1
York College and The Graduate Center, The American Museum of Natural History, The City University of New York, New York, NY, USA
2
Max-Delbrück Center for Molecular Medicine, Berlin, Germany
Neuronal circuits depend on the precise regulation of cell-surface receptors and ion channels. An
ongoing challenge in neuroscience research is deciphering the functional contribution of specific
receptors and ion channels using engineered modulators. A novel strategy, termed “tethered toxins”, was recently developed to characterize neuronal circuits using the evolutionary derived selectivity of venom peptide toxins and endogenous peptide ligands, such as lynx1 prototoxins.Herein, the discovery and engineering of cell-surface tethered peptides is reviewed, with particular attention given to their cell-autonomy, modular composition, and genetic targeting in different model organisms. The relative ease with which tethered peptides can be engineered, coupled with the increasing number of neuroactive venom toxins and ligand peptides being
discovered, imply a multitude of potentially innovative applications for manipulating neuronal circuits and tissue-specifi c cell networks, including treatment of disorders caused by malfunction of receptors and ion channels.
Understanding complex processes such as neuronal activity or cell signaling malfunctions that result in human disorders or diseases relies on extensive knowledge about the function, structure and precision of ion channels, receptors and modulators. As a result of this, ion channels are at present the third biggest target class in drug discovery; yet still remain underexploited as drug targets. Recent reviews describe the increasing interest in peptide venom toxins for the development of drug therapies directed towards ion channels and receptors (Blumenthal and Seibert, 2003
; Phui Yee et al., 2004
; Lynch et al., 2006
; Han et al., 2008
; Twede et al., 2009
). Specific areas in which peptide toxins have demonstrated their potential include Alzheimer’s disease (candoxin) (Nirthanan et al., 2002
), chronic pain (MVIIA) (Miljanich, 2004
) and myasthenic autoimmune response (α-Bgtx) (Drachman, 1981
; Mebs, 1989
). For instance, snake neurotoxins bind to nicotinic acetylcholine receptors (nAChRs) with affinities within the pico and nanomolar range (Chiappinelli, 1991
), which indicates that these would be among the best probes for investigating potential therapeutics that affect nAChR activity. The unique homologies of endogenous lynx1 prototoxins with venom toxins provided a biological scaffold for developing recombinant molecules to selectively modulate ion channels and receptors. Thus, based on the characteristics and mode of action of lynx1 cell-surface modulators, new classes of “tethered toxins” and “tethered ligands” were created as probes to characterize ion channels and receptors (Ibañez-Tallon et al., 2004
; Fortin et al., 2009
; Auer et al., 2009
; Stürzebecher et al., 2009). Tethering peptide toxins or ligands close to their point of activity in the cell plasma membrane provides a new approach to the study of cell networks and neuronal circuits, as it allows selective targeting of specific cell populations, enhances the working concentration of the ligand or blocker peptide, and permits the engineering of a large variety of t-peptides (e.g., including use of fluorescent markers, viral vectors and point mutation variants).
This focused review describes the identification of lynx-1 and related endogenous cell surface modulators, the development of the t-peptide technology, and the application of the t-peptide strategy to basic research, cell-based therapies, and drug discovery.
Cell-surface receptors and ion channels are modulated by a rich variety of peptide neurotransmitters, hormones and ligands, but there are few examples of membrane-anchored modulators in nature. The Ly6 superfamily which includes lynx1-and slurp-1 cholinergic modulators, elapid snake venom toxins, and Ly6 molecules of the immune system, constitutes a unique class of short proteins that are either tethered to the cell surface via a glycosylphosphatidylinositol (GPI) anchor, like lynx1, or secreted as venom toxins (Figure 1
). Members of this superfamily share the characteristic 8–10 cysteine motif that determines their compact three-finger structure. Examples of the GPI-anchor subgroup include molecules of the immune system such as CD59 (Davies et al., 1989
), ly6A-E (Rock et al., 1989
), ly6G (Mallya et al., 2006
), and ly6K (de Nooij-van Dalen et al., 2003
), the neuronal proteins lypd6 (Darvas et al., 2009
), ly6H (Dessaud et al., 2006
) and the urokinase plasminogen activator receptor (uPAR) (Blasi and Carmeliet, 2002
). Members lacking the GPI-anchor comprise the cholinergic modulators SLURP-1 (Adermann et al., 1999
; Chimienti et al., 2003
) and SLURP-2 (Tsuji et al., 2003
), cobra toxins, and other three finger venom toxins (Tsetlin, 1999
) (Figure 1
).
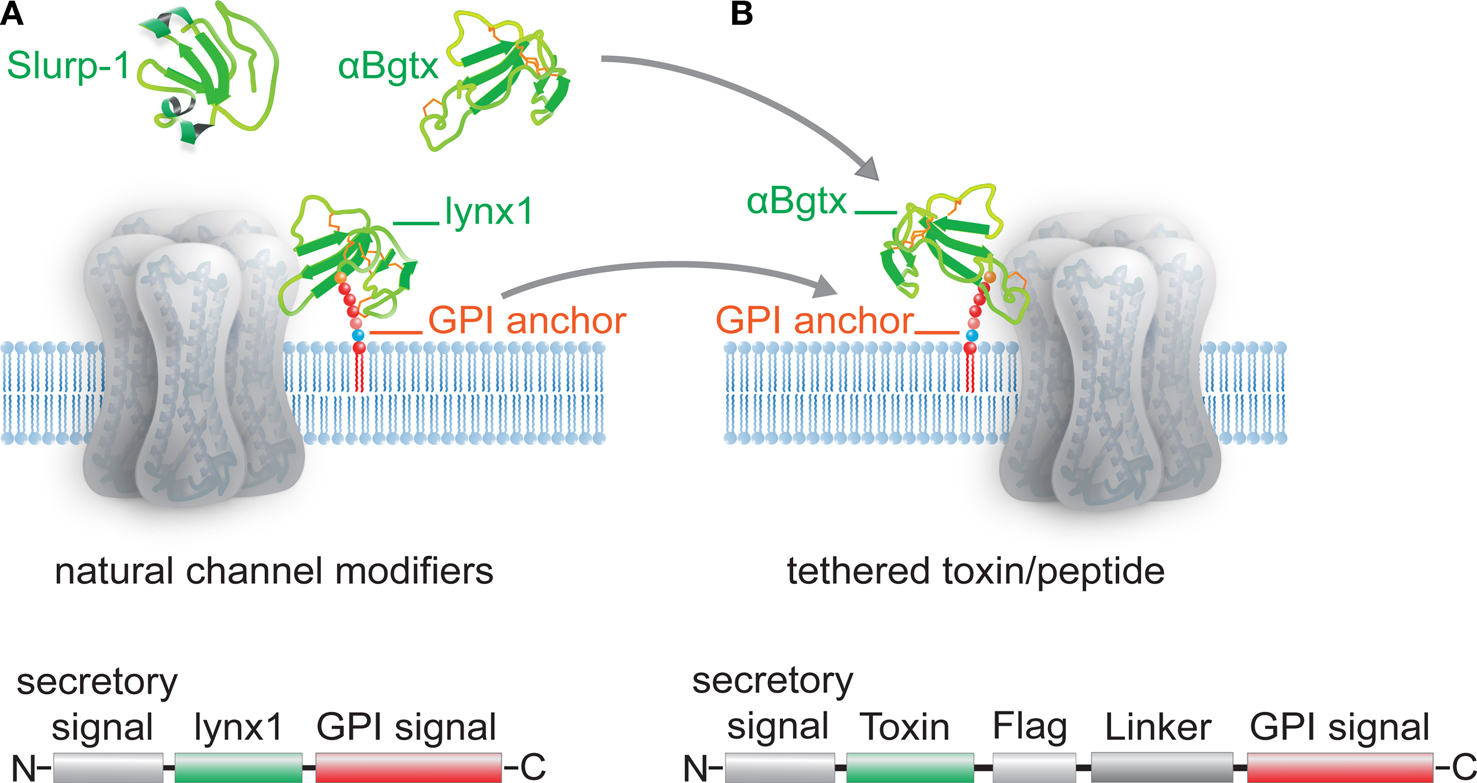
Figure 1. Schematic representation of Ly-6/uPAR channel modifiers and engineered tethered toxins (t-toxins). (A) Examples of the Ly-6/uPAR superfamily include soluble Slurp-1, snake α-bungarotoxin (αBgtx) and the GPI-anchored cell-membrane bound lynx1. The schematic below the drawing of the channel indicates the coding sequences associated with lynx1, namely an N-terminal secretory signal region, followed by the amino acid residues that correspond to the lynx1 peptide, and a C-terminal GPI anchor. (B) The structural homology of lynx1 with αBgtx gave rise to the tethered-peptide strategy of using the biological scaffold of lynx1 (secretory signal and GPI signal) to generate recombinant membrane-bound toxins and peptide ligands such as the illustrated t-αBgtx. The schematic below the drawing of the channel indicates the coding regions that were conserved from lynx1 (shown in A), and those that were altered to accommodate the αBgtx (the toxins sequence, flag tag, and linker regions).
Allosteric Modulation of nAChRs by Ly6 Superfamily Molecules
Lynx1 was the first identified member of the Ly6 superfamily capable of cell-surface modulatory action on a neurotransmitter receptor (Miwa et al., 1999
). Lynx1 assembles and colocalizes with nAChR in the brain (Ibañez-Tallon et al., 2002
) and in the lung (Sekhon et al., 2005
). nAChRs stably associated with lynx1 are less sensitive to their ligand agonists acetylcholine and nicotine, display more rapid desensitization, and show a shift in the distribution of channel openings toward a faster inactivating species with more uniform, larger amplitude currents (Ibañez-Tallon et al., 2002
). These findings, along with studies showing enhanced nicotine-mediated calcium influx and synaptic efficacy in lynx1 null mutant mice, are strong indicators that lynx1 is an allosteric modulator of nAChR function in vivo (Miwa et al., 2006
). Other lynx-like molecules recently identified have similar properties. Lynx2 and ly6H, which are GPI-anchored to the cell membrane, have been localized to central and peripheral neurons in mice (Dessaud et al., 2006
). Functional studies demonstrate that lynx2, but not ly6H, changes the agonist sensitivity and desensitization properties of nAChRs through direct association (Tekinay et al., 2009
). Consistent with lynx2 enrichment in neurons that participate in circuits controlling fear and anxiety, lynx2 null mice display increased anxiety-like behaviors due to enhanced nAChR activity (Tekinay et al., 2009
). Recently, a third allosteric modulator of nAChRs, named lypd6, has been identified in central neurons. Lypd6 is cell-membrane bound and selectively increases the calcium conductance of nAChRs (Darvas et al., 2009
). The lynx1-related secreted ligands SLURP-1 and SLURP-2, also act as neuromodulators of nAChRs and have been linked to skin disorders (Chimienti et al., 2003
; Arredondo et al., 2006
).
Ly6 Species Diversity
Lynx1-like molecules are well conserved across species, both in structure and function, suggesting the importance of cell-surface modulators of nicotinic receptors in nature. Examples of Ly6 superfamily species diversity include molecules found in C. elegans (Odr-2; Chou et al., 2001
), fireflies (Pr-lynx1; Choo et al., 2008
), Drosophila (Hijazi et al., 2009
) and chicken (recently identified prostate stem cell antigen PSCA; Hruska et al., 2009
). Pr-lynx1 and PSCA are of particular importance as Pr-lynx1 is the first modulator of nAChRs in an insect species (Choo et al., 2008
), and PSCA appears to prevent programmed cell death of neurons by antagonizing nAChRs (Hruska et al., 2009
). The lynx1-like family of allosteric modulators of nAChRs constitutes a unique example of cell-surface channel modifiers that have evolved for fine-tuning of neurotransmitter receptor function in vivo.
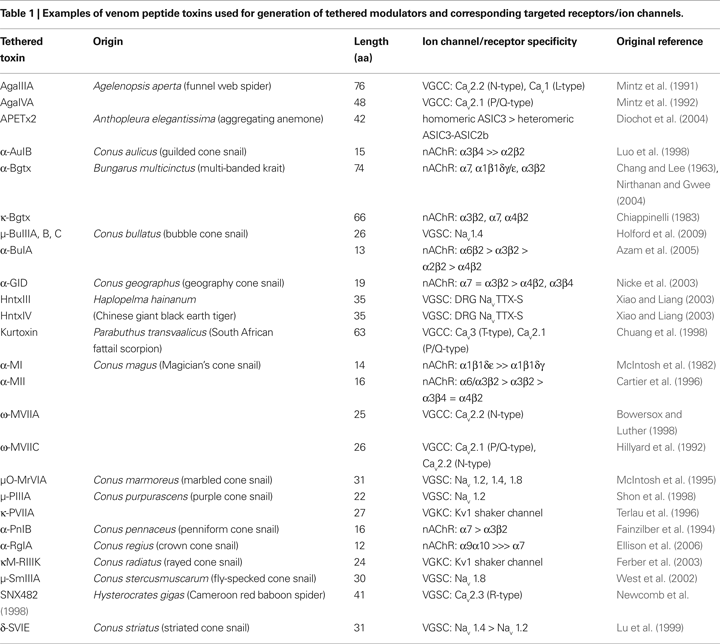
Venom peptide toxins from predatory organisms such as scorpions, snakes and marine cone snails are suitable as agents for use in engineering cell-type specific modulators. Neurotoxins isolated from animal venom are disulfide-rich peptides that have been used extensively to characterize the structure–activity relationships of specific ion channels (ligand or voltage-gated) and cell-surface receptors (Catterall, 1986
; Tsetlin, 1999
; Norton and Olivera, 2006
). α-bungarotoxin and other α-neurotoxins from Elapidae and Hydrophidae snakes were used to provide the first identification of nAChRs (Endo and Tamiya, 1991
; Hucho et al., 1996
; Léna and Changeux, 1998
). Similarly, α- and β-neurotoxins from scorpions have been broadly used to modify voltage-gated sodium channels (VGSC or Navs) by delaying inactivation (α) or shifting the membrane potential dependence (β) (Zuo and Ji, 2004
; Bosmans and Tytgat, 2007
). Neuropeptides from venomous marine cone snails (conotoxins) are prolific in their range and specificity for targeting various ion channels and receptors (Terlau and Olivera, 2004
; Gowd et al., 2008
; Han et al., 2009
). As shown in Table 1
, venom peptide toxins modulate a wide range of molecular targets. Peptide toxins vary in length from 12–30 residues in cone snails to 40–80 residues in toxins from scorpions and snakes. The relatively small size of these polypeptides, coupled with their structural integrity imposed by numerous disulfide bonds, have facilitated their use for investigating ion channels and receptors (Fontecilla-Camps et al., 1988
; Pashkov et al., 1988
; Tsetlin, 1999
; Zhang et al., 2006
).
Peptide Toxins Specific for nAChR and Voltage-Gated Ion Channels
α-Neuropeptides, which target nAChRs compete with cholinergic agonists and antagonists (Endo and Tamiya, 1991
) and are characterized by four to five disulfide bridges in snake and scorpion venoms (Possani et al., 1999
; Phui Yee et al., 2004
) or two to three disulfide bridges in cone snail toxins (Sine et al., 1995
; McIntosh et al., 1999
; Ellison et al., 2006
). α-neurotoxins that target nAChRs include α-Bgtx, MII and BuIA (Table 1
), while other α-neurotoxins slow the sodium current inactivation in excitable membranes (Couraud et al., 1982
). δ-Conotoxins, such as δ-SVIE, which bind to Navs also cause a delayed inactivation of sodium currents (Bulaj et al., 2001
). They share a cysteine framework with the structure C-C-CC-C-C, that is identical to that of μO-conotoxins, like MrVIA and MrVIB which inhibit Nav1.2, Nav1.4 and Nav1.8 (Terlau et al., 1996
; Daly et al., 2004
; Bulaj et al., 2006
). μ-Conotoxins contain a framework of CC-C-C-CC and are by far the best characterized of all the conotoxins that target Navs. Several μ-conotoxins such as PIIIA, SmIIIA, and potentially BuIIIA, inhibit Navs in a similar manner to tetrodotoxin (TTX), by binding to site I on the channel (Catterall, 2000
; Goldin, 2001
). Despite the ever growing number of discovered natural toxins, only a few κ-conotoxins have been characterized so far. κ-conotoxins bind to voltage-dependent potassium channels (VGKC or Kv) altering either the repolarization phase of action potentials or the resting membrane potential. The most commonly identified κ-conotoxin is PVIIA, which blocks Shaker K channels cloned from Drosophila (Naranjo, 2002
). Other gating modifiers of Kv channels are the phrixotoxins (PaurTX) I and II (Bosmans et al., 2006
). Blockers of voltage-gated calcium channels (VGCC or Cav) include agatoxin IIIA and IVA. The most prominent VGCC toxin antagonist is conotoxin MVIIA, a highly specific blocker of N-type calcium channels which has been developed as an analgesic for the treatment of chronic pain (Miljanich, 2004
; Staats et al., 2004
).
The high degree of specificity with which venom peptide toxins bind to voltage and ligand gated sodium (Nav), calcium (Cav), and potassium (Kv) ion channels, and receptors such as nAChRs, N-methyl-D-aspartate and G-protein coupled receptors (GPCRs), make them ideal for deciphering the connections between cell types, and for reversibly manipulating the activity of selective cell subtypes.
The unique functionality of endogenous lynx1-like cell-surface modulators provides a framework to use peptide toxins from predatory animals to manipulate ion channels and receptors in a novel manner. Peptide toxins that have been routinely employed for neuroscience research do not normally exist as cell-surface anchored molecules. Using the scaffold of the lynx1-like gene family, i.e., secretory signal and consensus sequences for GPI processing and recognition, it is possible to produce a series of tethered toxins (t-toxins) that are highly effective modifiers of neuronal activity (Ibañez-Tallon et al., 2004
). This design directs any bioactive peptide to the secretory pathway, where the signal sequence is cleaved and the GPI targeting sequence is substituted by a covalent bond to GPI, anchoring the peptide to the extracellular side of the plasma membrane of the cell in which it is expressed (Figure 1
B). Recombinant t-peptides have a modular framework consisting of a secretion signal, linkers, epitopes and/or fluorescent markers and membrane tethers (Figures 1
B and 2
A). The GPI-anchor targeting motif can be replaced with transmembrane (TM) domains fused with fluorescent marker genes (Figures 2
A,B) (Auer et al., 2009). So far, approximately 40 different chimeric t-toxins derived from the venom of several predatory animals have been cloned and their activity has been characterized on voltage and ligand-gated ion channels (Table 1
) (Ibañez-Tallon et al., 2004
; Hruska et al., 2007
; Wu et al., 2008
; Stürzebecher et al., 2009; Auer et al., 2009). Furthermore, the t-peptide strategy has also been successfully extended to other bioactive peptides, such as ligand peptides for constitutive activation of GPCRs (Choi et al., 2009
; Fortin et al., 2009
), illustrating the general applicability of this approach for cell-surface modulation of receptors.
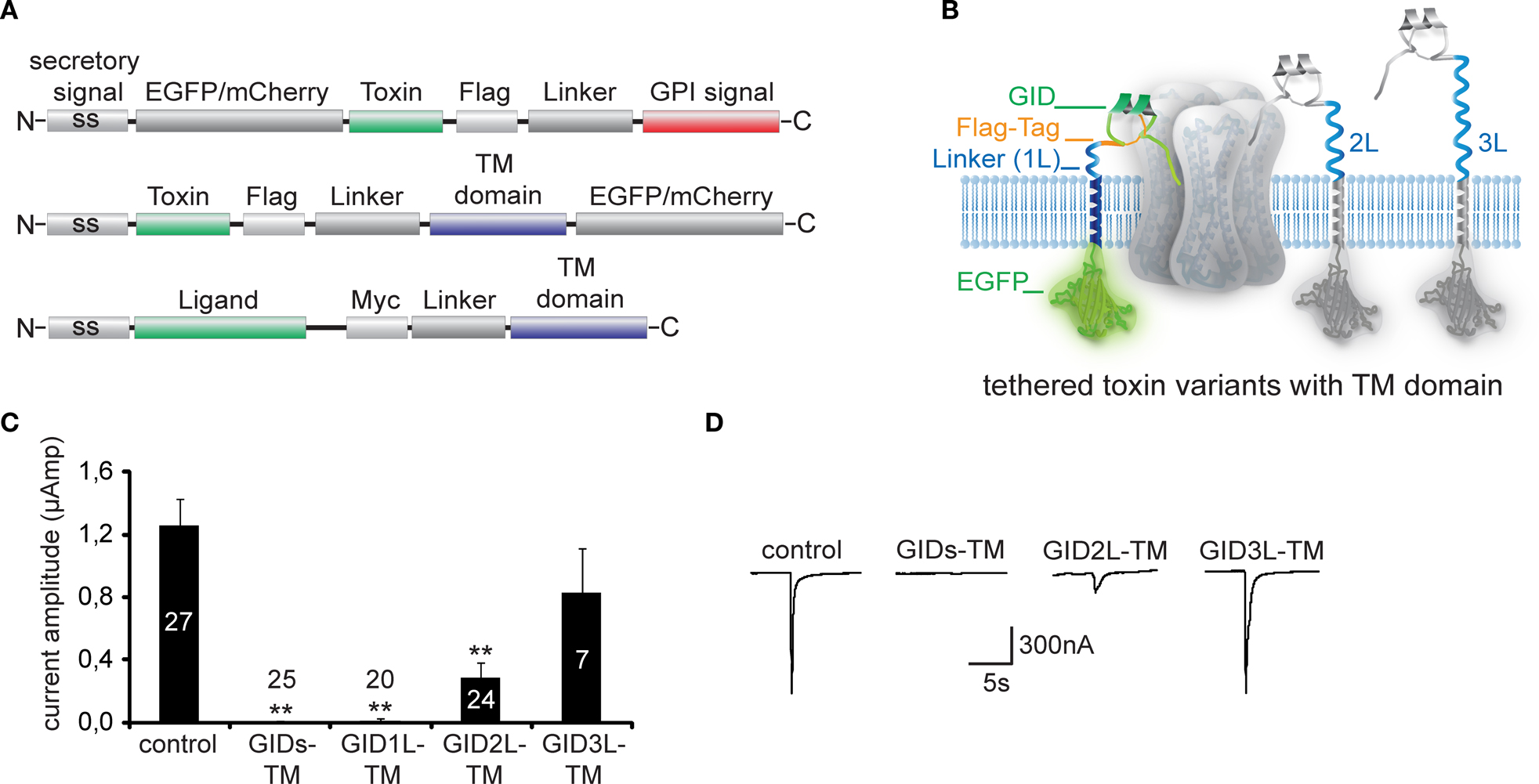
Figure 2. Modular architecture of membrane-tethered toxin and ligand peptides. (A) Illustration of tethered-peptide variants consisting of secretory pathway signal sequence (ss), toxin/ligand cassettes, fluorescence markers (EGFP or mCherry), epitopes for immunostaining (Flag-tag, Myc-tag), flexible linker regions, and distinct functional modules for membrane attachment (GPI-signal, transmembrane-domain TM). (B) Illustration of t-toxin carrying the nAChR-specific snail-toxin GID with varying linker lengths [short (s), 1L, 2L and 3L]. (C) Electrophysiological recordings in Xenopus laevis oocytes expressing α7 nAChR alone (control) or together with tethered-GID (t-GID) with increasing linker lengths. t-GID expression results in complete block of nicotine-induced α7 nAChR current for short (6 aa, GIDs-TM) and long (20 aa, GID1L-TM) linker variants, while longer linkers (GID2L-TM and GID3L-TM, 40 aa and 60 aa) lead to decreased blocking capability. Number of recorded cells displayed in/above columns. (D) Representative traces of electrophysiological recordings in (C) suggest an optimal distance of the GID peptide from the plasma membrane of 9–22 aa to achieve complete inhibition of α7 nAChR.
Components Critical to T-Peptide Synthesis
The biological activity of each tethered construct has to be individually evaluated and optimized per receptor/channel combination. While anyone with knowledge of cloning technology can construct a tethered-peptide, some forethought is required depending on how the t-peptide will be implemented. This is due to several factors including variability in the nature of the bioactive peptide and ion channel or receptor being targeted, and in the constraints imposed by the modular architecture of the tethers, linkers and epitope-tags. The vast amount of modular choices available for constructing tethered toxins point to its broad appeal and indicate the feasibility of tailor-made tethered manipulators for a wide range of different receptors/channels.
Amino acid composition of the bioactive peptide
Expression and functional assays have revealed that several elements are critical to achieve robust expression on the cell-surface and rotational flexibility for correct modulation of the t-toxin/peptide to the receptor or channel of interest. The affinity of the bioactive peptide for its cognate ion channel or receptor has to be taken into account. Toxins with a strong affinity are potentially more effective. It is also important to consider the composition and length of the peptides to be tethered, i.e., charges and hydrophobicity of the amino acid residues, number of cysteine bonds in the case of toxins, and existence of non-canonical residues, or terminal amidations. For example, substitution of hydroxylated or carboxylated amino acids with non-modified residues in highly post-translationally modified conotoxins, such as GVIA (Olivera et al., 1984
), RIIIK (Ferber et al., 2003
), PIIIA (Shon et al., 1998
) and GID (Nicke et al., 2003
), failed to yield satisfactory activity in their tethered constructs, except in the case of GID (see Figures 2
C,D). Most likely these variations in efficacy are dependent on the location of the posttranslational modification in the toxin sequence. In general, the makeup of the peptide or toxin to be tethered has to be taken into account, but it is not predictive of the expression and activity of its tethered form. MrVIA and MrVIB, which despite their high hydrophobicity content and difficulties to be chemically synthesized (Terlau et al., 1996
; Bulaj et al., 2006
), are well expressed at the cell-membrane, and functionally active when tethered (Ibañez-Tallon et al., 2004
; Wu et al., 2008
; Stürzebecher et al., 2009). Other examples are the tethered forms of α- and κ-bungarotoxins which were well expressed and functionally competent (Ibañez-Tallon et al., 2004
; Hruska et al., 2007
) despite being long peptides (68–82 aa), while shorter conotoxins such as SmIIIA (30aa) or MVIIC (26aa) were not heterologously expressed, possibly due to folding disturbances or high proportion of charged amino acids.
Distance of the linker region
Another relevant feature when designing t-peptide constructs is the linker sequence bridging the toxin peptide to the GPI anchor or TM domain (Figures 2
A,B). The distance of the t-toxin or t-peptide from the cell-surface has to be tailor-made for individual receptors and channels, and can be used for mapping active binding sites. Tethered constructs have been cloned using linkers consisting of glycine–asparagine repeats with lengths varying from 6 amino acids (short) to 20 amino acids (long), 40aa (2× long) or 60 aa (3× long) (Figure 2
B) (Ibañez-Tallon et al., 2004
). The longer flexible linker provides rotational freedom for the t-toxin to bind within the vestibule of voltage-gated channels (Ibañez-Tallon et al., 2004
), or for ligand peptides to reach their binding site, such as onto class B1 GPCRs (Fortin et al., 2009
). Experiments varying the length of the linker region of t-GID conotoxin indicate that a linker is necessary for inactivation of α7 nAChR currents. However, when the linker exceeds a certain length the inactivation is incomplete (Figures 2
B–D). Similarly, the tethered form of the neuropeptide pigment dispersing factor (t-PDF) requires a short linker for effective binding to its receptor (Choi et al., 2009
).
Choice of membrane tether: GPI vs. TM
The choice of membrane tether depends on the characteristics of the peptide as well as on the epitope-tags and markers to be used in combination (Figures 1
B and 2
A,B). GPI anchors, which are less bulky than TM domains, may facilitate the mobility of the t-peptide in close proximity to its receptor within the plasma membrane. If the toxin or peptide does not require a free N-terminus for interacting with its cognate receptor, GPI versions containing EGFP followed by the t-toxin may be used (Figure 2
). However, GPI anchors are susceptible to cleavage by endogenous phospholipases, such as PI-PLC and phospholipase D (Paulick and Bertozzi, 2008
). This has been suggested as a mechanism used by cells for selective and rapid release of certain GPI-anchored proteins at specific times. To avoid this potential problem, the GPI anchor can be replaced with a TM domain in t-toxins (Figure 2
). TM domains can be used to retain t-peptides at the cell-surface and link fluorescent markers to the cytoplasmatic side of the plasma membrane avoiding hindrances between them.
Tethered toxins and peptides can be used for very diverse applications pertaining to experimental animal physiology. Because of their mode of action at the cell-surface, membrane-anchored peptide molecules act only on ion channels and receptors present in the membrane of the cell that is expressing the t-toxin or t-peptide, and not on identical receptors present on neighboring cells that do not express the tethered construct (Ibañez-Tallon et al., 2004
). Several studies have shown that recombinant toxins as well as peptide ligands are not dispersed in solution and retain their high specificity for their cognate receptors, indicating that this approach can be used to restrict the site of neurotoxin or peptide ligand action to genetically targeted cells. For example, in vivo transgenic delivery of t-αBgtx in zebrafish using a muscle cell-specific promoter results in blockade of nAChR currents in muscle cells that express t-αBgtx but not in adjacent muscle fibers or in cells that express t-κBgtx, which has no activity on muscle-nAChRs (Ibañez-Tallon et al., 2004
). Similarly, experiments in chicken employing a viral system to transduce ciliary neurons have revealed that expression of t-αBgtx blocks calcium currents via nAChRs and prevents programmed cell-death of these neurons during early development (Hruska et al., 2007
). Further in vivo applications of the tethered toxin strategy have been carried out using transgenesis in Drosophila melanogaster. These studies have shown that cell-specific expression of the sodium channel toxin δ-ACTX-Hv1a in pacemaker clock neurons induces arrhythmicity (Wu et al., 2008
). Transgenic targeting of the same clock neurons with a tethered form of the PDF neuropeptide, which is normally rhythmically secreted by these neurons, constitutively activates its cognate GPCR, interfering with circadian control circuit (Choi et al., 2009
). These examples illustrate the possibilities for cell-specific targeting and cell-autonomous regulation of channels and receptors with genetically encoded tethered toxins. As venom toxins are established tools for dissecting ionic currents in many animal species, the tethered-toxin strategy allows cell specific functional analysis of ion channels and receptors in model organisms (e.g., zebrafish, flies, rats, large mammals) for which transgenic methodologies are commonly used but gene targeting strategies are yet not available.
Applications and Perspective Use of Cell-Surface Peptides to Basic Research, Cell-Based Therapies and Drug Discovery
Ion channels and receptors are involved in every physiological action from breathing to heart beating. Understanding the mechanics and functional activity of these macromolecular complexes is a grand challenge in science. The tethered-peptide method is one tool that has the potential to tackle certain aspects of this challenge, particularly in the area of cell-specific modulators (Figure 3
). Genetically encoded cell-surface modulators can be adapted to a wide range of applications due to their small size, amenability to point mutagenesis, and relative ease to be combined with fluorescent markers, viral and transgenic vectors, Cre-dependent and transcriptional-control elements, and subcellular targeting motifs. Inhibition or constitutive activation of ion channels and receptors can be attained in a cell-type specific manner depending on the selectivity of the neuroactive peptide or hormone.
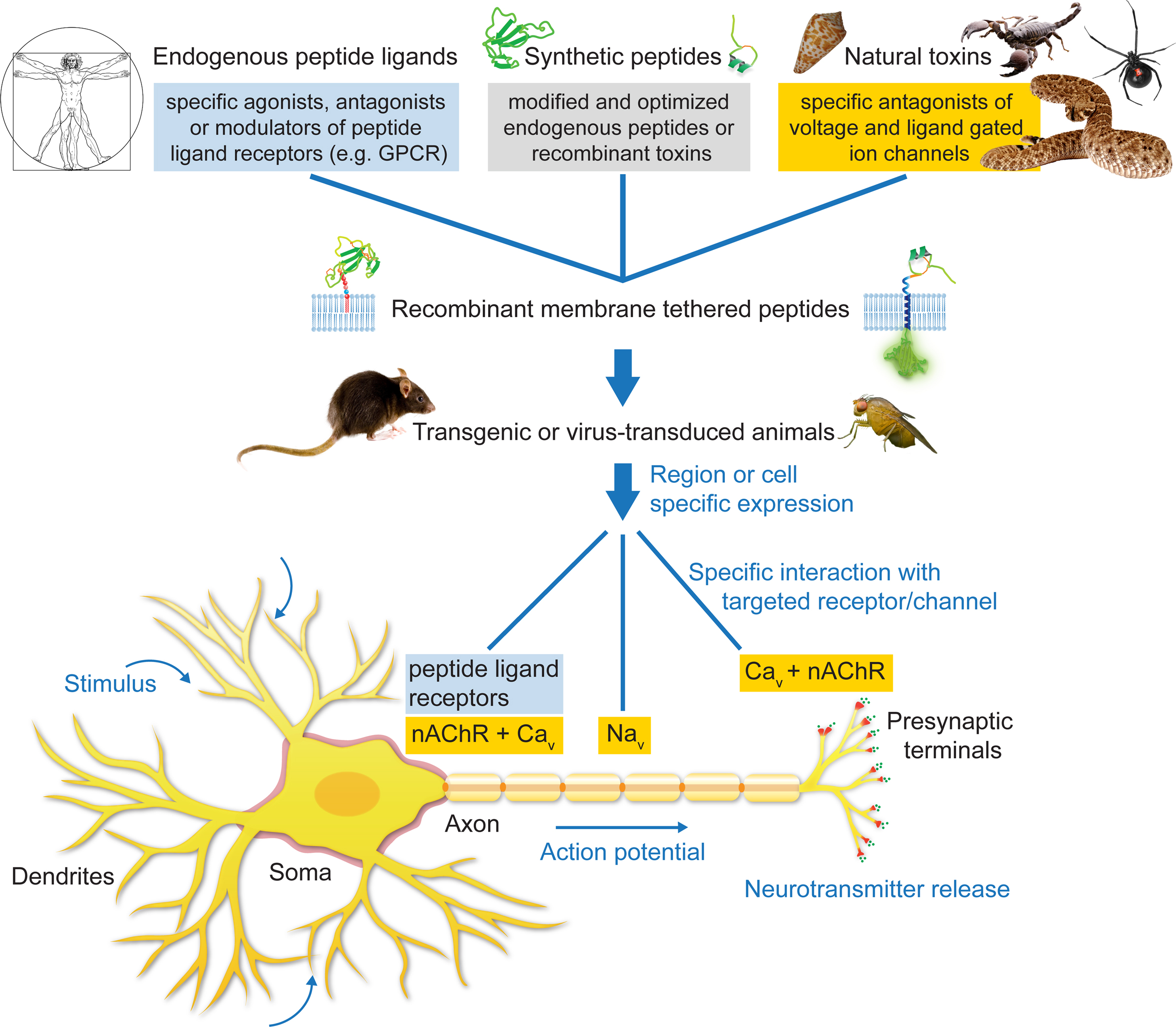
Figure 3. Applications of the tethered-peptide strategy. Endogenous peptide ligands, natural toxins, and synthetic, modified versions of ligands or toxins can be integrated into recombinant membrane-attached fusion constructs and applied in vitro in transfected or transduced cells in cell-culture, or in vivo in transgenic or virus-transduced animals. The t-peptide retains the specificity of the toxin/peptide ligand allowing controlled manipulation of distinct subtypes of ion channels and receptors in a given neuronal circuit without affecting other channels/receptors in the cell.
Neuronal Circuits: Dissecting Individual Ionic Currents
Manipulation of ion channels in specific neuronal populations within living animals can be achieved by transgenesis (Ibanez-Tallon et al., 2004
; Wu et al., 2008
). Similarly, viral methods can be employed to characterize the cellular functions mediated by specific ionic currents inactivated by delivery of t-toxins (Hruska et al., 2007
). Gene knockouts led to a wide range of studies to characterize the function of receptors and ion channels (Capecchi, 2005
). However, many receptors consist of multimeric assemblies of components, with common components frequently shared by functionally diverse receptor types, modifying any one gene can potentially compromise the function of every complex with which it is associated. For example, it has proven difficult to separate the contribution of Cav2.1 and Cav2.2 VGCC channels by targeted deletion of one or the other alpha subunits because of functional compensation of ionic currents (Inchauspe et al., 2004
; Takahashi et al., 2004
). As venom toxins inactivate specific ionic currents produced by a receptor or channel complex, the t-toxin strategy could be an alternative to prevent compensatory ionic currents that may occur upon gene-deletion of receptor subunits.
The cell-autonomous modulatory action of tethered peptides and their selectivity for cognate cell-surface molecules can be further exploited by directing t-peptide molecules to subcellular compartments within the neuron (Figure 3
). For instance, t-toxins could be directed to the axon initial segment where sodium channels are concentrated (Garrido et al., 2003
), or to the dendritic compartment (Lewis et al., 2009
). Further modifications of a number of optimized and novel tethered toxins for in vivo use will offer new possibilities for investigations regarding the physiology of neuronal circuits.
Channelopathies and Other Diseases
The tethered-peptide strategy represents a potential new avenue for the development of genetic therapies for chronic diseases caused by malfunction of ion channels and peptide ligand receptors. Several human disorders that affect nervous system functions have been traced to mutations in genes encoding ion channels or regulatory proteins (George, 2005
). These disorders, referred to as channelopathies, can be targeted by the tethered-peptide strategy when the disorder results in hyperactivity of the channel. Examples of hyperactive disorders include gain-of-function mutations in P/Q-type calcium channels, linked to familial hemiplegic migraine type 1 (Ophoff et al., 1996
; Tottene et al., 2009
), or mutations in neuronal nAChRs associated to autosomal dominant nocturnal frontal lobe epilepsy (Steinlein et al., 1995
; Klaassen et al., 2006
). One potential application would be to genetically introduce t-toxins into the corresponding mouse mutant models in a cell-specific manner to dissect the circuitry of the disease (Figure 3
). Conversely, activation of receptors with t-peptide ligands could be beneficial to control GPCRs in a cell-selective manner (Figure 3
). For instance, isoforms of glucagon-like and calcitonin-gene-related peptides are presently being used to regulate insulin release and bone remodeling in diabetes (Green and Flatt, 2007
) and osteoporosis (Hoare, 2005
). Similarly, feeding-regulation neuropeptides such as orexin or ghrelin (Shioda et al., 2008
) could be targeted to circuits involved in appetite control, or tethered opioid peptides could be directed to nociceptive neurons. With an ever-growing interest in identifying the potential of naturally occurring venom peptide toxins (Blumenthal and Seibert, 2003
; Han et al., 2008
; Twede et al., 2009
), as well as novel ligands for orphan GPCRs encrypted in the human proteome (Jiang and Zhou, 2006
; Shemesh et al., 2008
), an increasing number of peptide based therapies could be possible. Furthermore, parallel development on the safety of viral methods for genetic intervention will increase the number of diseases to which the t-peptide strategy is applicable.
Implications of Tethered Peptides for Drug Discovery
Ion channels and GPCRs are some of the biggest molecular drug targets yet presently remain underexploited in drug discovery efforts. Peptide toxins, which are highly effective modulators of ion channels and GPCRs, offer an intriguing opportunity for increasing the drug development pipeline. Specific areas in which peptide toxins have demonstrated their potential include chronic pain (Miljanich, 2004
) and myasthenic autoimmune response (Drachman, 1981
). A major drawback to the universal usage of peptide toxins in the development of therapeutics has been the scarcity of obtaining the venom product. To circumvent this, most toxins are synthesized chemically, but this too has significant problems, one being obtaining the correct disulfide scaffold with in vitro folding. To combat these synthesis hurdles several structural strategies and characterization methods have been developed (Munson and Barany, 1993
; Cuthbertson and Indrevoll, 2000
; Han et al., 2009
; Ueberheide et al., 2009
; Walewska et al., 2009
). However, even when the toxin is successfully synthesized, soluble toxins cannot be directed to single cell populations, are expensive, and have a limited time of application that makes their use in vivo problematic. The t-peptide strategy surmounts these limitations with the ability to recombinantly synthesize the toxins or peptide ligands in the cell itself, and co-express it with the molecular target (receptor or channel) to be screened. Such a cell-surface peptide tethering strategy can readily introduce point mutations to interconvert tethered agonists into antagonists. Several recent reports use the t-peptide technology to characterize point mutants of peptide hormones against class B1 GPCRs (Ibañez-Tallon et al., 2004
; Fortin et al., 2009
). In a similar manner, the t-peptide technology could be applied to screen gene libraries of t-peptides against specific membrane proteins by co-expression in the same cell. T-peptides with activating or blocking capabilities could be monitored with functional assays, i.e., calcium influx. This type of screen could be beneficial to block channels that are hyperactive in certain diseases, such as TRPP2, for which no natural toxins have yet been identified. These features make the t-peptide genetic approach a promising strategy for drug discovery and development of targeted therapeutics.
The t-peptide strategy is an innovative technique for manipulating neuronal circuits in order to dissect the specific biological roles of ion channels and cell-surface receptors both in vitro and in vivo. The studies presented here illustrate how the t-peptide approach can be used to increase cellular specificity of neuropeptides by restricting their actions to targeted cell types through membrane tethering. The t-peptide method is relatively easy to implement and has the potential to significantly impact neuroscience research and cell-based drug screening of membrane proteins for targeted therapeutics.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Helmholtz Gemeinschaft, and SFB 665. Mandë Holford acknowledges support from the Chemistry Division and Office of International Science and Engineering at the NSF (CHE 0610202).
Bulaj, G., Zhang, M., Green, B. R., Fiedler, B., Layer, R. T., Wei, S., Nielsen, J. S., Low, S. J., Klein, B. D., Wagstaff, J. D., Chicoine, L., Harty, T. P., Terlau, H., Yoshikami, D., and Olivera, B. M. (2006). Synthetic μO-conotoxin MrVIB blocks TTX-resistant sodium channel NaV1.8 and has a long-lasting analgesic activity. Biochemistry 45, 7404–7414.
de Nooij-van Dalen, A. G., van Dongen, G. A. M. S., Smeets, S. J., Nieuwenhuis, E. J. C., Stigter-van Walsum, M., Snow, G. B., and Brakenhoff, R. H. (2003). Characterization of the human Ly-6 antigens, the newly annotated member Ly-6K included, as molecular markers for head-and-neck squamous cell carcinoma. Int. J. Cancer 103, 768–774.
Han, T. S., Zhang, M., Walewska, A., Gruszczynski, P., Robertson, C. R., Cheatham, T. E., Yoshikami, D., Olivera, B. M., and Bulaj, G. (2009). Structurally minimized mu-conotoxin analogues as sodium channel blockers: implications for designing conopeptide-based therapeutics. Chem. Med. Chem. 4, 406–414.
Hruska, M., Keefe, J., Wert, D., Tekinay, A., Hulce, J., Ibanez-Tallon, I., and Nishi, R. (2009). Prostate stem cell antigen is an endogenous lynx1-like prototoxin that antagonizes alpha7 containing nicotinic receptors and prevents programmed cell death of parasympathetic neurons. J. Neurosci. (In press).
Newcomb, R., Szoke, B., Palma, A., Wang, G., Chen, X. H., Hopkins, W., Cong, R., Miller, J., Urge, L., Tarczy-Hornoch, K., Loo, J. A., Dooley, D. J., Nadasdi, L., Tsien, R. W., Lemos, J., and Miljanic, G. (1998). Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry 37, 15353–15362.
Nirthanan, S., Charpantier, E., Gopalakrishnakone, P., Gwee, M. C. E., Khoo, H., Cheah, L., Bertrand, D., and Kini, R. M. (2002). Candoxin, a novel toxin from Bungarus candidus, is a reversible antagonist of muscle (alphabetagammadelta) but a poorly reversible antagonist of neuronal alpha 7 nicotinic acetylcholine receptors. J. Biol. Chem. 277, 17811–17820.
Ophoff, R. A., Terwindt, G. M., Vergouwe, M. N., van Eijk, R., Oefner, P. J., Hoffman, S. M., Lamerdin, J. E., Mohrenweiser, H. W., Bulman, D. E., Ferrari, M., Haan, J., Lindhout, D., van Ommen, G. J., Hofker, M. H., Ferrari, M. D., and Frants, R. R. (1996). Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552.
Staats, P. S., Yearwood, T., Charapata, S. G., Presley, R. W., Wallace, M. S., Byas-Smith, M., Fisher, R., Bryce, D. A., Mangieri, E. A., Luther, R. R., Mayo, M., McGuire, D., and Ellis, D. (2004). Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial. J. Am. Med. Assoc. 291, 63–70.
Steinlein, O. K., Mulley, J. C., Propping, P., Wallace, R. H., Phillips, H. A., Sutherland, G. R., Scheffer, I. E., and Berkovic, S. F. (1995). A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 11, 201–203.
Tottene, A., Conti, R., Fabbro, A., Vecchia, D., Shapovalova, M., Santello, M., van den Maagdenberg, A. M. J. M., Ferrari, M. D., and Pietrobon, D. (2009). Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron 61, 762–773.