position C 878 are present both in the TCO vaccine and strain 81658 (JN542535), which may have been acquired from chickens vaccinated with TCO (22), and also in the USDA reference strain (JN542534), which showed very similar RFLP pattern when compared to the TCO (23). Presence of haplotype 1 in the field strains reported here may have been related to the finding by chance, of the TCO strain in recently vaccinated animals with a compromised immune state. Although we have no vaccine information related to the isolates of Buenos Aires (BA149-647 and 672), it is likely that animals from Entre Rios (ER 11-162) may have been vaccinated with the TCO vaccine.
Haplotype 2, which included the CEO vaccine, seems to be widely spread through the country. The origin of this haplotype in Argentina could probably be related with the introduction of the CEO vaccine. The use of the CEO vaccine has been banned in Argentina for the last 10 years, because of its associated reversion to virulence (1). However, it is likely that the CEO vaccine strain may have still been circulating since then, reverting to its virulent form to produce new outbreaks. However, such hypothesis requires further analysis, based on sequences of other ILTV genome regions, to test the homology of those field samples with the CEO strain.
Comparison of other ILTV field sequences here to other gJ sequences obtained from the Genebank suggests that the presence of certain nucleotides at positions 461 (T), 832 (G), and 894 (A), are exclusive to the Argentinian strains. Those changes are responsible for defining the circulating haplotypes 3, 5, and 4, respectively.
Although the analysis of the gJ sequence was not comprehensive enough to completely characterize circulating strains, differentiation of at least some of the circulating field strains was possible. Such characterization helped to identify and differentiate widespread from locally circulating strains. Haplotypes 4 and 5 were more prevalent in Entre Rios, compared to Buenos Aires. Moreover, most haplotype 4 and 5 cases were concentrated along a 46 km radius, on an area with high density of poultry farms located nearby a route (provincial route 23) frequently used by trucks used to move poultry. Data also suggest that both haplotypes have been circulating in Entre Rios since, at least, 2006 and 2008, respectively. That observation, in addition to the small, restricted area where those haplotypes coexist, suggests that some factors, like suppliers or shared workers, could be implicated in the transmission, maintenance, and high prevalence of these haplotypes in such limited region.
A remarkable drop of the frequency of haplotype 5 was detected during 2009–2011, followed by its recovery in 2012. Although, it is difficult to rule out that the reduction in incidence of haplotype 5 may have been due to selective under-reporting or under-detection during the study period, no data supports these explanations.
Alternative explanations may also include the reintroduction of the haplotype, after initial extinction, from an external source, or a variation on the virus virulence and transmissibility.
The analysis of deduced amino acid composition presented changes that affected not only the structure but also polarity of the virus, in some haplotypes. The possible effect of those changes on the gJ structure, also on its function, is unknown. Further functionality or structural studies on gJ would be necessary to analyze the relevance of those changes.
In the event of absence of complete or several partial sequences of ILTV strains, the methodology here may help to support the initial classification of ILTVs, and routine monitoring of changes in the pattern of prevalent haplotypes, which may ultimately help to assess the epidemiological situation of the disease in Argentina, and other regions endemically affected by the ILTV.
Nevertheless, taking into account, the reported allelic variation presented in other genomic regions (21) than the one studied here, further analysis of these regions would allow us to further differentiate field samples and compare them to circulating strains in other countries.
Author Contributions
GK and MC conceived and designed the study. CP, MR, and VO were responsible for most of PCR assay as well as data edition and the acquisition of field samples. GK, MC, AP, CP, FR, and VO contributed in data analysis. GK, MC, AP, and AV were responsible for data interpretation. The manuscript was written by MC, GK, AP, and also participated in the final revision of the manuscript as well as AV.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the sequencing service SIGYSA from INTA for technical assistance.
Funding
Funding for this study was provided by the projects PNBIO-1131032 and PNSA-1115056 awarded by the Instituto Nacional de Tecnologia Agropecuaria (INTA).
References
1. Guy J, Bagust T. Laryngotracheitis. 11th ed. In: Saif YM, editor. Diseases of Poultry. Iowa city: Iowa State Press, Blackwell Publishing Company (1999). p. 135–60.
2. Leib DA, Bradbury JM, Hart CA, McCarthy K. Genome isomerism in two alphaherpesviruses: herpesvirus saimiri-1 (herpesvirus tamarinus) and avian infectious laryngotracheitis virus. Arch Virol (1987) 93:287–94. doi:10.1007/BF01310982
3. Samberg Y, Cuperstein E, Benheim U, Arnovici I. The development of a vaccine against avian infectious laryngotracheitis. Immunizations of chickens with modified laryngotracheitis vaccine in the drinking water. Avian Dis (1971) 15(2):413–7. doi:10.2307/1588714
4. Gelenczei EF, Marty EW. Studies on a tissue-culture modifies infectious laryngotracheitis virus. Avian Dis (1964) 8:105–22. doi:10.2307/1587827
5. Kotiw M, Wilks CR, May JT. Differentiation of Infectious laryngotracheitis virus strains using restriction endonucleases. Avian Dis (1982) 26(4):718–30. doi:10.2307/1589858
6. Leib DA, Bradbury JM, Gaskell RM, Hughes CS, Jones RC. Restriction endonuclease patterns of some European and American isolates of avian infectious laryngotracheitis virus. Avian Dis (1986) 30:835–7. doi:10.2307/1590595
7. Andreasen JR, Glisson JR, Villegas P. Differentiation of vaccine strains and Georgia field isolates of infectious laryngotracheitis virus by their restriction endonuclease fragment patterns. Avian Dis (1990) 34:646–56. doi:10.2307/1591259
8. Keeler CL, Hazel JW, Hasting JE, Rosenberger JK. Restriction endonuclease analysis of delmarva field isolates of infectious laryngotracheitis virus. Avian Dis (1993) 37:418–26. doi:10.2307/1591668
9. Chang PC, Lee YL, Shien JH, Shieh HK. Rapid differentiation of vaccine strains and field isolates of infectious laryngotracheitis virus by restriction fragment length polymorphism of PCR products. J Virol Methods (1997) 66:179–86. doi:10.1016/S0166-0934(97)00050-5
10. Kirkpatrick NC, Mahmoudian A, O’Rourke D, Noormohammadi AH. Differentiation of infectious laryngotracheitis virus isolates by restriction fragment length polymorphic analysis of polymerase chain reaction products amplified from multiple genes. Avian Dis (2006) 50:28–34. doi:10.1637/7414-072205R.1
11. Neff C, Sudler C, Hoop RK. Characterization of western European field isolates and vaccines strains of avian infectious laryngotracheitis virus by restriction fragment length polymorphism and sequence analysis. Avian Dis (2008) 52:278–83. doi:10.1637/8168-110107-Reg.1
12. Chacon JL, Mizuma MY, Piantino Ferreira AJ. Characterization by restriction fragment length polymorphism and sequence analysis of field and vaccine strains of infectious laryngotracheitis virus involved in severe outbreaks. Avian Pathol (2010) 39(6):425–33. doi:10.1080/03079457.2010.516386
13. Kalendar R, Lee D, Schulman AH. FastPCR software for PCR, in silico PCR, and oligonucleotide assembly and analysis. Methods Mol Biol (2014) 1116:271–302. doi:10.1007/978-1-62703-764-8_18
14. Veits J, Mettenleiter TC, Fuchs W. Five unique open reading frames of infectious laryngotracheitis virus are expressed during infection but are dispensable for virus replication in cell culture. J Gen Virol (2003) 84(6):1415–25. doi:10.1099/vir.0.18926-0
15. Hall TA. BioEdit: a user-friendly biological sequence aligment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser (1999) 41:95–8.
16. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions specific gap penalties and weight matrix choice. Nucleic Acids Res (1994) 22:4673–80. doi:10.1093/nar/22.22.4673
17. Kulldorff M, Huang L, Konty K. A scan statistic for continuous data based on the normal probability model. Int J Health Geogr (2009) 8:58. doi:10.1186/1476-072X-8-58
18. Valdes-Donoso P, Mardones FO, Jarpa M, Ulloa M, Carpenter TE, Perez AM. Co-infection patterns of infectious salmon anaemia and sea lice in farmed Atlantic salmon, Salmo salar L., in southern Chile (2007–2009). J Fish Dis (2013) 36:353–60. doi:10.1111/jfd.12070
19. Junga I, Kulldorffb M, Richardc OJ. A spatial scan statistic for multinomial data. Stat Med (2010) 29(18):1910–8. doi:10.1002/sim.3951
20. Menendez KR, Garcia M, Spatz S, Tablante NL. Molecular epidemiology of laringotracheitis: a review. Avian Pathol (2014) 43(2):108–17. doi:10.1080/03079457.2014.886004
21. Choi EJ, La TM, Choi IS, Song CS, Park SY, Lee JB, et al. Genotyping of infectious laryngotracheitis virus using allelic variations from multiple genomic regions. Avian Pathol (2016) 45(4):443–9. doi:10.1080/03079457.2016.1155692
22. Spatz SJ, Volkening JD, Keeler CL, Kutish GF, Riblet SM, Boettger CM, et al. Comparative full genome analysis of four infectious laryngotracheitis virus (gallid herpesvirus-1) virulent isolates from the United States. Virus Genes (2012) 44:273–85. doi:10.1007/s11262-011-0696-3
Keywords: infectious laryngotracheitis virus, molecular characterization, epidemiology, spatial cluster analysis, glycoprotein J, Argentina
Citation: Craig MI, Rojas MF, van der Ploeg CA, Olivera V, Vagnozzi AE, Perez AM and König GA (2017) Molecular Characterization and Cluster Analysis of Field Isolates of Avian Infectious Laryngotracheitis Virus from Argentina. Front. Vet. Sci. 4:212. doi: 10.3389/fvets.2017.00212
Received: 03 April 2017; Accepted: 27 November 2017;
Published: 13 December 2017
Edited by:
Timothée Vergne, Institut de Recherche pour le Développement (IRD), FranceReviewed by:
Jean-Luc Guerin, Ecole Nationale Vétérinaire de Toulouse, FranceHelene Guis, Centre de coopération internationale en recherche agronomique pour le développement (CIRAD), France
Copyright: © 2017 Craig, Rojas, van der Ploeg, Olivera, Vagnozzi, Perez and König. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María I. Craig, craig.maria@inta.gob.ar;
Guido A. König, konig.guidoalberto@inta.gob.ar