Informational lesions: optical perturbation of spike timing and neural synchrony via microbial opsin gene fusions
1
MIT Media Lab, Massachusetts Institute of Technology, Cambridge, MA, USA
2
Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge, MA, USA
3
McGovern Institute, Massachusetts Institute of Technology, Cambridge, MA, USA
4
Koch Center for Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, USA
5
Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
Synchronous neural activity occurs throughout the brain in association with normal and pathological brain functions. Despite theoretical work exploring how such neural coordination might facilitate neural computation and be corrupted in disease states, it has proven difficult to test experimentally the causal role of synchrony in such phenomena. Attempts to manipulate neural synchrony often alter other features of neural activity such as firing rate. Here we evaluate a single gene which encodes for the blue-light gated cation channel channelrhodopsin-2 and the yellow-light driven chloride pump halorhodopsin from Natronobacterium pharaonis, linked by a ‘self-cleaving’ 2A peptide. This fusion enables proportional expression of both opsins, sensitizing neurons to being bi-directionally controlled with blue and yellow light, facilitating proportional optical spike insertion and deletion upon delivery of trains of precisely-timed blue and yellow light pulses. Such approaches may enable more detailed explorations of the causal role of specific features of the neural code.
It has long been debated to what extent synchronous or precisely-timed neural activity contributes to neural computation and behavior. Synchronous neural activity within or between brain regions has been observed during, or associated with, many brain functions including timing-dependent plasticity, global stimulus feature processing, visuomotor integration, emotion, working memory, motor planning, and attention (e.g., Gray et al., 1989
; Roelfsema et al., 1997
; Brivanlou et al., 1998
; Donoghue et al., 1998
; Steinmetz et al., 2000
; Fries et al., 2001
; Tallon-Baudry et al., 2001
; Froemke and Dan, 2002
; Perez-Orive et al., 2002
; Seidenbecher et al., 2003
; Courtemanche and Lamarre, 2004
; Buschman and Miller, 2007
), as measured with multielectrode recording, electroencephalography (EEG), magnetoencephalography (MEG), and local field potential (LFP) analysis. Furthermore, abnormal patterns of neural synchrony have been associated with a variety of neurological and psychiatric disorders such as Parkinson’s disease, epilepsy, autism, and schizophrenia (e.g., Bragin et al., 2002
; Uhlhaas and Singer, 2006
; Berendse and Stam, 2007
; Brown, 2007
; Huguenard and McCormick, 2007
; Orekhova et al., 2007
; Gonzalez-Burgos and Lewis, 2008
; Roopun et al., 2009
). Computationally, synchrony has been implicated in multiple processes, including grouping neurons into ‘cell assemblies’ that can more effectively represent information to downstream neural networks, acting as a clock for gating or multiplexing information, coordinating information flow within small neural networks, selecting stimuli for attention, and performing pattern recognition (e.g., Hopfield and Brody, 2001
; Friedrich et al., 2004
; Tiesinga and Sejnowski, 2004
; Borgers et al., 2005
, 2008
; Sohal et al., 2009
). However, determining the causal role of neural synchrony to neural computation and behavior has remained elusive, in part because selective perturbation of spike timing is difficult. In some specific systems, in which pharmacological or genetic strategies for selectively disrupting synchrony happened to be compatible with local cellular and network properties, pioneering attempts have been made to perturb spike timing without altering other aspects of neural coding such as spike rate (MacLeod and Laurent, 1996
; Bao et al., 2002
; Robbe et al., 2006
), but no generalized method for doing so exists.
Recently we and others have developed strategies for optically sensitizing neurons to being activated and silenced with different colors of light, by delivering the gene encoding for the blue light-gated cation channel channelrhodopsin-2 (ChR2) or the gene for the yellow-light driven chloride pump halorhodopsin from N. pharaonis (Halo/NpHR) to neurons, and illuminating them with pulses of light (Boyden et al., 2005
; Han and Boyden, 2007
; Zhang et al., 2007
). New neural activators and silencers continue to be developed, and these molecules have already begun to be applied to the study of neural dynamics, as investigators optically drive the activity of excitatory and inhibitory neurons and electrophysiologically characterize the resultant patterns (e.g., Boyden et al., 2009
; Cardin et al., 2009
; Han et al., 2009
; Sohal et al., 2009
; Talei Franzesi et al., 2009
). However, none of these experiments have attempted to isolate the distinct properties of synchrony and spike rate, both of which are modulated when neurons are activated or silenced by using either ChR2 or Halo by itself. Previously, we presented a strategy for perturbing spike timing without altering spike rate (Han and Boyden, 2007
), taking advantage of the fact that the spectral peaks for ChR2 activation and Halo activation are separated by over 100 nm (Figure 1
A), making it possible to express both molecules in the same neuron (Figure 1
B), thus conferring bi-directional control of the voltage of that neuron by blue and yellow light (Figures 1
C,D) (Han and Boyden, 2007
). As a consequence of this two-color bidirectional voltage control, precisely-timed blue and yellow light pulse trains, delivered to a neuron expressing both ChR2 and Halo, can be used to insert spikes into, and delete spikes from, the ongoing activity experienced by that neuron (Figure 1
E), resulting in precise perturbations of spike timing without changing overall spike rate (Figures 1
F,G). However, the delivery of two separate genes to precisely the same cell population can be a daunting proposition. Furthermore, it is important that ChR2 and Halo be expressed in a predictable proportion to one another, so that the insertion and deletion of spikes can be balanced; separate delivery of the two genes, or independent expression of the two genes, would not accomplish this. Recently we and others have presented gene fusions between ChR2 and Halo that enable them to be expressed in the same expression cassette, bridging ChR2 and Halo with an optimized form of the ‘self-cleaving’ 2A peptide sequence from foot-and-mouth disease virus or other picornaviruses (Han et al., 2008
; Tang et al., 2009
). The 2A sequence encourages a ribosomal skip between a glycine and proline within the 2A sequence, resulting in separate proteins being formed from the sequences on either side (Ryan et al., 1991
; Ryan and Drew, 1994
). In the Tang et al. paper, the first peer-reviewed presentation of opsin fusions using a 2A peptide bridge, the authors showed functional expression of both opsins in the same cell, in vivo, after delivery via AAV. We here quantitate the proportional expression of ChR2 and Halo in the same cell when they are expressed separated by a 2A bridge, and discuss the consequences for the specific difficult experimental goal of perturbing synchrony and precise spike timing. Such proteins will also be useful for exploring a large set of systems neuroscience experiments in which bi-directional control of the activity of a given neuron population is desired.
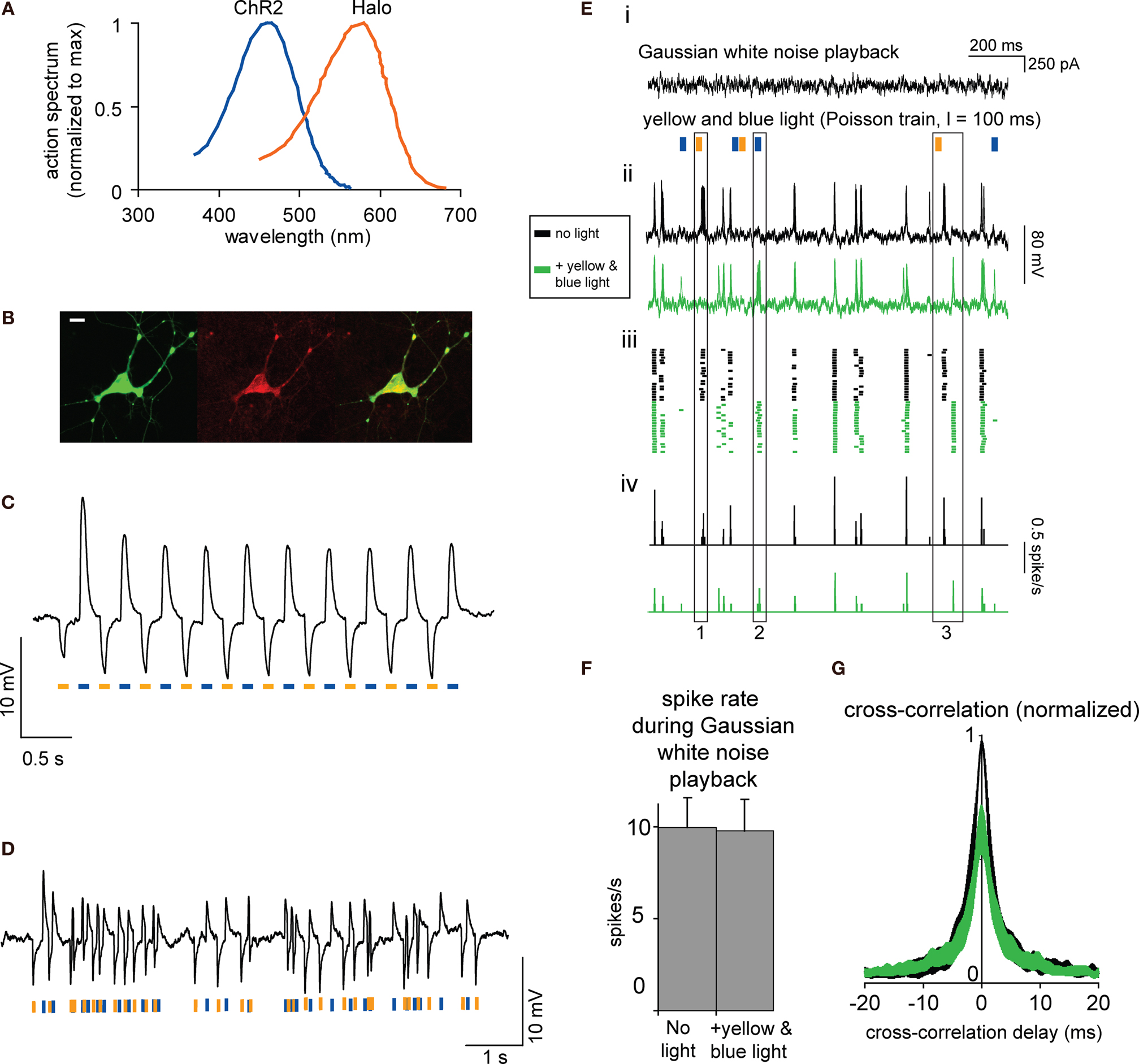
Figure 1. Schema for how to alter neural spike timing, using two-color illumination of neurons co-transfected with both ChR2 and Halo in a 1:1 ratio. Adapted from (Han and Boyden, 2007
). (A) Action spectrum for ChR2 (blue, adapted from Nagel et al., 2003
), overlaid with absorption spectrum for Halo (orange, adapted from Duschl et al., 1990
). Each spectrum is normalized to its own peak, for ease of comparison. (B) Co-expression of Halo-GFP (left) and ChR2-mCherry (middle) in a single neuron expressing both (right, overlay). Scale bar, 20 μm. (C) Hyperpolarization and depolarization events elicited in a single representative neuron, by two interleaved 2.5 Hz trains of yellow and blue light pulses (50 ms duration each), denoted by bars of respective coloration below the trace. (D) Hyperpolarization and depolarization events induced in a representative neuron by a Poisson train (mean inter-pulse interval λ = 100 ms) of alternating pulses of yellow and blue light (10 ms duration), denoted by bars of respective coloration below the trace. (E) Optical disruption of spike timing, without alteration of spike rate, for a representative current-clamped hippocampal neuron transfected simultaneously with ChR2 and Halo in vitro. Vertical boxes (labeled at bottom 1, 2, 3) highlight features referred to in the text of the Results. (i) Stimulus trace showing a subsegment of the somatically injected filtered Gaussian white noise current used in all these experiments (top), as well as of the Poisson train (mean inter-pulse interval λ = 100 ms) of alternating yellow and blue light pulses (bottom). (ii) 20-trace overlays of voltage responses to the somatically injected white noise current, either with no light (top, black traces) or with delivery of a Poisson train of yellow and blue light pulses (bottom, green traces). (iii) Spike raster plots for the traces shown in (Eii). (iv) Spike-timing histograms (bin size: 500 μs) for the rasters shown in (Eiii). (F) Spike rates of current-clamped hippocampal neurons transfected simultaneously with ChR2 and Halo in vitro (n = 7), injected with filtered Gaussian white noise current, either with no light (left) or with concurrent delivery of a Poisson train of yellow and blue light pulses (right). Plotted is mean ± standard error. (G) Cross-correlation between spike trains elicited by the same filtered Gaussian white noise current injection, played twice, when either both current injections were performed in the dark (black curve), or when one of the current injections was performed with concurrent delivery of a Poisson train of yellow and blue light pulses (green trace). Data is plotted as mean ± standard error (averaged across n = 7 neurons).
Cell Culture
All procedures involving animals were conducted in accordance with the National Institutes of Health Guide for the care and use of laboratory animals and approved by the Massachusetts Institute of Technology Animal Care and Use Committee. Hippocampal regions CA3-CA1 of postnatal day 0 Swiss Webster or C57 mice from Taconic or Jackson Labs were isolated and treated with trypsin (1 mg/ml) for ∼12 min. Digestion was stopped by Hanks solution supplemented with 20% fetal bovine serum and trypsin inhibitor (Sigma). Tissue was dissociated with Pasteur pipettes and centrifuged at 1000 rpm at 4°C for 10 min. Dissociated neurons were plated on glass coverslips pre-coated with Matrigel (BD Biosciences) at a rough density of approximately two to four hippocampi per 20 coverslips. Neurons were transfected using a commercially available calcium phosphate transfection kit (Invitrogen), at 3–5 days in vitro. GFP fluorescence was used to identify successfully-transfected neurons, indicating a net transfection efficiency of 1–10%. All images and electrophysiological recordings were made on 9–15 day-in-vitro neurons (approximately 6–10 days after transfection).
Molecular Biology
The ChR2GFP-2A-HaloYFP-N1 plasmid (‘ChR2-2A-Halo’) was constructed by first inserting the extended N-terminus 2A sequence (DNA sequence AAGAAACAGAAAATTGTGGCACCAGTGAAACAGACTTTGAATTTTGACCTTCTCAAGTTGGCGGGAGACGT
CGAGTCCAACCCTGGGCCC, translated peptide sequence KKQKIVAPVKQTLNFDLLKLAGDVESNPGP) into the BsrGI and NotI sites in the pEGFP-N1 vector, followed by inserting ChR2-GFP without stop codon into the KpNI and BsrGI sites in front of the 2A sequence, and then by inserting Halo-YFP without start codon into the XbaI and EcoRI sites downstream of the 2A sequence. The pEGFP-N1 vector drives gene expression from the CMV promoter.
Electrophysiology
Whole cell patch clamp recording was made on 9–15 day-in-vitro neurons using a Multiclamp 700B amplifier, connected to a Digidata 1440 digitizer (Molecular Devices) attached to a PC running pClamp 10. During recording, neurons were bathed in Tyrode’s solution containing (in mM) 150 NaCl, 2.4 KCl, 2 CaCl, 2 MgCl, 10 HEPES, 10 Glucose, 10 μM NBQX, 10 μM gabazine and 50 μM D-APV. Borosilicate glass (Warner) pipettes were filled with a solution containing (in mM) 130 K-Gluconate, 7 KCl, 2 NaCl, 1 MgCl2, 0.4 EGTA, 10 HEPES, 2 ATP-Mg, 0.3 GTP-Tris and 20 sucrose. Pipette resistance was ∼5 MΩ and the access resistance was 10–25 MΩ, which was monitored throughout the voltage-clamp recording.
Light-induced membrane photocurrents were induced by brief light pulses separated by periods in the dark, in neurons current-clamped or voltage-clamped, respectively. Light pulse trains were either input directly into pClamp software (Molecular Devices) or synthesized by custom scripts written in MATLAB (Mathworks), then played back through a Sutter DG-4 light source via a Digidata 1440 (Molecular Devices). Light was reflected into the sample off of a 700DCXRU (Chroma) dichroic in a Leica DMI6000B inverted microscope. In the DG-4, a yellow filter (Chroma, bandpass 575 ± 25 nm) was used to deliver light to activate Halo, and a GFP excitation filter (bandpass 470 ± 20 nm) was used to activate ChR2. Powers out the 40× objective were approximately 10 mW/mm2 in irradiance.
Data Analysis
Amplitude and timing data were analyzed using Clampfit 10 (Molecular Devices) and custom analysis scripts written in MATLAB.
We recently presented a demonstration of how one could use co-expression of separate ChR2 and Halo genes to support optical disruption of the timing of spikes, without significant alteration of spike rate (Figure 1
, adapted from Han and Boyden, 2007
). In review of this previously-published work (see Han and Boyden, 2007
for detailed results and methods): we cultured hippocampal neurons and then caused them to fire precisely-timed spike trains by patch clamping them in current-clamp mode, and repeatedly delivering to each neuron the same filtered Gaussian white noise current trace (see Figure 1
Ei, top, for a fragment thereof). Such noisy currents had been previously found to induce reliable spike trains in current-clamped neurons in acute cortical brain slices (Mainen and Sejnowski, 1995
). On some trials, we also illuminated the neuron with the Poisson train of alternating yellow and blue light pulses shown in Figure 1
D (see Figure 1
Ei, bottom). We found that when delivered alone, the filtered Gaussian white noise current trace indeed induced reliably-timed spike trains, across repeated trials (see Figure 1
Eii, top, for 20 overlaid traces, and Figure 1
Eiii, top, for corresponding spike rasters). When an optically-sensitized neuron was additionally driven by the Poisson train of yellow and blue light pulses, the neuron fired spikes with different timings than occurred in darkness, but the resultant spikes were nevertheless still similar across repeated trials of current injection + light illumination (see Figure 1
Eii, bottom, and Figure 1
Eiii, bottom, for overlaid traces and spike rasters respectively, for 20 trials; Figure 1
Eiv shows spike histograms). Inspection of this data shows that relative to the spike train elicited by current injection alone, the Poisson train of light pulses could sometimes abolish spikes that were previously reliable (vertical box 1 spanning Figures 1
Ei–iv), create new spikes which were not previously present (vertical box 2 spanning Figures 1
Ei–iv), or advance or delay the timing of specific spikes relative to their original timing in the dark (vertical box 3 spanning Figures 1
Ei–iv). We compared mean spike rates for neurons receiving the filtered Gaussian white noise current injection alone vs. with the additional Poisson light pulse train, and found no difference in spike rates for these two conditions (p > 0.90, t-test; n = 7 neurons; Figure 1
F), indicating that our optical intervention preserved spike rate. However, precise spike timing was altered significantly: cross-correlations of the spike trains elicited from the filtered Gaussian white noise current injection alone vs. with the additional Poisson light pulse train resulted in zero-lag cross-correlations that were on average 37% smaller than cross-correlations of pairs of spike trains elicited from the filtered Gaussian white noise current injection alone (p < 0.005, n = 7 neurons). This indicates that precise spike timing was indeed disrupted by the activation of Halo and ChR2, even while spike rate was preserved.
The strategy illustrated in Figure 1
centers around the idea of using blue light to push the voltage of a cell over spike threshold, and using yellow light to push the voltage of a cell under spike threshold. Critical to this strategy is the proportional expression of ChR2 and Halo in a predictable ratio to one another. Ideally, for every spike inserted, another will be deleted. A neuron that expresses high levels of both ChR2 and Halo will have a high probability of generating a spike in response to a blue light pulse, but will also have a high probability of losing a spike in response to a yellow light pulse. On the other hand, a neuron that expresses low levels of both ChR2 and Halo will have a low probability of generating a spike in response to a blue light pulse, but will also have a lower probability of losing a spike in response to a yellow light pulse. Thus to a first approximation, the strategy in Figure 1
should work to disrupt spike timing but not spike rate, independent of the net expression levels of ChR2 and Halo, as long as their proportion is kept constant. Of course, this approximation will break down if the scenario approaches an extreme: for example, a high-firing rate neuron may not be responsive to blue light pulses because of a ceiling effect on firing rate, but may be greatly affected by yellow pulses; on the other hand, a low-firing rate neuron may respond to blue light pulses with increased spiking, but may have few spikes to lose when yellow light pulses arrive. Thus it will be important to choose light pulse trains that deliver light pulses at appropriate rates given the neural population under investigation; the light pulse rate should scale with the firing rate of the neural population (and in principle, could even be adapted in real time during a physiology or behavior experiment). Two further comments should be made: given that ChR2 and Halo generate intrinsically different scales of currents (ChR2’s peak currents are an order of magnitude greater than Halo’s), the ratio of blue to yellow light powers may need to be adjusted to balance the current magnitudes against one another. Finally, in the intact brain in vivo, light powers will fall off with distance from the light source; however, the difference between blue and yellow light power attenuation in brain tissue is not very large over the short distances (∼1 mm) utilized for in vivo optical control scenarios, which are significantly less than the absorption length constant of visible light in tissue (Bernstein et al., 2008b
). Tiling the brain with arrays of LED- or laser-coupled optical fibers may allow for more even light distribution in the brain, than possible with just one inserted fiber (Bernstein et al., 2008a
; Zorzos et al., 2009
).
To express both ChR2 and Halo in a stoichiometric ratio, we evaluated a number of candidate strategies. Using two viruses to deliver the two genes could lead to significant variation of the ChR2/Halo ratio across cells. Placing an internal ribosome entry site (IRES) – a long sequence of several hundred base pairs, large compared to the space accorded in many kinds of viral vector – in between two genes often results in much lower expression of the second gene (levels 0–20% those of the first gene) (Mizuguchi et al., 2000
; Hennecke et al., 2001
; Yu et al., 2003
; Osti et al., 2006
). We decided upon a methodology using the ‘self-cleaving’ 2A sequence (Figure 2
A) from foot-and-mouth disease virus (Ryan et al., 1991
; Ryan and Drew, 1994
). When an mRNA encoding for ChR2-2A-Halo is being translated, a specific Gly-Pro bond in the 2A sequence would not be enduringly formed by the ribosome (a ‘ribosomal skip’), resulting in the separate translation of ChR2 and Halo into separate proteins. We utilized a variant of 2A with additional amino acids appended to the N-terminus which enhances cleavage from 90% to 96–99% (as assessed by an in vitro translation assay) (Donnelly et al., 2001a
,b
) (Figure 2
A, blue and green colored residues). The use of a different 2A peptide to co-express channelrhodopsin-2 and halorhodopsin within a single gene was recently published, including proof-of-principle electrophysiological data resulting from in vivo infection of neurons with the resulting virus (Tang et al., 2009
). Here, expression of the ChR2-2A-Halo gene by transfection in cultured hippocampal neurons resulted in excitatory and inhibitory photocurrents in response to blue and yellow light respectively. The photocurrents were smaller than would result from expression of either gene alone (Figure 2
C), in part because of overlap in the two molecules’ action spectra (Figure 1
A). Blue-light elicited photocurrents, measured in voltage clamp mode, were 157 ± 63 pA (n = 22 neurons; mean ± standard deviation), ∼30% of the value previously reported for raw ChR2 expression in cultured hippocampal neurons (Boyden et al., 2005
), and yellow-light elicited photocurrents were 40 ± 24 pA, ∼45% of the value previously reported for raw Halo expression in cultured hippocampal neurons (Han and Boyden, 2007
). These currents, nevertheless, are sufficient to cause a neuron near the threshold of spiking to fire an action potential or to be momentarily silent, and routinely resulted, in current-clamped neurons, in effective perturbations of membrane voltage by 5–10 mV (Figure 2
B). Plotting, for each recorded neuron, the blue-light elicited photocurrents vs. the yellow-light elicited photocurrents yielded a significant linear relationship with correlation coefficient r = 0.51 (R2 = 0.26, p < 0.02; Figure 2
D). Thus, the 2A expression vector here utilized was able to mediate proportional, functional expression of ChR2 and Halo in the same cell.
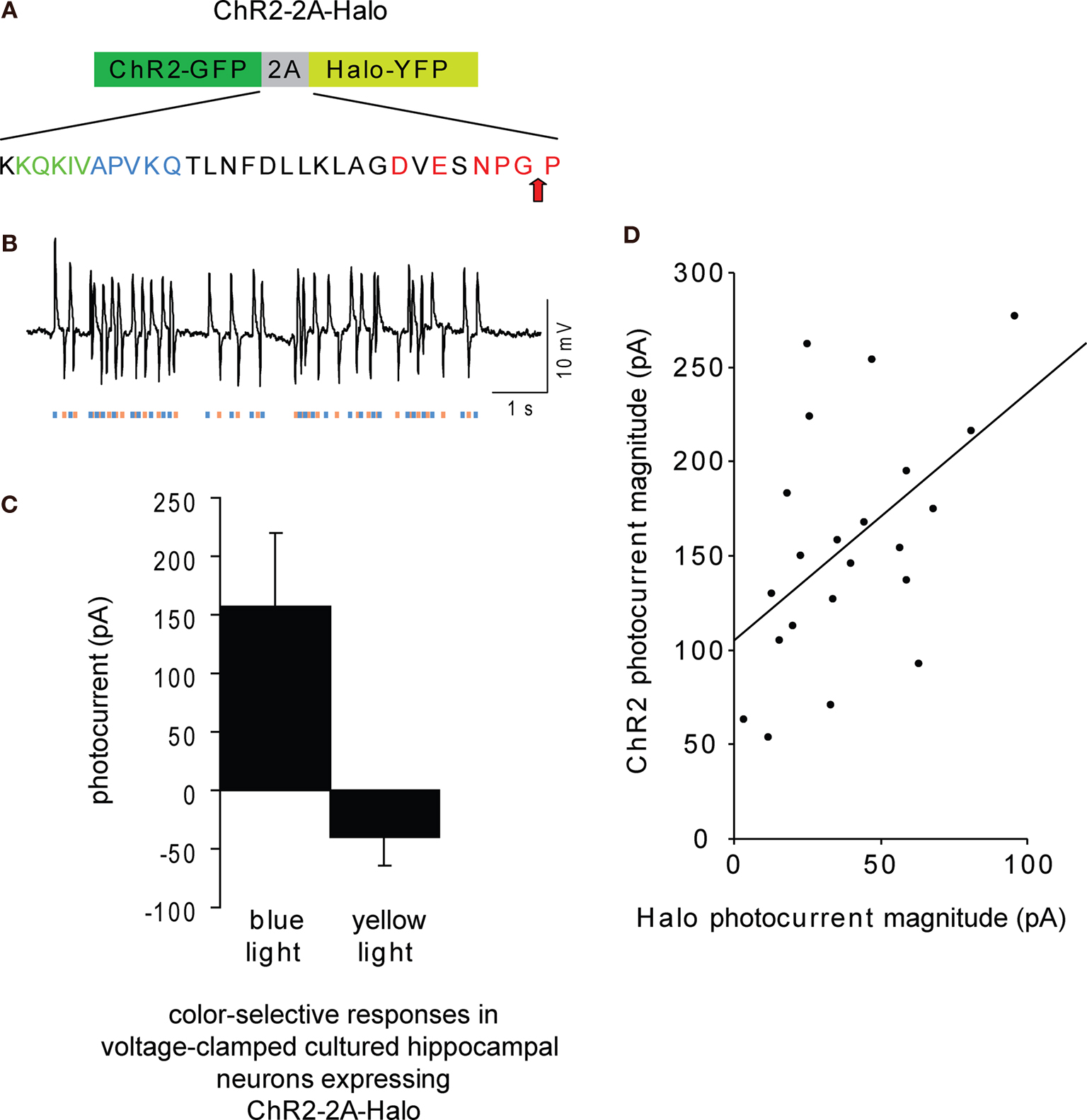
Figure 2. Gene fusion enabling proportional co-expression of ChR2 and Halo under a single promoter. (A) Schematic of the gene fusion ChR2-2A-Halo, highlighting the sequence of the 2A peptide here used. Red letters indicate consensus sequence for 2A-like ribosomal skip sequence (Donnelly et al., 2001a
; de Felipe et al., 2006
); blue letters indicate amino acids from the 1D peptide sequence that, when appended to the N-terminus of the 2A sequence, increase in vitro-translated protein cleavage from 90% to 96%, and green letters are part of a sequence from the 1D peptide sequence that boosts in vitro-translated protein cleavage to 99–100% levels (Donnelly et al., 2001a
,b
). (B) Hyperpolarization and depolarization events induced in a representative current-clamped hippocampal neuron in vitro, transfected with ChR2-2A-Halo, by a Poisson train (mean inter-pulse interval λ = 100 ms) of alternating pulses of yellow and blue light (10 ms duration), denoted by bars of respective coloration below the trace. (C) Peak photocurrents measured in voltage clamped ChR2-2A-Halo-expressing cultured hippocampal neurons under 1-s blue (left) or yellow (right) exposure (n = 22 neurons; bars represent mean ± standard deviation). (D) Plot of magnitude of blue light-elicited photocurrents vs. magnitude of yellow light-elicited photocurrents. Line shows linear regression fit to the plotted data.
What kinds of experiment are enabled by the use of linked microbial opsins for bi-directional control of the voltage of individual neurons? Many ‘obvious’ but much-desired experiments may be facilitated with use of this reagent. For example, by appending to ChR2 a myosin-binding domain (MBD) that preferentially targets it to cell bodies and dendrites (Lewis et al., 2009
), the resultant ChR2-MBD-2A-Halo could enable activation of a population of neurons when their cell bodies are illuminated with blue light, as well as silencing of specific projections when their axonal terminals are illuminated with yellow light. The ability to guarantee that a single population of neurons can be both activated and silenced is itself valuable, enabling testing of necessity and sufficiency of the same population of neurons, to a specific neural computation or behavior. However, it is useful to explore conceptually how this technology might be useful for probing complex questions concerning synchrony. We have, in Figure 3
, outlined in schematic form four protocols enabling perturbation of spike timing or synchrony, in order to study their contribution to circuit output. In all four cases, appropriately-timed blue and yellow light pulses are used to disrupt the timing of spikes in a region containing neurons sensitized to light with a 2A construct, thus altering the spike statistics. Because light will be delivered to all the neurons within a defined region, one caveat may apply: the optically-sensitized neurons in the region will be exposed to the same temporal pattern of blue and yellow light, and this common input may alter (e.g., increase) the synchrony between optically-sensitized neurons within the illuminated region; this may potentially complicate experiments where altered synchrony within a region is undesired, although again the use of multiple light sources to deliver multiple light patterns to a single region may ameliorate this issue.
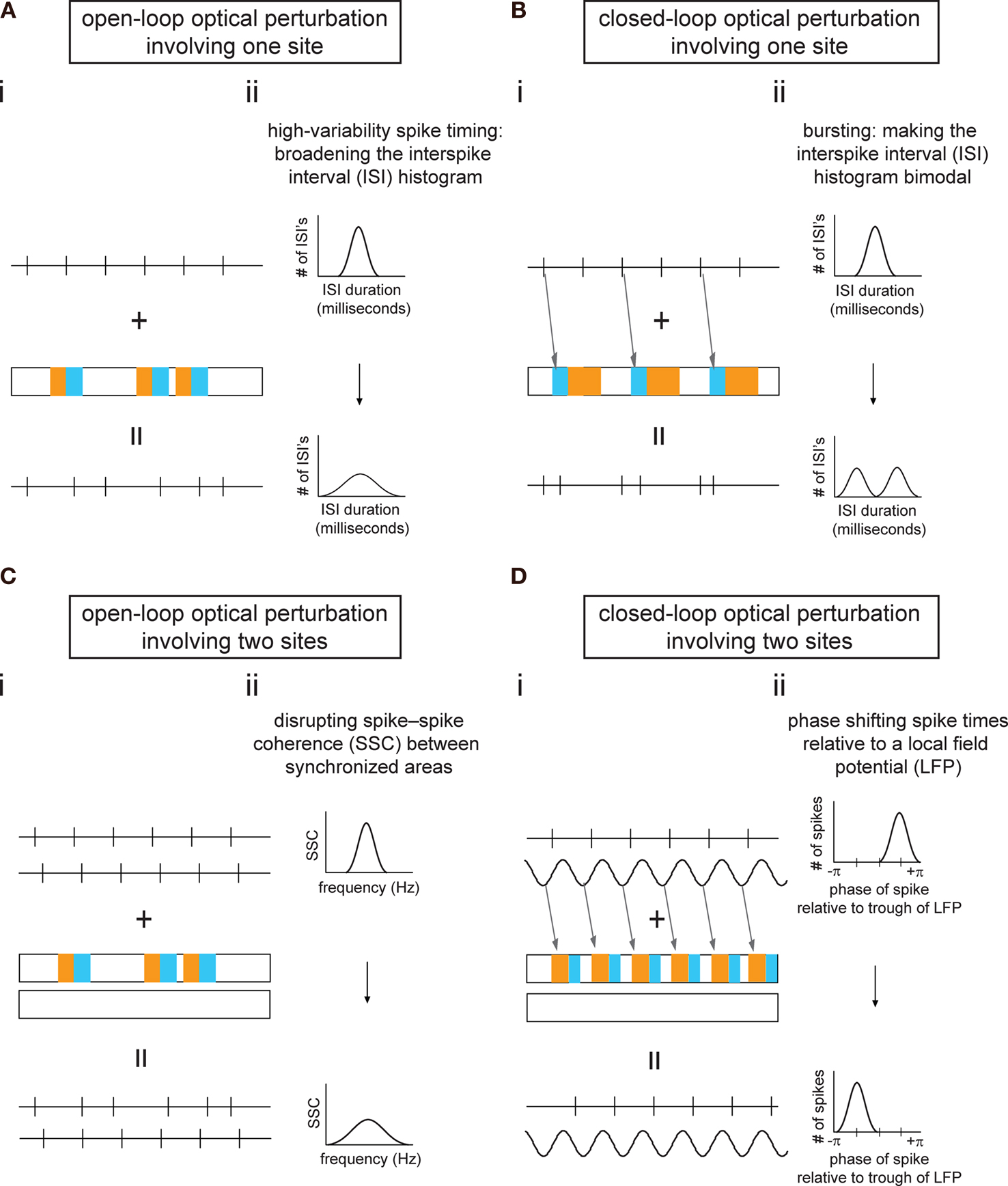
Figure 3. Synchrony- and spike timing-perturbation protocols, enabled by use of ChR2-2A-Halo. Each of the four panels (A–D) displays a schematic experimental protocol (i) along with the anticipated effect of the protocol on neural circuit dynamics (ii). An example is given for each of four kinds of experiment – (A) open-loop/one site, (B) closed-loop/one site, (C) open-loop/two sites, (D) closed-loop/two sites. Detailed descriptions of each panel follow. (A) Shows the use of Poisson train light pulse delivery to a site in which rhythmically-firing neurons express ChR2-2A-Halo. This protocol results in the interspike interval (ISI) becoming more variable than in the unilluminated state. (i) From top to bottom: spike train trace (for one example neuron) in dark; timeline of when yellow or blue light is delivered; spike train trace (for the example neuron) in light (e.g., the result of combining the original case, shown at top, with the delivery of light, shown in the middle). (ii) From top to bottom: ISI histogram in dark; ISI histogram in light. (B) Shows the use of optical perturbation in a closed-loop fashion, triggering blue-yellow pairs of light pulses off of particular recorded spikes in order to make the spike train more bursty. (i) From top to bottom: spike train trace in dark; timeline of when yellow or blue light is delivered (in this case, each blue-yellow pair is triggered by a spike; each trigger is indicated by an arrow); spike train trace under the closed-loop protocol. (ii) From top to bottom: ISI histogram in dark; ISI histogram under the closed-loop protocol. (C) Shows the use of optical perturbation in the style of (Ai), with light delivered to one site to disrupt the timing of activity at that site, relative to another site, which is left unilluminated. (i) From top to bottom: spike train traces recorded at two sites in the dark; timeline of when yellow or blue light is delivered to one of the sites while the other is kept in the dark; spike train traces in light. (ii) From top to bottom: spike-spike coherence (see Fries et al., 2008
for experimental examples of this measure) across the two areas plotted vs. spike frequency, measured in dark; spike-spike coherence across the two areas plotted vs. spike frequency, measured when one site is illuminated. (D) Shows the use of optical perturbation in a closed-loop fashion, triggering yellow-blue pairs of light pulses at one site, off of a feature of the local field potential (LFP) recorded at another site, which is left unilluminated. In this case, the trough of the LFP (bandpass filtered to yield a defined set of frequencies) at the second site serves as the trigger for delaying spikes at the first site. (i) From top to bottom: spike train trace and local field potential trace recorded at two separate sites; timeline of when yellow or blue light is delivered to the site from which spikes are recorded (in this case, each yellow-blue pair is triggered by the trough of the LFP at the second site; each trigger is indicated by an arrow); spike train trace and local field potential trace under the closed-loop protocol. (ii) From top to bottom: phase relationship between spikes recorded at the first site and local field potential troughs recorded at the second site, in the dark (e.g., see Pesaran et al., 2002
; Gregoriou et al., 2009
) for examples of this kind of data); phase relationship between spikes recorded at the first site and local field potential troughs recorded at the second site, under the closed-loop protocol.
One protocol is a direct generalization of the strategy outlined in Figure 1
, the delivery of a light pulse train to a brain region to increase interspike interval (ISI) variability, turning rhythmic neurons into irregularly-firing ones (Figure 3
A). This ‘informational lesion’ might enable the deletion of the information encoded by the precision of spiking of those cells, without eliminating all activity generated by those cells, and thereby causing a gross disruption of connected circuits (as might occur with a conventional lesion of the cells of interest). A slightly more complex version of this idea is illustrated in Figure 3
B, in which light pulses are precisely triggered, in a closed-loop fashion, upon the occurrence of particular recorded spikes, so that the neuronal firing rate becomes more bursty (but, once again, without changing overall spike rates). The ability to test the role of bursting in spike signaling may help resolve how the temporal integration properties of neural circuit elements contribute to neural computations. The 2A strategy can also be used to disrupt correlations between multiple regions that exhibit synchrony (Figure 3
C), making spike timing more variable in one region so that its activity correlation with activity in a second region decreases, or making spike timing more variable in one region to see if such activity causally influences the activity within the second region. In this way it might be possible to understand how regions communicate and process information together in a coordinated fashion. As a final example, the multi-site experiment can also be performed in a closed-loop fashion (Figure 3
D), triggering the delivery of a specific light pulse train to one region, upon a particular phase of the local field potential (LFP) recorded in a second region, and thus phase shifting spike times in the first region with respect to the LFP of the second. Many other possible protocols exist; the key advance enabled by the 2A construct in such protocols is the ability to lesion information or alter information in the brain, without grossly disrupting the spike rate, as happens with traditional lesions, some forms of pharmacology, or normal activation or silencing of neurons.
We here discuss the implications of gene fusions of ChR2 and Halo, linked by the ‘self-cleaving’ 2A peptide, capable of supporting cellular synthesis of both opsins in proportion to one another, towards enabling a set of neurons to be simultaneously sensitized to blue light activation and yellow-light silencing. As an example of computational neuroscience-driven molecular engineering, such reagents may enable novel kinds of perturbation, such as the disruption of spike timing in the absence of altering spike rate. The construct presented here is freely available from the nonprofit service Addgene (reagent ‘ChR2-2A-Halo’ at http://www.addgene.org/Edward_Boyden
) and is ready for use for transfection, gene gunning (Zhang et al., 2008
), transgenic implementation (Wang et al., 2007
), or electroporation (Petreanu et al., 2007
; Lagali et al., 2008
). The current construct, ChR2-2A-Halo yielded, when transfected into neurons, blue- and yellow-light elicited currents that were 30–40% of those obtained when ChR2 or Halo were expressed alone, respectively. Another paper that recently also disclosed the use of a 2A bridge to co-express ChR2 and Halo also showed currents that were smaller than those resulting from expression of individual opsins alone (Tang et al., 2009
). These are still useful currents, especially if the goal is to dither the activity of a neuron above and below spike threshold in order to add and subtract spikes (as opposed to strongly driving or inhibiting neurons); however, a question for any technology is whether the efficacy can be improved further. One solution would be to use stronger opsins to begin with, such as novel molecules like ChIEF (Lin et al., 2009
) and Arch (Chow et al., 2009a
,b
), which can mediate functionally higher currents than ChR2 and Halo, respectively. Other methods for expressing two genes also exist: for example, bi-directional promoters can lead to higher expression levels than can 2A, but at the expense of significantly more variability in the ratio of the protein levels resulting from each gene (e.g., see Fig. 3A in Amendola et al., 2005
), due to the greater chance for noise to creep in downstream of the stoichiometry-determining event.
The prospects for using optical control technologies for ultraprecise neuromodulation therapies in clinical settings are exciting, since precise activation and silencing of specific cell types may increase efficacy and reduce side effects of treatments relative to purely electrical methods. However, it is important to consider not just straightforward optical activation and silencing, but also complex perturbations such as desynchronization as here described, which could enable correction of neural dynamics abnormalities such as those found in neurological and psychiatric disorders like Parkinson’s and epilepsy. As pre-clinical studies of the safety and efficacy of optical control technology begin (Han et al., 2009
), it will be increasingly important to derive principles of neural control, prototyping novel therapies that take full advantage of the computational power of these new technologies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ESB acknowledges funding by the NIH Director’s New Innovator Award (DP2 OD002002-01), as well as by the NSF (0835878 and 0848804), the McGovern Institute Neurotechnology Award Program, the Department of Defense, NARSAD, the Alfred P. Sloan Foundation, the Jerry Burnett Foundation, the SFN Research Award for Innovation in Neuroscience, the MIT Media Lab, the Benesse Foundation, and the Wallace H. Coulter Foundation. XH acknowledges the Helen Hay Whitney Foundation and NIH 1K99MH085944. Thanks to Jennifer Raymond and Bob Desimone for discussions.
Bernstein, J., Ko, E., Strelzoff, A., Chan, S. C. Y., Gidwaney, V., Stickgold, E., Tentori, A. M., (Henninger, M. A., listed as first author since sponsor of poster), McConnell, J., Rodriguez, A., Monahan, P. A., Talei Franzesi, G., Han, X., Qian, X., and Boyden, E. S. (2008a). A scalable toolbox for systematic, cell-specific optical control of entire 3-D neural circuits in the intact mammalian brain. Soc. Neurosci. 493.4, UU25.
Bernstein, J. G., Han, X., Henninger, M. A., Ko, E. Y., Qian, X., Franzesi, G. T., McConnell, J. P., Stern, P., Desimone, R., and Boyden, E. S. (2008b). Prosthetic systems for therapeutic optical activation and silencing of genetically-targeted neurons. Proc. Soc. Photo Opt. Instrum. Eng. 6854, 68540H.
Boyden, E., Franzesi, G. T., Qian, X., Li, M., Han, X., Borgers, C., Kopell, N. J., Le Beau, F., and Whittington, M. A. (2009). Probing mechanisms of gamma rhythmogenesis with cell type-specific optical neural control. Front. Syst. Neurosci. Conference Abstract: Computational and Systems Neuroscience. doi: 10.3389/conf.neuro.3310.2009.3303.3299.
Wang, H., Peca, J., Matsuzaki, M., Matsuzaki, K., Noguchi, J., Qiu, L., Wang, D., Zhang, F., Boyden, E., Deisseroth, K., Kasai, H., Hall, W. C., Feng, G., Augustine, G. J. (2007). High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 104, 8143–8148.
Yu, X., Zhan, X., D’Costa, J., Tanavde, V. M., Ye, Z., Peng, T., Malehorn, M. T., Yang, X., Civin, C. I., and Cheng, L. (2003). Lentiviral vectors with two independent internal promoters transfer high-level expression of multiple transgenes to human hematopoietic stem-progenitor cells. Mol. Ther. 7, 827–838.