1
Biologie Cellulaire de la Synapse Normale et Pathologique, Institut National de la Santé et de la Recherche Médicale, Ecole Normale Supérieure, Paris, France
2
Institute of Neuroscience, Newcastle University, Newcastle upon Tyne, UK
3
The Wellcome Trust CR UK Gurdon Institute, University of Cambridge, Cambridge, UK
4
Laboratory of Molecular Neuro-Oncology, Howard Hughes Medical Institute, The Rockefeller University, New York, NY, USA
5
Medical Research Council Laboratory of Molecular Biology, Cambridge, UK
Nova proteins are neuron-specific RNA binding proteins targeted by autoantibodies in a disorder manifest by failure of motor inhibition, and they regulate splicing and alternative 3′ processing. Nova regulates splicing of RNAs encoding synaptic proteins, including the inhibitory glycine receptor α2 subunit (GlyRα2), and binds to others, including the GIRK2 channel. We found that Nova harbors functional NES and NLS elements, shuttles between the nucleus and cytoplasm, and that 50% of the protein localizes to the soma-dendritic compartment. Immunofluoresence and EM analysis of spinal cord motor neurons demonstrated that Nova co-localizes beneath synaptic contacts in dendrites with the same RNA, GlyRα2, whose splicing it regulates in the nucleus. HITS-CLIP identified intronic and 3′ UTR sites where Nova binds to GlyRα2 and GIRK2 transcripts in the brain. This led directly to the identification of a 3′ UTR localization element that mediates Nova-dependent localization of GIRK2 in primary neurons. These data demonstrate that HITS-CLIP can identify functional RNA localization elements, and they suggest new links between the regulation of nuclear RNA processing and mRNA localization.
Posttranscriptional regulation is thought to play a crucial role in generating diversity in the postsynaptic dendrite. This includes links between RNA splicing, RNA localization, RNA translation and synaptic formation, long term potentiation and synaptic plasticity (Schuman et al., 2006
; Sutton and Schuman, 2006
; Lin and Holt, 2007
; Richter and Klann, 2009
). In some cases specific RNA binding proteins (RNABPs) have been correlated with this regulation, including actions of FMRP (Bassell and Warren, 2008
), ZBP (Rodriguez et al., 2008
), Nova (Huang et al., 2005
; Ule and Darnell, 2006
), CPEB (Richter, 2007
) and others (Richter and Klann, 2009
). In other cases, regulation of RNA metabolism, such as activity-dependent alternative splicing and localization of NMDA receptor subunits (Ehlers et al., 1995
; Mu et al., 2003
), has been clearly defined, but the factors directly involved are only now beginning to be defined (Ule and Darnell, 2006
).
Studies in Drosophila and tissue culture cells suggest that splicing may be mechanistically coupled to mRNA localization in the cytoplasm (Pinol-Roma and Dreyfuss, 1992
; Luo and Reed, 1999
; Kataoka et al., 2000
; Le Hir et al., 2000
; Hachet and Ephrussi, 2004
). There is also emerging interest in the role of the 3′ UTR in RNA regulation in the synapse (Holt and Bullock, 2009
). Neurons have been found to generate preferentially long 3′ UTRs (Wang et al., 2008
) in a manner regulated by neuronal RNABPs (Licatalosi et al., 2008
), and these are potential sites for controlling RNA turnover or regulation by miRNAs in the synapse, as suggested by studies of Arc (Bramham et al., 2008
) or CaMKII synaptic RNAs (Ashraf et al., 2006
). However, an important unresolved question is whether information generated by regulation of alternative RNA processing is tagged at the time it is generated in the nucleus or whether the complexity of RNA generated there is handled independently in the synapse. One means of exploring this question is to ask whether the regulatory RNABPs regulating alternative RNA processing of specific transcripts also participate in their synaptic regulation.
Nova-1 and Nova-2 comprise a family of neuron-specific RNABPs targeted in a paraneoplastic syndrome in which inhibitory control of motor systems is affected (Musunuru and Darnell, 2001
; Darnell and Posner, 2003
; Darnell, 2006
). Nova proteins regulate alternative splicing in neurons (Jensen et al., 2000
; Polydorides et al., 2000
; Dredge and Darnell, 2003
; Dredge et al., 2005
; Ule et al., 2006
, 2005
; Licatalosi et al., 2008
). In vitro, Nova binds an intronic YCAY-rich element in GlyRα2 pre-mRNA upstream of the mutually exclusive exons 3A (E3A) and E3B (Buckanovich and Darnell, 1997
; Polydorides et al., 2000
), and in co-transfection minigene assays this leads to preferential utilization of E3A (Jensen et al., 2000
). It was subsequently recognized that Nova regulates numerous transcripts encoding synaptic proteins in the spinal cord and brain. Nova-null mice show defective splicing of inhibitory GlyRα2 receptors (Jensen et al., 2000
), GABAA receptors (Dredge and Darnell, 2003
), the Nova-1 transcript itself (Dredge et al., 2005
), and the neuron-specific isoform of agrin (Ruggiu et al., 2009
). More generally, a combination of genome-wide screens for Nova targets, including exon junction arrays (Ule et al., 2005
) and RNA-protein crosslinking (CLIP) methods (Ule et al., 2003
, 2005
; Jensen and Darnell, 2008
; Licatalosi et al., 2008
) coupled with additional biochemical and bioinformatic validation studies (Ule et al., 2006
) (reviewed in Licatalosi and Darnell, 2010
) have confirmed that Nova binds to YCAY-rich elements to regulate alternative splicing of a biologically coherent set of transcripts encoding synaptic proteins (Ule et al., 2005
; Ule and Darnell, 2006
).
Not all Nova-RNA targets are regulated at the level of alternative splicing. Nova is also able to bind 3′ UTR elements to regulate alternative polyadenylation (Licatalosi et al., 2008
), although other actions are less clear. For example, inhibitory responses to LTP (sIPSC of LTP) in the hippocampus were found to be dependent on Nova and on two of its target RNAs, encoding GIRK2 and GABAB receptors (Huang et al., 2005
), but the consequence of Nova binding to those transcripts is unknown. In motor neurons, Nova regulates Z+ agrin splicing, but rescuing this activity failed to restore the ability of motor neurons to innervate muscle, and suggested additional defects proximal to the motor axon (Ruggiu et al., 2009
).
The mounting evidence that nuclear RNA processing may also be coupled to RNA regulation in the cytoplasm, coupled with the detection of some Nova immunoreactivity in neuronal soma (Buckanovich et al., 1996
; Polydorides et al., 2000
), prompted us to investigate whether Nova might also act outside the nucleus. Here we show the existence of an abundant pool of cytoplasmic Nova. In tissue culture, Nova shuttles between the nucleus and cytoplasm, and it is required for RNA localization of the target GIRK2 mRNA in primary neurons. In vivo, the Nova target GlyRα2 mRNA is well documented to be transported to spinal motor neuron dendrites (Racca et al., 1997
, 1998
). We show that Nova is robustly detected in spinal motor neuron dendrites and, specifically, in inhibitory (gephyrin positive) synapses. Moreover, Nova co-localizes there with GlyRα mRNAs in spinal neurons, both in dendrites and beneath synapses, but is excluded from dendrites in the dorsal horn where GlyRα mRNAs do not extend into the dendritic tree (Racca et al., 1998
). Finally, biochemical analysis using CLIP identifies binding sites in the GIRK2 3′ UTR, and we find that Nova binding there is necessary for proper localization of GIRK2 mRNA in neuronal processes. Taken together, these data demonstrate that the splicing factor Nova and its nuclear RNA targets are also co-localized in dendrites close to synaptic contacts, suggesting a model in which the regulation of alternative splicing is coupled in cis to the expression of the same RNA in neuronal dendrites.
Nova Localization and Shuttling Between the Nucleus and Cytoplasm
To assess whether significant amounts of Nova protein are present in the brain outside of the nucleus, we performed Western blot analysis of nuclear and cytoplasmic fractions of mouse brain. Nova protein was readily detectable in both fractions, with the majority (∼60%, normalized to total protein) present in the nucleus (Figure 1
A), consistent with its role as a nuclear splicing factor and the high concentration of nuclear Nova evident by immunofluorescence (below). Interestingly, when we normalized our input by loading equal volumes of brain cytoplasm and nuclear fractions, a measure of the total amount of Nova present in each, we found that two thirds (68%) of total Nova protein is present in the cytoplasm (Figure 1
A). Immunofluorescence microscopy using anti-Nova antibodies confirmed an abundance of Nova immunoreactivity both within and outside of the nucleus (Figure 1
B). Taken together, these data demonstrate very significant amounts of Nova protein are present outside of the nucleus in mouse brain.
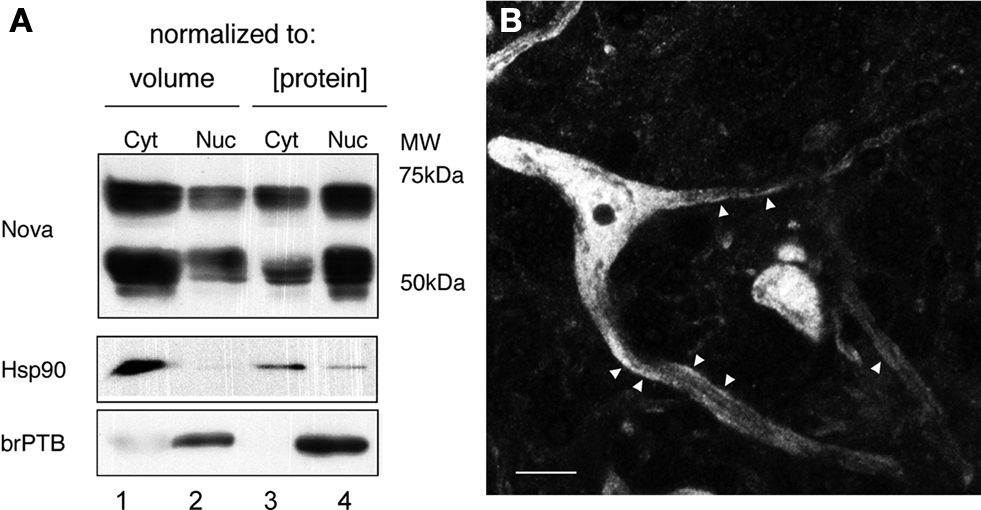
Figure 1. Subcellular distribution of Nova proteins. (A) Immunoblot analysis of Nova distribution in cytoplasmic and nuclear fractions from mouse brain (equal volumes (20 μl) of each fraction were loaded in lanes 1 and 2; equal protein amounts (50 μg) were loaded in lanes 3 and 4). Hsp90 is used as a cytoplasmic marker, and brPTB as a nuclear marker. The antibody used detects both Nova 1 (∼55 kD) and Nova-2 (∼75 kD) isoforms. (B) Nova signal is detected within the nucleus, somatic cytoplasm, and neurites of ventral horn spinal cord neurons. Within neurites, the signal is observed along the plasma membrane (arrowheads). Scale bar: 10 μm (B).
We asked whether Nova, like many RNABPs with this distribution, actively shuttles between the nucleus and cytoplasm. We assayed whether Nova endogenously expressed in a human neuroblastoma cell line (IMR-32) could shuttle into the nuclei of COS7 cells, an assay originally developed to document hnRNP-A1 shuttling (Pinol-Roma and Dreyfuss, 1992
). Four hours after fusion, with de novo protein synthesis blocked, Nova appeared in the COS7 cell nuclei (Figure 2
A). We repeated these experiments with fusions of another human neuroblastoma cell line [SK-N-BE(2)] and mouse 3T3 cells, again finding shuttling of endogenous Nova but not hnRNP-C1, a non-shuttling nuclear RNA binding protein (Figure 2
B). We also confirmed these results using an overexpressed Flag epitope-tagged Nova protein after transfection into HEK293 T cells and fusion to the neuroblastoma line N2A (data not shown). Taken together, these data indicate that, in tissue culture cells, Nova acts as a shuttling protein.
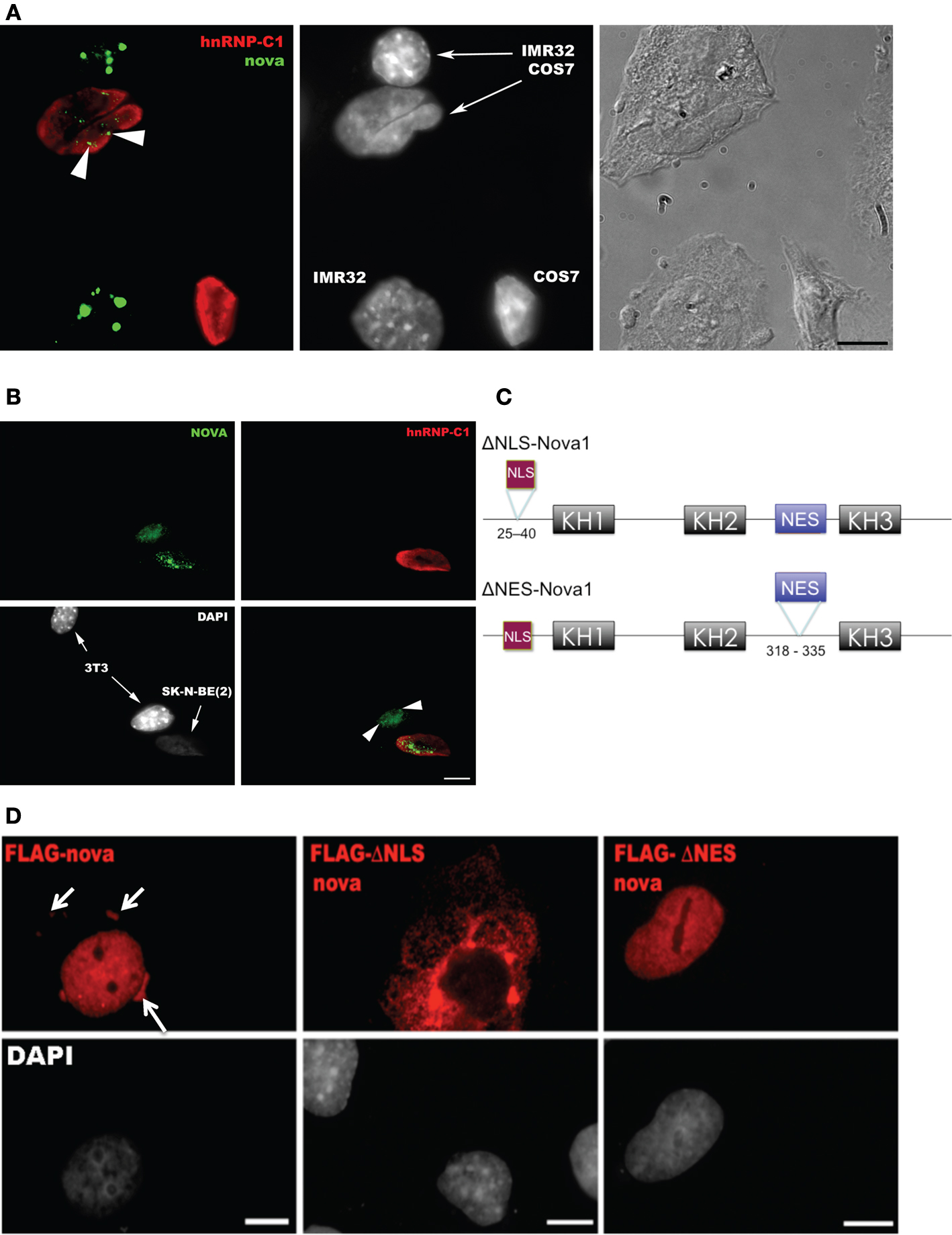
Figure 2. Nova proteins shuttle between the nucleus and cytoplasm. (A) IMR32 and COS7 cells were fused with PEG 3350, and anti-hnRNPC1 and anti-Nova antibodies were used to detect endogenous proteins. In this field one cell has been fused with COS7 (top; see phase contrast, right panel), and two unfused cells are evident (bottom); cell types can be distinguished with DAPI staining (middle panel). Nova proteins were detected in IMR32 and fused COS7 cells (arrowheads), but no signal in isolated COS7 cells. DAPI staining showed IMR32 cells and COS7 cells, respectively. (B) Shuttling of endogenous Nova from SK-N-BE(2) neuroblastoma cells into mouse NIH 3T3 cells; Nova, hnRNP-C12 and DAPI stains are shown as in (A). (C) Schematic of Flag-tagged Nova NLS and NES domains and deletion constructs generated. (D) COS7 cells were transfected with the indicated Flag-Nova1 plasmid constructs and stained with anti-flag antibody to visualize flag-Nova1 (top panels), and DAPI to visualize nuclei (bottom panels). Nova1 can be seen in the cytoplasm in cells transfected with the WT (left panel; arrows) but not in cells transfected with the ΔNES construct (right panel); in contrast the ΔNLS construct is largely excluded from nuclei (middle panel).
To investigate whether specific Nova protein domains regulate its subcellular localization, we examined the localization of Flag-tagged Nova constructs harboring deletions in either putative nuclear localization sequences (NLS) or nuclear export sequences (NES) (based on sequence homology with known motifs; Figure 2
C). Wild-type Flag-Nova was localized primarily to the nucleus of transfected COS7 cells, with some staining evident in the cytoplasm. In contrast constructs in which the putative NLS was deleted were localized in a reticular pattern in the cytoplasm, and constructs in which the putative NES was deleted were localized exclusively in the nucleus (Figure 2
D). These observations define distinct Nova domains that harbor NLS and NES activity.
These observations led us to examine Nova distribution in spinal motor neurons in greater detail. We found that the dendrites of most of the neurons in the intermediate zone and ventral horn were strongly immunopositive for Nova. Dendritic staining had a punctate pattern and tended to localize peripherally to the dendritic axial core and accumulate at dendritic branch points (Figure 1
B; see also Figures 4
A,B). The ultrastructural localization of Nova within neurons was investigated using pre- and post-embedding electron microscopy (EM) immunocytochemistry. EM confirmed the nuclear and cytoplasmic localization of Nova. Neuronal nuclei were heavily stained by Nova (Figure 3
A). Nuclear chromatin can be either highly condensed and scattered within the nucleoplasm or relaxed, depending on the neuronal subtype (Peters et al., 1991
). In neurons where the chromatin was relaxed (Figures 3
A,B), Nova signal was not associated with any identifiable intranuclear structures. In contrast, in nuclei where the chromatin was highly condensed, especially when apposed to the nuclear membrane, Nova immunoreactivity was concentrated over these chromatin-dense regions (Figure 3
C). In post-embedding experiments, Nova immunoreactivity was often observed near and within nuclear pores (Figure 3
C). In the somatic cytoplasm, the labeling was scattered and tended to be in proximity of or associated with cisternae of the endoplasmic reticulum. Nova immunoreactivity could also be observed close to postsynaptic specializations (Figures 3
A,D,E). Within dendrites, Nova was mainly peripheral along the plasma membrane, in the proximity of synaptic contacts (Figures 3
D,E).
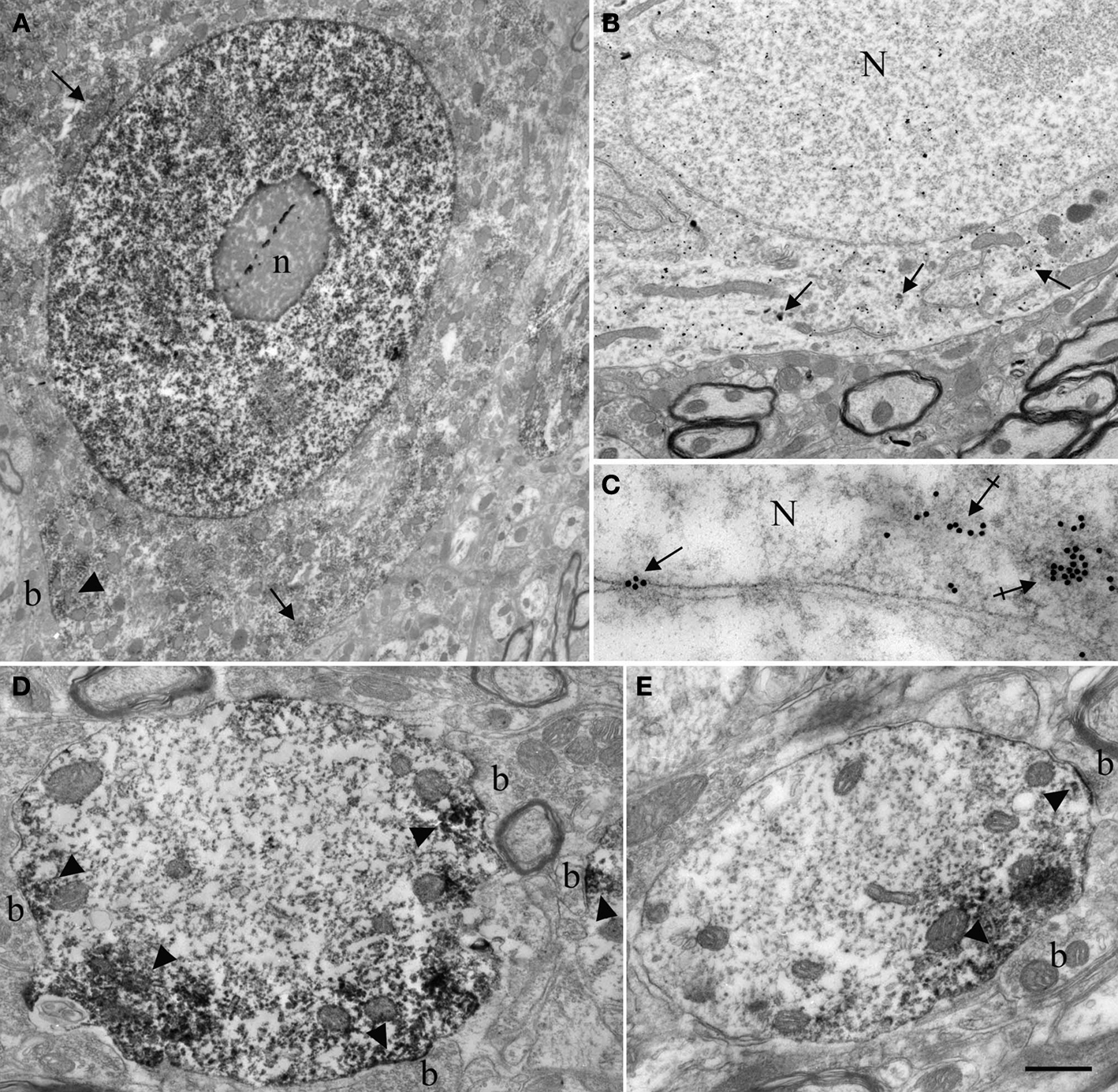
Figure 3. Ultrastructural localization of Nova in the somata and dendrites of ventral horn spinal cord neurons. (A,B) Micrographs of pre-embedding immunocytochemistry experiments. (C) Micrograph of a post-embedding immunocytochemistry experiment. (A) Distribution of Nova visualized with electron dense HRP reaction product (arrows). The nucleus is heavily stained but not the nucleolus (n). Presence of electron dense HRP reaction product (arrowhead) in front of a synaptic contact (b). (B) Gold particles associated with Nova are found within the nucleus (N), as well as within the somatic cytoplasm (arrows). (C) High magnification micrograph of the nuclear periphery. In spinal neuron nuclei (N), where the chromatin is condensed at the nuclear periphery in proximity of the inner side of the nuclear membrane, Nova (15 nm gold particles) accumulates over chromatin (crossed arrows) and can be found within nuclear pores (arrow). (D,E) Electron microscopic immunolabeling of Nova in dendrites (HRP reaction product) showing Nova tendency to accumulate peripherally to the dendritic center, and close to synapses (arrowheads). Axons are devoid of staining. Boutons (b). Scale bar: 4 μm (A); 2 μm (B); 0.3 μm (C); 1 μm (D,E).
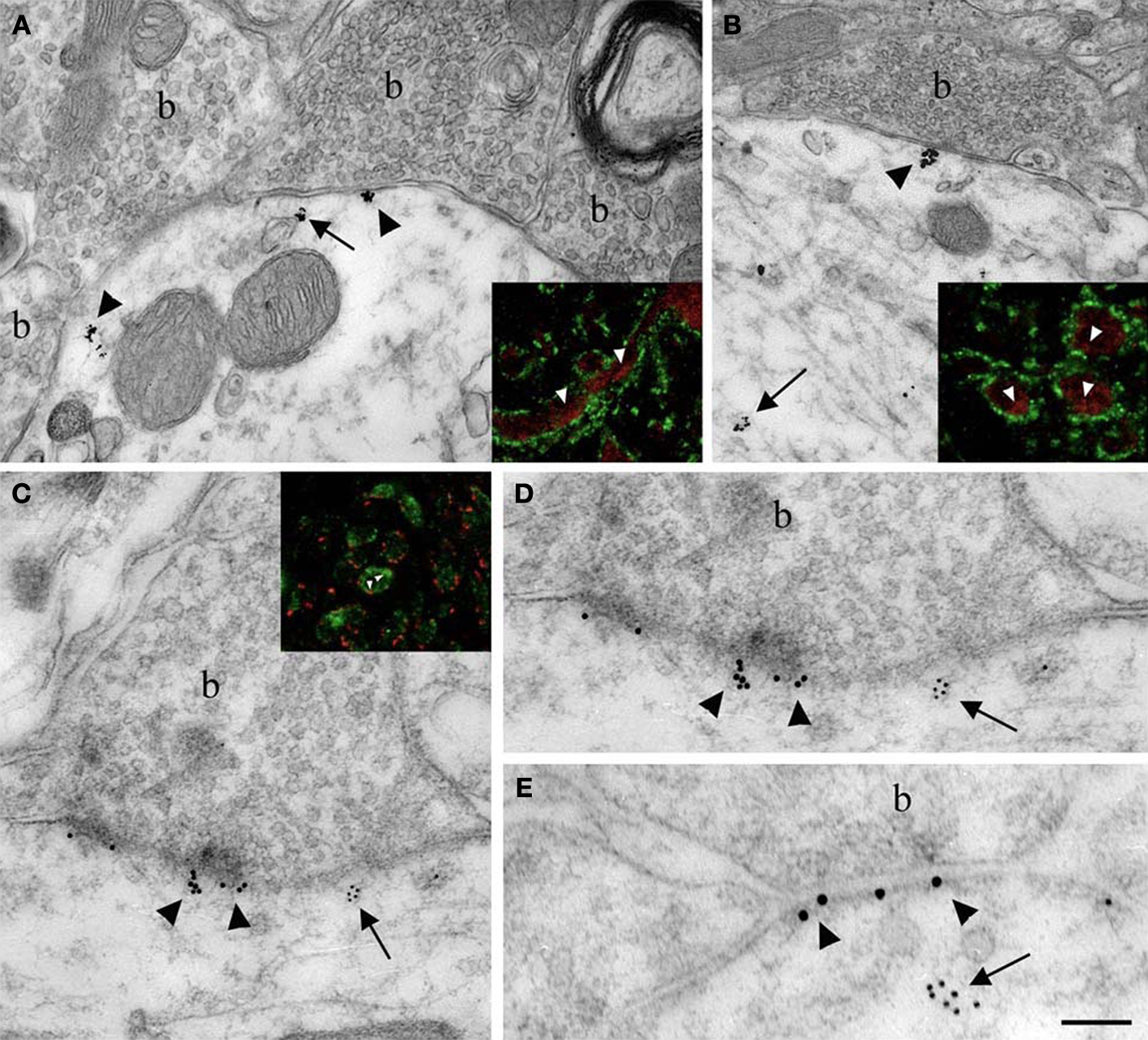
Figure 4. Localization of Nova at inhibitory synapses. (A,B) Nova signal detected by gold labeling (arrows) is observed in dendrite and opposed to boutons (b) at the dendritic periphery (arrowheads). Insets in (A) and (B), Double-immunofluorescence detection of Nova (in red) and synapsin (in green) show that Nova immunoreactivity is present opposite to synaptic contacts (arrowheads). (C,D,E) Simultaneous detection of Nova and gephyrin immunoreactivity within the postsynaptic cytoplasm. Nova immunoreactivity (10 nm gold particles; arrows) is found beneath postsynaptic regions where gephyrin immunoreactivity (15 nm gold particles; arrowheads) decorates and identifies inhibitory synapses. (D) High magnification of the postsynaptic cytoplasm of (C). Inset in (C), Double-immunofluorescence of Nova (in green) and gephyrin (in red) showed that Nova immunoreactivity is observed in proximity of gephyrin immunoreactivity synaptic contacts (arrowheads). Scale bar: 0.2 μm (A); 0.6 μm (B); 0.2 μm (C,E); 0.4 μm (D); 9 μm (insets in A,B); 6 μm (inset in C).
Nova is Present at Inhibitory Synapses
Immunoelectron microscopy for Nova (Figures 4
A,B) and double-immunofluorescence for Nova and synapsin (Figures 4
A,B insets), a presynaptic marker (Bloom et al., 1979
; De Camilli et al., 1979
, 1983
), confirmed the presence of Nova protein at synapses. To assess whether Nova protein was present at inhibitory synapses, we examined whether Nova and gephyrin reactivity co-localized. Gephyrin is considered a postsynaptic marker of inhibitory synapses (Triller et al., 1985
, 1987
; Altschuler et al., 1986
). Both immunofluoresence and immuno EM demonstrated that Nova immunoreactivity is present in the somato-dendritic cytoplasm of postsynaptic inhibitory differentiations identified by a strong labeling for gephyrin (Figures 4
C–E and inset).
Localization of Nova-RNA Targets
Nova binds to GlyRα2 pre-mRNA to regulate alternative splicing within the nucleus (Buckanovich et al., 1996
; Buckanovich and Darnell, 1997
; Jensen et al., 2000
). To examine whether Nova might play a role in the regulation of mRNAs within dendrites, we took advantage of the observation that GlyRα2 mRNA is present at inhibitory synapses and in dendrites (Racca et al., 1997
). Co-staining of spinal cord motor neurons for Nova proteins and GlyRα2 mRNA by EM and fluorescence microscopy revealed that Nova protein co-localizes with GlyRα2 mRNA in the dendrite (Figure 5
). GlyRα2 and GlyRα1 mRNA have previously been shown to co-localize (Racca et al., 1997
), and we found that Nova protein and GlyRα1 mRNA also co-localize (Figure 5
A). In the dorsal horn of the spinal cord, GlyRα2 mRNA transcripts do not translocate to dendrites, but are restricted to the neuronal somata (Racca et al., 1998
). Interestingly, in these neurons Nova immunoreactivity was restricted to somata as well (Figure 5
C).
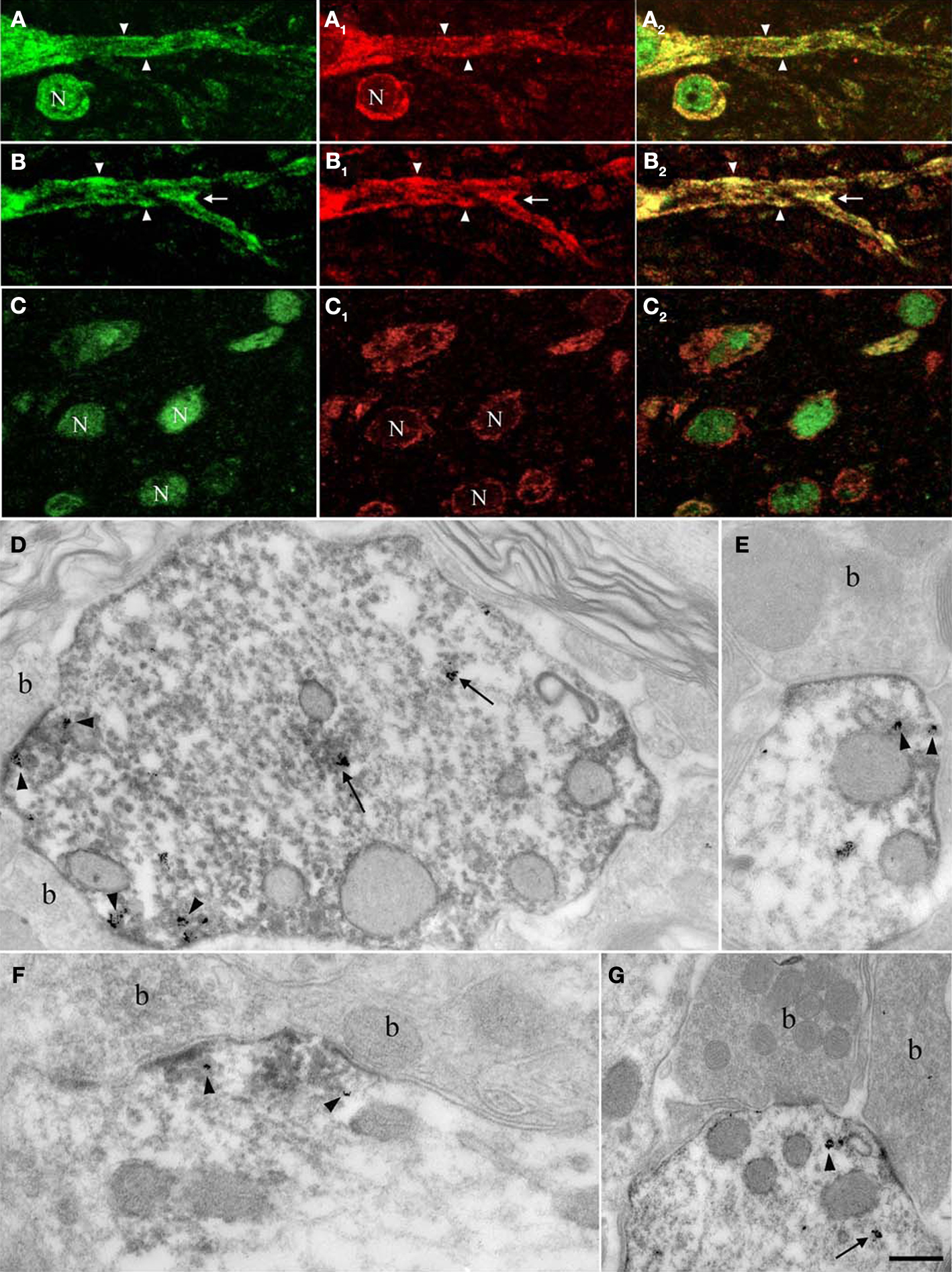
Figure 5. Nova and GlyRα2 mRNAs, its nuclear alternative splicing RNA target, colocalize outside the nucleus within dendrites. Fluorescence labeling for Nova immunoreactivity (in green; A,B,C) and ISH signal (in red) for GlyRα1 (A1) or GlyRα2 mRNAs (B1,C1). (A) Within the somatic and dendritic cytoplasm of ventral horn neurons, GlyRα1/2 (A1,B1) mRNAs and Nova (A,B) labeling co-localizes (in yellow; A1,B1). The labeling pattern of Nova mirrors that of GlyRα1/2 mRNAs. Both are also unevenly distributed within the dendritic cytoplasm and sometimes accumulate at the dendritic periphery (A,B; arrowheads) and branch points (B; arrows). Note that the signal corresponding to Nova protein or GlyRα1/2 mRNAs are separated in some areas. (C) in neurons of the dorsal horn, where GlyRα2 mRNA (C1) is restricted to the somatic cytoplasm, Nova immunoreactivity is detected in nuclei (N) and to a lesser extent in somatic cytoplasm (C) resulting in a less accentuated co-localization (C2). (D–G) Ultrastructural simultaneous detection of Nova immunoreactivity and GlyRα2 mRNAs. Nova (HRP immunolabeling) and GlyRα2 mRNA ISH signal (gold particles) colocalize within the dendritic cytoplasm (arrows) and in front of synaptic boutons (arrowheads, b). Note in (E,G) the association of Nova and mRNA signals with small cisternae. Scale bar: 15 μm (A–C); 0.2 μm (D–F); 0.35 μm (G).
Electron microscopy confirmed that Nova protein and GlyRα2 mRNA are co-localized within dendrites of spinal cord motor neurons. Here, as previously reported (Racca et al., 1997
), subsynaptic GlyRα mRNA was often detected in association with small cisternae (Figures 5
E,G) belonging to a protein synthesis-related subsynaptic apparatus (Gardiol et al., 1999
), and this mRNA again co-localized with Nova protein. GlyRα2 mRNA and Nova protein were also associated with postsynaptic differentiations in the vicinity of these minute subsynaptic cisternae, suggestive of a protein synthesis-related subsynaptic machinery, and consistent with the nature of GlyRα2 proteins as transmembrane proteins (Figures 5
D–G).
The spinal cord of Nova-null mice show defects in alternative splicing of RNAs encoding synaptic proteins (Ule et al., 2005
). To explore whether Nova-RNA targets are mislocalized in Nova-null neurons, we established primary neuronal cultures from E18.5 embryos of WT and Nova1/2 null (“DKO”) mice. Interestingly, although GlyRα2 transcripts are localized in spinal motor neurons in adult animals (Racca et al., 1997
, 1998
), both this localization and the level of transcript expression (Kuhse et al., 1991
; Malosio et al., 1991
) is developmentally regulated such that mRNA localization does not become evident until after the second postnatal week (C. Racca, unpublished data). Since Nova DKO mice die at birth (Ule et al., 2006
; Ruggiu et al., 2009
), Nova1 null animals die in the first 7–10 days postnatally (Jensen et al., 2000
) and Nova2 null animals usually die in the second postnatal week, we examined mRNA localization of a different Nova target.
We have previously shown that the ability of GIRK2 to mediate potentiation of slow inhibitory postsynaptic currents is Nova-dependent (Huang et al., 2005
). While the mechanism of Nova action on GIRK2 mRNA has not been determined, Nova CLIP data (Ule et al., 2003
) previously indicated that Nova directly binds to GIRK2 intronic pre-mRNA (intron 2). Moreover, GIRK2 mRNA has multiple isoforms, including at least three different alternative 3′ UTRs, which we confirmed by RT-PCR (data not shown). To address which, if any, of these isoforms Nova might directly bind to, we searched a robust map of Nova-RNA interactions in the brain developed using HITS-CLIP (Licatalosi et al., 2008
). This revealed a cluster of Nova CLIP tags (representing in vivo protein-RNA binding) within the distal-most 3′ UTR of GIRK2 mRNA (isoform GIRK2-1; see Materials and Methods), which was enriched in the Nova binding element YCAY (Figure 6
A). Additional upstream binding sites were also present in the GIRK2 transcript, most notably in the intronic region between the annotated GIRK2a-1 and GIRK2-1 3′ UTRs, and in the intron 2 site previously identified (∼187,000 nt upstream of the 3′ UTR). We confirmed that Nova was bound to the mature GIRK2 mRNA in neuronal cytoplasm by repeating CLIP in a cytoplasmic brain fraction (data not shown and Figure 6
A). The protein and transcript levels of GIRK2 remained unchanged in WT compared to Nova DKO brain (unpublished data). Taken together, these data suggested that Nova might act upon the GIRK2 3′ UTR element to regulate mRNA localization.
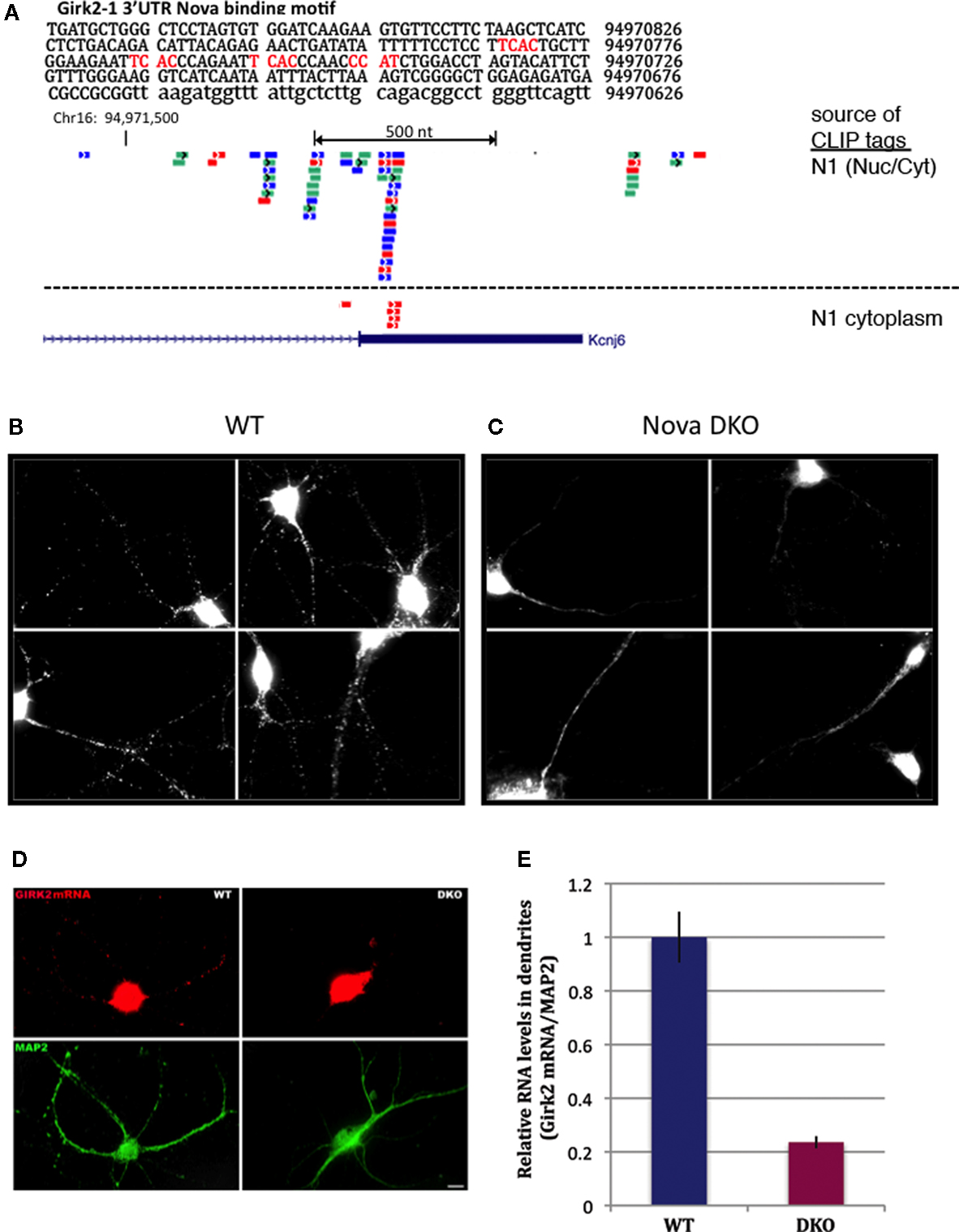
Figure 6. Nova regulates localization of the CLIP target GIRK2-1 mRNA isoform in dendrites. (A) Location of Nova1 CLIP tags in the 3′ UTR of the GIRK2-1 transcript. The RNA sequence of the GIRK2-1 3′ UTR in the region of the Nova binding site is shown, with YCAY elements highlighted in red. CLIP tags from mouse cortex (Nuc/Cyt) or cytoplasmic extracts of mouse brain (N1 cytoplasm) are shown separately. Nova tags are colored, with each color representing tags from a different mouse brain. Several other robust Nova CLIP target sites rich in YCAY elements were found in upstream sites, most notably in intron 2 ∼187,000nt upstream (see text). (B,C) Fluorescence ISH (FISH) using GIRK2-1 specific probes to detect mRNA in WT (B) or Nova DKO (C) primary cultured neurons obtained from E18.5 mouse cortex. No signal was seen with control ISH probes. (D) Comparison of GIRK2-1 FISH and MAP2 immunoreactivity in WT and Nova DKO primary neurons, as indicated. (E) Quantitation of fluorescence intensity from (D); MAP2 fluorescence signal was counted beginning 5–10 μm from the cell body and extended to the distal dendrites, and then the signal from GIRK2-1 was obtained from the same dendrite; at least 10 neurons were counted in each of three experiments. The results from a three independent experiments are plotted as the sum of the dendritic signal from GIRK2-1 (Cy3) divided by MAP2 (Cy5); error bars represent standard deviation (p < 0.05; Student’s t-test). The MAP2 signal alone was not significantly changed in WT vs. KO neurons (data not shown). Sense strand oligonucleotides were used as a negative control and showed no signal under identical conditions. Scale bar: 10 μm (D).
We therefore set out to examine localization of the GIRK2-1 isoform in neurons. We assessed GIRK2 mRNA localization using a set of Cy3-conjugated oligonucleotide probes directed at the GIRK2-1 3′ UTR extension in Nova WT and KO neurons. These probes did not yield a sufficient signal:noise to detect GIRK2 RNA isoforms in E18.5 brain sections (perhaps due to low expression of the GIRK2 and GIRK2-1 isoform; for example, as assessed by Affymetrix arrays of E18.5 mouse brain, the transcript normalized probe intensity for GIRK2 was 199, and for GIRK2-1 was ∼50, relative to a median of 708; data not shown). We therefore assessed GIRK2-1 mRNA localization in primary cultures of cortical neurons. These in situ hybridization (ISH) results demonstrated that GIRK2-1 mRNA extended from the cell soma into dendritic processes of WT neurons, where it was present in a punctate pattern. In contrast, in Nova DKO neurons, GIRK2 mRNA was restricted to the cell body and proximal dendrites, where it was present in a diffuse pattern (Figures 6
B,C). Quantitation of the fluorescence signal from GIRK2 mRNA in WT vs. Nova DKO dendrites, normalized to the immunofluorescence signal from MAP2, demonstrated ∼ 3-fold decrease in dendritic GIRK2 mRNA in DKO neurons (p < 0.05; Figures 6
D,E). We note also that mRNA appears to be more granular in WT compared to KO neurons; whether this relates to a role for Nova in mediating granule inclusion in dendrites for the localization of mRNAs it regulates is not known.
To address whether direct Nova binding to the GIRK2 3′ UTR (YCAY)4 element could localize the mRNA, we developed a reporter assay in N2A (neuroblastoma) tissue culture cells. We generated a construct encoding a destabilized d1EGFP (which has a short half life of ∼1 h, giving an image skewed toward detection of local protein expression) fused to a nuclear localization element (M9) and either a 208 nt fragment from the wild-type GIRK2 3′ UTR or a mutant 208 nt fragment which would abrogate Nova binding [in which the four YCAY repeats were mutated to YGUY; (Buckanovich and Darnell, 1997
; Lewis et al., 2000
); Figure 7
A]. Expression of d1EGFP outside the nucleus was used as an indirect measure of GIRK2-1 mRNA localization. Cells transfected with the WT YCAY construct showed a clear and quantifiable increase in d1EGFP localization to distal neurites, while d1EGFP expression in cells transfected with the mutant YGUY construct was largely restricted to the cell body (Figures 7
B,C), while the d1EGFP signal in the soma was not significantly changed. Interestingly, the T7-Nova signal also appeared more prominent in distal neurites in cells transfected with the WT YCAY construct, while the MAP2 signal was not changed, suggest that direct Nova binding to GIRK2 3′ UTR is necessary for proper localization of the mRNA.
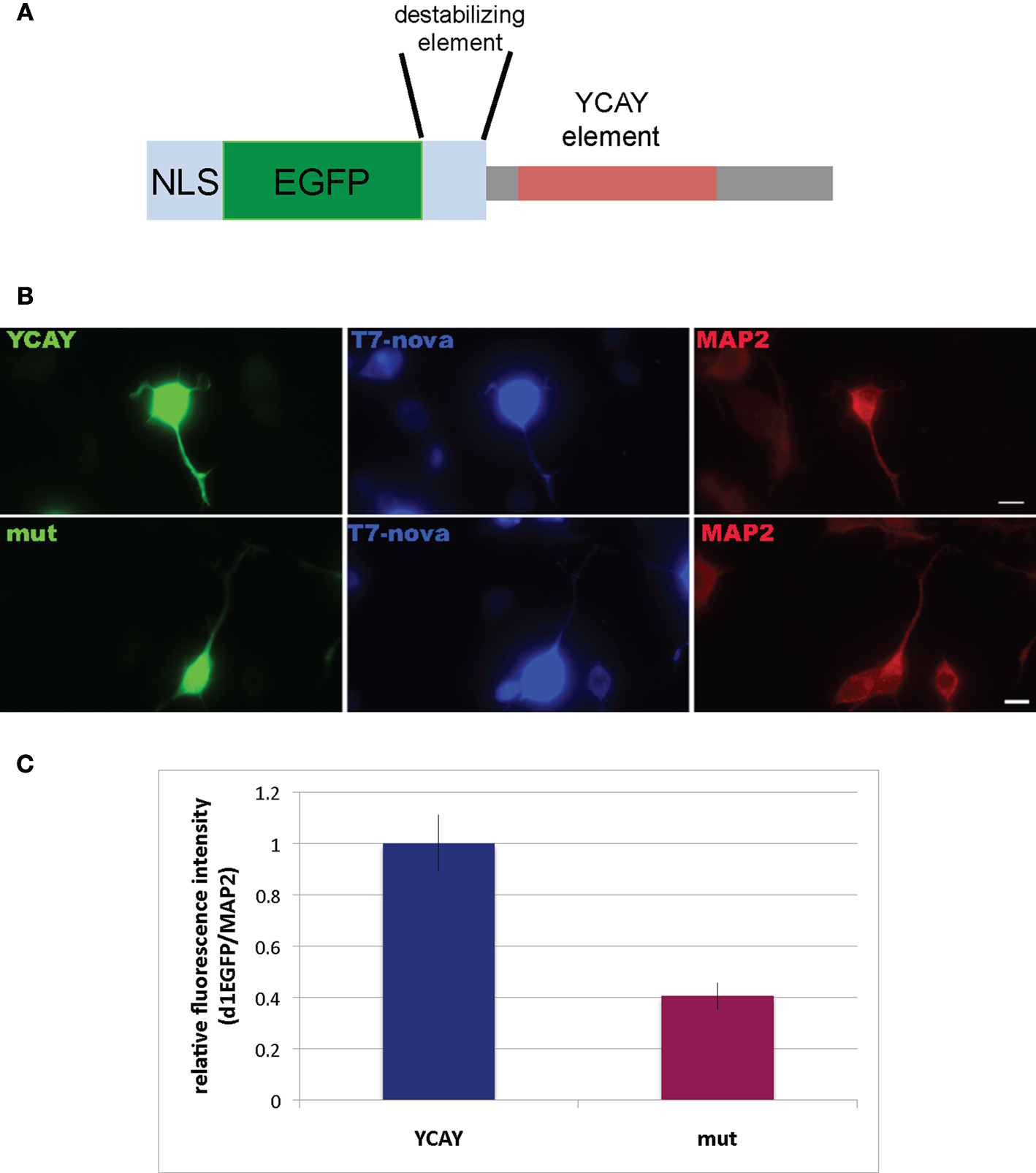
Figure 7. Binding of Nova to the GIRK2-1 YCAY element is necessary for proper localization. (A) Schematic of a reporter encoding destabilized d1EGFP with an M9 nuclear localization sequence (NLS) and the 3′ UTR GIRK2-1 YCAY element. An identical construct in which the four YCAY sequences were mutated to YGUY was also made. (B) Comparison of EGFP and MAP2 immunoreactivity in cells co-transfected with plasmid constructs encoding the YCAY or the mutant (“mut”) YGUY element, as indicated, and a construct expressing T7-tagged Nova. The WT Nova target mRNA (green channel, left panel) was localized in distal neurites in transfecting cells expressing the YCAY but not mutant GIRK2-1 element; T7-tagged Nova itself, detected with T7 antibody (middle panel) also appeared more localized in distal dendrites, while MAP2 (right panel) was unchanged in either condition. (C) Quantitation of EGFP fluorescence intensity from (B); Region of Interest (ROI) was chosen based on neurite in which MAP2 fluorescence signal was detected, beginning 5 μm from the cell body and extending outwards. The signal from d1EGFP was obtained from the same ROI; more than 10 cells were counted. The results are plotted as relative ratio of the sum of the neurite signal from d1EGFP (Cy2) divided by MAP2 (Cy3) in YCAY element or mutated constructs; error bars represent standard deviation (p < 0.05); results are from three independent experiments. Scale bar: 10 μm (B).
The action of Nova to regulate GlyRα2 pre-mRNA splicing and its co-localization with GlyRα2 mRNA suggests that protein may be bound to both GlyRα2 pre-mRNA and mature mRNA. We addressed this possibility by again examining Nova HITS-CLIP data (Licatalosi et al., 2008
). Figure 8
A illustrates the intronic region of GlyRα2 pre-mRNA previously shown in vitro and in minigene assays to mediate Nova-dependent inclusion of E3A (Buckanovich and Darnell, 1997
; Polydorides et al., 2000
). Remarkably, the intronic Nova binding motif (YCAY clusters) is surrounded by several Nova CLIP tags (Figure 8
A). In addition, examination of the 3′ UTR revealed several clusters of Nova CLIP tags, both around the stop codon and within the 3′ UTR itself (Figure 8
B). This co-incidence of tags in the intron and 3′ UTR also led us to re-examine intronic tags in GIRK2 pre-mRNA; an extremely robust tag cluster (identified in at least 17 different mouse CLIP experiments, from a variety of tissues, including neocortex, hippocampus and spinal cord) was identified harboring 13 YCAY elements within 1123 nucleotides (see Figure 6
legend). These data suggest a model in which intronic loading of Nova in the nucleus may couple regulation of pre-mRNA RNA metabolism (e.g. splicing) to 3′ UTR loading and regulation of mRNA expression (e.g. mRNA localization; Figure 9
).
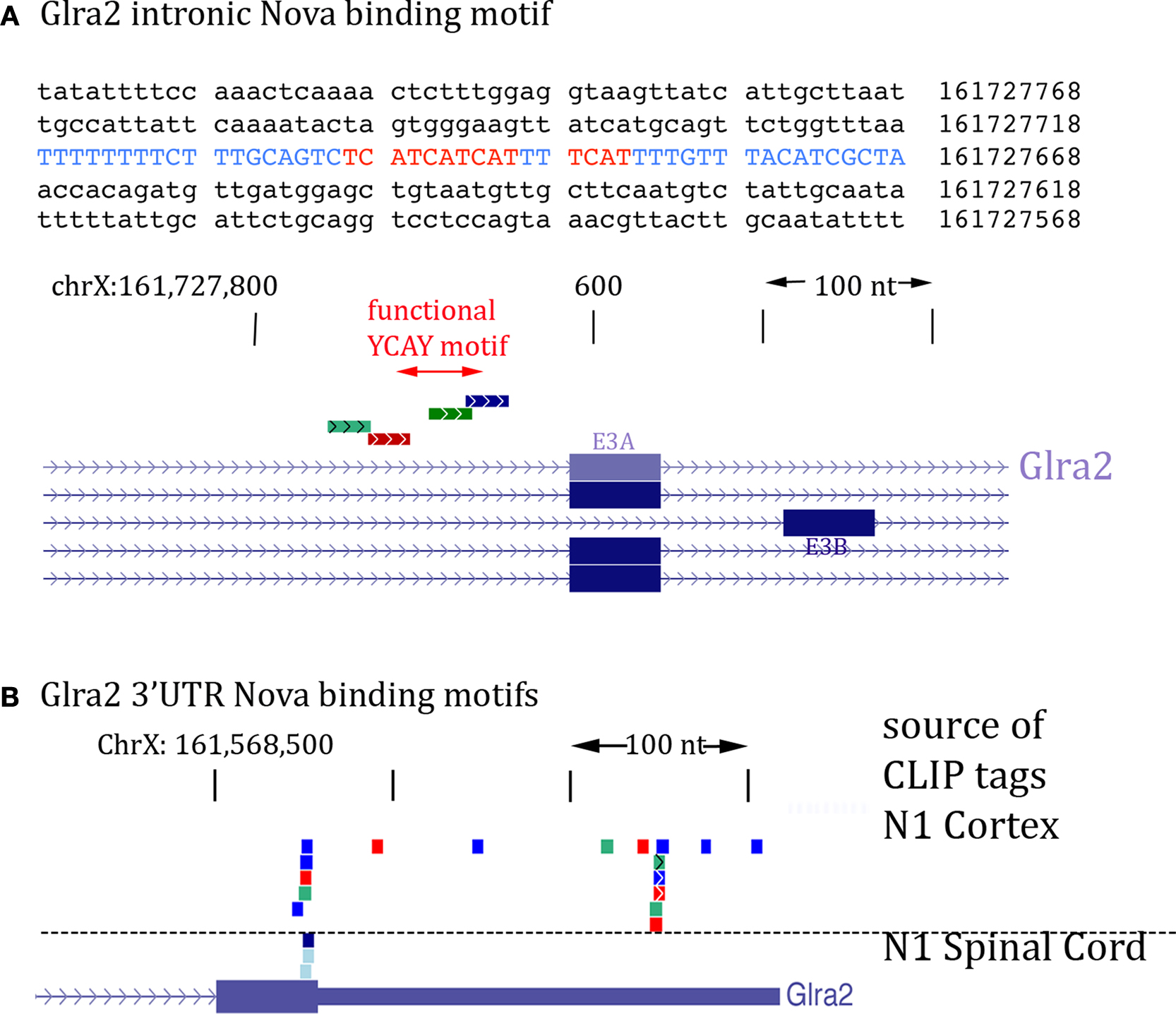
Figure 8. Location of Nova CLIP tags in the GlyRα2 transcript. (A) The sequence of the previously characterized Nova binding YCAY motif upstream of GlyRα2 exon 3A is shown (blue); mutation of these YCAY elements (red) abrogates E3A splicing (Buckanovich and Darnell, 1997
). Lower panel shows Nova HITS-CLIP tags in this intronic region. (B) Nova HITS-CLIP tags in the GlyRα2 3′ UTR. In (A,B) Nova tags are colored as in Figure 6
.
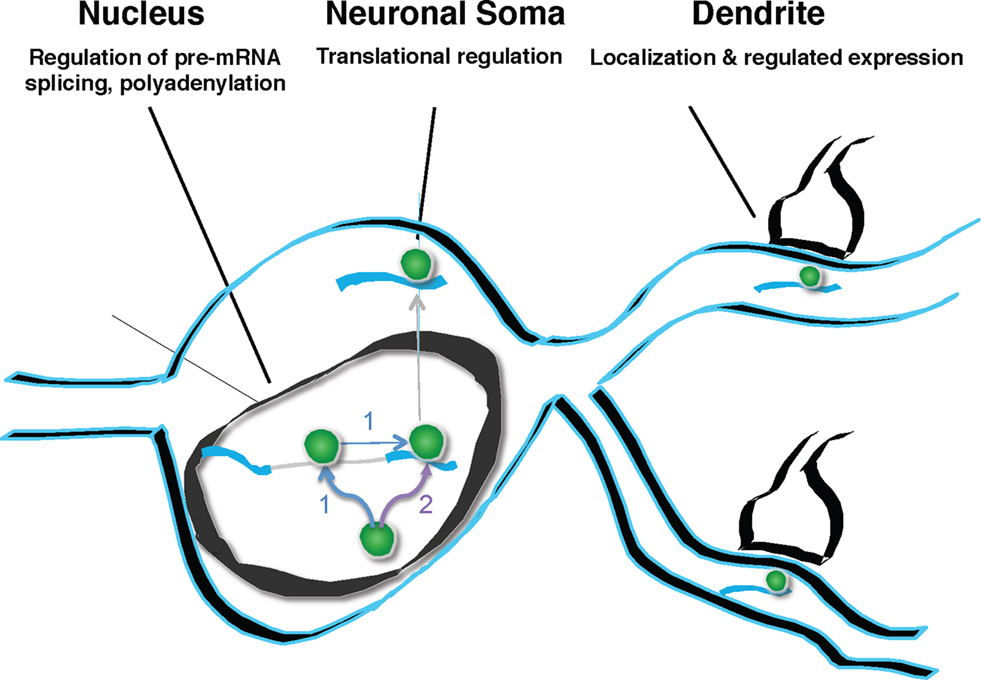
Figure 9. Model of Nova action. Nova binds to pre-mRNA in the nucleus. This binding can, but need not be intronic (pathway labeled “1”). If binding sites are in the region of alternatively spliced exons, Nova can mediate alternative splicing, according to the rules of a functional binding map, such that the position of Nova binding determines the outcome of alternative exon inclusion. Subsequently, Nova may multimerize in cis upon the same transcript (pathway 1), or may be independently deposited upon additional mRNA binding sites, such as those in the 3′ UTR (pathway labeled “2”). 3′ UTR binding sites may determine additional processing steps (such as mediating alternative polyadenylation), and mediate soma and dendritic mRNA localization (as in the case of GIRK2-1 and perhaps GlyRα2 mRNA). It is presumed, based for example on the studies of Singer and colleagues (Lawrence and Singer, 1986
; Kislauskis et al., 1997
; Rodriguez et al., 2008
), that such RNA localization would be coupled to the regulation of translation. In neurons, coupling pre-mRNA binding to mRNA localization and translation offers the possibility of linking information content generated in the nucleus (alternative splicing or polyadenylation) with differential gene expression in dendrites.
Nova functions as a neuron-specific alternative splicing factor (Jensen et al., 2000
; Dredge and Darnell, 2003
; Ule et al., 2003
; Dredge et al., 2005
; Licatalosi et al., 2008
), but we find here that the amount of Nova in the soma-dendritic compartment is at least as great as that in the nucleus. In ventral horn motor neurons, Nova proteins are localized within the nucleus, cytoplasm and dendrites and were often apposed to inhibitory synapses. Nova regulates the splicing of GlyRα2 mRNA (Buckanovich and Darnell, 1997
), a transcript known to be transported to the dendrites (Racca et al., 1997
), and we find here that Nova protein and GlyRα mRNA co-localize within the dendrite (Figure 5
), consistent with the finding that Nova binds directly to GlyRα pre-mRNA and 3′ UTR within the brain. In addition, Nova was required for proper localization of GIRK2 mRNA in primary neuronal cultures. Taken together, these observations suggest that the role of Nova in RNA regulation goes beyond its actions in the nucleus, such that it is able to act to coordinate nuclear RNA processing with local mRNA expression. Moreover, since Nova is critical for the normal physiology of motor neurons (Ruggiu et al., 2009
) and hippocampal neurons (Huang et al., 2005
), and at least in motor neurons, correction of a Nova-dependent Z+ agrin splicing defect failed to rescue the physiologic defect (paralysis) in Nova DKO mice, it is possible that such actions may play critical roles in CNS neurons.
Nova Proteins and the Inhibitory Synapse
Nova proteins are the neuron-specific target antigens in a paraneoplastic neurological disorder, Paraneoplastic Opsoclonus Myoclonus and Ataxia (POMA; Darnell and Posner, 2006
). Clinically, POMA symptoms suggest an impairment of CNS inhibition. Interestingly, these clinical observations correlate with the finding that Nova proteins bind to introns, exons and UTRs of many RNAs encoding components of the inhibitory synapse [e.g. subunits of the GlyR and GABAA and GABAB receptors, gephyrin, GIRK2 and others (Jensen et al., 2000
; Dredge and Darnell, 2003
; Ule et al., 2003
, 2005
; Ule and Darnell, 2006
; Licatalosi et al., 2008
)]. GlyRα2 subunit and gephyrin mRNAs encode components of the inhibitory glycinergic synapse (Moss and Smart, 2001
). GIRK2 encodes a G protein-activated inwardly rectifying K+ channel that mediates GABAB inhibition in a Nova-dependent manner. We have found a physiologically relevant action of Nova that appears to be specific to one alternatively processed GIRK2 isoform. Nova binds to a YCAY element in the GIRK2-1 3′ UTRs and localizes GIRK2-1 mRNA, but we found no evidence for Nova-dependent localization of other GIRK2 mRNA isoforms (data not shown). These data are most consistent with a model in which Nova binds to a 3′ UTR element to mediate mRNA localization, as observed for many other protein-RNA interactions (Kislauskis and Singer, 1992
; Bassell and Singer, 2001
; Palacios and St Johnston, 2001
; Martin and Ephrussi, 2009
).
More is known about the action of Nova on the RNAs encoding the GABAA and GlyRα2 transcripts, where specific YCAY binding sites have been mapped within introns that mediate Nova-dependent alternative exon usage (Buckanovich and Darnell, 1997
; Jensen et al., 2000
; Dredge and Darnell, 2003
). Here we provide convergent data demonstrating that Nova is present in the nucleus and, along with GlyRα mRNAs and gephyrin, within the dendrites and in the vicinity of inhibitory synapses in central neurons. These findings suggest that Nova might provide a means to couple nuclear RNA binding to the dendritic and subsynaptic localization of GlyRα2 mRNA. This could ultimately regulate the composition or number of glycine receptors at inhibitory synapses and, consequently, synaptic function. More generally, the localization of Nova in the inhibitory synapse predicts the possibility that the physiology of these synapses may show Nova-dependent features, perhaps reflected in the finding that inhibitory responses to LTP in the hippocampus are absent in Nova-null mouse brain (Huang et al., 2005
).
In the nucleus, Nova binding to GlyRα2 pre-mRNA leads to prevalent expression of GlyRα2A subunit mRNA (Jensen et al., 2000
). Both GlyRα2A and 2B subunit mRNAs are expressed prenatally in the CNS (Kuhse et al., 1990
, 1991
). After postnatal day 5 the expression of GlyRα2B subunit mRNA decreases, whereas the expression level of GlyRα2A subunit mRNA is not altered (Kuhse et al., 1991
). Interestingly, the localization of GlyRα subunit mRNAs to dendrites is also developmentally regulated and starts occurring after the second postnatal week (C. Racca, unpublished data). This observation meant that we focused our studies on adult animals, and were not able to study GlyRα2 subunit mRNA localization in Nova KO mice (which typically die in early postnatal life; Jensen et al., 2000
). Nonetheless, the data suggest that the main form of GlyRα2 subunit mRNA present in dendrites is likely to be GlyRα2A, the one also favored by Nova-mediated splicing. GlyRs containing either α2A or α2B subunit differ in their agonist sensitivity (Miller et al., 2004
), such that those containing α2B subunit are more sensitive to agonists (glycine, alanine and taurine) than are those containing α2A. Although yet to be proved, our data raise the possibility that neurons modulate their ligand sensitivity by local regulation, through Nova-mediated regulation of splicing, localization and/or expression of GlyRα2A mRNA.
Subsynaptic RNPs could be Assembled in the Nucleus in a Splicing-Dependent Manner
Nova proteins were detected in the nucleoplasm, nuclear pores, and within the somato-dendritic cytoplasm, co-localized with target mRNAs. Although functional Nova binding sites were originally identified as intronic elements, the protein also binds to exons and 3′ UTR regions of different RNAs (Darnell, 2006
; Licatalosi et al., 2008
). However, it has been unclear whether there is any relationship between these disparate binding sites. GlyRα mRNA is a robust transcript for addressing this question for two reasons. Nova binding sites in intron 3 were necessary to mediate alternative splicing in minigene assays (Buckanovich and Darnell, 1997
; Polydorides et al., 2000
). At the same time, GlyRα mRNAs are among the best documented dendritically localized transcripts (Racca et al., 1998
; Steward and Schuman, 2001
). The subcellular localization of Nova within dendrites of ventral horn but not dorsal horn spinal cord neurons parallels the dendritic localization of GlyRα subunit mRNAs (Figures 5
A–C). The co-localization of Nova protein, GlyRα2 and GIRK2 mRNA outside of the nucleus, together with HITS-CLIP data demonstrating direct Nova binding to GlyRα2 and GIRK2 intronic and 3′ UTR elements (Figures 6 and 8
) indicate a link between the nuclear and cytoplasmic actions of Nova. Nova interacts with each transcript both prior to nuclear processing, as pre-mRNA, and after processing and cytoplasmic transport.
One plausible scenario by which Nova regulates both GlyRα2 pre-mRNA splicing and GlyRα2 mRNA localization/expression would be via independent binding to intronic and 3′ UTR elements, as suggested for the localization of oskar mRNA in Drosophila oocytes (Hachet and Ephrussi, 2004
). Binding to both sites could also be linked within the nucleus, for example by co-deposition of Nova in cis on introns and 3′ elements of the same transcript. A more complex possibility invokes a direct link between the two, involving remodeling of the Nova/GlyRα2 RNA-protein complex. In this scenario, Nova would bind directly to intronic sequences to mediate splicing, and then remain associated with the mature GlyRα2 mRNA. Although HITS-CLIP detects binding to introns that are common to multiple processed mature mRNA forms, because CLIP efficiency is very low (only ∼1% of transcripts are cross-linked), it cannot distinguish whether Nova-RNA interactions are present on pre-mRNA transcripts or a subset that are subsequently processed (in either alternative exons or poly(A) sites) in a specific manner. In addition, we note that not all 3′ UTRs harboring Nova tags showed evidence of Nova-dependent localization (MAP1B, for example; data not shown), consistent with the idea that Nova may mediate a variety of actions upon the 3′ UTRs of different mRNAs.
Multimerization of RNABPs in cis is thought to be important in the interaction of Rev with the RRE stem-loop of HIV (Malim and Cullen, 1991
), and of hnRNP-A1 with the HIV-1 tat transcript to antagonize the action of some SR proteins (Zhu et al., 2001
). In these instances, recognition of single high affinity motifs is followed by lower affinity multimerization of the RNABPs in cis on target RNAs to provide biologically important deposition of RNABPs in the area of the initial high affinity binding site. Diffusion in cis along DNA is an analogous process clearly documented to be important in transcription factor dynamics, such that DNA binding proteins are believed to spend most of their time bound to DNA non-specifically searching for high affinity binding sites present in cis (Elf et al., 2007
; Gorman et al., 2007
; Gorman and Greene, 2008
; Visnapuu and Greene, 2009
).
Such diffusion and multimerization around binding sites may be considered for Nova based on several observations: clusters of YCAY elements are seen in both the GlyRα2 and GIRK2 intronic and 3′ UTR elements (Figures 6 and 8
), these are necessary for Nova-dependent action (splicing for GlyRα (Jensen et al., 2000
) and localization for GIRK2-1, Figure 7
), and clusters of binding elements may more generally be important to allow a sufficient concentration of RNABPs for effector action (Martin and Ephrussi, 2009
). Although the results in overexpression transfection studies (Figure 7
) were done with reporters that do not have introns, and therefore suggest that Nova binding the 3′ UTR can localize mRNA without prior intronic binding, the effects on localization were smaller than that quantitated for native GIRK2-1 RNA localization (Figure 6
), and do not preclude the possibility that intronic binding may precede localization in systems in which RNABP/RNAs are not overexpressed (i.e. in the nervous system). An interesting feature of a model in which Nova splicing is coupled to 3′ UTR deposition is that it predicts that Nova binding to pre-mRNA might be directly coupled both to generation of a specific spliced isoform (e.g. GlyRα2-E3A mRNA), and to its subsequent somato-dendritic localization and protein expression (Figure 9
). Assessment of this model will require careful biochemical mapping and coordination of key Nova binding sites from among the many identified intronic and 3′ UTR binding sites identified by HITS-CLIP (Licatalosi et al., 2008
).
Another, and not mutually exclusive factor that might influence Nova assembly with mature mRNA is through interaction with other RNABPs recruited onto the mature mRNA, as suggested for bicoid mRNA and several other localized RNAs (Arn et al., 2003
; Martin and Ephrussi, 2009
). Vegetal pole ribonucleoprotein particles assembled in Xenopus oocyte nuclei change upon export to the cytoplasm, where the RNA-protein complexes can recruit new RNABPs (Kress et al., 2004
). Such RNA-protein complexes are believed to be the substrates of mRNA localization and are assembled by the recruitment of trans-acting factors, including motor proteins and the interaction of RNABPs with cis-acting sequences often situated in the 3′ UTR regions. These cis-acting sequences are believed to be sufficient to mediate the localization of the mRNA (St Johnston, 2005
). This view has been challenged by the demonstration that, at least in the case of oskar, splicing and 3′ UTR sequences are independent requirements for mRNA localization (Hachet and Ephrussi, 2004
). Indeed, this and previous works (Hachet and Ephrussi, 2001
; Palacios et al., 2004
) suggest that cytoplasmic localization of an mRNA could be linked to the splicing-dependent deposition of RNABPs at the junction of the freshly assembled exons (Palacios, 2002
; Giorgi and Moore, 2007
). Hints of similar phenomenon are beginning to emerge from studies of mammalian ZBP RNABPs (ZBP1 with ZBP2, also known as KSRP), which are capable of localizing mRNAs harboring cis-acting sequences, and also interact with proteins implicated in regulating alternative splicing (Gu et al., 2002
; Rodriguez et al., 2008
). Thus the association of Nova with GlyR pre-mRNA may lead to deposition of factors that mark the mature mRNA with cis-acting RNABPs, presumably including Nova itself, that help direct the subsequent localization of the mRNA.
Nuclear/Cytoplasmic Fractionation Method
Brain tissue was Dounce homogenized in cold 10 mM HEPES (pH 7.9), 10 mM NaCl, 1.5 mM MgCl2, 0.2% Triton X-100, 10 mM NaF, protease inhibitors (Roche) and spun at 3000 × g for 3 min. Supernatant was collected as cytoplasmic fraction, and pellet was resuspended in 20 mM HEPES (pH 7.9), 25% glycerol, 1.5 mM MgCl2, 1.4 mM KCl, 0.2 mM EDTA, 0.5% NP-40, 10 mM NaF, protease inhibitors (Roche) and 5% DNase, incubated 5 min at 37°C, dialyzed against 1× PBS, pH 7.4, 1.5 mM MgCl2, 0.5% NP-40, 10 mM NaF, and collected as nuclear fraction. Both fractions were diluted to the same final volume and ultracentrifuged at 100,000 × g for 30 min.
Immunoblot Analysis
Equal volumes (20 μl) or equal total protein amounts (50 μg) were loaded onto 10% SDS-PAGE gels, and transferred to PVDF membranes (Millipore). Membranes were blocked and incubated with primary antibodies including rabbit anti-Nova (1:750) (Buckanovich and Darnell, 1997
), mouse anti-Hsp90 (1:1000; Transduction laboratories), and rabbit anti-brPTB (Polydorides et al., 2000
).
Oligonucleotide Probes
Oligonucleotide probes encoded: α1 residues 1050–1094 and 1096–1143 (Grenningloh et al., 1987
; Malosio et al., 1991
; Racca et al., 1997
, 1998
); α2 residues 1682–1726 and 1789–1810 (Kuhse et al., 1990
; Malosio et al., 1991
; Racca et al., 1997
, 1998
).
Primary Antibodies
Different antibodies, all giving the same pattern of staining (data not shown), were used at appropriate concentrations depending on the experiment to detect Nova epitopes. Affinity-purified rabbit anti-Nova (Yang et al., 1998
); human sera from POMA patients; affinity-purified human anti-Nova; mouse monoclonal antibody anti-gephyrin (Pfeiffer et al., 1984
); rabbit anti-synapsin (Bloom et al., 1979
; De Camilli et al., 1979
).
Specific antibodies and concentrations used in each figure are listed:
Figure 1
B: Single IC: anti-Nova human serum (1:1000); Goat anti-Human FITC (Jackson) 1:200
Figures 3
A,D,E: anti-Nova Rabbit Purified (1:750); Goat anti-Rabbit Biotinylated (Vector) 1:200
Figure 3
B: anti-Nova human serum (1:1000); Goat anti-Human Nanogold, 1 nm (Nanoprobrobes) 1:50
Figure 3
C: anti-Nova Rabbit serum (1:200); Goat anti-Rabbit, 15 nm Gold particle (British Biocell) 1:50
Figures 4
A,B: anti-Nova human serum (1:1000); Goat anti-Human Nanogold, 1 nm (Nanoprobrobes) 1:50
Figures 4
C–E: anti-Nova Rabbit serum (1:400); Goat anti-Rabbit, 10 nm Gold particle (British Biocell) 1:50 + anti-Gephyrin Mouse mAb (Boehringer Mannheim)1:100; Goat anti-Mouse, 15 nm Gold particle (British Biocell) 1:50
Figure 4
inset in A and B: anti-Nova human serum (1:1000); Goat anti-Human FITC (Jackson) 1:200 + anti-Synapsin Rabbit Serum 1:6000; Goat anti-Rabbit Cy3 (Jackson) 1:200
Figure 4
inset in C: anti-Nova Rabbit Purified (1:750); Goat anti-Rabbit FITC (Jackson) 1:200 + anti-Gephyrin Mouse mAb (Boeringher Mannheim) 1:100; Goat anti-Mouse Cy3 (Jackson) 1:200
Figures 5
A–C: anti-Nova Rabbit Purified (1:750); Goat anti-Rabbit FITC (Jackson) 1:200; and for ISH: Sheep anti-DIG (Boehringer Mannheim) 1:1000/donkey anti-Sheep Cy3 (Jackson) 1:200
Figures 5
D–G: anti-Nova human serum (1:200); Goat anti-Human Biotinylated (Vector) 1:200 and for ISH Sheep anti-DIG Nanogold, 1 nm (Nanoprobrobes) 1:50
Tissue Preparation
This study was performed in full accordance with the European Communities Council Directives (86/609/EEC) and the French national Committee (87/848) recommendations and the Rockefeller University Animal Care and Use Committee guidelines. Adult Sprague-Dawley rats (Janvier, France) were deeply anaesthetized with pentobarbital (60 mg/kg body weight, i.p.), and intracardially perfused. For fluorescent immunocytochemistry and in situ hybridization (ISH) animals were perfused with 4% paraformaldehyde (PFA) in phosphate buffer saline (0.1 M, pH 7.2; PBS). For EM immunocytochemistry and ISH animals were perfused with 4% PFA and 0.1% glutaraldehyde in PBS. Spinal cords were removed and postfixed in 4% PFA in PBS overnight at 4°C. Spinal cord sections were cut onto a vibratome and collected in PBS.
Fluorescent Immunocytochemistry on Spinal Cord Sections
Spinal cord 30 μm sections were rinsed in 50 mM NH4Cl in PBS for 15min, and permeabilized with 0.1% Triton X-100, 0.1% bovine gelatin in PBS for 10 min. The primary antibodies were incubated in the same buffer overnight at 4°C. Sections were then rinsed in PBS (3 × 10 min each) and incubated for 2 h at room temperature (RT) with the corresponding fluorescent secondary antibodies (carboxymethyl indocyanine (Cy3) or fluorescein isothiocyanate (FITC); Jackson ImmunoRes Labs (West Grove, PA, USA), in 0.1% bovine gelatin in PBS). After three washes in PBS (10 min each), sections were mounted on slides with Vectashield (Vector Lab.).
Fluorescent in situ Hybridization and Immunocytochemistry on Spinal Cord Sections
Fluorescent in situ hybridization (ISH) was as previously described (Racca et al., 1997
, 1998
). Digoxigenin labeled probes and Nova proteins were contemporaneously revealed in 100 mM Tris-HCl pH 7.5, 150 mM NaCl, 2% BSA, 0.3% Triton X-100, overnight, 4°C. The anti-digoxigenin and anti-Nova primary antibodies were detected by incubating sections with the appropriate secondary antibodies (in PBS, 2 h at RT). Each incubation was followed by three washes in PBS (10 min each). Finally, sections were mounted on slides with Vectashield (Vector Lab.)
Image Acquisition
The sections processed for fluorescent immunocytochemistry and ISH were observed with an epifluorescent Zeiss microscope, or a Leica confocal laser scanning microscope. All images presented here were obtained with the Leica confocal laser scanning microscope. For confocal images the background noise was reduced by applying a Gaussian filter to the optical sections.
Electron Microscopic Immunocytochemistry
100 μm thick vibratome sections were cryoprotected in 20% glycerol-20% sucrose in PBS, and permeabilized by freezing and thawing. Sections were collected in PBS, rinsed in 50 mM NH4Cl in PBS for 15 min, and 0.1% bovine gelatin in PBS for 10 min. The free-floating sections were incubated with the primary antibodies in 0.1% bovine gelatin in PBS, overnight, at 4°C. The following day, sections were rinsed three times in PBS (10 min each) and incubated with the secondary antibodies (in PBS-1% BSA for biotinylated antibodies, 2 h RT or in PBS-0.2% fish gelatin for gold antibodies, overnight 4°C). Biotinylated antibodies were revealed with the ABC Elite kit (in PBS, 1 h, RT; Vector Lab) and the peroxidase reaction was carried out in the presence of DAB and hydrogen peroxide (Sigma Fast, Sigma Aldrich). Nanogold-coupled antibodies were amplified as described (Trembleau et al., 1994
).
Electron Microscopic Pre-Embedding Non-Radioactive in situ Hybridization and Immunocytochemistry
Fifty micrometer sections were cryoprotected and permeabilized as for EM immunocytochemistry. Prehybridization and hybridization were as described above for fluorescent ISH. After the stringency washes, sections were rinsed in PBS and incubated in the primary antibody in 1% BSA in PBS, overnight, at 4°C. After three PBS rinses (10 min each), DIG molecules and the primary antibody were detected by gold- and biotin-coupled antibodies, respectively in 0.8% BSA, 0.2% Fish Gelatin in PBS, overnight, 4°C. After three PBS rinses (10 min each) sections were incubated in 4% PFA in PBS (10 min), rinsed three times in PBS (10 min each) and several times in cold distillated water. Gold-coupled sheep anti-DIG secondary antibodies were detected by a silver enhancement-gold toning protocol as described (Trembleau et al., 1994
). After three PBS rinses (10 min each), biotinylated antibodies were detected by peroxidase-DAB reaction as in classical immunocytochemical methods.
The sections processed for immunocytochemistry and ISH were dehydrated, osmicated and flat embedded in araldite (Fluka) resin. Ultrathin sections were prepared, mounted in copper grids and contrasted with uranyl acetate and lead citrate before examination under a Jeol CX II transmission electron microscope at 80 kV.
Electron Microscopic Post-Embedding Immunocytochemistry
Vibratome 200 μm thick sections were cryoprotected with 2 M sucrose in PBS, rapidly frozen in liquid propane, cryosubstituted and infiltrated with HM20 resin (Polysciences, Germany) in a cryosubstitution unit (AFS; Reichert). Polymerization was induced by UV light for 3 days, at −45°C. Ultrathin sections were prepared and mounted on formvar-coated 400 mesh nickel grids and processed for immunocytochemistry as follows. Sections were etched in a saturated sodium ethanolate solution for 2 s and rinsed in distilled H2O. After 10 min in 0.1% NaBH4, 50 mM Glycine in Tris Buffered Saline (TBS) (50 mM, pH 7.4, 50 mM TBS) the grids were rinsed by three washes of 50 mM TBS (10 min each). Grids were incubated in drops containing the primary antibodies in 50 mM TBS, overnight or 48 h at 4°C. Unbound primary antibodies were removed by 50 mM TBS washes and by incubating grids in drops of TBS (10 min) and 150 mM TBS (10 min, three times each). Primary antibodies were revealed by immunoglobulins coupled to calibrated gold particles (10 and 15 nm; British BioCell Int.) in Polyethylene Glycol 20000 (5 mg/ml PEG), 2% BSA in 150 mM TBS, 2 h at 37°C. The sections were then rinsed several times in 50 mM TBS, and stabilized in 2% glutaraldehyde in 50 mM TBS. Finally, after several washes in H2O, the grids were contrasted with uranyl acetate and lead citrate and examined under a Jeol CX transmission electron microscope at 80 kV.
Imaging Controls
Sense and random oligonucleotide probes and omission of any oligonucleotide or primary antibody or any single major step in the development of DAB reactions, fluorescent ISH and immunocytochemistry resulted in no labeling of any cells. All the oligonucleotide probes and antibodies used have been described and fully characterized previously (Malosio et al., 1991
; Sato et al., 1991
; Buckanovich et al., 1993
, 1996
; Racca et al., 1997
, 1998
). For double-fluorescence experiments, controls included either the independent omission of each single major step of the immunocytochemistry and/or ISH protocols, one at a time, or the replacement of the primary antibody by normal goat serum (Invitrogen; see Figure S1 in Supplementary Material).
Nova Clip
Nova cytoplasmic HITS-CLIP was performed as described previously (Licatalosi et al., 2008
), with minor modification. After the brain was UV-irradiated to covalently crosslink RNA-protein complexes, nuclear/cytoplasmic fractionation was performed as described above. In brief, extracts were then partially RNased to reduce the modal size of cross-linked RNA bound to Nova to ∼50 nt. Extracts were immunoprecipitated with a Nova1-specific antibody (Millipore), RNA linkers added with T4 RNA ligase, complexes purified by SDS-PAGE, protein removed with proteinase K, and RNA sequenced by reverse transcription and sequencing with an Illumina Bioanalyzer. One brain was utilized for Nova1 cytoplasmic CLIP which yielded total 1,504,346 tags, 815,172 of which were able to be mapped to the mouse genome and 48,518 of which mapped to unique locations. These unique tags were used for analysis of cytoplasmic Nova binding sites in GlyRα2 and GIRK2 transcripts. Additional CLIP experiments used rabbit anti-Nova antibody to perform CLIP as described (Licatalosi et al., 2008
), in biologic triplicate from mouse cortex (Figure 6
A) and in biologic duplicate from mouse spinal cord (Figure 8
B; Eom and Darnell, manuscript in preparation).
Nova HITS-CLIP binding sites were identified in the GIRK2 distal-most 3′ UTR (GIRK2A or GIRK2-1 isoform; NM_001025584.2) using the UCSC genome browser. Similarly, the GIRK2 intron 2 binding site identified (∼187,000 nt upstream of the 3′ UTR) was: (>mm9, chr16:95160755-95160878: CCATTCCTTC ACTATCCACA GCCCAAAAGC TAAGTCCTAA TCTCTGCATC TTAAAGACCA ATGTAAATGA CCCATACATC ATCACCACCA TCATCTTCAT CTTTGTCATC ACTGTCCTCT TCAT).
Heterokaryon Assays
HEK293-T or COS7/NIH3T3 cells were plated on gelatin coated coverslips. For shuttling of overexpressed Nova, transfection was performed before plating IMR-32/SK-N-BE(2) cells. Fusion of two different cells was induced by using 50% polyethylene glycol (PEG 3350; Sigma Aldrich) in water for 2 min. Cells were washed in PBS, and returned to medium containing 75 μg/ml cycloheximide for another 3 h incubation. Immunofluorescence was followed by fixation. HEK293-T, Neuro2a and COS7 cells were transfected with plasmid constructs by using Fugene6 (Roche) as described by manufacturer. After 24 h, the cells were fixed in PFA (4% in PBS) at RT for 15 min.
Constructs
T7-Nova1 have been described previously (Dredge and Darnell, 2003
). Flag-tagged ΔNLS-Nova1 and ΔNES-Nova1 were generated by PCR mutagenesis, deleting amino acids 25–40 and amino acids 318–335, respectively. GIRK2 YCAY elements were amplified by PCR and added into the pd1EGFP-N1 vector (Clontech). Mutations to YCAY elements were made by using QuickChange site-directed mutagenesis kit (Stratagene). In order to avoid effects from diffusion, the M9-NLS was added into vector.
GIRK2 YCAY primers are listed in Supplementary Material.
Immunofluorescence in Cultured Cells
For detection of proteins, we used monoclonal antibodies to T7 (Novagen), Flag (Sigma Aldrich) and hnRNP-C1 (generous gift from Dr. Piñol-Roma), and POMA serum for Nova proteins. All secondary antibodies were affinity-purified donkey antibodies to mouse or human IgG conjugated to a fluorochrome (Jackson Immunoresearch). Antibody incubations were for 1 h at RT in TBS with BSA (1%) and Triton X-100 (0.1%). Coverslips were mounted with Prolong gold antifade reagent (Invitrogen). Primary neuronal cultures were generated from E18.5 cortical neurons prepared as described (Eom et al., 2003
). Cells were fixed at 14 DIV for immunofluorescence staining with MAP2 monoclonal antibody (Sigma Aldrich) or FISH.
Fluorescence in situ Hybridization (Fish) in Cultured Cells
Five amino-modified oligonucleotide probes (approximately 50 nts each; see Supplementary Material) were designed to mouse GIRK2 mRNA and synthesized on a DNA synthesizer incorporating five amino-modified thymidines (C6dT), purified and chemically labeled using fluorophores (Cy3; Amersham) as described (http://www.singerlab.org/protocols
). In situ hybridization was completed as previously described (Eom et al., 2003
). Coverslips were mounted with Prolong gold antifade reagent (Invitrogen).
Quantitative Analysis
For image acquisition, exposure times and other settings were kept constant within the same experiment. In order to avoid bias in quantitation, regions of interest (ROI) were first chosen from fluorescence images of MAP2 (a dendritic marker) and were then transferred onto the fluorescent image of the same cell. The ROI was chosen 5–10 μm from the cell soma to specifically capture dendritic images. Total fluorescent intensities were collected from the same ROI for FISH, GFP or MAP2. A mask covering the dendrite was created based on MAP2 signals, and the fluorescent intensity signals of FISH or GFP was normalized to that of MAP2. In FISH analysis, the Cy5 channel was used for MAP2 analysis, Cy3 for FISH analysis. In GFP analysis, the Cy3 channel was used for MAP2 and Cy2 for GFP analysis. Quantitative analysis was performed by determining normalized fluorescence (pixel) intensities in neurites. Student’s t-test was applied to measure statistical significance. More than 10 cells were counted.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank members of our laboratories for discussion, and Yoshika Hayakawa-Yano for critical review of the manuscript. Help with post-embedding electron microscopy from P. Rostaing was greatly appreciated. We thank Dr. DeCamilli (Yale University Medical School) for the anti-synapsin antibody, and Dr. Piñol-Roma for the anti-hnRNP-C1 antibody. Claudia Racca and this work were initially supported by: an EC TMR Marie Curie Research Training Grant. This work was supported by grants from Association France Myopathies, Fondation pour la Recherche Médicale, Société de Secours des Amis des Sciences and Human Frontiers Science Programme (Alejandra Gardiol, Antoine Triller), and from the National Institutes of Health (R01 NS34389 and NS40955; Robert B. Darnell). Robert B. Darnell is an Investigator of the Howard Hughes Medical Institute.
The Supplementary Material for this article can be found online at http://www.frontiersin.org/neuralcircuits/paper/10.3389/neuro.04/005.2010/
De Camilli, P., Cameron, R., and Greengard, P. (1983). Synapsin I (protein I), a nerve terminal-specific phosphoprotein. I. Its general distribution in synapses of the central and peripheral nervous system demonstrated by immunofluorescence in frozen and plastic sections. J. Cell Biol. 96, 1337–1354.
Ruggiu, M., Herbst, R., Kim, N., Jevsek, M., Fak, J. J., Mann, M. A., Fischbach, G., Burden, S. J., and Darnell, R. B. (2009). Rescuing Z+ agrin splicing in Nova null mice restores synapse formation and unmasks a physiologic defect in motor neuron firing. Proc. Natl. Acad. Sci. U.S.A. 106, 3513–3518.