Department of Psychiatry and the Brain Research Centre, University of British Columbia, Vancouver, BC, Canada
Excitability of individual neurons dictates the overall excitation in specific brain circuits. This process is thought to be regulated by molecules that regulate synapse number, morphology and strength. Neuronal excitation is also influenced by the amounts of neurotransmitter receptors and signaling molecules retained at particular synaptic sites. Recent studies revealed a key role for PSD- 95, a scaffolding molecule enriched at glutamatergic synapses, in modulation of clustering of several neurotransmitter receptors, adhesion molecules, ion channels, cytoskeletal elements and signaling molecules at postsynaptic sites. In this review we will highlight mechanisms that control targeting of PSD-95 at the synapse, and discuss how this molecule influences the retention and clustering of diverse synaptic proteins to regulate synaptic structure and strength. We will also discuss how PSD-95 may maintain a balance between excitation and inhibition in the brain and how alterations in this balance may contribute to neuropsychiatric disorders.
Neuronal excitability relies on the summation of excitatory and inhibitory signals (Li et al., 2006
; Schummers, 2002
; Voytenko and Galazyuk, 2006
), a process regulated by the number of excitatory vs. inhibitory (E/I) contacts received by a single neuron. Excitatory synaptic transmission in the brain is predominantly mediated by the neurotransmitter glutamate, while inhibitory transmission is mediated mainly by the neurotransmitter gamma-amino butyric acid (GABA). Synapses that contain each respective neurotransmitter differ in their composition and structure. The postsynaptic compartment of excitatory synapses is characterized by an electron-dense region, referred to as the postsynaptic density (PSD), attributable to the high density of neurotransmitter receptors and associated molecules at these sites. Although inhibitory postsynaptic sites lack PSDs, they also consist of complex protein matrices. Differences in the morphology of excitatory and inhibitory synapses also exist (Boeckers, 2006
; Craig et al., 2006
). The majority of glutamatergic synapses are found on bulbous protrusions known as spines, whereas most GABAergic synapses are formed on the dendritic shaft, as well as on the soma and proximal axonal regions (Fujiyama et al., 2002
; Hafidi and Hillman, 1997
; Knott et al., 2002
).
The level of excitation in the brain is kept in check through inhibitory control exerted by GABA neurons (Gulledge et al., 2005
). In this manner the neurons strike a “balance” that allows for neurons to become activated and convey discrete synaptic signals, while preventing excessive excitation. Yet this balance can be disturbed by even small changes in the level of inhibition, resulting in epileptiform activity (Chagnac-Amitai and Connors, 1989
). The relative contribution of molecular and cellular components to E/I balance of each brain region may vary. For example, high levels of intrinsic inhibition in cerebellar circuits are balanced by mainly excitatory afferents from various brain regions (Bower, 2002
), whereas intrinsic excitatory feedback loops in the hippocampus are modulated by interneurons to avoid hypersynchronicity and epileptiform activity (Levinson and El-Husseini, 2005a
; Mann et al., 2005
). Additionally, astrocytes have been implicated in modulating the excitation of both glutamatergic and GABAergic neuronal cells by sequestering and releasing glutamate (Schousboe, 2003
).
Homeostasis is a form of regulation of neuronal excitability that maintains system function at a set point, maintaining a constant output while exposed to changing inputs. Homeostatic control has been suggested to regulate neuronal function by modulating synaptic efficacy, synaptic strength, and membrane excitability (Baines, 2005
; Corner and Ramakers, 1992
; Davis and Goodman, 1998
; Leslie et al., 2001
; Mee et al., 2004
; Ramakers et al., 1990
, 1994
; Turrigiano and Nelson, 2004
; Turrigiano et al., 1998
; Van Den Pol et al., 1996
). This compensatory process occurs over a time scale of 2–3 days, ensuring that discrete synaptic events are not lost, but rather the system “set point” is readjusted. The strength of individual synapses is modulated to compensate for changes in other synapses, on the same neuron, that would otherwise alter overall cellular activity. These changes reflect a scaling up or down of synaptic activity, and as a result, homeostasis is often referred to as “synaptic scaling” (Figure 1
). By modifying multiple synapses to the same extent, relative differences in individual synapses produced by long lasting changes in synaptic plasticity, such as long term potentiation (LTP) or long term depression (LTD), can be maintained (for review see Turrigiano and Nelson, 2004
).
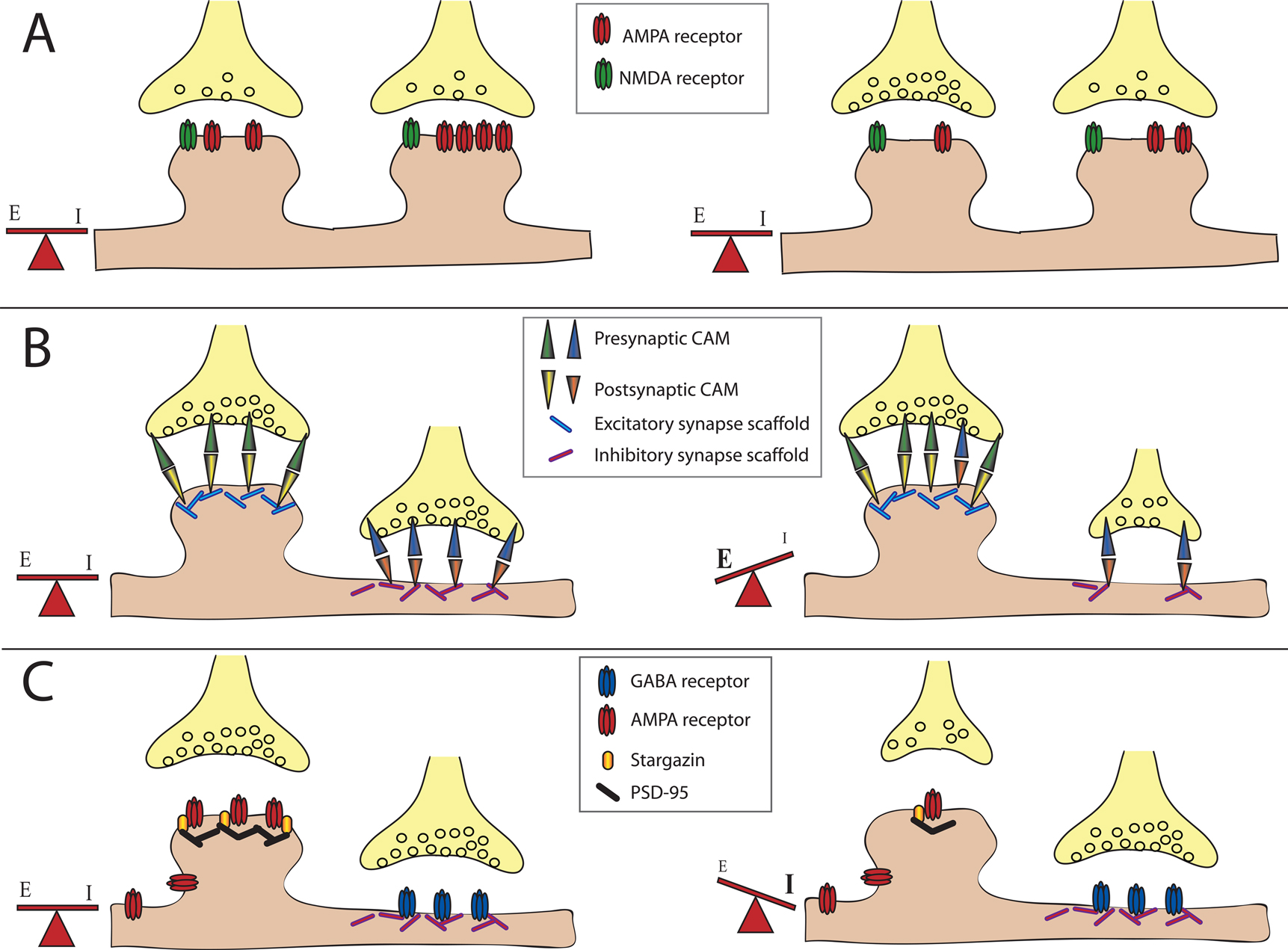
Figure 1. Balancing excitation and inhibition. The majority of excitatory neurotransmitter receptors are presented on bulbous structures known as spines whereas inhibitory neurotransmitter receptors are usually present on postsynaptic sites formed on the dendritic shaft. (A) Homeostatic mechanisms maintain balance. In this example of synaptic scaling an increase in synaptic input results in a decrease in the number of neurotransmitter receptors present at particular synapses to balance changes in synaptic strength. (B) Scaffolding molecules regulate retention of cell adhesion molecules at excitatory vs. inhibitory contacts. Scaffolding molecules retain specific adhesion molecules at either excitatory or inhibitory synapses. An example illustrating a loss of a scaffolding molecule that controls retention of adhesion molecules at an inhibitory synapses may cause a reduction in the number of adhesion molecules retained at this site, and a corresponding shift of these adhesion molecules to excitatory synapses. This results in a shift in the E/I balance towards enhanced excitation.(C) PSD-95 regulates retention of AMPA-type glutamate receptors at excitatory synapses. AMPA receptors directly associate with stargazin molecules, which in turn associate with PSD-95. Reduction in PSD-95 levels at the synapse reduces AMPA receptor retention. This results in a shift in the E/I balance towards decreased excitation, or increased inhibition.
Homeostatic mechanisms can influence both excitatory (Turrigiano et al., 1998
) and inhibitory synaptic transmission (Kilman et al., 2002
; Maffei et al., 2006
; Marty et al., 1996
; Rutherford et al., 1997
). For instance, blocking neuronal activity by treating cultured cortical neurons with TTX for 48 hours produces an increase in the amplitude of miniature excitatory postsynaptic currents, likely caused by homeostatic increase in postsynaptic sensitivity to glutamate (Turrigiano et al., 1998
). Treatments with AMPAkines, compounds that modulates AMPA receptor activity, upregulate AMPA receptor function during short treatment periods, but neurons display homeostatic adaptations following longer treatments, manifested by a decrease in AMPA receptor subunit expression as well as a decrease in the scaffolding proteins SAP 97 and GRIP1 (Jourdi et al., 2005
). Conversely, chronic exposure to ethanol potentiates GABAA receptors causing reduced neuronal activity and resulting in a heterosynaptic homeostatic increase in NMDA receptor density to compensate for the decrease in excitation. In addition, there is a corresponding increase in PSD-95 expression and dendritic spine size under these conditions (Carpenter-Hyland and Chandler, 2006
). Global changes in the amounts of multiple postsynaptic proteins following chronic activation or inhibition
of neuronal activity have also been reported (Colledge et al., 2003
; Ehlers, 2003
;El-Husseini Ael et al., 2002
; Sala et al., 2003
). Changes in synaptic activity produce a coordinated change in PSD composition including changes in the levels of receptors, scaffolding proteins, and signaling proteins. Thus, it is likely that a compensatory increase in synaptic activity is modulated by removal of glutamate receptors and associated proteins to control overall neuronal excitability.
Thus, homeostatic regulation of synaptic efficacy is a mechanism to maintain appropriate E/I balance. However, altered expression of specific molecules that control synapse maturation and strength have been recently proposed to influence the E/I synaptic balance in the brain. This review will focus on the contribution of PSD-95 to excitation, examining how it interacts with additional molecular components that influence excitatory and inhibitory synapse maturation, and ultimately how it affects various mechanisms for regulating this process. Finally, we will discuss implications of altered E/I ratio in cognitive disorders including autism, epilepsy, and schizophrenia.
The clustering of scaffolding molecules at contact sites is thought to play a role in the retention of specific neurotransmitter receptors at a particular synapse type. At the PSD, there are multiple families of proteins that serve as scaffolds upon which the assembly of neurotransmitter receptors, signal transduction components, and adhesion molecules occurs. A major scaffolding molecule localized at the PSD of excitatory glutamatergic synapses is the postsynaptic density protein PSD-95 (Figure 2
). PSD-95 is a member of the membrane-associated guanylate kinase (MAGUK) family, which contains 3 PDZ domains (domain first discovered in PSD-95/Dlg/ZO1 proteins), a Src-homology-3 (SH3) domain or WW motif (two conserved tryptophan residues), and a region homologous to yeast guanylate kinase (GK region) (Cho et al., 1992
; Kim and Sheng, 2004
). In yeast, guanylate kinase is involved in protein phosphorylation, but in the MAGUKs this region is catalytically inactive (Olsen and Bredt, 2003
). PSD-93, synapse associated protein (SAP)-102, and SAP-97 are other MAGUKs that exist at excitatory synaptic sites (Kim and Sheng, 2004
).
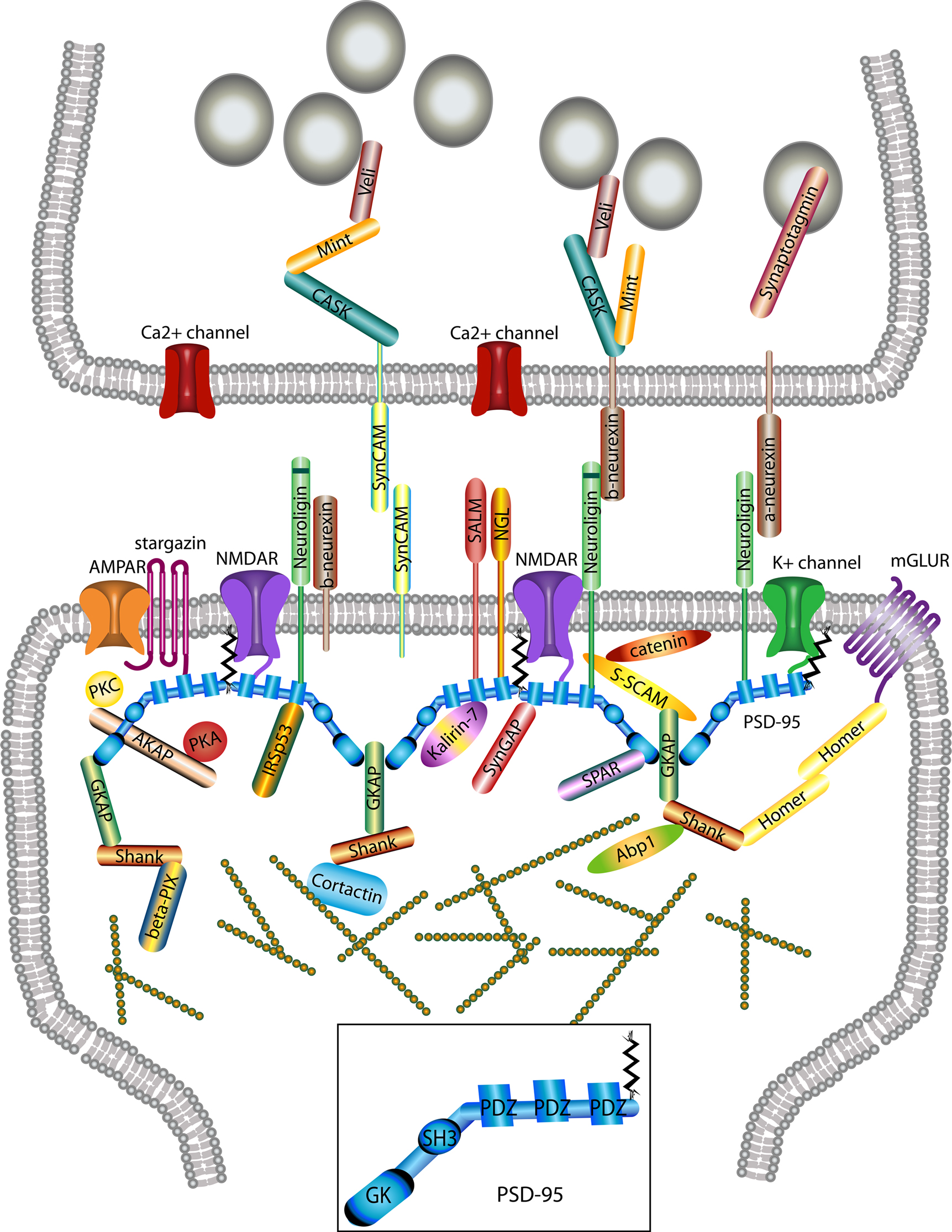
Figure 2. Macromolecular PSD-95 complex. The molecular organization of glutamatergic synapses is presented, but only major molecules associated with PSD-95 are shown. The various molecules portrayed regulate synapse function, morphology, trafficking and localization of adhesion molecules and neurotransmitter receptors.
Several other PDZ domain-containing proteins localized at the synapse have been identified that share some functional aspects with MAGUKs, being involved in protein assembly at the synapse. PICK1, a single PDZ domain molecule (Staudinger et al., 1997
), and GRIP/ABP, which contains multiple PDZ domains (Dong et al., 1997
), modulate trafficking of the AMPA-type glutamate receptor subunit GluR2. With multiple PDZ domains, in conjunction with additional protein interaction domains, MAGUK proteins are capable of forming a complex network of proteins localized to particular cellular regions. PDZ domains typically mediate interactions with the C-termini of various proteins typically detected at regions of cell-cell contact (Cho et al., 1992
; Craven and Bredt, 1998
; Kim and Sheng, 2004
). This localization in neurons suggests an important role for these PDZ proteins in forming and modulating synaptic contacts. The SH3 and GK domains also mediate interactions of PSD-95 with several other scaffolding molecules. For instance, the GK domain interacts with GKAP (Kim et al., 1997
; Takeuchi et al., 1997
), AKAP 79/150 (Colledge et al., 2000
), and microtubule-associated protein 1A (Brenman et al., 1998
), whereas the SH3 domain associates with the tyrosine kinase Pyk2 (Seabold et al., 2003
), and is also involved in intramolecular interaction with the GK domain of PSD-95 (McGee and Bredt, 1999
).
Interestingly, a different set of scaffold molecules seems to regulate receptor clustering at inhibitory synapses. A key component of the postsynaptic structure of inhibitory synapses is the scaffolding protein gephyrin. Gephyrin is not a neuron-specific protein (Feng et al., 1998
), but nevertheless plays a significant role in the development and functioning of both GABA and glycine synapses in neurons. At glycinergic synapses, gephyrin binds to both glycine receptors and to the cytoskeletal protein tubulin. In its capacity as a scaffolding protein, gephyrin is necessary for clustering of glycine receptors (Kirsch et al., 1993
; Levi et al., 2004
). GABAA receptors, particularly the gamma2 and gamma3 subunits, are important for postsynaptic clustering of gephryin and for synapse stabilization (Baer et al., 1999
, 2000
;Dumoulin et al., 2000
; Essrich et al., 1998
; Fischer et al., 2000
; Meier and Grantyn, 2004
; Schweizer et al., 2003
). While gephyrin is not necessary for the formation of GABA synapses (Dahan et al., 2003
), overexpression of a specific gephryin mutant induces inhibitory synapse formation, while reducing the formation of excitatory synapses, thus reducing the E/I ratio (Lardi-Studler et al., 2007
). Thus, differential recruitments of PDZ proteins at excitatory synapses and gephyrin to inhibitory contacts are potentially critical steps in modulating neurotransmitter receptor clustering and stability at contact sites, and in turn synaptic strength.
Numerous studies hint at an important role for PSD-95 multimerization in regulation of protein trafficking and ion channel clustering. PSD-95 homomultimerizes through its N-terminal region allowing for an expanded scaffolding region in the PSD (Hsueh et al., 1997
). This multimerization is dependant on two N-terminal cysteine residues (Hsueh and Sheng, 1999
). Conjugation of palmitate, a 16-carbon fatty acid, to proteins by thioesterification of cysteines provides a mechanism for membrane targeting. Importantly, the two N-terminal cysteines of PSD-95 are palmitoylated, enabling association with postsynaptic membranes, protein multimeriztion, and clustering of cell surface receptors and ion channels (Christopherson et al., 2003
; Craven et al., 1999
; El-Husseini et al., 2000a
,c
; Topinka and Bredt, 1998
). Palmitoylation is a feature particular to PSD-95 and PSD-93, but not to SAP-102 and SAP-97 (El-Husseini et al., 2000c
). This altered palmitoylation promotes differential regulation of ion channel clustering and synaptic clustering of the MAGUK molecules.
Protein palmitoylation also controls dynamic recruitment and removal of PSD-95 at the synapse, a process which influences retention of AMPA receptors (El-Husseini et al., 2002
). For instance, blocking palmitoylation reduces the membrane clustering of PSD-95 and AMPA receptors, and reduces the amplitude of AMPA receptor-mediated currents. PSD-95 regulates retention of AMPA receptors at the synapse through coupling to stargazin, a transmembrane protein that associates directly with AMPA receptors (Figure 1
C) (Bats et al., 2007
; Chen et al., 2000
; Chetkovich et al., 2002b
; Dakoji et al., 2003
; El-Husseini et al., 2000a
, 2002
;Fukata et al., 2005
; Schnell et al., 2002
; Tomita et al., 2003
, 2005
). By regulating a dynamic pool of AMPA receptors through its interaction with stargazin, and palmitate-mediated recycling of PSD-95 at the synapse, PSD-95 has a direct role in regulating synaptic strength. Consistent with these findings, in vivo two-photon microscopy, performed by Gray et al. (2006)
revealed dynamic cycling of PSD-95 in the developing neocortex, demonstrating that PSD-95 is localized at individual synapses for short periods, but becomes more stable as the animal ages. This study also revealed that the size, shape, and activity of neighboring synapses influence exchange of PSD-95 between synapses. An interesting implication of this observation is that synapses compete with each other for scaffolding proteins. This dynamic distribution provides a mechanism for regulating the amount of scaffolding proteins and associated glutamate receptors at individual synapses. As activation of a glutamatergic synapse is reduced, redistribution of PSD-95 and AMPA receptors provides a possible means to alter synaptic strength. However, the mechanism by which the exchange of PSD-95 between synapses is controlled remains unclear. One potential mechanism that controls
PSD-95 exchange between synaptic sites is regulated cycling of palmitate on PSD-95. Candidate enzymes that mediate the transfer of palmitate to PSD-95 have been recently identified, however the identity of enzymes that regulate removal of palmitate remain unknown (Fukata et al., 2004
; Huang et al., 2004
). Future studies are needed to clarify whether neuronal activity may control the dynamics of PSD-95 at the synapse by manipulating the activity of enzymes that control addition and removal of palmitate.
PSD-95 function and localization is also regulated by phosphorylation at its N-terminus, mediated by cyclin-dependant kinase 5 (cdk5) (Morabito et al., 2004
). This phosphorylation inhibits PSD-95 multimerization and PSD-95-mediated K+ channel clustering. Based on the reciprocal effects of phosphorylation and palmitoylation on PSD-95 function and localization, it is conceivable that these two forms of modification negatively regulate each other. Additional experiments directly comparing the effects of these modifications on PSD-95 and other proteins will be necessary to confirm this possibility.
Alternative splicing of MAGUKs is an additional factor influencing protein localization and function. Alternative splicing of PSD-95 at the N-terminus yields the PSD-95beta isoform which includes a L27 protein interaction motif replacing the palmitoylated motif that is expressed in the PSD-95alpha isoform (Chetkovich et al., 2002a
). This alternative form does not cluster with potassium channels when expressed in heterologous cells, but it does efficiently target to synapses in neurons. Similarly, alternative splicing of SAP-97 affects synaptic targeting and alters protein interactions. These additional amino acids spliced into the “hook” domain, producing the beta isoform, are necessary for SAP-97-mediated enhancement of AMPA receptor recruitment and function (Rumbaugh et al., 2003
). The majority of endogenous PSD-95 is the alpha isoform and the majority of endogenous SAP-97 is the beta isoform (Schluter et al., 2006
). Interestingly, the effects of the alpha isoforms are regulated independently of neuronal activity, while the beta isoform actions are activity-dependent. Thus structural and posttranslational modifications significantly alter MAGUK localization and effects on synaptic maturation, and specifically modifications of the N-terminal regions regulate PSD-95- and SAP-97-mediated effects on AMPA receptor function and neuronal excitation.
PSD-95 was initially identified as a binding partner of the NR2 subunit of NMDA-type glutamate receptors (Kornau et al., 1995
), suggesting the involvement of PSD-95 in NMDA receptor clustering. However, recent evidence points to an important role for PSD-95 in regulation of NMDA receptor activity and signaling, rather than clustering at the synapse (Kornau et al., 1995
). PSD-95 has been shown to influence surface expression of NMDA receptors and receptor desensitization (Li et al., 2003
; Lin et al., 2004
, 2006
; Roche et al., 2001
; Sans et al., 2000
). These studies support a role for NMDA receptor-PSD-95 interaction in glutamate-mediated signaling processes. In contrast, a role for PSD-95 in regulating AMPA receptor insertion and retention at the synapse has been documented by numerous studies. The first evidence indicating a direct role for PSD-95 in regulating AMPA receptor clustering at the synapse came from analysis performed on cultured hippocampal neurons overexpressing PSD-95 over a period of several days. In these experiments exogenous PSD-95 was shown to enhance AMPA receptor recruitment and excitatory synaptic responses (El-Husseini et al., 2000b
). In contrast, overexpression of PSD-95 did not influence NMDA receptor clustering in developing hippocampal neurons (El-Husseini et al., 2000b
). These findings were surprising since direct coupling of PSD-95 to AMPA receptors was not documented. Later, studies revealed that coupling of PSD-95 influences AMPA receptor trafficking and clustering through association with stargazin and related family members known as TARPs (Chen et al., 2000
, 2003
;Dakoji et al., 2003
; Schnell et al., 2002
). Several other studies showed that manipulation of PSD-95 levels over short and long time periods also influences AMPA receptor retention and activity at the synapse (Ehrlich and Malinow, 2004
; Ehrlich et al., 2007
; Nakagawa et al., 2004
; Schluter et al., 2006
). More recent studies showed that PSD-95 influences AMPA receptor retention at the synapse by restricting diffusion of the stargazin-AMPA receptor complex at synaptic membranes (Bats et al., 2007
; Schnell et al., 2002
). Consistent with these observations, mice lacking PSD-95 show normal NMDA receptor clustering and function, but reduced AMPA receptor function (Beique et al., 2006
; Ehrlich et al., 2007
; Elias et al., 2006
; Migaud et al., 1998
).
In addition to modulating ion channel clustering and function, PSD-95 has been shown to interact with numerous molecules that modulate synapse morphology (Figure 2
). These include synaptic Ras GTPase-activating protein, which regulates the Ras-extracellular signal-regulated kinase pathway (Kim et al., 1998
), kalirin-7, a guanine nucleotide exchange factor that regulates spine morphology (Penzes et al., 2001
), and the receptor tyrosine kinase ErbB4 (Garcia et al., 2000
). Association of PSD-95 with GKAP through the GK domain of PSD-95 may influence the recruitment of Shank (SH3 and ankyrin repeat-containing), a molecule coupled to the actin binding protein cortactin and the metabotropic glutamate receptor interacting protein Homer to control spine morphology (Kim et al., 1997
; Naisbitt et al., 1999
; Satoh et al., 1997
; Takeuchi et al., 1997
). Shank multimers assemble in large, sheet-like structures, and may thus serve as a platform foundation for many PSD structures (Baron et al., 2006
). Experiments using fluorescence recovery after photobleaching techniques to determine the turnover rates of scaffolding proteins in the PSD demonstrate that GKAP and Shank have faster recovery rates than PSD-95 (Kuriu et al., 2006
). This finding suggests that these proteins have differential synaptic mobility and that PSD-95 may be more stable at the synapse over short time periods. Additionally, this study found that disruption of PSD-95 membrane localization does not alter GKAP recovery dynamics in mature cultured neurons. In young neurons, PSD-95, GKAP, and Shank can be detected as a preformed complex delivered to both existing synapses and nascent neuronal contacts (Gerrow et al., 2006
). These preformed clusters are capable of recruiting functional presynaptic components. Indeed, knockdown of PSD-95 caused a corresponding reduction of GKAP and Shank clusters. These results further support an important role for PSD-95 in assembly of a postsynaptic scaffold complex involved in synapse maturation.
While GKAP and Shank are both abundant proteins, the level of PSD-95 is five times greater than each of these scaffolds in the PSDs of rat forebrain, and 10 times more abundant than NMDA receptors (Cheng et al., 2006
; Peng et al., 2004
). This molar excess of PSD-95 is indicative of its interactions with a vast number of proteins. Interestingly, it was recently reported that PSD-95 also interacts with the MAGUK scaffolding protein SAP97 (Cai et al., 2006
), thereby increasing the complexity of PSD-95-associated scaffolding complexes in the PSD. It is quite evident that regulation of scaffolding complexes directly affects the function and membrane expression of glutamate receptors. Thus, in addition to PSD-95, associated scaffold molecules are most likely key players in regulating excitatory synapse function, and the coordinated action of these molecules may tightly control neuronal excitability.
A new exciting development stems from studies on the significance of coupling of PSD-95 to adhesion molecules, in particular, members of the neuroligin family. In rodents, there are four genes that code for neuroligins (1, 2, 3, 4), while in humans, five genes have been identified (1, 2, 3, 4, 4Y) (Bolliger et al., 2001
; Craig and Kang, 2007
; Dean and Dresbach, 2006
; Ichtchenko et al., 1995
, 1996
;Irie et al., 1997
; Lise and El-Husseini, 2006
; Nguyen and Sudhof, 1997
). Neuroligin-1, the prototype member of the neuroligin family was identified as a binding partner of PSD-95 and has been shown to cluster at excitatory sites. Thus a role for neuroligins in excitatory synapse induction and development was proposed (Scheiffele et al., 2000
; Song et al., 1999
). However, unexpectedly, expression of neuroligins in cultured hippocampal neurons was shown to influence maturation of both excitatory and inhibitory presynaptic contacts (Levinson et al., 2005
; Prange et al., 2004
). Consistent with a role for neuroligins in the maturation of both excitatory and inhibitory contacts, knockdown of neuroligins in cultured neurons reduces synapse number and mainly reduces inhibitory synapse activity (Chih et al., 2005
). Further studies showed that neuroligins 1, 3, and 4 are predominately localized to excitatory synapses, while neuroligin 2 is enriched at inhibitory contacts (Chih et al., 2005
; Graf et al., 2004
; Levinson et al., 2005
; Prange et al., 2004
; Song et al., 1999
; Varoqueaux et al., 2004
).
The ability of neuroligins and their presynaptic partners neurexins to induce synapses and protein assembly at both excitatory and inhibitory contacts suggested an attractive model for controlling the number of newly formed synapses (Chih et al., 2005
; Graf et al., 2004
). However, recent evidence from mice lacking neuroligins indicates that neuroligins are not necessary for the induction of the majority of synapses, but rather for synapse maturation and activity (Varoqueaux et al., 2006
). This suggests that additional adhesion molecules may have compensated for the loss of neuroligins, or that neuroligins are involved primarily in certain aspects of synaptic development other than synapse induction. Although the precise role of neuroligins in vivo remains unclear, studies on mice lacking neuroligin 1–3 hints to altered E/I ratio, particularly in the region that controls respiratory function. Additional examinations reveal that the influence of neuroligins on synapse maturation is an activity-dependant process (Chubykin et al., 2007
). Furthermore, this activity-dependence specifically modulates neuroligin 1 at excitatory synapses and neuroligin 2 at inhibitory synapses, in agreement with the endogenous localization of these molecules.
The mechanisms that control accumulation of neuroligins at a particular synaptic site are thought to modulate synapse maturation, strength and plasticity. One mechanism that controls retention of neuroligins to particular synapse types is alternative splicing. Neuroligins have sites for alternative splicing at two conserved locations, referred to as sites A and B, in the extracellular domains (Ichtchenko et al., 1996
; Lise and El-Husseini, 2006
). The locations of the splice sites are conserved, but the amino acid inserts are different for each neuroligin. Studies conducted on neuroligin 1 and neuroligin 2 have determined that the A site insert in each molecule increases neuroligin association with inhibitory synapses and the B site insert increases neuroligin 1 association with excitatory synapses (Chih et al., 2006
). Emerging evidence suggest that intracellular mediated interactions also influence retention of neuroligins at the synapse. The C-terminal tails of neuroligins interact with PSD-95 through PDZ-dependent interactions (Irie et al., 1997
). Thus, PSD-95 may play a role in regulating the E/I synaptic ratio by modulating neuroligns’ localization and function at particular synapses (Craig and Kang, 2007
; Dalva et al., 2007
; Gerrow and El-Husseini, 2006
; Levinson and El-Husseini, 2005a,b
; Lise and El-Husseini, 2006
). Indeed, PSD-95 overexpression enriches neuroligin 1, with the B site exon, at excitatory synapses and also results in a shift in the localization of neuroligin 2, containing the A site exon, from inhibitory to excitatory synapse (Levinson et al., 2005
; Prange et al., 2004
). Manipulation of neuroligins localization at the expense of inhibitory contacts suggests that PSD-95 may influence the E/I synaptic ratio (Figure 1
B). Indeed, an increase in the E/I synaptic current was observed in neurons overexpressing PSD-95, whereas knockdown of PSD-95 reduced the E/I ratio (Levinson et al., 2005
; Prange et al., 2004
). These findings suggest that the stoichiometry of PSD-95 and neuroligin family members regulates the strength of synapses, directly affecting E/I balance (Levinson and El-Husseini, 2005a
,b
). Similar experiments performed with scaffolding proteins typically localized at inhibitory synapses will be instrumental in testing this model. Furthermore, assessment of neuroligin distributions in mice lacking PSD-95 will be important to clarify whether similar changes in the redistribution of neuroligins occur in vivo.
Additional evidence exists to suggest that synapses may compete for adhesion molecule complexes to regulate the type and strength of synapses formed. For instance, when GABA innervation is absent in isolated hippocampal pyramidal neurons, gephyrin mistargets to glutamatergic synapses (Rao et al., 2000
), possibly due to a lack of appropriate inhibitory synapse adhesion molecules and scaffolding proteins. Furthermore, overexpression of gephyrin increases GABA receptor clustering while reducing the number of PSD-95 clusters (Lardi-Studler et al., 2007
). Candidate molecules that potentially compete with PSD-95 to regulate adhesion molecule retention at the synapse include synaptic scaffolding molecule (S-SCAM) which has a structure similar to PSD-95, and interacts with neuroligins at both excitatory and inhibitory synapses (Iida et al., 2004
; Sumita et al., 2007
). Recent studies focusing on S-SCAM reveal that neuroligin, S-SCAM, and PSD-95 form a ternary complex, and suggest that S-SCAM is anchored at the synapse by beta-catenin, thus influencing accumulation of neuroligins and PSD-95 at synapses. Since S-SCAM is localized at both excitatory and inhibitory synapses, it is possible that this scaffolding molecule is a competitor of PSD-95 for neuroligin 2 binding. In this way it could sequester neuroligin 2 at inhibitory synapses in physiologically “balanced” situations. Additional studies are needed to clarify the developmental recruitment patterns of these proteins and whether they indeed compete for retention of these adhesion molecules at the synapse.
Neuroligins bind through their extracellular N-termini to neurexins, neuron-specific cell adhesion molecules present at presynaptic terminals. Neurexins bind directly to CASK, a PDZ domain-containing protein homologous to PSD-95. Through this interaction, it is thought that neurexins are coupled to the vesicle fusion machinery via the tripartite complex containing CASK, Mint-1 and Veli (Biederer and Sudhof, 2000
; Borg et al., 1998
; Butz et al., 1998
; Hata et al., 1996
). Thus the interaction of neuroligins with neurexins may activate an array of molecular responses leading to structural reorganization of the presynaptic compartment. Surprisingly, a recent discovery by Taniguchi et al. (2007)
demonstrates that neurexins are also expressed at postsynaptic sites. Similar to presynaptic forms, these postsynaptic neurexins interact with neuroligins, but this cis interaction inhibits the transsynaptic association of neuroligin with presynaptic neurexins, subsequently reducing synaptic density. The interplay between neurexins, neuroligins, PSD-95, and glutamate receptors may provide an interesting mechanism to direct synapse-specific functional and structural alterations.
Additional adhesion molecules associated with PSD-95 are emerging as potential modulators of E/I balance. Netrin-G CAMs are a family of adhesion molecules expressed preferentially in the CNS. A Netrin-G ligand (NGL) has recently been reported, the binding of which to Netrin G CAM is important for axon outgrowth (Lin et al., 2003
). Recently, NGL-2 has been shown to directly associate with the first two PDZ domains of PSD-95 (Kim et al., 2006
), inducing presynaptic differentiation and clustering of excitatory postsynaptic proteins, and increasing the number of excitatory synapses. Thus NGL-2 may work synergistically with neuroligins to affect the E/I ratio. Another recently discovered family of adhesion molecules that associate with PSD-95 and NMDA receptors consists of the synaptic adhesion-like molecule (SALM) (Wang et al., 2006
). SALM1 expression recruits PSD-95 and NMDA receptors to synapses through PDZ-dependant mechanisms and increases the number of excitatory synapses. Accordingly, knockdown reduces the number of spines as well as the frequency of spontaneous miniature excitatory postsynaptic currents (Ko et al., 2006
). Considering the existence of several families of adhesion molecules with redundant function at the synapse, it is likely that crosstalk between these complexes is important for controlling synaptic balance in vivo. The cell adhesion molecule ADAM22 (A Disintegrin And Metalloprotease) also interacts with PSD-95 through a PDZ-mediated mechanism forming a complex with stargazin (Fukata et al., 2006
). The soluble, secreted ligand of ADAM22, LGI1, increases AMPA receptor-mediated currents through postsynaptic mechanisms. It is not yet known if ADAM22 affects synapse formation and maturation. Further overexpression and knockdown studies are necessary to determine the effect of ADAM22 on synapse type, number, and ultimately the E/I balance.
As discussed earlier, altered expression of PSD-95 influences recruitment of AMPA receptors at the synapse, thereby modulating synaptic strength (El-Husseini et al., 2000b
; Schnell et al., 2002
). These results suggest that PSD-95, through modulation of AMPA receptor retention at the synapse, may also regulate synaptic plasticity. Indeed, PSD-95 overexpression occludes LTP, possibly due to saturation of receptor signaling, and enhances LTD, perhaps as a result of increasing the number of AMPA receptors available for internalization (Ehrlich and Malinow, 2004
; Stein et al., 2003
). Complementary to these findings, mice that do not express PSD-95 show decreased AMPA receptor currents and enhanced LTP (Beique et al., 2006
; Ehrlich et al., 2007
; Elias et al., 2006
). Thus, the levels of PSD-95, as well as its association with AMPA receptors, not only modulate neuronal excitation, but also synaptic plasticity.
It is well established that alterations in synaptic plasticity, such as LTP and LTD, also require phosphorylation and dephosphorylation of AMPA receptor subunits (Barria et al., 1997
; Kameyama et al., 1998
; Lee et al., 2000
; McDonald et al., 2001
). The SH3 and GK domains of PSD-95 bind to AKAP 79/150, a scaffold molecule coupled to the protein kinases PKA and PKC, as well as the Ca2+-dependant protein phosphatase calcineurin at the synapse (Carr et al., 1992
). Thus, it appears that AKAP 79/150 brings many of these kinases and phosphatases necessary for mediating these changes into proximity of AMPA receptors, producing compartmentalized regulatory complexes. It has recently been discovered that during LTD, not only are AMPA receptors dephosphorylated, but also that AKAP 79/150 is redistributed to the cytoplasm, segregating it from the membrane (Smith et al., 2006
). This change in localization suggests that, via its ability to serve as a scaffold for enzymes necessary for modulating synaptic strength and neuronal excitation, controlling the anchoring of AKAP 79/150 itself serves a potential regulatory role in modulating these processes. In conjunction with this, induction of NMDA receptor-dependent LTD causes ubiquitin-mediated removal of PSD-95 from the synapse. Mutations that prevent ubiquitination of PSD-95 inhibit AMPA receptor internalization following NMDA receptor activation (Colledge et al., 2003
). This regulation of PSD-95 by protein degradation provides a mechanism to disassemble the large scaffolding complex present at the PSD, and to indirectly modulate glutamate receptors, thus altering synaptic strength. A direct comparison between the time courses for PSD-95 and AKAP 79/150 activity-dependant removal from synaptic membranes would be useful to determine the precise sequence of events, and to determine if the removal of one type of scaffold is necessary for removal of the other.
LTD-induced alterations in AMPA receptor membrane expression require actin depolymerization (Morishita et al., 2005
). Conversely, LTP-induced alterations in synaptic composition require actin polymerization (Krucker et al., 2000
; Lin et al., 2005
). Changes in the recruitment and/or assembly of PSD-95/GKAP/Shank/cortactin complex may provide a mechanism for mediating the structural changes associated with changes in synaptic plasticity. Lisman and Raghavachari (2006)
recently proposed an interesting model to account for synaptic changes associated with LTP. They suggest that during LTP, “hyperslots” of postsynaptic proteins are inserted into the synapse. Since these hyperslots are capable of binding AMPA receptors, it is intriguing to consider PSD-95 and stargazin as components of the hyperslot complex. This model also predicts that presynaptic changes in neurotransmitter release result from retrograde signaling, possibly through adhesion molecules, from the newly incorporated hyperslots. Indeed, PSD-95-neuroligin-neurexin association fulfills this role as a retrograde signal to increase the probability of neurotransmitter release (Futai et al., 2007
), providing an additional mechanism for PSD-95 to affect cellular excitation.
The imbalances between excitation and inhibition observed in the brains of patients with many neurological disorders demonstrate the importance of mechanisms involved in maintaining proper E/I balance in normal brain function. An increase in the E/I synaptic ratio may result in enhanced neuronal excitability, associated with increased seizure susceptibility. Epilepsy is a heterogeneous disorder with multiple moluculare causes, characterized by repetitive occurrence of pathological patterns of synchronous neuronal activity (Behrens et al., 2007
; Stafstrom, 2006
; Stief et al., 2007
). In a simplified model, seizure activity can be triggered by an increase in excitation, or a decrease in inhibition that causes an overall increase in excitatory activation of the brain (Brenner, 2004
; Clancy and Kass, 2004
; McCormick and Contreras, 2001
; Stafstrom, 2006
; Ziburkus et al., 2006
). PSD-95 has been implicated in modulating seizure activity, as many epileptic patients show increased levels of PSD-95-NMDA receptor complexes (Ying et al., 2004
).
Schizophrenia is another complex neuropsychiatric disorder that could be affected by altered E/I synaptic ratio (Konradi and Heckers, 2003
; Krystal et al., 1994
; Lewis and Levitt, 2002
; Lewis and Moghaddam, 2006
; Moghaddam, 2003
). Enhanced PSD-95-ErbB4 coupling has been detected in patients with schizophrenia (Hahn et al., 2006
). In other cases of schizophrenia, reduction in GABA production and signaling in the prefrontal cortex has been observed, leading to a reduction in the inhibitory tone imposed on glutamatergic neurons in this region. However, it is not clear whether this is a cause or an effect of schizophrenia (Lewis and Moghaddam, 2006
; Lewis et al., 2005
;Sawaguchi et al., 1988
, 1989
; Wassef et al., 2003
). Changes in PSD-95 levels have also been associated with Fragile X syndrome, an inherited form of mental retardation (Tsiouris and Brown, 2004
). This disease is caused by silencing of a single gene (FMR1) that codes for the Fragile X mental retardation protein (FMRP), a protein involved in mRNA translation, and for which expression is normally induced by mGluR activation (Feng et al., 1997
; Li et al., 2001
; Pieretti et al., 1991
; Weiler et al., 1997
). Evidence suggests that FMRP is involved in mGluR-mediated synaptic plasticity (Huber et al., 2002
; Laggerbauer et al., 2001
; Weiler et al., 1997
), and activation of mGluR increases translation of PSD-95 through an FMRP-mediated mechanism (Todd and Malter, 2002
; Todd et al., 2003
). Mice mutant for FMRP display brain-region specific imbalances of multiple excitatory and inhibitory neurotransmitters (Gruss and Braun, 2004
), and are prone to auditory-induced seizures that are not caused by generally enhanced excitation (Chen and Toth, 2001
). Fragile X therefore represents a disease state that can result from altered E/I balance, but is not exclusive to glutamate and GABA circuitry.
Autism, a pervasive developmental disorder with a strong genetic link, is another disorder that can be induced by altered E/I balance (Deonna and Roulet, 2006
; Polleux and Lauder, 2004
; Trevathan, 2004
). A recent model of autism suggests that altered neuronal excitability, resulting from synaptic imbalance, underlies this disorder (Andres, 2002
; Rubenstein and Merzenich, 2003
). Although specific genes implicated in autism have not been firmly identified, mutations in two members of the neuroligin family of cell adhesion molecules have been identified in multiple patients (Chih et al., 2004
; Chubykin et al., 2005
; Comoletti et al., 2004
; Jamain et al., 2003
; Laumonnier et al., 2004
). However, since recent genetic screens suggest that mutations in NLGs are rather rare in autistic patients (Gauthier et al., 2005
; Yan et al., 2005
), it is more likely that autism may result from abnormal expression of a diverse set of genes encoding synaptic proteins with related functions in synaptic development and activity. In support of this possibility, mutations in the scaffolding protein Shank (Durand et al., 2007
), as well as neurexin (Feng et al., 2006
), have recently been associated with autism. Interestingly, a mouse model that expresses the neuroligin 3 mutation associated with some cases of autism has recently been developed (Tabuchi et al., 2007
). This model presents a gain of function mutation that results in enhanced inhibition, supporting the hypothesis that perturbing the function of neuroligins can produce an imbalance in the E/I ratio. In addition to the previously mentioned disorders, Rett syndrome, Parkinson’s disease, and Tourette’s syndrome are also characterized by alterations in E/I balance, and are caused by various alterations in molecular, cellular, and neuronal circuitry. Recent findings indicate that deletion of the scaffolding molecule SAP90/PSD95-associated protein 3 (SAPAP3) produces behaviors characteristic of obsessive-compulsive disorder, including compulsive grooming and increased anxiety (Welch et al., 2007
). Thus disruption of multiple proteins in the PSD network can produce various neuro/psychological phenomena. The wide-ranging disorders mentioned here have a significant impact on affected individuals, families, and society, and as such, greater understanding of the role of E/I balance in these illnesses is necessary for effective treatment possibilities.
PSD-95 is also implicated in behavioral and synaptic plasticity associated with psychostimulant drug addiction. For example, animal models of chronic psychostimulant abuse display reduced levels of PSD-95 (Yao et al., 2004
). Recent studies reveal a direct interaction between the N-sterminal region of PSD-95 and the dopamine D1 receptor (Zhang et al., 2007
). This interaction reduces the amount of D1 receptor on the surface, reducing dopamine-mediated excitation of medium spiny neurons and cortical pyramidal neurons. Furthermore, PSD-95 knockout animals demonstrate enhanced D1-mediated responses. The authors hypothesize that interactions between D1 receptors, PSD-95, and NMDA receptors provide a mechanism for simultaneous regulation of surface expression, and also prevents excessive positive feedback generated between the two receptors. Taken together, these findings suggest that psychostimulants can alter the balance between dopaminergic and glutamatergic signaling at individual spines by altering receptor/scaffold complexes.
Recent research into the role of PSD-95 in recruitment of glutamate receptors and adhesion molecules at the synapse has brought much insight into the basic principles governing assembly of protein complexes at glutamatergic synapses. It is evident from recent studies that scaffolding molecules such as PSD-95 play prominent roles in the assembly of ion channels and associated proteins at excitatory synaptic sites. Recent in vitro data also revealed that PSD-95 may influence the retention of particular adhesion molecules at excitatory contacts at the expense of inhibitory synapses. Thus it is attractive to propose that these molecules act as the molecular sensors that control the balance between excitation and inhibition.
In conclusion, the progress that has been made during the last few years will lead to a more complete understanding of synaptic balance in the years to come. However, considering the existence of numerous molecules that regulate this process, future studies are needed to address mechanisms that control cross-talk between various scaffold molecules and adhesive systems to define the contribution of each system in the control of E/I synaptic strength and brain function. Further understanding of the mechanisms that govern assembly of proteins at other types of neuronal contacts, such as GABAergic, dopaminergic and serotonergic synapses, may help uncover other mechanisms that influence cross talk between adhesion and scaffolding molecules at a particular synapse type and in turn E/I synapse number and strength. Manipulation of the expression of several of the newly discovered proteins at early stages of neuronal development may help us uncover how alteration in the expression of these molecules influence the E/I ratio and how this might lead to the development of neuropsyhiatric illness.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This manuscript is dedicated in loving memory to Alaa El-Husseini. We would like to thank K. Gerrow for assistance with Illustrator and J. Levinson for review of the manuscript.
Cheng, D., Hoogenraad, C. C., Rush, J., Ramm, E., Schlager, M. A., Duong, D. M., Xu, P., Wijayawardana, S. R., Hanfelt, J., Nakagawa, T., et al. (2006). Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell Proteomics 5, 1158–1170.
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., Nygren, G., Rastam, M., Gillberg, I. C., Anckarsater, H., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27.
Huang, K., Yanai, A., Kang, R., Arstikaitis, P., Singaraja, R. R., Metzler, M., Mullard, A., Haigh, B., Gauthier-Campbell, C., Gutekunst, C. A., et al. (2004). Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron 44, 977–986.
Jourdi, H., Lu, X., Yanagihara, T., Lauterborn, J. C., Bi, X., Gall, C. M., and Baudry, M. (2005). Prolonged positive modulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors induces calpain-mediated PSD-95/Dlg/ZO-1 protein degradation and AMPA receptor down-regulation in cultured hippocampal slices. J. Pharmacol. Exp. Ther. 314, 16–26.
Krystal, J. H., Karper, L. P., Seibyl, J. P., Freeman, G. K., Delaney, R., Bremner, J. D., Heninger, G. R., Bowers Jr, M. B., and Charney, D. S. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry. 51, 199–214.
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M. P., Raynaud, M., Ronce, N., Lemonnier, E., Calvas, P., et al. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557.
Levinson, J. N., Chery, N., Huang, K., Wong, T. P., Gerrow, K., Kang, R., Prange, O., Wang, Y. T., and El-Husseini, A. (2005). Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J. Biol. Chem. 280, 17312–17319.
Sumita, K., Sato, Y., Iida, J., Kawata, A., Hamano, M., Hirabayashi, S., Ohno, K., Peles, E., and Hata, Y. (2007). Synaptic scaffolding molecule (S-SCAM) membrane-associated guanylate kinase with inverted organization (MAGI)-2 is associated with cell adhesion molecules at inhibitory synapses in rat hippocampal neurons. J. Neurochem. 100, 154–166.
Weiler, I. J., Irwin, S. A., Klintsova, A. Y., Spencer, C. M., Brazelton, A. D., Miyashiro, K., Comery, T. A., Patel, B., Eberwine, J., and Greenough, W. T. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. U.S.A. 94, 5395–5400.