1
Dominick P. Purpura Department of Neuroscience, Albert Einstein College of Medicine, Bronx, NY, USA
2
Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA, USA
3
Faculty of Medicine, Department of Experimental Medicine and Biochemical Sciences, University of Rome “Tor Vergata”, Rome, Italy
4
Faculty of Medicine, Developmental and Molecular Genetics Section (VIB11), Catholic University of Leuven, Leuven, Belgium
Localization of mRNAs to dendrites and local protein synthesis afford spatial and temporal regulation of gene expression and endow synapses with the capacity to autonomously alter their structure and function. Emerging evidence indicates that RNA binding proteins, ribosomes, translation factors and mRNAs encoding proteins critical to synaptic structure and function localize to neuronal processes. RNAs are transported into dendrites in a translationally quiescent state where they are activated by synaptic stimuli. Two RNA binding proteins that regulate dendritic RNA delivery and translational repression are cytoplasmic polyadenylation element binding protein and fragile X mental retardation protein (FMRP). The fragile X syndrome (FXS) is the most common known genetic cause of autism and is characterized by the loss of FMRP. Hallmark features of the FXS include dysregulation of spine morphogenesis and exaggerated metabotropic glutamate receptor-dependent long term depression, a cellular substrate of learning and memory. Current research focuses on mechanisms whereby mRNAs are transported in a translationally repressed state from soma to distal process and are activated at synaptic sites in response to synaptic signals.
Synaptic plasticity or long-lasting alterations in the efficacy of synaptic connections between two neurons has been proposed to be the cellular substrate of learning and memory (Malinow and Malenka, 2002
; Bredt and Nicoll, 2003
; Collingridge et al., 2004
; Neves et al., 2008
). Persistent changes in synaptic efficacy are thought to involve at least two distinct phases – an early phase (on the order of minutes to hours) that is independent of new protein synthesis and a more long-lasting late phase (on the order of hours to days) that is dependent upon new protein synthesis (Bramham and Wells, 2007
; Bramham, 2008
; Richter and Klann, 2009
). The first mechanism typically involves activity-dependent alterations in the phosphorylation and number of postsynaptic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPA receptor) and N-methyl D-aspartate receptors (NMDA receptor), which are delivered to the postsynaptic membrane via exocytosis and removed via endocytosis (Collingridge et al., 2004
; Lau and Zukin, 2007
; Neves et al., 2008
; Holtmaat et al., 2009
; Newpher and Ehlers, 2009
). The second more long-lasting mechanism, postulated to underlie long-lasting memory storage, involves local or ‘on-site’ protein synthesis in dendrites by translational machinery strategically localized in and around synaptic sites (Bramham and Wells, 2007
; Bramham, 2008
; Richter and Klann, 2009
). The identities of the newly synthesized proteins that are required to maintain altered synaptic efficacy are, however, as yet unclear.
Considerable evidence indicates that alterations in synaptic strength are locked-in by alterations in structural remodeling. These morphological changes include induction of new dendritic spines, enlargement of spines already extant, and the splitting of single spines into two or more functional synapses (Bourne and Harris, 2008
; Holtmaat et al., 2009
; Newpher and Ehlers, 2009
). More recently, studies involving spine long-term potentiation (LTP), in which single spines are activated via photolysis of caged glutamate and imaged by means of two-photon microscopy, demonstrate that LTP is accompanied by enlargement of dendritic spines (Matsuzaki et al., 2004
; Harvey and Svoboda, 2007
; Harvey et al., 2008
; Lee et al., 2009
).
mRNA translation, a complex process involving the participation of many proteins and RNAs, is generally divided into three phases: initiation, elongation, and termination. Although each of these steps serves as a point of regulation to control the amount of protein that is produced, initiation is by far the most important and the salient features of this segment are important to keep in mind when considering how synapse stimulation induces changes in the translational apparatus.
Initiation begins with the formation of a ternary complex comprised of GTP, met-tRNA, and the initiation factor eIF2 (Pestova et al., 2007
). Together with additional initiation factors (eIF3, eIF5, eIF1, and eIF1A), the ternary complex associates with the 40S ribosomal subunit to form a 43S pre-initiation complex (Chaudhuri et al., 1999
; Majumdar et al., 2003
). In mammals, this large bolus of factors is recruited to the 5′ end of the mRNA through interactions with the initiation factors eIF4G and eIF3; at this point, it is referred to as the 48S pre-initiation complex (Lamphear et al., 1995
). The DExH box protein DHX29, a putative helicase, is also a required component of this complex for the translation of mRNAs with highly structured 5′ UTRs (Pisareva et al., 2008
). Proper positioning of the complex on the 5′ end requires the m7G cap-binding complex consisting of eIF4E, eIF4G and eIF4A, which together are called eIF4F (Gingras et al., 1999
). With the aid of the initiation factors eIF1, eIF1A, and DHX29, the 48S complex then scans the mRNA in the 3′ direction until it encounters an AUG initiation codon in the correct context, generally the first AUG, where it is joined by the 60S subunit to begin polypeptide elongation (Pestova et al., 1998
; Pestova and Kolupaeva, 2002
; Pisareva et al., 2008
).
The association between eIF4E, the cap binding factor, and eIF4G, is often regulated upon synaptic activity. The protein 4E BP can bind eIF4E and prevent eIF4G from joining with eIF4E, thus abrogating cap-dependent translation. Phosphorylation of 4E-BP either directly or indirectly by the kinase mammalian Target of Rapamycin (mTOR) causes it to dissociate from eIF4E, thereby liberating the factor to bind both the cap and eIF4G and initiate translation (Gingras et al., 1999
).
A diversity of synaptic signals regulates the translation of dendritic RNAs. Such localized stimuli include those that trigger synapse formation, pruning, neurite outgrowth/collapse, activity-dependent synaptic plasticity and injury-induced axonal regeneration. A critical regulator of activity-dependent protein synthesis in dendrites is metabotropic glutamate receptors (mGluRs). mGluRs are G protein-coupled receptors enriched at excitatory synapses throughout the brain where they act to regulate glutamatergic neurotransmission (Nakanishi, 1994
; Pin and Duvoisin, 1995
; Conn and Pin, 1997
). Group I mGluRs (mGluR1/5) activate phospholipase C, leading to mobilization, and the extracellular signal-regulated kinase (ERK)–mitogen-activated protein kinase (MAPK) pathway through which they modulate signal-to-nucleus communication. Signaling by mGluR1/5 is critical to synaptic circuitry formation during development and is implicated in forms of plasticity including LTP (Aiba et al., 1994
), long-term depression (LTD) (Aiba et al., 1994
; Oliet et al., 1997
; Palmer et al., 1997
; Huber et al., 2001
; Bear et al., 2004
) associative learning (Aiba et al., 1994
) and cocaine addiction (Kenny and Markou, 2004
). mGluR1/5 elicit synapse-specific modifications in synaptic strength and spine morphology by stimulating rapid local translation of dendritic mRNAs including Fmr1, encoding fragile X mental retardation protein (FMRP) (Greenough et al., 2001
). Expression of mGluR-LTD at Schaffer collateral to CA1 pyramidal cell synapses is mediated by persistent internalization of AMPARs and in adolescent wild-type (WT) mice requires de novo protein synthesis (Huber et al., 2000
; Snyder et al., 2001
) that is dependent on the mTOR (Hou and Klann, 2004
). The protein synthesis dependent is both age and region-specific in that CA1 mGluR-LTD in neonates (Nosyreva and Huber, 2005
) and mGluR-LTD at cortical synapses (Desai et al., 2006
) require neither local protein synthesis nor FMRP function.
Mammalian Target of Rapamycin is a central regulator of cell growth and proliferation, autophagy, ribosome biogenesis, and translation (Hay and Sonenberg, 2004
; Klann and Dever, 2004
; Sarbassov et al., 2005
) and a critical signaling pathway downstream of group I mGluRs. In neurons, components of the mTOR signaling cascade are present at synapses and are thought to influence synaptic plasticity via regulation of local protein synthesis (Tang and Schuman, 2002
). mTOR is activated in dendrites by stimulation of group I mGluRs (Klann and Dever, 2004
) and is required for mGluR-LTD at Schaffer-collateral to CA1 synapses (Hou and Klann, 2004
; Banko et al., 2006
; Antion et al., 2008
). Growing evidence indicates that dysregulation of mTOR is associated with human diseases, including cancer, diabetes, and autism (Jaworski and Sheng, 2006
; Sabatini, 2006
; Dann et al., 2007
).
The fragile X syndrome (FXS) is the most common heritable form of mental retardation (Chiurazzi et al., 2003
; Jacquemont et al., 2007
; Bassell and Warren, 2008
) as well as the most common known genetic cause of autism (Fryns et al., 1984
). FXS is caused by loss-of-function mutations in the FMRP, which is encoded by the FMR1 gene located on the X chromosome. In humans, the FXS typically results from expansion of a CGG repeat sequence in the 5′-untranslated region and silencing of the FMR1 gene (Feng et al., 1995
; Garber et al., 2006
). Patients with the FXS exhibit a wide-range of neurological deficits including cognitive impairment, seizures, emotional instability, sleep disorders, attention deficits, autonomic dysfunction, and autism (Hagerman and Hagerman, 2002
; O’Donnell and Warren, 2002
; Chiurazzi et al., 2003
; Jin and Warren, 2003
). FMRP, the gene product of the FMR1 gene, is an RNA binding protein that associates with many mRNAs, some of which encode proteins important for neuronal development and plasticity. FMRP controls activity-dependent dendritic mRNA localization (Dictenberg et al., 2008
), stability (Brown et al., 2001
; Miyashiro et al., 2003
; D’Hulst et al., 2006
; Zalfa et al., 2007
; Zhang et al., 2007
) and translational efficiency of dendritic mRNAs in response to stimulation of mGluRs (Weiler et al., 1997
; Todd et al., 2003
; Muddashetty et al., 2007
; Westmark and Malter, 2007
; Napoli et al., 2008
).
FMRP is detected in cell bodies, in dendritic shafts and branch points (Ferrari et al., 2007
), and at the base of synaptic spines and spine heads (Feng et al., 1997
). FMRP mRNA is translated near synapses in response to neurotransmitter activation (Weiler et al., 1997
); moreover, different models of experience-dependent plasticity such as whisker stimulation (Todd and Mack, 2000
), visual experience (Gabel et al., 2004
) and exposure to enriched environment (Irwin et al., 2005
) also promote FMRP mRNA translation.
Mice lacking FMRP (Bakker et al., 1994
) exhibit abnormalities in dendritic spine morphology (Bagni and Greenough, 2005
), cognitive deficits (O’Donnell and Warren, 2002
), exaggerated, protein synthesis-dependent LTD at Schaffer collateral to CA1 pyramidal cell synapses in adolescent WT mice, but protein synthesis-independent in neonatal WT mice (Nosyreva and Huber, 2005
) and in Fmr1 KO mice (Huber et al., 2002
; Koekkoek et al., 2005
; Hou et al., 2006
; Nosyreva and Huber, 2006
; Zhang et al., 2009
), and decreased LTP in the cortex (Li et al., 2002
; Chuang et al., 2005
; Larson et al., 2005
; Zhao et al., 2005
). Recent findings indicate that dysregulation of mTOR signaling in the hippocampus is an important functional link between overactivated mGluR signaling, aberrant protein synthesis, and exaggerated mGluR-LTD in Fmr1 KO mice (Sharma et al., 2009
). Several neurotransmitter receptors are dysregulated in the absence of FMRP. For example, the Fmr1 KO mouse exhibits aberrant numbers and/or activity of mGluR5 (Giuffrida et al., 2005
; Dolen et al., 2007
), NMDA (Gabel et al., 2004
; Desai et al., 2006
) and AMPA (Pilpel et al., 2009
; Schuett et al., 2009
) receptor signaling.
A hallmark feature of the FXS in humans, also observed in the Fmr1 KO mouse, is that of synaptic spine dysmorphogenesis. Spines on hippocampal and cortical neurons of Fmr1 KO mice exhibit enhanced density and are thinner and longer than those of age-matched WT mice, resembling an immature morphology. These observations indicate that FMRP normally controls processes involved in synapse maturation, stabilization, and elimination (Bagni and Greenough, 2005
). Electrophysiology experiments using cultured neurons indicate a delay in establishing synaptic connections during a brief window in brain development (Braun and Segal, 2000
). In the somatosensory cortex, although spine dysmorphogenesis persists into adulthood (Galvez and Greenough, 2005
; Restivo et al., 2005
), it can be rescued if the animals (mice) are exposed to enriched environmental conditions (Restivo et al., 2005
; Meredith et al., 2007
).
Synaptic activity in the brain can trigger long-lasting changes in synaptic strength; i.e., LTP and LTD. Early in development, LTP seems to be important for retaining nascent synapses, whereas LTD is more directly involved in activity-guided synapse elimination. The combination of these two phenomena contributes to the essential brain function of learning and memory. Impaired synaptic activity almost certainly results in impaired mental development (Vaillend et al., 2008
). New proteins that contribute to re-arrangements and re-assembling of synapse are made locally from mRNAs that are trafficked to dendrites (Steward and Levy, 1982
). Local changes in synaptic efficacy are protein synthesis dependent (Steward and Schuman, 2003
; Martin and Zukin, 2006
; Lin and Holt, 2008
); synaptic activity also promotes an increase in the number of polysomes at spines, presumably reflecting increased ‘on-site’ local synthesis of proteins critical to alterations in synaptic efficacy and structural remodeling of spines (Ostroff et al., 2002
).
In neurons, FMRP is packaged in ribonucleoprotein (RNP) granules, some components of which have been identified (Ohashi et al., 2002
; Brendel et al., 2004
; Kanai et al., 2004
; Ling et al., 2004
; Villace et al., 2004
; Dictenberg et al., 2008
; Napoli et al., 2008
). FMRP granules contain several proteins involved in translation (Pur alpha and beta, hnRNPU, Staufen, EF1alpha, PAPB1, eIF4E, ribosomal proteins), cytoskeleton dynamics (tubulin, tau, actin, Rac1, CYFIP), and the movement of molecules and organelles (dynein, myosins and kinesins). Importantly, αCAMKII and Arc, targets of FMRP that are critical to synaptic plasticity and known to be synthesized de novo in response to neuronal activity, are detected in these granules (Kanai et al., 2004
). FMRP-containing RNP granules are transported along microtubules in dendrites (Antar et al., 2004
; Ferrari et al., 2007
). These data strongly support a role for FMRP in dendritic delivery of RNA and regulation of local translation in specific synaptic compartments.
The molecular mechanism(s) by which FMRP controls translation has been investigated by assessing the polysome/mRNP distribution of the FMRP-containing complex. Unfortunately, there is little consensus regarding the results of these experiments. Initially, FMRP was shown to co-sediment entirely with polyribosomes or even larger granules (Ohashi et al., 2002
; Ceman et al., 2003
; Khandjian et al., 2004
; Stefani et al., 2004
) or to co-sediment essentially equally with polysomes and mRNPs (Feng et al., 1997
; Brown et al., 2001
; Ohashi et al., 2002
). Other laboratories found that FMRP co-sedimented mainly with the mRNP fraction (Siomi et al., 1996
; Ishizuka et al., 2002
; Zalfa et al., 2003
; Hock et al., 2007
; Napoli et al., 2008
). These findings are summarized in Figure 1
. While this apparent discrepancy could arise owing to differences in methodologies, brain region or cell type, a recent study by Bagni and colleagues shows that FMRP regulates synaptic translation at the level of initiation (Napoli et al., 2008
). These authors showed that FMRP inhibits translation initiation through an interacting factor, CYFIP1/Sra1, which binds the cap binding factor eIF4E. Interestingly, CYFIP1 binds eIF4E via a novel domain with a characteristic ‘reverse L shaped’ structure that is also assumed by the canonical eIF4E-binding motif (Marcotrigiano et al., 1999
). The importance of CYFIP1 in the FMRP-regulatory circuit was demonstrated by an increased level of proteins encoded by known FMRP target mRNAs upon reduction of CYFIP1 in neurons. Furthermore, the Brain Cytoplasmic RNA 1, another FMRP binding partner, increases the affinity of FMRP for the CYFIP1-eIF4E complex in the brain (Napoli et al., 2008
).
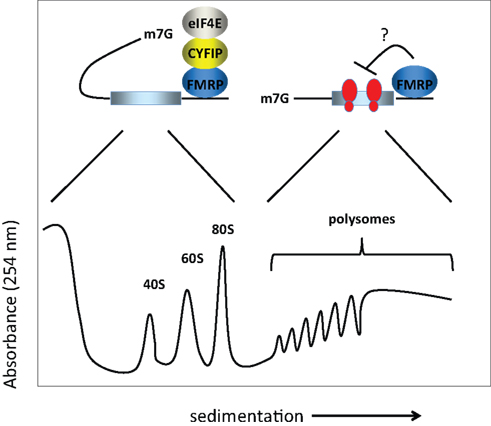
Figure 1. Model for FMRP activity. FMRP has been reported to sediment in both the RNP fractions (i.e., less the 80S monosome) as well as with polysomes in sucrose gradients. In the RNP fractions, FMRP is associated with CYFIP1 and the cap binding factor eIF4E. Here, we show that CYFIP1 inhibits translational initiation by binding initiation factor eIF4E and preventing assembly of eIF4E with eIF4G. In the polysome fractions, FMRP may inhibit protein synthesis by inhibiting polypeptide elongation (i.e., by retarding the rate at which ribosomes move along the mRNA). Thus, FMRP may inhibit translation at two distinct steps, initiation and elongation. 40S and 60S are the ribosomal subunits, 80S is the monosome or complex of the two subunits, and m7G is the cap, a modified nucleotide containing a methyl tag, present at the 5′ end of the mRNA.
Translation in neurons is regulated by synaptic activity. In cultured neurons, protein synthesis can be activated by several synaptic stimuli such as BDNF and DHPG. BDNF stimulates translation at synapses via the mTOR and ERK–MAPK pathways, and likely involves the modulation of translational initiation. Moreover, BDNF activates the translation of Arc and αCaMKII mRNAs (Yin et al., 2002
; Schratt et al., 2004
; Napoli et al., 2008
), two FMRP targets (Zalfa et al., 2003
; Park et al., 2008
). Stimulation of neurons by BDNF or DHPG promotes dissociation of CYFIP1 from eIF4E at synapses, an event associated with initiation of protein synthesis. Thus, FMRP-dependent translational repression in brain is mediated, at least in part, by CYFIP1 (Napoli et al., 2008
). Activity dependent changes in FMRP phosphorylation is observed in dendrites and show a temporal correlation with the translational profile of select FMRP target transcripts (Narayanan et al., 2007
). Post-translational modifications of FMRP and or CYFIP1 may account for such reversible control of local translation. Finally, the temporal activity of FMRP seems also to be regulated by mGluR signaling. mGluR-LTD induces a transient, translation-dependent increase in FMRP that is rapidly reversed through degradation by the ubiquitin-proteasome pathway (Hou et al., 2006
). Synaptic stimulation also affects the proteasome pathway. NMDA receptor-dependent synaptic stimulation promotes a redistribution of proteasomes from dendritic shafts to synaptic spines and thus provides a mechanism for local protein degradation (Bingol and Schuman, 2006
). Synaptic proteasomal activity seems to affect a large number of synaptic proteins including specific postsynaptic scaffolds whose turnover after ubiquitination is enhanced. Interestingly, the long-term activity-dependent increases of CaMKII and PSD-95, encoded by two mRNAs that are translationally repressed by FMRP (Zalfa et al., 2003
; Hou et al., 2006
; Muddashetty et al., 2007
; Zalfa et al., 2007
) requires proteasome-mediated degradation (Ehlers, 2003
). These findings suggest an interplay between mRNA localization and FMRP degradation that is integrated with neuronal activity and directly regulated by the ubiquitin-proteasome system.
Fragile X mental retardation protein key target mRNAs encode proteins involved in synaptic structure and function (Bagni and Greenough, 2005
; Bassell and Warren, 2008
). Among them an interesting target candidate is Arc/Arg 3.1 (Zalfa et al., 2003
; Park et al., 2008
). Arc/Arg 3.1 mRNA translation is rapidly up-regulated following mGluR stimulation, an effect that is not detected in the absence of FMRP. Arc/Arg 3.1 mRNA synaptic translation has been proposed to be critical for synapse-specific LTD (Park et al., 2008
). Thus Arc/Arg 3.1 reduces the number of GluR2/3 receptors leading to a decrease in AMPAR-mediated synaptic currents (Rial Verde et al., 2006
). Consequently, Arc/Arg 3.1 may be one on the ‘key LTD’ synaptic proteins whose upregulation would lead to the impaired hippocampal mouse LTD observed in FMR1 KO mice. Bear et al. (2004)
proposed that a net loss of synaptic AMPA and NMDA receptors, possibly due to an increased internalization mediated by one or several LTD proteins, would contribute to a weakened spine and possibly the dysmorphogenesis observed in the FXS.
Fragile X mental retardation protein can specifically recognize its mRNA targets in multiple ways: directly via RNA structures such as G-quartets and/or G-rich and/or U-rich sequences, via BC1 RNA, or microRNAs (Bagni and Greenough, 2005
; Bassell and Warren, 2008
). It is unclear whether the alternative binding motifs or non-coding RNAs are specific for different messengers or, rather, function as alternative modes to target the same mRNAs during development or synaptic response. Further work is needed to understand the functional consequences of impaired local protein synthesis in the developing brain and how this correlates with the autistic and FXS phenotypes.
The CPEB is an RNA binding protein that recognizes target RNAs via specific sequences in their 3′-untranslated regions. CPEB promotes dendritic delivery of mRNAs along microtubules (Huang et al., 2003
) and polyadenylation-induced translation of mRNAs in response to synaptic activity (Wu et al., 1998
; Huang et al., 2002
; Shin et al., 2004
; Du and Richter, 2005
) (Figure 2
depicts key features of CPEB-controlled translation). In neurons, CPEB localizes to the cell body and postsynaptic sites and, within dendrites, is packaged in RNP granules that localize near synapses. In neurons CPEB targets a number of mRNAs that encode proteins critical to synaptic plasticity (Du and Richter, 2005
; Pique et al., 2008
). Disruption of CPEB stunts the development of dendritic arbors and prevents neurons from integrating into the functional visual system of Xenopus laevis (Bestman and Cline, 2008
). Moreover, loss of CPEB activity in mice prevents the formation of some forms of synaptic plasticity and interferes with memory formation (Alarcon et al., 2004a
; Berger-Sweeney et al., 2006
; McEvoy et al., 2007
; Miniaci et al., 2008
). CPEB knockout mouse exhibit reduced theta burst-induced LTP at Schaffer collateral to CA1 pyramidal cell synapses of the hippocampus (Alarcon et al., 2004a
). In addition, these mice exhibit a deficit in extinction of memory, a phenomenon whereby behavioral responses diminish and eventually become extinct when there is no reinforcement of the memory (Berger-Sweeney et al., 2006
; McEvoy et al., 2007
; Miniaci et al., 2008
). Although extinction requires the formation of new memories, the underlying mechanisms by which it occurs are probably distinct from those of memory acquisition and consolidation (Abel and Lattal, 2001
). These data suggest an important role for CPEB in linking local protein synthesis and morphological plasticity to synaptic plasticity.
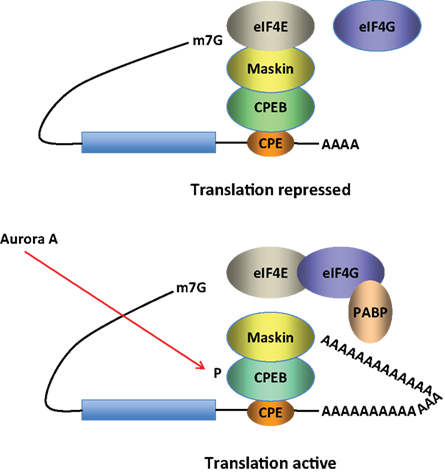
Figure 2. Model for CPEB activity. Some mRNAs contain a cytoplasmic polyadenylation element (CPE), which is bound by CPEB. CPEB also interacts with Maskin (or Neuroguidin, a functionally related protein), which in turn interacts with the cap (m7G) binding protein eIF4E. Such mRNAs are translationally inactive because Maskin inhibits the association of eIF4E and eIF4G, another initiation factor that helps recruit the 40S ribosomal subunit to the 5′ end of the mRNA. Cues such as NMDA receptor activation stimulate the kinase Aurora A, which phosphorylates CPEB, an event that causes poly(A) tail elongation. poly(A) binding protein (PABP) binds the newly elongated poly(A) tail and recruits eIF4G. The PABP-eIF4G dimer helps to displace Maskin from eIF4E, allowing eIF4G to bind eIF4E and initiate translation.
Zearfoss et al. identified growth hormone (GH) as one protein whose level is reduced ∼10-fold in the CPEB KO hippocampus (Zearfoss et al., 2008
). GH mRNA contains no binding sites for CPEB, and both GH mRNA and pre-mRNA are reduced in the KO versus WT hippocampus, suggesting that an mRNA encoding a transcription factor that regulates GH gene expression is under the control of CPEB. Indeed, CPEB controls the synthesis of c-jun, which regulates the expression of the GH gene. GH also induces LTP in hippocampal slices; GH-induced LTP, like electrical stimulation-elicited LTP, is reduced at CA1 synapses of the CPEB KO mouse. Moreover, LTP induced by GH or theta burst stimulation is reduced if polyadenylation is inhibited by the drug cordycepin (3′-deoxyadenosine), which terminates adenylate polymers. These and other results suggest that GH acts in both autocrine and paracrine fashion to regulate plasticity through CPEB-mediated control of c-jun translation.
The CPEB is also present in invertebrates, where it may act as a polyglutamine-containing prion-like factor to control plasticity; in Aplysia sensory neurons where CPEB RNA expression has been disrupted by an antisense oligonucleotide, long-term facilitation is not maintained (Si et al., 2003b
). The isoform of Aplysia neuronal CPEB differs from the vertebrate CPEB noted above in that it contains a high concentration of glutamine residues. Polyglutamine can be found in proteins that resemble a prion, an infectious agent consisting of self-reproducing protein. The authors (Si et al., 2003a
,b
) suggested that this CPEB isoform could assume a prion-like structure following synaptic experience and form an indelible synaptic tag. If true, then CPEB, rather than the proteins derived from CPEB-regulated translation, would comprise a tag that distinguishes experienced from naive synapses. Si et al. (2003a)
demonstrated that Aplysia CPEB resembled a prion in that it was protease resistant and had a fast sedimentation rate in sucrose gradients. More compelling evidence came from experiments in transduced yeast, where Aplysia CPEB assumed two forms: one that aggregated (i.e., prion-like) and one that did not. Very unexpectedly, the aggregated form of CPEB bound to RNA and converted the nonaggregated form into an aggregated form. Such epigenetic inheritance is a hallmark of prion formation and lead the authors to opin that synaptic activity could cause neuronal Aplysia CPEB to become prion-like and stimulate the translation of RNAs, cause it to alter its substrate specificity, or release some mRNAs from a translationally dormant state. Si et al. (2003a)
also suggested that once it becomes prion-like, CPEB would need no further stimulation to maintain activity.
Vertebrates contain three additional CPEB-like genes, all of which are expressed in the brain (Theis et al., 2003
). Two of these CPEB-like proteins contain some polyglutamine but they do not have a strong affinity for the CPEB binding site and do not regulate polyadenylation (Huang et al., 2006
). Although the relationship between vertebrate CPEB proteins and prions, if any, remains to be determined, a Drosophila CPEB isoform, Orb2, contains polyglutamine and is important for long term memory; when the polyglutamine is deleted, long term memory is disrupted (Keleman et al., 2007
). While this study did not examine prion formation, or even mRNA translation, it does point to the glutamine stretch of the CPEB isoform as being potentially important for memory formation.
Synaptic plasticity is a key phenomenon in the development and function of neuronal networks. Several studies provide strong evidence linking mRNA trafficking and dendritic protein synthesis to synaptic plasticity. Relatively little is known about how these processes are interrelated, including coupling of synaptic signalling to the translational machinery, selective translation of specific mRNA at synapses, and specific delivery of newly transcribed mRNAs to activated synaptic sites. Two molecules that connect receptor signalling to local protein synthesis in spines are CPEB and FMRP. CPEB localizes to postsynaptic sites where it regulates translation of proteins critical to synaptic plasticity (Du and Richter, 2005
; Pique et al., 2008
), remodeling of dendritic arborization and organization of the functional visual system (Bestman and Cline, 2008
). CPEB KO mice exhibit deficits in synaptic plasticity and memory formation (Alarcon et al., 2004b
; Berger-Sweeney et al., 2006
; McEvoy et al., 2007
; Miniaci et al., 2008
). These findings suggest an important role for CPEB in linking local protein synthesis and dendritic remodeling to synaptic plasticity. Similarly, FMRP localizes to synapses where it regulates translation of proteins critical to synaptic plasticity and structural remodeling. Fmr1 KO mice exhibit abnormal dendritic spines, cognitive deficits, aberrant synaptic plasticity in different areas of the brain, and a reduced number of mGluR and AMPA receptors. Pharmacological or genetic manipulations that attenuate activity of group I mGluRs (Dolen et al., 2007
) or exposure to an enriched environment increases AMPAR subunit abundance in the visual cortex (Restivo et al., 2005
; Meredith et al., 2007
) and rescues, at least in part, the hallmark behavioral and synaptic abnormalities of the fragile X phenotype. Dissection of the molecular mechanisms underlying these rescue experiments represent a starting point in our understanding of FMRP function and of the linkage between neuronal activity, receptor activation, and local protein synthesis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O’Donnell, W. T., Tenenbaum, S. A., Jin, X., Feng, Y., Wilkinson, K. D., Keene, J. D., Darnell, R. B., and Warren, S. T. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487.
Giuffrida, R., Musumeci, S., D’Antoni, S., Bonaccorso, C. M., Giuffrida-Stella, A. M., Oostra, B. A., and Catania, M. V. (2005). A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J. Neurosci. 25, 8908–8916.
Holtmaat, A., Bonhoeffer, T., Chow, D. K., Chuckowree, J., De Paola, V., Hofer, S. B., Hubener, M., Keck, T., Knott, G., Lee, W. C., Mostany, R., Mrsic-Flogel, T. D., Nedivi, E., Portera-Cailliau, C., Svoboda, K., Trachtenberg, J. T., and Wilbrecht, L. (2009). Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat. Protoc. 4, 1128–1144.
Irwin, S. A., Christmon, C. A., Grossman, A. W., Galvez, R., Kim, S. H., DeGrush, B. J., Weiler, I. J., and Greenough, W. T. (2005). Fragile X mental retardation protein levels increase following complex environment exposure in rat brain regions undergoing active synaptogenesis. Neurobiol. Learn. Mem. 83, 180–187.
Koekkoek, S. K., Yamaguchi, K., Milojkovic, B. A., Dortland, B. R., Ruigrok, T. J., Maex, R., DeGraaf, W., Smit, A. E., VanderWerf, F., Bakker, C.E., Willemsen, R., Ikeda, T., Kakizawa, S., Onodera, K., Nelson, D. L., Mientjes, E., Joosten, M., De Schutter, E., Oostra, B. A., Ito, M., and De Zeeuw, C. I. (2005). Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in fragile X syndrome. Neuron 47, 339–352.
McEvoy, M., Cao, G., Llopis, P. M., Kundel, M., Jones, K., Hofler, C., Shin, C., and Wells, D. G. (2007). Cytoplasmic polyadenylation element binding protein 1-mediated mRNA translation in Purkinje neurons is required for cerebellar long-term depression and motor coordination. J. Neurosci. 27, 6400–6411.
Napoli, I., Mercaldo, V., Boyl, P. P., Eleuteri, B., Zalfa, F., De Rubeis, S., Di Marino, D., Mohr, E., Massimi, M., Falconi, M., Witke, W., Costa-Mattioli, M., Sonenberg, N., Achsel, T., and Bagni, C. (2008). The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134, 1042–1054.
Ohashi, S., Koike, K., Omori, A., Ichinose, S., Ohara, S., Kobayashi, S., Sato, T. A., and Anzai, K. (2002). Identification of mRNA/protein (mRNP) complexes containing Puralpha, mStaufen, fragile X protein, and myosin Va and their association with rough endoplasmic reticulum equipped with a kinesin motor. J. Biol. Chem. 277, 37804–37810.
Park, S., Park, J. M., Kim, S., Kim, J. A., Shepherd, J. D., Smith-Hicks, C. L., Chowdhury, S., Kaufmann, W., Kuhl, D., Ryazanov, A. G., Huganir, R. L., Linden, D. J., and Worley, P. F. (2008). Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83.
Weiler, I. J., Irwin, S. A., Klintsova, A. Y., Spencer, C. M., Brazelton, A. D., Miyashiro, K., Comery, T. A., Patel, B., Eberwine, J., and Greenough, W. T. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. U.S.A. 94, 5395–5400.