 Hannah Gaimster1*
Hannah Gaimster1* Lisa Chalklen1
Lisa Chalklen1 Mark Alston2
Mark Alston2 John T. Munnoch1
John T. Munnoch1 David J. Richardson1
David J. Richardson1 Andrew J. Gates1
Andrew J. Gates1 Gary Rowley1
Gary Rowley1- 1School of Biological Sciences, University of East Anglia, Norwich, UK
- 2Earlham Institute (formerly The Genome Analysis Centre), Norwich, UK
Nitrous oxide (N2O) is a stable, ozone depleting greenhouse gas. Emissions of N2O into the atmosphere continue to rise, primarily due to the use of nitrogen-containing fertilizers by soil denitrifying microbes. It is clear more effective mitigation strategies are required to reduce emissions. One way to help develop future mitigation strategies is to address the currently poor understanding of transcriptional regulation of the enzymes used to produce and consume N2O. With this ultimate aim in mind we performed RNA-seq on a model soil denitrifier, Paracoccus denitrificans, cultured anaerobically under high N2O and low N2O emitting conditions, and aerobically under zero N2O emitting conditions to identify small RNAs (sRNAs) with potential regulatory functions transcribed under these conditions. sRNAs are short (∼40–500 nucleotides) non-coding RNAs that regulate a wide range of activities in many bacteria. Hundred and sixty seven sRNAs were identified throughout the P. denitrificans genome which are either present in intergenic regions or located antisense to ORFs. Furthermore, many of these sRNAs are differentially expressed under high N2O and low N2O emitting conditions respectively, suggesting they may play a role in production or reduction of N2O. Expression of 16 of these sRNAs have been confirmed by RT-PCR. Ninety percent of the sRNAs are predicted to form secondary structures. Predicted targets include transporters and a number of transcriptional regulators. A number of sRNAs were conserved in other members of the α-proteobacteria. Better understanding of the sRNA factors which contribute to expression of the machinery required to reduce N2O will, in turn, help to inform strategies for mitigation of N2O emissions.
Introduction
Nitrous oxide is a potent greenhouse gas with an approximate 300 fold greater radiative potential per molecule than carbon dioxide. In addition to this, it has been described as the biggest single cause of depletion of ozone over the Arctic (Ravishankara et al., 2009). Emissions of N2O are continuing to increase every year by approximately 0.25% and once released into the atmosphere it remains stable for around 150 years. The major source (around 70%) of this atmospheric loading of N2O is from agriculture, mainly from the use of nitrogen-containing fertilizers by soil microbes. Collectively, these features make N2O an important target for mitigation strategies (Richardson et al., 2009).
N2O is produced as an intermediate in the sequential reduction of nitrate () to di-nitrogen (N2), via nitrite (), nitric oxide (NO), and N2O, in a process known as denitrification (Zumft and Kroneck, 2006). Reduction of N2O to N2 by soil microbes is the major biological route for removal of N2O (Pomowski et al., 2011). This reaction is catalyzed by a N2O reductase, NosZ. However, the increasing emission of N2O implies that NosZ is not always able to carry out this removal step in balance with the earlier steps in the denitrification pathway that form N2O. It follows that any transcriptional regulation that represses the expression of nosZ will in turn reduce the degradation of N2O and lead to net emission. Despite the pivotal importance in N2O mitigation, transcriptional regulation of NosZ, and other key enzymes involved in denitrification is poorly understood.
Paracoccus denitrificans is a soil dwelling member of the α-proteobacteria and is well-studied as a biochemical model for denitrification. The P. denitrificans genome encodes biochemical apparatus to switch between aerobic and anaerobic respiration and to utilize a range of electron donors in a modular respiratory network. The genome P. denitrificans was sequenced in 2006 and utilizing this, work by our laboratory began to unpick the transcriptome of P. denitrificans, cultured under a range of environmentally relevant conditions by using microarrays. This work revealed that the nos genes, and therefore N2O reduction, are strongly regulated by copper availability (Sullivan et al., 2013). A recent estimate suggested that 20% of Europe’s arable lands are biologically copper deficient (Alloway, 2008). In the presence of limited copper, only basal levels of nosZ expression are observed, whereas optimal copper concentrations lead to much higher levels of expression of nosZ. This results in transient accumulation of N2O in P. denitrificans grown in a limited copper media, whereas P. denitrificans grown in an optimal copper media do not accumulate N2O. This work therefore highlighted an abiotic factor, copper availability, in inducing global changes in gene expression regarding N2O emissions (Sullivan et al., 2013). Using the conditions described in this study, we sought to identify and understand if any sRNAs were transcribed and therefore could be playing a role in this key process.
Bacterial sRNAs are an emerging class of regulatory RNAs which are ∼40–500 nucleotides in length. These molecules are found in numerous species of bacteria and until relatively recently, sRNAs were largely an unknown and unexplored area of research. Work by various groups has shown sRNAs can modulate numerous physiological mechanisms and pathways, reviewed in Storz et al. (2011). sRNAs can target either proteins or mRNA transcripts. If the target of a sRNA is a protein, these sRNAs can be further categorized into two distinct groups, the trans-acting, and cis-acting sRNAs (Gottesman and Storz, 2011). Trans-acting sRNAs are defined as those encoded within the intergenic regions of a bacterial genome and act on target RNAs located in distinct locations across the rest of the genome. Trans-acting sRNAs can have less than perfect complementarity to their targets and as a result sometimes require a RNA chaperone, Hfq, to facilitate nucleotide binding (Moll et al., 2003). Conversely, cis-acting sRNAs originate from the antisense strand of an ORF and sometimes have direct regulatory influence on that particular ORF, though this is not true for all cis-acting sRNAs. The recent introduction of RNA-Seq technologies and associated bioinformatic tools has now made the analysis of bacterial transcriptomic data considerably more extensive and efficient.
In this work a combination of RNA-seq alongside bioinformatic approaches were used to gain an insight into the sRNA landscape of P. denitrificans was cultured anaerobically under high N2O and low N2O emitting conditions, and aerobically under zero N2O emitting conditions. The aim of this study is to understand the global sRNA profile in P. denitrificans as sRNAs could potentially be a valid target to reduce N2O emissions.
Materials and Methods
Bacterial Strains, Growth Media, and Conditions
Paracoccus denitrificans was grown in a defined minimal medium which contained 29 mmol/L Na2HPO4, 11 mmol/L KH2PO4, 10 mmol/L NH4Cl, 0.4 mmol/L MgSO4, and supplemented with 30 mmol/L sodium succinate, 20 mmol/L NaNO3, and 2 mL/L Vishniac and Santer trace elements solution [130 mmol/L EDTA, 7.64 mmol/L ZnSO4, 25 mmol/L MnCl2, 18.5 mmol/L FeSO4,0.89 mmol/L (NH4)6Mo7O24, 6.4 mmol/L CuSO4,6.72 mmol/LCoCl2, 37.4 mmol/L CaCl2] (Crutzen et al., 2008).
For high N2O emitting culture conditions, CuSO4 was omitted from the trace elements solution as in Sullivan et al. (2013). Anaerobic batch cultures (200 mL) inoculated with 1% (v/v) of stationary phase cells that had been pre-grown in minimal media. Vessels used were 250 mL Duran bottles with screw-cap lids and gas-tight silicone septa. Cultures were sparged with N2 for 10 min to impose an anaerobic environment and incubated statically at 30°C. For zero N2O emitting conditions, aerobic conditions were created by using 50 ml of media in a 250 ml flask and shaking at 200 rpm at 30°C.
Measurement of N2O Levels
Headspace gas samples (3 mL) were taken using a 5 mL gas-tight syringe (Hamilton) and stored in 3 mL pre-evacuated screw cap EXETAINER® vials (Labco). N2O gas samples were analyzed by GC through injection of a 50 μL sample into a Clarus 500 gas chromatographer (PerkinElmer) with an electron capture detector and Elite-PLOT Q [DVB Plot column, 30 m × 0.53 mm ID, carrier: N2, make-up: 95% (v/V) argon/5% (v/v) methane]. Standards of N2O [5, 100, 1,000, 5,000, and 10,000 ppm (Scientific and Technical Gases)] were used to quantify N2O levels. Total N2O amounts were calculated by applying Henry’s Law constant for N2O at 30°C, KH cc of 0.5392. Values of N2O (in micromoles) were multiplied by two to adjust values to micromole N in the form of N2O (N⋅N2O), this takes into account of the stoichiometry of N in N2O.
RNA Extraction
For RNA extraction, 30 mL of mid exponential phase cells (OD600 ≈ 0.4) was added to 12 mL of ice-cold 95% ethanol/5% phenol (pH = 4.3) (v/v) solution, and incubated on ice for 30 min to stabilize RNA and prevent degradation. RNA was isolated, using the Trizol method according to the protocol described in (Kröger et al., 2012). Trace DNA contamination was removed using Turbo DNA-free DNase (Ambion) and this was confirmed by PCR amplification of RNA samples using MyFi DNA polymerase (Bioline) according to the manufacturer’s instructions. RNA was quantified spectrophotometrically using a Nanodrop 2000 (Thermo Scientific), and integrity of RNA samples was analyzed using an Experion Automated Electrophoresis platform (Bio-Rad) using RNA StdSens chips (Bio-Rad) according to the manufacturer’s instructions.
Library Preparation and Sequencing
Library preparation and sequencing were performed by Vertis Biotechnology AG, Germany. Briefly, the total RNA samples were split into two, one was enriched for the small RNA fractions < 200 nt (s) specifically using the RNeasy MinElute Cleanup Kit (Qiagen). Ribosomal RNA molecules were depleted from both samples using the Ribo-Zero rRNA Removal Kit for bacteria (Epicenter). The rRNA depleted RNA fractions were first treated with Tobacco Acid Pyrophosphatase (TAP, Epicenter). Afterward, oligonucleotide adapters were ligated to the 5′ and 3′ ends of the RNA samples. First-strand cDNA synthesis was performed using M-MLV reverse transcriptase and the 3′ adapter as primer. The resulting cDNAs were amplified by PCR using a high fidelity DNA polymerase. The cDNA was purified using the Agencourt AMPure XP kit (Beckman Coulter Genomics) and was analyzed by capillary electrophoresis. The cDNA pool was sequenced on an Illumina NextSeq 500 system using 75 bp read length.
Identification and Analysis of sRNAs
Raw reads were trimmed then and aligned to the P. denitrificans genome (Genbank numbers: CP000489.1, CP000490.1 and CP000491.1). Bam files for each condition were converted to strand-specific wig files to allow viewing in IGB, alongside the annotated P. denitrificans genome. Both raw (fastaq files) and processed data (wig files) are available on the GEO database (Series record number: GSE85362)
Expression levels of each gene in the genome under each condition from the non sRNA enriched sample as RPKM = Reads Per Kb exon (contig) per Million mapped reads (Mortazavi et al., 2008) were also determined and so these can be directly compared to each other.
Candidate sRNAs were identified manually using wig files for the sRNA enriched fraction using the IGB browser as small (<200 bp) transcripts expressed from intergenic regions or antisense to characterized ORFs. In order to obtain normalized expression intensities of the read coverage depth for the sRNAs, the number of reads for the sRNA was normalized relative to the total number of reads in the library for each condition. Mfold was used to predict candidate sRNA secondary structure1 (Zuker, 2003). The nearest Rho-independent terminator to each sRNA was identified from the TransTerm2 (Kingsford et al., 2007). Potential gene targets for each sRNA were identified using TargetRNA23 (Kery et al., 2014). A single biological replicate for each condition was used, as in the approach used by McClure et al. (2014).
RT-PCR Validation of sRNAs
The method used was that described by Khoo et al. (2012). Briefly, purified RNA was reverse transcribed into cDNA with an oligo(dT)18 primer using RevertAid First Strand cDNA Synthesis Kit (Fermentas). cDNA was used as the template for PCR using MyFi polymerase together with primers that were designed based on the sequences of sRNA candidates (Supplementary Table S2). Amplified products were analyzed by 3% agarose gel electrophoresis with GeneRulerTM Low Range DNA Ladder (ThermoFisher scientific) run in parallel. PCR products were purified with the QIAquick Gel Purification Kit (Qiagen, Germany) and confirmed by Sanger sequencing (Eurofins).
Results
Identification of 167 Putative sRNAs in P. denitrificans
Paracoccus denitrificans was grown to exponential phase (16 h, OD600 ≈ 0.4), under 3 different conditions, high N2O emitting anaerobic, low N2O emitting anaerobic and zero N2O emitting aerobic conditions. Different N2O conditions were established by growing P. denitrificans under different copper and oxygen regimes as described in Sullivan et al. (2013). This previous work showed that P. denitrificans grown anaerobically in a low copper media emitted approximately 1–2 mM N2O, whereas P. denitrificans grown anaerobically in an optimal copper media did not accumulate N2O. Therefore culturing P. denitrificans under these conditions provides a way to analyze the sRNA landscape of P. denitrificans under high and low N2O emitting conditions respectively. Furthermore, this earlier paper performed a transcriptomic analysis using a DNA microarray under the same high N2O emitting anaerobic and low N2O emitting anaerobic conditions. We were therefore able to subsequently compare our RNA-seq data to the gene expression changes previously reported. The additional condition of zero N2O emitting aerobically grown cultures allowed us to assess the effect of oxygen availability on sRNA expression in P. denitrificans.
To validate our culture conditions, N2O levels emitted from the different regimes were measured. The high N2O anaerobic culture produced 1.8 mM N2O and the low N2O anaerobic culture produced 0.05 mM N2O. This is in good agreement with the previous report by Sullivan et al. (2013) and provided a solid platform for RNA isolation from the 3 cultures. The RNA samples were split, one was enriched for sRNAs specifically and used for sRNA identification while the other was not enriched for sRNAs and was instead used to provide genome wide expression data. This resulted in roughly 20 million 75 bp reads for each culture condition. Under high N2O emitting anaerobic conditions, expression of nosZ was at 10 RKPM whereas under low N2O emitting anaerobic conditions expression of nosZ was at 120 RPKM. Therefore, expression of nosZ was approximately 12 fold lower than under high N2O emitting anaerobic conditions than under low N2O emitting anaerobic conditions. This was also consistent with the results as reported by Sullivan et al. (2013).
Candidate sRNAs were then conservatively identified from the reads obtained from the sRNA enriched condition manually using the IGB browser. A sRNA was called when a clearly enriched peak of <200 bp of at least 100 reads was expressed from an intergenic regions or antisense to characterized ORFs. Hundred and sixty seven sRNAs were identified across the whole genome as shown in Figure 1. Eighty four of these sRNAs were intergenic and 83 were antisense to ORFs. These were distributed across the entire genome with 110 on chromosome 1, 38 on chromosome 2 and 18 on the plasmid. A selection of 16 putative sRNAs (which were subsequently verified as being expressed) are shown in Table 1, with all the 167 sRNAs listed in Supplementary Table S1.
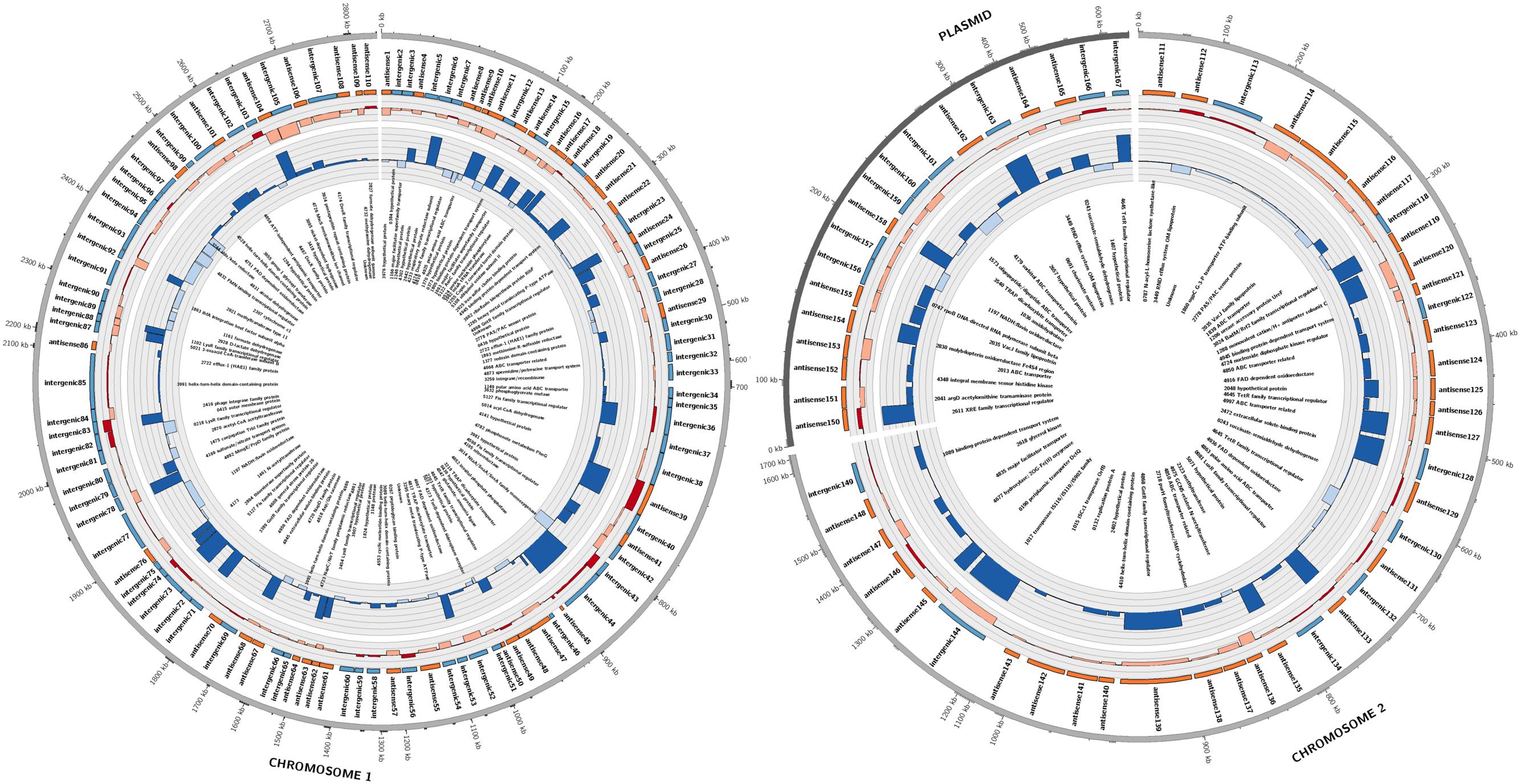
FIGURE 1. Summary of sRNAs identified in the P. denitrificans transcriptome in high N2O (anaerobic), low N2O (anaerobic) and zero N2O emitting (aerobic) conditions. Outer-to-inner rings: position in the P. denitrificans chromosome 1, chromosome 2 or plasmid; sRNA name; sRNA relative size and location, color-coded according to intergenic (blue) or antisense to ORF (orange) positions: sRNA expression level, color coded as increased expression in high N2O anaerobic compared to low N2O anaerobic (dark red), or lower expression in high N2O anaerobic compared to low N2O anaerobic (pale red), increased expression in low N2O anaerobic compared to zero N2O aerobic (dark blue) or lower expression in low N2O anaerobic compared to zero N2O aerobic (pale blue), with each ring representing increments of 2 log2-fold units of differential expression; predicted target for sRNA, Gene identifier (pden) number is included along with gene name when known. Note: for spacing purposes the gene names for predicted targets for 5 sRNAs on chromosome 1 could not be included, 4173 TonB-dependent receptor, 4861 ABC transporter related, 4986 ATP-NAD/AcoX, kinase 0810 solute-binding protein, 5071 hypothetical protein.
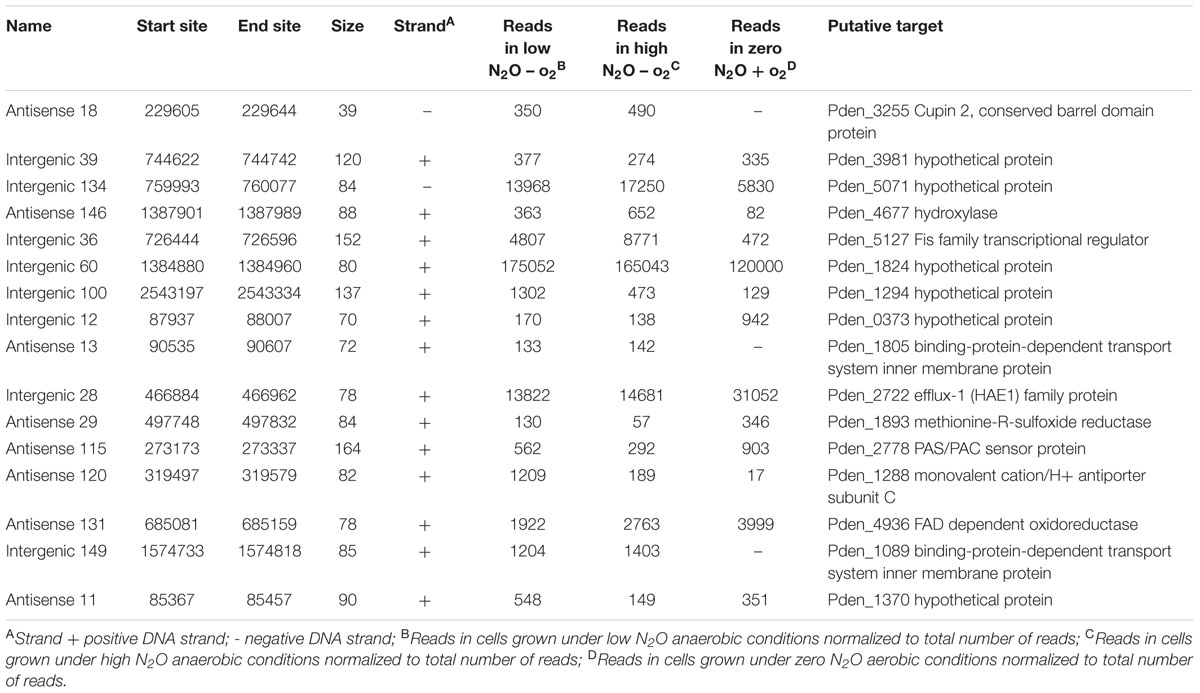
TABLE 1. The 16 sRNAs confirmed by RT-PCR as expressed in the P. denitrificans genome.
Confirmation of Expression of 16 sRNAs
To independently confirm the presence and size of a selection of sRNAs predicted by RNA-seq we used RT-PCR (Pánek et al., 2008; Khoo et al., 2012; Panda et al., 2015; Kwenda et al., 2016). Briefly, RNA was reverse transcribed into cDNA. The cDNA produced was used as the template for PCR together with primers that were designed based on the relevant sequences of sRNA candidates (contained in Supplementary Table S2). Of 40 predicted sRNAs tested, 16 (40%) gave positive results of the expected size as shown in Figure 2. These PCR products were subsequently verified by Sanger sequencing. This proportion of successful validation of sRNAs (40%) fits well with other published data where validation is often successful around 40–50% of the time (examples include Kröger et al., 2012) where 60 new sRNAs were identified, of which 29 were confirmed (48%) and Khoo et al. (2012), where 15 were tested with 8 being verified (53%).
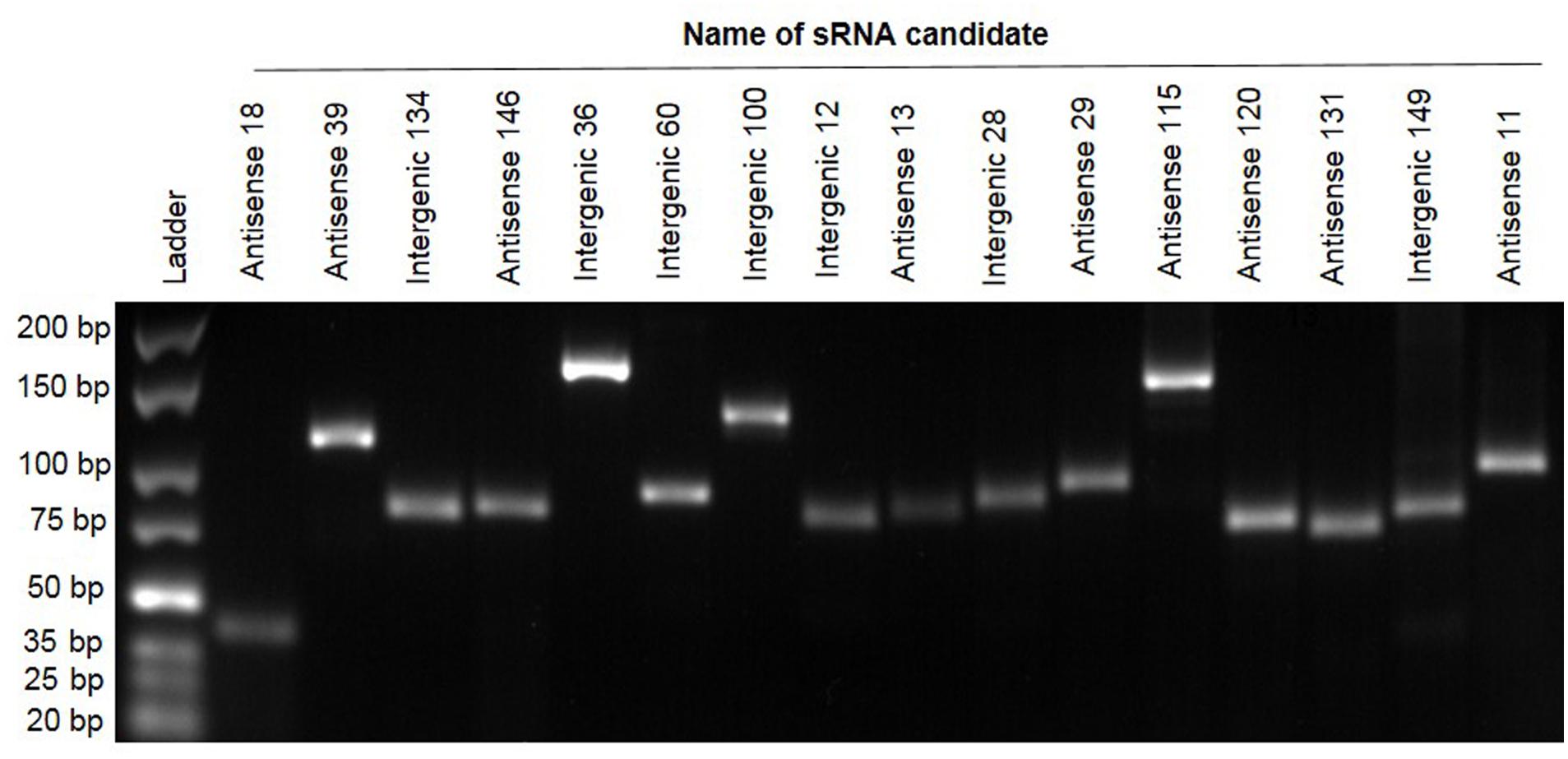
FIGURE 2. Reverse transcriptase polymerase chain reaction validation of 16 sRNA candidates. Electrophoresis of PCR products of 16 sRNAs on 3% agarose gel. Lane 1: GeneRuler Ultra Low Range DNA Ladder, Lanes 2–17. Confirmed sRNA candidates.
sRNAs in P. denitrificans Are Highly Structured and Are Predicted to Have a Wide Range of Targets
In order to gain more insight into the sRNAs identified various online tools were used to provide further information. Supplementary Table S1 provides all the information for all 167 sRNAs, but figures contained within this paper focus solely on the 16 confirmed sRNAs above for brevity. The nearest Rho-independent terminator to each sRNA was identified using TransTerm (Kingsford et al., 2007). The secondary structure of the sRNAs was predicted using Mfold with default parameters set (Zuker, 2003). All of the sRNAs were shown to have significant predicted secondary structure, with the predicted structures for the 16 confirmed sRNAs shown in Figure 3. Furthermore, 90% of the sRNAs (151/167 total) were predicted to form highly structured molecules including more than one hairpin loop. This is important as it shows that many of the sRNAs here have the potential to form complex conformations similar to those commonly associated with many other directly acting RNA transcripts, including known bacterial sRNAs.
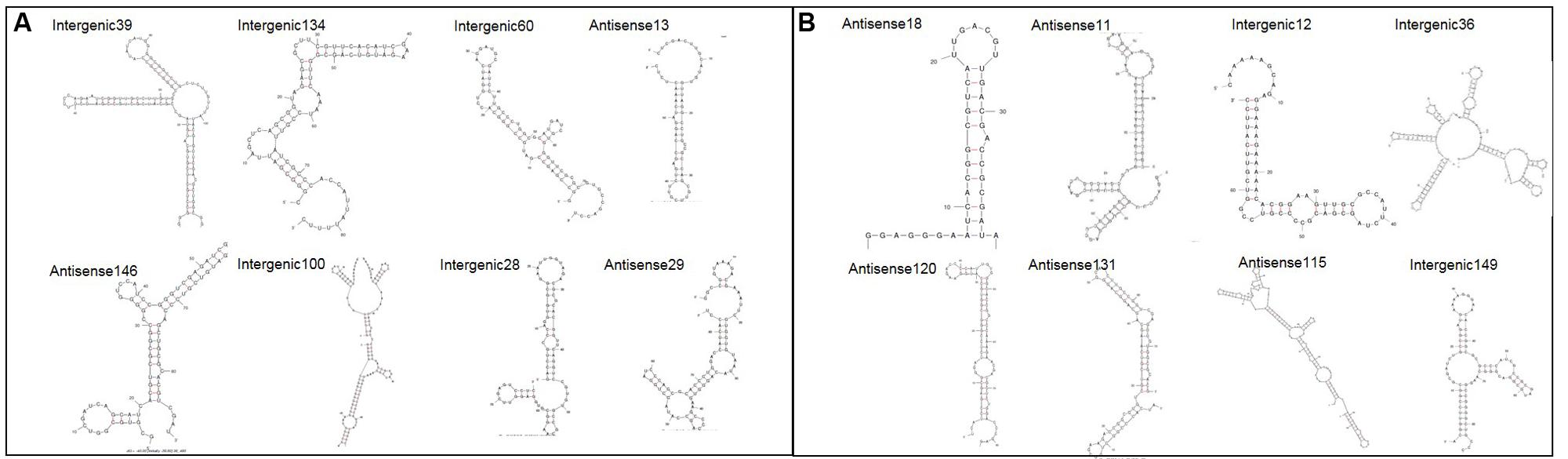
FIGURE 3. Predicted secondary structures for 16 confirmed sRNAs. The secondary structure for each confirmed sRNA was predicted using Mfold (Zuker, 2003). (A) Shows sRNAs which have putative homologues in other closely related bacteria and (B) shows sRNAs which have no putative homologues.
Putative gene targets were predicted using TargetRNA (Kery et al., 2014) the three top targets (i.e., most energetically favorable) included in Supplementary Table S1 (with the top target highlighted in red). When we consider the top 3 targets for each sRNA, the most commonly predicted targets were transcriptional regulators such as the Xre, Fis and TetR families. These were predicted as targets in 118/167 sRNAs, the largest class by far. Another class of targets which was predicted in 100/167 of the sRNAs were transporters including metal and ABC transporters. There were also many cases where predicted targets included hypothetical proteins or proteins of unknown function (133/167 sRNAs). This information could potentially be helpful in eventually assigning a function to these unknown proteins.
Conservation and Homolog Identification of P. denitrificans sRNAs
In order to see if any similar sRNAs had previously been identified, we searched the bacterial sRNA databases sRNATarBase (Cao et al., 2010) and BSRD (Li et al., 2013) using the BLAST options. We were initially surprised to see that when the 16 sRNAs from Table 1 were input no sRNA homologues were identified in either database. However, these databases contain sRNAs identified from previous studies which have typically been performed in Gram-negative γ-proteobacteria such as Escherichia coli and Salmonella. On reflection, given the substantial difference in GC content between these organisms, it is perhaps not that surprising that no homologues to these putative P. denitrificans sRNAs were found. However it is possible that although the sequences of the sRNAs may have diverged significantly to prevent detection by sequence alignment alone, there may be structural conservation which would not be detected by this method.
In further analysis, the sequence conservation of novel sRNAs in other bacteria was investigated using BLASTn and the results shown in Table 2. A BLASTn comparison of each sequence was performed to all sequenced bacterial genomes (E-value = 10-6, word = 11). Only hits with nucleotide identity higher than 60% combined with coverage between query and subject sequence higher than 80% were considered to be conserved. Some 8/16 confirmed sRNAs (antisense 39, intergenic 134, intergenic 60, antisense 13, antisense 146, intergenic 100, intergenic 28 and antisense 29) were found to have conserved sequences in other α-proteobacteria, mainly in the Rhodobacteraceae family. It is expected that these conserved sRNAs may play a conserved role in such closely related species.
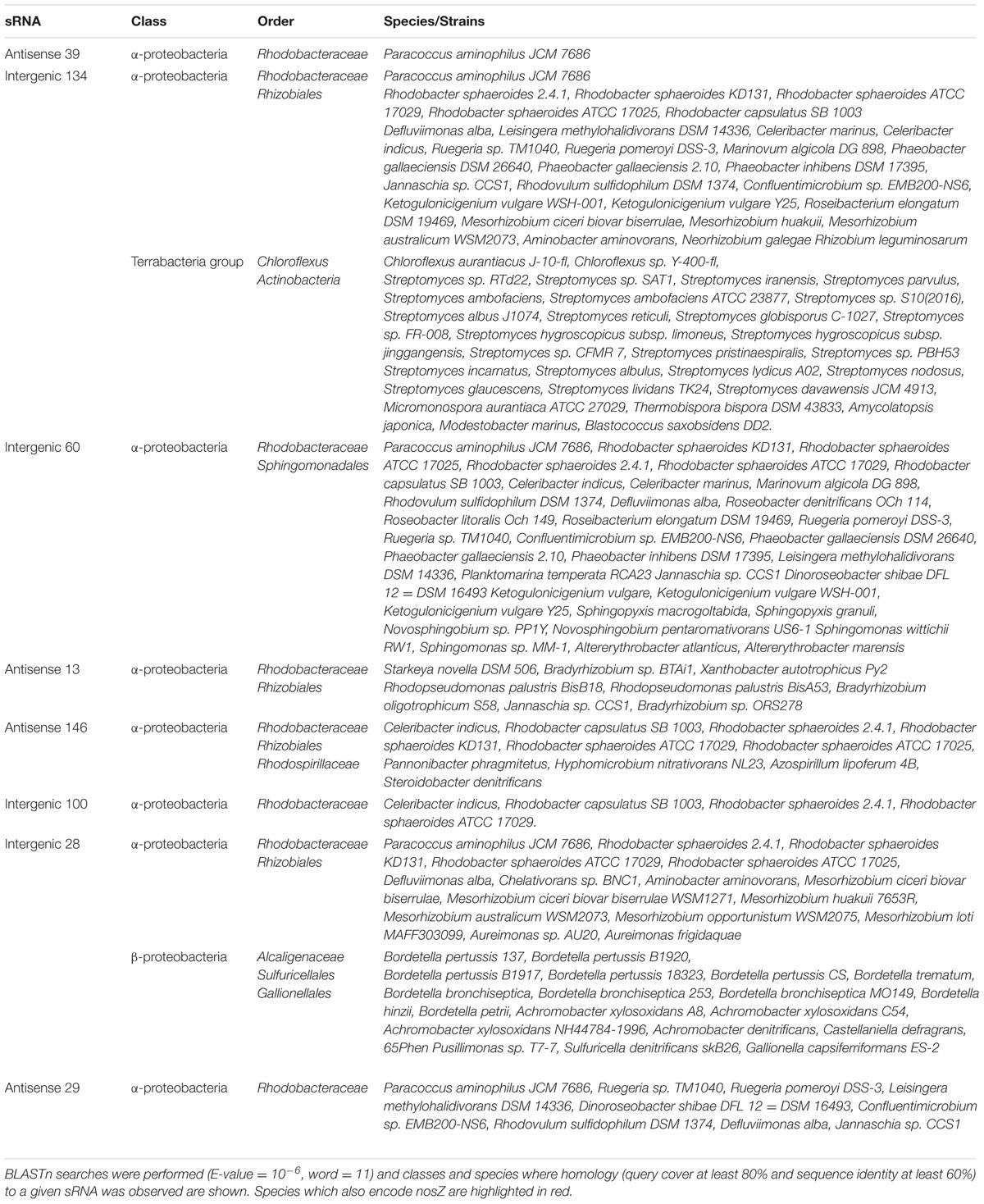
TABLE 2. Conservation of the sequences of sRNAs across different orders, classes, and species of proteobacteria.
The remaining eight confirmed sRNAs (intergenic 36, intergenic 12, antisense 29, antisense 11, antisense 115, antisense 120 and intergenic 149) showed no sequence homology to any other bacteria. Therefore, it seems likely that most of the sRNAs identified in our approach may be specific to closely related Rhodobacterales bacteria with some being species specific to P. denitrificans.
However, because the sequences of the sRNAs are conserved in other bacteria, this is not to say they are true sRNA candidates in other bacteria. In order to assign putative homologues to the 16 confirmed P. denitrificans sRNAs BLASTn searches of the sRNAs were performed as before, but in addition to this the genomic context of the sRNA was taken into account and the sequences aligned to the sRNA. The results are shown in Figure 4. Only intergenic sRNAs which had previously been confirmed as expressed were used for this analysis. For several sRNAs, intergenic 134, intergenic 60, intergenic 29 and intergenic 39, potential homologues with high sequence identity were found in genomes of bacterial species affiliated to the Rhodobacterales. Two of these sRNAs, intergenic 134 and intergenic 100, shared the same genetic context as well as significant sequence similarity with a putative sRNA in R. sphaeroides.

FIGURE 4. Multiple sequence alignment of putative homologues to confirmed intergenic sRNAs in P. denitrificans. +, indicates sRNA present in the same genetic context as in P. denitrificans, i.e., same gene up or downstream and (+) indicates sRNA present, but genetic context not conserved.
Differential Expression of sRNAs under High and Low N2O Emitting Conditions
It was clear that many of the sRNAs were expressed at different levels under the experimental conditions used (Supplementary Table S1 includes expression values for all 167 sRNAs under all three conditions). For example, as shown in Table 1, sRNA intergenic 100 is expressed threefold higher under low N2O emitting conditions compared to under high N2O emitting conditions. Interestingly, analysis of the entire dataset showed 59/167 (35%) sRNAs were differentially expressed by twofold higher or lower (i.e., a ratio either < 0.5 or > 2 between high N2O emitting conditions and low N2O emitting conditions). Seven sRNAs showed a larger than fivefold change in expression between the two conditions (i.e., a ratio either < 0.2 or > 5 between conditions). A further 3 sRNAs showed a greater than 10-fold change in expression between the two conditions (i.e., a ratio either < 0.1 or > 10) between conditions). However, we acknowledge that future work is required to determine the significance of this differential expression.
Despite this, it is expected that the different levels of expression under different conditions will likely reflect the role of the sRNA in P. denitrificans physiology, with the 59 sRNAs showing increased expression under high N2O potentially playing a key role in the response to denitrification and N2O emissions.
Differential Expression of sRNAs under Aerobic Zero N2O and Anaerobic Low N2O Conditions
From the conditions used it was also possible to compare the expression of sRNAs under aerobic zero N2O emitting conditions and anaerobic low N2O conditions. For example, as shown in Table 1 sRNA intergenic 36 was expressed most highly under low N2O emitting anaerobic conditions, and showed an 18 fold reduction in expression under zero N2O emitting aerobic conditions. Analysis of all 167 sRNAs showed that 51/167 (31%) sRNAs were differentially expressed by 2 fold higher or lower (i.e., a ratio either < 0.5 or > 2 between low N2O emitting anaerobic conditions and zero N2O emitting aerobic conditions). Sixteen sRNAs showed a larger than fivefold change in expression between the two conditions (i.e., a ratio either < 0.2 or > 5 between conditions). A further 55 sRNAs showed a greater than 10-fold change in expression between the two conditions (i.e., a ratio either < 0.1 or > 10) between conditions). This analysis comes with the same caveat as before, that future work is required to determine the significance of this differential expression.
However, we do suggest it is likely that the different levels of expression under different conditions will likely reflect the role of the sRNA in P. denitrificans, with the 106 sRNAs showing increased expression under low N2O emitting anaerobic conditions potentially playing a key role in the response to anaerobic conditions specifically.
Discussion
It has already been well established that various environmental factors including pH, aeration and metal availability affects production of N2O at an enzymatic level (Richardson et al., 2009). Despite this we lack detailed knowledge of the effects these factors can play at the transcriptional level. Recently, using microarrays, our laboratory demonstrated that copper availability affects transcription of the genes to produce enzymes required for N2O production in the model denitrifier P. denitrificans. This work provides a significant advance in understanding the N2O relevant transcriptional factors by identifying the sRNA landscape of P. denitrificans under high N2O and low N2O emitting conditions. Here we have shown that many sRNAs are expressed differentially under these conditions, suggesting a potential role for sRNAs in N2O production and/or reduction.
This work has revealed the expression of 16 sRNAs in P. denitrificans, which is the first description, to our knowledge, of sRNA expression in P. denitrificans. We foresee that the data provided in Supplementary Table S1, which contains information on all 167 sRNAs found will become a useful resource for the P. denitrificans and N-cycling community.
The putative target genes for many of our identified sRNAs included genes encoding products involved in transcriptional regulation such as the TetR family of regulators, which may act globally. This is consistent with other studies where global regulators are subject to regulation by multiple Hfq-dependent sRNAs in other species of bacteria (Lee and Gottesman, 2016). In addition to this many predicted targets of the sRNAs included proteins involved in transport. Interestingly this was the most commonly predicted target of sRNAs in the marine bacterium Ruegeria pomeroyi (Rivers et al., 2016). R. pomeroyi is closely related to P. denitrificans and it is possible that regulation of transporters may be a conserved role for sRNA across these related species. Also, many sRNAs identified here were predicted to interact with proteins of currently unknown function. A recent review compared predicted targets compared with true biological targets for organisms such as E. coli when many experimentally determined targets are known and concluded that there was a high number of true biological targets with relatively low scores from predictions (Pain et al., 2015). Nevertheless, it is useful to predict potential targets sRNAs as we have done here as this may help guide future research. However, it is clear that the only way to confirm a sRNA-target interaction is by experimental validation. Therefore, future work will concentrate on characterizing the functional roles and targets for putative sRNAs described here, particularly those we believe may function in the regulation of N2O production and/or reduction. It will also be interesting to see if any sRNAs are Hfq-dependent in the same way previously described sRNAs are in other bacterial species (Storz et al., 2011). P. denitrificans is predicted to encode an Hfq protein, which shows 95% sequence identity to R. sphaeroides Hfq (e value 5 × 10-57) and 54% sequence identity to P. aeruginosa Hfq (e value and 1 × 10-28) respectively (shown in Supplementary Figure S1). It has been shown that many sRNAs in these bacteria are indeed Hfq-dependent (Sonnleitner et al., 2006; Berghoff et al., 2011) so it seems likely that some P. denitrificans sRNAs could also be Hfq-dependent.
Half of the 16 confirmed sRNAs are conserved in other species in the α-proteobacteria in classes such as the Rhodobacteraceae and Rhizobiales. One of these eight sRNAs, intergenic 28, also had sequence homology to members of the β-proteobacteria in Alcaligenaceae, Sulfuricellales and Gallionellales. Interestingly, conserved hits in the Alcaligenaceae family included members of the Bordetella genus. These included various strains of the host restricted human pathogens B. pertussis and B. bronchiseptica, but also the environmental strain B. petrii. B. petrii has been isolated from various environmental niches such as river sediment and polluted soil and it has been suggested that it represents an evolutionary link between free-living environmental bacteria and the host-restricted obligate pathogenic Bordetella (Gross et al., 2008). It seems possible that this sRNA might therefore be an example of a genetic element found in soil dwelling bacteria such as P. denitrificans and B. petrii which is still retained in pathogens.
Also of note was sRNA intergenic 134 which has sequence homology to bacterial classes in the Terrabacteria group, Chloroflexus and Actinobacteria. This is intriguing as the terrabacteria are an evolutionary distinct clade to the hydrobacteria clade which include the α-proteobacteria. However, Actinobacteria are primarily soil dwelling organisms so it is possible that this sRNA plays an important role in the adaption to the soil environment of Actinobacteria and P. denitrificans.
The remaining 50% of the confirmed sRNAs have no sequence conservation to other related bacteria. This suggests that some P. denitrificans sRNAs are species specific while others are conserved in other closely related bacteria. Such an observation is consistent with the conservation seen between other species (Gottesman and Storz, 2011).
As these sRNAs were identified under high and low N2O emitting conditions respectively, we wanted to see if the sRNAs were present in bacterial species which had the ability to reduce N2O to N2 by encoding nosZ. The sequence of P. denitrificans nosZ was used as a query sequence for a BLASTn search (E value = 10-6, word = 11). Only hits with nucleotide identity higher than 60% combined with coverage between query and subject sequence higher than 80% were considered to be conserved and species which encoded nosZ are highlighted in Table 2. Seven out of the eight sRNAs have conserved sequences in several species which also encode nosZ. This may suggest that as the sRNA sequence and nosZ are found in the same species many times, nosZ expression could potentially be controlled by sRNAs.
Furthermore, this work focused on identifying classical intergenic and antisense to ORF sRNAs exclusively but there is increasing evidence that other classes of sRNAs exist in bacteria. sRNAs that exist intragenically or at 3′ and 5′ ends of ORFs have been validated (Vogel et al., 2003; Chao et al., 2012; Kröger et al., 2012). Further analysis of the data produced in this study will help show if this is the case in P. denitrificans, which could potentially increase the number of sRNAs in this organism significantly.
The long term goal of this work is to produce a compendium of transcriptional regulation information on denitrification in P. denitrificans as a model organism for this process. Better understanding of the intrinsic factors, such as sRNAs, which contribute to transcription of the N2O machinery will, in turn, help to inform strategies for mitigation of N2O emissions. More generally, this work, along with the work of many other laboratories in identifying a wide range of novel bacterial sRNAs, very much suggests that the prevalence and various roles of bacterial sRNAs are only just beginning to be appreciated.
Author Contributions
HG designed experiments, acquired data, wrote, revised and approved final manuscript. LC acquired data, drafted manuscript and approved final manuscript. JM interpreted data, drafted manuscript and approved final manuscript. MA acquired data, drafted manuscript and approved final manuscript. AG designed experiments, drafted, revised and approved final manuscript. DR designed experiments, drafted, revised and approved final manuscript. GR designed experiments, wrote, drafted, revised and approved final manuscript.
Funding
This work was funded by the Biotechnology and Biological Sciences Research Council (United Kingdom) (BB/L022796/1)
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01806/full#supplementary-material
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
BLAST, basic local alignment search tool; cDNA, complementary deoxyribonucleic acid; GC, gas chromatography; IGB, integrated genome browser; mRNA, messenger ribonucleic acid; N2O, nitrous oxide; RNA, ribonucleic acid; ORF, open reading frame; RT-PCR, reverse transcriptase polymerase chain reaction; sRNA, small ribonucleic acid.
Footnotes
- ^ http://unafold.rna.albany.edu/?q=mfold
- ^ http://transterm.cbcb.umd.edu/
- ^ http://tempest.wellesley.edu/btjaden/TargetRNA2/
References
Berghoff, B. A., Glaeser, J., Sharma, C. M., Zobawa, M., Lottspeich, F., Vogel, J., et al. (2011). Contribution of Hfq to photooxidative stress resistance and global regulation in Rhodobacter sphaeroides. Mol. Microbiol. 80, 1479–1495. doi: 10.1111/j.1365-2958.2011.07658
Cao, Y., Wu, J., Liu, Q., Zhao, Y., Ying, X., Cha, L., et al. (2010). sRNATarBase: A comprehensive database of bacterial sRNA targets verified by experiments. RNA 16, 2051–2057. doi: 10.1261/rna.2193110
Chao, Y., Papenfort, K., Reinhardt, R., Sharma, C. M., and Vogel, J. (2012). An atlas of Hfq-bound transcripts reveals 3’ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J. 3120, 4005–4019. doi: 10.1038/emboj.2012.229
Crutzen, P. J., Mosier, A. R., Smith, K. A., and Winiwarter, W. (2008). N2O release from agro-biofuel production negates global warming reduction by replacing fossil fuels Atmos. Chem. Phys. 8, 389–395.
Gottesman, S., and Storz, G. (2011). Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 3:12. doi: 10.1101/cshperspect.a003798
Gross, R., Guzman, C. A., Sebaihia, M., dos Santos, V. A., Pieper, D. H., Koebnik, R., et al. (2008). The missing link: Bordetella petrii is endowed with both the metabolic versatility of environmental bacteria and virulence traits of pathogenic Bordetella. BMC Genomics 9:449. doi: 10.1186/1471-2164-9-449
Kery, M. B. F. M., Livny, J., and Tjaden, B. (2014). TargetRNA2: identifying targets of small regulatory RNAs in bacteria. Nucleic Acids Res. 42, 124–129. doi: 10.1093/nar/gku317
Khoo, J. S., Chai, S. F., Mohamed, R., Nathan, S., and Firdaus-Raih, M. (2012). Computational discovery and RT-PCR validation of novel Burkholderia conserved and Burkholderia pseudomallei unique sRNAs. BMC Genomics 13(Suppl. 7):S13. doi: 10.1186/1471-2164-13-S7-S13
Kingsford, C. L., Ayanbule, K., and Salzberg, S. L. (2007). Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 8:R22. doi: 10.1186/gb-2007-8-2-r22
Kröger, C., Dillon, S. C., Cameron, A. D., Papenfort, K., Sivasankaran, S. K., Hokamp, K., et al. (2012). The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. U.S.A. 109, 1277–1286. doi: 10.1073/pnas.1201061109
Kwenda, S., Gorshkov, V., Ramesh, A. M., Naidoo, S., Rubagotti, E., Birch, P. R., et al. (2016). Discovery and profiling of small RNAs responsive to stress conditions in the plant pathogen Pectobacterium atrosepticum. BMC Genomics 12:47. doi: 10.1186/s12864-016-2376-0
Lee, H. J., and Gottesman, S. (2016). sRNA roles in regulating transcriptional regulators: Lrp and SoxS regulation by sRNAs. Nucleic Acids Res. 44, 6907–6923. doi: 10.1093/nar/gkw358
Li, L., Huang, D., Cheung, M. K., Nong, W., Huang, Q., and Kwan, H. S. (2013). BSRD: a repository for bacterial small regulatory RNA. Nucleic Acids Res. 41, 233–238. doi: 10.1093/nar/gks1264
McClure, R., Tjaden, B., and Genco, C. (2014). Identification of sRNAs expressed by the human pathogen Neisseria gonorrhoeae under disparate growth conditions. Front. Microbiol. 2014:5456. doi: 10.3389/fmicb.2014.00456
Moll, I., Leitsch, D., Steinhauser, T., and Bläsi, U. (2003). RNA chaperone activity of the Sm-like Hfq protein. EMBO Rep. 4, 284–289. doi: 10.1038/sj.embor.embor772
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Pain, A., Ott, A., Amine, H., Rochat, T., Bouloc, P., and Gautheret, D. (2015). An assessment of bacterial small RNA target prediction programs. RNA Biol. 12, 509–513. doi: 10.1080/15476286.2015
Panda, G., Tanwer, P., Ansari, S., Khare, D., and Bhatnagar, R. (2015). Regulation and RNA-binding properties of Hfq-like RNA chaperones in Bacillus anthracis. Biochim. Biophys. Acta 1850, 1661–1668. doi: 10.1016/j.bbagen.2015.03.016
Pánek, J., Bobek, J., Mikulík, K., Basler, M., and Vohradský, J. (2008). Biocomputational prediction of small non-coding RNAs in Streptomyces. BMC Genomics 9:217. doi: 10.1186/1471-2164-9-217
Pomowski, A., Zumft, W. G., Kroneck, P. M., and Einsle, O. (2011). N2O binding at a [4Cu:2S] copper-sulphur cluster in nitrous oxide reductase. Nature 477, 234–237. doi: 10.1038/nature10332
Ravishankara, A. R., Daniel, J. S., and Portmann, R. W. (2009). Nitrous Oxide (N2O): the dominant ozone-depleting substance emitted in the 21st century. Science 326, 123–125. doi: 10.1126/science.1176985
Richardson, D., Felgate, H., Watmough, N., Thomson, A., and Baggs, E. (2009). Mitigating release of the potent greenhouse gas N2O from the nitrogen cycle - could enzymic regulation hold the key? Trends Biotechnol. 27, 388–397. doi: 10.1016/j.tibtech.2009.03.009
Rivers, A. R., Burns, A. S., Chan, L. K., and Moran, M. A. (2016). Experimental identification of small non-coding RNAs in the model marine bacterium Ruegeria pomeroyi DSS-3. Front. Microbiol. 29:380. doi: 10.3389/fmicb.2016.00380
Sonnleitner, E., Schuster, M., Sorger-Domenigg, T., Greenberg, E. P., and Bläsi, U. (2006). Hfq-dependent alterations of the transcriptome profile and effects on quorum sensing in Pseudomonas aeruginosa. Mol. Microbiol. 59, 1542–1558. doi: 10.1111/j.1365-2958.2006.05032.x
Storz, G., Vogel, J., and Wassarman, K. M. (2011). Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 43, 880–891. doi: 10.1016/j.molcel.2011.08.022
Sullivan, M. J., Gates, A. J., Appia-Ayme, C., Rowley, G., and Richardson, D. J. (2013). Copper control of bacterial nitrous oxide emission and its impact on vitamin B12-dependent metabolism. Proc. Natl. Acad. Sci. U.S.A. 110, 19926–19931. doi: 10.1073/pnas.1314529110
Vogel, J., Bartels, V., Tang, T. H., Churakov, G., Slagter-Jäger, J. G., Hüttenhofer, A., et al. (2003). RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res. 31, 6435–6443. doi: 10.1093/nar/gkg867
Zuker, M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415. doi: 10.1093/nar/gkg595
Keywords: sRNA, regulation, denitrification, soil, NosZ, nitrous oxide
Citation: Gaimster H, Chalklen L, Alston M, Munnoch JT, Richardson DJ, Gates AJ and Rowley G (2016) Genome-Wide Discovery of Putative sRNAs in Paracoccus denitrificans Expressed under Nitrous Oxide Emitting Conditions. Front. Microbiol. 7:1806. doi: 10.3389/fmicb.2016.01806
Received: 03 August 2016; Accepted: 27 October 2016;
Published: 14 November 2016.
Edited by:
Graeme W. Nicol, L’Université de Lyon, FranceReviewed by:
James Moir, University of York, UKMohd Firdaus Raih, Universiti Kebangsaan Malaysia, Malaysia
Copyright © 2016 Gaimster, Chalklen, Alston, Munnoch, Richardson, Gates and Rowley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannah Gaimster, h.gaimster@uea.ac.uk