1
Department of Biotechnology and Biosciences, University of Milano-Bicocca, Milan, Italy
2
Center for Nanomedicine and Tissue Engineering, A.O. Ospedale Niguarda Ca’ Granda, Milan, Italy
3
San Raffaele Scientific Institute and University, Milan, Italy
4
Cardiff School of Biosciences, Cardiff University, Cardiff, UK
Since its discovery almost three decades ago, the secreted neurotrophin brain-derived neurotrophic factor (BDNF) has been firmly implicated in the differentiation and survival of neurons of the CNS. More recently, BDNF has also emerged as an important regulator of synaptogenesis and synaptic plasticity mechanisms underlying learning and memory in the adult CNS. In this review we will discuss our knowledge about the multiple intracellular signalling pathways activated by BDNF, and the role of this neurotrophin in long-term synaptic plasticity and memory formation as well as in synaptogenesis. We will show that maturation of BDNF, its cellular localization and its ability to regulate both excitatory and inhibitory synapses in the CNS may result in conflicting alterations in synaptic plasticity and memory formation. Lack of a precise knowledge about the mechanisms by which BDNF influences higher cognitive functions and complex behaviours may constitute a severe limitation in the possibility to devise BDNF-based therapeutics for human disorders of the CNS.
The ability to store and recall information is one of the most amazing capacities of higher organisms. In recent years, our understanding of the basic molecular and cellular mechanisms underlying cognitive processing and behaviour has considerably advanced, providing not only the basis for future therapy of memory disorders, but also for slowing down normal age-related cognitive decline in humans. Studies of learning and memory in animal models have identified a number of gene products that are necessary for these processes. Among them is brain-derived neurotrophic factor (BDNF). BDNF was first purified from the mammalian brain based on its survival-promoting action on dorsal root ganglion cells (Barde et al., 1982
), and was classified as the second member of the neurotrophin family of growth factors, after nerve growth factor (NGF; Cohen et al., 1954
). Neurotrophins are required for the development of the nervous system of vertebrates, and the family also includes neurotrophin-3 (NT3), neurotrophin-4/5, neurotrophin-6 and neurotrophin-7. Among neurotrophins, BDNF, and its major receptor TrkB, has the most abundant and widespread expression in the developing and adult mammalian brain (Murer et al., 2001
). Likewise, the action of this neurotrophin in the adult CNS is now the most extensively studied, probably because it has been shown to have a critical role in long-term potentiation (LTP), a form of synaptic plasticity which is still widely considered a cellular model of long-term memory (LTM) formation (Bliss and Collingridge, 1993
; Martin et al., 2000
).
Activity-Dependent BDNF Regulation and Release
The structural organization of the Bdnf gene has recently been reviewed for mouse and rat (Liu et al., 2006
; Aid et al., 2007
), and for human (Liu et al., 2005
). Its structure and regulation is complex: a total of 9 promoters produce 24 different transcripts, all of them translated into an identical mature dimeric protein, suggesting a multilevel regulation of expression (Figure 1
). Indeed, BDNF promoters are individually recruited to direct the tissue-specific expression of the different BDNF transcripts. Moreover, specific exon-containing transcripts are differentially regulated by a variety of stimuli, which regulate neuronal activity such as physical exercise, seizures, ischemia, osmotic stress, and antidepressant treatment. It is thought that the different exons may have different functions particularly with relation to the intracellular targeting and the availability of BDNF by controlling translation and/or stability. Protein non-coding antisense transcripts are expressed from the human BDNF gene locus. These may function as another level of complexity in the regulation of BDNF gene expression in vivo.
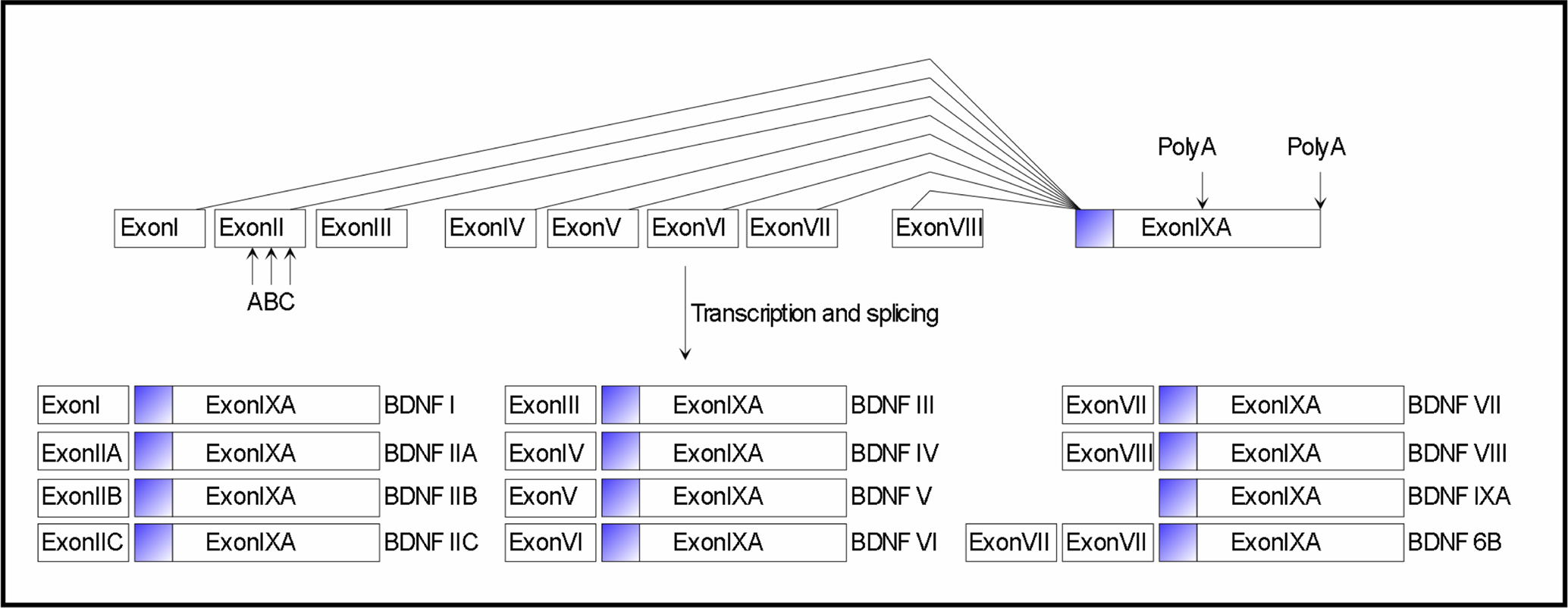
Figure 1. Mouse and rat Bdnf gene structure and transcripts. Exons are indicated by boxes. Filled box in exon IXA indicates the coding region of the Bdnf gene. Lines indicate splice variants. Arrows indicate within-exon splice sites and alternative polyadenylation sites. The Bdnf gene is transcribed from different promoters, immediately preceding each of the 5′ exons (exons I–VIII), so that each full-length transcript contains a unique 5′-exon and a common 3′-exon (exon IXA) that encodes the BDNF protein. Transcript BDNF6B results from splicing events that incorporate exons VII, VIII and IXA.
BDNF protein is synthesized as a precursor, pre-proBDNF protein, resulting after cleavage in a 32-kDa proBDNF protein. ProBDNF is either proteolytically cleaved intracellularly by enzymes like furin or pro-convertases and secreted as the 14 kDa mature BDNF (mBDNF), or secreted as proBDNF and then cleaved by extracellular proteases, such as metalloproteinases and plasmin, to mBDNF (reviewed in Lessmann et al., 2003
). The extent of intracellular and extracellular processing of proBDNF is not exactly clear, but proBDNF is less efficiently processed by intracellular proteases compared to other neurotrophins and secretion of proBDNF with respect to mBDNF seems to prevail (Mowla et al., 2001
). Nevertheless, both proBDNF and mBDNF are preferentially sorted and packaged into vesicles of the activity-regulated secretory pathway. ProBDNF is not an inactive precursor of BDNF, but rather it is a signalling protein in its own right (see below). ProBDNF is released in the immature and mature CNS in an activity dependent manner (Mowla et al., 2001
; Yang et al., 2009
; Figure 2
). The intracellular localization of BDNF is predominantly somatodendritic but it is also enriched in the dendrites, where it is also synthesized from mRNA in close proximity to spines (reviewed in Tongiorgi et al., 1997
; Tongiorgi, 2008
). BDNF is present in pre- and postsynaptic compartments and it can undergo both retrograde and anterograde transport. Moreover, BDNF can act via autocrine and paracrine mechanisms, depending on the site of cell surface receptors through which it signals (reviewed in Murer et al., 2001
). The activity-regulated release of BDNF can occur via three mechanisms dependent on the site of release: (i) Ca2+ influx-dependent release from postsynaptic sites, which is mediated by Ca2+ influx through ionotropic glutamate receptors and voltage gated Ca2+-channels (Hartmann et al., 2001
), (ii) Ca2+ influx-dependent release from presynaptic sites (Balkowiec and Katz, 2002
), and (iii) Ca2+ influx-independent release that relies on Ca2+ release from intracellular stores (Griesbeck et al., 1999
). Neuronal activity also regulates the transport of BDNF mRNA and protein into dendrites (reviewed in Tongiorgi et al., 1997
; Tongiorgi, 2008
), and these mechanisms are considered to be responsible for the ability of locally translated BDNF to modulate synaptic transmission and synaptogenesis (reviewed in Lu and Figurov, 1997
). Evidence of the importance for the regulated trafficking of BDNF to cognitive function comes from the only single nucleotide polymorphism (SNP) identified in the human BDNF gene, Val66Met (Egan et al., 2003
). This SNP consists in the substitution of Met for Val at position 66 in the pro-region of BDNF which not only alters the trafficking, distribution and activity-dependent release of BDNF from neurons but also results in memory impairments in rodent models and in an increased susceptibility towards disorders such as depression, bipolar disorder and eating disorder in humans carrying the mutation (Chen et al., 2004
).
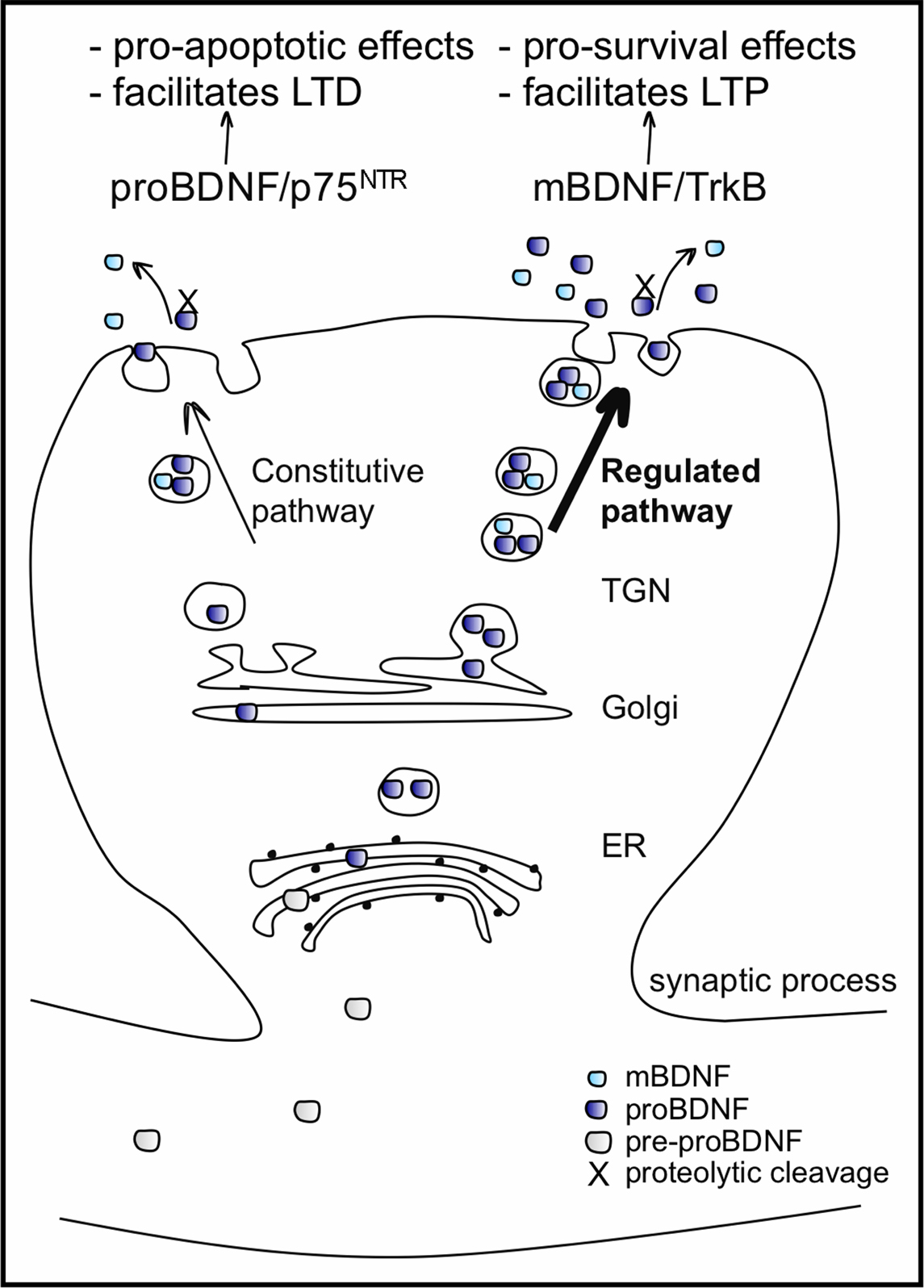
Figure 2. BDNF processing, packaging and secretion in neurons. BDNF is synthesized as a pre-proBDNF protein, which has its pre-sequence cleaved off in the endoplasmic reticulum (ER). The resulting 32-kDa proBDNF moves, via the Golgi apparatus, into the trans-Golgi network (TGN) where two kinds of secretory vesicles are generated: those of the constitutive secretory pathway and those of the regulated pathway, whose secretion is activity-dependent. ProBDNF packaged in both types of vesicles is either proteolytically cleaved and secreted as 14-kDa mBDNF, or secreted as proBDNF and cleaved by extracellular proteases. The extent of the intra and extracellular processing of proBDNF is not exactly clear, but secretion of the proBDNF predominates. Both proBDNF and mBDNF are preferentially packaged into vesicles of the regulated secretory pathway. Once released, proBDNF binds preferentially to pan neurotrophin receptor p75NTR and mBDNF binds preferentially to both pre-and post-synaptic TrkB receptors, activating different intracellular secondary messenger cascades and affecting distinct cellular responses.
BDNF-Dependent Signalling Pathways and their Targets
BDNF binds and activates, both pre- and postsynaptically, two different transmembrane receptor proteins: the tropomyosin related kinase TrkB receptor with high affinity, and the pan neurotrophin receptor p75NTR with low affinity (see Figure 3
). At present, virtually all the synaptic effects of BDNF are attributed to TrkB activation. However, there is evidence that proBDNF binds p75NTR preferentially (Teng et al., 2005
), which has distinct functional consequences (see below). BDNF can also bind TrkB splice variants lacking the tyrosine kinase domain required for downstream signalling, which are found mainly, but not exclusively, in glial cells (Klein et al., 1990
). Therefore, the binding of BDNF to these TrkB variant receptors can act as a dominant-negative inhibitor of BDNF signalling by forming heterodimers with the full-length TrkB, and by internalizing BDNF to function as a clearance receptor (Haapasalo et al., 2002
). ProBDNF, p75NTR and truncated TrkB isoforms can therefore be considered as negative regulatory mechanisms of the canonical BDNF–TrkB association, with consequent effects for synaptic plasticity and perhaps learning and memory.
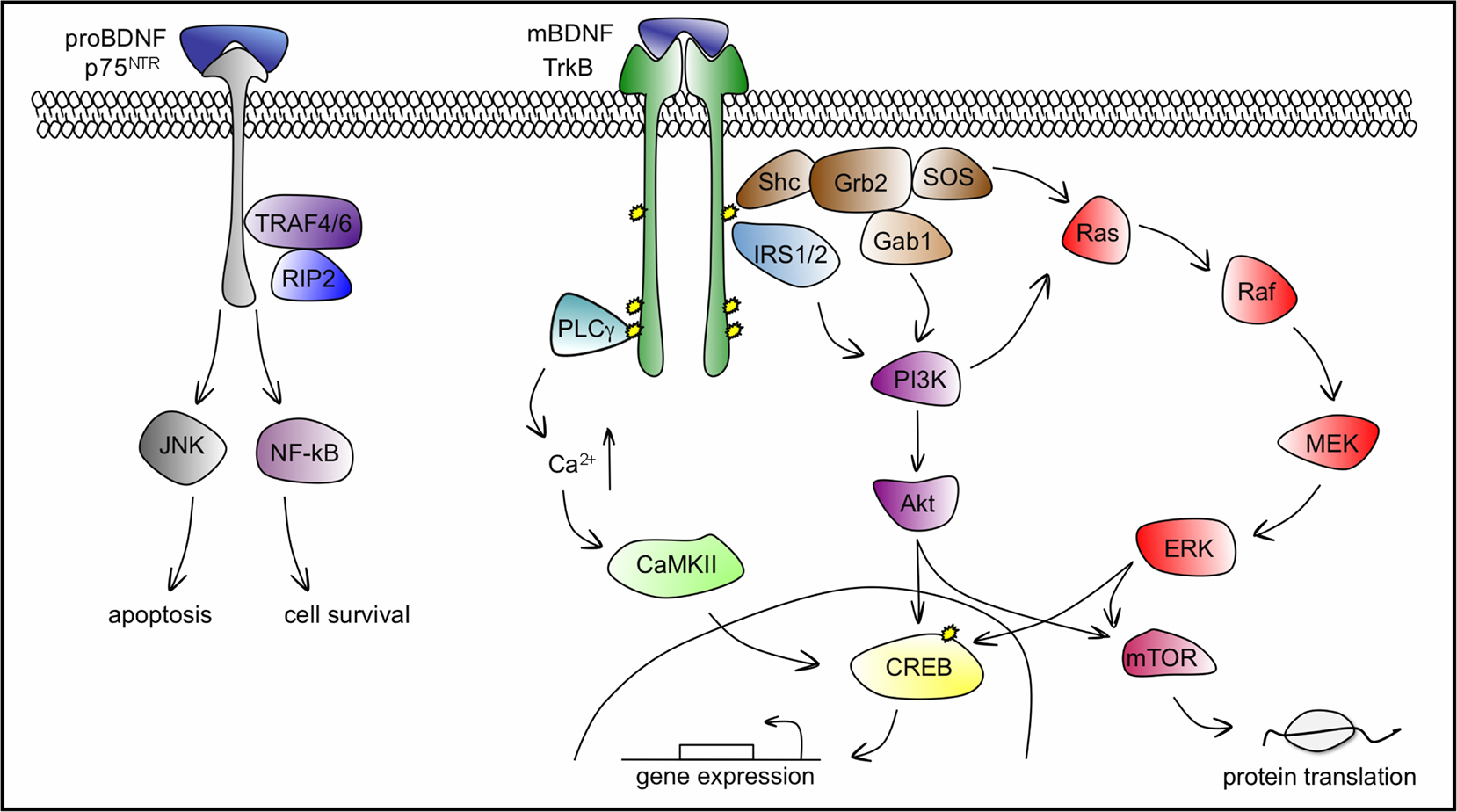
Figure 3. BDNF–TrkB and BDNF–p75NTR signalling pathways. BDNF binds TrkB with high affinity to induce its dimerization and autophosphorylation of tyrosine residues in the cytoplasmic kinase domain that serve as docking sites for effector molecules and trigger the activation of three main signalling pathways: PLCγ, PI3K and ERK cascades, which ultimately lead to the phosphorylation and activation of the transcription factor CREB that mediates transcription of genes essential for the survival and differentiation of neurons. The recruitment of PLCγ increases intracellular Ca2+ levels and leads to the activation of CaMKII to phosphorylate CREB. PI3K can be activated via the Shc/Grb2/SOS complex through Gab1 and by IRS1/2. Lipid products generated by the activated PI3K, the phosphatidylinositides, bind and activate protein kinase Akt, upstream of CREB. The ERK cascade can be activated both by the Shc/Grb2/SOS complex and by PI3K. ERK phosphorylation leads directly to CREB phosphorylation. Both Akt and ERK activate mTOR, responsible for enhanced translation initiation. BDNF binds p75NTR with low affinity, leading to apoptosis through the JNK cascade or cell survival through the NF-kB cascade. PLCγ, phospholipase Cγ; PI3K, phosphatidylinositol 3-kinase; ERK, extracellular signal-regulated kinase; CaMKII, calcium-calmodulin dependent kinase; Shc, src homology domain containing; Grb2, growth factor receptor-bound protein 2; SOS, son of sevenless; Gab1, Grb-associated binder 1; IRS1/2, insulin receptor substrates 1/2; CREB, cAMP-calcium response element binding protein; Ras, GTP binding protein; Raf, Ras associated factor; MEK, MAP/Erk kinase; mTOR, mammalian target of rapamycin; TRAF4/6, tumour necrosis factor receptor associated factor 4/6; RIP2, receptor interacting protein 2; JNK, c-Jun N-terminal kinase; NF-kB, nuclear factor k B.
TrkB activation by BDNF follows the general scheme for receptor tyrosine kinases and initiates three major cascades of signalling pathways: phospholipase Cγ (PLCγ), phosphatidylinositol 3-kinase (PI3K), and the well-characterized cascade governed by extracellular signal-regulated kinases (ERK), member of the mitogen-activated protein kinase (MAPK) family (reviewed in Segal, 2003
; Figure 3
). TrkB receptor activation by BDNF results in its dimerization and in the autophosphorylation of specific tyrosine residues in its cytoplasmic kinase domain. Transphosphorylation of Y785 recruits PLCγ. Activation of this pathway is directly implicated in a rise of intracellular Ca2+ via its release from intracellular stores, and in the activation of the Ca2+-calmodulin dependent kinase, CaMKII. The elevation of intracellular Ca2+ is one of the most important biochemical outcomes of BDNF signalling in the postsynaptic cell. Especially exciting evidence indicates that Ca2+-regulated mRNA translation occurs locally at postsynaptic sites (Wu et al., 1998
; Aakalu et al., 2001
). This mechanism provides a means for the rapid and accurate expression of activity-induced gene products such as BDNF at activated synapses. Moreover, the CaMKII activated transcription factor CREB has been shown to recognize CRE and a CaRE regulatory elements in the Bdnf gene, activating its transcription (reviewed in West et al., 2002
). Thus, BDNF can regulate its own expression via activation of CaMKII signalling.
The transphosphorylation of TrkB Y490 upon BDNF binding permits the association of SH2 (src homology-type 2) linker proteins such as shc (src homology domain containing) and insulin receptor substrate-1 and -2 (IRS2, IRS2). This event leads to the activation of the PI3K and ERK signalling pathways. Src sequentially recruits an intermediary protein Grb2 and the guanine nucleotide exchange factor SOS, initiating the GTP loading and activation of Ras and the activation of the Raf, MEK and ERK kinase cascade. Phosphorylated ERK translocates to the nucleus to activate transcription factors such as CREB to regulate gene expression. Grb2 can also recruit another intermediary binding protein, Gab1 to activate PI3K and the downstream kinase, Akt (also known as protein kinase B). BDNF can also activate the PI3K pathway via a direct interaction between IRS1/IRS2 and PI3K (Yamada et al., 1997
). The PI3K pathway was shown to mediate the protective effects of BDNF in several neuronal cell types in vitro, including hippocampal neurons (Zheng and Quirion, 2004
). Ras was the first neurotrophin-activated signalling protein shown to mediate neuronal survival (reviewed in Kaplan and Miller, 2000
), which can involve Ras-mediated activation of PI3K, however, Ras-dependent neuronal survival is also mediated by the ERK pathway. BDNF was also shown to facilitate local translation of proteins in dendrites by activation of mammalian target of rapamycin (mTOR) via PI3K (Schratt et al., 2004
). Both mTOR and ERK are able to regulate the assembly of the eIF4e complex and phosphorylation of S6K1, both contributing to an enhanced mRNA translation initiation at active synapses (reviewed in Klann and Dever, 2004
; Bramham and Wells, 2007
). The differential activation and role of these cascades in neuronal survival will likely depend on the cell type and the involvement of specific physiological or pathological stimuli.
The activation of p75NTR by BDNF initiates both prosurvival NF-kB and pro-apoptotic Jun kinase signalling cascades. p75NTR-mediated survival involves the activation of the NF-kB pathway via the association of p75NTR with tumour necrosis factor receptor associated factor 4/6 (TRAF4/6) and receptor interacting protein-2 (RIP2, reviewed in Chao, 2003
; Figure 3
).
BDNF elicits rapid effects on synaptic transmission and membrane excitability primarily via activation of these signalling pathways. BDNF affects synaptic transmission by acting at pre- and postsynaptic sites. It can induce the presynaptic release of glutamate and GABA through TrkB–ERK-mediated phosphorylation of synapsin (Jovanovic et al., 2000
). At least for glutamatergic synapses, the BDNF-induced enhancement in transmission is mediated by an increase in the number of docked vesicles at the active zones of synapses (Tyler and Pozzo-Miller, 2001
). Postsynaptically, BDNF also rapidly modulates excitatory and inhibitory transmission by altering the activation kinetics of glutamatergic NMDA receptors and inhibitory GABA receptors (reviewed in Rose et al., 2004
). While the inhibition of GABA transmission in the adult brain occurs via TrkB–PLCγ signalling, the potentiation of NMDA receptor responses and enhancement in Ca2+ influx is meditated by a novel mechanism in which the src-family tyrosine kinase Fyn, activated by TrkB, directly phosphorylates the NR2B subunit. BDNF also upregulates surface expression of AMPA receptors by inducing their rapid surface translocation to increase excitatory transmission (Narisawa-Saito et al., 2002
; Itami et al., 2003
), an effect that requires ERK signalling (Li and Keifer, 2009
; Figure 4
).
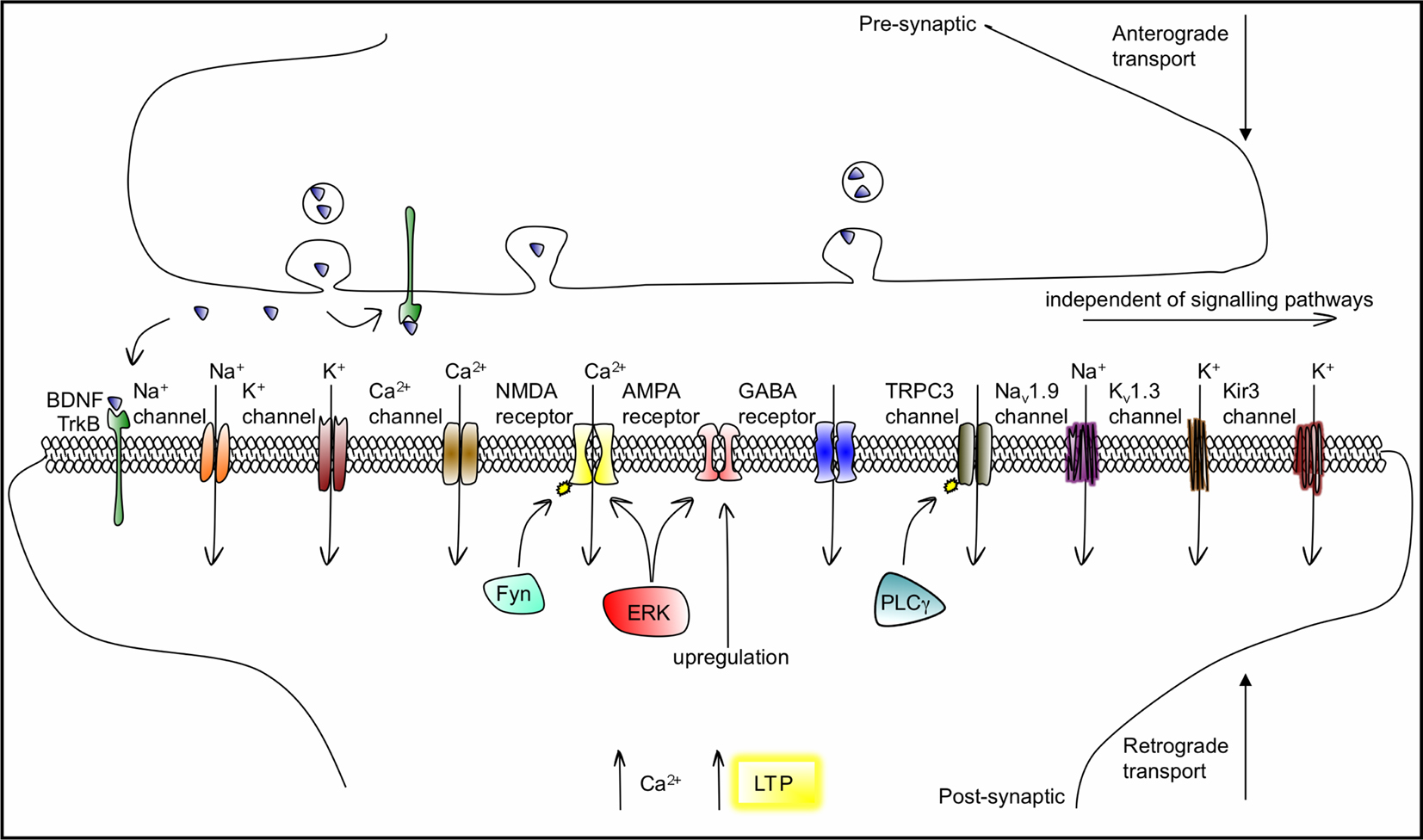
Figure 4. BDNF/TrkB actions on ligand-gated, voltage-gated and second-messenger-gated ion channels, which mediate fast and slow synaptic transmission in neurons. BDNF is transported anterogradely and retrogradely and can activate TrkB receptors both pre- and postsynaptically. The association of BDNF with TrkB modulates or activates ion channels including Na+, Ca2+ and K+ channels, within a range of seconds to minutes, through intracellular signalling cascades. TRPC3 is a non-selective cation channel that needs to be phosphorylated by TrkB to open, via PLCγ, a process also acting in the range of minutes. BDNF enhances glutamatergic neurotransmission by increasing open probability of NMDA (by promoting its phosphorylation, via Fyn-dependent and Fyn-independent mechanisms) and by upregulating AMPA expression. ERK signalling is involved in both NMDA and AMPA gating. In the millisecond range, BDNF/TrkB can directly gate the Nav1.9 Na+, the Kv1.3 K+ and the Kir3 K+ ion channels. The resulting depolarizations contribute to the facilitation of the induction of LTP.
The modulation of membrane excitability by BDNF occurs through a range of mechanisms, as outlined in Figure 4
; via TrkB activation of PLCγ and the subsequent alteration in the activity of the nonselective cation TRPC3 channel (Li et al., 1999
), the gating of the Na+ channel Nav1.9 by an unknown BDNF–TrkB mechanism (Blum et al., 2002
), and the increased gating probability of the voltage-gated potassium channel Kv1.3 (Tucker and Fadool, 2002
) and of the G protein-gated potassium channel Kir3 (Ippolito et al., 2002
). In addition, the effects of BDNF on neuronal excitability may also be in part mediated by the regulation of Arc/Arg3.1 activity. BDNF regulates the expression of the immediate early gene Arc/Arg3.1 (Yin et al., 2002
; Ying et al., 2002
). The functions of Arc/Arg3.1 are not as yet fully understood. It is known that Arc/Arg3.1 interacts with, and regulates, the stabilization of the neuronal cytoskeleton and it can also regulate the postsynaptic trafficking of endosomes, particularly important for the expression of surface AMPA receptors at excitatory synapses (reviewed in Bramham et al., 2008
). Both of these cellular processes are crucial to both basal synaptic transmission and synaptic plasticity. It thus appears that BDNF displays the properties of a classical neurotransmitter: presynaptic synthesis and vesicular storage, activity-dependent release, activation and targeting of specific postsynaptic receptors and fast ion channel gating (reviewed in Altar and DiStefano, 1998
).
The role of multiple signalling mechanisms in learning and memory has been well documented, however the exact role that BDNF plays modulating these signalling pathways during learning remains to be elucidated. For example, mutant mice where the PLCγ binding site of TrkB has been disrupted show impaired hippocampal LTP and associative learning, demonstrating the importance of PLCγ signalling in hippocampal plasticity mechanisms (Minichiello et al., 2002
; Gartner et al., 2006
; Gruart et al., 2007
). In addition, pivotal roles have already been shown for the ERK pathway (Orban et al., 1999
; Sweatt, 2004
; Thomas and Huganir, 2004
), the PI3K–Akt pathway (Lin et al., 2001
) and the transcription factor CREB (Barco et al., 2003
) in synaptic plasticity and learning and memory. Although there is no direct evidence for BDNF activation of the NF-kB pathway in synaptic plasticity and LTM, a few studies have shown a contribution of the NF-kB signalling pathway in initial memory consolidation (Yeh et al., 2002
; Freudenthal et al., 2005
; Merlo et al., 2005
). Thus, BDNF can activate multiple signalling pathways that may act in a concerted fashion to regulate downstream cellular effects necessary for synaptic plasticity and memory formation. The interaction between each of these intracellular pathways has to be further resolved, but the extent to which each of them is activated and selective biological responses initiated is likely to depend on the levels of BDNF and TrkB, the temporal pattern of BDNF stimulation and whether the signalling is activated pre- or postsynaptically.
There are multiple feedback systems that control the activity of BDNF. In addition to being able to increase its own transcription via a CREB mediated mechanism (Finkbeiner et al., 1997
), BDNF can also increase the surface expression of TrkB (Haapasalo et al., 2002
). Furthermore, it can regulate its own release (Canossa et al., 1997
). These properties are likely to contribute to the reinforcement and stabilization of synaptic connections. Indeed, administration of exogenous BDNF itself can produce a form of synaptic potentiation that has all the hallmarks of LTP (reviewed in Bramham and Messaoudi, 2005
). Nevertheless, prolonged exposure to BDNF induces a negative feedback loop by depleting TrkB receptors on the neuronal surface and resulting in long-term receptor desensitization to BDNF (Frank et al., 1996
). Therefore, BDNF has a unique role in coupling neuronal activity to structural and functional properties of neuronal circuits.
In contrast to these rapid effects on synaptic transmission and membrane excitability, BDNF ultimately mediates slower cellular events. It is well established that, similar to other neurotrophins, BDNF promotes the differentiation, growth, target innervation and survival of neurons during the development of the central and peripheral nervous systems (Poo, 2001
; Huang and Reichardt, 2003
). However, much less is known about analogous functions in the adult brain. The importance of BDNF-mediated signalling for maintaining the survival and dendritic complexity of selective populations of neurons in the adult brain, including excitatory glutamatergic cortical neurons, has been established (Ghosh et al., 1994
; Mamounas et al., 1995
; Hu et al., 2005
). However, the precise effects of BDNF on neuronal morphology are cell and layer specific. For example, BDNF promotes dendritic arborization of cortical neurons in layers IV, but inhibits dendritic arborization in layer VI (McAllister et al., 1995
, 1997
). Whilst BDNF overexpression in transgenic mice results in increases in dendritic complexity in the hippocampal dentate gyrus (Tolwani et al., 2002
). The effects of BDNF to promote dendritic complexity are likely to be mediated by the activation of TrkB, while activation of p75NTR negatively regulates dendritic morphology (Zagrebelsky et al., 2005
). With regards to cell survival in the adult brain, BDNF-mediated signalling prevents apoptosis of hippocampal and cerebellar granule cells (Minichiello and Klein, 1996
; Alcantara et al., 1997
). It is also a positive regulator of neurogenesis in the subgranular zone of the hippocampal dentate gyrus (Sairanen et al., 2005
; Scharfman et al., 2005
; Henry et al., 2007
; Young et al., 2007
).
BDNF and LTP
LTP is the best studied form of synaptic plasticity and is considered as a cellular correlate of learning and memory. It is defined as an activity induced sustained increase in synaptic strength. The induction of LTP is associated with the activation of a large number of signalling cascades, including the ones activated by BDNF. Kafitz et al. (1999)
showed that low concentrations of BDNF causes membrane depolarization of hippocampal, cortical and cerebellar neurons within a few milliseconds, leading to the firing of an action potential. This was a remarkable finding because, until then, only classic neurotransmitters had been found to have such a rapid effect on the membrane potential of neurons. Since then, substantial evidence has accumulated to indicate a critical role for BDNF in LTP induction, not only at hippocampal synapses at both the Schaffer collateral → CA1 synapse (Kang and Schuman, 1995
) and in the dentate gyrus (Messaoudi et al., 2002
), but also in the visual cortex (Akaneya et al., 1997
).
Initially, in vitro studies showed that exogenous BDNF promoted the induction of LTP in young hippocampal slices (Figurov et al., 1996
), and rapidly enhanced the frequency of miniature excitatory postsynaptic currents in solitary neurons (Taniguchi et al., 2000
). Conversely, LTP was attenuated in slices pre-treated with function-blocking BDNF antibodies or the fusion protein TrkB-IgG, a molecular scavenger of endogenous BDNF (Figurov et al., 1996
; Kang et al., 1997
). Additional evidence for the role of BDNF in LTP was provided by studies with transgenic mice. Induction of LTP at the Schaffer collateral → CA1 synapse was severely impaired in two independent lines of BDNF null mutant mice. Similarly, cortical LTP impairment was observed in heterozygous BDNF mice of a third independently generated mutant (Bartoletti et al., 2002
). Crucially, treatment of hippocampal slices from these transgenic mice with recombinant BDNF protein completely reversed deficits in LTP and significantly improved deficits in basal synaptic transmission at the Schaffer collateral /CA1 synapse (Korte et al., 1995
; Patterson et al., 1996
; Pozzo-Miller et al., 1999
). These “rescue” experiments indicated that BDNF was the key mediator of LTP induction.
BDNF is essential for late phase LTP (L-LTP), which lasts at least 8 h after tetanization (Korte et al., 1998
). L-LTP depends on both gene transcription and protein synthesis, and requires cAMP signalling and CREB (Kang et al., 1997
; Korte et al., 1998
). The conversion of proBDNF into mBDNF by protease tissue plasminogen activator (tPA)-activated plasmin is essential for hippocampal L-LTP (Pang et al., 2004
). The application of mBDNF was sufficient to rescue L-LTP when protein synthesis was inhibited; suggesting that BDNF activated TrkB signalling is the key mechanism for L-LTP induction. Indeed, the concomitant blockade of TrkB signalling at pre- and postsynaptic sites impaired the induction of L-LTP (Gartner et al., 2006
). The role of BDNF in L-LTP extends beyond the process of induction and is required for the maintenance L-LTP (Barco et al., 2005
). The activation of cAMP signalling may function to trigger the release of BDNF and the insertion of TrkB receptor into the membrane in BDNF-dependent L-LTP (Patterson et al., 2001
). L-LTP is also associated with the activation of the ERK1/2 kinases, their translocation to the nucleus and the subsequent activation of CREB (Patterson et al., 2001
). This may provide the mechanism that leads to the increase in BDNF mRNA expression in the hippocampus 2–4 h after application of tetanic stimulation (Castren et al., 1993
; Dragunow et al., 1993
). It remains to be seen whether delayed increases in BDNF release, TrkB insertion and BDNF expression contribute to the maintenance of L-LTP.
LTM Formation
The role of BDNF in learning and memory has been established by investigations in in vivo rodent models. BDNF mRNA expression was found to be increased in the hippocampus of rats following training in the Morris water maze (MWM; Kesslak et al., 1998
), radial arm maze (Mizuno et al., 2000
), passive avoidance (Ma et al., 1998
) and contextual fear conditioning (Hall et al., 2000
). Thus consistently indicating that the regulation of BDNF activity is a correlate of hippocampal learning in vivo. Interestingly, BDNF protein presents its highest expression in the hippocampus, neocortex, cerebellum, striatum and amygdala (Dugich-Djordjevic et al., 1995
; Kawamoto et al., 1996
), all key brain areas responsible for cognitive functions. Its regulation by learning also extends to these brain regions. For example, increases in BDNF mRNA and protein levels and TrkB phosphorylation were found in the amygdala following fear conditioning (Rattiner et al., 2004a
). Furthermore, intra-hippocampal BDNF administration improved performance in MWM (Cirulli et al., 2004
), and pre-training infusions of anti-BDNF antibodies caused an impairment in MWM (Mu et al., 1999
) and in passive avoidance (Alonso et al., 2002
). Likewise, infusion of anti-BDNF antibodies into the parietal cortex impaired inhibitory avoidance by blocking CREB activation (Alonso et al., 2005
). These studies strongly suggest that BDNF has an essential role in the consolidation of LTM in a wide range of behavioural protocols in wild-type animals.
Differential regulation of specific exon-containing BDNF transcripts has been associated with the consolidation of LTM, particularly exon I, IV and VI variants (Rattiner et al., 2004b
; Chhatwal et al., 2006
; Ou and Gean, 2007
; Lubin et al., 2008
). Furthermore, the changes in the expression of specific BDNF transcripts was associated with altered chromatin structure (Lubin et al., 2008
), and the binding of active (phosphorylated) CREB (Ou and Gean, 2007
) in the promoters proximal to these specific exons. In these latter studies, the administration of a NMDA receptor antagonist and a CREB decoy not only prevented the change in the expression of learning-associated BDNF variants but also attenuated the learning. This strongly suggests a casual relationship between NMDA receptor- and CREB-dependent mechanisms in regulating the expression of specific BDNF variants essential for LTM formation. Although all transcripts of BDNF are translated into an identical protein, some variants can be targeted to different cellular compartments, including dendrites for local translation at active synapses (reviewed in Tongiorgi, 2008
). This “spatial code” for the delivery of BDNF mRNA to specific sites may be a key mechanism for regulating the local availability of BDNF to control cellular and synaptic events that underlie learning and memory.
In the 1990s, the advent of the technology to genetically modify mice, including the deletion (knockout, KO) or overexpression of BDNF and TrkB, made it possible to determine a specific role for these gene products in synaptic plasticity and animal behaviour. New advances in these techniques now allow the inducible and reversible gene targeting in selected brain regions (conditional mutagenesis), the transgenic rescue of disrupted genes and the single-cell recording of hippocampal place cells in mutant mice. Early studies using KO animal models with global reductions of BDNF, revealed that BDNF null mutant mice die within 2 days after birth and only a small fraction live for 2–4 weeks (Ernfors et al., 1994
). Although the CNS of these mutants shows no gross structural abnormalities, they displayed symptoms of nervous system dysfunction and had substantially reduced numbers of cranial and spinal sensory neurons. Heterozygous BDNF KO mice do not exhibit these abnormalities and have a normal life span, although they do present a serotonergic dysfunction (Lyons et al., 1999
). LTP was significantly reduced in the CA1 region of the hippocampus of surviving homozygous mice and heterozygous KO mice (Ernfors et al., 1994
). Heterozygous KO mice showed learning deficits in the MWM (Linnarsson et al., 1997
) and in contextual fear conditioning (Lu et al., 2004
).
TrkB KO mice showed a more severe phenotype than BDNF KO mice and the few mice that were able to survive up to 3 weeks displayed neuronal deficiencies in the CNS and PNS (Klein et al., 1993
), and an increased number of apoptotic central neurons especially in the dentate gyrus (Alcantara et al., 1997
). More recently, the generation of conditional BDNF KO mice circumvented the problem of postnatal lethality and of developmental effects, since gene disruption occurs only at defined time points and allows for some regional specificity of gene deletion. One conditional model, with the inducible loss of BDNF in forebrain regions of adult mice, showed impaired contextual fear conditioning and hippocampal LTP (Monteggia et al., 2004
). Another conditional BDNF mutant (Emx-BDNF-KO), lacking BDNF from the early embryonic development specifically in forebrain regions, fail to learn the MWM task, although they also presented with mild obesity, shortened lifespan, increased inter-male aggressiveness and infertility which are indicative of more widespread brain dysfunction (Gorski et al., 2003
). More recently, using lentiviral delivery of CRE recombinase into the dorsal hippocampus of adult mice floxed at the BDNF locus, mice with a site-specific deletion of BDNF were generated. These mice showed an impairment in MWM and novel object recognition tests (Heldt et al., 2007
). A conditional TrkB-CRE mutant mouse was generated, in which the deletion TrkB is restricted to the forebrain and occurred only postnatally. These mice showed severe deficits in hippocampus-dependent learning tasks such as MWM and eight-arm radial maze, and hippocampal LTP, but exhibited normal learning in the simple passive avoidance task (Minichiello et al., 1999
). However, these mice also displayed a range of non-mnemonic behaviours that would directly influence their performance in the memory tasks.
The inherent problems in producing transgenic animals that consistently result in changes in behaviour that mask cognitive performance can be overcome using the administration of antisense oligonucleotides or RNA interference (RNAi) to prevent the translation of BDNF locally. The continuous intracerebroventricular infusion of antisense BDNF oligonucleotides in rats, with significant reduction of BDNF mRNA and protein levels in the hippocampus, resulted in impaired spatial learning in radial arm maze (Mizuno et al., 2000
) and reduced LTP (Ma et al., 1998
). Infusions of antisense cDNA acutely into the hippocampus that prevents the translation of endogenous BDNF prevented the consolidation of contextual fear conditioning, an effect that was reversed by co-administration of mBDNF (Lee et al., 2004
). These latter investigations, which had the advantage of molecular technologies for manipulating BDNF levels in a regional and time-limited manner, clearly demonstrate BDNF has an essential and causal role in LTM processes. One similar study using RNAi with small interfering RNAs (siRNA) directed against BDNF mRNA, has shown that BDNF synthesis was necessary for phrenic long-term facilitation, a form of serotonin-dependent synaptic plasticity in the spinal cord (Baker-Herman et al., 2004
). The success of this technique to test the role of BDNF in synaptic plasticity in vivo opens the way for extending the use of RNAi technology to the study of learning and memory in whole animal models.
BDNF has recently been shown to have an additional role in the maintenance of LTM after acquisition. Using two different hippocampal-dependent learning tasks, contextual fear conditioning and inhibitory avoidance, administration of BDNF-blocking antibodies during a restricted time window around 12 h after training blocked memory retention (Bekinschtein et al., 2007a
). During this same time-window, mBDNF rescued the effect of protein synthesis inhibition on retention (Bekinschtein et al., 2008
). This novel finding suggests that BDNF in the rat hippocampus is necessary and sufficient for the stabilization of recurrent rounds of consolidation-like protein synthesis-dependent processes essential for persistence of LTM.
The development of transgenic mice particularly those overexpressing BDNF, has also added new and contradictory information concerning the relationship between BDNF, LTP and learning and memory. Mice overexpressing the full-length TrkB receptor have improved cognitive skills but attenuated LTP (Koponen et al., 2004
), and mice overexpressing a truncated form of the TrkB receptor show a memory deficit in the MWM but normal LTP (Saarelainen et al., 2000
). These studies indicate that the requirement for TrkB-mediated signalling in LTM and LTP are different. Transgenic mice overexpressing BDNF under the β-actin promoter presented a 30–40% increase in the level of BDNF protein in the brain and revealed a LTP disruption in the CA1 area and a significant memory deficit in the passive avoidance test (Croll et al., 1999
). This corresponded roughly to the same effect of attenuating BDNF activity. Similarly, more restricted overexpression of BDNF (two- to threefold depending on the structure) to the postnatal forebrain resulted in learning and memory impairments in both passive avoidance, eight-arm radial maze and MWM (Cunha et al., 2009
). These studies suggest that BDNF may negatively regulate learning and memory and that too much as well as too little BDNF can impair LTM and LTP. These observations in transgenic mice overexpressing BDNF may reflect a developmental shift in the balance between inhibitory and excitatory transmission in the brain. Indeed, mutant mice overexpressing BDNF in the forebrain show an acceleration in the maturation of GABAergic innervation and inhibition in the visual cortex (Huang et al., 1999
). This idea is supported by evidence that heterozygous BDNF KO mice show several deficits in interneuron maturation, including the altered expression of hallmark calcium binding proteins and peptide neurotransmitters (Jones et al., 1994
). However, a general role for BDNF in modulating inhibitory circuitry is unknown. We here propose a model for the role of BDNF in modulating excitatory and inhibitory transmission in the brain and its functional consequences in learning and memory (Figure 5
).
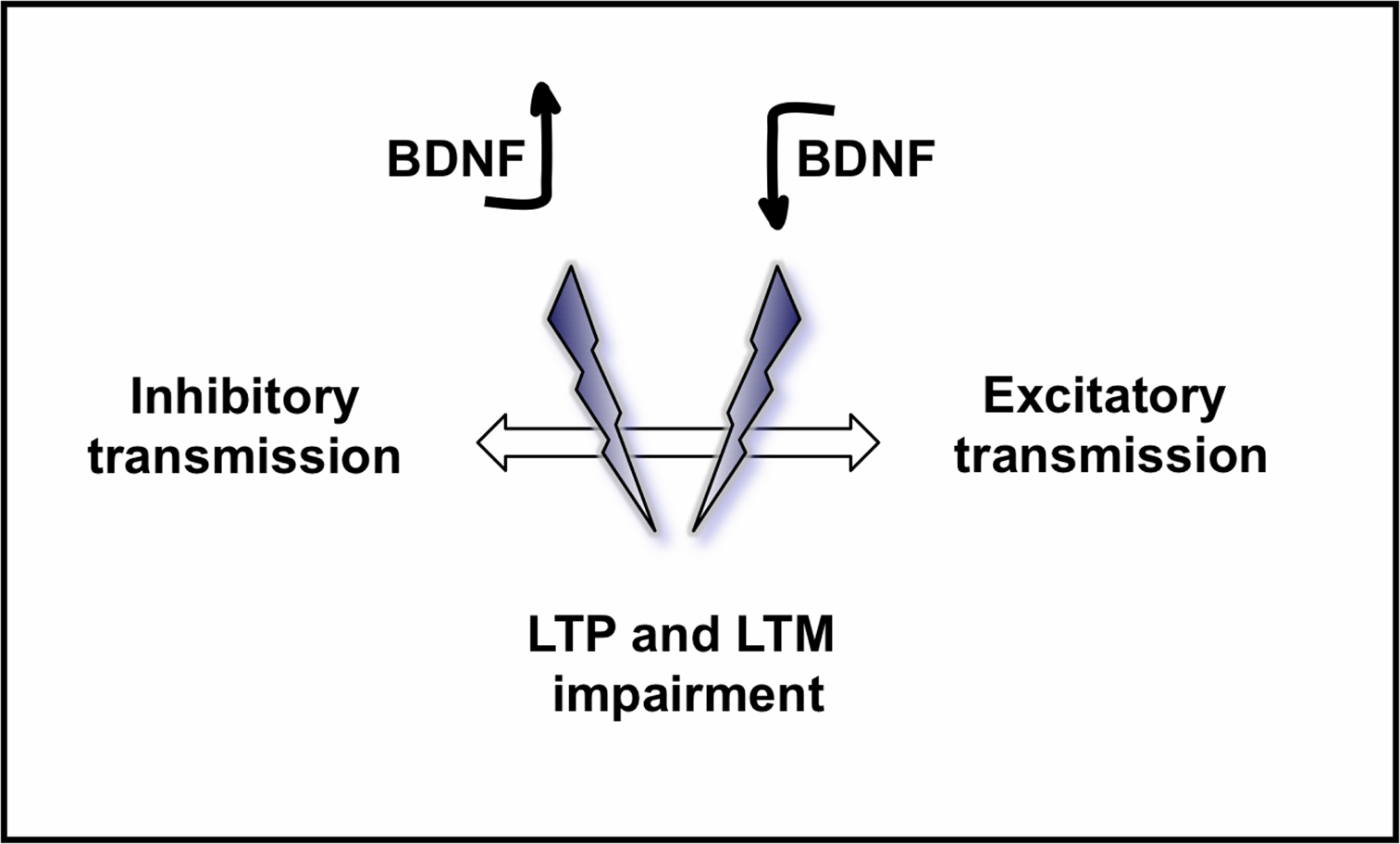
Figure 5. Model proposed for the role of BDNF on excitatory and inhibitory circuits in the brain and its consequences on LTP and LTM. While a physiological amount of BDNF in the normal brain has been demonstrated to have positive effects on learning and memory, both an increased level of BDNF, and a decreased level of BDNF may disrupt the equilibrium between inhibitory and excitatory neurotransmission in the brain, leading to a loss of synaptic refinement and consequently impairing LTP, learning and memory.
A functional association between synaptic activity and inhibitory signalling mediated by BDNF was reported over a decade ago. In cultured neurons from the visual cortex, the blockade of spontaneous activity reduced the number of GABAergic neurons, as well as the magnitude and frequency of spontaneous inhibitory postsynaptic currents and reduced the GABA-mediated inhibition of pyramidal cells, effects that were reversed by BDNF application (Rutherford et al., 1997
). In addition, BDNF increased the frequency of miniature postsynaptic currents at both excitatory → excitatory neurons and excitatory → inhibitory neurons (Schinder et al., 2000
). Thus, BDNF has an important function in controlling and regulating the balance between excitatory and inhibitory activity to maintain network function. In the adult neocortex, BDNF (via TrkB) controls the coordinated regulation of excitatory pyramidal cells and inhibitory interneurons, permitting scaling of synaptic responses to stabilize network activity (Rutherford et al., 1997
; Desai et al., 1999
). How exactly increases in glutamate and GABA release are integrated during learning and memory processes is still to be determined, but BDNF signalling may modulate an autoregulatory circuit between excitatory pyramidal cells and inhibitory interneurons, where specific signalling pathways are preferentially activated in time and in selective neuronal subtype. This kind of autoregulatory homeostasis (Turrigiano, 2007
) may have an important contribution to metaplasticity, the so-called plasticity of plasticity by which neural activity at one point in time can change the ability of cells to undergo plasticity subsequently (Abraham, 2008
). The link between metaplasticity and learning is not clear. However, there is evidence that learning induces alterations (histone acetylation and DNA methylation events) in the structure of the nuclear DNA-histone chromatin complex in gene promoter regions, important for regulating gene transcription, to directly affect new learning (Levenson et al., 2004
; Miller and Sweatt, 2007
). This presents a molecular mechanism by which changes in behaviour are brought about by experience. The recent observation that chromatin remodelling following BDNF-dependent learning causes changes in the expression of exon-specific BDNF variants (Lubin et al., 2008
), suggests not only that BDNF plays a role in the metaplasticity of memory but that selective variants may be preferentially involved.
BDNF and LTM Reconsolidation and Extinction
Although evidence has accumulated to show that BDNF has a critical role in the consolidation of LTM and LTP, few studies have addressed its contribution to other forms of LTM processes and plasticity, which include the reconsolidation and extinction of established LTM, and long-term depression (LTD).
Reconsolidation is the protein synthesis-dependent restabilization of the labile, active memory trace initiated by retrieval (reviewed in Nader, 2003
; Alberini, 2005
). It is viewed as a constructive mechanism for updating information content of an established memory. Initial studies indicated that unlike consolidation, the reconsolidation of contextual fear memory was not dependent on mBDNF in the hippocampus (Lee et al., 2004
). As mentioned before, mBDNF is generated by the proteolytic cleavage of the precursor, proBDNF, by the tPA-mediated activation of plasmin (Seidah et al., 1996
). Accordingly, the retrieval of contextual fear memory under conditions that favour reconsolidation were not correlated with alterations in the levels of proBDNF or mBDNF activity in CA1 (Barnes and Thomas, 2008
), despite evidence that proBDNF is released in an activity dependent manner (Yang et al., 2009
). Hence, BDNF has no apparent role in reconsolidation. In added proof, the updating (strengthening by additional learning events) of contextual fear memory relies on molecular processes supporting reconsolidation rather than consolidation (Lee, 2008
).
Extinction occurs when a conditioned stimulus (CS) is presented without the reinforcement of a biologically salient unconditioned stimulus (US), and manifests as a weakening of the conditioned response. It is currently considered to be the generation of a new memory about a CS, a so-called CS–no US association (Bouton and Sunsay, 2003
). Several studies have suggested a role for BDNF signalling in extinction. Mice with a hippocampus-specific deletion of BDNF induced in adulthood show impaired extinction of aversive memory (Heldt et al., 2007
). Furthermore, the extinction of amygdala-dependent fear memory was dependent on TrkB activation in this region (Chhatwal et al., 2006
). A recent study has indicated that decreased proteolysis of BDNF was correlated with the extinction of hippocampal-dependent memory: extinction of contextual fear memory upregulated the levels of hippocampal proBDNF with respect to mBDNF, and inhibition of tPA activity in the hippocampus potentiated extinction (Barnes and Thomas, 2008
). The same study indicated that the proteolysis of BDNF regulated the consolidation of the CS–US association, however the proteolysis of proBDNF was a requirement for consolidation. These data indicate that formation of new memories relies on the regulation of proBDNF proteolysis, but the cleavage of proBDNF to mBDNF strengthens consolidation but attenuates extinction.
Similarly, opposing cellular actions of mBDNF and proBDNF have been described for synaptic plasticity (Lu et al., 2005
). As mentioned above, the cleavage of proBDNF to mBDNF by tPA is essential for LTP in the hippocampus (Pang et al., 2004
), whilst proBDNF-mediated signalling facilitates LTD in the hippocampus via the activation of the p75NTR neurotrophin receptor (Woo et al., 2005
). Evidence that hippocampal-dependent extinction is mediated by an increased proBDNF/mBDNF ratio may suggest that the synaptic and molecular events underlying extinction closely resemble LTD. The close correlation between the control of synaptic memory and the consolidation and extinction of LTM may additionally indicate that different forms of synaptic plasticity model distinct memory processes.
Permissive and instructive roles of BDNF in mediating synaptic plasticity in the CNS are well established (reviewed in Bramham and Messaoudi, 2005
). The activity-driven and persistent increase in synaptic efficacy, the hallmark of LTP, is accompanied by ultrastructural changes in dendritic spines at excitatory glutamatergic synapses (Lee et al., 1980
; Chang and Greenough, 1984
; Geinisman et al., 1993
; Harris et al., 2003
). These changes include the formation of new spines (Engert and Bonhoeffer, 1999
; Maletic-Savatic et al., 1999
; Toni et al., 1999
) and increases in the number of glutamatergic AMPA receptors in the dendritic spines (Isaac et al., 1995
; Liao et al., 1995
; Durand et al., 1996
). The rapid increase in synapse area after LTP inducing stimulation results in changes in synapse shape and receptor availability, thus augmenting neurotransmission (Chen et al., 2007
). Likewise, hippocampal-dependent learning is associated with similar synaptic rearrangements (Leuner et al., 2003
; Knafo et al., 2004
) as well as AMPA receptor trafficking into synapses in CA1 (Hu et al., 2007
). BDNF can increase the number of dendritic spines in CA1 (Tyler and Pozzo-Miller, 2001
; Alonso et al., 2004
). Similarly BDNF overexpression increase dendritic complexity in hippocampal dentate gyrus (Tolwani et al., 2002
). These data suggest that BDNF released in an activity-dependent manner (Aicardi et al., 2004
) modulates the number and shape of dendritic spines in mature hippocampal neurons (Zagrebelsky et al., 2005
). It will be interesting to see in future studies whether alterations in dendritic complexity mediated by BDNF are associated with learning and memory. LTP induced increases in spine density are themselves associated with rapid changes in stabilization of the actin cytoskeleton by the formation of polymerized actin, F-actin (Lin et al., 2005
). It has been recently reported that endogenous BDNF released during LTP induction in CA1 promotes F-actin formation in potentiated synapses by phosphorylation and attenuation of the activity of ADF/cofilin which normally acts to cleave polymerized actin (Rex et al., 2007
). Although there are no studies directly investigating LTM yet, a recent study has shown that exposure to an enriched-environment, a form of experience-dependent learning, results in the phosphorylation of cofilin in the postsynaptic spines in CA1 and in a concomitant increase in their size (Fedulov et al., 2007
). Native BDNF-induced increases in spine density are also dependent on the insertion of the nonselective cation channel, TRPC3 (Amaral and Pozzo-Miller, 2007
). Interestingly, for classical conditioning in turtles, BDNF is required for AMPA receptor trafficking into activated synapses (Li and Keifer, 2008
). TRPC3 channels may therefore emerge as novel effectors of BDNF-mediated dendritic remodelling, plasticity and LTM.
It is apparent that there is a correlation between the level of neurogenesis in the dentate gyrus and hippocampal-dependent memory (Zhao et al., 2008
), and that new granule cells become selectively recruited into the existing hippocampal networks with learning (Kee et al., 2007
). Furthermore it may be that co-ordinated apoptosis and neurogenesis mechanisms are especially important for memory formation, at least for some forms of hippocampal-dependent learning (Dupret et al., 2007
). However, it is not clear whether hippocampal neurogenesis is causal for LTM. There is no direct evidence for the involvement of BDNF in learning associated neurogenesis. Nevertheless, exposure to an enriched environment and exercise elevated the levels of BDNF in the hippocampus, increased neurogenesis and improved learning (Olson et al., 2006
). The exact mechanisms by which apoptosis and neurogenesis facilitate memory formation and an exact role for BDNF in these two cellular processes remains to be fully understood.
The control of BDNF mRNA translation in dendrites may be particularly important for LTM. Memory is not only represented by an altered synaptic efficiency, it is also sparsely distributed across a neuronal and synapses network (Wilson and McNaughton, 1993
; Guzowski et al., 1999
; Repa et al., 2001
; Perez-Orive et al., 2002
; Rumpel et al., 2005
). The regulated release of BDNF and the BDNF-mediated translation at local synapses would modulate synaptic activity and neural connectivity specifically at active synapses, which ultimately form the distributed memory “engram”. BDNF rapidly initiates the translation of several dendritically localized mRNAs associated with LTP and spine morphogenesis, including CaMKII, the GluR1 AMPA receptor and Arc/Arg3.1 (Yin et al., 2002
; Schratt et al., 2004
; Kanhema et al., 2006
). In addition to the ERK and PLCγ pathways, signalling through PI3K, Akt and downstream mTOR has been particularly implicated in regulating the activity of the translational machinery in response to BDNF–TrkB signalling (reviewed in Soule et al., 2006
). mTOR contributes to persistent forms of LTP and LTD (Kelleher et al., 2004
). It is also necessary for the consolidation (Bekinschtein et al., 2007b
; Myskiw et al., 2008
; Belelovsky et al., 2009
) and reconsolidation (Blundell et al., 2008
) of aversive memory. At least for inhibitory avoidance, activation of mTOR depends on hippocampal BDNF (Slipczuk et al., 2009
).
BDNF has emerged as an important mediator of synaptic plasticity. It not only plays a role in LTP and LTD but it is perhaps the key instructor for plasticity-related processes underlying LTM. This is particularly exemplified by “rescue” studies where the addition of BDNF restores learning after endogenous depletion of BDNF. The key role for BDNF in LTM is further underscored because regulation of BDNF but not other members of the neurotrophin family is associated with LTM formation (Hall et al., 2000
; Rattiner et al., 2004a
; Hennigan et al., 2009
). Nevertheless, other neurotrophins including NGF and NT3, in a similar manner to BDNF, may have a more specific role in the altered mnemonic processes associated with neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases, by promoting the survival of specific neuronal populations affected in these conditions (Williams et al., 1989
; Chen and Tonegawa, 1997
; Tong et al., 2004
; Gu et al., 2009
). To complicate the matter, consistent evidence now indicates that BDNF, especially when overexpressed, may preferentially stimulate inhibitory pathways and negatively affecting learning, thus considerably limiting its potential therapeutic use. Moreover, we still do not know what are the cellular processes necessary for the maintenance of LTM that are instructed by BDNF after learning. Nor whether separate BDNF-mediated processes are central to the persistence of memory after recall. A new generation of sophisticated genetic tools, including cell specific siRNA and overexpression studies, targeting individual BDNF variants and components of the downstream BDNF–TrkB and BDNF–p75NTR signalling cascades, under precise spatial and temporal control within specific brain regions, are now necessary to unravel the complex roles of this fascinating and pleiotropic molecule in learning and memory processes.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge Fundação para a Ciência e a Tecnologia, Portugal, which, through the POCTI 2010 from the European Science Foundation, has financed research leading to the writing of this review (fellowship SFRH/BD/9625/2002 to Carla Cunha) and also the MJ Fox Foundation for Parkinson’s Research, the Parkinson’s Disease Society of the UK and the Italian Ministry of Health (to Riccardo Brambilla).
Bartoletti, A., Cancedda, L., Reid, S. W., Tessarollo, L., Porciatti, V., Pizzorusso, T., and Maffei, L. (2002). Heterozygous knock-out mice for brain-derived neurotrophic factor show a pathway-specific impairment of long-term potentiation but normal critical period for monocular deprivation. J. Neurosci. 22, 10072–10077.
Chen, Z. Y., Patel, P. D., Sant, G., Meng, C. X., Teng, K. K., Hempstead, B. L., and Lee, F. S. (2004). Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J. Neurosci. 24, 4401–4411.
Croll, S. D., Suri, C., Compton, D. L., Simmons, M. V., Yancopoulos, G. D., Lindsay, R. M., Wiegand, S. J., Rudge, J. S., and Scharfman, H. E. (1999). Brain-derived neurotrophic factor transgenic mice exhibit passive avoidance deficits, increased seizure severity and in vitro hyperexcitability in the hippocampus and entorhinal cortex. Neuroscience 93, 1491–1506.
Egan, M. F., Kojima, M., Callicott, J. H., Goldberg, T. E., Kolachana, B. S., Bertolino, A., Zaitsev, E., Gold, B., Goldman, D., Dean, M., Lu, B., and Weinberger, D. R. (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112, 257–269.
Gartner, A., Polnau, D. G., Staiger, V., Sciarretta, C., Minichiello, L., Thoenen, H., Bonhoeffer, T., and Korte, M. (2006). Hippocampal long-term potentiation is supported by presynaptic and postsynaptic tyrosine receptor kinase B-mediated phospholipase Cgamma signaling. J. Neurosci. 26, 3496–3504.
Griesbeck, O., Canossa, M., Campana, G., Gartner, A., Hoener, M. C., Nawa, H., Kolbeck, R., and Thoenen, H. (1999). Are there differences between the secretion characteristics of NGF and BDNF? Implications for the modulatory role of neurotrophins in activity-dependent neuronal plasticity. Microsc. Res. Tech. 45, 262–275.
Kanhema, T., Dagestad, G., Panja, D., Tiron, A., Messaoudi, E., Havik, B., Ying, S. W., Nairn, A. C., Sonenberg, N., and Bramham, C. R. (2006). Dual regulation of translation initiation and peptide chain elongation during BDNF-induced LTP in vivo: evidence for compartment-specific translation control. J. Neurochem. 99, 1328–1337.
Koponen, E., Voikar, V., Riekki, R., Saarelainen, T., Rauramaa, T., Rauvala, H., Taira, T., and Castren, E. (2004). Transgenic mice overexpressing the full-length neurotrophin receptor trkB exhibit increased activation of the trkB-PLCgamma pathway, reduced anxiety, and facilitated learning. Mol. Cell. Neurosci. 26, 166–181.
Liu, Q. R., Walther, D., Drgon, T., Polesskaya, O., Lesnick, T. G., Strain, K. J., de Andrade, M., Bower, J. H., Maraganore, D. M., and Uhl, G. R. (2005). Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 134, 93–103.
Lyons, W. E., Mamounas, L. A., Ricaurte, G. A., Coppola, V., Reid, S. W., Bora, S. H., Wihler, C., Koliatsos, V. E., and Tessarollo, L. (1999). Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc. Natl. Acad. Sci. U.S.A. 96, 15239–15244.
Narisawa-Saito, M., Iwakura, Y., Kawamura, M., Araki, K., Kozaki, S., Takei, N., and Nawa, H. (2002). Brain-derived neurotrophic factor regulates surface expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors by enhancing the N-ethylmaleimide-sensitive factor/GluR2 interaction in developing neocortical neurons. J. Biol. Chem. 277, 40901–40910.
Pozzo-Miller, L. D., Gottschalk, W., Zhang, L., McDermott, K., Du, J., Gopalakrishnan, R., Oho, C., Sheng, Z. H., and Lu, B. (1999). Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. J. Neurosci. 19, 4972–4983.
Teng, H. K., Teng, K. K., Lee, R., Wright, S., Tevar, S., Almeida, R. D., Kermani, P., Torkin, R., Chen, Z. Y., Lee, F. S., Kraemer, R. T., Nykjaer, A., and Hempstead, B. L. (2005). ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 25, 5455–5463.
Yamada, M., Ohnishi, H., Sano, S., Nakatani, A., Ikeuchi, T., and Hatanaka, H. (1997). Insulin receptor substrate (IRS)-1 and IRS-2 are tyrosine-phosphorylated and associated with phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor in cultured cerebral cortical neurons. J. Biol. Chem. 272, 30334–30339.
Ying, S. W., Futter, M., Rosenblum, K., Webber, M. J., Hunt, S. P., Bliss, T. V., and Bramham, C. R. (2002). Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J. Neurosci. 22, 1532–1540.