 Jie-Yin Chen†
Jie-Yin Chen† Xiao-Feng Dai
Xiao-Feng Dai- Laboratory of Cotton Disease, Institute of Food Science and Technology, Chinese Academy of Agricultural Sciences, Beijing, China
Verticillium wilt, caused by the Verticillium dahliae phytopathogen, is a devastating disease affecting many economically important crops. Previous studies have shown that the exoproteome of V. dahliae plays a significant role in this pathogenic process, but the components and mechanisms that underlie this remain unclear. In this study, the exoproteome of V. dahliae was induced in a cotton-containing C’zapek-Dox (CCD) medium and quantified using the high-throughput isobaric tag technique for relative and absolute quantification (iTRAQ). Results showed that the abundance of 271 secreted proteins was affected by the CCD medium, of which 172 contain typical signal peptides generally produced by the Golgi/endoplasmic reticulum (ER). These enhanced abundance proteins were predominantly enriched in carbohydrate hydrolases; 126 were classified as carbohydrate-active (CAZymes) and almost all were significantly up-regulated in the CCD medium. Results showed that CAZymes proteins 30 and 22 participate in pectin and cellulose degradation pathways, corresponding with the transcription levels of several genes encoded plant cell wall degradation enzyme activated significantly during cotton infection. In addition, targeted deletion of two pectin lyase genes (VdPL3.1 and VdPL3.3) impaired wilt virulence to cotton. This study demonstrates that the V. dahliae exoproteome plays a crucial role in the development of symptoms of wilting and necrosis, predominantly via the pathogenic mechanisms of plant cell wall degradation as part of host plant infection.
Introduction
Verticillium wilt, caused by the phytopathogen Verticillium dahliae, is an extremely destructive disease affecting hundreds of dicotyledonous plant species. This disease is particularly hard to eradicate and manage because the survival structures produced by the wilt pathogen remain viable in soil for more than two decades, persistently threatening crops (Fradin and Thomma, 2006; Klosterman et al., 2009). Similar to other known pathogens of this type, V. dahliae colonizes and proliferates in the vascular (xylem) system of a plant, disrupting water-conduction, and leading to a series of characteristic symptoms including wilting leaves, stunting, necrosis, and vein clearing (Klosterman et al., 2009). Typically, V. dahliae enters the host plant either via the root or at the sites of lateral root formation (Bishop and Cooper, 1983), before traversing the endodermis, which normally acts as a physical barrier to pathogen infection (Schnathorst, 1981; Vallad and Subbarao, 2008). When the pathogen reaches the vascular tissue, hyphae start to bud and form conidia which are carried by the xylem fluid and trapped at vessel end walls before germinating and penetrating adjacent vessel elements to continue colonization and start another infection cycle (Cooper and Wood, 1975; Bishop and Cooper, 1983). The pathogen produces toxic or elicitor-like substances as well as hydrolytic enzymes that facilitate its colonization and proliferation in the unique plant vascular system environment (Fradin and Thomma, 2006; Klosterman et al., 2009). Given this background, the V. dahliae exoproteome, including secreted proteins, is thought to play an important role in determining pathogenicity (Fradin and Thomma, 2006).
The arsenal of proteins secreted by plant pathogens have been the focus of recent studies as these modulate interactions between microbes and their hosts. The pathogen exoproteome, comprising multiple pathogenic factors, is likely to be involved in the infection process (Rep, 2005; Kamoun, 2006; Bent and Mackey, 2007; Thomma et al., 2011). Examples include LysM effectors from the tomato leaf mold fungus Cladosporum fulvum and wheat leaf blotch fungus Mycosphaerella graminicola (de Jonge et al., 2010; Marshall et al., 2011), necrosis and ethylene-inducing-like protein (NLP) genes from the Verticillium wilt fungus V. dahliae (Zhou et al., 2012; Santhanam et al., 2013), and small cysteine-rich proteins (SCRPs) from the vascular wilt fungus Fusarium oxysporum f. sp. Lycopersici and the rice blast fungus Magnaporthe oryzae (Lievens et al., 2009; Valent and Khang, 2010). A number of studies have focused on the exoproteome using proteomic and genomic techniques (Spanu, 2012; Girard et al., 2013) as this is thought to play an important role in pathogenesis. Investigations include studies on Fusarium spp. (Phalip et al., 2005; Paper et al., 2007; Ravalason et al., 2012), Magnaporthe oryzae (Wang et al., 2011; Jung et al., 2012), Xanthomonas oryzae (Wang et al., 2013), and Botrytis cinerea (González-Fernández et al., 2014). A deletion mutant of the Verticillium transcription regulator Vta2, which controls expression of 125 secreted proteins, failed to colonize plants and induce disease symptoms, indicating a role in pathogenicity (Tran et al., 2014), and the genome of two strains of the vascular wilt fungus V. dahliae, VdLs.17 and JR2, isolated from lettuce and tomato, respectively, have been sequenced (Klosterman et al., 2011; de Jonge et al., 2013). Comparative genomic analyses have also shown that V. dahliae has markedly increased secretion levels of pathogenic factors, including LysM effectors, NLPs, and an Ave1 virulence factor (Klosterman et al., 2011; de Jonge et al., 2012, 2013; Zhou et al., 2012; Santhanam et al., 2013; Klimes et al., 2015), in agreement with previous reports that the purified exoproteome of this pathogen can induce chlorosis and necrosis on the leaves of susceptible plants (Buchner et al., 1982, 1989; Nachmias et al., 1985; Meyer et al., 1994; Mansoori et al., 1995; Davis et al., 1998). These results all suggest that V. dahliae requires the presence of exoproteome virulence factors in order to infect host plants.
Hydrolases in the exoproteome are considered important for the generation of disease symptoms and pathogenesis, especially the enzymes involved in plant cell wall degradation (King et al., 2011; Glass et al., 2013; Kubicek et al., 2014). Indeed, genomic analysis has demonstrated that the expansion of PCWDEs is closely related to pathogenicity. This is especially true in the case of pectinases and cellulases, which participate in plant cell wall (pectin and cellulose) degradation (Lévesque et al., 2010; Ma et al., 2010; Ospina-Giraldo et al., 2010; Amselem et al., 2011; Klosterman et al., 2011). In V. dahliae, the pathogenic role of several PCWDEs has been identified; disruption of sucrose non-fermenting 1 (VdSNF1), which regulates catabolic repression, resulted in the reduced expression of a large number of PCWDE genes and in severely impaired virulence to plants (Tzima et al., 2011). Similarly, a knockout mutant in the glucosyltransferase homolog gene VDAG_02071 resulted in drastically reduced disease symptoms in Nicotiana benthamiana compared with the wild-type strain (Klosterman et al., 2011), while deletion of the VdSSP1 gene, the product of which is a secretory protein involved in pectin and starch degradation, reduced virulence levels on cotton (Liu Y. et al., 2013). These results therefore suggest that V. dahliae utilizes several CWDEs to infect host plants. Previous research has also shown that pectinolytic enzymes in the exoproteome of Verticillium spp. play a critical role in pathogenesis; these include polygalacturonase, pectate lyase, and pectinesterase, which induce necrosis of plant tissues in vitro and cause wilt symptoms (Wang and Keen, 1970; Mussel and Strause, 1972; Cooper and Wood, 1980; Huang and Mahoney, 1999). A comparative genomic analysis showed that Verticillium spp. increased pectinolytic enzyme activities to accelerate plant cell wall degradation, especially polysaccharide lyase (PL) and glycoside hydrolase (GH) gene families (Klosterman et al., 2011). Thus, PCWDEs in the exoproteome of V. dahliae, especially pectinases and cellulases, likely play a crucial role in pathogenesis.
The iTRAQ method, coupled with liquid chromatography-tandem mass spectrometry (LC–MS/MS), has previously been utilized in proteomics analysis. The iTRAQ technique facilitates the quantification of protein concentrations in biological systems and has been applied, for example, to Trichoderma reesei (Adav et al., 2013), Phanerochaete chrysosporium (Adav et al., 2012), Aspergillus fumigatus (Liu D. et al., 2013), and Arthrobotrys oligospora (Liang et al., 2013). However, although a number of Verticillium spp. proteomes have been reported (Mandelc et al., 2009, 2013; El-Bebany et al., 2010; Mandelc and Javornik, 2015), no data are available on the exoproteome associated with the interactions between V. dahliae and host plants. Thus, in this study, the nutritional components of the unique in vitro CCD medium were used to evaluate the induced secretome of V. dahliae. The pathogenic function of the induced secretome was analyzed, and its components were identified. The pathogenic function of proteins was analyzed by knocking out their encoding genes. This study provides insights into the mechanism by which the proteins secreted by V. dahliae facilitate its adaption to the unique environment of the plant vascular system.
Materials and Methods
Strain Cultivation and Exoproteome Extraction
The high virulence defoliating strain of V. dahliae, Vd991, was used for exoproteome analysis. This strain was grown on a potato dextrose agar (PDA) medium for 7 days to enable the production of conidia, before these were harvested by washing the plate with sterilized water. The resulting suspension was then filtered through gauze to remove mycelia and conidia concentration was adjusted to 5 × 106 conidia/mL. The CCD medium was prepared by sowing a susceptible cultivar of Gossypium hirsutum L., ‘Junmian No.1’, in sterilized soil at 28°C under a photoperiod of 14 h light/10 h dark for 3 weeks. Subsequently, whole plants without leaves were harvested and ground into powder in liquid nitrogen, and the CCD medium was prepared using a C’zapek-Dox (CD, i.e., 2 g/L NaNO3, 0.5 g/L MgSO4-7H2O, 0.5 g/L KCl, 100 mg/L 14 FeSO4-7H2O, and 1 g/L K2HPO4, pH 7.2) solution supplemented with 1% (w/v) cotton tissue powder.
In order to induce the exoproteome, 5 mL (5 × 106 conidia/mL) of prepared Vd991 conidia were cultured on the CCD medium and CD medium. Three replicates of this preparation were incubated at 25°C and centrifuged at 180 rpm. Under normal conditions, conidia were incubated in the original CD medium supplemented with 1% (w/v) sucrose but without the 1% (w/v) cotton tissue powder; at the same time, two media were cultured in the same conditions but without inoculating Vd991 conidia to create a control group. After 5 days of incubation, culture suspensions were filtered through four layers of gauze and the fungal biomass was further removed via centrifugation. Supernatants were collected and the solution was filtered through a 0.22 μm membrane (Millipore, Temecula, CA, USA). The Avant purification system (GE Healthcare, Piscataway, NJ, USA) was used for exoproteome enrichment; the filtered solution was enriched using a Hitrap TM Capto SP/Q column (GE Healthcare, New Jersey, USA), desalted using sephasex G75 (GE Healthcare, Piscataway, NJ, USA). Finally, the purified exoproteome was freeze-dried before being resolved in sterilized water. Exoproteome concentration was then determined using 2-D Quant kit assay reagents (GE Healthcare, Piscataway, NJ, USA) following the manufacturer’s instructions.
Wilting Activity Determination
A small amount (1.0 μg) of purified exoproteome protein was dissolved in 20.0 μL sterilized water in a 0.5 mL microcentrifuge tube in order to determine wilting activity. To do this, G. hirsutum L., ‘Junmian No.1’ cotyledons containing petiole were collected from 2-week-old seedlings and were inserted directly into the microcentrifuge tube containing the exoproteome solution. When all this solution was absorbed by the cotyledons, 2 mL of sterilized water was added to the tube and it was placed in a box with a transparent cover to maintain appropriate moisture. Cotyledon phenotypes were investigated after 72 h at 25°C using an illumination incubator. Four treatments were carried out for the wilting activity assay, including the induced exoproteomes purified from the CCD medium, the CD medium, and the blank control of the two types. In each treatment, 15 cotton cotyledons were detected.
iTRAQ Labeling, Spectrometric Analysis, and Data Processing
Samples containing 100 μg of protein were digested using Trypsin Gold (Promega, Fitchburg, WI, USA) for 16 h at 37°C to generate peptides. Subsequent to digestion, these peptides were dried by vacuum centrifugation, and resuspended in 0.5 M TEAB. Desalted peptides were then labeled with iTRAQ reagents (Applied Biosystems, Carlsbad, CA, USA) following the manufacturer’s instructions (Zieske, 2006). Thus, three repeats of extracted proteins in the CD medium were labeled with reagents 113, 114, and 115, while the fractions in the CCD medium were labeled with reagents 116, 117, and 118, respectively. Three technical repeats of each treatment were performed, and the peptide mixtures were pooled and dried by vacuum centrifugation. These pooled iTRAQ-labeled peptide mixtures were fractionated into ten portions using strong cationic exchange chromatography and desalted in a nanobored C18 column with a picofrit nanospray tip (i.e., 365 μm i.d × 15 cm, 5 μm particles, 150 À pore size) (Phenomenex Inc., Torrance, CA, USA).
Data acquisition was performed using time-of-flight (TOF)-MS in a TripleTOF5600 MS system (AB SCIEX, Concord, ON, Canada) fitted with a Nanospray III source (AB SCIEX) and using a pulled quartz tip as the emitter (New Objectives, Woburn, MA, USA). Data were acquired using an ion spray voltage of 2.5 kV, a curtain gas of 30 PSI, a nebulizer gas of 15 PSI, and an interface heater temperature of 150°C. The MS was operated with a resolving power of greater than or equal to 30,000 full width at maximum (FWHM) for TOF scans. Instrument data files (.wiff) for fractions were transformed to.mgf files using MS convertion (AB SCIEX, Concord, ON, Canada) before being merged into one.mgf.
Raw files were processed using the Mascot computational proteomics platform version 2.3.01. Fragmentation spectra were searched against the Vd991 genome encoding protein database (9,818 genes) maintained in our laboratory with fragment mass tolerances set to 20 mmu and with up to one missed cleavage. For database searches, carbamidomethylation of cysteine was set as a fixed modification, while the oxidation of glutamine and protein N-terminal acetylation were chosen as variable modifications. Both peptide and protein identifications were filtered at 1% false discovery rates so proteins identified with at least two unique peptides were independent of peptide score.
Exoproteome Annotation
Secretory proteins were annotated on the basis of subcellular localization, performed using the WoLF PSORT software (fungi model) to identify putative extracellular proteins (Horton et al., 2007). Signal peptides and signal peptide cleavage sites of putative extracellular proteins were predicted using the SignalP software (version 4.1; D-Score cut-off set to 0.500) (Petersen et al., 2011). All putative extracellular proteins with signal peptides were then analyzed for the presence of transmembrane domains using the TMHMM 2.0 software (Krogh et al., 2001), while those containing just a signal peptide but lacking transmembrane domains, as predicted by either of the two software packages, were identified as secreted proteins. Non-classical secretory proteins (NCSPs) were predicted using the SecretomeP software (Bendtsen et al., 2004), and annotation of putative CAZymes was performed using the Hidden Markov Model (HMM)-based routine of the CAZymes database (Cantarel et al., 2009). Significant hits compared to this database were identified using the set of putative CAZymes in the BLAST software (i.e., e-value < 1e-5, similarity >30%) (Altschul et al., 1997), subsequently used to increase accuracy of annotation. The CAZymes involved in plant cell wall degradation were then classified using methods outlined in previous studies (Battaglia et al., 2011; Goodwin et al., 2011), while the homologues of known pathogenicity-related genes were predicted using the Pathogen-Host Interactions (PHI) database (version 3.6)1 (Winnenburg et al., 2008). Proteins comprised of less than 400 amino acids and containing up to four cysteine residues were designated as SCRPs, and pathways were annotated using the Putative functional annotations were interrogated to known databases using BLASTP to identify the best homologues, including the eggNOGs (Powell et al., 2012), InterProScan (Jones et al., 2014), and Gene Ontology (GO). The significance of GO catalog for induced exoproteome was identified using a Fisher’s Exact Test (filtered with FDR ≤ 0.05). The Web Gene Ontology Annotation Plot (WEGO) software was used to retrieve GO annotations of unigenes for describing biological processes, molecular functions and cellular components (Ye et al., 2006). Kyoto Encyclopedia of Genes and Genomes (KEGG) database implemented in the BlastP software (Kanehisa et al., 2012).
Agrobacterium-Mediated Targeted Gene Deletion
In order to generate the deletion constructs of PL3 genes, flanking sequences were obtained from Vd991 genomic DNA. Thus, the hygromycin element, which consists of the TrpC-promoter, the hygromycin phosphotransferase gene, and the Nos-terminator, was amplified in our laboratory with Hyg-F and Hyg-R primers from the pCT-Hyg vector. These three fragments were fused into one via fusion PCR while nest PCR was carried out to obtain the amplicon, which included the upstream fragment, hygromycin elements, and the downstream fragment. Amplified products were then cloned into a pGKO2-Gateway vector (Khang et al., 2005) as previously described (Liu Y. et al., 2013). All the primers used for vector construction are listed in Supplementary Table S7; the recombinant vectors for the deletion mutant were transferred into Agrobacterium tumefaciens AGL-1 for fungal transformation.
The A. tumefaciens-mediated transformation of V. dahliae was carried out for the homolog recombination and complementary transformants as previously described (Mullins and Kang, 2001). Homologous recombination transformants were selected on the PDA medium (i.e., potato, 200 g/L, glucose, 20 g/L, agar, 15 g/L) with 200 μg/mL of cefotaxime, 50 μg/mL of hygromycin, and 200 μg/mL of 5-fluoro-2′-deoxyuridine. Single spore isolation was performed for all transformants with positive ones verified using PCR.
Pathogenicity Assays
Pathogenicity assays were performed on cotton seedlings when the first euphylla was developed; 5 × 106 conidia/mL suspensions in sterile water were prepared, and six pots of cotton seedlings with three repeats were prepared for each transformant. Each pot containing five cotton seedlings was fully immersed in a new tray previously immersed in a 15 mL conidial suspension. Seedlings were then grown and maintained at 28°C under a 14 h light/10 h dark photoperiod, and disease symptom phenotype was investigated 3 weeks after inoculation.
Gene Expression Analysis
To determine the in planta expression of PL3 genes, cotton seedlings were root inoculated with 5 × 106 conidia/mL of a V. dahliae suspension. Whole plants were harvested at 3, 5, 7, 9, and 14 days post inoculation (dpi) and flash frozen in liquid nitrogen for RNA extraction using a Total RNA Miniprep Kit (Axygen, Tewksbury, MA, USA) and cDNA synthesis using a FastQuant cDNA Reverse Transcriptase Kit (TianGen, Beijing, China). Quantitative reverse transcription-PCR (qRT-PCR) was performed to identify expression levels of PCWDE genes affected by T-DNA insertion using the FastFire qPCR premix, SYBR Green (TianGen, Beijing, China), and relative gene expression levels were calculated using the 2-ΔΔCT method (Livak and Schmittgen, 2001) with the β-tubulin gene (VDAG_10074, VdLs.17) as an internal control. All primers are listed in Supplementary Table S3; real-time PCR conditions comprised an initial 94°C denaturation step for 10 min, followed by 40 cycles at 94°C for 15 s and at 60°C for 1 min.
Results
The Induced V. dahliae Exoproteome Displays Cytotoxic Activity on Cotton
Strain Vd991 of V. dahliae, isolated from a G. hirsutum host, is a highly virulent strain that causes Verticillium wilt in cotton (Figure 1A). To evaluate the cytotoxic activity of these secreted proteins, wilting activity of an exoproteome purified from a culture suspension was evaluated on cotton cotyledons. Utilizing sucrose as the sole carbon source in the CD medium, the exoproteome of V. dahliae exhibited little cytotoxic activity on cotton leaves, which showed few wilting spots (Figure 1B). However, the induced exoproteome of V. dahliae purified from modified medium containing the CCD medium caused serious chlorosis and necrosis of cotton leaves (Figure 1B). These results indicate that the induced exoproteome possesses significant cytotoxic activity and causes symptoms similar to those induced by inoculation with V. dahliae strains.
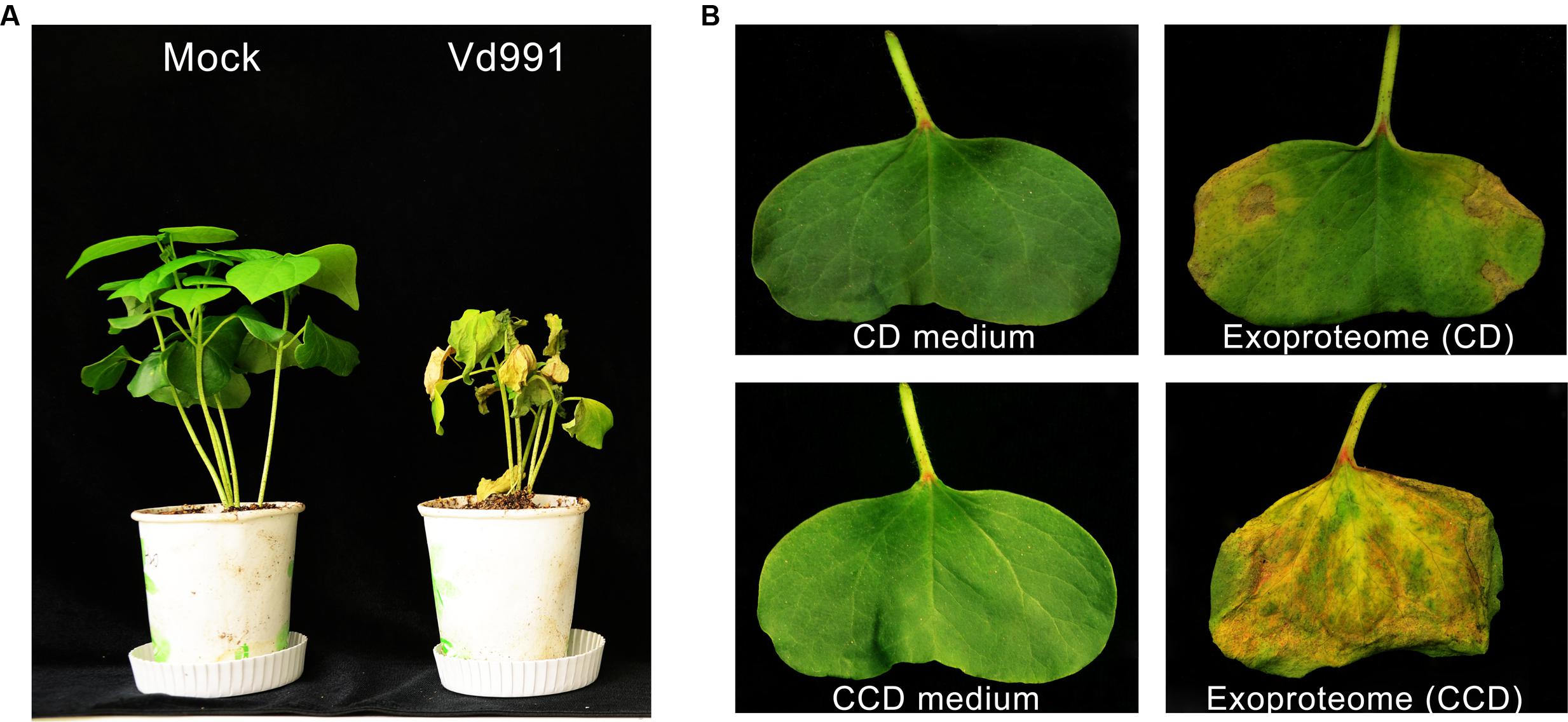
FIGURE 1. Cytotoxic activity of the induced exoproteome of the V. dahliae strain Vd991. (A) Virulence detection of Vd991 infection on cotton. Two-week-old seedlings of susceptible cotton of G. hirsutum L., ‘Junmian No.1’ were inoculated with sterile water (Mock), wild-type (WT) V. dahliae. The disease symptom phenotype was investigated 3 weeks after inoculation. (B) Validation of the cytotoxic activity of the Vd991 exoproteome on cotton leaves. CD medium, C’zapek-Dox medium; CCD medium, cotton-containing C’zapek-Dox medium. Wilting activity of an exoproteome purified from a culture suspension was evaluated on cotton cotyledons. The CD medium and CCD medium were used as the negative controls.
Identification of the Induced Exoproteome Components of V. dahliae
A total of 1,881 suspected proteins were identified using a concatenated target and decoy strategy with Vd991 genome sequences (9,818 protein-encoding sequences) (Supplementary Figure S1; Supplementary Table S1). Using cut-off values based on unique alignment peptides ≥2, FDR < 0.01, and 1.5-fold change, 536 proteins were quantified by iTRAQ using three biological replicates. Next, two-way ANOVA of fold changes (model: fold change = medium conditions + replicates + system error) in the three biological replicates and filtering of regulated proteins on the basis of a P-value < 0.00001 allowed us to identify 325 proteins with highly significant fold change abundances in the induced exoproteome compared to those from the original CD medium (Supplementary Figure S1; Supplementary Table S2). Of the 325 differentially expressed proteins, abundances of 230 were higher in the CCD medium compared with that which used sucrose as the carbon source (Supplementary Table S2). These results suggest that the V. dahliae pathogens up-regulate the expression of most exoproteome components to adapt to the cotton plant tissue environment.
The Induced Exoproteome Is Predominantly Produced by the Golgi/Endoplasmic Reticulum (ER) Secretory System in V. dahliae
Although the exoproteome in this study was purified from culture filtrate, some intracellular proteins were likely present due to cytolysis during cultivation. This was confirmed by evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG) annotation of the induced exoproteome which shows intracellular localization of several proteins, 54 of which are involved in information storage and processing, including ribosomal proteins and eukaryotic translation initiation factors (Table 1; Supplementary Table S3). This result also indicates that a small number of intracellular proteins were present in the induced exoproteome due to normal cytolysis of V. dahliae.

TABLE 1. Secretory analysis of inducing exoproteome by bioinformatics.
The subcellular localization of identified proteins was predicted using multiple software tools. Results showed that the proteins predicted to be extracellular via WoLF-PSORT and to harbor a signal peptide by SignalP4.1, but lacking a transmembrane domain as predicted by TMHMM2.0, were secreted proteins generally from the Golgi/ER systems. With the exception of the 54 intracellular proteins discussed above, 172 of the 271 with affected abundances were identified as secreted (Table 1; Supplementary Figure S1; Supplementary Table S4). However, because some extracellular proteins do not contain a signal peptide, these were subjected to SecretomeP analysis to identify NCSPs. In this way, 15 proteins lacking a transmembrane domain were identified as NCSPs by filtering those with NN scores > 0.5 using the SecretomP software (Table 1; Supplementary Figure S1; Supplementary Table S4). In total, 69% (187/271) of predicted proteins in the induced exoproteome were identified as secreted (Table 1). The ratio of typical secreted proteins containing a signal peptide was significantly higher than NCSPs in the induced exoproteome (Table 1), indicating that secreted proteins of V. dahliae were produced mainly by the Golgi/ER system. In addition, 31% of the remaining identified proteins (84) were not predicted by bioinformatic analysis to be secretory (Table 1; Supplementary Figure S1), suggesting that the V. dahliae pathogen possesses as yet unknown secretory mechanisms.
Function of the Induced Exoproteome in V. dahliae Pathogenicity
Previous genomic studies on Verticillium spp. suggest a gene family expansion such that encoded secreted proteins are involved in plant cell wall degradation, a process which likely facilitates proliferation of the fungi in the unique environment of the plant vascular system (Klosterman et al., 2011). Thus, to identify factors related to pathogenicity in the induced exoproteome, the function of proteins present at enhanced abundance was analyzed via annotation using several databases. Results show that of the 271 proteins that have enhanced abundance in the induced exoproteome, at least 74 are predicted to be involved in carbohydrate transport and metabolism based on eggNOG annotation (Figure 2A). Indeed, GO annotation showed that 160 proteins occurring at enhanced abundance are involved in molecular catalytic function (Figure 2B), including 95 that are enriched in hydrolase activity, principally the hydrolysis of O-glycosyl compounds (Figure 2C). These results therefore suggest that the induced exoproteome plays a crucial role in V. dahliae carbohydrate metabolism in the CCD medium.
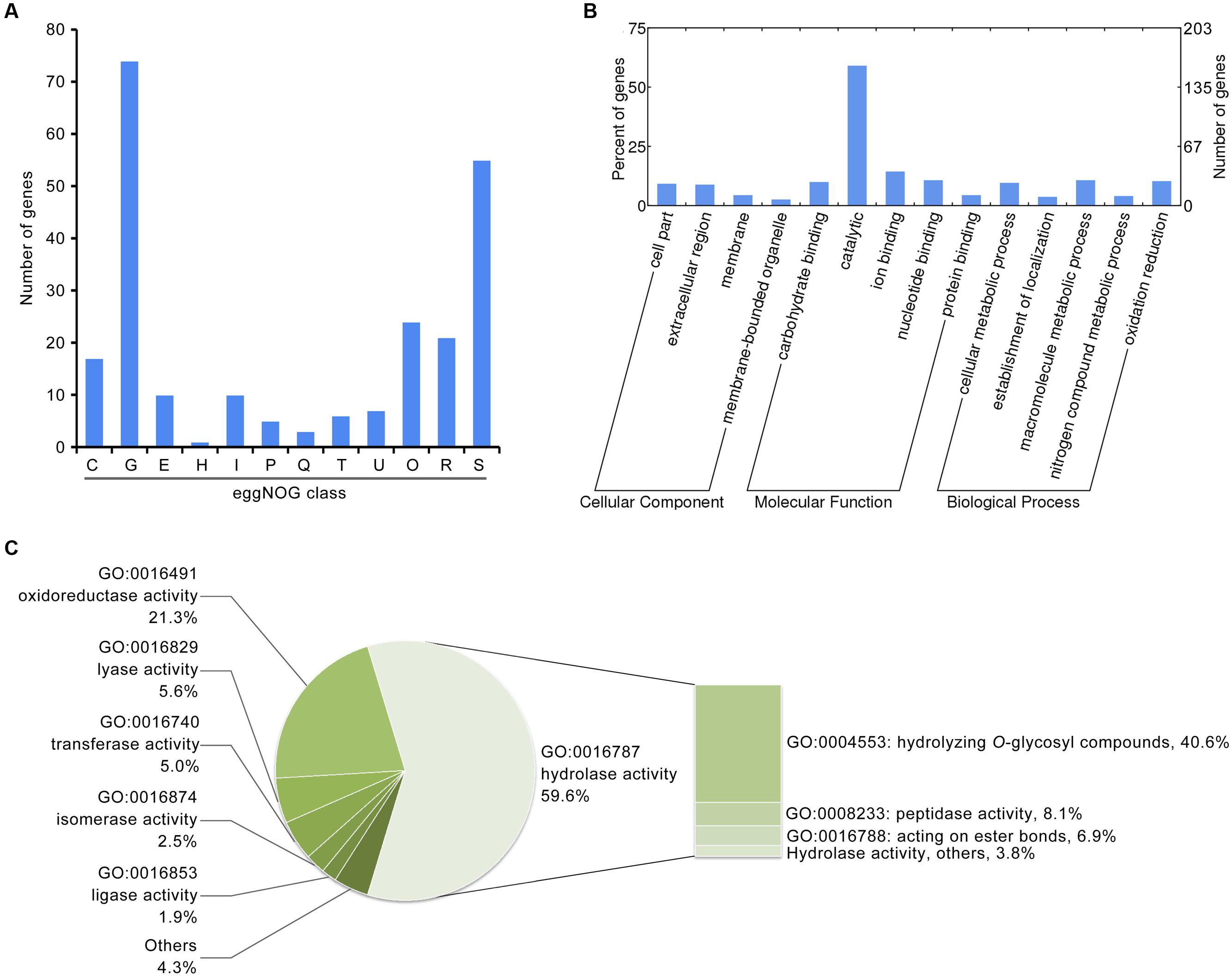
FIGURE 2. Functional annotation of the V. dahliae induced exoproteome. (A) Functional annotation of the induced exoproteome using the eggNOG database. The alphabet in x-axis represents the eggNOG class. T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; O, posttranslational modification, protein turnover, chaperones; C, energy production and conversion; G, carbohydrate transport and metabolism; E, amino acid transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport and catabolism; R, general function prediction only; S, function unknown. (B) GO annotation of the V. dahliae induced exoproteome. The significance of GO annotations for induced exoproteome was identified using a Fisher’s Exact Test (FDR ≤ 0.05), and functional clusters was performed using WEGO software. (C) Function catalog of the induced exoproteome by catalytic class (GO:0003824).
In addition, several enhanced abundance proteins in the induced exoproteome are biologically involved with reactive oxygen species, and 17 of these are clustered in energy production and conversion according to eggNOG annotation (Figure 2B). Overall, 34 proteins are oxidoreductase active, according to the GO annotation (Figure 2C). Partially secreted proteins are referred to as effectors, and 16 hypothetical proteins (i.e., without homologue function) belonging to the small cysteine-rich protein family (< 400 amino acids, ≥4 cysteine residues; Supplementary Table S5) were identified in the induced exoproteome. These probably function to facilitate pathogen colonization by modulating host biochemistry and physiology. In addition, the abundance of known effectors NLPs, were significantly up-regulated 9.05 ± 2.04 and 3.75 ± 0.17 fold in the CCD medium, respectively (Supplementary Table S5). These results suggest that the functions of these enhanced abundance proteins in the induced exoproteome are closely associated with the pathogenicity of V. dahliae to cotton.
The Induced Exoproteome Is Significantly Associated with Plant Cell Wall Degradation
The induced exoproteome is enriched in hydrolase activity, principally O-glycosyl compounds (Figure 2C), which implies involvement in carbohydrate metabolism. To confirm this hypothesis, further functional annotation of the proteins identified in the induced exoproteome was performed using the CAZymes database (Yin et al., 2012). In total, 126 of the 271 enhanced abundance proteins were recognized as CAZymes, 105 of which contained only one functional domain (CAZymes module) (Supplementary Table S6). More than half of these proteins (n = 72) contained a GH module, which facilitates hydrolysis and/or rearrangement of glycosidic bonds. In addition, 24 proteins contained carbohydrate-binding modules (CBM), which promote the hydrolase activity of carbohydrate hydrolase (Figure 3A; Supplementary Table S6), while 13, 15, and 24 proteins exhibited PL, carbohydrate esterase, and auxiliary activities (Figure 3A), respectively. However, proteins containing a glycosyltransferase module, which mediates the formation of glycosidic bonds, were not identified in the induced exoproteome, and of the CAZymes in the induced exoproteome, almost all (112 out of 126) were typical secreted forms (i.e., harboring a signal peptide), 28 were SCRPs, and 49 were homologous to known pathogenic factors in the PHI database, of which 16 have previously been reported to influence the virulence of other microbial pathogens according to the PHI database annotation (Figure 3B).
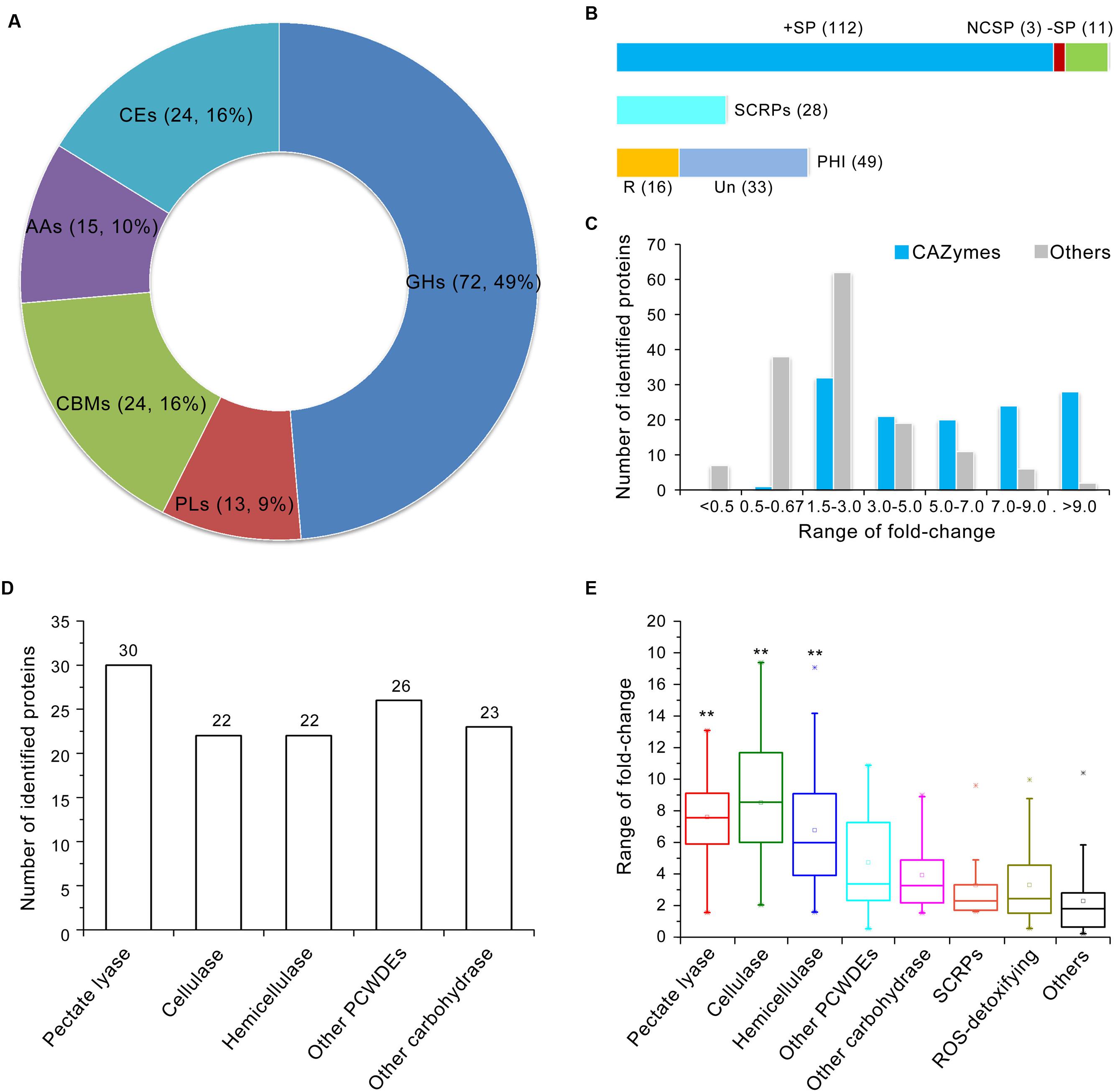
FIGURE 3. Annotation of CAZymes in the V. dahliae induced exoproteome. (A) CAZymes catalog of the induced exoproteome. GHs, glycoside hydrolases; PLs, polysaccharide lyases; CBMs, carbohydrate-binding modules; AAs, auxiliary activities; CEs, carbohydrate esterases. Two numbers in bracket represents the quantity and ratio of the total CAZymes. (B) Analysis of the potential pathogenic function of CAZymes. The upper bar is the secretory characteristic of CAZymes, the bar in blue, red and green color represent the CAZymes contains typical signal peptide, non-classical secretory proteins, and without signal peptide, respectively; the middle bar represents CAZymes belongs to SCRPs; the bottom bar represents CAZymes belongs to PHI protein, the bar in yellow and watchet represents the CAZymes homolog to PHI that reduced (R) virulence and unaffected (UN) pathogenicity, respectively. (C) Distribution of abundance variation of CAZymes in the induced exoproteome. The blue and gray bar represents the quantity of identified CAZymes and other identified proteins among different variation range in induced exoproteome, respectively. (D) Statistics showing the number of identified proteins involved in plant cell wall degradation in the induced exoproteome. (E) Abundance variations of identified proteins involved in cell wall degradation. The range of fold-change was displayed by the Box whisker plot. In each box plot, the central point represents the median, the rectangle gives the interval between the 25 and 75% percentiles, and the whisker indicates the range, the color asterisk (∗) represents the extreme outlier. Significant of abundance variations in pectate lyase, cellulase, and hemicellulose to other proteins were identified using a Student’s t-test with a significance threshold of 0.01(∗∗).
On the basis of abundance variations of proteins identified in the induced exoproteome, CAZyme regulation patterns differed significantly. For example, abundance of almost all CAZymes was significantly up-regulated in the induced exoproteome, with the exception of one protein containing the CBM50 domain which was reduced in abundance (VEDA_09538) (Figure 3C; Supplementary Table S6). Results show that 28 of the 30 identified proteins with a greater than ninefold enhanced abundance were CAZymes (Figure 3C); the functional clustering of these proteins suggests that they are mainly involved in plant cell wall degradation as 30, 22, and 22 are related to pectin, cellulose, and hemicellulose degradation, respectively (Figure 3D). In addition, CAZymes abundance associated with plant cell wall degradation was significantly greater than that of the other identified proteins (Figure 3E). Those involved in the degradation of pectin and cellulose were considerably up-regulated, more than 75% at least five times in the CCD medium (Figure 3E). These results strongly suggest that enhanced plant cell wall degradation activity plays an important role in the CCD medium because of the presence of cotton cell wall components, and that this mechanism promotes the germination of the V. dahliae pathogen and its proliferation in xylems.
The Induced Exoproteome Is Involved Mainly in Pectin and Cellulose Degradation
To overcome the plant cell wall barrier, phytopathogenic fungi produce enzymes that degrade polymers, particularly pectin and cellulose. Results show that of the CAZymes related to pectin degradation, 30 were up-regulated in the CCD medium, including 13 CAZymes module types (e.g., PL1, PL3, and GH28) (Supplementary Figure S2). Similarly, 22 identified proteins, including nine CAZymes module types (e.g., GH3, GH5, GH7, and CBM1) involved in cellulose degradation were also up-regulated in the CCD medium (Supplementary Figure S2B). Interestingly, abundances of CAZymes involved in pectin and cellulose degradation were also up-regulated in the CCD medium; proteins 26 and 18, respectively, were up-regulated by more than fivefold (Supplementary Figure S2). These results strongly suggest that CAZymes associated with the degradation of pectin and cellulose play a crucial role in V. dahliae germination and proliferation in the CCD medium.
Pathway annotation shows that at least 32 up-regulated CAZymes are involved in starch and sucrose metabolism (ko:00500)2, required for the degradation of plant cell walls (Figure 4). Indeed, of the proteins involved in pectin degradation, the abundance of 15 which mediate the degradation of pectin to pectate and onward to D-galacturonate or 4-(4-deoxy-α-D-gluc-4-enuronosyl)-D-galacturonate were strongly up-regulated in the CCD medium (Figure 4). Similarly, the abundance of 10 proteins, most of which are involved in the degradation of cellulose to cellobiose and then onward to β-D-glucose, were also significantly up-regulated in the CCD medium (Figure 4), as were the abundances of proteins involved in the metabolism of other plant cell wall components (e.g., 1,3-β-glucan, and starch) (Figure 4). Thus, the V. dahliae pathogen recruits CAZymes to participate in plant cell wall degradation (cotton tissue) by breaking down pectin and cellulose.
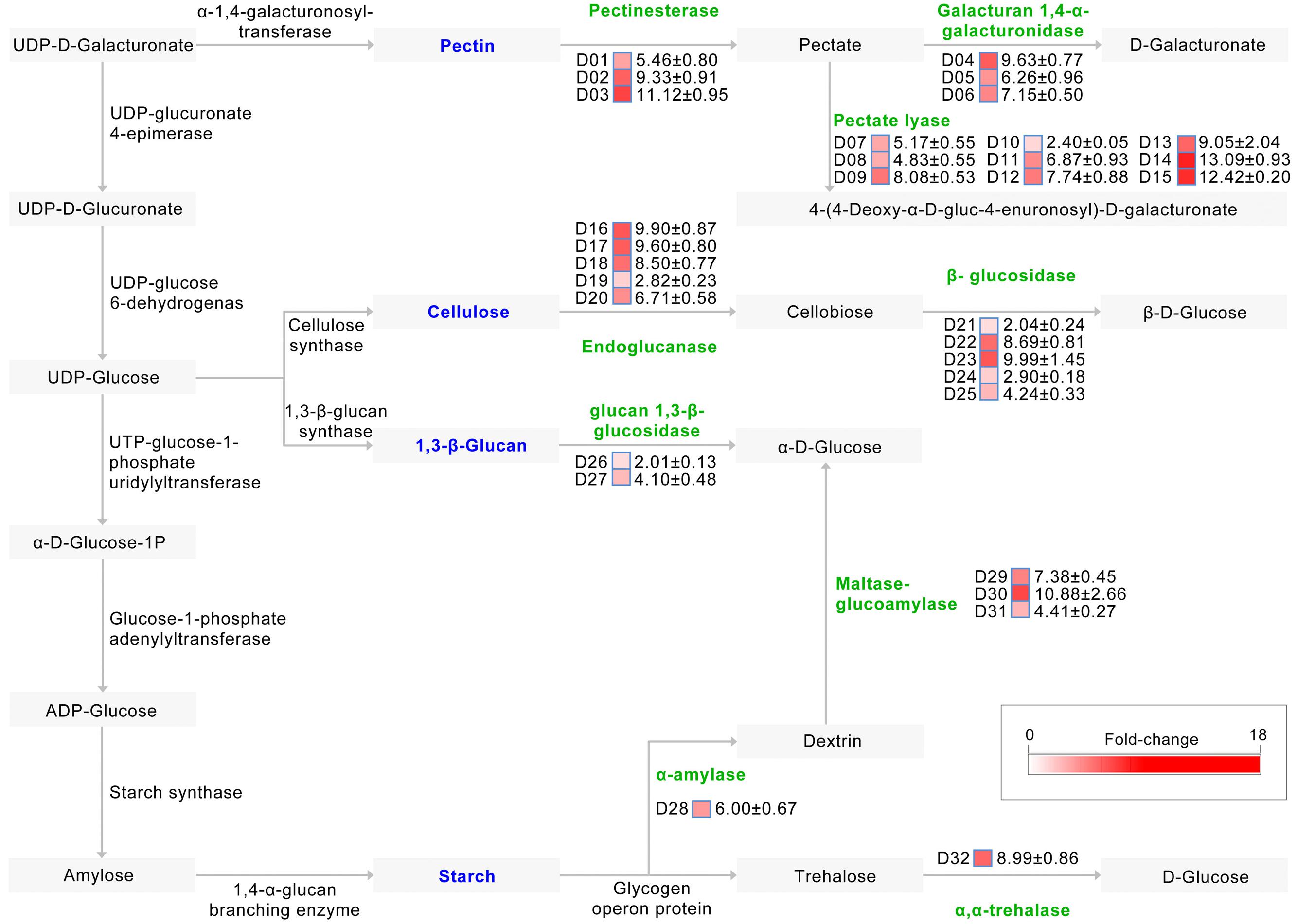
FIGURE 4. Induced exproteome involved in plant cell wall degradation. Identified proteins were matched to a reference pathway using the KEGG database, while the plant cell wall degradation pathway was drawn according to the starch and sucrose metabolism pathways (ko:00500). D01 to D32 represents 32 proteins involve in plant cell wall degradation, the fold change (original value) of identified proteins were presented by the color blocks from white (zero fold-change) to red (the maximum fold-change).
The Role of Identified Proteins in V. dahliae Virulence
To evaluate the pathogenic function of PCWDEs in V. dahliae, expression levels of several involved in pectin and cellulose degradation were analyzed. Results show that transcription levels of PCWDEs involved in pectin and cellulose degradation are markedly increased during V. dahliae infection of susceptible cotton plants (Figures 5A,B), while real-time PCR showed that these levels increased markedly around three dpi, and that the expression of many remained high at 5–7 dpi (Figures 5A,B). These periods are generally considered to be the key initial germination and proliferation stages of cotton infection.
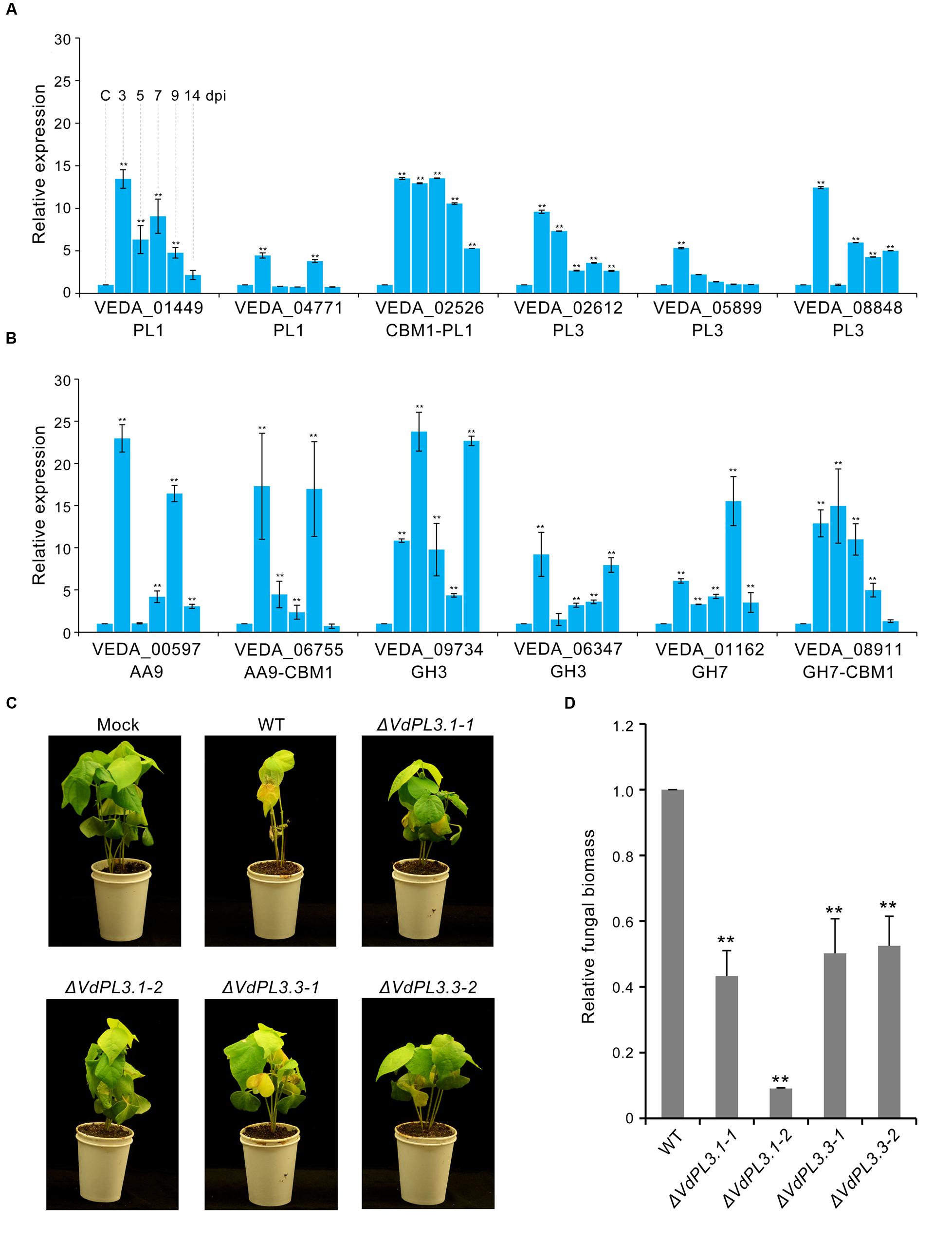
FIGURE 5. Virulence function analysis of identified proteins in the exoproteome. (A) Expression of genes involved in pectin degradation in V. dahliae infection on cotton roots determined via qRT-PCR. The control (capital letter C) shows the expression level of genes in a mixture of non-inoculated conidial and cotton root tissue; the housekeeping gene β-tubulin (VDAG_10074) was used as an endogenous control. Error bars show standard error, ∗∗ denotes statistical significance (P ≤ 0.01) of different time points compared to control using an unpaired Student’s t-test. (B) Expression of genes involved in cellulose degradation by V. dahliae. (C) Phenotypes of cotton seedlings inoculated with VdPL3.1 and VdPL3.3 gene-deletion strains. Two-week-old seedlings of susceptible cotton, Gossypium hirsutum L., ‘Junmian No.1,’ were inoculated with sterile water (Mock), wild-type (WT) V. dahliae, and the two independent VdPL3.1 and VdPL3.3 gene-deletion strains. (D) Fungal biomass of the gene-deletion strain on cotton was determined using qRT-PCR. Error bars represent standard error, and ∗∗ denotes statistical significance (P ≤ 0.05) of gene-deletion strains compared to WT using an unpaired Student’s t-test.
In order to further verify the role of PCWDEs in V. dahliae pathogenesis, the PL3 subfamily, involved in pectin degradation, was subjected to further analysis. This subfamily exhibits pectate lyase activity, cleaving poly-1, 4-α-D-galacturonan (pectate, the backbone of pectin) to 4-(4-deoxy-α-D-gluc-4-enuronosyl)-D-galacturonate at the non-reducing end (Figure 4). The results of this study show that the abundances of three PL3 genes (i.e., VEDA_02612, VEDA_05899, and VEDA_08848, denoted VdPL3.1–VdPL3.3) were significantly up-regulated 6.87 ± 0.93, 13.09 ± 0.93, and 12.42 ± 0.20 times, respectively, in the CCD medium (Supplementary Figure S2A). These results suggest that PL3 genes thus play an important role in the infection of cotton.
In order to confirm the pathogenic functions of PCWDEs in the induced exoproteome, targeted replacement of the three PL3 genes (i.e., VdPL3.1–VdPL3.3) by a hygromycin resistance cassette through homologous recombination was pursued, and gene deletion was verified by PCR (Supplementary Figure S3), of which two were used for further analysis in this study. Pathogenicity analysis of these three strains showed that the ΔVdPL3.1 and ΔVdPL3.3 mutants were significantly decreased in virulence, leading to a reduction in stunting and no obvious disease symptoms in susceptible cotton compared with the wild-type strain (Figure 5C). Unexpectedly, although the transcription level of another PL3 gene, VdPL3.2, was also significantly induced during cotton infection, the deletion strain, ΔVdPL3.2, exhibited virulence similar to that seen in the wild-type strain (Supplementary Figure S4). This result is likely due to the fact that V. dahliae encodes other CAZymes that can complement the pectate lyase function of VdPL3.2. Similarly, only a double mutant in PL3 genes encoding pectate lyase A and D (i.e., pelA and pelD) in Nectria hematococca led to drastically reduced pathogenicity to Pisum sativum (Rogers et al., 2000). We conclude that CAZymes in the induced exoproteome of V. dahliae, including the pectin lyases VdPL3.1 and VdPL3.3, are involved in plant cell wall degradation and play a crucial role in the pathogenesis of this fungus.
Discussion
The pathogen V. dahliae is the causal agent of plant Verticillium wilt disease, causing billions of dollars in annual crop losses (Fradin and Thomma, 2006; Klosterman et al., 2009). Previous studies have shown that the exoproteome, including toxins, effectors, and PCWDEs, likely plays a critical role in V. dahliae pathogenesis (Fradin and Thomma, 2006). To date, several secreted proteins have been identified using traditional separation and purification techniques and have been demonstrated to be associated with disease symptoms in susceptible host plants (Buchner et al., 1982, 1989; Nachmias et al., 1985; Meyer et al., 1994; Mansoori et al., 1995; Davis et al., 1998). However, pathogenic factors in the V. dahliae exoproteome have so far not been reported. In this study, we show that the V. dahliae exoproteome from an CCD medium culture caused more serious wilting and necrosis in cotton cotyledon leaves compared with that from an original CD medium utilizing sucrose as a carbon source. In addition, results show that the impaired cotton phenotype was similar to that of leaves infected by V. dahliae inoculation (Figure 1), suggesting production of several virulence factors in the CCD medium. Thus, identification of the components of the induced exoproteome using iTRAQ combined with LC-MS/MS could facilitate determination of the mechanisms underlying infection of cotton by V. dahliae.
Although several previous studies have reported that crude extracts of the V. dahliae exoproteome exhibit virulence to host plants (Buchner et al., 1982; Mansoori et al., 1995; Yuan et al., 2006), active components remain unknown. In this study, 325 proteins showed significant changes in abundance in the CCD medium compared with those in the original CD utilizing sucrose as a carbon source (Table 1; Supplementary Table S2). Of these, 69% were secreted and 63% were typical secreted proteins containing a signal peptide (Table 1), suggesting protein secretion by V. dahliae occurs mainly via the Golgi/ER system. In addition, because 31% of these proteins lacked a signal peptide and were not classified as NCSPs (independent of the Golgi/ER), this suggests that V. dahliae may possess as yet unknown secretory mechanisms.
Secreted effectors facilitate colonization of the host plant by pathogens as they modulate host biochemistry and physiology (Stergiopoulos and de Wit, 2009). Previous research has shown that several kinds of effectors are conserved among phytopathogens, including LysM, NLPs, and Ave1 (de Jonge et al., 2012, 2013; Zhou et al., 2012; Santhanam et al., 2013), while others have been described simply as SCRPs with no known function. Our results show that the abundances of 28 SCRPs were significantly up-regulated in the induced exoproteome (Figure 3B), suggesting that they likely play an important role in V. dahliae pathogenesis. In addition, abundances of two NLP proteins, NLP1 and NLP2 (Zhou et al., 2012; Santhanam et al., 2013), were also significantly enhanced (9.05 ± 2.04 and 3.75 ± 0.17 times, respectively) in the induced exoproteome (Supplementary Table S5), while one LysM effector (VEDA_00253) was also up-regulated 5.16 ± 0.50 times following culture in the CCD medium. These results suggest that secreted effectors are important adaptions of V. dahliae to the cotton vascular system environment, likely protecting this pathogen from the deleterious effects of host defenses.
A few previous studies have reported that hydrolases are important to the pathogenicity of V. dahliae (Puhalla and Bell, 1981; Pegg and Brady, 2002). For example, hydrolases involved in carbohydrate metabolism were shown to be affected following VdSNF1 deletion, leading to failed colonization and the induction of disease symptoms on tomato and eggplant (Tzima et al., 2011). In this study, the functional clustering of up-regulated secretory proteins showed that the number of hydrolases significantly increased in the CCD medium (Figure 2). Previous transcriptome analyses have also demonstrated increased hydrolase expression during the interaction between V. dahliae and its host plant (Xiong et al., 2014, 2015), and this has also been reported in other phytopathogens (Lysøe et al., 2011; Mathioni et al., 2011; O’Connell et al., 2012). Carbohydrate enzymes are necessary for the growth of pathogenic fungi in the presence of low carbohydrate levels, as well as possibly also for the penetration of plant roots by soil-borne fungi gaining access to the xylem (Kubicek et al., 2014). One previous comparative genomics study reported that the V. dahliae genome encodes a greater number of CAZymes than is the case in other fungi, suggesting an enhanced capacity to degrade plant cell walls (Klosterman et al., 2011). Indeed, analysis of the secretome of the closely related vascular wilt pathogen V. alboatrum in a simulated xylem fluid showed that carbohydrate hydrolases were more abundant in lethal strains than in others (Mandelc and Javornik, 2015). Thus, as shown here, to adapt to the conditions of the CCD medium, V. dahliae produced numerous extracellular proteins that participate in carbohydrate metabolism, most of which are significantly up-regulated in the presence of low nutrient levels (Figure 3). Our results confirm that carbohydrate hydrolases play a crucial role in the virulence of vascular phytopathogens by facilitating their evasion of host defenses. This is necessary for the successful growth of phytopathogens in carbohydrate-poor environments, facilitates fungal colonization, and the production of survival structures in plant tissue.
The main cell wall components in dicotyledonous plants include pectic polysaccharides, cellulose microfibrils, hemicelluloses, and glycoproteins (Carpita and Gibeaut, 1993). This primary cell wall functions as a barrier to prevent the penetration of pathogens and infection (Hématy et al., 2009). To overcome this barrier, pathogens produce PCWDEs that degrade polysaccharides and obtain nutrients (King et al., 2011). Genomic sequencing has revealed that the pectate lyase (i.e., PL1, PL3, and PL9), rhamnogalacturonan lyase (i.e., PL4 and PL11), and CBM1 families of enzymes, which mediate the cleavage of pectin and cellulose during infections, are increased in abundance in V. dahliae (Klosterman et al., 2011), associated with enhanced plant cell wall degradation. In this study, analysis of the expression of PCWDE genes in planta demonstrates their involvement in V. dahliae pathogenicity (Figure 5A); highest expression occurred at 3 dpi, when the fungus entered the root xylem vessels and proliferated into the vascular system (Gold and Robb, 1995; Heinz et al., 1998; Chen et al., 2004).
Several pectinolytic enzymes (i.e., polygalacturonase, pectate lyase, and pectinesterase) are known to be capable of necrotizing plant tissues in vitro and causing wilt symptoms in vivo (Mussel and Strause, 1972; Cooper and Wood, 1980; Huang and Mahoney, 1999). Thus, to confirm the pathogenicity of these pectin-lytic enzymes, the functions of three pectate lyase genes were evaluated using a genetic knockout experiment. However, although the VdLs.17 strain encodes for 11 PL3 genes all of which potentially encode secreted proteins likely involved in pectin degradation (Klosterman et al., 2011), few previous studies have evaluated the role of these genes in V. dahliae pathogenicity. Results presented here show that these three PL3 genes were markedly up-regulated in the induced exoproteome as well as in planta after V. dahliae infection (Figure 5A). Indeed, the virulence to cotton plants of strains containing the VdPL3.1 and VdPL3.3 deletions were markedly reduced (Figures 5C,D). Our findings suggest that PCWDEs function as important virulence factors in V. dahliae pathogenesis.
Conclusion
The V. dahliae exoproteome purified using the CCD medium caused serious wilting and necrosis in cotton cotyledons. A number of potential pathogenicity-related factors were identified in the exoproteome via iTRAQ combined with LC-MS/MS. Protein abundance data suggest that pathogenicity-related factors in the exoproteome are significantly up-regulated, particularly CAZymes involved in carbohydrate metabolism. Further analysis indicates a marked increase in PCWDE levels in the exoproteome, which participate in pectin and cellulose degradation. Indeed, PL3 deletion mutants exhibited reduced virulence against cotton, which suggests that some PCDWEs in the induced exoproteome of V. dahliae function as virulence factors. Our findings strongly suggest that hydrolytic enzymes in the induced exoproteome of V. dahliae are closely related to plant cell wall degradation, and thus likely play a crucial role in both host colonization and proliferation in the unique plant vascular system environment.
Author Contributions
XD conceived the study and designed all experiments, while JC performed the data analysis and interpretation, HX performed the pathogenicity characterization experiment and wrote the manuscript, YG and DZ performed the target gene deletion, and LL and YB performed the gene expression analysis. All authors have read, commented on, and approved the manuscript.
Funding
This work was supported by CAAS (an Agricultural Science and Technology Innovation Program grant to XD), the Special Public Welfare Industry Research on Agriculture (201503109), the Major State Basic Research Development Program of China (973 Program) (2011CB100705), the China Natural Scientific Foundation (No. 31200113), the China Major Projects for Transgenic Breeding (2011ZX08005).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We gratefully acknowledge for Prof. Seogchan Kang sharing the pGKO2 vector.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01709/full#supplementary-material
Footnotes
References
Adav, S. S., Chao, L. T., and Sze, S. K. (2013). Protein abundance in multiplexed samples (PAMUS) for quantitation of Trichoderma reesei secretome. J. Proteomics 83, 180–196. doi: 10.1016/j.jprot.2013.03.023
Adav, S. S., Ravindran, A., Cheow, E. S., and Sze, S. K. (2012). Quantitative proteomic analysis of secretome of microbial consortium during saw dust utilization. J. Proteomics 75, 5590–5603. doi: 10.1016/j.jprot.2012.08.011
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Amselem, J., Cuomo, C. A., van Kan, J. A., Viaud, M., Benito, E. P., Couloux, A., et al. (2011). Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 7:e1002230. doi: 10.1371/journal.pgen.1002230
Battaglia, E., Benoit, I., van den Brink, J., Wiebenga, A., Coutinho, P. M., Henrissat, B., et al. (2011). Carbohydrate-active enzymes from the zygomycete fungus Rhizopus oryzae: a highly specialized approach to carbohydrate degradation depicted at genome level. BMC Genomics 12:38. doi: 10.1186/1471-2164-12-38
Bendtsen, J. D., Jensen, L. J., Blom, N., Von Heijne, G., and Brunak, S. (2004). Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 17, 349–356. doi: 10.1093/protein/gzh037
Bent, A. F., and Mackey, D. (2007). Elicitors, effectors, and R genes: the new paradigm and a lifetime supply of questions. Annu. Rev. Phytopathol. 45, 399–436. doi: 10.1146/annurev.phyto.45.062806.094427
Bishop, C. D., and Cooper, R. M. (1983). An ultrastructural study of root invasion in three vascular wilt diseases. Physiol. Plant Pathol. 22, 15–27. doi: 10.1016/S0048-4059(83)81034-0
Buchner, V., Burstein, Y., and Nachmias, A. (1989). Comparison of Verticillium dahliae -produced phytotoxic peptides purified from culture fluids and infected potato stems. Physiol. Mol. Plant Pathol. 35, 253–269. doi: 10.1016/0885-5765(89)90055-6
Buchner, V., Nachmias, A., and Burstein, Y. (1982). Isolation and partial characterization of a phytotoxic glycopeptide from a proteinlipopolysaccharide complex produced by a potato isolate of Verticillium dahliae. FEBS Lett. 138, 261–264. doi: 10.1016/0014-5793(82)80456-0
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The Carbohydrate-Active enZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, 233–238. doi: 10.1093/nar/gkn663
Carpita, N. C., and Gibeaut, D. M. (1993). Structural models of primary cell walls in flowering plants: consistency of molecular structure with the physical properties of the walls during growth. Plant J. 3, 1–30. doi: 10.1111/j.1365-313X.1993.tb00007.x
Chen, P., Lee, B., and Robb, J. (2004). Tolerance to a non-host isolate of Verticillium dahliae in tomato. Physiol. Mol. Plant Pathol. 64, 283–291. doi: 10.1007/s00425-008-0840-z
Cooper, R. M., and Wood, R. K. S. (1975). Regulation of synthesis of cell wall degrading einzymes by Verticillium albo-atrum and Fusarium oxysporum f. sp. lycopersici. Physiol. Plant Pathol. 5, 135–156. doi: 10.1016/0048-4059(75)90017-X
Cooper, R. M., and Wood, R. K. S. (1980). Cell wall degrading enzymes of vascular wilt fungi. III. Possible involvement of endo-pectin lyase in Verticillium wilt of tomato. Physiol. Plant Pathol 16, 285–300. doi: 10.1016/0048-4059(80)90043-0
Davis, D. A., Low, P. S., and Heinstein, P. (1998). Purification of a glycoprotein elicitor of phytoalexin formation from Verticillium dahliae. Physiol. Mol. Plant Pathol. 52, 259–273. doi: 10.1006/pmpp.1998.0150
de Jonge, R., Bolton, M. D., Kombrink, A., van den Berg, G. C., Yadeta, K. A., and Thomma, B. P. (2013). Extensive chromosomal reshuffling drives evolution of virulence in an asexual pathogen. Genome Res. 23, 1271–1282. doi: 10.1101/gr.152660.112
de Jonge, R., van Esse, H. P., Kombrink, A., Shinya, T., Desaki, Y., Bours, R., et al. (2010). Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science 329, 953–955. doi: 10.1126/science.1190859
de Jonge, R., van Esse, H. P., Maruthachalam, K., Bolton, M. D., Santhanam, P., Saber, M. K., et al. (2012). Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc. Natl. Acad. Sci. U.S.A. 109, 5110–5115. doi: 10.1073/pnas.1119623109
El-Bebany, A. F., Rampitsch, C., and Daayf, F. (2010). Proteomic analysis of the phytopathogenic soilborne fungus Verticillium dahliae reveals differential protein expression in isolates that differ in aggressiveness. Proteomics 10, 289–303. doi: 10.1002/pmic.200900426
Fradin, E. F., and Thomma, B. P. H. J. (2006). Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol. Plant Pathol. 7, 71–86. doi: 10.1111/j.1364-3703
Girard, V., Dieryckx, C., Job, C., and Job, D. (2013). Secretomes: the fungal strike force. Proteomics 13, 597–608. doi: 10.1002/pmic.201200282
Glass, N. L., Schmoll, M., Cate, J. H., and Coradetti, S. (2013). Plant cell wall deconstruction by ascomycete fungi. Annu. Rev. Microbiol. 67, 477–498. doi: 10.1146/annurev-micro-092611-150044
Gold, J., and Robb, J. (1995). The role of the coating response in Craigella tomatoes infected with Verticillium dahliae, races 1 and 2. Physiol. Mol. Plant Pathol. 47, 141–157. doi: 10.1006/pmpp.1995.1048
González-Fernández, R., Aloria, K., Valero-Galván, J., Redondo, I., Arizmendi, J. M., and Jorrín-Novo, J. V. (2014). Proteomic analysis of mycelium and secretome of different Botrytis cinerea wild-type strains. J. Proteomics 97, 195–221. doi: 10.1016/j.jprot.2013.06.022
Goodwin, S. B., M’barek, S. B., Dhillon, B., Wittenberg, A. H., Crane, C. F., Hane, J. K., et al. (2011). Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 7:e1002070. doi: 10.1371/journal.pgen.1002070
Heinz, R., Lee, S. W., Saparno, A., Nazar, R. N., and Robb, J. (1998). Cyclical systemic colonization in Verticillium-infected tomato. Physiol. Mol. Plant Pathol. 52, 385–396. doi: 10.1006/pmpp.1998.0163
Hématy, K., Cherk, C., and Somerville, S. (2009). Host-pathogen warfare at the plant cell wall. Curr. Opin. Plant Biol. 12, 406–413. doi: 10.1016/j.pbi.2009.06.007
Horton, P., Park, K. J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587. doi: 10.1093/nar/gkm259
Huang, L. K., and Mahoney, R. R. (1999). Purification and characterization of an endopolygalacturonase from Verticillium albo-atrum. J. Appl. Micriobiol. 86, 145–156. doi: 10.1016/0003-9861(70)90193-1
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Jung, Y. H., Jeong, S. H., Kim, S. H., Singh, R., Lee, J. E., Cho, Y. S., et al. (2012). Secretome analysis of Magnaporthe oryzae using in vitro systems. Proteomics 12, 878–900. doi: 10.1002/pmic.201100142
Kamoun, S. (2006). A catalogue of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 44, 41–60. doi: 10.1146/annurev.phyto.44.070505.143436
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., and Tanabe, M. (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, 109–114. doi: 10.1093/nar/gkr988
Khang, C. H., Park, S. Y., Lee, Y. H., and Kang, S. A. (2005). Dual selection based, targeted gene replacement tool for Magnaporthe grisea and Fusarium oxysporum. Fungal Genet. Biol. 42, 483–492. doi: 10.1016/j.fgb.2005.03.004
King, B. C., Waxman, K. D., Nenni, N. V., Walker, L. P., Bergstrom, G. C., and Gibson, D. M. (2011). Arsenal of plant cell wall degrading enzymes reflects host preference among plant pathogenic fungi. Biotechnol. Biofuels 4, 4–16. doi: 10.1186/1754-6834-4-4
Klimes, A., Dobinson, K. F., Thomma, B. P., and Klosterman, S. J. (2015). Genomics spurs rapid advances in our understanding of the biology of vascular wilt pathogens in the genus Verticillium. Annu. Rev. Phytopathol. 53, 181–198. doi: 10.1146/annurev-phyto-080614-120224
Klosterman, S. J., Atallah, Z. K., Vallad, G. E., and Subbarao, K. V. (2009). Diversity, pathogenicity, and management of Verticillium species. Annu. Rev. Phytopathol. 47, 39–62. doi: 10.1146/annurev-phyto-080508-081748
Klosterman, S. J., Subbarao, K. V., Kang, S., Veronese, P., Gold, S. E., Thomma, B. P., et al. (2011). Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens. PLoS Pathog. 7:e1002137. doi: 10.1371/journal.ppat.1002137
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kubicek, C. P., Starr, T. L., and Glass, N. L. (2014). Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 52, 427–451. doi: 10.1146/annurev-phyto-102313-045831
Lévesque, C. A., Brouwer, H., Cano, L., Hamilton, J. P., Holt, C., Huitema, E., et al. (2010). Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol. 11:R73. doi: 10.1186/gb-2010-11-7-r73
Liang, L., Wu, H., Liu, Z., Shen, R., Gao, H., Yang, J., et al. (2013). Proteomic and transcriptional analyses of Arthrobotrys oligospora cell wall related proteins reveal complexity of fungal virulence against nematodes. Appl. Microbiol. Biotechnol. 97, 8683–8692. doi: 10.1007/s00253-013-5178-1
Lievens, B., Houterman, P. M., and Rep, M. (2009). Effector gene screening allows unambiguous identification of Fusarium oxysporum f. sp. lycopersici races and discrimination from other formae speciales. FEMS Microbiol. Lett. 300, 201–215. doi: 10.1111/j.1574-6968.2009.01783.x
Liu, D., Li, J., Zhao, S., Zhang, R., Wang, M., Miao, Y., et al. (2013). Secretome diversity and quantitative analysis of cellulolytic Aspergillus fumigatus Z5 in the presence of different carbon sources. Biotechnol. Biofuels 6:149. doi: 10.1186/1754-6834-6-149
Liu, Y., Chen, J. Y., Wang, J. L., Li, L., Xiao, H. L., Adam, S. M., et al. (2013). Molecular characterization and functional analysis of a specific secreted protein from highly virulent defoliating Verticillium dahliae. Gene 529, 307–316. doi: 10.1016/j.gene.2013.06.089
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using Real-Time quantitative PCR and the 2-ΔΔCt method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lysøe, E., Seong, K. Y., and Kistler, H. C. (2011). The transcriptome of Fusarium graminearum during the infection of wheat. Mol. Plant Microbe Interact. 24, 995–1000. doi: 10.1094/MPMI-02-11-0038
Ma, L. J., van der Does, H. C., Borkovich, K. A., Coleman, J. J., Daboussi, M. J., Di Pietro, A., et al. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373. doi: 10.1038/nature08850
Mandelc, S., and Javornik, B. (2015). The secretome of vascular wilt pathogen Verticillium albo-atrum in simulated xylem fluid. Proteomics 15, 787–797. doi: 10.1002/pmic.201400181
Mandelc, S., Radisek, S., Jamnik, P., and Javornik, B. (2009). Comparison of mycelial proteomes of two Verticillium albo-atrum pathotypes from hop. Eur. J. Plant Pathol. 125, 159–171. doi: 10.1007/s10658-009-9467-6
Mandelc, S., Timperman, I., Radišek, S., Devreese, B., Samyn, B., and Javornik, B. (2013). Comparative proteomic profiling in compatible and incompatible interactions between hop roots and Verticillium albo-atrum. Plant Physiol. Biochem. 68, 23–31. doi: 10.1016/j.plaphy.2013.03.017
Mansoori, B., Milton, J. M., and Smith, C. J. (1995). Isolation and partialpurification of a phytotoxin related to pathogenic Verticillium species. J. Phytopathol. 143, 33–36. doi: 10.1016/j.plaphy.2013.03.017
Marshall, R., Kombrink, A., Motteram, J., Loza-Reyes, E., Lucas, J., Hammond-Kosack, K. E., et al. (2011). Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiol. 156, 756–769. doi: 10.1104/pp.111.176347
Mathioni, S. M., Beló, A., Rizzo, C. J., Dean, R. A., and Donofrio, N. M. (2011). Transcriptome profiling of the rice blast fungus during invasive plant infection and in vitro stresses. BMC Genomics 12:49. doi: 10.1104/pp.111.176347
Meyer, R., Slater, V., and Dubery, L. A. (1994). A phytotoxic proteinlipopolysccharide complex produced by Verticillium dahliae. Phytochemistry 35, 1449–1453. doi: 10.1016/S0031-9422(00)86872-7
Mullins, E. D., and Kang, S. (2001). Transformation: a tool for studying fungal pathogens of plants. Cell Mol. Life Sci. 58, 2043–2052. doi: 10.1007/PL00000835
Mussel, H. W., and Strause, B. (1972). Characterization of two polygalacturonases produced by Verticillium albo-atrum. Can. J. Biochem. 50, 625–632. doi: 10.1139/o72-086
Nachmias, A., Buchner, V., and Burstein, Y. (1985). Biological and immunochemical characterization of a low molecular weight phytotoxin isolated from a protein-lipopolysaccharide complex produced by a potato isolate of Verticillium dahliae Kleb. Physiol. Plant Pathol. 26, 43–55. doi: 10.1016/0048-4059(85)90029-3
O’Connell, R. J., Thon, M. R., Hacquard, S., Amyotte, S. G., Kleemann, J., Torres, M. F., et al. (2012). Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 44, 1060–1065. doi: 10.1038/ng.2372
Ospina-Giraldo, M. D., Griffith, J. G., Laird, E. W., and Mingora, C. (2010). The CAZyome of Phytophthora spp.: a comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genomics 11:525. doi: 10.1186/1471-2164-11-525
Paper, J. M., Scott-Craig, J. S., Adhikari, N. D., Cuomo, C. A., and Walton, J. D. (2007). Comparative proteomics of extracellular proteins in vitro and in planta from the pathogenic fungus Fusarium graminearum. Proteomics 7, 3171–3183. doi: 10.1002/pmic.200700184
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 8, 785–786. doi: 10.1038/nmeth.1701
Phalip, V., Delalande, F., Carapito, C., Goubet, F., Hatsch, D., Leize-Wagner, E., et al. (2005). Diversity of the exoproteome of Fusarium graminearum grown on plant cell wall. Curr. Genet. 48, 366–379. doi: 10.1007/s00294-005-0040-3
Powell, S., Szklarczyk, D., Trachana, K., Roth, A., Kuhn, M., Muller, J., et al. (2012). eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 40, D284–D289. doi: 10.1093/nar/gkr1060
Puhalla, J. E., and Bell, A. A. (1981). “Genetics and biochemistry of wilt pathogens,” in Fungal Wilt Diseases of Plants, eds M. E. Mace, A. A. Bell, and C. H. Beckman (New York, NY: Academic Press), 146–192.
Ravalason, H., Grisel, S., Chevret, D., Favel, A., Berrin, J. G., Sigoillot, J. C., et al. (2012). Fusarium verticillioides secretome as a source of auxiliary enzymes to enhance saccharification of wheat straw. Bioresour. Technol. 114, 589–596. doi: 10.1016/j.biortech.2012.03.009
Rep, M. (2005). Small proteins of plant-pathogenic fungi secreted during host colonization. FEMS Microbiol. Lett. 253, 19–27. doi: 10.1016/j.femsle.2005.09.014
Rogers, L. M., Kim, Y. K., Guo, W., González-Candelas, L., Li, D., and Kolattukudy, P. E. (2000). Requirement for either a host- or pectin-induced pectate lyase for infection of Pisum sativum by Nectria hematococca. Proc. Natl. Acad. Sci. U.S.A. 97, 9813–9818. doi: 10.1073/pnas.160271497
Santhanam, P., van Esse, H. P., Albert, I., Faino, L., Nürnberger, T., and Thomma, B. P. H. J. (2013). Evidence for functional diversification within a fungal NEP1-like protein family. Mol. Plant Microbe Interact. 26, 278–286. doi: 10.1094/MPMI-09-12-0222-R
Schnathorst, W. C. (1981). “Life cycle and epidemiology of Verticillium,” in Fungal Wilt Diseases of Plants, eds M. E. Mace, A. A. Bell, and C. H. Beckman (New York, NY: Academic Press), 81–111. doi: 10.1016/B978-0-12-464450-2.50009-7
Spanu, P. D. (2012). The genomics of obligate (and nonobligate) biotrophs. Annu. Rev. Phytopathol. 50, 91–109. doi: 10.1146/annurev-phyto-081211-173024
Stergiopoulos, I., and de Wit, P. J. (2009). Fungal effector proteins. Annu. Rev. Phytopathol. 47, 233–263. doi: 10.1146/annurev.phyto.112408.132637
Thomma, B. P., Nürnberger, T., and Joosten, M. H. (2011). Of PAMPs and effectors: the blurred PTI-ETI dichotomy. Plant Cell 23, 4–15. doi: 10.1105/tpc.110.082602
Tran, V. T., Braus-Stromeyer, S. A., Kusch, H., Reusche, M., Kaever, A., Kühn, A., et al. (2014). Verticillium transcription activator of adhesion Vta2 suppresses microsclerotia formation and is required for systemic infection of plant roots. New Phytol. 202, 565–581. doi: 10.1111/nph.12671
Tzima, A. K., Paplomatas, E. J., Rauyaree, P., Ospina-Giraldo, M. D., and Kang, S. (2011). VdSNF1, the sucrose nonfermenting protein kinase gene of Verticillium dahliae, is required for virulence and expression of genes involved in cell-wall degradation. Mol. Plant Microbe Interact. 24, 129–142. doi: 10.1094/MPMI-09-09-0217
Valent, B., and Khang, C. H. (2010). Recent advances in rice blast effector research. Curr. Opin. Plant Biol. 13, 434–441. doi: 10.1016/j.pbi.2010.04.012
Vallad, G. E., and Subbarao, K. V. (2008). Colonization of resistant and susceptible lettuce cultivars by a green fluorescent protein-tagged isolate of Verticillium dahliae. Phytopathology 98, 871–885. doi: 10.1094/PHYTO-98-8-0871
Wang, M. C., and Keen, N. T. (1970). Purification and characterization of endopolygalacturonase from Verticillium albo-atrum. Arch. Biochem. Biophys. 141, 749–757. doi: 10.1016/0003-9861(70)90193-1
Wang, Y., Kim, S. G., Wu, J., Huh, H. H., Lee, S. J., Rakwal, R., et al. (2013). Secretome analysis of the rice bacterium Xanthomonas oryzae (Xoo) using in vitro and in planta systems. Proteomics 13, 1901–1912. doi: 10.1002/pmic.201200454
Wang, Y., Wu, J., Park, Z. Y., Kim, S. G., Rakwal, R., Agrawal, G. K., et al. (2011). Comparative secretome investigation of Magnaporthe oryzae proteins responsive to nitrogen starvation. J. Proteome Res. 10, 3136–3148. doi: 10.1021/pr200202m
Winnenburg, R., Urban, M., Beacham, A., Baldwin, T. K., Holland, S., Lindeberg, M., et al. (2008). PHI-base update: additions to the pathogen host interaction database. Nucleic Acids Res. 36, D572–D576. doi: 10.1093/nar/gkm858
Xiong, D., Wang, Y., Ma, J., Klosterman, S. J., Xiao, S., and Tian, C. (2014). Deep mRNA sequencing reveals stage-specific transcriptome alterations during microsclerotia development in the smoke tree vascular wilt pathogen, Verticillium dahliae. BMC Genomics 15:324. doi: 10.1186/1471-2164-15-324
Xiong, D., Wang, Y., and Tian, C. (2015). Transcriptomic profiles of the smoke tree wilt fungus Verticillium dahliae under nutrient starvation stresses. Mol. Genet. Genomics 290, 1963–1977. doi: 10.1007/s00438-015-1052-4
Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297. doi: 10.1093/nar/gkl031
Yin, Y., Mao, X., Yang, J., Chen, X., Mao, F., and Xu, Y. (2012). dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, W445–W451. doi: 10.1093/nar/gks479
Yuan, H. Y., Yao, L. L., Jia, Z. Q., Li, Y., and Li, Y. Z. (2006). Verticillium dahliae toxin induced alterations of cytoskeletons and nucleoli in Arabidopsis thaliana suspension cells. Protoplasma 229, 75–82. doi: 10.1007/s00709-006-0154-6
Zhou, B. J., Jia, P. S., Gao, F., and Guo, H. S. (2012). Molecular characterization and functional analysis of a necrosis- and ethylene-inducing, protein-encoding gene family from Verticillium dahliae. Mol. Plant Microbe Interact. 25, 964–975. doi: 10.1094/MPMI-12-11-0319
Keywords: Verticillium dahliae, exoproteome, CAZymes, plant cell wall degradation enzyme, pectinases
Citation: Chen JY, Xiao HL, Gui YJ, Zhang DD, Li L, Bao YM and Dai XF (2016) Characterization of the Verticillium dahliae Exoproteome Involves in Pathogenicity from Cotton-Containing Medium. Front. Microbiol. 7:1709. doi: 10.3389/fmicb.2016.01709
Received: 26 August 2016; Accepted: 12 October 2016;
Published: 28 October 2016.
Edited by:
Vijai Kumar Gupta, National University of Ireland, Galway, IrelandReviewed by:
Gerardo Díaz-Godínez, Autonomous University of Tlaxcala, MexicoHao-Xun Chang, University of Illinois, USA
Copyright © 2016 Chen, Xiao, Gui, Zhang, Li, Bao and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Feng Dai, daixiaofeng_caas@126.com
†These authors have contributed equally to this work.