Dual transneuronal tracing in the rat entorhinal-hippocampal circuit by intracerebral injection of recombinant rabies virus vectors
1
Division of Systems Neuroscience, Tohoku University Graduate School of Life Sciences, Tohoku, Japan
2
Kavli Institute for Systems Neuroscience and Centre for the Biology of Memory, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
3
Department of Anatomy and Neurosciences, VU University Medical Center, Amsterdam, The Netherlands
Dual transneuronal tracing is a novel viral tracing methodology which employs two recombinant viruses, each expressing a different reporter protein. Peripheral injection of recombinant pseudorabies viruses has been used as a powerful method to define neurons that coordinate outputs to various peripheral targets of motor and autonomic systems. Here, we assessed the feasibility of recombinants of rabies virus (RV) vector for dual transneuronal tracing in the central nervous system. First, we examined whether two different RV-vectors can double label cells in vitro, and showed that efficient double labeling can be realized by infecting targeted cells with the two RV-vectors within a short time interval. The potential of dual transneuronal tracing was then examined in vivo in the entorhinal-hippocampal circuit, using the chain of projections from CA3 pyramidal cells to CA1 pyramidal cells and subsequently to entorhinal cortex. Six days after the injection of two RV-vectors into the left and right entorhinal cortex respectively, double-labeled neurons were observed in CA3 bilaterally. Some double-labeled neurons showed a Golgi-like labeling. Dual transneuronal tracing potentially provides a powerful and sensitive method to study issues such as the amount of convergence and divergence within and between circuits in the central nervous system. Using this sensitive technique, we established that single neurons in CA3 are connected to the entorhinal cortex bilaterally with only one synaptic relay.
Neurotropic viruses, such as α-herpesviruses (herpes simplex virus type 1 and pseudorabies virus) and rhabdoviruses (rabies virus), move from neuron to neuron via synapses and self-amplify through replication in infected neurons. These features make these viruses a powerful tool to reveal the hierarchical connectivity in complex neuronal circuits (for reviews, see Aston-Jones and Card, 2000
; Kelly and Strick, 2000
; Kuypers and Ugolini, 1990
; Loewy, 1998
; Norgren and Lehman, 1998
). Recently, a novel viral tracing methodology called dual viral transneuronal tracing has been introduced by employing two recombinants of pseudorabies virus Bartha (PRV-Bartha) engineered to express unique reporter proteins (Banfield et al., 2003
; Cano et al., 2004
; Enquist and Card, 2003
; Jansen et al., 1995
; Kim et al., 1999
; Song et al., 2005
; Ter Horst, 2000
). With such an approach, different neuronal circuits can be visualized separately, and the divergent projections of a single neuron as an element in two neuronal circuits can be detected by the co-existence of the two different reporter proteins. By peripheral injection into muscles or organs in rodents, this method has been effective in defining neurons controlling multiple muscles or neurons that coordinate the outputs to various peripheral targets of the motor and autonomic systems (Billig et al., 2000
, 2001
; Kerman et al., 2003
, 2006
; Krout et al., 2003
; Ueyama et al., 1999
). With central application, dual transneuronal tracing will provide the potential to examine the complicated connections of multiple neuronal circuits in the central nervous system. PRV-Bartha is to be preferred over other α-herpesviruses since it is less likely to induce neuronal degeneration and therefore it will not infect anatomically non-connected nearby cells. However, when applied at a high titer, neuronal death and subsequent unintended infection may occur (Loewy et al., 1991
). PRV-Bartha also reportedly infects glial cells and it does not seem to infect primates (Loewy, 1998
; Ugolini, 1995
). Therefore, an alternative viral vector, such as rabies virus (RV) is to be preferred for applications in the central nervous system. RV is a nonsegmented negative-strand RNA-virus which belongs to the genus Lyssavirus of the family Rhabdoviridae. RV selectively infects neurons, not glial cells. Moreover, RV does not cause cellular lysis even when applied at a high titer, and therefore local spread of the viruses from the infected neurons into nearby cells is limited. Furthermore, unlike PRV-Bartha, RV can infect primates in addition to rodents (Kelly and Strick, 2000
; Nassi and Callaway, 2007
; Ugolini, 1995
). Like PRV-Bartha, RV transports transsynaptically to higher order neurons in a time dependent manner. These features make RV an ideal transneuronal tracer not only in rodents but also in primates.
Recently, we developed a recombinant RV-vector (rHEP5.0-CVSG) which acts as a potential tool for selective gene delivery in the central nervous system. This newly developed vector infects neurons from axon terminals and effectively propagates transsynaptically and retrogradely. Furthermore, infection with this viral strain, which expresses monomeric red fluorescent protein (mRFP), produces Golgi-like labeling of neurons in vivo by immunostaining against mRFP (Inoue et al., 2004
). These results indicate that, by inserting unique reporter proteins, this newly developed vector can be used as a potential neuroanotomical tool for dual transneuronal tracing. The goal of the present study is to examine the utility of dual viral tracing with recombinant RV-vectors. With this purpose in mind, we engineered several recombinant RV-vectors expressing different reporter proteins. We examined the critical condition for efficient double labeling in vitro with the use of cell cultures. In order to assess the reliability of this novel methodology in vivo, we chose the hippocampal region, whose afferent and efferent connections are well-known (for an overview, see Witter and Amaral, 2004
). Briefly, areas of the hippocampal region, including the dentate gyrus, areas CA3, CA1 and the entorhinal cortex, exhibit a series of connections that are ideal to test the potential of our viral vectors for single and double labeling. Projections arising in CA3 are relatively confined, mainly targeting the lateral septum and CA1, bilaterally. The bilateral projections from CA3 to CA1, also referred to as Schaffer collaterals, are overall symmetrical and the terminal density from the two sides are comparable. Moreover, these bilateral projections appear to arise from single cells in CA3 (Ishizuka et al., 1990
; Laurberg and Sorensen, 1981
; Li et al., 1994
; Swanson and Cowan, 1977
; Swanson et al., 1978
, 1980
). Furthermore there are no direct projections from the CA3 pyramidal cells to the entorhinal cortex (Kerr et al., 2007
; see also Witter and Amaral, 2004
). In contrast, CA1 pyramidal cells project to the entorhinal cortex and with the exception of those arising from the most dorsal tip of CA1, these projections are strictly ipsilateral (van Groen and Wyss, 1990
). On the basis of this known anatomy, it may be likely inferred that a single CA3 cell may be able to influence the entorhinal cortex bilaterally, with only one CA1 synapse in between (Figure 1
). This inferred connectivity has however not been established and our new dual viral tracing technique would be perfectly suited to confirm this connectional chain by injecting one of the two different vectors in the entorhinal cortex on either side. Since no apparent differences in axonal lengths or retrograde transport velocity between the two CA1 to entorhinal pathways have been established, and the bilateral CA3 to CA1 projections are symmetrical, originating from single neurons, we expect that possible variations in the progression of the virus infections from the two injected areas in medial entorhinal cortex (MEC) on either side are minimal. These features make the CA3 pyramidal cells, and their transsynaptic connectivity with the entorhinal cortex an appropriate model to test the possibility of dual viral tracing in the central nervous system. The results show that dual transneuronal tracing is feasible and that neurons identified in this manner may exhibit Golgi-like staining comparable to that seen in single transneuronal tracing. The latter feature contributes to the power of this approach to unravel complex circuitry in the central nervous system.
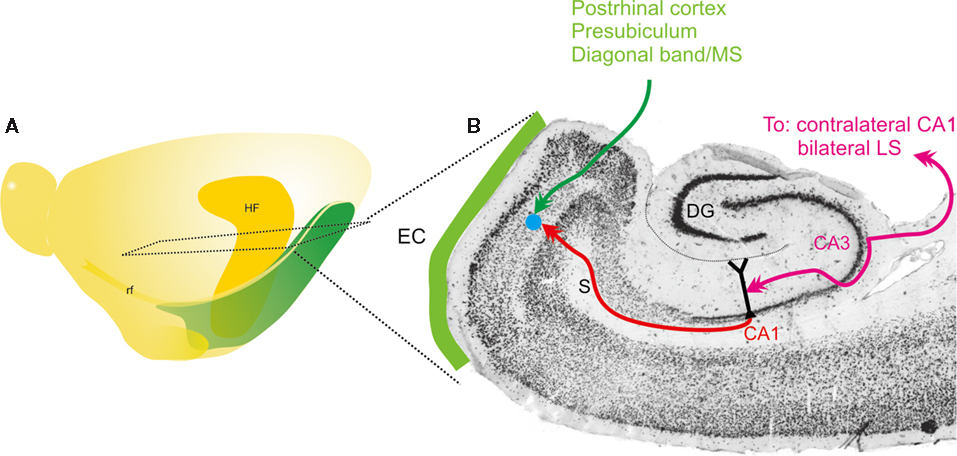
Figure 1. Schematic representation of main connectivity of the CA3-to-CA1-to-entorhinal connections. (A) Schematic drawing of the left hemisphere of the rat brain indicating the position and extend of the hippocampal formation (HF) and entorhinal cortex. Also indicated is the level of the horizontal section, represented in (B). (B) Horizontal section of the rat hemisphere illustrating the entorhinal cortex (EC), some of its main inputs from postrhinal cortex, presubiculum and diagonal band/medial septum (MS) in green. In purple, the bilateral connectivity of CA3 to CA1 and the lateral septum (LS) is indicated. Red indicates the ipsilateral projection from CA1 to entorhinal cortex. The blue dot schematically illustrates the deposit of the RV-vector.
Viruses
The construction of the RV full-length cDNA vector (pHEP5.0-CVSG) was described previously (Inoue et al., 2004
). Three recombinant RV-vectors were newly generated for dual viral tracing by inserting transgenes: the LacZ gene which encodes for β-galactocidase (β-gal) or the gene for green fluorescent protein (GFP) variants, into the multiple insertion site of the virus vector genome (Figure 2
). To create a vector which expresses either β-gal or the yellow fluorescent protein named Venus (Nagai et al., 2002
), a PCR fragment of LacZ cDNA (Invitrogen, USA) and Venus cDNA containing the Sbf I site, open reading frame, and Sac II were amplified and inserted into the same site of pHEP5.0-CVSG, respectively. The resulting plasmids were designated pHEP5.0-CVSG-β-gal and pHEP5.0-CVSG-Venus.
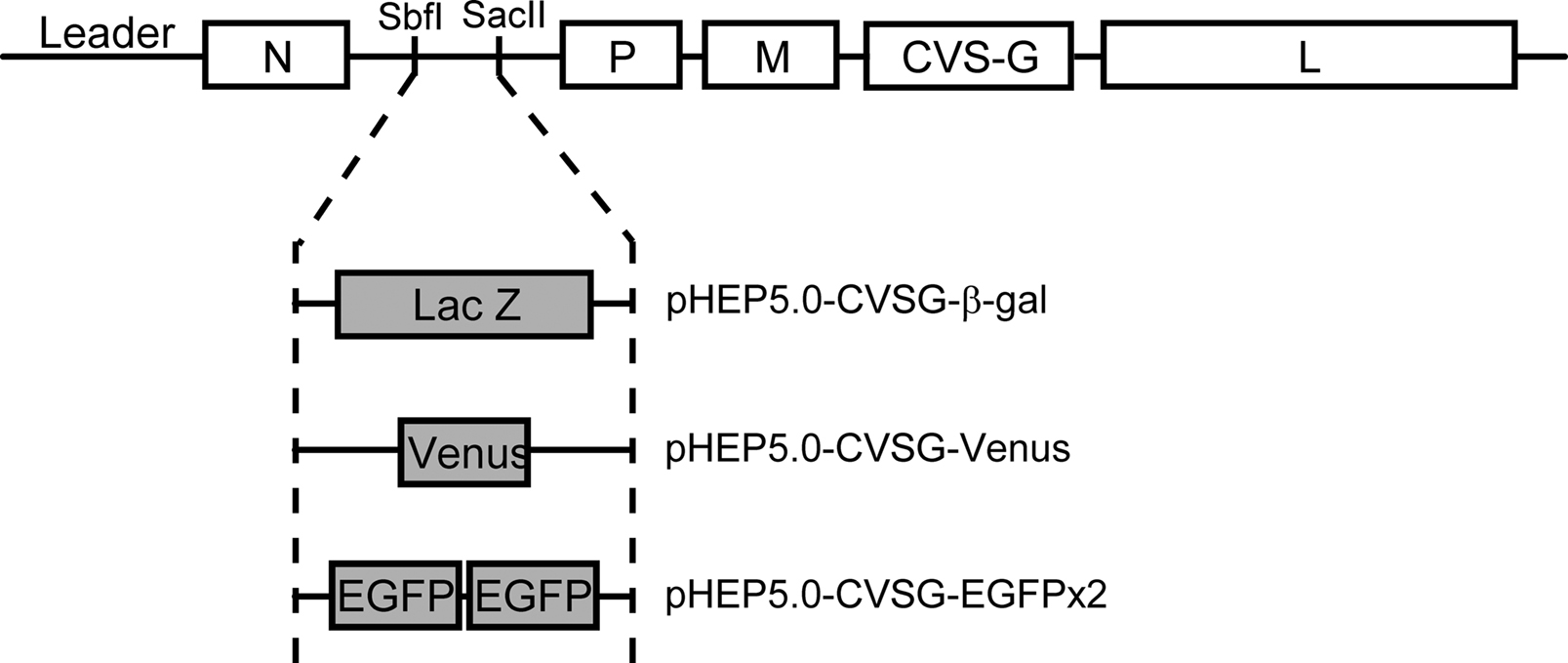
Figure 2. Schematic diagram showing the genomes of the recombinant rabies virus vectors. The recombinant rabies virus vector was based on the HEP-Flury strain with the exception of the G gene which was derived from the CVS strain. Three vectors were generated from this vector by inserting different reporter genes encoding either β-gal, Venus or enhanced green fluorescent protein (EGFP), into the N-P intergenic sequence which contains Sbf I and Sac II site.
To create a vector which highly expresses EGFP, two EGFP genes were inserted into pHEP5.0mi-CVSG, which has two transcriptional units between the N gene and P gene region. This vector was generated by inserting the following synthesized sequence, 5′-CCTGCAGGATTTGCGGCCGCATAAGCTAGCAAAAAAACTAACACCGTACGTTTTCCGCGG-3′ (Sbf I site, Not I site, poly A signal, transcription start signal, BsiW I site, Sac II site underlined in order), into the multiple insertion site of the pHEP5.0-CVSG. Two sets of PCR fragments of EGFP cDNA (Clontech, USA), one containing the Sbf I site, open reading frame, and the Not I site, and the other containing the BsiW I site, open reading frame, and the Sac II site, were amplified and inserted into the multiple insertion site of pHEP5.0mi-CVSG, resulting in pHEP5.0-CVSG-EGFPx2.
The recombinant virus was rescued by using mouse neuroblastoma cells of A/J mouse origin (NA) as described previously (Inoue et al., 2003
). The rescued virus generated from the pHEP5.0-CVSG-β-gal, pHEP5.0-CVSG-Venus, and pHEP5.0-CVSG-EGFPx2 were designated rHEP5.0-CVSG-β-gal, rHEP5.0-CVSG-Venus, and rHEP5.0-CVSG-EGFPx2, respectively. To determine the viral titer, we conducted direct florescent test using NA cells, as described elsewhere (Wiktor et al., 1984
). A viral suspension was kept in small aliquots at −80°C. Each aliquot was thawed in a safety cabinet before each experiment.
Viral Infection: In Vitro
NA cells were plated on glass coverslips in a 35-mm dish and maintained in minimum essential medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) at 37°C. To evaluate the ability of the rabies virus vectors to infect a cell in which an ongoing infection exists, two vectors expressing different reporter proteins, rHEP5.0-CVSG-β-gal and rHEP5.0-CVSG-Venus, were applied to the same culture dish at different time intervals. First, 300 μl of rHEP5.0-CVSG-Venus (1.0 × 107 focus-forming units (FFU)/ml) was applied. After a certain time delay (0 h, 2 h, 6 h, 12 h), 300 μl of rHEP5.0-CVSG-β-gal (1.0 × 107 FFU/ml) was applied. One hour after each infection, cells were washed and 2 ml of MEM was added in order to remove the medium containing virus vectors. A similar series of experiments was conducted applying the viruses in a reversed order.
Immunocytochemistry and Analysis
36 hours after the second virus infection, cells were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 45 min at 4°C, washed three times for 5 min with PBS, and then immersed in PBS containing 5% Goat Serum and 0.1% Triton X-100 for an hour at room temperature. Cells were then incubated overnight at 4°C with anti-β-gal, rabbit IgG (1:1500; Cappel, Aurora, OH, USA) and anti-GFP, mouse IgG (1:750; Molecular Probes, Eugene, OR, USA) diluted in the same blocking solution. The primary antibody was aspirated and cells were washed and permeabilized in PBS containing 0.1% Triton X-100 (PBT). Cells were then incubated in Cy3-conjugated anti-rabbit, goat IgG (1:300; Jackson ImmunoResearch, West Grove, PA, USA) and Cy2-conjugated anti-mouse, goat IgG (1:200; Jackson ImmunoResearch) diluted in PBT for 2 h at room temperature. The cells were then washed three times with PBS, and coverslips were adhered to the glass slides with VectaMount AQ Aqueous Mounting Medium (Vector Laboratories, Burlingame, CA, USA).
Fluorescence was studied with the use of an inverted microscope (Zeiss Axiovert 200M; Carl Zeiss, Thornwood, NY, USA) equipped with a laser scanning unit (Zeiss LSM 5 Pascal; Carl Zeiss). All images were captured at a fixed Z-level, under the same optical conditions. GFP labeled neurons were pseudocolored green and cells containing β-gal were pseudocolored magenta. From the overlap of green and magenta, double-labeled neurons appeared white. Double-labeled neurons were determined using Zeiss software as well as by overlaying confocal images in Adobe Photoshop version 6.0 (Adobe Systems, San Jose, CA, USA). The number of labeled neurons was counted, and the percentage of double-labeled cells from cells labeled by the firstly applied RV-vector was determined under each condition.
Surgical Procedures and Viral Injection
A total of 22 young adult male Wistar rats weighing 200–230 g were used in this study. All experiments with injections of recombinant rabies virus vectors were carried out in a special laboratory (biosafety level 2) designated for in vivo infectious experiments. All experiments were conducted according to the Guidelines of the National Institutes of Health and the Guidelines for Animal Care and Use, published by our institute.
Rats were deeply anaesthetized with sodium pentobarbital (50 mg/kg, i.p.), and were mounted in the stereotaxic frame. The skull was exposed, and a small burr hole was drilled above the injection site at 8.4 mm posterior to bregma, and 4.8 mm lateral (based on Paxinos and Watson, 1986
). The injection was made at a depth of 3.4 mm below the cortical surface by means of a glass micropipette (tip diameter = 50 μm) connected to a 1 μl Hamilton microsyringe.
In 11 rats, 400–600 nl of rHEP5.0-CVSG-β-gal (1.0 × 107 FFU ml) was injected into the MEC. In another 11 rats, 400–600 nl of rHEP5.0-CVSG-β-gal (1.0 × 107 FFU/ml) and 300–400 nl of rHEP5.0-CVSG-EGFPx2 (1.0 × 107 FFU/ml) was injected into the left and right MEC, respectively. Each virus was injected with 1% of pontamine sky blue in order to determine the infection sites. After the injection at the rate of 20 nl per minute, the pipette was left in place for an additional 15 min before withdrawal. After all injections were completed, the wound was sutured, and the animal was monitored for recovery from anesthesia and returned to its home cage. Throughout the survival times, the rats were kept inside a small safety cabinet.
Immunohistochemistry and Analysis
After survival periods of 3–6 days, the animals were deeply anaesthetized with sodium pentobarbital (100 mg/kg, i.p.). In order to stain β-gal labeled neurons with X-gal staining, rHEP5.0-CVSG-β-gal injected animals were transcardially perfused with 0.9% NaCl in 0.1 M phosphate buffer (PB; pH 7.4) followed by a mixed solution of 2% paraformaldehyde and 0.2% glutaraldehyde in 0.1 M PB (this fixative is preferred since high concentrations of paraformaldehyde lowers the sensitivity of β-gal). Animals with dual viral injections were transcardially perfused and fixed with 10% sucrose in 0.1 M PB followed by 4% paraformaldehyde in 0.1 M PB (this fixative is used to avoid the induction of high levels of autofluorescence by glutaraldehyde). The brains were removed from the skull, postfixed in the same fresh fixative as was used for the perfusion for 4 h at 4°C and then cryoprotected in PB containing 30% sucrose for at least 48 h at 4°C. The brains were coronally sectioned at 50 μm on a freezing microtome, and were collected in four equally spaced series for processing.
In order to visualize β-gal labeled neurons, one series of sections was processed for x-gal staining. Floating sections were washed with rinse buffer (0.9% NaCl, 2 mM MgCl2, 100 mM HEPES, pH 7.4, 0.01% sodium deoxycholate, 0.1% Triton-100), and then incubated overnight in x-gal staining solution (5 mM potassium ferricyanide and 5 mM potassium ferrocyanide, and 1 mg/ml x-gal dissolved in rinse buffer) at 31°C. After washes in PBS, the sections were mounted onto gelatin-coated glass slides, air-dried, dehydrated in ethanol, cleared in xylene, and coverslipped with mounting medium (Mount Quick; Cosmo Bio, Tokyo, Japan). The second series of sections were Nissl stained with thionin. Sections were examined under an Olympus Provis AX70 microscope (Olympus, Tokyo, Japan) and photographed with a FUJIX digital camera HC-2500 and Photograb-2500 software (Fujifilm, Tokyo, Japan). The cytoarchitectonic borders were established by comparing dark field images and the adjacent Nissl stained sections. The numbers of β-gal labeled neurons in CA1, CA3, the septum, and the diagonal band were counted from 15 sections (400 μm apart) in rats with survival period of 3.5 and 6 days.
A double labeling immunofluorescence procedure similar to that used for the in vitro study was conducted to visualize β-gal and GFP labeled neurons by using anti-β-gal, rabbit IgG (1:1500; Cappel) and anti-GFP, mouse IgG (1:750; Molecular Probes) as primary antibodies, and Cy3-conjugated anti-rabbit, goat IgG (1:300; Jackson ImmunoResearch) and Cy2-conjugated anti-mouse, goat IgG (1:200; Jackson ImmunoResearch) as secondary antibodies. Fluorescent neurons were imaged at a fixed z-level using a laser scanning confocal unit (Zeiss LSM 5 Pascal; Carl Zeiss) coupled to a Zeiss Axiovert 200M. Double-labeled neurons were determined by overlaying pseudo-colored high-magnification images of each reporter protein in Adobe Photoshop software. The cytoarchitectonic borders were established from the adjacent Nissl stained sections. The numbers of single- and double-labeled neurons in the bilateral CA3 were counted from three regularly spaced sections at a 400 μm interval. The numbers of labeled neurons in the three sections were totaled and the percentage of double-labeled neurons out of the total labeled numbers was determined.
Double Labeling with RV-Vectors In Vitro
There is a significant problem in dual viral tracing owing to the nature of viruses, called viral interference. This viral specific phenomenon is defined as an interference mechanism that is established after primary infection, preventing the infected cell from being superinfected by the same or a closely related virus. This may yield false negative results in case of double labeling studies. In order to examine the effect of viral interference on double labeling, we applied two virus vectors, rHEP5.0-CVSG-β-gal and rHEP5.0-CVSG-Venus, to the same culture dish at different time intervals. Figure 3
A shows photomicrographs of labeled cells, firstly infected by rHEP5.0-CVSG-Venus, followed by rHEP5.0-CVSG-β-gal. When both vectors were applied simultaneously, approximately 75% of the cells labeled by Venus were double-labeled (Figure 3
B). However, the number of double-labeled cells decreased as the time interval between the first and second infection increased. At a 2 h interval, the percentage of double-labeled cells decreased to 51.6%, and when the delay between the first and second infection was 12 h, only 16.3% were double-labeled. The decrease in the percentage of double-labeled cells was not so severe when the order of the virus infections was reversed, and in that case, 65.5% of cells were still double-labeled after 6 h of delay. Although these results suggest that there are differences between the two RV-vectors in that rHEP5.0-CVSG-β-gal has a longer time window for successful double labeling than rHEP5.0-CVSG-Venus, both viruses showed efficient double-labeling when the two RV-vectors infect the cell with only a short interval in between.
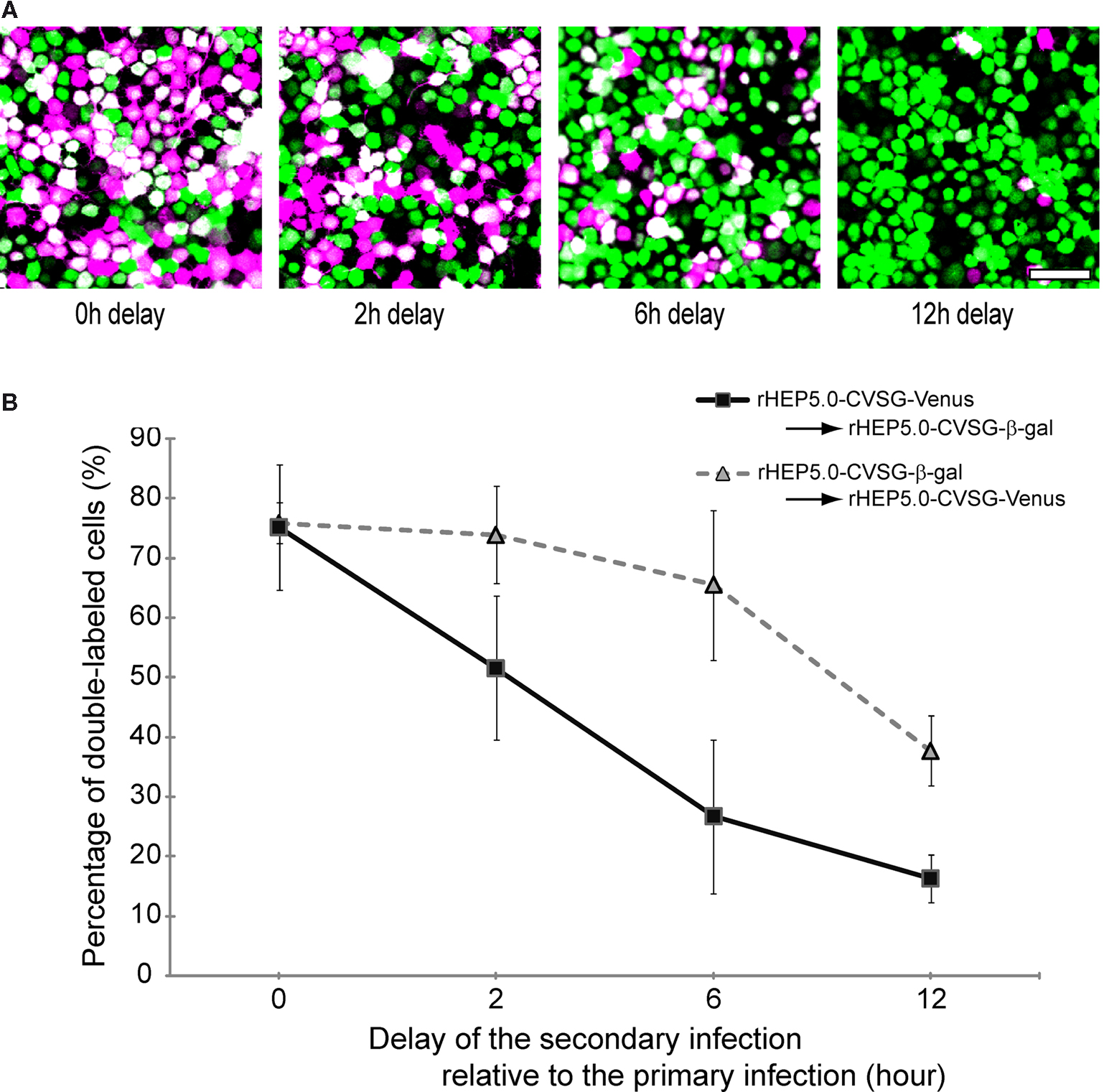
Figure 3. Double labeling of cultured cells following infection of two viruses at different time intervals. (A) Fluorescence photomicrographs of NA cells labeled by rHEP5.0-CVSG-Venus (green) and rHEP5.0-CVSG-β-gal (magenta). Cultured cells were primary infected with rHEP5.0-CVSG-Venus, and after a certain time delay (0 h, 2 h, 6 h, 12 h), further infected with rHEP5.0-CVSG-β-gal. Double-labeled neurons are shown in white. (B) The percentage (mean + SD) of double-labeled neurons among the cells labeled by the firstly applied RV-vector was determined for each set of conditions. NA cells were infected with rHEP5.0-CVSG-Venus prior to rHEP5.0-CVSG-β-gal (squares), or infected with rHEP5.0-CVSG-β-gal prior to rHEP5.0-CVSG-Venus (triangles). Note that the rate of double-labeled neurons decreases as the time delay gets longer. Scale bar = 50 μm in (A).
Temporal Pattern of Labeling with RV-Vector
Prior to double labeling, we made single injections to examine the transneuronal transport of the RV-vector along the multisynaptic pathway from CA3 to MEC (Figure 4
A). The vector rHEP5.0-CVSG-β-gal was injected into MEC and the distribution of labeled neurons was examined at different post-inoculation survival times. The survival times of the animals were 3.5 days (N = 1), 4 days (N = 3), 5 days (N = 2) and 6 days (N = 5). In all animals (N = 11), the injection site, as indicated by the presence of pontamine sky blue (Figure 4
B) was centered in MEC or involved the border region between MEC and the parasubiculum.

Figure 4. Temporal pattern of labeled neurons following injection of r5.0-CVSG-β-gal in the MEC, visualized using X-gal staining. Distribution of retrogradely labeled neurons after 3.5 days (C–F,H) and 6 days (G,I) of infection. (A) Schematic representation of the relevant circuitry of the hippocampus formation. The colored panel indicates the injection site. (B) Photomicrograph of Nissl-stained section with a representative injection site in the MEC. The arrow indicates the injection site marked by pontamine sky blue. (C–E) Labeled cells observed in the postrhinal cortex, presubiculum, and the diagonal band. (F) Only a few labeled neurons were observed in the lateral septum after 3.5 days of survival times (also note the labeled cells in the diagonal band). (G) Six days after infection, the number of labeled cells has increased slightly in the lateral septum, whereas numbers in the diagonal band and medial septum have increased significantly. (H) Ipsilateral CA1 neurons are labeled 3.5 days post-infection; no contralateral labeling is present. (I) After 6 days survival time, CA3 pyramidal cells are labeled bilaterally. DB, diagonal band; LS, lateral septum; MEC, medial entorhinal cortex; MS, medial septum; PaS parasubiculum; POR, postrhical cortex; PrS, presubiculum. Scale bars = 1000 μm in (B), (G) [applies to (F)], (I) [applies to (H)], 200 μm in (E) [applies to (C,D)].
Samples of 3.5, 4, and 6 days post-infection were compared to ascertain the time dependent transneuronal labeling of RV-vectors. To assess the overall infectious capacity of the RV-vector we analyzed first order labeling in the main afferent structures of the MEC. After 3.5 or 4 days, labeled cells were seen in postrhinal cortex, presubiculum, and in the diagonal band (Figures 4
C–E). Also in the entorhinal cortex, labeled cells were seen. These findings are in line with previously published results of retrograde tracing of entorhinal inputs (Kerr et al., 2007
). No labeled cells were observed in the medial prefrontal cortex, and olfactory cortices, known to provide only weaker inputs to MEC, and only sparse labeling was seen in the retrosplenial cortex (not illustrated). After 3.5 days, β-gal labeled neurons were already seen in CA1 (Figure 4
H) and subiculum (not illustrated). Most of the cells in CA1 were located in the pyramidal cell layer exhibiting the typical morphology of pyramidal cells. Occasionally, labeled cells were seen in stratum oriens and stratum radiatum. Six days after infection, labeled second order neurons are present in a number of brain areas, including the medial septum (Figure 4
G). The overall number and spread of infected entorhinal neurons increased with longer survival times.
An important aim of this study is to assess the feasibility of dual transneuronal labeling, for which we have chosen the bilateral projection of CA3 to CA1. Since it is well established that CA3 projections not only target CA1 but also project to the lateral septum bilaterally (Witter and Amaral, 2004
), we assessed labeling in the latter structure as well in order to determine which intermediate pathway mediated the second order infection of CA3 neurons. After 3.5 days of injection, although a lot of neurons were labeled in CA1 region, only a few β-gal labeled neurons were observed in the lateral septum and CA3 (Figures 4
F,H, Table 1
). Since there are no direct projections from the CA3 and the lateral septum to MEC (Risold and Swanson, 1997
; Witter and Amaral, 2004
), this labeling is considered to result from transneuronal labeling, representing second order neurons. At 6 days post-injection, high numbers of ipsi- and contralateral CA3 neurons were labeled (Figure 4
I). In lateral septum, second order labeling increased slightly, compared to 3.5 and 4 days post-infection (compare Figures 4
F,G). These data strongly suggests that the dramatic increase in the number of labeled CA3 pyramidal cells bilaterally is mostly due to transsynaptic transport, not by way of the lateral septum, but via CA1 and the Schaffer collaterals that terminate there.

Dual Transneuronal Tracing with RV-Vectors
In order to produce stronger labeling from infected neurons, we used rHEP5.0-CVSG-EGFPx2 as a pair with rHEP5.0-CVSG-β-gal for dual viral tracing instead of rHEP5.0-CVSG-Venus. rHEP5.0-CVSG-EGFPx2 is also based on rHEP5.0-CVSG, but has two EGFP genes inserted in the N-P intergenic sequence. Thus, this vector shows higher expression level of GFP than rHEP5.0-CVSG-Venus for YFP, especially in vivo (data not shown).
In 11 animals, one of the two vectors, rHEP5.0-CVSG-β-gal and rHEP5.0-CVSG-EGFPx2, was injected into MEC of the left and the other into MEC of the right hemisphere (Figure 5
A), and the animal was perfused and fixed after 6 days. Successful labeling by the two RV-vectors was observed in bilateral CA3 in two cases (Table 2
). In the remaining cases (9 out of 11 rats), sparse or no β-gal labeling was observed in CA3 after 6 days of survival, although numerous EGFP labeled neurons were observed in the bilateral CA3 region. Even in CA1, there were fewer β-gal labeled neurons compared to EGFP labeled neurons. The lack of β-gal labeled neurons in CA3, likely due to low levels of first order infection in CA1, is the main reason for the low percentage of double-labeled animals (2 of 11 rats).
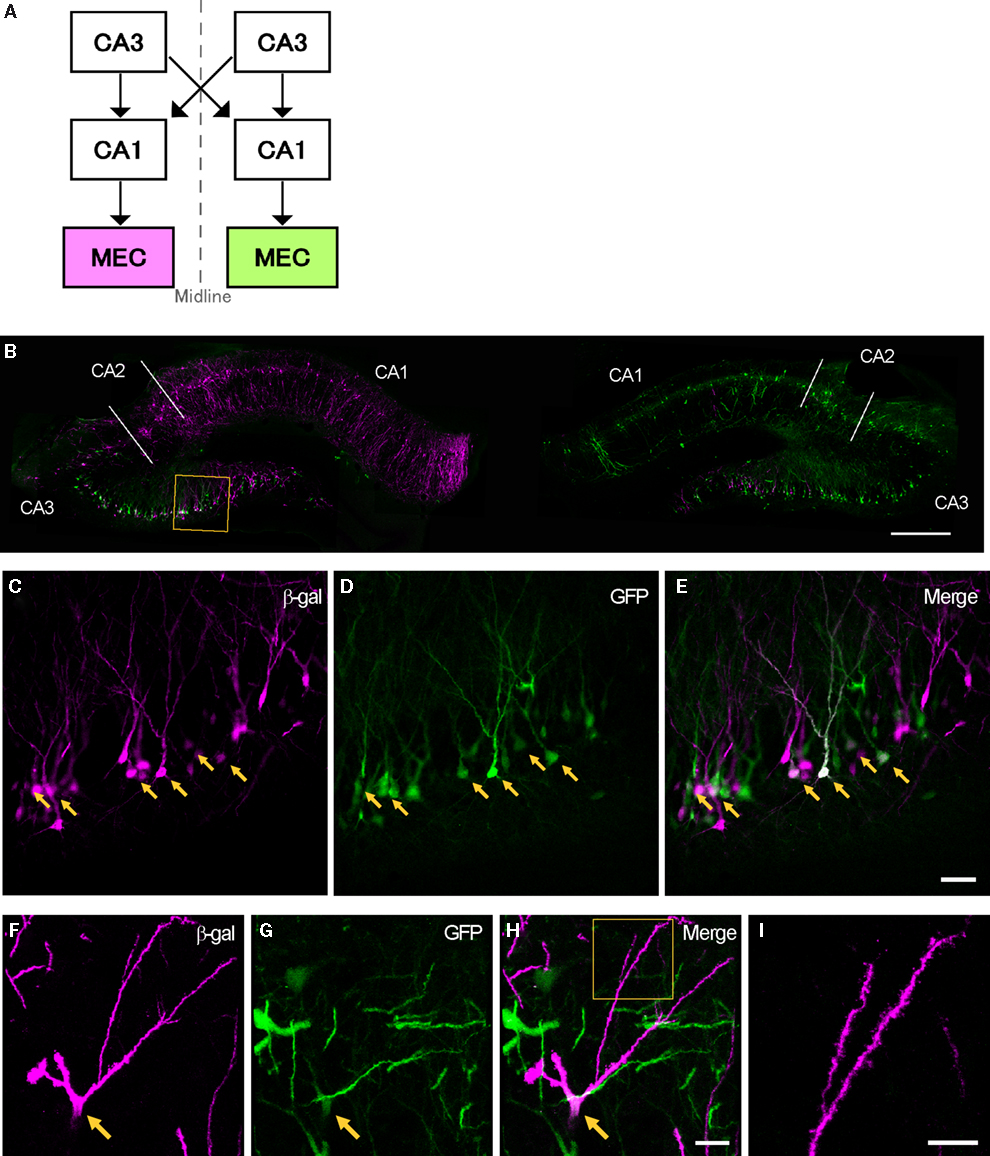
Figure 5. Transsynaptic double labeling of neurons in the hippocampus after 6 days following bilateral injections of two RV-vectors into MEC. (A) Schematic representation of the relevant circuitry in the hippocampal formation. The colored panels show the injection site (green: rHEP5.0-CVSG-EGFPx2, magenta: rHEP5.0-CVSG-β-gal). (B) Fluorescence micrograph showing the distribution of labeled neurons in the dorsal hippocampus. In field CA1, neurons are labeled only by the virus vector injected into the ipsilateral MEC, whereas β-gal (magenta) and GFP (green) labels intermingle in CA3 region. Note that double-labeled neurons are observed in CA3. (C–E) Labeled CA3 pyramidal cells taken from the box in (B). Neurons labeled by both β-gal (C) and GFP (D) can be seen in white in the merged image (E). Arrows indicate double-labeled neurons. (F–H) Photomicrographs demonstrating Golgi-like labeling of a double-labeled CA3 pyramidal cell. High magnification image shows that the dendrites and their spines can also be visualized in the double-labeled neuron. MEC, medial entorhinal cortex. Scale bar = 500 μm in (B), 50 μm in (E) [applies to (C,D)], 20 μm in (H) [applies to (F,G)], 10 μm in (I).
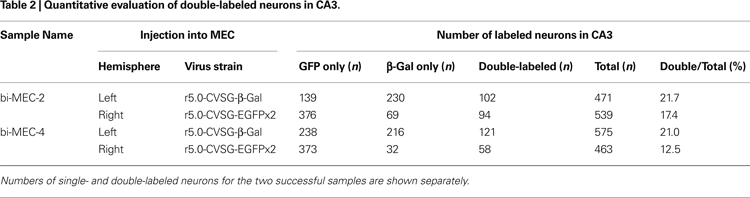
In the two successful experiments, CA1 neurons were predominantly labeled by the vector which was injected into the ipsilateral MEC, whereas β-gal and GFP labels intermingle in CA3 bilaterally (Figure 5
B). Although the number of EGFP labeled neurons exceeded β-gal labeled neurons in the CA3 region ipsi- and contralateral to each injection site, some CA3 pyramidal cells were double-labeled (Table 2
). Figure 5
C–E present high magnification photomicrographs of representative single- and double-labeled CA3 neurons. Strong signals were observed in the cell bodies and dendrites of a large number of the infected neurons. In some double-labeled neurons, the level of expression of at least one of the two proteins was such as to provide a Golgi-like labeling of that neuron (Figures 5
F–I). Occasionally, we observed labeled axons in our preparations (not illustrated).
Our newly developed RV-vector, rHEP5.0-CVSG, shows neuron specific retrograde infection similar to that reported for the Challenge Virus Standard (CVS) strain, which has been generally used for tracing experiments (Inoue et al., 2004
). Here we demonstrated that this vector can efficiently infect a variety of neurons located in main afferent areas of the entorhinal cortex. Moreover, efficient transsynaptic labeling of a number of second order populations was observed as well. We specifically focused on the transsynaptical infection of CA3 pyramidal cells from CA1 cells via the Schaffer collaterals, and showed that dual viral labeling in CA3 by bilateral entorhinal injections is possible. This makes RV-vectors powerful tools to carry out transsynaptic retrograde double labeling experiments, allowing to study relationships between various circuits that together form larger brain systems. This vector, rHEP5.0-CVSG, when subsequently immunostained, showed a complete Golgi-like labeling in some double-infected neurons by at least one of the two reporter proteins. Combining this method with anterograde tracing techniques (Wouterlood et al., 2002
, 2007
) will provide the means to study the afferent connectivity of single neurons characterized by two of its efferent targets. This can be a major new tool to study complex neuronal networks.
The use of viral tracers for dual tracing has its inherent problems. First, there are limitations to the use of our RV-vector, similar to those in case of CVS strain of RV, in that some cell types, such as granule cells in the dentate gyrus, cannot be infected (Jackson and Reimer, 1989
). Note that PRV-Bartha cannot infect CA3 pyramidal cells through synapses of the Schaffer collaterals (Sik et al., 2006
). Second, one cannot exclude the possibility of false negative results owing to virus interference. Depending on the virus family, this inhibition mechanism may occur at various stages of the viral life cycle, including surface binding, endocytosis, and subsequent replication steps (Lee et al., 2005
; Simon et al., 1990
; Singh et al., 1997
; Whitaker-Dowling et al., 1983
). Since the potential block at each level occurs at different time points, the degree of virus interference changes over time after the primary infection. Our results demonstrated the effect of virus interference on double labeling in an in vitro preparation, and showed that the efficiency of the second infection decreases in a time dependent manner. A similar result has also been reported in case of double labeling of neurons using PRV recombinants, showing that the time window for successful double labeling using PRV recombinants is also small (Banfield et al., 2003
). Thus, in order to realize efficient double labeling using viral tracers, two viruses must infect a neuron within a few hours. To achieve this, we need to take into consideration the possible differences in speed between the different RV vectors.
The propagation speed of RV is determined by several factors within the infection pathway: (1) number of crossing synapses; (2) strength of the synaptic connection; and (3) length of axonal transport (Lycke and Tsiang, 1987
; Ugolini, 1995
). In dual tracing, differences between these factors within the two circuits may influence or even prevent effective dual infection. Therefore, in order to assess the potential for dual transsynaptic labeling, we used the symmetrical circuit arising from the CA3 region, thus reducing the possible variations in the progression of infection to a minimum. Previous studies on dual viral tracing using PRV recombinants assessed the importance of using isogenic strains of viruses with similar virulence and rate of transport and replication in order to achieve successful dual tracing (Banfield et al., 2003
; Cano et al., 2004
; Jansen et al., 1995
; Kim et al., 1999
; Ter Horst, 2000
). Therefore, in this study, we used recombinants based on the same vector, rHEP5.0-CVSG. Even so, in the in vitro study, we noted a difference between the two RV-vectors, rHEP5.0-CVSG-β-gal and rHEP5.0-CVSG-Venus, in that the former has a longer time window for successful double labeling than the latter (Figure 3
B). In line with our previous report (Inoue et al., 2004
), we suggest that the length of the inserted foreign gene has an effect on the replication speed. Therefore, rHEP5.0-CVSG-β-gal which contains a longer foreign gene (LacZ gene; 3 Kb) than rHEP5.0-CVSG-Venus (Venus gene; 0.7 Kb) will replicate more slowly. This slow rate of replication may have suppressed the time dependent increase of the progression of viral interference in rHEP5.0-CVSG-β-gal infected cells. Differences between the two RV-vectors in the efficacy of infection were also observed in the in vivo study. where rHEP5.0-CVSG-β-gal labeled much less neurons than rHEP5.0-CVSG-EGFPx2 (two EGFP genes; 1.4 Kb) in CA1 and in CA3. This difference is one likely reason for the low percentage of successful double labeling. However, we cannot exclude that the infection of rHEP5.0-CVSG-β-gal has been prohibited by the preinfection of the faster spreading rHEP5.0-CVSG-EGFPx2. Because of the differences in the infection properties, the use of these RV-vectors is not the most optimal in order to assess the connections of two very similar neural circuits such as used in this study. However, this approach allowed us to document the differences in infection properties in vivo. Combining specific properties of these vectors with known differences in pathway lengths or strengths may yield a very efficient tool to detect the connectional interactions between two circuits.
Since viral interference is a characteristic feature of viruses, we cannot completely rule out false negative results. There may be neurons in CA3 that have a multisynaptic projection to both of the injection sites but are still only single labeled. Therefore, we have to realize that quantitative analysis is difficult in dual viral tracing since the result may underestimate the real number of collateralized neurons. Moreover, we did not systematically analyze the possible relationship between injected viral volume and number of first order labeled cells. However, we can conclude that those neurons that express both reporter proteins undoubtedly have an anatomical connection, mediated by at least one synaptic contact, to both of the originally infected target areas and such characterized neurons can be the subject of subsequent detailed anatomical and or in vitro electrophysiological studies.
The organization of neuronal connectivity of the hippocampal formation has been explored in detail using various tracing techniques, including a variety of anterograde and retrograde tracing techniques (Ishizuka et al., 1990
; Laurberg and Sorensen, 1981
; Li et al., 1994
; Swanson et al., 1978
, 1980
; see also Witter and Amaral, 2004
for a review). However, these conventional tracers are not suitable for tracing chains of connected neurons due to the inability to cross synapses. In order to analyze multisynaptic neuronal circuits, several experiments with injections at distinct synaptic stations must be conducted (Naber et al., 2001
). Such an approach may require large numbers of animals, and it does not unequivocally show that neurons indeed form a chain of connections linking together two targets with only one synaptic intermediary in between. In view of the known connectivity, CA3 labeling with both vectors is most likely mediated through synapses of the CA3 Schaffer collaterals onto first order infected CA1 neurons. Alternative routes through the lateral septum are unlikely in view of the reported sparseness or even absence of direct projections from the lateral septum to the MEC (present data, see also Risold and Swanson, 1997
). It is also unlikely that CA3 neurons might be labeled as a result of primary infection of interneurons in CA1, since most of our first order CA1 neurons showed the typical morphology of pyramidal cells. We did observe occasional labelling in CA1 of cells outside the pyramidal cell layer already after 3.5 days, indicating first order labeling of presumably GABA-ergic neurons. This is in line with previous reports (see Cenquizca and Swanson, 2007
for review). Second order labeling of interneurons in CA1 is likely to occur as well, but we have not systematically assessed this. It is however unlikely, in view of the established time needed to cross a single synapse, that the observed labeling in CA3 after 6 days is a third order phenomenon. We therefore conclude that single neurons in CA3 are connected to the entorhinal cortex bilaterally, with only one synaptic relay at the level of CA1 cells. This finding adds to our understanding of the complex interplay between the left and right hippocampal region. Although the functional relevance of this interaction is still poorly understood, it has been implicated in the ability of rats to segregate relevant associations among spatial stimuli from irrelevant stimuli (Wesierska et al., 2005
). In view of the known organization of this connectional chain, the present finding was not unexpected, but in fact, ours is the first paper that directly demonstrates this transsynaptic connection from individual CA3 neurons to MEC bilaterally. This clearly illustrates the promising potential of dual transneuronal viral tracing to unravel the complex connectivity of neural network.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Atsushi Miyawaki (Riken, Japan) for providing cDNA of Venus. This work was supported by Grant-in-Aid for Scientific Research on Priority Areas (Integrative Brain Research) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Wouterlood, F. G., van Haeften, T., Blijleven, N., Perez-Templado, P., and Perez-Templado, H. (2002). Double-label confocal laser-scanning microscopy, image restoration, and real-time three-dimensional reconstruction to study axons in the central nervous system and their contacts with target neurons. Appl Immunohistochem. Mol. Morphol. 10, 85–95.