 Li Liu1,2†
Li Liu1,2† Jinwen Tian1†Caiyi Lu1†Xi Chen1Yicheng Fu1Bin Xu1
Jinwen Tian1†Caiyi Lu1†Xi Chen1Yicheng Fu1Bin Xu1 Chao Zhu1Yanmei Sun1Yu Zhang1Ying Zhao1
Chao Zhu1Yanmei Sun1Yu Zhang1Ying Zhao1 Yang Li1*
Yang Li1*- 1Department of Cardiology, General Hospital of People's Liberation Army, Beijing, China
- 2The Third Department of Internal Medicine, Beijing Municipal Corps Hospital of Chinese People's Armed Police Force, Beijing, China
Mutations in hERG cause long QT syndrome type 2 which is characterized by a prolonged QT interval on electrocardiogram and predisposition to life-threatening ventricular tachyarrhythmia, syncope, and sudden death. hERG-G572S induces trafficking defects of hERG channel protein from Golgi to the plasma membrane and results in a dominant negative suppression of hERG current density. As an accessory β subunit, KCNE2 promotes hERG migration from Golgi to cellular membrane. In this study, we investigated the rescue effect of KCNE2 in a G572S mutation of hERG. Transfection was performed into HEK293 cells. Patch clamp technique, western blotting analyses and confocal microscopic examination were used. Results showed that KCNE2 had a significantly enhanced effect on G572S mutation current. The increase of current was largest at KCNH2:KCNE2 of 1:3. Confocal images showed co-expressing G572S and KCNE2 could cause a substantial up-regulated membrane protein (155 kDa) expression. Expression of membrane protein accumulated markedly with increasing ratio of KCNH2:KCNE2. G572S defective mutant could be restored by both KCNE2 and lower temperature (27°C), which suggested that the lower temperature could be the favorable circumstances for the rescue function of KCNE2. In this study, we successfully set up “the action potential” on the HEK 293 cells by genetically engineered to express Kir2.1, Nav1.5, and Kv11.1, wherein on reaching over an excitation threshold by current injection. The results suggested that KCNE2 could shorten action potential duration which was prolonged by G572S. These findings described electrophysiological characteristics of the LQT2 syndrome mutation KCNH2-G572S and regulation by accessory protein KCNE2, and provided a clue about LQT2 and relative rescue mechanism.
Introduction
Human hereditary long QT syndrome (LQTS) is a heterogeneous cardiac repolarization disorder characterized by a prolonged QT interval on the surface electrocardiogram (Schwartz et al., 2012). The human-ether-a-go-go-related gene (hERG or KCNH2) encodes the pore-forming alpha subunit of K+ channel that resembles the rapid component of delayed rectifier current (IKr) in cardiac myocytes (Warmke and Ganetzky, 1994; Sanguinetti et al., 1995; Trudeau et al., 1995). Mutations in hERG cause long QT syndrome type 2 (LQT2) (Sanguinetti et al., 1995; Schweigmann et al., 2014), which is characterized by a prolonged QT interval on electrocardiogram and predisposition to life-threatening ventricular tachyarrhythmia, syncope and sudden death (Saenen and Vrints, 2008).
In 1999, Abbott et al. described that MiRP1 formed stable assemblies with hERG and the resulting channel complex had functional attributes like those of native, cardiac IKr channels. There are discrepancies among investigators in describing the effects of KCNE2 on hERG currents (Abbott et al., 1999; Mazhari et al., 2001; Weerapura et al., 2002; Eldstrom and Fedida, 2011). Previous studies showed that KCNE2 could modulate single channel conductance (Abbott et al., 1999), gating kinetics (Zhang et al., 2001), the rate of protein degradation (Zhang et al., 2012), regulate hERG endocytosis, alter some aspects of hERG pharmacology (Dupuis et al., 2005) and promote hERG migration from Golgi to cellular membrane as an accessory β subunit (Um and McDonald, 2007).
It is estimated that at least 80% of missense mutations in KCNH2 result in defects in assembly and hence trafficking of functional channels to the plasma membrane (Anderson et al., 2006; Walker et al., 2007). The considerable work has been undertaken looking at pathways of hERG trafficking and the role of chaperones in detecting misfolded mutant proteins (Thomas, 2003). To our knowledge, hERG-G572S induces trafficking defects of hERG channel protein from Golgi to the plasma membrane and results in a dominant negative suppression of hERG current density. hERG-G572S channel subunits can coassemble with WT subunits, and this results in retention of WT subunits within the ER. In silico modeling it is found that G572S would cause a significant prolongation of AP duration (Zhao et al., 2009).
As mentioned above, we speculated KCNE2 could have some effects on hERG-G572S. This study sought to research electrophysiological characteristics of KCNH2-G572S and regulation of KCNE2 on KCNH2-G572S mutation.
Materials and Methods
Site-Directed Mutagenesis and Transfection into HEK293 Cells
The G572S mutation was introduced into the KCNH2 cDNA by directed mutagenesis (QuickChange Site-Directed Mutagenesis kit, Stratagene). The Wild-type (WT) and mutant SCN5A were also inserted in frame into a pcDNA3.1 vector plasmid (Invitrogen) to be expressed. Sequence analysis was used to confirm the presence of the mutation. WT and mutant of KCNH2 were transiently transfected in Human embryonic kidney (HEK)-293 cells. HEK293 cells were transiently transfected with 1.2 μg of the KCNH2 construct, either WT or G572S, using Lipofectamin 2000 (Life Technologies, Gaithersburg, MD, USA) according to the manufacturer's protocol. The green fluorescent protein (GFP) gene was as a reporter gene in co-transfection HEK293 cells with plasmid. After 6 h, the transfection medium was replaced with regular medium. Transfected HEK293 cells were cultured for 48–72 h. Only cells exhibiting green fluorescence were separated by enzymatic treatment and seeded in plastic Petri dishes bottomed with a coverslip. Using the patch-clamp technique, currents were recorded.
Patch Clamp Recording
hERG currents were recorded at room temperature using the whole-cell patch-clamp configuration with the MultiClamp 700B amplifier (Axon Instruments). Data were sampled at 10 kHz and filtered at 5 kHz subsequently for analysis (Digidata 1440A, Axon Instruments). Recording pipettes were pulled from borosilicate capillary tubes by using P-97 programmable patch micropipette horizontal puller (Sutter Instruments). Micropipette resistance ranged from 2.0 to 5.5 MΩ when filled with the internal solution and immersed in the external solution. To minimize voltage clamp errors, series resistance (Rseries) was compensated by <2.0 MΩ and usually ≥80% compensation was achieved in all experiments. The membrane capacitance was compensated by approximately 80~90% of their initial value. The internal solution contained (in mM): K aspartate 85, KCl 45, Na pyruvate 5, K2ATP 3, MgCl2 4, EGTA 10, HEPES 10, and glucose 11, adjusted to pH 7.2 with KOH. Cells were perfused with an external (bath) solution containing (in mM): NaCl 140, CaCl2 1, MgCl2 1, KCl 4, HEPES 10, and glucose 5, adjusted to pH 7.4 with NaOH.
To record the INa, cells were bathed in a solution that contained (in mM) NaCl 40, CaCl2 2, CsCl 5, MgCl2 1.2, HEPES 10, and glucose 5, adjusted to pH 7.4 with CsOH. The glass pipettes were filled with a solution of (in mM) CsCl 60, caesium aspartate 80, EGTA 11, HEPES 10, Na2ATP 5, at pH 7.2, adjusted with CsOH. To avoid current drift of INa, sodium ion concentration of external (bath) solution was set at 40 uM, room temperature was kept at 20°C, micropipette of 0.5 μm-inner diameter was used, and micropipette resistance ranged from 0.5 to 1 MΩ when filled with the internal solution and immersed in the external solution (Sokolov et al., 2013).
To record AP, cells were bathed in a solution that contained (in mM) NaCl 137, KCl 5.9, CaCl2 2.2, MgCl2 1.2, glucose 14, and HEPES 10. The pH of the solution was adjusted to 7.4 with NaOH. The pipette solution for whole-cell recordings contained (in mM) KCl 140, MgCl2 4, HEPES 10, Na2ATP 2, and EGTA 0.05. The pH of the pipette solution was adjusted to 7.2 with KOH (Fujii et al., 2013).
Western Blotting Analyses
Total homogenates were obtained from HEK293 cells using standard methods (Zhou et al., 1998). Blots were probed with primary antibodies against Kv11.1. Chemiluminescent detection was performed with substrate reagents from Pierce Biotechnology. Densitometric analysis was performed with Image for Windows software (V. Beta 4.0.2; Scion). The expression level of Kv11.1 protein was monitored using Western blotting analyses. HEK293 cells expressing KCNH2-G572S were lysed, and the total proteins (TP) were extracted. Moreover, the total protein membrane (PM) was purified using a plasma membrane protein extraction kit according to the manufacturer's instructions (Biovision, Inc. USA). TP or PM (150 μg) per sample was separated by 10% SDS-PAGE and blotted onto nitrocellulose membrane (Stratagene, La Jolla, CA). Subsequently, hERG protein was detected using primary antibodies against Actin (Affinity Reagents), the specific polyclonal rabbit anti-hERG antibody (Santa Cruz Biotechnology, CA) and goat anti-rabbit Alexa Fluor 700 (Molecular Probes, Eugene, OR, dilution 1:2000). Densitometry and the Scion Image Software (Scion, Frederick, MD) were used to quantify the band densities. All data were normalized against Actin (n = 3).
Confocal Microscopy Examination
HEK293 cells were transfected with different hERG plasmids (pcDNA3-WT-hERG, pcDNA3-G572S-hERG and pcDNA3-KCNE2). Forty eight hours later, HEK 293 cells were fixed in 4% paraformaldehyde, treated with 0.1% Triton X-100, and blocked with 3% bovine serum albumin (BSA) at room temperature. These cells were then stained with rabbit polyclonal anti-hERG (1:50 dilution, Alomone, Israel) and mouse monoclonal anti-KCNE2 (1:100 dilution, Abcam, MA, USA) at 4°C overnight, followed by incubation with Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 555 goat anti-mouse IgG at 37°C for 2 h. Stained cells were examined under a confocal microscope (FV10i, Olympus, Japan) for subcellular location of hERG protein.
The Assay System for Action Potential
To investigate the effect of G572S-hERG mutation on action potential duration, we built a cell system in which sodium (Nav1.5) channels, inward rectifier potassium channel (Kir2.1) and hERG potassium channels were co-expressed according to Fujii reported (Fujii et al., 2012). HEK293 cells with stable expression of SCN5A cDNA (Nav1.5) were transfected with 2.0 μg hERG cDNA (Kv11.1) and 2.0 μg KCNJ2 cDNA (Kir2.1) according to Lipofectamine method (Life Technologies, Gaithersrburg, MD, USA) suggested by the manufacturer. CD8 cDNA was co-transfected to be used as a reporter gene (EBo-pCD vector, American Type Culture Collection). CD8-positive cells identified using Dynabeads (Dynal, M-450 CD8) were patch-clamped 48–72 h after transfection.
Data Analysis
Data are expressed as means ± S.E. Voltage protocols and data analysis were done with pCLAMP 10.2 (Axon Instruments) and Origin 6.1 (Microcal Software). Student's t-test was used for statistical analysis between the experimental groups (two-tailed). For multiple group comparisons, statistical significance was determined by ANOVA. With a ANOVA followed by a Student-Newman-Keuls (S-N-K) post hoc test, significance between any two groups was evaluated. P-values lower than 0.05 were considered statistically significant. The mid-point of activation was obtained using a Boltzmann function where G = [1 + exp(V1/2-V) /k]−1, where V1/2 and k are the mid-point and the slope factor, respectively, and G = I/(V-Vrev) where Vrev is the reversal potential and V is the membrane potential. The steady state of inactivation was also fitted using the Boltzmann equation. With a two-exponential function, the time constants of inactivation were assessed. All data were fitted using a nonlinear least-squares minimization method.
Results
Effect of KCNE2 on KCNH2-G572S Currents
The step and tail currents of KCNH2 elicited upon from a holding potential of −90 mV, the membrane potential was stepped from −50 to +60 mV for 2000 ms, and then repolarized to −40 mV for 3000 ms to elicit outward tail current. The standard protocol was applied with a start-to-start interval of 15 s. First of all, to define the functional changes of the mutation G572S, whole-cell currents of WT or G572S mutant channels were recorded respectively, without KCNE2. Currents recorded from WT channel showed the typical hERG current traces: relatively slow activation and deactivation with an inwardly rectifying profile. Compared with WT, tail current densities of G572S significantly decreased. After co-transfecting with KCNE2, tail current densities of G572S and WT were both significantly increased. At +60 mV, tail current densities of WT and G572S before and after co-transfecting with KCNE2 were 65.4 ± 3.6 vs. 110.4 ± 7.5 pA/pF of WT and 10.2 ± 1.2 vs. 89.4 ± 6.4pA/pF of G572S (n = 10, P < 0.01). We found that KCNE2 had a more significantly enhanced effect on G572S mutation current than on WT (Figures 1A,B). To test the effects of KCNE2 quantity, we used the different ratio of 3:1, 1:1, and 1:3 of KCNH2:KCNE2 in the experiment. The results showed the current was largest at KCNH2-G572S:KCNE2 of 1:3 (Figure 1C).
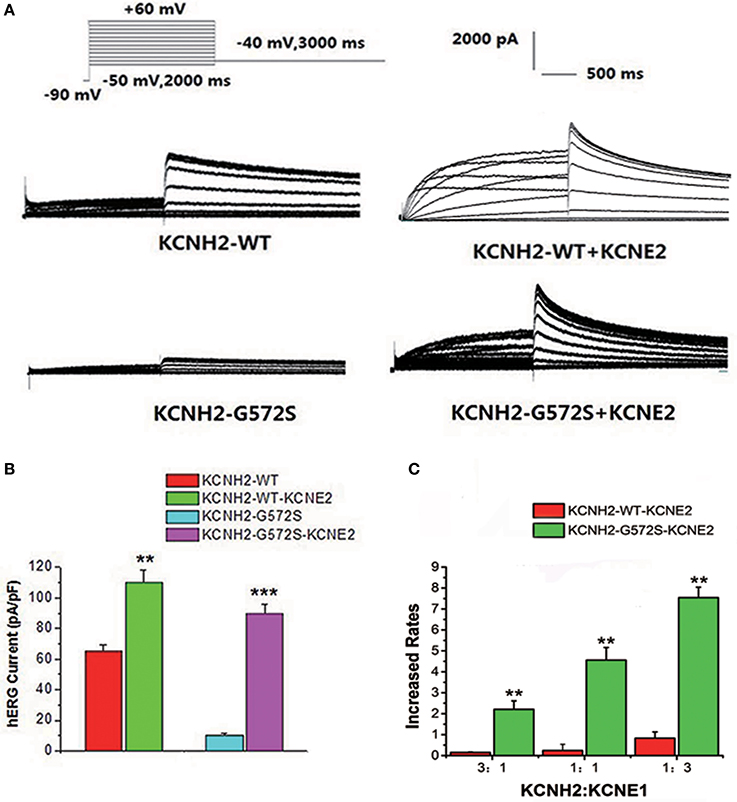
Figure 1. Effect of KCNE2 on KCNH2-G572S current. Compared with WT, tail current of G572S is significantly decreased. After co-transfecting KCNH2 and KCNE2, tail currents of G572S and WT are significantly increased (A). A more significant enhancement of mutation current is found than WT by KCNE2 at test potential of +60 mV. After co-transfecting with KCNE2, tail current densities of G572S and WT were both significantly increased. KCNE2 had a more significantly enhanced effect on G572S mutation current densities than on WT (B). Due to the different proportion of KCNH2: KCNE2, the currents are different, furthermore, current is the largest at KCNH2: KCNE2 of 1:3. n = 10, **P < 0.01 and ***P < 0.001 (C).
Effect of KCNE2 on Current-Voltage Relationship Curve of KCNH2-G572S Currents
As shown in Figure 2A, I-V relationship curve for step currents of G572S was significantly lower than that of WT between −10 and +20 mV, indicating there was a more obvious reduction in G572S amplitudes compared to in WT. When co-expressing KCNE2, G572S and WT currents were both significantly enhanced. As shown in Figure 2B, I-V relationship curve for tail current of G572S was lower than that of WT over +10 mV of test potentials and enhancement of G572S currents by KCNE2 was more prominent than that of WT (n = 10, p < 0.01).
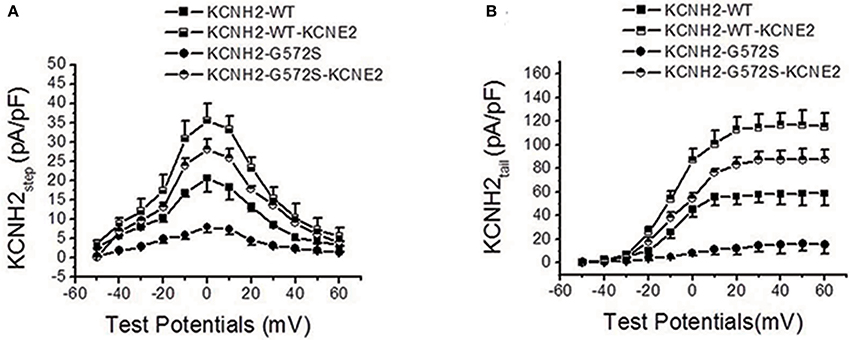
Figure 2. Effect of KCNE2 on current-voltage relationship curve of KCNH2-G572S current. I-V relationship curve for step currents of G572S is lower than that of WT between −10 and +20 mV of test potentials and G572S and WT currents are significantly enhanced by KCNE2 (A). I-V relationship curve for tail current of G572S is lower than that of WT over +10 mV of test potentials and increase of G572S currents (n = 10, P < 0.001) by KCNE2 is more significant than WT (n = 10, P < 0.01) (B).
Effect of KCNE2 on Gating Kinetics of KCNH2-G572S Currents
The steady-state activation was assessed using standard tail current analysis. Cells were depolarized to potentials in the range −60 to +30 mV (4 s) and tail current was recorded at −120 mV. The steady-state inactivation currents for G572S and WT were measured with the following protocol: Channels were inactivated under double-pulse model with condition pulse depolarization +20 mV (1 s) and following short test pulses to potentials ranging from −120 to +20 mV (15 ms, 10 mV-increments) that was applied to recover the channels from inactivation. Returning to the holding potential of +20 mV evoked large outward inactivating currents. Tail current data were normalized to the maximum current value and fitted with Boltzmann function. Either V1/2 or k of steady-state activation and inactivation of G572S mutant were similar to that of WT, and the parameters of steady-state activation and inactivation do not change after co-transfecting KCNE2 and KCNH2. Fast time constants of deactivation of G572S and WT were similar either with or without KCNE2. Slow time constants of deactivation of G572S and WT with co-expressed KCNE2 were shorter at range of test potentials (Figure 3).
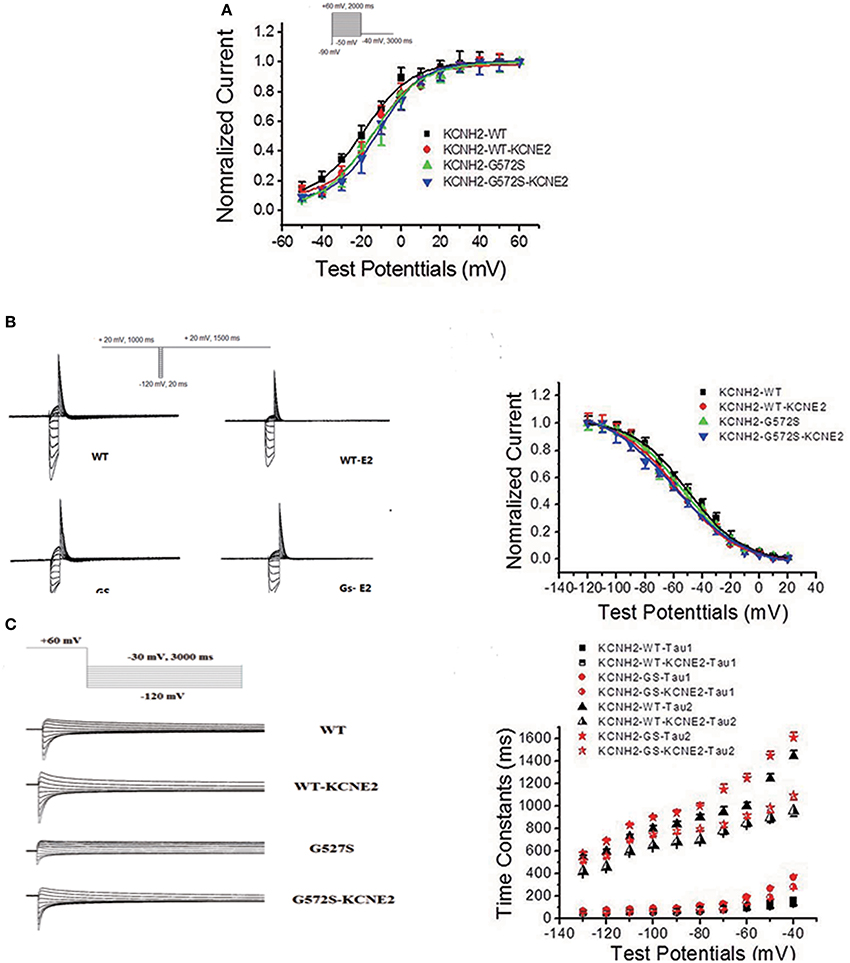
Figure 3. Effect of KCNE2 on gating kinetics of KCNH2-G572S current. Tail current data were normalized to the maximum current value and fitted with Boltzmann function. Either V1/2 or k of steady-state activation and inactivation of G572S mutant is similar to that of WT and the parameters steady-state activation and inactivation do not change after co-transfecting KCNE2 and KCNH2 (A,B). Fast time constants of deactivation of G572S and WT are similar either with or without KCNE2. Slow time constants of deactivation of G572S and WT with co-expressed KCNE2are shorter at range of test potentials (C).
Effect of KCNE2 on Protein Expression of KCNH2-G572S Channel
To further investigate the regulation of KCNE2 on G572S mutation, subcellular localization of mutant proteins in the cell surface was examined. Confocal images were taken from the HEK293 cells expressing WT, G572S, and G572S-KCNE2, respectively. The images showed the localization of hERG protein on cell membrane. Cells expressing WT had a marked surface membrane localization of hERG; whereas the cells expressing G572S apparently had a reduced fluorescent intensity on membrane surface. When co-transfecting HEK293 cells with G572S and KCHE2, the fluorescent intensity on membrane surface increased (Figure 4A).
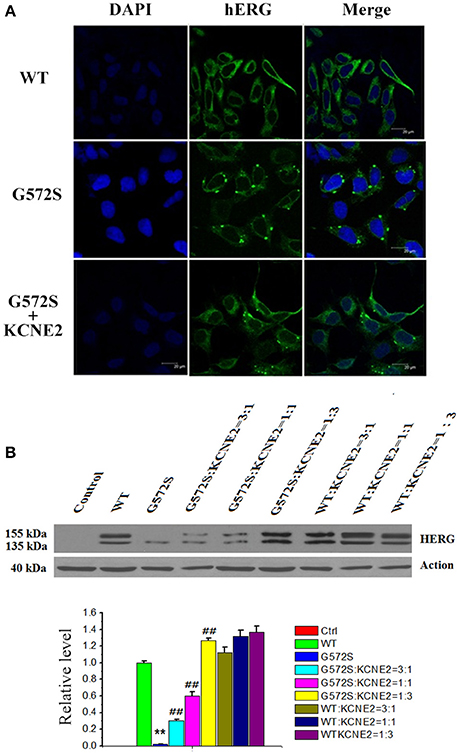
Figure 4. Rescue effect of KCNE2 on protein expression of KCNH2-G572S channel. Confocal images were taken from the HEK293 cells expressing WT, G572S, and G572S-KCNE2, respectively. The immunofluorescence images showed the localization of hERG protein on cell membrane. The hERG-GFP labeling on the cell membrane was shown in green, while the chromatin-DAPI was shown in blue (A). Western blot analysis of the expression of KCNH2-G572S and KCNH2-WT channel protein was performed in transfected HEK293 cells. G572S channel mature protein fraction in the region of the cell membrane (155 kDa) is markedly decreased than that of WT, but when co-transfecting KCNE2, the membrane protein (155 kDa) expression substantially increased. Due to the different proportion of KCNH2: KCNE2, the membrane protein (155 kDa) expression are different, furthermore, the expression is the largest at KCNH2: KCNE2 of 1:3. n = 10, **P < 0.01, vs. WT group; ##P < 0.01, vs. G572S group (B).
WT channel protein on western blot was present in both immature core-glycosylated form of 135 kDa that was localized to the ER and mature complexly glycosylated form of 155 kDa that was inserted into the cell membrane (Zhou et al., 1998; Ficker et al., 2003). To investigate the expression of hERG protein of G572S mutation, western blotting analyses of the different fractions were performed. Figure 4B showed that in the case of WT, co-transfecting with KCNE2 increased the expression of hERG protein on the plasma membrane (155 kDa) without affecting the total protein content in whole cell homogenates. Compared with WT, G572S channel mature protein fraction in the region of the cell membrane (155 kDa) was markedly decreased while non-mature protein (135 kDa) had no change. Co-expressing G572S and KCNE2 could cause a substantial up-regulated membrane protein (155 kDa) expression. Furthermore, expression of membrane protein accumulated markedly with increasing ratio of KCNH2:KCNE2. This result showed there was more membrane protein (155 kDa) expression at KCNH2-G572S:KCNE2 of 1:3 in current study. Protein expressing both of KCNH2-WT:KCNE2 of 3:1and KCNH2-WT:KCNE2 of 1:1 were also increased, however, the increase rate was smaller than KCNH2-WT:KCNE2 of 1:3. The result showed that KCNE2 maybe had rescue effect on trafficking dysfunction of G572S. This experiment was repeated three times (P < 0.01) (Figure 4B).
Rescue Effects of KCNE2 on G572S Mutation under the Condition of Low Temperature
It has been reported that there is temperature-dependent rescuing effect in some trafficking-deficient mutations of hERG protein, such as R752W, G601S, and N470D. Low temperature culturing of cells has been reported to increase the surface expression of many hERG mutants. To examine whether this temperature-dependent rescuing effect worked for G572S mutant, representative western blot analysis of hERG protein expression was performed at 37°C and 27°C, respectively. It was obtained that matured protein (155 kDa) of G572S mutant were 31.6% at 27°C and 17.2% at 37°C. This result showed that exposure of cells at 27°C restored the matured protein (155 kDa) expression of G572S, compared with at 37°C. With co-expression of KCNE2, matured protein (155 kDa) of G572S mutant were increased to 92.5% at 27°C and 68.4% at 37°C, respectively. These results suggested that lower temperature could be the favorable circumstance for the rescue function of KCNE2 (Figure 5).
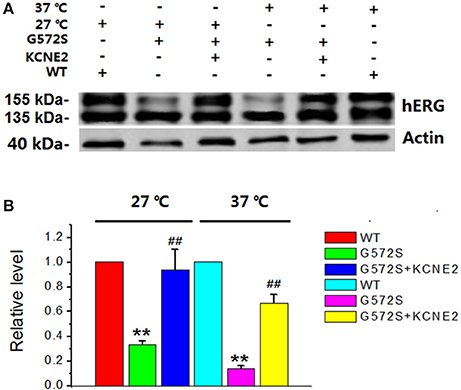
Figure 5. Rescue effects of KCNE2 on G572S mutation at lower temperature. Representative western blot analysis of the hERG protein expression was performed at 37°C and 27°C with/without KCNE2, respectively (A). The matured protein of G572S was increased at 27°C compared with at 37°C, which was inferred by the appearance of the 155-kDa protein in western blot analysis. Furthermore, co-transfecting G572S with KCNE2 led to another substantial increase of membrane protein expression both at 37° and 27°C. (n = 3) **P < 0.01, vs. WT group; ##P < 0.01, vs. G572S group (B).
Building of “The Action Potential (AP)” by Co-Transfecting SCN5A, KCNJ2, and KCNH2 into HEK293 Cells
HEK 293 cell line was utilized as a proof-of-concept unexcitable somatic cell source based on its low levels of endogenous membrane currents, uniform shape and growth, and extensive use as a heterologous expression system for studies of ion channel function (Thomas and Smart, 2005). We chose three basic components of action potential. KCNJ2 (Kir2.1,) channel maintained the resting membrane potential, SCN5A (Nav1.5) channel mainly contributed 0 phase depolarzion of AP, and KCNH2 (Kv11.1) had critical role in repolarization of AP. To verify subcellular localization of Nav1.5, Kir2.1, and Kv11.1 proteins, distribution of three channels was detected by confocal microscopy. Images demonstrated that Nav1.5, Kir2.1, and Kv11.1 proteins were stable co-expressed on the HEK293 cell membrane (Figure 6A). To assess whether these proteins expressing on cell membrane were functional, we recorded INa, IK1, and IKr currents in voltage clamp mode using whole-cell patch-clamp technique respectively (Figure 6B). Three currents were recorded, which indicated co-transfection or co-expression was successful. Subsequently, we applied current injection ranged from 900 to 1500 pA for 5 ms to stimulus HEK293 cell. AP was found on the oscilloscope with a shorter AP duration with 9.2 ± 0.2 ms of 50% repolarization of APD (APD50) and 49.5 ± 2.7 ms of 90% repolarization of APD (APD90). Action potential amplitudes (APA) were 108.5 ± 3.4 mV and resting membrane potentials (RMP) were −70.1 ± 1.2 mV (Figure 6C).
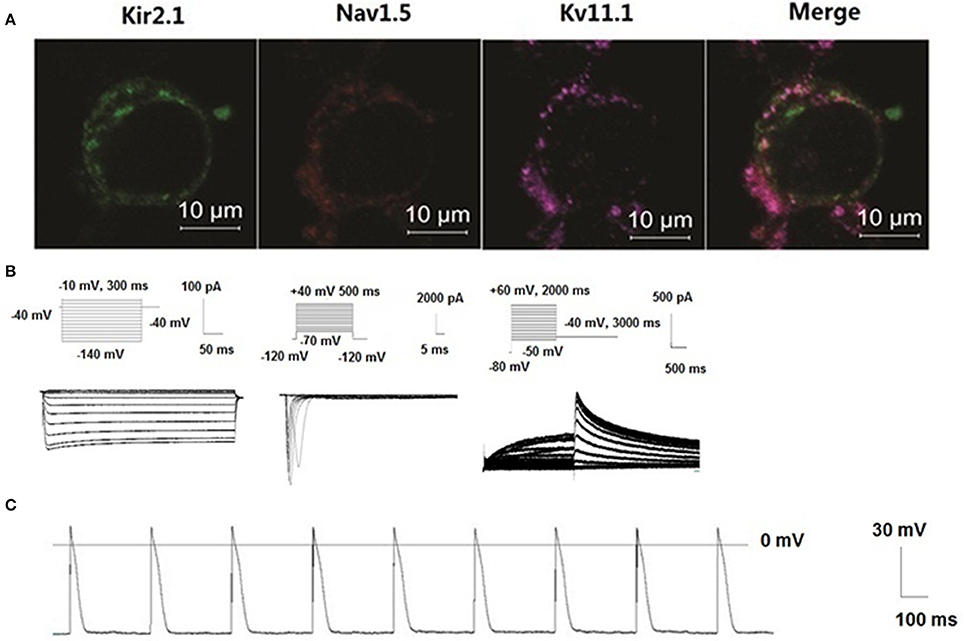
Figure 6. New building of the action potential by co-transfecting Nav1.5, Kir2.1, and KCNH2 into HEK293 cells. Distribution of three channels was detected by confocal microscopy. Images demonstrated that Nav1.5, Kir2.1, and Kv11.1 proteins were stable co-expressed on the HEK293 cell membrane (A). Schematic representation of the voltage-clamp protocol and the representative Nav1.5, Kir2.1, and KCNH2 current traces (B). Action potentials were structured by Nav1.5, Kir2.1, and Kv11.1 channel porteins in the HEK293 cells (C).
Rescue Effect of KCNE2 on Action Potential Composed of KCNH2-G572S
To demonstrate the contribution of KCNE2 on shortening the APD prolonged by G572S, we established Kir2.1+Nav1.5+Kv11.1-G572S HEK293 cell line, and AP was recorded with/without co-expressing KCNE2. Figure 7A showed the two representative AP traces and visually, KCNE2 could shorten the APD which was prolonged by G572S. APD90 of G572S and WT were 85.7 ± 4.5 ms and 49.5 ± 2.7 ms respectively. APD90 of G572S after co-transfecting with KCNE2 were shortened to 56.2 ± 3.3 ms (n = 10, P < 0.01). This suggested that KCNE2 could rescue APD prolonged by G572S. Furthermore, different ratio of 3:1, 1:1, and 1:3 of KCNH2:KCNE2 was used in the experiment. The most significantly rescue effect was at KCNH2: KCNE2 of 1:3 which was in accord with the effect of KCNE2 on G572S mutation current. (n = 10, P < 0.01, Figures 7B,C).
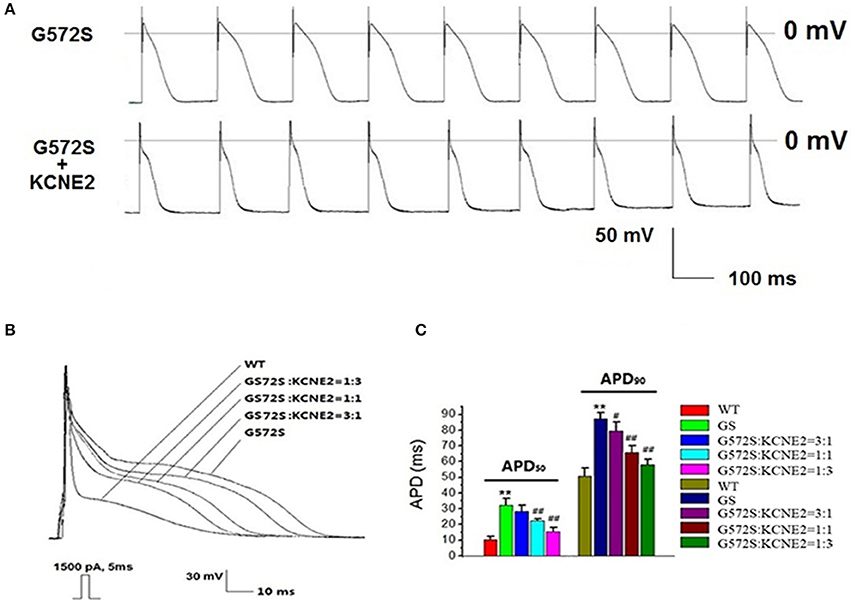
Figure 7. Rescue effect of KCNE2 on action potential composed of KCNH2-G572S. New building action potential was recorded by using clamp-patch technique (A). It was found that prolongation of APD50 and APD90 of G572S mutant were both significantly shortened by KCNE2. Due to the different proportion of KCNH2: KCNE2, this rescue effect by KCNE2 were different, furthermore, the most significantly rescue effect was at KCNH2: KCNE2 of 1:3. (n = 10), **P < 0.01, vs. WT group; #P < 0.05, vs. G572S group; ##P < 0.01, vs. G572S group (B,C).
Discussion
In this experiment, we first found KCNE2 could significantly increase the tail current densities of KCNH2-G572S mutation. Previous experiments have shown that KCNH2-G572S mutation led to loss of channel function, with a significantly lower current and a markedly prolongation of action potential duration and QT interval in electrocardiogram (Splawski et al., 2000; Napolitano et al., 2005; Tester et al., 2005). Our results suggest that KCNE2 restores the channel function, increases both WT and G572S current and shortens the APD prolonged by G572S.
Protein processing and trafficking may play an important and regulated role in controlling channel characteristics (Geva and Schuldiner, 2014). To our knowledge, there are two main reasons why the loss of function happen following channel gene mutant (Sanguinetti et al., 1996; Thomas, 2003): (1). Change of the gating dynamics causes a decline current; (2). The trafficking defect of channel protein to the cell membrane gives rise to down-regulation of the membrane channel proteins and reduces current. hERG channels are translated and initially glycosylated at the endoplasmic reticulum (ER, molecular weight of hERG protein is 135 kDa) (Petrecca et al., 1999). After folding properly with the assistance of chaperones, hERG channels are trafficking to the Golgi for secondary glycosylation (molecular weight of hERG protein is 155 kDa) and then further trafficking to the membrane (Gong et al., 2002; Smith et al., 2011). In this study, our results showed that the cells expressing G572S apparently had a reduced fluorescent intensity and maturation membrane protein (155 kDa) expression on membrane surface. These rusults suggested that G572S maybe led to channel protein migration defects. We have focused on the physical protein-protein association and trafficking of KCNE2 and hERG. After co-transfection with KCNE2, membrane protein expression of G572S mutation and hERG currents densities are increased significantly. The small proteins encoded by KCNE2 have been proposed as accessory subunits for the hERG channel (Abbott et al., 1999). So, it is supported the concept that assembly of KCNE proteins with K+ channels happens early in the biosynthetic process, probably in the ER (Shi et al., 1996; Krumerman et al., 2004; McKeown et al., 2008). KCNE2 can promote the Kv11.1 channel protein to traffic the cell surface and expression of membrane protein (Um and McDonald, 2007). This may partly explain the rescue mechanism of KCNE2 on G572S mutations in this study.
Mutations in different locations of the hERG potassium-channel gene are associated with different levels of risk for arrhythmic cardiac events in LQT2, with the greatest risk related to mutations in the critical pore region of the channel (Moss et al., 2002). G572S is a hERG pore domain mutation which locates in the short linker between the fifth transmembrane domain and the 5-P α-helix of the pore domain. The placement of the inactivation helix (between the S5- and pore helix) is based on functional studies. Anderson et al. speculated that the location of amino acid substitutions in putative Kv11.1-helical or-sheet domains appears to correlate with the trafficking-deficient and mutations disrupted Kv11.1 channel biogenesis (Anderson et al., 2006). Based on this speculation, we hypothesize that KCNE2 propably correct the change of the protein space structure caused by G572S, and this correction could stabilize the channel protein in configurations that facilitate proper trafficking.
As we have known, at lower temperatures, an increased ER retention time might make an improved channel folding. Additionally, a reduced incubation temperature could reverse the mutant's effects on hERG currents according to inhibition of proteasomal degradation and therefore increase the surface expression of these hERG mutants (Thomas et al., 2003). At physiological temperatures mutant channels cannot pass quality control processes of the ER and do not reach the plasma membrane. At reduced temperatures, protein folding might be altered and ultimately permitted exit from the ER (Ficker et al., 2000). It has been reported that low temperature could correct hERG defective protein trafficking, thus increase the surface expression of some LQT2-causing mutants, such as G601S, K28E, N407D, and A422T (Zhou et al., 1999; Gong et al., 2006; Chen et al., 2007; Guo et al., 2012). G572S has a dominant negative trafficking defect and a severe phenotype (Zhao et al., 2009). In our research, matured protein expression of G572S was found to be restored by lower incubation temperature, which is similar to those mutations with trafficking defect mentioned above. This result supported G572S was likely a temperature-sensitive mutation localized to the transmembrane domain. Based on our observation, KCNE2 induced a more effectively up-regulation of matured protein (155 kDa) expression of G572S at lower temperature, which has not been reported before. However, the molecular mechanism responsible for this phenomenon remains to be analyzed in the future.
In the study about mechanism of ion channel diseases, it is indispensable to evaluate the effect of mutation channels on the action potential. However, subculture of the myocardial cell isolation is difficult. Other difficulties lie in cellular toxicity of transfection reagents and low transfection efficiency. Besides, low differentiation rate of induced pluripotent stem cells (iPS) limits to its application (Müller et al., 2013). Although computer simulation can better reflect the effects of mutations channel of action potential (ten Tusscher et al., 2004), it is unable to objectively reflect the real situation. It was reported that the HEK 293 cells genetically engineered to stably express both SCN5A, KCNJ2, much like primary excitable cells, could develop membrane excitability. wherein on reaching an excitation threshold by current injection, they reproducibly fired an “all-or-none” AP (Kirkton and Bursac, 2011). We chose resting potential current of IK1, depolarization current of INa and major repolarization current of IKr to build an action potential and then a typical sample of myocardial cell-like action potential was recorded. However, it seems to be different from the classic AP. The possible reason might be a lack of calcium channel which contributes to the repolaration plateau of action potential.
By using the above system, we found hERG-G572S was a loss of function mutation which could lead a reduction of hERG current densities and could prolong APD of HEK 293 cell line established in our research. However, with KCNE2 co-expression, hERG current density increased and the APD was significantly shortened. This assay system which was used to evaluate function of hERG channel genetic mutations in our research is a useful tool because of its simplicity and high cost performance.
The findings in this study described electrophysiological characteristics of the LQT2 syndrome mutation KCNH2-G572S and regulation by accessory protein KCNE2. The study provided a clue about LQT2 and relative rescue mechanism. It might prompt a certain treatment of KCNE2 on LQT2.
Author Contributions
All authors contributed to this work significantly. LL, contributed to study design, performed site-directed mutagenesis, transfection, and patch clamp experiments independently, interpreted results of experiments, drafted manuscript, prepared figures. JT, performed western blotting experiments, analyzed data, interpreted results of experiments, drafted manuscript. CL, performed confocal microscopy examination, interpreted results of experiments, revised the manuscript. XC, YF, BX, CZ, participated in interpreting results of experiments, edited the manuscript. YS, YUZ, YIZ, revised the manuscript. YL, corresponding author, contributed to conception and design of research, analyzed, and interpreted data of experiments, prepared figures, drafted and revised the manuscript, and accounted for all aspects of the work. All authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the grants from the National Natural Science Foundation of China (No: 81170177, 81100215, 81373835, and 30770901).
References
Abbott, G. W., Sesti, F., Splawski, I., Buck, M. E., Lehmann, M. H., Timothy, K. W., et al. (1999). MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97, 175–187.
Anderson, C. L., Delisle, B. P., Anson, B. D., Kilby, J. A., Will, M. L., Tester, D. J., et al. (2006). Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 113, 365–373. doi: 10.1161/CIRCULATIONAHA.105.570200
Chen, M. X., Sandow, S. L., Doceul, V., Chen, Y. H., Harper, H., Hamilton, B., et al. (2007). Improved functional expression of recombinant human ether-a-go-go (hERG) K+ channels by cultivation at reduced temperature. BMC Biotechnol 7:93. doi: 10.1186/1472-6750-7-93
Dupuis, D. S., Klaerke, D. A., and Olesen, S. P. (2005). Effect of β-adrenoceptor blockers on human ether-a-go-go-related gene (HERG) potassium channels. Basic Clin. Pharmacol. Toxicol. 96, 123–130. doi: 10.1111/j.1742-7843.2005.pto960206.x
Eldstrom, J., and Fedida, D. (2011). The voltage-gated channel accessory protein KCNE2: multiple ion channel partners, multiple ways to long QT syndrome. Expert Rev. Mol. Med. 13, e38. doi: 10.1017/S1462399411002092
Ficker, E., Dennis, A. T., Wang, L., and Brown, A. M. (2003). Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ. Res. 92, e87–e100. doi: 10.1161/01.RES.0000079028.31393.15
Ficker, E., Thomas, D., Viswanathan, P. C., Dennis, A. T., Priori, S. G., Napolitano, C., et al. (2000). Novel characteristics of a misprocessed mutant HERG channel linked to hereditary long QT syndrome. Am. J. Physiol. Heart Circ. Physiol. 279, H1748–H1756.
Fujii, M., Hayashi, K., Ohya, S., Yamamura, H., and Imaizumi, Y. (2013). New screening system for selective blockers of voltage-gated K(+) channels using recombinant cell lines dying upon single action potential. J. Pharmacol. Sci. 123, 147–158.
Fujii, M., Ohya, S., Yamamura, H., and Imaizumi, Y. (2012). Development of recombinant cell line co-expressing mutated Nav1.5, Kir2.1, and hERG for the safety assay of drug candidates. J. Biomol. Screen. 17, 773–784. doi: 10.1177/1087057112442102
Geva, Y., and Schuldiner, M. (2014). The back and forth of cargo exit from the endoplasmic reticulum. Curr. Biol. 24, R130–R136. doi: 10.1016/j.cub.2013.12.008
Gong, Q., Anderson, C. L., January, C. T., and Zhou, Z. (2002). Role of glycosylation in cell surface expression and stability of HERG potassium channels. Am. J. Physiol. Heart Circ. Physiol. 283, H77–H84. doi: 10.1152/ajpheart.00008.2002
Gong, Q., Jones, M. A., and Zhou, Z. (2006). Mechanisms of pharmacological rescue of trafficking-defective hERG mutant channels in human long QT syndrome. J. Biol. Chem. 281, 4069–4074. doi: 10.1074/jbc.M511765200
Guo, J., Zhang, X., Hu, Z., Zhuang, Z., Zhu, Z., Chen, Z., et al. (2012). A422T mutation in HERG potassium channel retained in ER is rescurable by pharmacologic or molecular chaperones. Biochem. Biophys. Res. Commun. 422, 305–310. doi: 10.1016/j.bbrc.2012.04.153
Kirkton, R. D., and Bursac, N. (2011). Engineering biosynthetic excitable tissues from unexcitable cells for electrophysiological and cell therapy studies. Nat. Commun. 2, 300. doi: 10.1038/ncomms1302
Krumerman, A., Gao, X., Bian, J. S., Melman, Y. F., Kagan, A., and McDonald, T. V. (2004). An LQT mutant minK alters KvLQT1 trafficking. Am. J. Physiol. Cell. Physiol. 286, C1453–C1463. doi: 10.1152/ajpcell.00275.2003
Mazhari, R., Greenstein, J. L., Winslow, R. L., Marbán, E., and Nuss, H. B. (2001). Molecular interactions between two long-QT syndrome gene products, HERG and KCNE2, rationalized by in vitro and in silico analysis. Circ. Res. 89, 33–38.
McKeown, L., Swanton, L., Robinson, P., and Jones, O. T. (2008). Surface expression and distribution of voltage-gated potassium channels in neurons (Review). Mol. Membr. Biol. 25, 332–343. doi: 10.1080/09687680801992470
Moss, A. J., Zareba, W., Kaufman, E. S., Gartman, E., Peterson, D. R., Benhorin, J., et al. (2002). Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation 105, 794–799.
Müller, M., Seufferlein, T., Illing, A., and Homann, J. (2013). Modelling human channelopathies using induced pluripotent stem cells: a comprehensive review. Stem Cells Int. 2013:496501. doi: 10.1155/2013/496501
Napolitano, C., Priori, S. G., Schwartz, P. J., Bloise, R., Ronchetti, E., Nastoli, J., et al. (2005). Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA 294, 2975–2980. doi: 10.1001/jama.294.23.2975
Petrecca, K., Atanasiu, R., Akhavan, A., and Shrier, A. (1999). N-linked glycosylation sites determine HERG channel surface membrane expression. J. Physiol. 515(Pt 1), 41–48. doi: 10.1111/j.1469-7793.1999.041ad.x
Saenen, J. B., and Vrints, C. J. (2008). Molecular aspects of the congenital and acquired Long QT Syndrome: clinical implications. J. Mol. Cell Cardiol. 44, 633–646. doi: 10.1016/j.yjmcc.2008.01.006
Sanguinetti, M. C., Curran, M. E., Spector, P. S., and Keating, M. T. (1996). Spectrum of HERG K+-channel dysfunction in an inherited cardiac arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 93, 2208–2212. doi: 10.1073/pnas.93.5.2208
Sanguinetti, M. C., Jiang, C., Curran, M. E., and Keating, M. T. (1995). A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81, 299–307.
Schwartz, P. J., Crotti, L., and Insolia, R. (2012). Long-QT syndrome: from genetics to management. Circ. Arrhythm. Electrophysiol. 5, 868–877. doi: 10.1161/CIRCEP.111.962019
Schweigmann, U., Biliczki, P., Ramirez, R. J., Marschall, C., Takac, I., Brandes, R. P., et al. (2014). Elevated heart rate triggers action potential alternans and sudden death. translational study of a homozygous KCNH2 mutation. PLoS ONE 9:e103150. doi: 10.1371/journal.pone.0103150
Shi, G., Nakahira, K., Hammond, S., Rhodes, K. J., Schechter, L. E., and Trimmer, J. S. (1996). β subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron 16, 843–852.
Smith, J. L., McBride, C. M., Nataraj, P. S., Bartos, D. C., January, C. T., and Delisle, B. P. (2011). Trafficking-deficient hERG K(+) channels linked to long QT syndrome are regulated by a microtubule-dependent quality control compartment in the ER. Am. J. Physiol. Cell Physiol. 301, C75–C85. doi: 10.1152/ajpcell.00494.2010
Sokolov, S., Peters, C. H., Rajamani, S., and Ruben, P. C. (2013). Proton-dependent inhibition of the cardiac sodium channel Nav1.5 by ranolazine. Front. Pharmacol. 4:78. doi: 10.3389/fphar.2013.00078
Splawski, I., Shen, J., Timothy, K. W., Lehmann, M. H., Priori, S., Robinson, J. L., et al. (2000). Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102, 1178–1185.
ten Tusscher, K. H., Noble, D., Noble, P. J., and Panfilov, A. V. (2004). A model for human ventricular tissue. Am. J. Physiol. Heart Circ. Physiol. 286, H1573–H1589. doi: 10.1152/ajpheart.00794.2003
Tester, D. J., Will, M. L., Haglund, C. M., and Ackerman, M. J. (2005). Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2, 507–517. doi: 10.1016/j.hrthm.2005.01.020
Thomas, D. (2003). Defective protein trafficking in hERG-associated hereditary long QT syndrome (LQT2): molecular mechanisms and restoration of intracellular protein processing. Cardiovasc. Res. 60, 235–241. doi: 10.1016/j.cardiores.2003.08.002
Thomas, D., Kiehn, J., Katus, H. A., and Karle, C. A. (2003). Defective protein trafficking in hERG-associated hereditary long QT syndrome (LQT2): molecular mechanisms and restoration of intracellular protein processing. Cardiovasc. Res. 60, 235–241. doi: 10.1016/j.cardiores.2003.08.002
Thomas, P., and Smart, T. G. (2005). HEK293 cell line: a vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 51, 187–200. doi: 10.1016/j.vascn.2004.08.014
Trudeau, M. C., Warmke, J. W., Ganetzky, B., and Robertson, G. A. (1995). HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269, 92–95.
Um, S. Y., and McDonald, T. V. (2007). Differential association between HERG and KCNE1 or KCNE2. PLoS ONE 2:e933. doi: 10.1371/journal.pone.0000933
Walker, V. E., Atanasiu, R., Lam, H., and Shrier, A. (2007). Co-chaperone FKBP38 promotes HERG trafficking. J. Biol. Chem. 282, 23509–23516. doi: 10.1074/jbc.M701006200
Warmke, J. W., and Ganetzky, B. (1994). A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. U.S.A. 91, 3438–3442. doi: 10.1073/pnas.91.8.3438
Weerapura, M., Nattel, S., Chartier, D., Caballero, R., and Hébert, T. E. (2002). A comparison of currents carried by HERG, with and without coexpression of MiRP1, and the native rapid delayed rectifier current. Is MiRP1 the missing link? J. Physiol. 540, 15–27. doi: 10.1113/jphysiol.2001.013296
Zhang, M., Jiang, M., and Tseng, G. N. (2001). minK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as β subunit of cardiac transient outward channel? Circ. Res. 88, 1012–1019. doi: 10.1161/hh1001.090839
Zhang, M., Wang, Y., Jiang, M., Zankov, D. P., Chowdhury, S., Kasirajan, V., et al. (2012). KCNE2 protein is more abundant in ventricles than in atria and can accelerate hERG protein degradation in a phosphorylation-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 302, H910–H922. doi: 10.1152/ajpheart.00691.2011
Zhao, J. T., Hill, A. P., Varghese, A., Cooper, A. A., Swan, H., Laitinen-Forsblom, P. J., et al. (2009). Not all hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant negative trafficking defect and a severe phenotype. J. Cardiovasc. Electrophysiol. 20, 923–930. doi: 10.1111/j.1540-8167.2009.01468.x
Zhou, Z., Gong, Q., and January, C. T. (1999). Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J. Biol. Chem. 274, 31123–31126. doi: 10.1074/jbc.274.44.31123
Keywords: long QT syndrome type 2, KCNH2, KCNE2, action potential, low temperature, trafficking
Citation: Liu L, Tian J, Lu C, Chen X, Fu Y, Xu B, Zhu C, Sun Y, Zhang Y, Zhao Y and Li Y (2016) Electrophysiological Characteristics of the LQT2 Syndrome Mutation KCNH2-G572S and Regulation by Accessory Protein KCNE2. Front. Physiol. 7:650. doi: 10.3389/fphys.2016.00650
Received: 05 September 2016; Accepted: 12 December 2016;
Published: 27 December 2016.
Edited by:
Harold H. Zakon, University of Texas at Austin, USAReviewed by:
Carmen Valenzuela, Alberto Sols Biomedical Research Institute (CSIC), SpainNúria Comes, University of Barcelona, Spain
Copyright © 2016 Liu, Tian, Lu, Chen, Fu, Xu, Zhu, Sun, Zhang, Zhao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Li, liyangbsh@163.com
†These authors have contributed equally to this work.